
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous photo-induced polymerization and surface modification by microfluidic spinning to produce functionalized polymer microfibers: the effect of their surface modification on cell adhesion†
Wasif
Razzaq
ab,
Christophe A.
Serra
a,
Candice
Dussouillez
c,
Naji
Kharouf
d,
Irene Andrea
Acuña Mejía
a,
Antoine
Kichler
c and
Delphine
Chan-Seng
 *a
*a
aUniversité de Strasbourg, CNRS, Institut Charles Sadron UPR 22, F-67000 Strasbourg, France. E-mail: delphine.chan-seng@ics-cnrs.unistra.fr
bDepartment of Materials, National Textile University, Sheikhupura Road, Faisalabad 37610, Pakistan
cUniversité de Strasbourg, CNRS, Laboratoire de Conception et Applications des Molécules Bioactives UMR 7199, 3Bio Team, F-67000 Strasbourg, France
dINSERM, UMR_S 1121 Biomaterials and Bioengineering, 1 rue Eugène Boeckel, 67000 Strasbourg, France and Université de Strasbourg, Faculté de Chirurgie Dentaire, 8 rue Sainte Elisabeth, 67000 Strasbourg, France
First published on 10th November 2023
Abstract
Functionalized polymer fibers were prepared by microfluidic spinning involving simultaneous photopolymerization and surface modification. A capillary-based microfluidic device was used with two miscible coaxially co-flowing phases to afford polymer fibers by the photopolymerization of poly(ethylene glycol) diacrylate present in the core phase and the surface modification of the fibers thanks to the presence of molecules (i.e., thiol and amine groups) reactive towards acrylate groups in the sheath phase. The use of molecules with higher functionality in thiol groups or higher concentration of these molecules increased the number of functional groups present at the surface of the fibers, while an increase of the flow rate of the sheath phase decreased it. The modification of the surface properties of the fibers was demonstrated by contact angle measurements showing differences in wetting properties and by incubation with RAW264.7 macrophages exhibiting a significant increase in cell adhesion for the thiol-modified microfibers.
1. Introduction
Polymers have been extensively used in advanced applications due to their flexible nature and capability to be tailored to specific needs. The effectiveness and performances of polymer materials mainly depend on their bulk and surface properties.1,2 However, the surface properties of polymer materials sometimes limit their use due to the disparity between the required and acquired surface properties as most polymer materials are inert in nature and usually have low surface energy.3 The main goal of surface functionalization is to control the surface properties (i.e., chemical, physical, and tribological properties) of polymer materials and to implement new functionalities without changing their bulk properties4 for applications in biomedicine,5 microfluidics,6 water treatment,7 energy storage,8 and textiles.9 Their wettability, adhesion, chemical resistance, bio-inertness, and barrier properties, which are critical in many of these applications, are thus aimed to be improved.10 Surface modification can be classified into two main categories: (i) physical modification, including simple physical adsorption of molecules and layer-by-layer assembly, and (ii) chemical modification such as the attachment of specific molecules on the surface by covalent bond formation, surface polymerization, and plasma treatment.11–13 Chemical surface modification is generally more promising due to attachment through a covalent bond which eliminates the desorption risk and ensures long-term stability.12 Chemical surface modification approaches could involve multiple steps including the production of the substrate (e.g., flat surface, fiber, and particle), activation of its surface, and attachment of specific molecules14 which could be time-consuming and increase the cost of producing these materials.Micro- and nanofibers have gained significant attention over the last few decades due to their features such as a large surface area-to-volume ratio, diverse morphologies, ability to be folded, the possibility of weaving into three-dimensional structures, and excellent mechanical properties. These high-performance characteristics of fibers open the way for them to be extensively used in many applications such as biomedicine, tissue engineering, fiber optics, sensors, wearable electronics, and water treatment.15–18 Surface modification of fibers made of natural and synthetic polymers has been considered for example to enhance their compatibilization in a polymer matrix for the development of fiber-reinforced composite materials19 and to mimic the structure of the extracellular matrix for the elaboration of scaffolds in tissue engineering.20 In the latter case, ligands such as biotin21 and the RGD (arginine–glycine–aspartic acid) motif22,23 have been covalently attached to polymer fibers by thio–maleimide coupling and by alkyne–azide cycloaddition reactions, respectively, to promote cell adhesion through recognition by integrins and other cellular receptors. Another approach consists in modifying the surface of polymer fibers by immersion in a solution of dopamine to form a polydopamine cell-attaching shell around the polymer fiber.24,25 However, the modification of their surface is always considered post-production of the fibers. The need for processes in which surface modification could be done simultaneously with the production of the fibers would be groundbreaking and profitable.
Besides melt spinning, wet spinning, and electrospinning, microfluidics is an emerging interdisciplinary technique for the continuous production of microfibers26 by systematic manipulation of multiple phases (miscible or immiscible) in a microchannel.27 One of the main advantages of microfluidics is the possibility to use a wide range of solidification methods such as ionic crosslinking, chemical crosslinking, solvent evaporation, non-solvent-induced phase separation, and photopolymerization.28 While melt spinning,29 wet spinning,30 draw spinning,31 macromolecular assembly,32 and electrospinning33 rely on the physical solidification of existing polymers, microfluidic spinning through photopolymerization opens new perspectives in the field since monomers are used as starting materials. UV-induced photopolymerization is an interesting choice to produce hydrogel-based microparticles34 and microfibers.35 Studies have been recently reported in the literature about producing and controlling the diameters and morphologies of fibers and particles using microfluidics.36 But to the best of our knowledge, no work has described yet the use of microfluidics for the simultaneous production and surface modification of microfibers and particles. Herein, we report a microfluidic approach for the rapid in-process surface modification of polymer microfibers with molecules bearing multiple thiol groups ((SH)x). A capillary-based microfluidic device was used to produce the microfibers by photopolymerization using two co-flowing miscible fluids (core and sheath phases) in a similar manner to that previously reported (Fig. 1).35,37,38 (SH)x was introduced in the sheath phase to promote the reaction at the interface of the two phases with acrylate groups from the core phase. After investigating the effect of various parameters such as the polyfunctionality of the thiol-based molecule, its concentration in the sheath phase, and the flow rate of the sheath phase (Φs) on the functionalization of the microfibers, the work was then extended to molecules with multiple amine groups for the surface modification of microfibers and to microparticles also produced by microfluidics. As fibers can be used to prepare scaffolds in tissue engineering,39,40 these fibers were studied after incubation with RAW264.7 cells to determine their capacity (or not) to promote cell adhesion.
