
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancement of replacement lithography by combination of photocleavable groups with ultrashort thiolates†‡
Christian
Fischer
,
Florian
Born
and
Andreas
Terfort
 *
*
Institute of Inorganic and Analytical Chemistry, Goethe University Frankfurt, Max-von-Laue-Straße 7, 60438 Frankfurt am Main, Germany. E-mail: aterfort@chemie.uni-frankfurt.de
First published on 22nd March 2024
Abstract
The radiation-induced replacement lithography of self-assembled monolayers (SAMs) is one of the most flexible patterning techniques in nanotechnology as it not only permits a localized substitution, but also a variation in chemistry when applied in a sequential manner. While it typically proceeds by weakening of the molecule–substrate interactions, we present here an approach in which the molecules within the SAM are cleaved, leaving behind a SAM consisting of ultrashort molecules (thioglycolic acid), which are labile enough to be efficiently replaced with a different kind of molecule. The key of this process was the introduction of a photocleavable ortho-nitrobenzyl (ONB) group carrying hexyl groups, which result in primary SAMs stable enough to withstand the carefully chosen replacement conditions. The primary SAMs were characterized by ellipsometry, infrared reflection spectroscopy (IRRAS) and contact angle goniometry, showing a somewhat surprising conformation of the SAM constituents in which the molecular dipole moment is arranged parallel to the surface. This causes the system carrying only one hexyl group to be more stable than the one with two hexyl groups. Upon irradiation with light of 365 nm, the former molecule becomes easily exchanged by (deuterated) dodecanethiol, as could be quantified by IRRAS, with an exchange yield of >80% at an area dose of 48 J cm−2. As a proof of principle, irradiation was performed with differently patterned masks, demonstrating the viability of the method for lithography.
1. Introduction
Self-assembled monolayers, SAMs for short, are a unique molecular type of surface modifiers, which allow for a large variety of interfacial properties. The structure of the precursor molecules determines the surface properties of the SAM through intermolecular interactions and the functionality at the outer surface of the SAMs.1,2 The range of modifiable properties ranges from wettability3,4 to electronic properties like work functions, charge carrier injection or transport properties, often used in molecule-based electronics,5–8 photovoltaics,9,10 thermoelectricity11,12 or triboelectricity13 applications. Furthermore, SAMs are also used for the patterning of surfaces either using micro-contact printing14 or (nano)lithography.15,16 The latter operates with different types of radiation like UV,17 X-ray18 or electron beams,16 with the patterns either being formed by the application of photomasks18,19 or by the use of sequential patterning techniques.16,17 Basically, SAMs can be used either as negative or positive resists. In the former case, the stability of the monolayers becomes increased, which typically occurs through crosslinking, e.g. by UV-induced polymerization of diine groups or by electron-beam induced dehydrogenative coupling between aromatic units.15,20–22 While this cross-linking opens the opportunity for exciting new applications, such as formation of nanomembranes,23 the possibility for changing surface functionalities is quite limited.15 In contrast, positive resist behavior opens the opportunity for the removal and/or replacement of the irradiated parts of the SAM or e.g. metal-complex based photoresists,24 permitting either classical follow-up steps like localized etching25,26 or the insertion of new functionalities by binding of new SAM-forming molecules.27,28 For this replacement, typically the binding of the SAM constituents to the substrate is weakened e.g. by photo-induced oxidation using atmospheric dioxygen.29,30 This nevertheless requires very high area doses as well as high photon energies (hard UV light), which is inconvenient and sometimes requires specialized equipment.15 It has been demonstrated that dedicated photocleavable groups can be used to achieve selective bond scissions at much lower photon energies and fluxes due to their relatively high absorption cross sections at the respective wavelengths. This already has been applied for the modification of the tail groups of SAMs, which has been used for lithography31–34 as well as localized protein immobilization35,36 and the construction of sensors37 after further chemical modifications. So far, this strategy has been exclusively applied to systems in which the backbone of the SAM remained on the surfaces. This limits the scope of the approach as only chemical modifications at the tail groups can be made. We therefore decided to check if the concept of photocleavable groups might be combined with the complete exchange of the remaining SAM backbones. As it is known that the stability of SAMs scales with the length of the carbon chains of their constituents,38–40 the idea was to use an as-short-as-possible backbone, which can be replaced easily with other, longer molecules, see Fig. 1a. The base of our system is thioglycolic acid (TG), which should be readily exchanged with other precursors, as previous studies have shown that SAMs of alkane carboxylic acids with short chains are not very stable.38 Its carboxyl terminus can be easily protected with a photocleavable group, for which we chose the well-established ortho-nitrobenzene (ONB) group,41,42 which offers robustness and high chemical stability in combination with mild release conditions using UV light of 365 nm wavelength. Furthermore, the group is slim enough to fit into the structure of a densely packed SAM. To increase the stability of the SAM prior to irradiation, alkyl chains can be introduced, which – like in the simple alkanethiol SAMs – contribute van der Waals interactions within the SAM. As a compromise between this stabilization and solubility, we choose the hexyl group, either on both oxygen atoms or in combination with a methyl group (see Fig. 1b). The combination of these elements (TG group, ONB group, alkyl chains) leads to the design of precursors 1 and 2. As a control for the role of the ONB group, precursor 3 was designed, which basically possesses the same structure as precursor 1, but without the nitro group, which is the center piece for the photocleaving properties (compare Fig. 1b).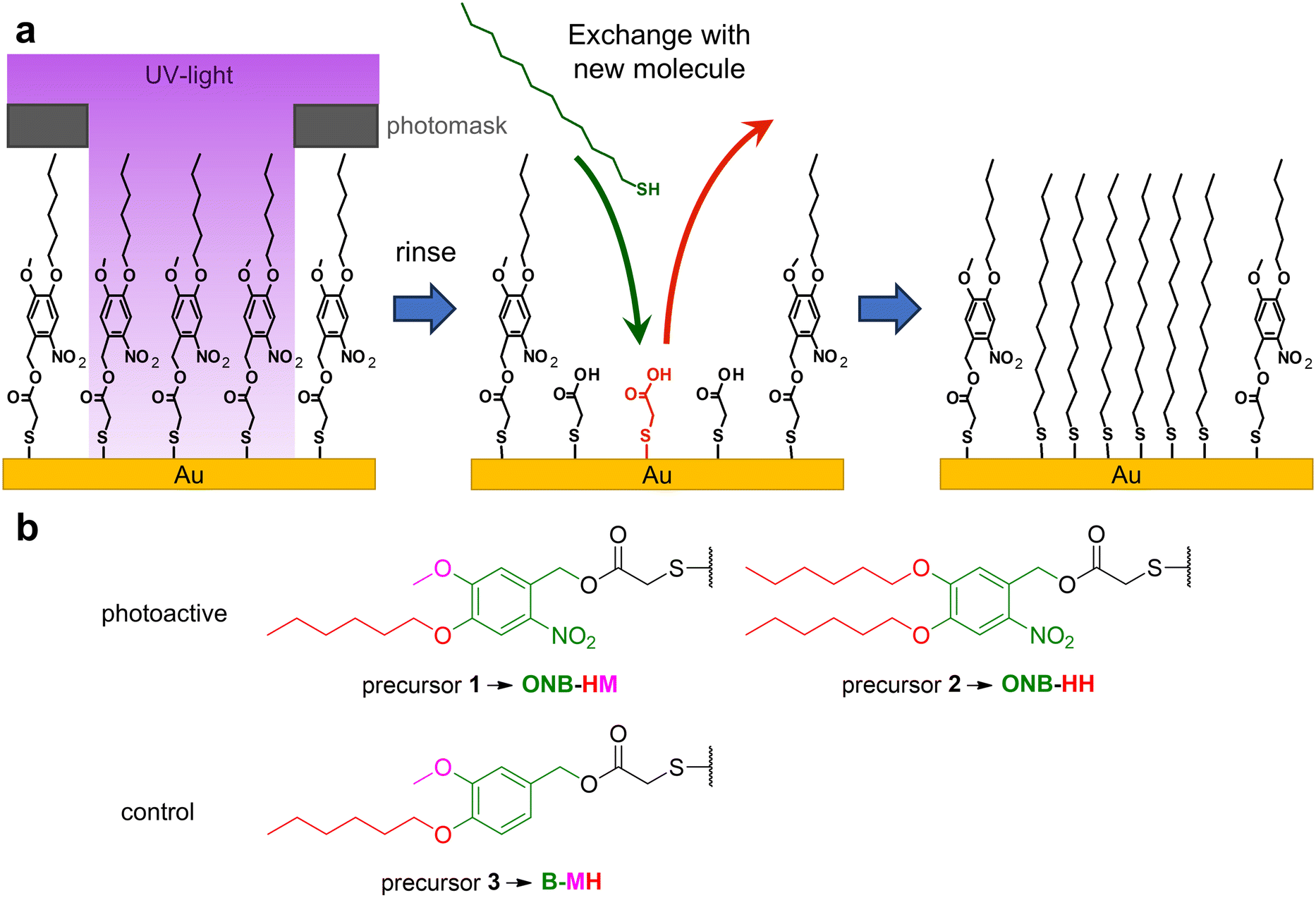 | ||
| Fig. 1 a Concept for the application of photoactive SAMs with ultrashort backbones, which after irradiation lead to the formation of the weakly bound thioglycolic acid (TG) SAMs, which in turn should be replaced by a new SAM precursor (here: dodecanethiol (C12)). b Overview of the naming scheme of the SAMs that are formed from precursors 1, 2 and 3, respectively. The parts responsible for the abbreviations are highlighted: ortho-nitrobenzyl (ONB, green) and benzyl (B, green) with methoxy (M, purple) and hexyloxy (H, red) in the IUPAC numbering order resulting in the respective abbreviations. | ||
2. Experimental section
Chemicals
