
High-precision stable isotope measurements of tungsten and molybdenum in single sample aliquots combined with optimized separation for mixed double spikes†
Teruhiko
Kashiwabara
 *a,
Yusuke
Fukami
ab,
Sayuri
Kubo
a,
Ayako
Watakabe
a,
Minako
Kurisu
a,
Satoshi
Tokeshi
c,
Tsuyoshi
Iizuka
d and
Katsuhiko
Suzuki
a
*a,
Yusuke
Fukami
ab,
Sayuri
Kubo
a,
Ayako
Watakabe
a,
Minako
Kurisu
a,
Satoshi
Tokeshi
c,
Tsuyoshi
Iizuka
d and
Katsuhiko
Suzuki
a
aJapan Agency for Marine-Earth Science and Technology (JAMSTEC), 2-15 Natsushimacho, Yokosuka, Kanagawa 237-0061, Japan. E-mail: teruhiko-kashiwa@jamstec.go.jp
bDepartment of Chemistry, Gakushuin University, Mejiro, Tokyo 171-8588, Japan
cMarine Works Japan Ltd., 3-54-1, Oppamahigashi, Yokosuka, Kanagawa 237-0063, Japan
dDepartment of Earth and Planetary Science, Graduate School of Science, The University of Tokyo, Hongo, Bunkyo-ku, Tokyo 113-003, Japan
First published on 30th April 2024
Abstract
A key driver to develop stable tungsten (W) isotope geochemistry is its unique relationship with molybdenum (Mo). Here, we establish a combined double-spike (DS) method for W (180W–184W spike) and Mo (97Mo–100Mo spike) to perform simple, efficient, and robust isotope measurements of these two chemically analogous elements in single sample aliquots. Based on previous column chemistry, we optimized two-stage anion-exchange procedures to remove matrix elements, particularly the critical interference of Ta and Hf on 180W, and to collect sharply separated W and Mo fractions. The obtained recoveries are quantitative for both elements, and their purities are sufficiently high to achieve high-precision measurements comparable to previous DS measurements of individual elements. The reproducibility of our isotope measurements for in-house standard solutions (2SD) was ±0.02‰ for δ186W and ±0.03‰ for δ98Mo. We applied our method to 27 geological reference materials including 10 igneous rocks (AGV-2, JA-3, JR-1, JB-1, JB-1a, JB-2, JB-3, W-2a, TDB-1, and WGB-1), 9 sediments (Nod-A-1, Nod-P-1, JMn-1, JMS-1, JMS-2, CRM7302-a, HISS-1, MESS-4, and PAC-3), and 8 sedimentary and metasedimentary rocks (SDC-1, SDO-1, SBC-1, SCO-1, SCO-2, JSL-1, JSL-2, and IOC-1) to produce a comprehensive data set. The data set confirmed the accuracy of our measurements and expanded the reference materials available for interlaboratory comparisons of δ186W and δ98Mo. The data set also indicates potential pitfalls in sample preparations for particular sample types and shows several variations of W and Mo isotopes possibly related to low-/high-temperature geochemical processes. Our new method, plus the reference data set, will facilitate the development of stable isotope geochemistry for W and Mo.
1 Introduction
Recent interest in isotope studies of tungsten (W) has extended to its stable isotope fractionation in terrestrial and extraterrestrial systems.1–4 While previous W isotope studies have primarily focused on the short-lived 182Hf–182W decay system to study planetary accretion and core formation,5,6 the mass-dependent fractionation of W isotopes is an emerging tool that can potentially constrain a wide range of geochemical processes in crust/mantle systems and low temperature environments.7–13 In particular, stable W isotopes on Earth's surface are expected to fractionate to a higher degree during low temperature processes,14,15 and the W isotope compositions of seawater and some sediments exhibit significant deviations from those of mantle and crustal rocks.7,9,12,16 These initial observations of stable W isotope variations on Earth's surface have served as a basis to discuss the material cycling and processes related to weathering on the land surface, the elemental budget in the ocean, and magmatic cycles in subduction zones.8,12,17An important driver for the future development of stable W isotope geochemistry is this element's unique relationship with molybdenum (Mo). The Mo isotope system is one of the most successful examples in the past two decades of growing non-traditional stable isotope geochemistry,18,19 where its mass-dependent variations have provided well-established proxies, particularly for the oxygenation of Earth's oceans and atmosphere, and its applications have still been extending to solid Earth sciences and other areas including ore deposits, oil, and anthropogenic tracing.20–24 This wide application of Mo isotopes stems from their rich redox and coordination chemistry, both of which tend to drive significant isotope fractionations.18,25 Interestingly, many of the chemical properties of W (e.g. ionic radius and charge) are very similar to those of Mo, but slightly different reactivities in terms of both redox and coordination chemistry are known to cause distinct Mo/W ratios in some reservoirs including seawater,26,27 sediments,28,29 and biological systems.30,31 This similar but not identical chemistry of W could also cause potential isotope fractionations during various geochemical processes,32,33 which, in combination with Mo isotopes, could improve our understanding of the Earth system.
However, determining the W and Mo isotope ratios of single geological samples involves several significant challenges. First, W isotope measurements require special attention to obtain reliable isotope data because the W isotope variations in natural systems are still very small due to the small relative mass differences. The current applications of Mo isotopes stem from a number of tests for its analytical protocols including purification, mass-bias correction, and choice of standard materials, which have improved Mo isotope measurements in terms of precision, robustness, simplicity, efficiency, etc.34–43 In contrast, agreement has been reached for W isotope measurements among several different labs only for limited USGS reference materials (AGV-2, BHVO-2, Nod-A-1, and Nod-P-1), which are now commercially unavailable.3,4,17,44,45 Given the widespread interest in W isotopes, as for Mo isotopes, W isotope compositions should be measured for a wide range of reference materials with different chemical compositions to evaluate the validity of analytical protocols used in different labs including future studies.
The second challenge is to get efficiency in a series of preparations and measurements of the two elements, because two-element measurements in general require samples that are twice as large and involve twice the time and effort. The combined use of multi-element isotopes undoubtedly has advantages to constrain natural processes because the individual isotopes could add different information on the same targets depending on their own chemistry.46–51 However, in many cases, this could lead to the loss of precious samples and increased effort because individual elements must be purified via different chemical procedures to avoid matrix effects in the measurements. In particular, W and Mo are both soluble oxyanions in oxic aqueous phases, and their abundances are generally low in rocks and sediments.52,53 Thus, large amounts of samples have to be processed in a series of chemical treatments, which could also induce potential artifacts such as incomplete digestion, recovery, and contamination.42 Furthermore, their separate digestions are susceptible to chemical and/or sample heterogeneities, which could preclude accurate comparisons of the small isotope variations of the two elements.