 | ||
| Fig. 1 Simultaneous production of polymer microfibers by photopolymerization and their surface modification with thiol or amine groups: (a) capillary-based microfluidic device used with (b) highlight of the reactions occurring at the interface of the two phases (illustrated with molecules with thiol groups) and (c) the structure of molecules with multiple thiols and amines used. | ||
2. Results and discussion
2.1 Surface modification of microfibers with molecules having multiple thiol groups
The core phase (Φc) producing the polymer microfiber was composed of poly(ethylene glycol) diacrylate (PEGDA, with a molecular weight of 400 g mol−1) as a difunctional monomer (80 v%) and Irgacure 369 as a photoinitiator (3 w/v%) in ethanol (20 v%). The sheath phase (Φs) consisted of a viscous phase of poly(ethylene glycol) (PEG, molecular weight of 300 g mol−1) in which molecules with multiple thiol groups ((SH)x with x being the number of thiol groups present in the molecule) were dissolved. The flow rates of the sheath (Qs) and core phases (Qc) were 350 μL min−1 and 1.5 μL min−1, respectively. When the core phase came out of the capillary, a co-axial flow of the core and sheath phases was established for which both fluids were in contact at the interface. Upon irradiation at 365 nm, PEGDA photopolymerized in the core phase, while coupling reactions41,42 occurred at the interface of both phases between acrylate groups from the core phase and thiol groups present in the sheath phase. As the flow was considered laminar in microfluidics (Reynolds number (Re) = 0.26), only mixing by molecular diffusion was expected to take place. However, for the given experiments, the characteristic length (5 cm) and characteristic time (8 s for the lowest velocity) of the core phase to reach the UV arrangement were so short that the diffusion of the molecules with thiol groups into the bulk of the core phase was almost negligible, supporting the hypothesis that the thiol–acrylate reaction would mainly take place at the interface.3,6-Dioxa-1,8-octanedithiol, trimethylolpropane tris(3-mercaptopropionate), and pentaerythritol tetra(3-mercaptopropionate) were used as molecules bearing two, three, and four thiol groups ((SH)2, (SH)3, and (SH)4, Fig. 1), respectively. These molecules with multiple thiol groups were dissolved in the sheath phase at a fixed molar concentration in either thiol groups or molecules. The simultaneous photopolymerization and surface modification were conducted upon irradiation of the co-flowing phases at 365 nm and the unreacted (SH)x and residual PEG were removed from the surface of the fibers by washing them successively with water, ethanol, and acetone. The amount of free thiol groups present at the surface of fibers was determined using a fixed mass of fibers (10 mg) and by Ellman's test, which is commonly used to detect and quantify free thiol groups (Fig. 2a, yellow solution due to the product formed by the reaction of free thiol with 5,5′-dithio-bis-(2-nitrobenzoic acid) used in Ellman's test). The presence of sulfur atoms was also confirmed by energy-dispersive spectroscopy (EDS) measurements (ESI, Fig. S1b†). As the minimum number of thiol groups involved in the attachment of (SH)x on the fiber was one, the other thiol groups could either form a covalent bond with the fiber or remain as free thiol groups. The probability of having free thiol groups increased with the number of thiol groups in (SH)x, which was indeed the result we observed (Fig. 2b). The non-linearity of the correlation between the number of thiol groups used in the feed and that on the fibers was attributed to the possibility of forming disulfide linkages as the surface density of thiol groups increased.