All solvents and chemicals were purchased from commercial suppliers in varying qualities and used as received unless otherwise stated. A full list can be found in the ESI.‡Synthesis of SAM precursors
The synthesis of SAM precursors 1, 2 and 3 started either from vanillin or from 3,4-dihydroxybenzaldehyde following the pathway shown in Scheme 1 and is described in full detail in the ESI.‡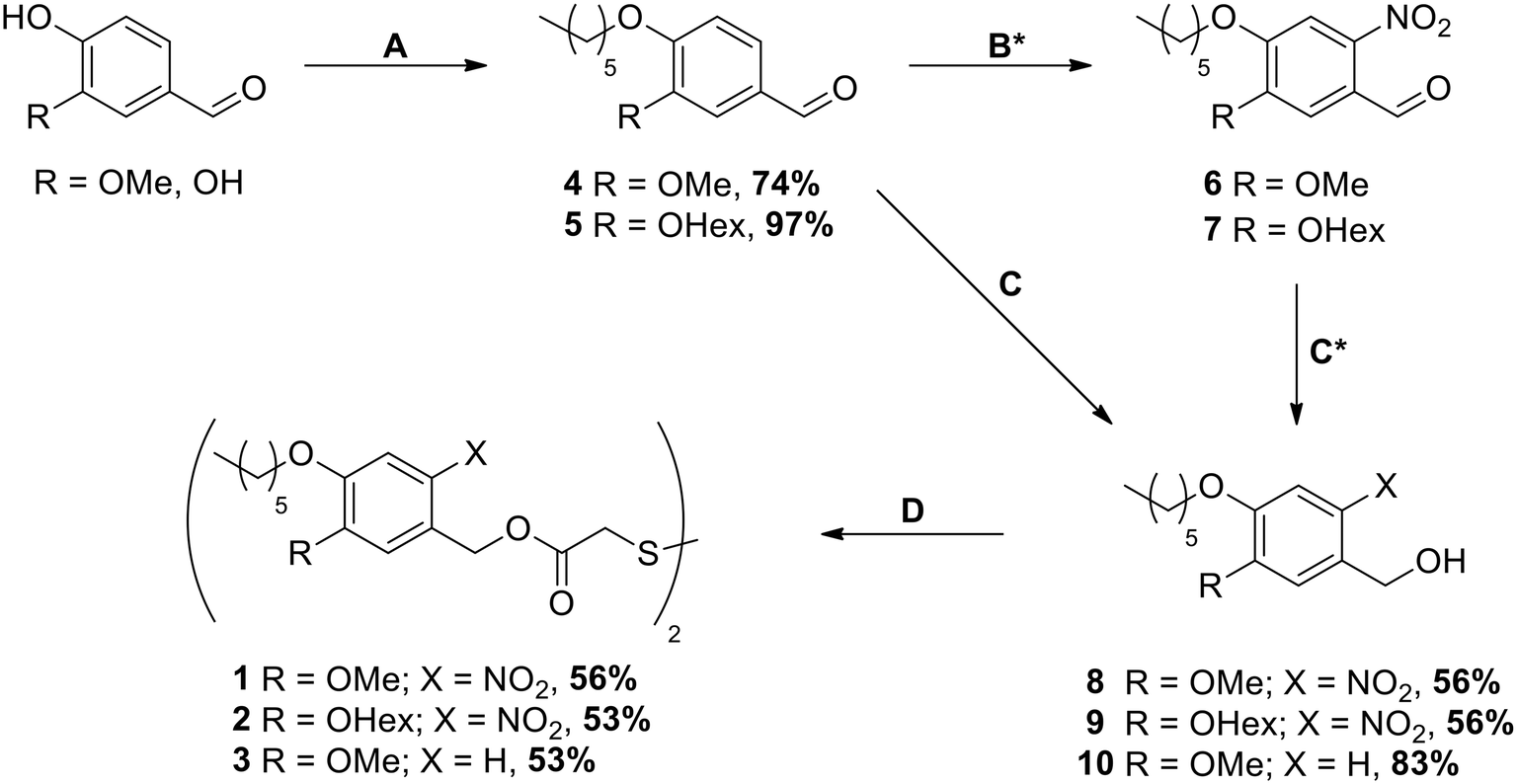 | ||
| Scheme 1 Synthesis of SAM precursors 1, 2 and 3 starting from vanillin or 3,4-dihydroxy-benzaldehyde. A 1-Bromohexane, DMF, 60 °C, 72 h; B 65% HNO3, AcOH, 60 °C, 24 h; C NaBH4, abs. THF, 0 °C, 4 h; D DIAD, PPh3, dithiodiglycolic acid, THF, 70 °C, 3 h. * Reactions B and C were carried out without purification of the intermediates; yields of compounds 8 and 9 were calculated over both reaction steps. | ||
Substrates
The gold substrates were prepared in-house by thermal evaporation of 5 nm Ti (adhesive layer) followed by 200 nm Au onto commercial 6-inch Si(100) wafers in ultra-high vacuum (UHV). The resulting films were polycrystalline, exposing mostly (111) orientated surfaces of individual crystallites with a measured RMS roughness (AFM) of 2.6 ± 0.9 nm across the substrates.Monolayer preparation
Substrates were freshly cut into 1 cm × 1 cm pieces, rinsed with ethanol (p.a. grade) and cleaned in hydrogen plasma43 (0.5 mbar) for 2 min. For ellipsometry, larger substrates (1 cm × 2 cm) were used, and the substrate parameters were measured at this stage. The SAMs of compounds 1, 2 and 3 were prepared by immersion of the freshly prepared substrates in solutions of the respective precursors in spectroscopic grade acetonitrile (0.2 mM, 24 h, room temperature). Subsequently, the SAMs were thoroughly washed with acetonitrile and dried in a stream of nitrogen. In addition, hexadecanethiolate (C16) and perdeuterated dodecanethiolate (C12-D) SAMs were prepared as references using gold substrates from the same wafers as the samples. Characterization took place immediately after the preparation.Ellipsometry
Measurements were carried out with a Sentech SE 400 ellipsometer using a He–Ne laser at a wavelength of 632.8 nm and a beam diameter of 1–2 mm at an angle of incidence of 70° with respect to the surface normal. For each substrate, four different spots were measured. The extinction coefficients of the SAMs were assumed to be zero and the real part of the refractive indices was set to 1.45 for all measurements.IR spectroscopy
All measurements were carried out using a Thermo Scientific Nicolet 6700 FT-IR spectrometer with a narrow-band mercury–cadmium–telluride semiconductor detector. The optical pathway was purged with dried and CO2-free air continuously. SAMs were analyzed using a Smart SAGA unit with a resolution of 4 cm−1 operating with p-polarized light at an angle of incidence of 80° relative to the surface normal, while bulk substances were characterized using a Smart Performer unit (attenuated total reflection, ATR). Background spectra of a C12-D SAM on Au(111) were recorded every three measurements except for the exchange experiments, for which the spectra were measured against a C16 SAM on Au(111) as a background to determine the content of C12-D deposited during the exchange experiment. 256 scans per sample were averaged and the resulting spectra mildly baseline-corrected. For band assignment, density-functional theory (DFT) calculations were carried out at the BP86 (ref. 44–46) level using the def2-SVP47 basis set with the ORCA 4.0 (ref. 48) program. Molecular geometries were optimized prior to the calculation of IR spectra using the same software package.Contact angle/wetting properties
Three substrates per molecule were measured with a contact angle goniometer (OPTREL) using the static sessile drop method with water as a working fluid. To prevent evaporation of the droplets during the measurements, the measuring chamber was equilibrated with water vapor 1 h prior to the experiments. For each substrate, three independent droplets were measured.Exchange experiments without irradiation
All exchange experiments were carried out with 1 cm × 1 cm substrates prepared as stated previously (see Monolayer preparation). After the initial analysis (IRRA spectroscopy and ellipsometry), the SAMs were placed in an 0.1 mM exchange solution of C12-D (ethanol, p.a. grade, degassed) for 5 and 30 minutes, as well as 1, 2, 5 and 20 hours. After removal from the solution, the samples were rinsed with ethanol (p.a. grade) and blown dry by a stream of nitrogen. Then, characterization with IRRA spectroscopy was performed to determine the extent of the exchange reaction. The spectra were measured using a SAM of C16 as a background and analyzed using the band area tool of the OMNIC software. The obtained band areas for the selected IR bands were compared to either the initially measured IRRA spectra before the exchange reaction, or in the case of the C12-D bands the average band area of the reference between 2250 cm−1 and 2050 cm−1. For the other SAMs, the area between 1490 cm−1 and 1600 cm−1 was used for analysis.Exchange experiments with irradiation
For each of the samples, the IRRA spectrum was recorded before they were transferred into a nitrogen-filled chamber and irradiated. For irradiation, UV light of 365 nm (UV-LED 5 W, see ESI‡ for full data, Table S4) was used at an energy density of 100 mW cm−2 for 1, 2, 4 and 8 minutes, resulting in a total irradiation energy of 6, 12, 24 and 48 J cm−2, respectively. Afterwards, the samples were rinsed three times with ethanol (p.a. grade) and blown dry by a stream of nitrogen, before immersion in the exchange solutions for 5 minutes. Further handling of the samples was carried out the same way as stated in the first set of experiments (see Exchange experiments without irradiation).Patterning experiments and scanning electron microscopy (SEM)
Two masks were used for the experiments, one with a line pattern with different line widths of 200 μm, 151 μm and 114 μm, and a second one with a sharkskin pattern with the same line widths. The patterns were designed using the AutoCAD software and printed in high resolution (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 dpi) on a mylar film (Zitzmann GmbH, Germany), which is transparent for UV and visible light. Samples for etching were prepared according to the general sample preparation with a size of 1 × 2 cm. After deposition of the SAM, the samples were irradiated under a nitrogen atmosphere for 8 min at 100 mW cm−2 with a photomask directly on top of the substrate. After irradiation, the samples were rinsed with ethanol (p.a. grade) and analyzed with an Amray 1920 ECO SEM (SEMTech Solutions, Inc., Billerica, MA) operating at 2 keV with a scintillation detector (Everhart-Thornley). Some of the samples were placed into a freshly prepared ferri/ferrocyanide etching solution25 consisting of KOH (1.0 M), K2S2O3 (0.1 M), K3Fe(CN)6 (0.01 M) and K4Fe(CN)6 (0.001 M) in distilled water (18.2 MΩ) for 25 minutes, before being rinsed with distilled water (18.2 MΩ) and ethanol (p.a. grade). The samples were then inspected with a VHX-500F digital microscope with a VH-Z00R lens (KEYENCE) and again analyzed by SEM.