Recently, Tsujisaka et al. (2019)44 made an important contribution to compare W and Mo isotope data in single sample aliquots. This work has particular significance in (i) presenting the efficient separation of the two chemically analogous elements in the same procedure to compare their isotope data from identical aliquots, and (ii) expanding the available data sets of W and Mo isotopes by measuring 12 reference materials to allow further tests of analytical protocols in different labs. However, their method could expect further improvements in terms of simplicity and robustness. First, their separation protocols, a combination of chelating resin extraction using a pump system and subsequent anion exchange, are operationally complex and require specific setups to deal with a wide range of samples.16,44 Second, standard-sample bracketing with external corrections using Ru and Re in their measurements, which were chosen as a consequence of their separation, could still suffer from potential artificial mass fractionations during sample preparation and instrumental analysis. Third, they actually showed a significant discrepancy between their results obtained with and without using a desolvating nebulizer; they suggested that such a device should not be used with their external correction method, although it is commonly used to enhance analytical sensitivity.
Here, we propose the simultaneous application of a double spike (DS) technique to W and Mo isotope measurements by optimizing previous separation procedures for the two elements. The DS method provides a critical advantage for obtaining reliable isotope data that are accurately corrected for mass fractionations during chemical treatment and measurements by spiking with two artificially enriched isotopes of the same elements.54 Compared with the external correction by element doping and standard-sample bracketing, additional practical benefits include the following: (i) the DS method is not dependent on a priori assumption of perfect matrix matching of samples to standards and quantitative recovery in the chemical treatment; (ii) natural fractionations can be resolved from instrumental mass bias in exactly the same analyses without time-consuming analysis of a larger number of standards; (iii) the method can also provide concentrations to a precision hardly achievable by other methods.41,55 Thus, if the DS method could be successfully applied to W and Mo in a single procedure, isotope measurements of the two elements in the same sample could be markedly improved in terms of simplicity and robustness.
The aim of this study is to establish a simple, efficient, and robust method for highly precise and accurate measurements of W and Mo isotopes in single sample aliquots. We optimized two-stage anion-exchange procedures to combine the DS methods of W and Mo isotopes, where their separations from the matrix were carefully evaluated and controlled to obtain four isotopes that are free from spectral overlaps, respectively. Then, we applied the method to determine W and Mo isotope compositions in 27 geological reference materials, where some of the isotope data for W, Mo, or both are presented for the first time. The comprehensive data sets obtained here demonstrate the utility of our method and extend the basis for interlaboratory comparisons of the measurements of W and Mo isotopes. Our new method, plus the reference data set, will contribute to clarifying how W isotopes behave in the Earth system relative to Mo isotopes, and facilitate the development of the stable isotope geochemistry of W and Mo.
2 Experimental
2.1 Preparation of 180W–184W and 97Mo–100Mo double spikes
The precision of the DS technique greatly depends on the spike compositions and the spike–sample ratios.56,57 We chose 180W–184W for the W double spike and 97Mo–100Mo for the Mo double spike with 183W and 186W, and 95Mo and 98Mo for their inversions, respectively. This choice of the W spike provides the lowest analytical errors and a wide range of optimal spike/sample ratios in theoretical calculations using the double spike toolbox56 (Fig. S1†); in addition, the exclusion of 182W allows the determination of mass-dependent W isotope variations from a single measurement while avoiding the mass-independent variations of radiogenic 182W. However, the low natural abundance of 180W (∼0.12%) can cause several difficulties (i) in precise and accurate determination of 180W in the reference material (NIST SRM3163) during spike calibration, (ii) in obtaining a pure 180W spike, and (iii) due to serious isobaric interference from 180Hf (35.08%) and 180Ta (0.012%) for natural samples. Therefore, efficient chemical purification of 180W is critically important, and was carefully examined in this study. For Mo, we followed the common choice of the Mo spike in many previous studies because it allows low error magnification during the inversion, availability of those isotopes at a high purity, and is the least affected by isobaric interference.35,41,42Single spikes of 180W, 184W, 97Mo, and 100Mo were purchased from Oak Ridge National Laboratory (ORNL, USA) in the forms of metal (batch no. 128482 for 184W; 159791 for 97Mo; 4085882 for 100Mo) and oxide (batch no. 128101 for 180W) powders. About 10 mg powders were weighed separately in pre-cleaned PFA bottles (Savillex, USA); then, the metals were digested in 2 mL of 35% H2O2 (TAMA pure-AA-10 grade, Tama Chemical Co. Ltd, Japan) and the oxide was digested in 2 mL of 20% NH3 (TAMA pure-AA-100 grade, Tama Chemical Co. Ltd, Japan). The solutions were evaporated to complete dryness at 90 °C, re-dissolved in 2 M HNO3–0.5 M HF, and agitated for 30 min in an ultrasonic bath. These single spike solutions were gravimetrically mixed to prepare the double spike solutions of W and Mo, with solution proportions of 71.14% 180W and 28.86% 184W, and 37.32% 97Mo and 62.68% 100Mo, following the optimal 180W/184W and 97Mo/100Mo ratios calculated using the double spike toolbox.56 The prepared double spike solutions were evaporated to dryness at 90 °C, re-dissolved in 2 M HNO3–0.5 M HF, and agitated for 30 min in an ultrasonic bath four times. These treatments were repeated twice to ensure the complete equilibrium of isotopic exchange in the solutions. Finally, the double spike solutions were stored in 2 M HNO3–0.5 M HF following Krabbe et al. (2017).3
The isotopic compositions and concentrations of the double spikes were calibrated relative to NIST 3163 solution (lot no. 080331) for W and NIST 3134 solution (lot no. 130418) for Mo by multicollector inductively coupled plasma mass spectrometry (MC-ICP-MS). The measured isotope ratios of the NIST standard solutions were normalized to 0.92767 for 186W/184W58 and to 1.00313 for 100Mo/97Mo41 using the exponential law. The accuracy of the double spikes and their robustness to over-/under-spiking were evaluated by measuring mixtures of DS and NIST standard solutions at various DS/NIST ratios from 0.1 to 10 (p = DS/(NIST + DS) ≈ 0.1–1). The δ186/184W and δ98/95Mo values obtained for all the different mixtures were identical to those of an optimal DS/sample ratio (0.822 for W and 0.936 for Mo) within analytical uncertainties (Fig. S2†), indicating that the calibrations of the double spikes are accurate over a wide range of DS/sample ratios.