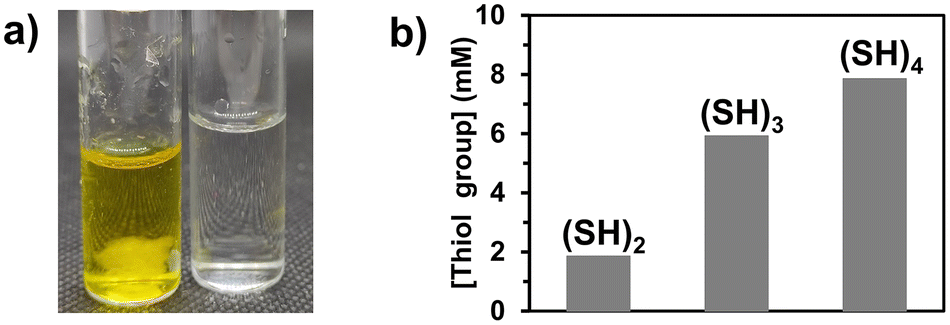 | ||
| Fig. 2 Surface modification of microfibers with thiol groups: (a) image highlighting the presence of thiol groups on the fibers (left, yellow color due to the formation of 2-nitro-5-thiobenzoic acid by the reaction of the free thiol groups with 5,5′-dithio-bis-(2-nitrobenzoic acid)) as compared to a solution without fibers (control, right) using the Ellman's test and (b) influence of the number of thiol groups per molecule (15 mol% of (SH)x in the sheath phase) on the concentration of free thiol groups present at the surface of the fibers. | ||
We then focused our study on the impact of the operating parameters choosing (SH)3 as the molecule with multiple thiol groups as it causes less difficulties when characterizing the fibers produced ((SH)2 could potentially lead to a low concentration of free thiol groups and thus poor detection, while (SH)4 may reach saturation due to the high number of thiol groups). The control of the quantity of thiol groups attached onto the fibers was evaluated by varying the concentration of (SH)3 in the sheath phase from 1 to 20 mol% while keeping all other operating parameters constant. The concentration of free thiol groups at the surface of the fiber increased linearly as the concentration of (SH)3 increased in the sheath phase (Fig. 3a), which was expected. Indeed, with increasing thiol concentration in the sheath phase, the availability of thiol molecules for thiol-acrylate bonds was higher, which thus led to a higher number of free thiol groups. The fibers were characterized by scanning electron microscopy (SEM, Fig. 3b) for morphology and size assessment. As the concentration of (SH)3 used in the sheath phase was increased, the diameter or width of the fibers increased (81 μm, 114 μm, and 161 μm when using 1, 5, and 10 mol% of (SH)3 in the sheath phase, respectively) which could indicate the formation of a thicker shell due to a higher crosslinking propensity of the thiol groups, leading to disulfide linkages as the concentration of (SH)3 increased. The fibers obtained with 1 mol% (SH)3 were generally homogeneous with a relatively smooth surface, while for higher content of (SH)3, the fibers were not always cylindrical and could exhibit irregularities as depicted in the inset of the SEM images. The differences in the shapes of the fibers (cylindrical vs. flattened) could be attributed to the misalignment of the capillary in the outlet tubing (i.e., ideally coaxially aligned in the center of the outlet tubing) and could be affected by vibrations or disturbances. The irregular surface on fibers has been previously reported as being dependent on the environmental conditions used, notably the relative humidity,43 but it could also be related to the diffusion capacity of ethanol present in the core phase through the bulk of the polymer fiber and the shell created around the fiber through the formation of disulfide linkages between the thiol molecules during the surface modification step.
 | ||
| Fig. 3 Effect of the concentration of (SH)3 used in the sheath phase on (a) the quantity of free thiol groups at the surface of PEGDA microfibers as determined by Ellman's test (fiber mass = 10 mg, flow rate of the sheath phase = 350 μL min−1, flow rate of the core phase = 1.5 μL min−1) and (b) the surface integrity of the microfibers imaged by SEM with a zoom at higher magnification on the fiber surface to show its texture in the insets. | ||
The flow rate of the sheath phase was expected to influence the coupling reaction as follows: increasing this flow rate would lower not only the time of contact between the thiol groups in the sheath phase and the acrylate ones in the core phase, but also the residence time in the UV arrangement at higher flow rates. The flow rate of the sheath phase was varied from 300 to 700 μL min−1 while keeping the flow rate of the core phase constant at 2 μL min−1 and maintaining a concentration of (SH)3 in the sheath phase at 10 mol%. The quantity of free thiol groups present on the microfibers decreased linearly as the flow rate of the sheath fluid increased as expected (Fig. 4).
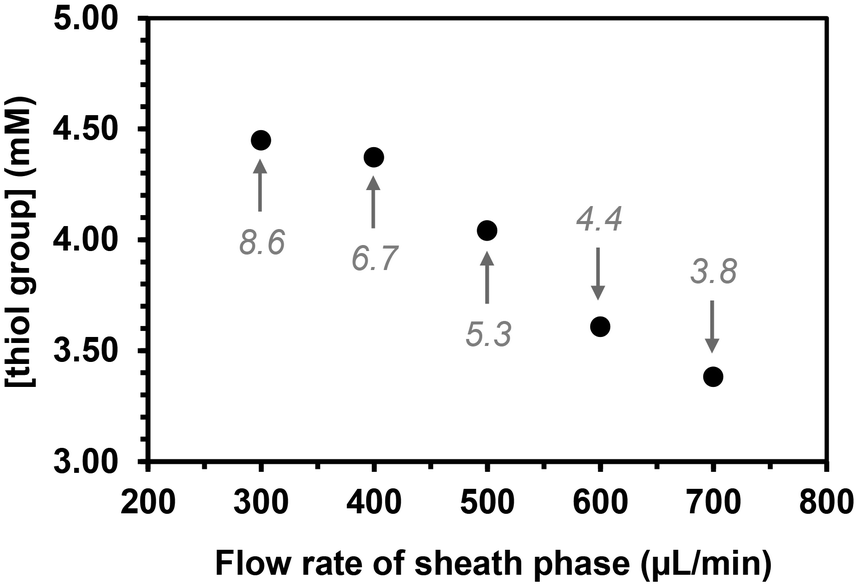 | ||
| Fig. 4 Effect of the flow rate of the sheath phase (10 mol% (SH)3) on the quantity of free thiol groups present at the surface of PEGDA microfibers (flow rate of the core phase = 2 μL min−1). The values in italics are the average time in seconds for a molecule present in the core phase to travel from the capillary exit to the end of the UV arrangement. | ||
The fibers were then produced under three different flow rates of the sheath phase (300, 350, and 450 μL min−1) using the same quantity of (SH)3 (17 mol%) in the sheath phase for 15 min. Ellman's test was performed for the total amount of fibers obtained for each flow rate at the fixed time to determine the quantity of free thiol groups. It was assumed that each (SH)3 molecule would engage one thiol group to react with an acrylate group to participate in the functionalization of the fibers, while the other two thiol groups remained as free thiol groups. After calculating the volume of sheath fluid used in 15 min, the quantity of thiol groups was calculated for that volume. The percentage of thiol groups attached to the fiber surface (SH%) was estimated by dividing the number of moles of thiol groups present at the surface of the fibers as determined by Ellman's test by the number of moles of thiol groups initially present in the sheath phase. The percentage of attachment and hence the efficiency of the process slightly decreased when increasing the flow rate of the sheath phase (Fig. 5) with the highest attachment percentage at 300 μL min−1 (lowest flow rate applied) determined as 82.5%. This could be due to the high flow rate and velocity of the fluid reducing the residence time in the UV arrangement, thus leading to a lower probability of thiol–acrylate reactions.