000 dpi) on a mylar film (Zitzmann GmbH, Germany), which is transparent for UV and visible light. Samples for etching were prepared according to the general sample preparation with a size of 1 × 2 cm. After deposition of the SAM, the samples were irradiated under a nitrogen atmosphere for 8 min at 100 mW cm−2 with a photomask directly on top of the substrate. After irradiation, the samples were rinsed with ethanol (p.a. grade) and analyzed with an Amray 1920 ECO SEM (SEMTech Solutions, Inc., Billerica, MA) operating at 2 keV with a scintillation detector (Everhart-Thornley). Some of the samples were placed into a freshly prepared ferri/ferrocyanide etching solution25 consisting of KOH (1.0 M), K2S2O3 (0.1 M), K3Fe(CN)6 (0.01 M) and K4Fe(CN)6 (0.001 M) in distilled water (18.2 MΩ) for 25 minutes, before being rinsed with distilled water (18.2 MΩ) and ethanol (p.a. grade). The samples were then inspected with a VHX-500F digital microscope with a VH-Z00R lens (KEYENCE) and again analyzed by SEM.
3. Results and discussion
3.1. Synthesis
The synthesis was carried out starting from vanillin for compounds 1 and 3 and from 3,4-dihydroxybenzaldehyde for compound 2 (see Scheme 1). After the introduction of the alkyl chains via the Williamson ether synthesis, both compounds 4 and 5 were nitrated. In contrast to the literature-known procedures for similar molecules,49,50 using only HNO3 resulted in low yields due to poor solubility of the compounds in HNO3 or water. The use of acetic acid (AcOH) as a co-solvent was therefore crucial for the yield of the reaction, as it significantly improved the solubility of compounds 4 and 5. The purification of nitrated compounds 6 and 7 turned out to be difficult; we therefore directly reduced the crude mixtures using NaBH4, allowing the easy separation of compounds 8 and 9 from side products of both reactions via recrystallisation or column chromatography. As a control for our later irradiation experiments, we also synthesized compound 10 by directly reducing compound 4 without nitration. With the photocleavable ONB groups at hand, in the last step we used the Mitsunobu reaction to attach these protecting groups onto dithiodiglycolic acid, forming the precursors for the respective SAMs. With this route, we were able to prepare 1, 2 and 3 in similar yields, which allowed us to investigate their SAM formation and irradiation behaviors.3.2. Ellipsometry
While most SAMs typically are deposited from ethanol, we had to choose a different solvent in this work, as precursors 1, 2 and 3 were almost insoluble in this solvent. Deposition experiments from tetrahydrofuran resulted in SAMs with a high degree of contamination as detected by IRRAS. Therefore, acetonitrile was used as the solvent for the deposition, as all molecules are easily soluble in acetonitrile and no contamination of the SAMs could be observed (see IR spectroscopy). Taking into account a similar molecular length of 2.0 nm (including the Au–S bond), the ellipsometry measurements suggest the formation of monolayers in all three cases, with the highest value being found for ONB-HM with 1.82 ± 0.17 nm. The values for ONB-HH and B-MH SAMs were somewhat lower, with layer thicknesses of 1.55 ± 0.13 nm and 1.54 ± 0.12 nm, respectively. As ellipsometric measurements are a convolution of refractive indices and layer thicknesses, the apparently higher thickness of the ONB-MH SAM might be a result of an increased refractive index, as the nitro group typically increases the refractive index of a material (here: in comparison to B-MH), while the additional hexyl group in ONB-HH reduces the refractive index. To learn more about the orientation and order of the different molecular parts, the SAMs were further investigated by IR spectroscopy.3.3. IR spectroscopy
Not surprisingly, the IR spectra for all three molecules are quite similar within the bulk series (as recorded by ATR) and the SAM series (IRRA). In particular, the spectra of ONB-HM and ONB-HH are nearly identical, while the spectra of B-MH showed slight differences due to the missing NO2 group (Fig. 2). For an in-depth analysis, the spectra were also calculated by DFT, and vibrations and their transition dipole moments were assigned to the different signals (see ESI,‡ Fig. S13–S16 and Tables S1–S3).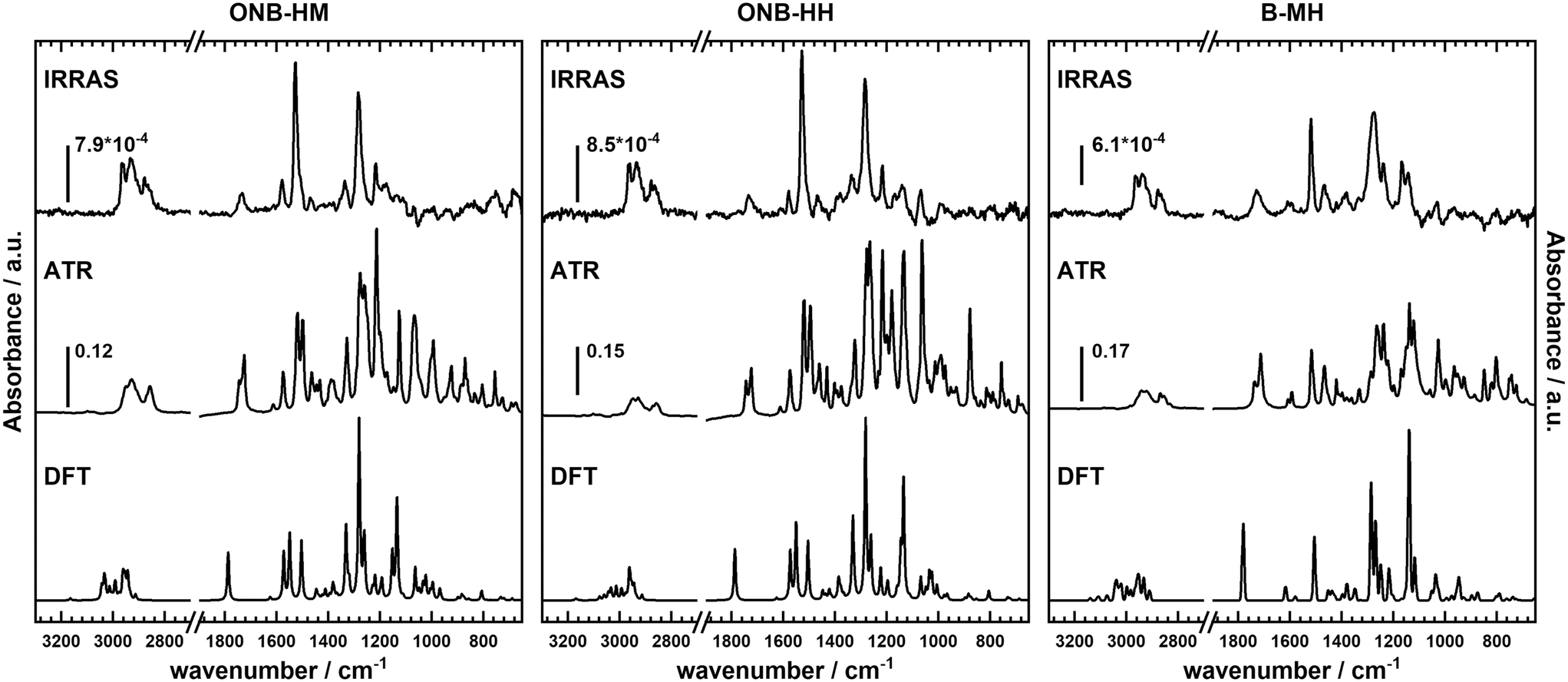 | ||
| Fig. 2 Representation of the IRRA, ATR and calculated spectra (from top to bottom) of ONB-HM, ONB-HH and B-MH, respectively. The ATR spectra were obtained from the respective disulfides (precursors 1, 2 and 3); therefore, they show peak splitting for certain vibrations due to synchronization between vibrations in the two parts of the symmetric disulfides. | ||
SAMs on metal surfaces show distinct weakening or enhancement of vibrations in the IRRA spectra (surface selection rules),51,52 as vibrations with a transition dipole moment (TDM) parallel to the surface normal are enhanced, while those with a TDM perpendicular to the surface normal and therefore parallel to the surface are extinguished. A comparison between ATR and IRRA spectra therefore allows for an analysis of the orientation of the molecular groups on the surface.51,52 For the following discussion, we additionally define two more axes, A and B (Fig. 3), along which most of the TDMs found in our series of molecules can be oriented.