2.2 Geological reference materials
We examined 27 geological reference materials provided by the United States Geological Survey (USGS), the Geological Survey of Japan (GSJ), the National Research Council Canada (NRC), the National Metrology Institute of Japan (NMIJ), and the Canadian Certified Reference Materials Project of the Canada Centre for Mineral and Energy Technology (CANMET-CCRMP). The materials include 10 igneous rocks (andesites: AGV-2 and JA-3; rhyolite: JR-1; basalts: JB-1, JB-1a, JB-2, and JB-3; diabase: W-2a and TDB-1; gabbro: WGB-1), 9 sediments (manganese nodules: Nod-A-1, Nod-P-1, and JMn-1; marine sediments: JMS-1, JMS-2, CRM7302-a, HISS-1, MESS-4, and PAC-3), and 8 sedimentary and metasedimentary rocks (mica schist: SDC-1; shales: SDO-1, SBC-1, SCO-1, and SCO-2; slates: JSL-1 and JSL-2; iron ore: IOC-1). Sample descriptions including collection sites, processing, and chemical information are provided in reference data from each provider and the literature cited in this paper.2.3 Sample digestion
After drying overnight at 110 °C, powdered samples were weighed in a 15 mL PFA vessel (Savillex, USA) depending on the W or Mo concentration, and adequate amounts of the 180W–184W and 97Mo–100Mo double spikes were added to adjust the spike/sample ratios to 0.822 for W and 0.936 for Mo, respectively.56 Because the Mo/W ratios are mostly larger than 1 (as shown in Section 3.4), the minimum amount of the samples required for digestion was determined based on the W concentration to collect more than ca. 200 ng of W after spiking. Most of the samples weighed 5–300 mg with the DS added and were digested and split into three solutions to run chemical separation and measurements as replicates. Some samples containing <0.3 μg g−1 W (HISS-1, JB-2, W-2a, and TDB-1) or Mo (SDC-1) were digested as three individual solutions (300–500 mg aliquots). The samples were first digested with a mixture of 0.4 mL HCl (30 vol%), 4 mL HF (38 vol%), and 3 mL H2O2 (35 vol%) at 50 °C for 13 h, in which HCl, HF, and H2O2 are effective to digest the matrices of Fe oxides, SiO2, and Mn oxides, respectively. Then, the solutions were evaporated to dryness at 90 °C, followed by sequential addition of 2 mL HNO3 (68 vol%), 0.5 mL HCl (30 vol%), 7 mL ultrapure water, and 0.1 mL HF (38 vol%). We paid attention to this sequence to avoid the potential loss of W or Mo by precipitation of fluoride. After agitation in an ultrasonic bath for 30 min, the digested samples were heated at 90 °C in closed vessels for more than 12 h to reach isotope equilibrium in the solution, and were then evaporated to dryness at 90 °C. Some samples still showed the presence of blackish organic materials; these remaining materials were decomposed in a mixture of 500 μL HClO4 and 600 μL HNO3 by heating at 160 °C for more than one night, and were then evaporated to complete dryness at 195 °C. Finally, the samples were dissolved in 10 mL of 0.6 M HF–0.14% H2O2, agitated four times in an ultrasonic bath for 30 min, and then filtered through a membrane filter of 0.2 μm pore size (ADVANTEC, Japan) to obtain the solution in 0.6 M HF–0.14% H2O2 for subsequent chemical separation procedures. The final solutions using H2O2 were prepared less than 12 h before the separation procedures to avoid variation caused by H2O2 in the elution processes through, probably, bubble formation in the column, as mentioned in several previous studies.59–612.4 Chemical separation
A two-stage separation procedure using an anion-exchange resin (AG-1 X8, 200–400 mesh, Biorad, USA) was developed based on the previous single-step procedure of Irisawa and Hirata (2006)62 as a guide (Table 1). Because some studies using an AG1-X8 resin reported relatively high blanks of Mo,42,63 the resin was carefully washed sequentially with 1 M NH4NO3, 5 M HNO3–5 M HF, and 4 M HCl–5 M HF and rinsed with ultrapure water between each wash. Each step was left to stand overnight and was repeated three times; then, the resin was stored in 4 M HCl. Before the separation procedure, about 2 mL resin was charged in a polyethylene column (2 mL bed volume with 39 mm height, Bio-Rad, USA) and cleaned successively with 10 mL of 2 M HF–4 M HCl, 10 mL of 4 M HNO3, and 10 mL of MQ water. Then, conditioning of the resin was conducted by loading 5 mL of 2 M HF.| Step | Regents | Volume/mL |
|---|---|---|
| Cleaning | 2 M HF–4 M HCl | 10 |
| 4 M HNO3 | 10 | |
| Pure water | 10 | |
| Conditioning | 2 M HF | 5 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||
| First column | ||
| F11: sample loading | 0.6 M HF–0.14% H2O2 | 10 |
| F21: removal of matrix | 1 M HCl–0.6% H2O2 | 15 |
| F31: removal of matrix | 4 M HCl | 7.5 |
| F41: elution of Mo and W | 4 M HNO3 | 20 |
|
||
| Second column | ||
| F12: sample loading | 0.6 M HF–0.14% H2O2 | 10 |
| F22: removal of matrix | 1 M HCl–0.6% H2O2 | 12 |
| F32: removal of matrix | 0.5 M HF–4 M HCl | 5 |
| F42: elution of W | 1 M HF–6 M HCl | 20 |
| F52: elution of Mo | 0.1 M HF–4 M HNO3 | 10 |
The first column was designed to recover W and Mo in the same fraction and separate them from unwanted elements (Table 1, F11–F14). The sample solution (10 mL) in 0.6 M HF–0.14% H2O2 was loaded onto the resin with a 2.5 mL step (F11). Then, the matrix and other unwanted elements including Fe, Mn, Ti, Zr, Nb, Ru, Hf, and Ta were removed by sequentially loading 15 mL of 1 M HCl–0.6% H2O2 (F21) and 7.5 mL of 4 M HCl (F31). After the removal of the matrix elements, Mo and W were eluted in the same fraction with 20 mL of 4 M HNO3 (F41). Organic materials from the resin in the obtained Mo and W solution were carefully decomposed by the addition of 400 μL HClO4 and heating at 160 °C for 12 h to dryness, followed by the further addition of 200 μL HNO3 and 200 μL HClO4 and heating at 190 °C for 5 h to remove them completely. The procedures using HClO4 and its complete dryness were necessary to obtain high recovery of Mo and W, as mentioned in several previous studies.62,64 The resulting sample cakes were re-dissolved in 0.6 M HF–0.14% H2O2 and were agitated for 30 min in an ultrasonic bath to prepare the solution for the subsequent column procedure. Again, the procedure using H2O2 was conducted less than 12 h before the second separation procedure.