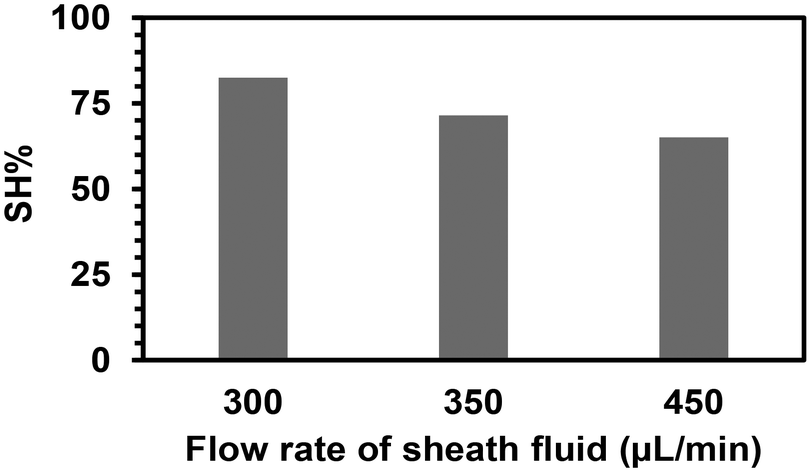 | ||
| Fig. 5 Efficiency of the covalent attachment of (SH)3 on the microfibers relative to the quantity used in the sheath phase for different flow rates of the sheath phase. | ||
The Young's modulus was determined by tensile tests as 6 ± 2 MPa for both unmodified and thiol-modified fibers. According to the literature, myogenesis (i.e., the formation of skeletal muscular tissue) is usually favored when fibers have a modulus of 5–15 MPa. This is encouraging for the potential use of these fibers in tissue engineering.44
2.2 Extension to molecules with multiple amine groups and to microparticles
Surface modification of polymer fibers was also conducted similarly by replacing (SH)x with hexamethylenediamine ((NH2)2) or tris(2-aminoethyl)amine ((NH2)3) as molecules with multiple amine groups (Fig. 1). The presence of primary amines on the prepared fibers was confirmed using the Kaiser test, a colorimetric test for the qualitative identification of primary amines. The solutions turned blue, indicating the formation of Ruhemann's complex formed due to the presence of primary amines (ESI, Fig. S2†). However, nitrogen atoms were not detected by EDS measurement (ESI, Fig. S1c†), suggesting that the amount of amine groups attached to the fiber was low. This could be explained by the reaction differences as the kinetics of acrylate groups to react with amines is slower than that with thiols. These conditions to modify the surface of the fibers with amine groups did not seem to have a strong impact on the appearance of the fibers. The fibers obtained using 10 mol% of (NH2)3 were characterized by SEM (Fig. 6) showing homogeneous fibers with a diameter slightly higher (105 μm) than those produced in the absence of molecules with multiple amine or thiol groups (93 μm) and a relatively smooth surface. | ||
| Fig. 6 SEM images of a) unfunctionalized microfibers as the control and b) amine-functionalized fibers obtained using 10 mol% of (NH2)3. | ||
To show the versatility of the proposed strategy, surface-functionalized spherical polymer microparticles were prepared. As the use of immiscible phases in the microfluidic process is a prerequisite for particle formation due to surface or interfacial forces, Miglyol 812 (medium-chain triglycerides employed as a pharmaceutical excipient) was used as the sheath phase instead of PEG, while the core phase remained the same as for fiber preparation. (SH)3 was added at 10 mol% to the sheath phase, and particles were produced using flow rates of 200 μL min−1 for the sheath phase and 2 μL min−1 for the core phase. Particles were obtained upon UV irradiation at 365 nm of the core phase droplets that were collected and washed multiple times with ethanol and acetone to get rid of the residual Miglyol and unreacted thiol molecules from the particle surface. The microparticles were homogeneous in size with a diameter of 220 μm (ESI, Fig. S3†). Ellman's test was performed to confirm the successful attachment of thiol molecules to the surface of the microparticles.