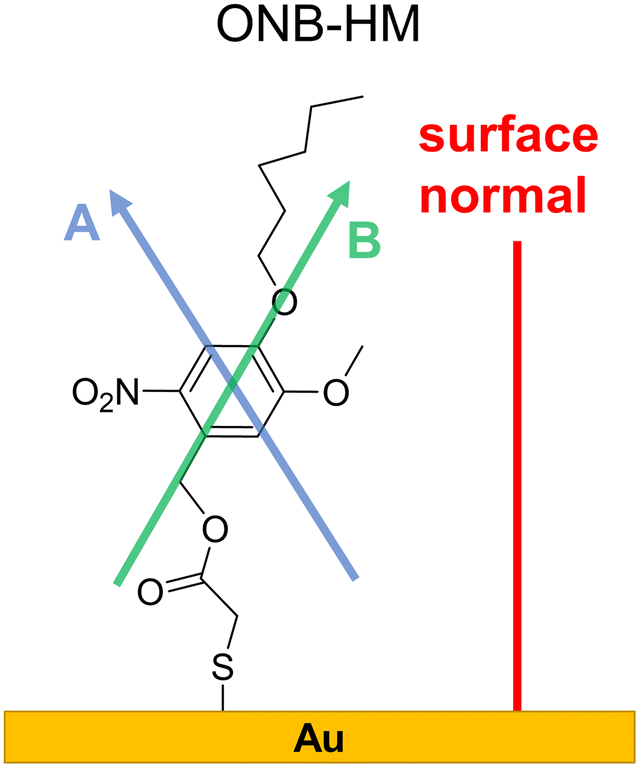 | ||
| Fig. 3 Surface conformation of the ONB-HM molecules in the SAM as deduced from the IR analysis. The shown axes A, passing through the two H atoms directly attached to the aromatic ring, and B, through the benzyl–CH2-group and the oxygen atom of the hexyl ether group, serve as orientation references for the TDMs discussed in the IR analysis (see Table S1‡). | ||
For the ONB-MH SAMs, the out-of-plane vibrations of the aromatic ring at 877 cm−1 are extinguished in the IRRA spectra suggesting that the ring plane of the aromatic system is parallel to the surface normal. Also, the CO stretching vibration at 1736 cm−1 is weakened compared to the ATR spectra indicating an orientation with a larger angle relative to the surface normal. By comparison to the calculated TDMs of the vibrations, this suggests an orientation as shown in Fig. 3. Surprisingly, the NO2 vibrations at 1581 cm−1 (asymmetric, anti-phase, TDM ∥-A axis) and 1528 cm−1 (asymmetric, in-phase, TDM ∥-B axis) are only slightly weakened, while the vibration at 1335 cm−1 (symmetric, TDM ⊥-B axis) is even weaker than the other two NO2 vibrations, which suggests the depicted orientation almost perpendicular to the surface normal. This unexpected orientation of the nitro group and thus the benzene ring might be explained by constructive dipole–dipole interactions within the SAM, as they have already been described for fluorine-containing systems.53 This should have consequences for the order of the alkyl groups, which can be deduced from the position of the asymmetric stretching vibration of the CH2-groups.54,55 This vibration is found at 2935 cm−1 that is indicative of alkyl chains with a high degree of gauche conformations, which goes in line with alkyl chains that cannot attain optimal van der Waals interactions when being stretched out.
The general behavior of these vibrations in the ONB-HH SAMs is nearly identical, suggesting a similar orientation of the benzene ring and the nitro moiety (see Fig. S14 and Table S2 in the ESI‡), which leads to even more conformational disorder in the alkyl part (signal at 2933 cm−1). The resulting increased spatial demand of the molecule might be an alternative explanation for the lower film thicknesses observed in the ellipsometry measurements.
The situation is less clear for the B-MH SAMs due to the missing strong NO2 vibrations. The CO stretching vibration shows a similar weakening compared to the other two molecules, with the aromatic out-of-plane vibrations also extinguished in the IRRA spectra, suggesting an upright orientation of the aromatic ring. The other significant vibrations of this molecule, being the aromatic CC stretching vibrations (1593 cm−1, TDM ⊥-B axis; 1518 cm−1, TDM ⊥-A axis; 1471 cm−1, TDM ⊥-A axis) and the CO vibrations of the alkoxy groups (1274 cm−1, TDM ⊥-A axis; 1235 cm−1, TDM ∥-A axis), exhibit some strong weakening (1593 cm−1, 1471 cm−1, 1235 cm−1), while other vibrations are only slightly influenced (1518 cm−1, 1274 cm−1, see Table S3 and Fig. S16‡). The observed effects in the ellipsometry measurements are probably due to the mentioned differences of the refractive indices as mentioned before.
3.4. Wetting properties
The interface between all three SAMs and water droplets during the water contact angle (WCA) measurements is primarily formed by the alkyl chain part of the molecules. Even though the chains themselves are partly unordered according to the IR analyses, more polar groups are still embedded in the backbone of the SAM; so, a mostly hydrophobic behavior is expected. Therefore, C12-D was chosen as a reference SAM with a WCA of 113°, which is typical for well-ordered, all-trans configured alkane thiolate SAMs on gold surfaces.56 The ONB-HM and BMH SAMs exhibited WCAs of around 85°, suggesting only a slightly hydrophobic surface structure, while ONB-HH showed a slightly higher WCA of 98° (Table 1).| C12-D | ONB-HM | ONB-HH | B-MH | |
|---|---|---|---|---|
| Contact angle | 113° ± 2° | 88° ± 4° | 98° ± 4° | 83° ± 3° |
Although both B-MH and ONB-HM possess the C6 alkyl chain and a methoxy group at the SAM–liquid interface, the wetting properties are similar to those of the methoxy terminated alkane thiolate SAM (85° for HS-C11-OMe57), with the effect of the hexyl group on the wetting properties being diminished due to the unordered conformation of the hexyl chain caused by the particular orientation of the aromatic units. This increased wetting due to molecular disorder has been reported for mixed SAMs of alkane thiolates with different chain lengths in close proximity.58–60 This effect is weaker in the case of the ONB-HH SAM, where the two hexyl chains on one hand are disordered but nevertheless can better cover up the heteroatoms of the molecule.58 Thus, the ONB-HH SAM shows a WCA in between the ones reported for ethoxy- and propoxy-terminated alkane thiolate SAMs (96° and 104° for HS-C11-OEt and HS-C11-OPr, respectively).57
3.5. Exchange experiments
After making sure that the molecules reliably form monolayers, the main goal of the project – the facilitated exchange reaction after photochemical cleavage – could be pursued. As it has been well-known that even densely packed SAMs are prone to exchange reactions during prolonged exposure to solutions of a second kind of thiol,53,61–63 our native monolayer systems were investigated for their stability against exchange with perdeuterated dodecanethiol (C12-D) (Fig. 4). This molecule was chosen as it shows characteristic signals in the IR spectra, which do not interfere with those of the new molecules. In these experiments, both ONB-HM and B-MH behaved similarly, with a certain exchange within the first 4 hours before leveling out at around 65% of the remaining content for B-MH and 48% for ONB-HM after 20 hours in the C12-D exchange solutions. The content of the C12-D molecules in the SAM increases at the same rate as the desorption is happening, suggesting a one-to-one exchange of the molecules in the SAM. This is further supported by calculating the sum of, for example, the ONB-HM content and the C12-D content, which in all cases is approximately 100%. The observed differences likely result from different packing densities of the SAMs.In contrast to this, the ONB-HH SAM becomes exchanged much faster, resulting in a sharp drop to only 40% remaining content after 4 hours and being nearly completely replaced after 20 hours. This highlights a significantly lower stability of the ONB-HH SAM, which makes this SAM unsuitable for the following irradiation experiments, as even after 5 minutes of exchange only 60% of the molecules are left in place compared to the 86% and 90% of the ONB-HM and B-MH SAMs, respectively. For this reason, the following irradiation experiments were carried out without the SAMs formed from ONB-HH, using only the ONB-HM and BMH SAMs.