The second column was designed to recover W and Mo in the different fractions (Table 1, F12–F52). The sample loading and matrix removal procedures are mostly the same as for the first column, which serves for further purification of Mo and W from the remaining matrix elements. The important difference from the first column is the use of HF from F32 to F52, where the addition of HF serves to separate W from Mo sharply in the subsequent fractions, and also to remove Ta by retaining it on the resin in the presence of HF.63 Then, W was eluted with 20 mL of 1 M HF–6 M HCl (F42), and Mo was subsequently eluted with 10 mL of 0.1 M HF–4 M HNO3 (F52). Again, organic materials from the resin were carefully decomposed by HClO4 additions (400 μL for F42 and 200 μL for F52) and heating at 160 °C for 12 h to dryness. After the further addition of 200 μL HNO3 and 200 μL HClO4, the solutions were completely evaporated by heating at 190 °C for 5 h; then, the sample cake of the W fraction was re-dissolved in 2% HNO3–0.05% HF and that of the Mo fraction was re-dissolved in 2% HNO3. The sample solutions were agitated for 30 min in an ultrasonic bath and diluted to 50 ng g−1 for isotope measurements.
2.5 Mass spectrometry
Isotope measurements were conducted by MC-ICP-MS (Neptune Plus, Thermo Fisher Scientific, Germany) at the Japan Agency for Marine-Earth Science and Technology (JAMSTEC). The RF power was 1200 W and the flow rates of the cooling, auxiliary, and nebulizer gases were adjusted to 16, 0.8, and 0.8 L min−1, respectively. The samples were introduced into the plasma using a self-aspirating PFA nebulizer with an aspiration rate of ca. 100 μL min−1 connected to an Aridus II desolvating system (Teledyne CETAC Technologies, USA). The flow rates of the sweep Ar gas and additional N2 gas were controlled to maximize the signal intensities for W and Mo. Measurements were performed in low-resolution mode. Faraday cups with 1011 (or 1012) Ω resistor amplifiers were assigned as shown in Table 2. On-peak background subtraction was performed using beam intensities that were measured by introducing blank solutions of 2% HNO3–0.05% HF for W and 2% HNO3 for Mo before performing the sample run. These background measurements consist of a single block of 40 cycles with an integration time of 4.2 s. The mass-bias-corrected isotope ratios were obtained by solving the DS equations iteratively using the Newton–Raphson method following Rudge et al. (2009).56| Faraday cup | L4 | L3 | L2 | L1 | C | H1 | H2 | H3 | H4 |
|---|---|---|---|---|---|---|---|---|---|
| Amplifier (Ω) | 1012 | 1011 | 1011 | 1011 | 1011 | 1011 | 1011 | 1011 | 1011 |
| Monitored mass for W | 178 Hf | 180 W | 181 Ta | 182 W | 183 W | 184 W | 186 W | 188 Os | 189 Os |
| Isobars | 180Hf, 180Ta | 184Os | 186Os | ||||||
| Molecular interferents | 179HfH | 181TaH | 182WH | 183WH | 185ReH | ||||
| Natural abundance (%) | |||||||||
| W | 0.12 | 26.50 | 14.31 | 30.64 | 28.43 | ||||
| Hf | 27.28 | 35.08 | |||||||
| Ta | 0.01 | 99.99 | |||||||
| Os | 0.02 | 1.59 | 13.24 | 16.15 | |||||