2.3 Surface properties of thiol- and amine-modified microfibers
The change in the surface properties of (SH)3- and (NH2)3-modified microfibers compared to unmodified PEGDA microfibers was demonstrated by contact angle measurements using the Wilhelmy method. Unmodified PEGDA microfibers had a contact angle of 32° which complied with the literature value45 for hydrogels prepared with PEGDA having a molecular weight of 400 g mol−1 (Fig. 7a). (SH)3-modified microfibers (10 mol%) exhibited a contact angle of 74° indicating a significant decrease in hydrophilicity (Fig. 7b), which was consistent with the fact that (SH)3 was practically insoluble in water. (NH2)3-modified microfibers showed an intermediate value of contact angle (49°, Fig. 7c) which was almost close to the value found in the literature for (NH2)3-modified surfaces.46 The presence of thiol or amine groups on the surface of the fibers decreased the hydrophilicity of PEGDA fibers, showing a change in wetting properties after surface modification. | ||
| Fig. 7 Contact angle measurement of PEGDA microfibers using the Wilhelmy method for the (a) unmodified, (b) (SH)3-modified, and (c) (NH2)3-modified microfibers. The fibers were prepared using the following conditions: Qs = 350 μL min−1, Qc = 1.5 μL min−1, concentration of (SH)3 and (NH2)3 in the sheath phase = 10 mol%. | ||
2.4 Effect of the surface modification of the microfibers on cell adhesion
An important aspect of tissue engineering approaches is the development of scaffolds to which cells can attach, proliferate, and differentiate.47–49 Fibrous scaffolds are one of the most promising materials because they can enable guided cell growth, alignment, and migration.50–52 The newly developed PEGDA-based fibers were investigated for their ability to promote cell adhesion by light microscopy and SEM analysis (Fig. 8 and ESI, Fig. S4 and S5†). The results showed that murine RAW264.7 macrophages were unable to adhere to non-modified PEGDA microfibers, while a few cells could adhere to the amine-modified fibers (Fig. S3†). (SH)3-modified microfibers were the ones with the best cell adhesion results, especially those prepared with 10 mol% of (SH)3 in the sheath (Fig. 8c). Among the known parameters affecting cell adhesion, surface roughness53 and wettability54 were the ones showing a different behavior for the (SH)3-modified fibers when compared to the unmodified and (NH2)3-modified ones, which could explain our results. After 26 h of incubation, light microscopy already showed cellular adhesion with the fibers modified by 10% thiol (ESI, Fig. S5†). These results were confirmed at 48 h by SEM showing macrophages attached, and sometimes even spread and flattened onto the thiol-microfibers (Fig. 8c). Next, the biocompatibility of the neutral and 10%-modified fibers was evaluated. Therefore, we added increasing amounts of non-modified and 10% thiol-modified fibers to the RAW cells and determined cell viability after 48 h. To determine the number of living cells a CellTiter-Glo 2.0 Cell viability assay was conducted. The results show that there is indeed a dose-dependent cytotoxicity but the percentage of cell viability remains relatively good (approx. 65%) even with the highest quantity of fibers (ESI, Fig. S6†). Notably, the thiol-modified fibers do not induce higher cytotoxicity than the neutral material, indicating that the photoinitiator was successfully removed. Finally, we investigated whether other cells could also adhere to the thiol-modified fibers. Therefore, using the same experimental conditions as with the macrophages, we added cells from a human colon cancer (HCT116 cell line) to the material. We found that these cells could also adhere to the 10% thiol-modified fibers although to a lesser extent than the RAW cells (ESI, Fig. S7†). | ||
| Fig. 8 SEM images of thiol-modified microfibers prepared using (a) 1 mol%, (b) 5 mol%, and (c) 10 mol% of (SH)3 in the sheath phase and incubated with RAW264.7 cells for 48 h. | ||
Taken together, these results show that the surface properties of PEGDA-based scaffolds (i.e. functionalization) play a crucial role in interactions with cells. It is noteworthy that one batch of fibers produced with 1 mol% of (NH2)3 also presented surface irregularities (as those found with the 10% (SH)3 fibers) without inducing cell adhesion, showing that this does not influence cell adhesion. Cell adhesion may thus be influenced by the wetting properties induced by the functionalization of the fibers with thiol groups. On the other hand, our results are not unexpected since numerous publications have reported the presence of thiol-reactive groups on the cell surface which can not only be used to facilitate uptake of maleimide-modified nanoparticles55 or thiol-reactive compounds56 but also to favor cell adhesion.57
3. Conclusions
A microfluidic approach using a capillary-based device was applied to produce polymer fibers and introduce functional groups at their surface in a rapid one-step process. The functional groups introduced at the surface of the crosslinked PEGDA fibers were thiol and amine groups that affected the surface properties of the fibers according to their concentration present at the fiber surface that was tuned through the operating conditions. This process was shown to be also applicable to obtain microparticles with thiol groups at their surface. The potential of the functionalized microfibers was illustrated by evaluating the ability of macrophages to adhere to them showing here the superiority of the thiol-functionalized fibers as compared to the unmodified and amine-modified ones. Further modification of the fibers by conjugating, for example, an RGD motif,58–60 could be in the future an interesting strategy to further improve the cell adhesion of the prepared thiol-modified microfibers. This work introduced a strategy without post-production modification which could reduce the cost and the time needed to obtain functionalized fibers. This paves the way for the preparation of a wide range of polymer fibers of different nature and with the desired functionality present on their surface in an easy and fast process for a wide range of applications including biosensors and membranes.4. Experimental
4.1. Materials
Poly(ethylene glycol) diacrylate (PEGDA, Mn = 400 g mol−1, Polysciences), 3,6-dioxa-1,8-octaedithiol ((SH)2, >97.0%, TCI), trimethylolpropane tris(3-mercaptopropionate) ((SH)3, >85.0%, TCI), pentaerythritol tetra(3-mercaptopropionate) ((SH)4, >90.0%, TCI), hexamethylenediamine ((NH2)2, 98.0%, Sigma Aldrich), tris(2-aminoethyl)amine ((NH2)3, 96.0%, Sigma Aldrich), ethanol (99.9%, Carlo Erba), Irgacure 369 (Ciba), 5,5′-dithiobis(2-nitrobenzoic acid) (Ellman's reagent, TCI), sodium phosphate monobasic (Fluka analytical), Miglyol 812 (Caelo), sodium phosphate dibasic (Sigma Aldrich), L-cysteine hydrochloride monohydrate (>99.0%, TCI), ethylenediaminetetraacetic acid (EDTA), PEG300 (Sigma Aldrich), and Dulbecco's phosphate-buffered saline (PBS, modified without calcium chloride and magnesium chloride, liquid, sterile-filtered, suitable for cell culture, Sigma-Aldrich) were used as received.4.2. Synthesis of the fibers