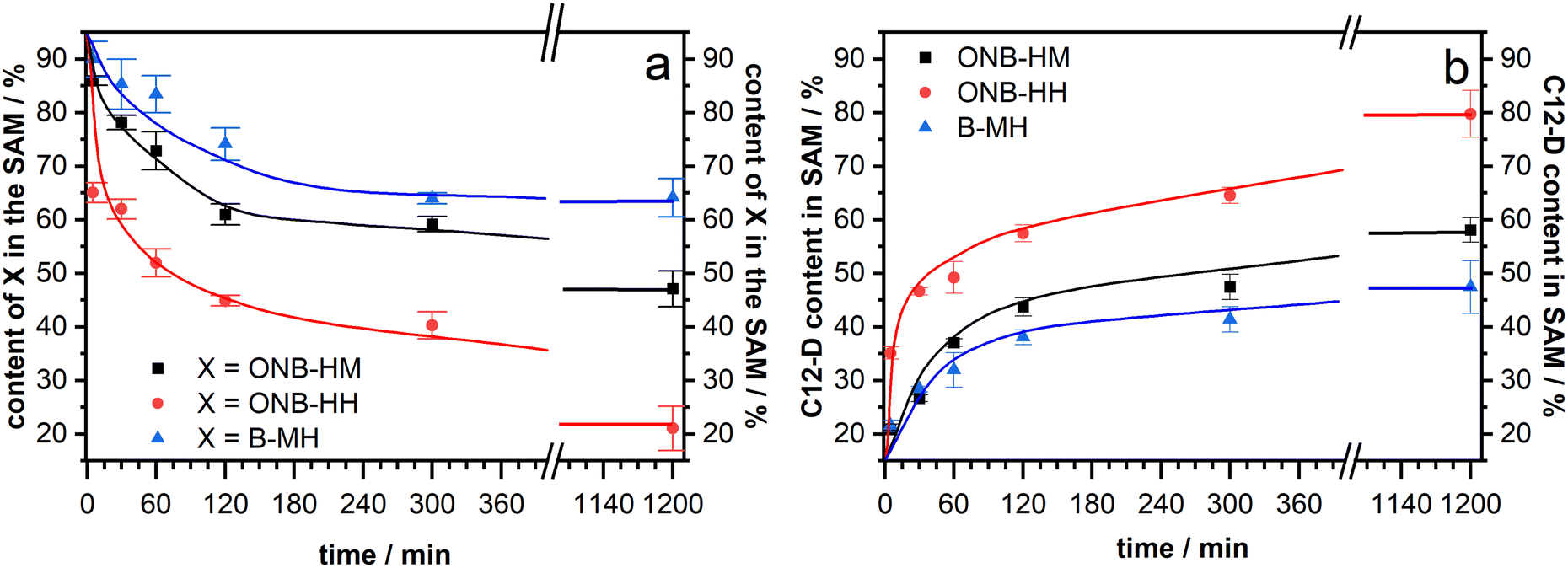 | ||
| Fig. 4 SAM composition during the exchange experiments without irradiation, starting with a single component SAM of molecule X (ONB-HM, ONB-HH or B-MH), which becomes exchanged by C12-D. (a) Remaining content of molecule X in the SAMs. (b) Content of C12-D in the SAM after exchange. The shown lines are guides for the eye and not fitted data. | ||
3.6. Irradiation experiments
The cleavage reaction of the ONB groups proceeds at wavelengths much longer than typically reported for the irradiation-induced exchange of thiolate SAMs. We recorded the UV spectra of precursors 1 and 3 (Fig. 5). Non-nitrated 3 (precursor for B-MH) showed an absorption band at 280 nm, while for 1 (→ ONB-HM) two major absorption bands with maxima at 346 nm and 300 nm were found. This is in good agreement with the literature-known absorption bands of ONB-groups and irradiation wavelengths typically in the range of 350 to 370 nm.42 We therefore chose a wavelength of 365 nm for the selective cleavage and substitution of the ONB-HM monolayer. For the irradiation process, an LED with the corresponding peak wavelength was chosen (see emission spectrum (blue curve) in Fig. 5), as this kind of light source provides almost monochromatic light with no stray radiation at lower wavelengths, which might induce competing processes, or higher wavelengths, which might heat up the layer system.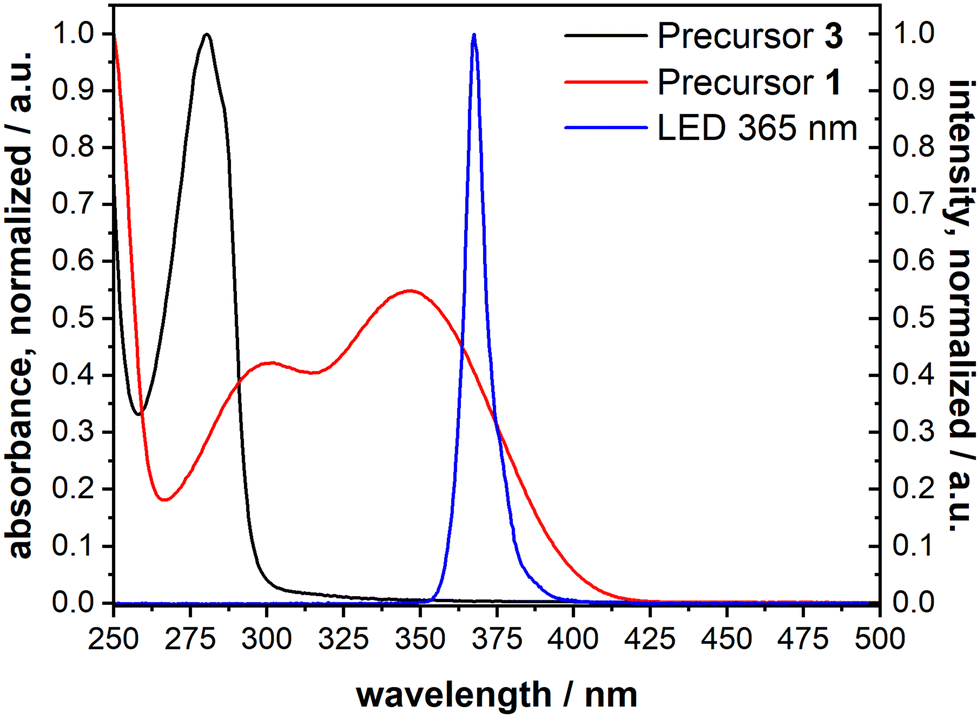 | ||
| Fig. 5 UV-vis absorption spectra of 1 (→ ONB-HM, red) and 3 (→ B-MH, black) measured in acetonitrile, as well as the emission spectrum of the chosen LED with a peak wavelength of 365 nm (blue). | ||
Samples of both ONB-HM and B-MH SAMs were irradiated with 6, 12, 24 and 48 J cm−2 and then exposed to C12-D solutions for 5 minutes, before analysis of the resulting SAM compositions by IRRA spectroscopy (see Fig. 6). The B-MH SAMs only showed a minor influence of the irradiation on the extent of the exchange, with a remaining content of 80% after the highest dose of 48 J cm−2 and subsequent exchange with C12-D (as compared to 90% without irradiation). This result was expected, as this molecule does not bear a nitro group, and may only be released by the irradiation-induced cleavage of the Au–S bond by oxidation, for which the cross section at 365 nm is very low.27,29,64 Although the samples were irradiated in a nitrogen atmosphere to mitigate this oxidation process, there might still be some oxygen contamination left due to the transfer process of the substrate, leading to this oxidation reaction.
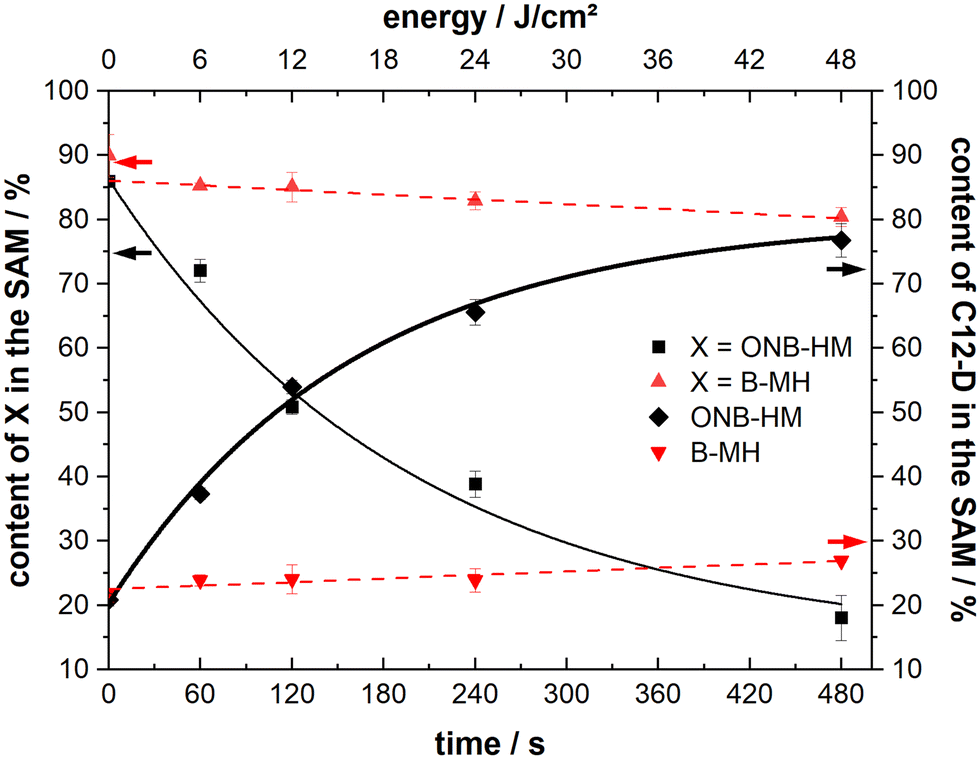 | ||
| Fig. 6 Remaining content of molecule X (ONB-HM or B-MH) as well as C12-D content in the SAMs after irradiation for the given times and exchange for 5 minutes. The given energy correlates to the irradiation times as the light flux constantly remained at 100 mW cm−2 for all experiments. | ||
In contrast to this, the ONB-HM SAMs show a very pronounced dose-dependent decrease in the remaining content in the SAM, while the corresponding content of the exchange molecule C12-D shows a dose-dependent increase. This continues to a point where the ONB-HM SAM after 48 J cm−2 has been nearly completely replaced by C12-D (remaining ONB-HM: 18% and C12-D: 77%). Although the data suggest that the exchange can be driven further, it looks like that the exchange cannot be driven to completeness, as previous studies on similar molecules have reported that a reduction of the NO2 group can also occur under irradiation, leading to the deactivation of some molecules, which then remain on the surface.32
During the irradiation process, a slight heating of the substrate (<40 °C) was observed that might influence the results of the measurement due to known thermal degradation of SAMs.65 We therefore performed another set of experiments with the monolayer-covered substrates: 1) by heating them in an oven at 60 °C for eight minutes in a nitrogen atmosphere (equivalent to the longest irradiation experiments). 2) By irradiating them with a wavelength of 450 nm to ensure no or only minimal photocleavage, while still being in the UV-absorption range of the gold substrate (simulating the substrate heating due to light absorption). In both experiments, the extent of the following exchange reaction was the same – within the margin of error – as for the non-irradiated samples (about 85% of the ONB-HM SAM remaining). Therefore, all the observed increases for the exchange of ONB-HM after irradiation with light of 365 nm are due to the photocleavage and not a result of other degradation processes.