| Monitored mass for Mo | 91 Zr | 92 Mo | 94 Mo | 95 Mo | 96 Mo | 97 Mo | 98 Mo | 99 Ru | 100 Mo |
| Isobars | 94Zr | 96Zr, 96Ru | 98Ru | 100Ru | |||||
| Molecular interferents | 40Ar55Mn | 40Ar56Fe | 40Ar57Fe | 40Ar58Fe | |||||
| Natural abundance (%) | |||||||||
| Mo | 14.53 | 9.15 | 15.84 | 16.67 | 9.60 | 24.39 | 9.82 | ||
| Zr | 11.22 | 17.15 | 17.38 | 2.80 | |||||
| Ru | 5.54 | 1.87 | 12.76 | 12.60 | |||||
For the W isotope measurements, a Ni jet-sampler cone and a Ni X-skimmer cone were used. The flow rates of the sweep Ar and additional N2 gases were adjusted to ca. 5.5 L min−1 and 1 mL min−1, where the typical instrumental sensitivity was 800–1100 V per 1 μg g−1. The signal intensities of 180W, 182W, 183W, 184W, and 186W were collected in static mode with 178Hf, 181Ta, 188Os, and 189Os to correct the isobaric interference of 180Hf and 180Ta on 180W, 184Os on 184W, and 186Os on 186W, respectively. Data were acquired in five blocks of 20 cycles with an integration time of 4.2 s. After each sample run, a washout was performed with 1 M HNO3–0.5 M HF for 11.2 min followed by 2% HNO3–0.05% HF for 14 min. The δ186/184W values of the samples were calculated relative to the NIST SRM 3163 standard solution (lot no. 140606) as:
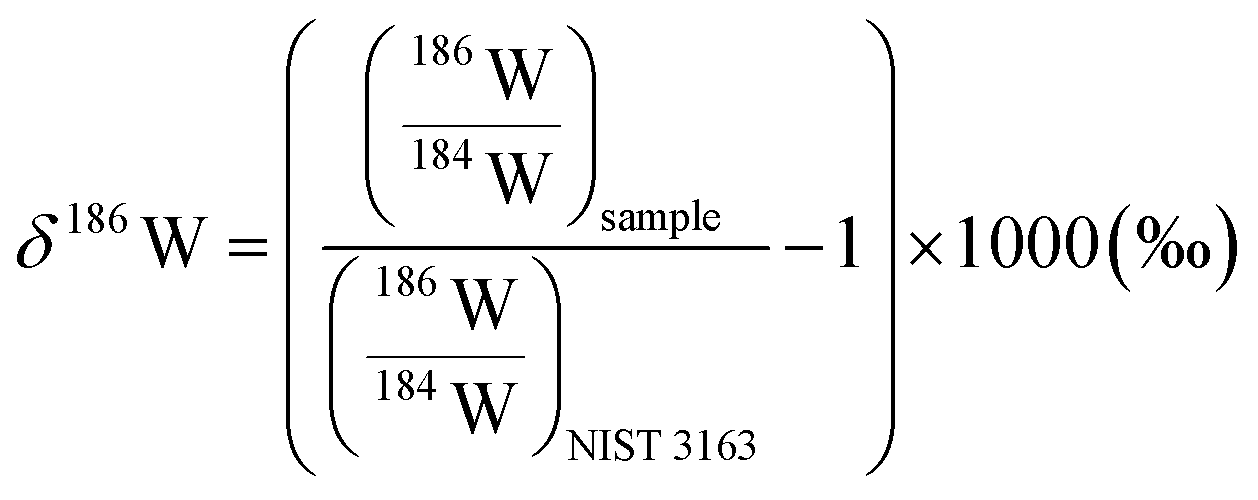
For the Mo isotope measurements, a Ni normal-sampler cone was used in combination with a Ni X-skimmer cone. The flow rates of the sweep Ar and additional N2 gases were adjusted to ca. 4.9 L min−1 and 2 mL min−1, and the typical instrumental sensitivity was 900–1300 V per 1 μg g−1. Data were acquired in five blocks of 16 cycles with an integration time of 4.2 s. The masses of 92Mo, 94Mo, 95Mo, 96Mo, 97Mo, 98Mo, and 100Mo were collected in static mode with monitoring of 91Zr and 99Ru to check for potential interference of 98Ru, 100Ru, 94ZrH, and 96ZrH. After each sample run, a washout was performed with 1 M HNO3–0.5 M HF for 8.4 min, followed by 2% HNO3 for 19.6 min. The δ98/95Mo values were calculated relative to the NIST 3134 SRM standard solution (lot no. 130418) as follows:
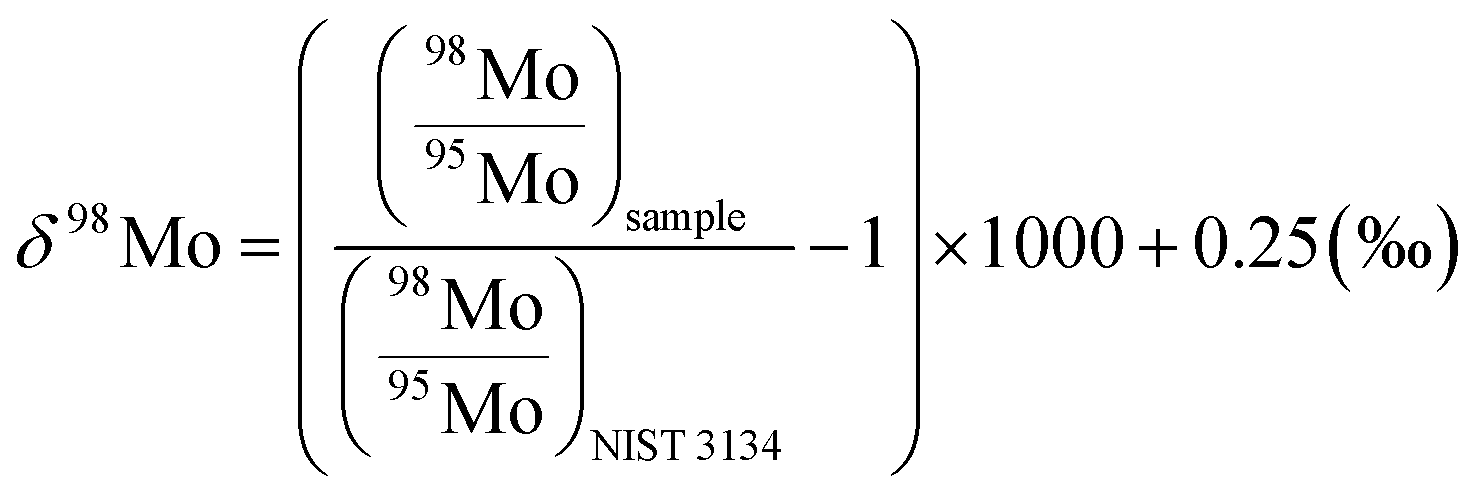
Among the previous efforts35,65 to deal with the absence of universal reference materials to express the Mo isotope ratio, we followed the definition by Nägler et al. (2014)40 because this definition meets the recommendation by IUPAC66 to use NIST SRM3134 as an anchor point for δ98/95Mo and also allows direct comparison with previous data with the canonical δ98Mo values of seawater (+2.3‰) and marine ferromanganese oxides (−0.7‰) by defining its δ98Mo value as + 0.25‰.
Instrumental calibrations were conducted by measuring the spiked in-house standard solutions of W from Kanto Chemicals (lot no. 104K9519) and Mo from Spex (lot no. 25-55MOY) relative to their NIST solutions above. Repeated measurements of these solutions over analytical sessions provided an external reproducibility of δ186W = 0.00 ± 0.03‰ (2SD, n = 51) for NIST SRM3163, δ186W = 0.08 ± 0.02‰ (2SD, n = 9) for Kanto Chemicals, δ98Mo = 0.00 ± 0.04‰ (2SD, n = 31) for NIST SRM 3134, and δ98Mo = −0.39 ± 0.03‰ (2SD, n = 6) for Spex solutions.