The core phase was composed of poly(ethylene glycol) diacrylate (PEGDA, Mn = 400 g mol−1) (80 v%), ethanol (20 v%), and Irgacure 369 (3 w/v%). The sheath phase was composed of poly(ethylene glycol) (PEG300) and a specific concentration of a compound with multiple thiol or amine groups. The core and sheath phases were injected thanks to syringe pumps at different combinations of sheath flow rates and core flow rates. Both phases came in contact at the exit of the capillary and the jet of the core phase spontaneously formed and was surrounded by the sheath fluid. The polymerized fibers were collected and washed many times with water, ethanol, and acetone to remove residual PEG300 as well as unreacted thiols or amines from the surface of the fibers, and then they were dried under ambient conditions overnight.4.3. Ellman's test
4.4. Kaiser test
The Kaiser test was used for the detection of free primary amine groups63 after surface modification of the fibers with (NH2)2 and (NH2)3. The fibers modified with amine molecules and ninhydrin solution were put in a capped glass vial and heated at 90 °C for 15 min. The color of the solution turned purple or blue (depending on the number of free amines) confirming the presence of free amine groups on the surface of the fibers (Fig. 7a).4.5. Mechanical measurements
Tensile tests were performed on an Instron ElectroPuls E3000 tensile machine equipped with a 10 N load cell. A strain rate of 0.01 s−1 was used and tests were carried out at room temperature.4.6. Contact angle measurements
Contact angle measurements were performed to verify the change in wetting properties at the surface of the fibers resulting from the covalent attachment of thiol- or amine-based compounds. The measurements were performed using a Biolin tensiometer at room temperature by adapting the Wilhelmy plate method. The fiber was vertically suspended with one end fixed outside the water and the other end attached to a light metallic piece to keep the fiber straight. The angles were measured during the immersion and withdrawal. The reported angle is the average of five values measured for the contact angle.4.7. Cell culture
RAW264.7 is a murine macrophage cell line obtained from ATCC (TIB-71). This cell line was maintained in Dulbecco's modified Eagle's medium (DMEM) high glucose (SIGMA, D0819-500ML) containing 5% FBS (Gibco, 42Q2455K) in an incubator at 37 °C, 80% humidity and 5% CO2. The human colorectal carcinoma cell line HCT116 was cultured at 37 °C under a humid atmosphere (5% CO2) in RPMI 1640 (Roswell Park Memorial Institute) supplemented with 2 mM L-glutamine, 100 units per mL penicillin, 100 μg mL−1 streptomycin and 10% fetal calf serum (FCS).4.8. Scanning electron microscopy
The unmodified, thiol- and amine-modified microfibers, in absolute ethanol, were transferred to a 24-well plate and washed with PBS. 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RAW264.7 cells per well were then plated with each microfiber and incubated for 48 h. After incubation, the microfibers and the adhered cells were transferred to a new 24-well plate to be washed gently with PBS. The samples were fixed as described in a previous study,64 by using a solution of 0.05 M glutaraldehyde in 4% cacodylate buffer for 2 h. Subsequently, the specimens were rinsed using a 4% cacodylate buffer three times, 5 min each, and subsequently dehydrated in a graded series of ethanol (35%, 50%, 70%, 95%, and 100%) for 3 min each. Finally, they were dried using the drying agent hexamethyldisilazane (HMDS). The samples were transferred from 100% ethanol into a 1:1 solution of HMDS for 10 min, and then transferred into 100% HMDS twice, 10 min each. All specimens were sputter-coated with gold–palladium (20/80) using a Hummer JR sputtering device (Technics, CA, USA). The samples were observed at a magnification of 500× with a working distance of 10 mm and a 10 kV acceleration voltage of the electrons through a scanning electron microscope (SEM, Quanta 250 FEG scanning electron microscope, FEI Company, Eindhoven, The Netherlands). EDX analysis was performed by using a magnification of 5000× during an acquisition time of 50 s and a working length of 10 mm to attain the spectrum of chemical elements present on the surface. Pd and Au are presented in the chemical analysis due to the coating procedure before SEM observation.
000 RAW264.7 cells per well were then plated with each microfiber and incubated for 48 h. After incubation, the microfibers and the adhered cells were transferred to a new 24-well plate to be washed gently with PBS. The samples were fixed as described in a previous study,64 by using a solution of 0.05 M glutaraldehyde in 4% cacodylate buffer for 2 h. Subsequently, the specimens were rinsed using a 4% cacodylate buffer three times, 5 min each, and subsequently dehydrated in a graded series of ethanol (35%, 50%, 70%, 95%, and 100%) for 3 min each. Finally, they were dried using the drying agent hexamethyldisilazane (HMDS). The samples were transferred from 100% ethanol into a 1:1 solution of HMDS for 10 min, and then transferred into 100% HMDS twice, 10 min each. All specimens were sputter-coated with gold–palladium (20/80) using a Hummer JR sputtering device (Technics, CA, USA). The samples were observed at a magnification of 500× with a working distance of 10 mm and a 10 kV acceleration voltage of the electrons through a scanning electron microscope (SEM, Quanta 250 FEG scanning electron microscope, FEI Company, Eindhoven, The Netherlands). EDX analysis was performed by using a magnification of 5000× during an acquisition time of 50 s and a working length of 10 mm to attain the spectrum of chemical elements present on the surface. Pd and Au are presented in the chemical analysis due to the coating procedure before SEM observation.
4.9. Cell viability
The unmodified and 10% thiol-modified fibers in absolute ethanol were transferred to a 24-well plate and washed with PBS. 150000 RAW264.7 cells per well were then incubated with increasing amounts of fibers and incubated for 48 h. To determine the effect of fibers on cell viability, a CellTiter-Glo 2.0 Cell viability assay (#9241, Promega) was conducted. This kit is based on cell metabolic activity and quantified ATP release after cell lysis, which is directly proportional to the measured luminescence. A volume of CellTiter-Glo reagent was added to each well at a ratio of 1:1 (v/v) and the plate was agitated for 3 min to induce cell lysis and ATP release. The supernatants were then transferred into a white 96-well plate after another 15 min of incubation at room temperature (and protected from light). Measurement of luminescence was performed with a microplate reader (Safas Monaco SP2000, Genius 5801). The results were expressed by the subtraction of background luminescence from the medium control alone and the expression of viability relative to non-treated controls (considered as 100% viability).