3.7. Patterning experiments
The selective activation by light opens the opportunity for localized reactions, by applying either localized beams or masks. As a proof of concept, we employed photomasks with either simple line patterns or sharkskin patterns. Using these, ONB-HM SAMs were irradiated with a dose of 48 mW cm−2 to achieve almost complete exchange. Unfortunately, the exchanged areas could not be visualized neither by optical microscopy nor by SEM, presumably due to very similar surface properties. As the photoreaction in its first step results in a carboxylic-acid terminated surface, it could be expected that this surface would show a different work function and thus show a contrast to the ONB-HM surface in the SEM. Indeed, in some but not all samples this contrast could be attained (see Fig. 7a). We therefore decided to enhance this contrast by etching away the part of the gold surface that was only covered by the ultrashort SAM, as this should be much less stable against Au etchants than the hydrophobic ONB-HM SAM. Indeed, this resulted in samples with clear contrasts both in optical microscopy and SEM (see Fig. 7b–d). The patterns of the masks became well reproduced with clear and straight edges. Additional analysis by SEM clearly revealed that the etched patterns were somewhat wider than the spacings in the mask (e.g. 158 μm vs. 151 μm), which might be either due to stray light under the mask because of imperfect contact or by under etching of the protective ONB-HM SAM (or both). As these experiments served as a proof of concept that patterning with these molecules is possible, we concluded our experiments at this point and did not put more effort in optimizing the photolithographic process.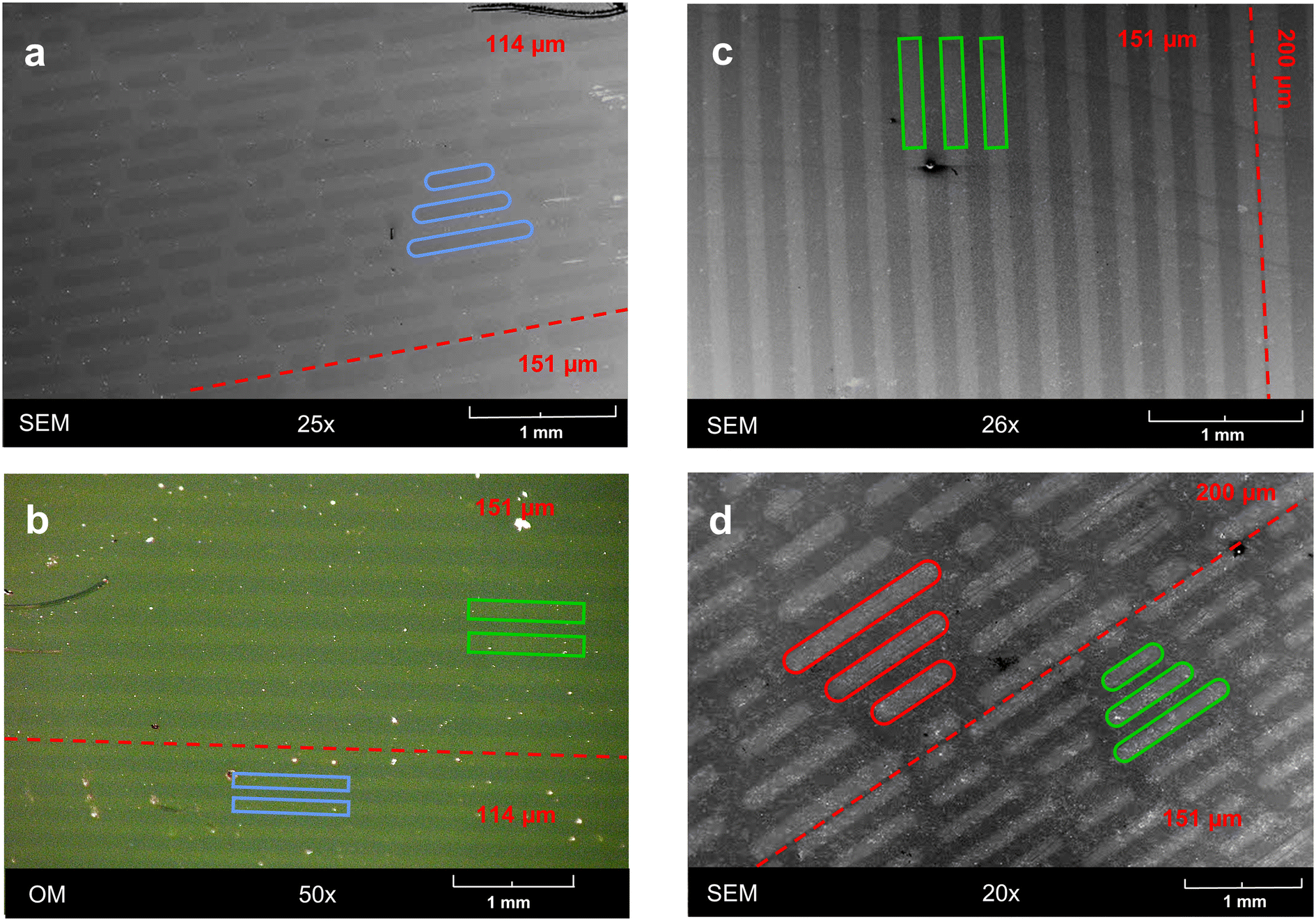 | ||
| Fig. 7 Micrographs of ONB-HM SAMs irradiated through a photomask. a) SEM image of a surface that was only rinsed after irradiation. The pattern results from different work functions. b) Optical micrograph of a surface after selective etching of the irradiated areas. c) and d) SEM images of samples with two different patterns (c: line pattern, d: sharkskin pattern) after etching. Borders between different pattern sizes are indicated by a dotted line. For clearness, some of the structures are highlighted in different colors. The numbers indicate the width of the patterns. | ||
4. Conclusions
In an effort to facilitate the SAM-based replacement lithography process,16,18 we introduced photocleavable groups into the precursor molecules, which upon cleavage would result in a monolayer of an ultrashort molecule, thioglycolic acid, that is known to be easily replaceable by longer thiols. For this, two molecules bearing hexyl chains, (ONB-HM)21 and (ONB-HH)22, were synthesized and their SAMs were characterized by ellipsometry, IRRA spectroscopy and contact angle measurements. The data suggest that the conformation of the molecules puts the nitro groups parallel to the surface, presumably to maximize dipole–dipole interactions between the molecules in the SAM. As a result, the hexyl groups become disordered, but nevertheless provide a more or less hydrophobic surface comparable to the ones of alkoxy-terminated SAMs.57Investigations of the exchange reaction with C12-D without prior irradiation of the SAMs (ONB-HM, ONB-HH) revealed a slow exchange for ONB-HM and a (unexpectedly) faster one for ONB-HH, disqualifying the latter for further experiments. Irradiation experiments at 365 nm showed that the envisioned exchange reaction can indeed be driven almost to completeness (>80%) with an area dose of 48 J cm−2. By using a similar molecule, which does not bear a nitro group, B-MH, it could be demonstrated that this replacement process is indeed driven by the photochemical reaction at the ONB group, as the exchange of the latter SAMs is almost not influenced by irradiation. The observation that the exchange reaction with C12-D results in a 1:1 substitution supports the idea of densely packed monolayers in both cases, ONB-HM and B-MH.
The demonstration of the patterning capability of the ONB-HM SAM was carried out by irradiation of the SAM through a photomask. While the exchange reaction did not provide enough contrast in the applied microscopies, the irradiated surface without the exchange reaction permitted the visualization in SEM. Using the ONB-HM SAM as an etch resist, the patterns could be enhanced, making them now visible in SEM and optical micrographs.
Compared to previous studies17 using the direct UV photooxidation of the sulfur anchoring group, which achieved only about 60% transformation at a dose of 200 J cm−2, the new system attains an exchange of >80% at a significantly lower dose of 48 J cm−2, clearly demonstrating the advantage of photocleavable groups in replacement lithography.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
FCI within the Verband der Chemischen Industrie. C. F. and A. T. thank the Fonds der Chemischen Industrie (FCI) for providing a PhD stipend.References
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Self-Assembled Monolayers of Thiolates on Metals as a Form of Nanotechnology, Chem. Rev., 2005, 105(4), 1103–1170, DOI:10.1021/cr0300789.
- C. Vericat, M. E. Vela, G. Benitez, P. Carro and R. C. Salvarezza, Self-Assembled Monolayers of Thiols and Dithiols on Gold: New Challenges for a Well-Known System, Chem. Soc. Rev., 2010, 39(5), 1805, 10.1039/b907301a.