3 Results and discussion
3.1 Elution profiles
Fig. 1a and b show elution profiles for a manganese nodule (JMn-1, 10 mg) with some interfering elements (1 μg each of Nb, Ru, Hf, and Ta) added. During the loading of the sample solution (F11), major elements such as Mn and Fe directly passed through the column. In this step, W and Mo and many interfering elements (Ti, Zr, Nb, Hf, and Ta) were strongly retained on the resin by complexing with HF and/or H2O2.59,61,67–69 Large parts of the interfering elements other than Ta were eluted by flushing with 10 mL of 1 M HCl–0.6% H2O2 in F21. This molarity of HCl was effective in removing a significant amount of the remaining Fe in the column,62 although a higher molarity of HCl could be more effective to elute some elements such as Nb and Ta based on their partition coefficients.63,68 Instead, we found that the addition of another 5 mL of the same solution in F21 provided critical improvement for the removal of Ta. Further removal of the matrix was performed in F31 by flushing with 4 M HCl. This step was slightly modified from the use of 5 mL of 4 M HCl–0.1 M HF in Irisawa and Hirata (2006)62 because no use of HF in this fraction was effective in eluting the remaining high field strength elements (HFSEs) while retaining Mo on the resin.63,70 Nevertheless, the HCl solutions in F31 could relatively mobilize W on the resin,70 so this flushing was limited to 7.5 mL to suppress the breakthrough of W. Then, W and Mo were eluted together with 4 HNO3 and collected (F41).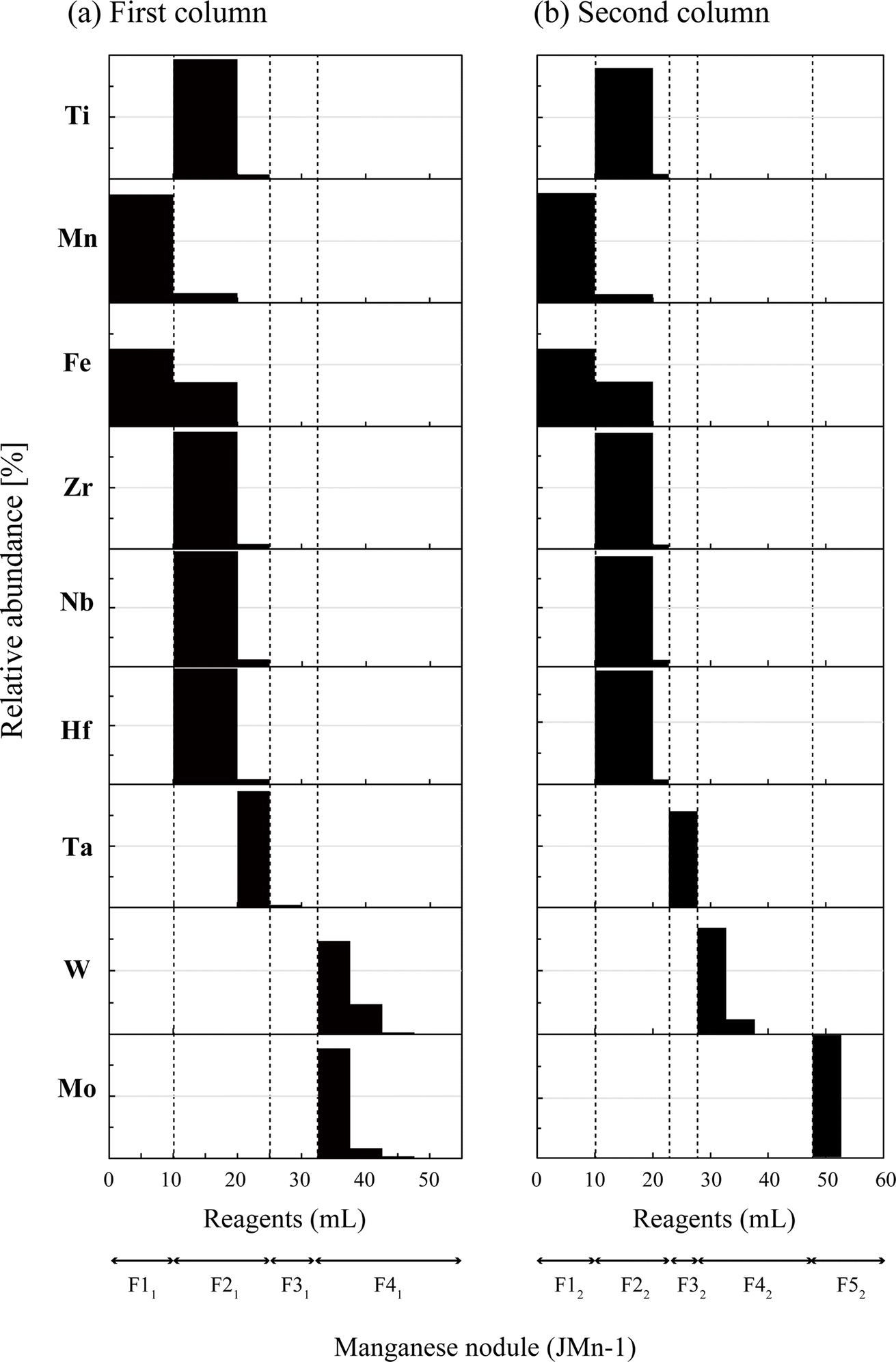 | ||
| Fig. 1 Elution profiles obtained using an anion exchange resin (AG1-X8) for the solution of a manganese nodule (JMn-1, 10 mg) with added interfering elements (1 μg each of Nb, Ru, Hf, and Ta): (a) the first-column and (b) the second-column procedures. The elution of Ru is not shown because it was completely removed during digestion and evaporation procedures. | ||
The second column (Fig. 1b) was tested using a newly prepared JMn-1 solution with the same interfering elements added to clearly illustrate the elution profiles. As with the first column (Fig. 1a), matrix elements including Ti, Mn, Fe, Zr, Nb, Hf, and Ta were eluted sequentially with F12–F32 (Fig. 1b). Differences from the first column are (i) the smaller volume of 1 M HCl–0.6% H2O2 (F22, 12 mL) relative to F21 (15 mL) and (ii) replacement of 7.5 mL of 4 M HCl (F31) with 5 mL of 4 M HCl–0.5 M HF (F32). Both modifications were consequences of the optimization to promote the subsequent separation of W and Mo based on their different affinities to the resin.63,68 However, these modifications reduced the Ta elution in F32 to ca. 80%, whereas the elution was more than 94% in F21 in the first column. Further removal of Ta from the W and Mo fractions was achieved by adding HF in F42 and F52 because Ta is strongly retained on the resin in the presence of HF.63 Then, we finally collected the sharply separated W fraction (F42) and the following Mo fraction (F52) as purified individual fractions.
We also tested the elution profiles for two other samples (Fig. S3A:† AGV-2 and B: an artificially mixed solution) and found that both showed profiles consistent with those of the manganese nodule (Fig. 1). These results indicate that our separation method works similarly irrespective of the markedly different sample matrices, which could be advantageous in terms of (i) dealing with a wide range of geological samples and (ii) the capability of processing total samples on the column. This robustness for the matrix is derived from the simple repetition of the anion-exchange separation, where the majority of the matrix elements pass directly through the column without strong interaction with the resin, in contrast to the cation-exchange separation, for which sufficient ion exchange sites must be present to accommodate the matrix ions to be removed.71–74
Our development of the W separation procedure in Irisawa and Hirata (2006)62 provides some consequent benefits for obtaining pure W and Mo fractions. First, the HF/H2O2-based W separation also works for the effective separation of Mo from Fe because of their different solution chemistries. This procedure could solve a common difficulty in dealing with Fe-rich samples by the traditional HCl-based Mo separation using an anion-exchange resin, where Mo and Fe show similar elution.21,34 Second, the addition of H2O2 and HClO4 in the procedures promotes the loss of Ru and Os as volatiles,74 which could otherwise be critical isobaric interferents for DS measurements of Mo and W. We found the complete loss of Ru and Os in the final F42 and F52 fractions by using HClO4 in a series of chemical procedures.