Author contributions
Wasif Razzaq: conceptualization, methodology, investigation, visualization, and writing; Christophe Serra: conceptualization and supervision; Candice Dussouillez: investigation; Naji Kharouf: investigation; Irene Andrea Acuña Mejía: investigation; Antoine Kichler: supervision and writing; Delphine Chan-Seng: conceptualization, supervision, visualization, and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. R. is thankful to the Higher Education Commission Pakistan for his Ph.D. scholarship. This work at the Interdisciplinary Institute HiFunMat, as part of the ITI 2021–2028 program of the University of Strasbourg, CNRS and Inserm, was supported by IdEx Unistra (ANR-10-IDEX-0002) and SFRI (STRAT'US project, ANR-20-SFRI-0012) under the framework of the French Investments for the Future Program. The authors thank Wiebke Drenckhan, Leandro Jacomine, and the PLAMICS platform at the Institut Charles Sadron for the access to the equipment used respectively for contact angle measurement and the scanning electron microscope (characterization of the microparticles) and MiNaMec (Micro Nano Mécanique) platform for conducting the mechanical tests.References
- P. Fabbri and M. Messori, in Modification of Polymer Properties, ed. C. F. Jasso-Gastinel and J. M. Kenny, William Andrew Publishing, 2017, pp. 109–130, DOI:10.1016/B978-0-323-44353-1.00005-1.
- N. Nady, M. C. R. Franssen, H. Zuilhof, M. S. M. Eldin, R. Boom and K. Schroën, Desalination, 2011, 275, 1–9 CrossRef CAS.
- M. Ozdemir, C. U. Yurteri and H. Sadikoglu, Crit. Rev. Food Sci. Nutr., 1999, 39, 457–477 CrossRef CAS PubMed.
- H. Dong and T. Bell, Surf. Coat. Technol., 1999, 111, 29–40 CrossRef CAS.
- J. M. Goddard and J. H. Hotchkiss, Prog. Polym. Sci., 2007, 32, 698–725 CrossRef CAS.
- J. Zhou, A. V. Ellis and N. H. Voelcker, Electrophoresis, 2010, 31, 2–16 CrossRef CAS PubMed.
- D. J. Miller, D. R. Dreyer, C. W. Bielawski, D. R. Paul and B. D. Freeman, Angew. Chem., Int. Ed., 2017, 56, 4662–4711 CrossRef CAS PubMed.
- Y. Niu and H. Wang, ACS Appl. Nano Mater., 2019, 2, 627–642 CrossRef CAS.
- S. Luo and W. J. Van Ooij, J. Adhes. Sci. Technol., 2002, 16, 1715–1735 CrossRef CAS.
- Y. H. Ding, M. Floren and W. Tan, Biosurf. Biotribol., 2016, 2, 121–136 CrossRef CAS PubMed.
- E. Ruckenstein and Z. F. Li, Adv. Colloid Interface Sci., 2005, 113, 43–63 CrossRef CAS PubMed.
- W. Sun, W. Liu, Z. Wu and H. Chen, Macromol. Rapid Commun., 2020, 41, 1900430 CrossRef CAS PubMed.
- H. Amani, H. Arzaghi, M. Bayandori, A. S. Dezfuli, H. Pazoki-Toroudi, A. Shafiee and L. Moradi, Adv. Mater. Interfaces, 2019, 6, 1900572 CrossRef.
- S. D. Katti, R. Vasita and K. Shanmugam, Curr. Top. Med. Chem., 2008, 8, 341–353 CrossRef PubMed.
- J.-D. Liu, X.-Y. Du and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 25089–25096 CrossRef CAS PubMed.
- T. Cui, J. Yu, Q. Li, C.-F. Wang, S. Chen, W. Li and G. Wang, Adv. Mater., 2020, 32, 2000982 CrossRef CAS PubMed.
- Y. Zhao, X.-g. Hu, S. Hu and Y. Peng, Biosens. Bioelectron., 2020, 166, 112447 CrossRef CAS PubMed.
- A. Abrishamkar, A. Nilghaz, M. Saadatmand, M. Naeimirad and A. J. deMello, Biomicrofluidics, 2022, 16, 061504 CrossRef CAS.
- J. Cruz and R. Fangueiro, Procedia Eng., 2016, 155, 285–288 CrossRef CAS.
- A. M. Jordan, V. Viswanath, S.-E. Kim, J. K. Pokorski and L. T. J. Korley, J. Mater. Chem. B, 2016, 4, 5958–5974 RSC.
- D. A. Boyd, A. R. Shields, J. Naciri and F. S. Ligler, ACS Appl. Mater. Interfaces, 2013, 5, 114–119 CrossRef CAS PubMed.
- S.-E. Kim, J. Wang, A. M. Jordan, L. T. J. Korley, E. Baer and J. K. Pokorski, ACS Macro Lett., 2014, 3, 585–589 CrossRef CAS PubMed.
- M. Cavanaugh, E. Silantyeva, G. Pylypiv Koh, E. Malekzadeh, W. D. Lanzinger, R. K. Willits and M. L. Becker, J. Funct. Biomater., 2019, 10, 24 CrossRef CAS PubMed.
- S. H. Ku and C. B. Park, Biomaterials, 2010, 31, 9431–9437 CrossRef CAS PubMed.
- S. R. Nielsen, F. Besenbacher and M. Chen, Phys. Chem. Chem. Phys., 2013, 15, 17029–17037 RSC.
- Y. Jun, E. Kang, S. Chae and S.-H. Lee, Lab Chip, 2014, 14, 2145–2160 RSC.
- L. Cai, F. Bian, H. Chen, J. Guo, Y. Wang and Y. Zhao, Chem, 2021, 7, 93–136 CAS.