- S. Lee, A. Puck, M. Graupe, R. Colorado, Y.-S. Shon, T. R. Lee and S. S. Perry, Structure, Wettability, and Frictional Properties of Phenyl-Terminated Self-Assembled Monolayers on Gold, Langmuir, 2001, 17(23), 7364–7370, DOI:10.1021/la0111497.
- R. Colorado and T. R. Lee, Wettabilities of Self-Assembled Monolayers on Gold Generated from Progressively Fluorinated Alkanethiols, Langmuir, 2003, 19(8), 3288–3296, DOI:10.1021/la0263763.
- D. Boudinet, M. Benwadih, Y. Qi, S. Altazin, J.-M. Verilhac, M. Kroger, C. Serbutoviez, R. Gwoziecki, R. Coppard, G. Le Blevennec, A. Kahn and G. Horowitz, Modification of Gold Source and Drain Electrodes by Self-Assembled Monolayer in Staggered n- and p-Channel Organic Thin Film Transistors, Org. Electron., 2010, 11(2), 227–237, DOI:10.1016/j.orgel.2009.10.021.
- A. Vilan, D. Aswal and D. Cahen, Large-Area, Ensemble Molecular Electronics: Motivation and Challenges, Chem. Rev., 2017, 117(5), 4248–4286, DOI:10.1021/acs.chemrev.6b00595.
- M. Gärtner, E. Sauter, G. Nascimbeni, A. Petritz, A. Wiesner, M. Kind, T. Abu-Husein, M. Bolte, B. Stadlober, E. Zojer, A. Terfort and M. Zharnikov, Understanding the Properties of Tailor-Made Self-Assembled Monolayers with Embedded Dipole Moments for Interface Engineering, J. Phys. Chem. C, 2018, 122(50), 28757–28774, DOI:10.1021/acs.jpcc.8b09440.
- Y. Liu, M. Zeplichal, S. Katzbach, A. Wiesner, S. Das, A. Terfort and M. Zharnikov, Aromatic Self-Assembled Monolayers with pentafluoro-Λ6-Sulfanyl (−SF5) Termination: Molecular Organization and Charge Transport Properties, Nano Res., 2023, 16(5), 7991–8002, DOI:10.1007/s12274-022-5350-8.
- F. Ali, C. Roldán-Carmona, M. Sohail and M. K. Nazeeruddin, Applications of Self-Assembled Monolayers for Perovskite Solar Cells Interface Engineering to Address Efficiency and Stability, Adv. Energy Mater., 2020, 10(48), 2002989, DOI:10.1002/aenm.202002989.
- S. Y. Kim, S. J. Cho, S. E. Byeon, X. He and H. J. Yoon, Self-Assembled Monolayers as Interface Engineering Nanomaterials in Perovskite Solar Cells, Adv. Energy Mater., 2020, 10(44), 2002606, DOI:10.1002/aenm.202002606.
- S. Park, H. Kang and H. J. Yoon, Structure–Thermopower Relationships in Molecular Thermoelectrics, J. Mater. Chem. A, 2019, 7(24), 14419–14446, 10.1039/C9TA03358K.
- J. Jang, P. He and H. J. Yoon, Molecular Thermoelectricity in EGaIn-Based Molecular Junctions, Acc. Chem. Res., 2023, 56(12), 1613–1622, DOI:10.1021/acs.accounts.3c00168.
- S.-H. Shin, Y. E. Bae, H. K. Moon, J. Kim, S.-H. Choi, Y. Kim, H. J. Yoon, M. H. Lee and J. Nah, Formation of Triboelectric Series via Atomic-Level Surface Functionalization for Triboelectric Energy Harvesting, ACS Nano, 2017, 11(6), 6131–6138, DOI:10.1021/acsnano.7b02156.
- B. D. Gates, Q. Xu, J. C. Love, D. B. Wolfe and G. M. Whitesides, Unconventional Nanofabrication, Annu. Rev. Mater. Res., 2004, 34(1), 339–372, DOI:10.1146/annurev.matsci.34.052803.091100.
- A. Turchanin, M. Schnietz, M. El-Desawy, H. H. Solak, C. David and A. Gölzhäuser, Fabrication of Molecular Nanotemplates in Self-Assembled Monolayers by Extreme-Ultraviolet-Induced Chemical Lithography, Small, 2007, 3(12), 2114–2119, DOI:10.1002/smll.200700516.
- A. Terfort and M. Zharnikov, Electron-Irradiation Promoted Exchange Reaction as a Tool for Surface Engineering and Chemical Lithography, Adv. Mater. Interfaces, 2021, 8(10), 2100148, DOI:10.1002/admi.202100148.
- Y. L. Jeyachandran, N. Meyerbröker, A. Terfort and M. Zharnikov, Maskless Ultraviolet Projection Lithography with a Biorepelling Monomolecular Resist, J. Phys. Chem. C, 2015, 119(1), 494–501, DOI:10.1021/jp510809a.
- M. Zharnikov and M. Grunze, Modification of Thiol-Derived Self-Assembling Monolayers by Electron and x-Ray Irradiation: Scientific and Lithographic Aspects, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2002, 20(5), 1793–1807, DOI:10.1116/1.1514665.
- M. Zharnikov, A. Shaporenko, A. Paul, A. Gölzhäuser and A. Scholl, X-Ray Absorption Spectromicroscopy Studies for the Development of Lithography with a Monomolecular Resist, J. Phys. Chem. B, 2005, 109(11), 5168–5174, DOI:10.1021/jp040649g.
- D. N. Batchelder, S. D. Evans, T. L. Freeman, L. Haeussling, H. Ringsdorf and H. Wolf, Self-Assembled Monolayers Containing Polydiacetylenes, J. Am. Chem. Soc., 1994, 116(3), 1050–1053, DOI:10.1021/ja00082a028.
- H. Menzel, M. D. Mowery, M. Cai and C. E. Evans, The Effect of Spacer Length on the Polymerization of Diacetylenes in Sams on Gold Surfaces, Macromol. Symp., 1999, 142(1), 23–31, DOI:10.1002/masy.19991420105.
- Y.-H. Chan, J.-T. Lin, I.-W. P. Chen and C. Chen, Monolayers of Diphenyldiacetylene Derivatives: Tuning Molecular Tilt Angles and Photopolymerization Efficiency via Electrodeposited Ag Interlayer on Au, J. Phys. Chem. B, 2005, 109(41), 19161–19168, DOI:10.1021/jp0529366.
- A. Turchanin and A. Gölzhäuser, Carbon Nanomembranes from Self-Assembled Monolayers: Functional Surfaces without Bulk, Prog. Surf. Sci., 2012, 87(5–8), 108–162, DOI:10.1016/j.progsurf.2012.05.001.
- G. Lim, K. Lee, S. Choi and H. J. Yoon, Organometallic and Coordinative Photoresist Materials for EUV Lithography and Related Photolytic Mechanisms, Coord. Chem. Rev., 2023, 493, 215307, DOI:10.1016/j.ccr.2023.215307.
- Y. Xia, X.-M. Zhao, E. Kim and G. M. Whitesides, A Selective Etching Solution for Use with Patterned Self-Assembled Monolayers of Alkanethiolates on Gold, Chem. Mater., 1995, 7(12), 2332–2337, DOI:10.1021/cm00060a023.
- Y. Xia, X.-M. Zhao and G. M. Whitesides, Pattern Transfer: Self-Assembled Monolayers as Ultrathin Resists, Microelectron. Eng., 1996, 32(1–4), 255–268, DOI:10.1016/0167-9317(95)00174-3.
- N. Ballav, T. Weidner and M. Zharnikov, UV-Promoted Exchange Reaction as a Tool for Gradual Tuning the Composition of Binary Self-Assembled Monolayers and Chemical Lithography, J. Phys. Chem. C, 2007, 111(32), 12002–12010, DOI:10.1021/jp0726788.
- P. Burgos, M. Geoghegan and G. J. Leggett, Generation of Molecular-Scale Compositional Gradients in Self-Assembled Monolayers, Nano Lett., 2007, 7(12), 3747–3752, DOI:10.1021/nl072180h.
- D. A. Hutt and G. J. Leggett, Influence of Adsorbate Ordering on Rates of UV Photooxidation of Self-Assembled Monolayers, J. Phys. Chem., 1996, 100(16), 6657–6662, DOI:10.1021/jp952734h.
- C. Zhou and A. V. Walker, UV Photooxidation of a Homologous Series of n -Alkanethiolate Monolayers on GaAs (001): A Static SIMS Investigation, J. Phys. Chem. C, 2008, 112(3), 797–805, DOI:10.1021/jp075863u.
- K. Critchley, J. P. Jeyadevan, H. Fukushima, M. Ishida, T. Shimoda, R. J. Bushby and S. D. Evans, A Mild Photoactivated Hydrophilic/Hydrophobic Switch, Langmuir, 2005, 21(10), 4554–4561, DOI:10.1021/la046851s.
- K. Critchley, L. Zhang, H. Fukushima, M. Ishida, T. Shimoda, R. J. Bushby and S. D. Evans, Soft-UV Photolithography Using Self-Assembled Monolayers, J. Phys. Chem. B, 2006, 110(34), 17167–17174, DOI:10.1021/jp0630370.
- K. Critchley, R. Ducker, J. P. Bramble, L. Zhang, R. J. Bushby, G. J. Leggett and S. D. Evans, Photo-Deprotection Patterning of Self-Assembled Monolayers, J. Exp. Nanosci., 2007, 2(4), 279–290, DOI:10.1080/17458080701675780.