3.2 Recovery, purity, and procedural blank
The total recovery of W and Mo through the series of procedures was almost 100% for the three standards (JMn-1, AGV-2, and mixed solution) (Table 3). In contrast, the matrix elements were significantly reduced at each separation step, and they were finally reduced to 10−1 to 10−5% relative to their initial weights (Fig. 2, Tables S1 and S2†). The first column effectively removed most of the matrix elements relative to W and Mo, but the removal of Fe may not have been sufficient for manganese nodules (JMn-1) because Fe was still comparable to W and Mo in the collected F41 solution. The second column was effective for further purification, and the relative concentrations of matrix elements in the final W and Mo fractions (F42 and F52) were reduced to X/W < 10−4 to 10−6 and X/Mo < 10−3 to 10−7, respectively (Fig. 2, Tables S1 and S2†).| W [%] | Mo [%] | |
|---|---|---|
| JMn-1 | 99.6 ± 6.7 | 101.3 ± 7.7 |
| AGV-2 | 102.3 ± 7.2 | 99.2 ± 6.9 |
| Artificial solution | 97.8 ± 7.4 | 100.9 ± 6.8 |
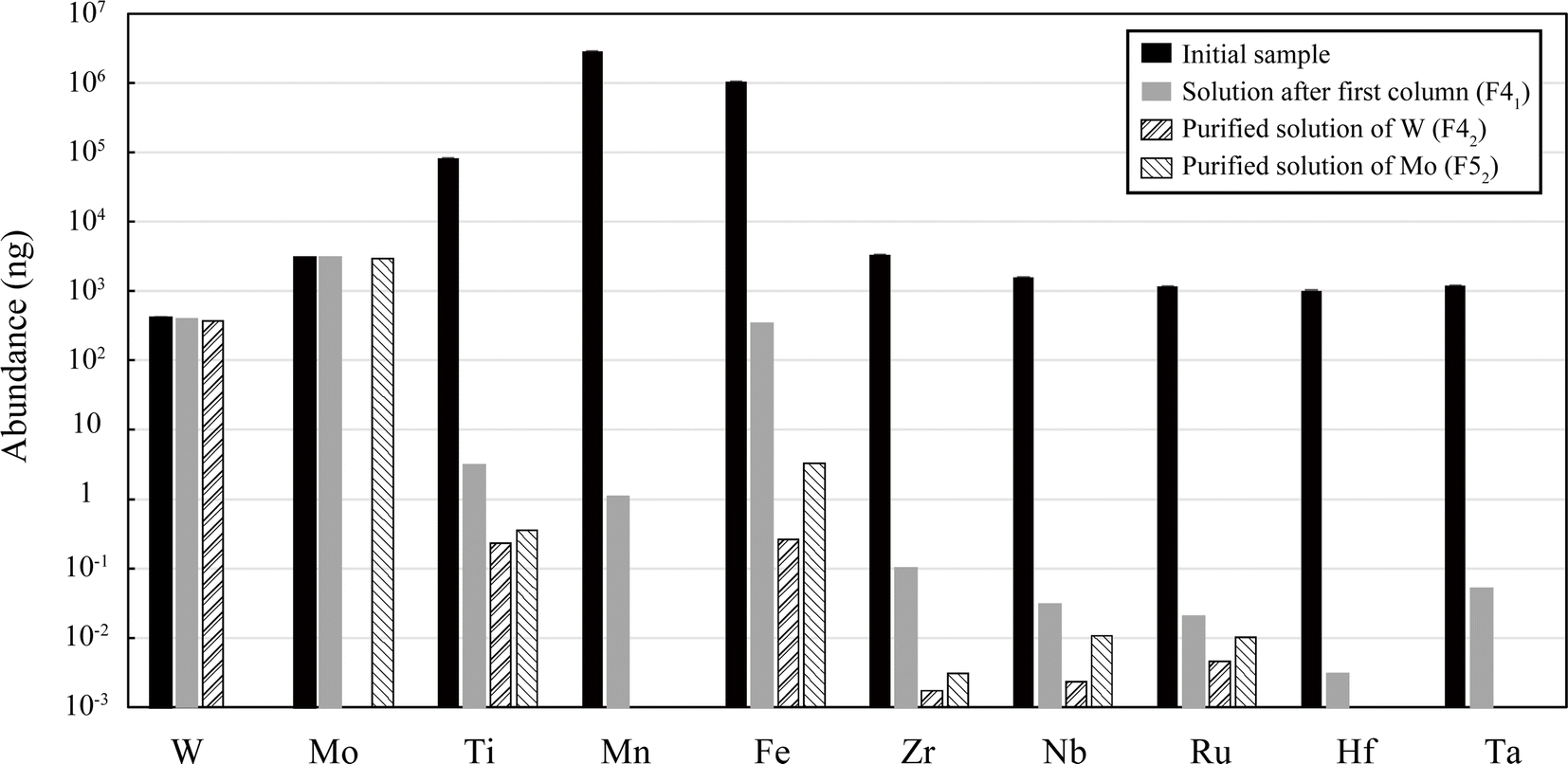 | ||
| Fig. 2 Amounts of matrix elements in a solution of manganese nodule (JMn-1, 10 mg) with added interfering elements (1 μg each of Nb, Ru, Hf, and Ta) and in the solutions after each separation step using AG1-X8. | ||
The recovery yields of W and Mo achieved in this method seem to be higher relative to those obtained in other DS studies, where the priority was given in general to ensure high-purity rather than recovery yield.1,3,4,12,17,45 We found that some steps are important to achieve quantitative recovery in our procedures. First, the use of HClO4 to decompose the resin-derived organic materials prevents the loss of W during the dry-down procedures. Some W studies inferred the partial loss of W through the evaporation step because of (i) co-precipitation with Ca/Mg-fluorides, (ii) formation of insoluble W species, and (iii) adsorption on HFSE hydroxides or organic materials.2,61,75 However, we obtained high recovery when HClO4 was completely dried and attention was paid to avoid sample loss due to static electricity, as mentioned in previous studies.62 Second, H2O2 should be used carefully, because it can have complex effects on the column chemistry of W and Mo via (i) promoting their dissolution, (ii) increasing their distribution coefficient to the resin, and (iii) forming air bubbles within the column and slowing the elution process.59,61 We confirmed that the variation in recovery yields caused by H2O2 can be minimized by controlling the timing of sample preparation prior to loading on the column.