- X.-Y. Du, Q. Li, G. Wu and S. Chen, Adv. Mater., 2019, 31, 1903733 CrossRef CAS.
- R. Hufenus, Y. Yan, M. Dauner and T. Kikutani, Materials, 2020, 13, 4298 CrossRef CAS PubMed.
- A. Rohani Shirvan, A. Nouri and A. Sutti, Eur. Polym. J., 2022, 181, 111681 CrossRef CAS.
- M. M. Denn, Ann. Rev. Fluid Mech., 1980, 12, 365–387 CrossRef CAS.
- Y. Loo, S. Zhang and C. A. E. Hauser, Biotechnol. Adv., 2012, 30, 593–603 CrossRef CAS.
- G. C. Rutledge and S. V. Fridrikh, Adv. Drug Delivery Rev., 2007, 59, 1384–1391 CrossRef CAS PubMed.
- B. K. Pullagura, S. Amarapalli and V. Gundabala, Colloids Surf. A, 2021, 608, 125586 CrossRef CAS.
- W. Razzaq, C. A. Serra, L. Jacomine and D. Chan-Seng, J. Taiwan Inst. Chem. Eng., 2022, 132, 104215 CrossRef CAS.
- G. Luo, L. Du, Y. Wang, Y. Lu and J. Xu, Particuology, 2011, 9, 545–558 CrossRef CAS.
- W. Razzaq, C. Serra and D. Chan-Seng, Chem. Commun., 2022, 58, 4619–4622 RSC.
- W. Razzaq, C. A. Serra and D. Chan-Seng, Eur. Polym. J., 2022, 174, 111321 CrossRef CAS.
- Y. Zhang, X. Liu, L. Zeng, J. Zhang, J. Zuo, J. Zou, J. Ding and X. Chen, Adv. Funct. Mater., 2019, 29, 1903279 CrossRef.
- L. Tian, J. Ma, W. Li, X. Zhang and X. Gao, Macromol. Biosci., 2023, 23, 2200429 CrossRef CAS PubMed.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- H. Fashandi and M. Karimi, Polymer, 2012, 53, 5832–5849 CrossRef CAS.
- J. Dolgin, S. N. Hanumantharao, S. Farias, C. G. Simon and S. Rao, Fibers, 2023, 11, 39 CrossRef CAS.
- H. Zhang, L. Wang, L. Song, G. Niu, H. Cao, G. Wang, H. Yang and S. Zhu, J. Appl. Polym. Sci., 2011, 121, 531–540 CrossRef CAS.
- L. Shen, J. Zuo and Y. Wang, J. Membr. Sci., 2017, 537, 186–201 CrossRef CAS.
- G. Chen, T. Ushida and T. Tateishi, Macromol. Biosci., 2002, 2, 67–77 CrossRef CAS.
- M. Jafari, Z. Paknejad, M. R. Rad, S. R. Motamedian, M. J. Eghbal, N. Nadjmi and A. Khojasteh, J. Biomed. Mater. Res., Part B, 2017, 105, 431–459 CrossRef CAS PubMed.
- D. Howard, L. D. Buttery, K. M. Shakesheff and S. J. Roberts, J. Anat., 2008, 213, 66–72 CrossRef CAS PubMed.
- K. Wang, L. Liu, J. Xie, L. Shen, J. Tao and J. Zhu, ACS Appl. Mater. Interfaces, 2018, 10, 1566–1574 CrossRef CAS PubMed.
- J. Xie, H. Shen, G. Yuan, K. Lin and J. Su, Mater. Sci. Eng., C, 2021, 120, 111787 CrossRef CAS PubMed.
- T. L. Jenkins and D. Little, npj Regener. Med., 2019, 4, 15 CrossRef PubMed.
- S. Di Cio and J. E. Gautrot, Acta Biomater., 2016, 30, 26–48 CrossRef CAS PubMed.
- H. Fan and Z. Guo, Biomater. Sci., 2020, 8, 1502–1535 RSC.
- A. Kichler, J. S. Remy, O. Boussif, B. Frisch, C. Boeckler, J. P. Behr and F. Schuber, Biochem. Biophys. Res. Commun., 1995, 209, 444–450 CrossRef CAS PubMed.
- G. Gasparini, G. Sargsyan, E.-K. Bang, N. Sakai and S. Matile, Angew. Chem., Int. Ed., 2015, 54, 7328–7331 CrossRef CAS PubMed.
- B. Le-Vinh, C. Steinbring, N.-M. Nguyen Le, B. Matuszczak and A. Bernkop-Schnürch, ACS Appl. Mater. Interfaces, 2023, 15, 40304–40316 CrossRef CAS PubMed.
- S. L. Bellis, Biomaterials, 2011, 32, 4205–4210 CrossRef CAS PubMed.
- S. E. D'Souza, M. H. Ginsberg and E. F. Plow, Trends Biochem. Sci., 1991, 16, 246–250 CrossRef PubMed.
- U. Hersel, C. Dahmen and H. Kessler, Biomaterials, 2003, 24, 4385–4415 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- https://assets.fishersci.com/TFS-Assets/LSG/manuals/MAN0011216_Ellmans_Reag_UG .
- E. Kaiser, R. L. Colescott, C. D. Bossinger and P. I. Cook, Anal. Biochem., 1970, 34, 595–598 CrossRef CAS PubMed.
- N. Kharouf, S. Sauro, A. Eid, J. Zghal, H. Jmal, A. Seck, V. Macaluso, F. Addiego, F. Inchingolo, C. Affolter-Zbaraszczuk, F. Meyer, Y. Haikel and D. Mancino, J. Funct. Biomater., 2023, 14, 9 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lp00032j |
| This journal is © The Royal Society of Chemistry 2024 |