- P. Prompinit, A. S. Achalkumar, X. Han, R. J. Bushby, C. Wälti and S. D. Evans, Improved Photoreaction Yields for Soft Ultraviolet Photolithography in Organothiol Self-Assembled Monolayers, J. Phys. Chem. C, 2009, 113(52), 21642–21647, DOI:10.1021/jp907950c.
- S. Xia, M. Cartron, J. Morby, D. A. Bryant, C. N. Hunter and G. J. Leggett, Fabrication of Nanometer- and Micrometer-Scale Protein Structures by Site-Specific Immobilization of Histidine-Tagged Proteins to Aminosiloxane Films with Photoremovable Protein-Resistant Protecting Groups, Langmuir, 2016, 32(7), 1818–1827, DOI:10.1021/acs.langmuir.5b04368.
- S. Chen and L. M. Smith, Photopatterned Thiol Surfaces for Biomolecule Immobilization, Langmuir, 2009, 25(20), 12275–12282, DOI:10.1021/la9017135.
- K. Drexler, J. Smirnova, M. Galetskaya, N. Schweizer, G. Gauglitz and U. E. Steiner, Optical Detection of Photorelease Kinetics on Gold and Glass Surfaces Using Streptavidin-Coupled Biotinylated Photolabile Protecting Groups for Nucleosides, ChemPhysChem, 2017, 18(20), 2890–2898, DOI:10.1002/cphc.201700714.
- Z. Dai and H. Ju, Effect of Chain Length on the Surface Properties of ω-Carboxy Alkanethiol Self-Assembled Monolayers, Phys. Chem. Chem. Phys., 2001, 3(17), 3769–3773, 10.1039/b104570a.
- L. Srisombat, A. C. Jamison and T. R. Lee, Stability: A Key Issue for Self-Assembled Monolayers on Gold as Thin-Film Coatings and Nanoparticle Protectants, Colloids Surf., A, 2011, 390(1–3), 1–19, DOI:10.1016/j.colsurfa.2011.09.020.
- B. Singhana, A. C. Jamison, J. Hoang and T. R. Lee, Self-Assembled Monolayer Films Derived from Tridentate Cyclohexyl Adsorbates with Alkyl Tailgroups of Increasing Chain Length, Langmuir, 2013, 29(46), 14108–14116, DOI:10.1021/la401899q.
- A. P. Pelliccioli and J. Wirz, Photoremovable Protecting Groups: Reaction Mechanisms and Applications, Photochem. Photobiol. Sci., 2002, 1(7), 441–458, 10.1039/b200777k.
- P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Photoremovable Protecting Groups in Chemistry and Biology: Reaction Mechanisms and Efficacy, Chem. Rev., 2013, 113(1), 119–191, DOI:10.1021/cr300177k.
- K. Raiber, A. Terfort, C. Benndorf, N. Krings and H.-H. Strehblow, Removal of Self-Assembled Monolayers of Alkanethiolates on Gold by Plasma Cleaning, Surf. Sci., 2005, 595(1–3), 56–63, DOI:10.1016/j.susc.2005.07.038.
- J. P. Perdew, Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33(12), 8822–8824, DOI:10.1103/PhysRevB.33.8822.
- J. P. Perdew, Erratum: Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34(10), 7406–7406, DOI:10.1103/PhysRevB.34.7406.
- A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38(6), 3098–3100, DOI:10.1103/PhysRevA.38.3098.
- F. Weigend, Accurate Coulomb-Fitting Basis Sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8(9), 1057, 10.1039/b515623h.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA Quantum Chemistry Program Package, J. Chem. Phys., 2020, 152(22), 224108, DOI:10.1063/5.0004608.
- M. Bakthadoss and A. Devaraj, Diastereoselective Construction of Highly Functionalized Tetrahydroquinolinoisoxazole Scaffolds via Intramolecular Nitrone Cycloaddition, Tetrahedron Lett., 2015, 56(25), 3954–3960, DOI:10.1016/j.tetlet.2015.05.004.
- K. K. Behara, Y. Rajesh, Y. Venkatesh, B. R. Pinninti, M. Mandal and N. D. P. Singh, Cascade Photocaging of Diazeniumdiolate: A Novel Strategy for One and Two Photon Triggered Uncaging with Real Time Reporting, Chem. Commun., 2017, 53(68), 9470–9473, 10.1039/C7CC04635A.
- R. G. Greenler, Infrared Study of Adsorbed Molecules on Metal Surfaces by Reflection Techniques, J. Chem. Phys., 1966, 44(1), 310–315, DOI:10.1063/1.1726462.
- H. A. Pearce and N. Sheppard, Possible Importance of a “Metal-Surface Selection Rule” in the Interpretation of the Infrared Spectra of Molecules Adsorbed on Particulate Metals; Infrared Spectra from Ethylene Chemisorbed on Silica-Supported Metal Catalysts, Surf. Sci., 1976, 59(1), 205–217, DOI:10.1016/0039-6028(76)90301-0.
- C. Fischer, S. Das, Q. Zhang, Y. Liu, L. Weinhardt, D. O’Hagan, M. Zharnikov and A. Terfort, Lateral Dipole Moments Induced by All-Cis-pentafluorocyclohexyl Groups Cause Unanticipated Effects in Self-Assembled Monolayers, Nano Res., 2023, 16, 11030–11041, DOI:10.1007/s12274-023-5818-4.
- R. G. Snyder, H. L. Strauss and C. A. Elliger, Carbon-Hydrogen Stretching Modes and the Structure of n-Alkyl Chains. 1. Long, Disordered Chains, J. Phys. Chem., 1982, 86(26), 5145–5150, DOI:10.1021/j100223a018.
- R. A. MacPhail, H. L. Strauss, R. G. Snyder and C. A. Elliger, Carbon-Hydrogen Stretching Modes and the Structure of n-Alkyl Chains. 2. Long, All-Trans Chains, J. Phys. Chem., 1984, 88(3), 334–341, DOI:10.1021/j150647a002.
- Y.-S. Shon and T. R. Lee, Chelating Self-Assembled Monolayers on Gold Generated from Spiroalkanedithiols, Langmuir, 1999, 15(4), 1136–1140, DOI:10.1021/la981568c.
- P. E. Laibinis, C. D. Bain, R. G. Nuzzo and G. M. Whitesides, Structure and Wetting Properties of ω-Alkoxy-n-Alkanethiolate Monolayers on Gold and Silver, J. Phys. Chem., 1995, 99(19), 7663–7676, DOI:10.1021/j100019a054.
- E. B. Troughton, C. D. Bain, G. M. Whitesides, R. G. Nuzzo, D. L. Allara and M. D. Porter, Monolayer Films Prepared by the Spontaneous Self-Assembly of Symmetrical and Unsymmetrical Dialkyl Sulfides from Solution onto Gold Substrates: Structure, Properties, and Reactivity of Constituent Functional Groups, Langmuir, 1988, 4(2), 365–385, DOI:10.1021/la00080a021.
- Y.-S. Shon, S. Lee, S. S. Perry and T. R. Lee, The Adsorption of Unsymmetrical Spiroalkanedithiols onto Gold Affords Multi-Component Interfaces That Are Homogeneously Mixed at the Molecular Level, J. Am. Chem. Soc., 2000, 122(7), 1278–1281, DOI:10.1021/ja991987b.
- R. R. San Juan and T. B. Carmichael, Formation of Self-Assembled Monolayers with Homogeneously Mixed, Loosely Packed Alkyl Groups Using Unsymmetrical Dialkyldithiophosphinic Acids, Langmuir, 2012, 28(51), 17701–17708, DOI:10.1021/la303966z.
- J. B. Schlenoff, M. Li and H. Ly, Stability and Self-Exchange in Alkanethiol Monolayers, J. Am. Chem. Soc., 1995, 117(50), 12528–12536, DOI:10.1021/ja00155a016.
- H. Kang, Y. Kim, I. Choi, R. Chang and W.-S. Yeo, Determination of Self-Exchange Rate of Alkanethiolates in Self-Assembled Monolayers on Gold Using Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry, Anal. Chim. Acta, 2014, 843, 38–45, DOI:10.1016/j.aca.2014.07.028.
- P. E. Laibinis, M. A. Fox, J. P. Folkers and G. M. Whitesides, Comparisons of Self-Assembled Monolayers on Silver and Gold: Mixed Monolayers Derived from HS(CH2)21X and HS(CH2)10Y (X, Y = CH3, CH2OH) Have Similar Properties, Langmuir, 1991, 7(12), 3167–3173, DOI:10.1021/la00060a041.
- E. Cooper and G. J. Leggett, Influence of Tail-Group Hydrogen Bonding on the Stabilities of Self-Assembled Monolayers of Alkylthiols on Gold, Langmuir, 1999, 15(4), 1024–1032, DOI:10.1021/la980795b.
- E. Delamarche, B. Michel, H. Kang and Ch. Gerber, Thermal Stability of Self-Assembled Monolayers, Langmuir, 1994, 10(11), 4103–4108, DOI:10.1021/la00023a033.
Footnotes |
| † Dedicated to George M. Whitesides on the occasion of his 85 birthday. |
| ‡ Electronic supplementary information (ESI) available: Description of the synthesis procedure; NMR spectra; full analysis of the IR data; additional contact angle goniometry data. See DOI: 10.1039/d3lf00248a |
| This journal is © The Royal Society of Chemistry 2024 |