On the other hand, the purities of the final W and Mo fractions obtained using this method seem to be sufficiently high, because they are comparable to the results obtained in previous DS studies of individual elements.3,4,45 In particular, Hf and Ta must be reduced to Hf/W < 10−4 and Ta/W < 10−2, respectively, because the isobaric interference of 180Hf and 180Ta on 180W is serious for DS measurements using the 180W spike.1,4,76 These requirements were sufficiently achieved by our method (Ta/W and Hf/W < 10−6; Table S2†). The purity of the Mo fraction also seems to be high enough because Zr, Ru, and Fe were all reduced to X/Mo ≈ 10−3 to 10−6. The effective removal of specific elements such as Ta and Ru, which did not receive sufficient attention in previous studies, is critically important for the application of the DS method to W and Mo isotopes.44,63
We obtained procedural blanks in a series of procedures without geological samples: they were for 0.04 ± 0.01 ng for W and 0.25 ± 0.08 ng for Mo (2SD, n = 10). These values are less than 0.5% of W and Mo in the samples at maximum because we collected at least 50 ng of W and Mo in the purified solutions. We confirmed that the majority of the blanks for W and Mo were, on one hand, derived from the resin; therefore, the careful washing prior to the storage and the separation procedures, as described in the Experimental section, is effective in reducing their contributions. On the other hand, we sometimes found significant blanks for Mo (up to 50 ng) derived from Teflon vials (Savillex, USA), even after repeated careful washing. Some studies reported high blanks of W for Teflon vials,74,77 but this was not observed here. In any case, it is necessary to watch the blank contribution of Teflon vials for Mo (and W) to pick up low-blank vials for the experiments.
3.3 Validation of the procedures from isotope measurements
We examined a series of our procedures in terms of instrumental requirements by performing isotope measurements. First, we compared δ186W (Fig. 3A) and δ98Mo (Fig. 3B) among (a) chemically unprocessed NIST solutions with individual additions of the W or Mo DS, (b) the processed solutions of (a), and (c) processed solutions with mixed additions of the W and Mo DS, and confirmed that the isotope ratios of the three solutions were identical within error. We also checked the consistency of secondary standard solutions between (d) unprocessed solutions with individual additions of the W or Mo DS and (e) processed solutions with mixed additions of the W and Mo DS. These agreements demonstrate that (i) potential isotope fractionations during the chemical separation and the measurement were properly corrected by the DS method, and (ii) the mixing of the W and Mo DS does not alter the isotope ratios in the presence of a small impurity of Mo in the W DS or vice versa, although they are chemically analogous elements.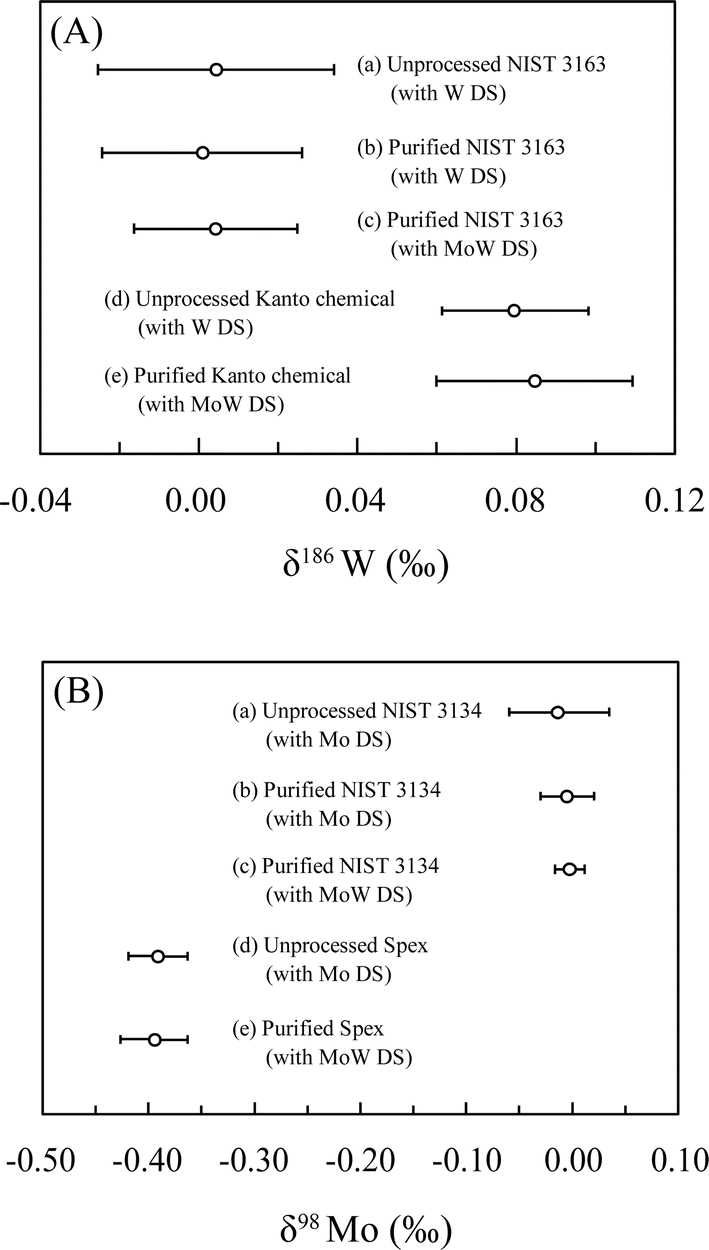 | ||
| Fig. 3 Comparison of (A) δ186W and (B) δ98Mo values of standard solutions to test the effects of the chemical separation and the mixing of the W and Mo DS. Standard solutions used for W were NIST 3163 and in-house Kanto Chemical solutions; those for Mo were NIST 3134 and in-house Spex solutions. Error bars are 2 SD from repeated measurements of the same solutions (n = 3). | ||
We examined the effects of spectral/non-spectral interference on the isotope measurements by analyzing NIST solutions doped with some key elements in various concentration ratios (Fig. 4). For the W-DS measurements, special care must be taken with respect to isobaric interference on 180W (180Hf and 180Ta), 184W (184Os), and 186W (186Os). In our test, we obtained correct values for the NIST solution for Hf/W < 10−6 and Ta/W < 10−3 without any corrections for their interference, and the corrections worked well up to Hf/W < 10−4 and Ta/W < 10−2 (Fig. 4a and b), as suggested by previous studies.1,4,76 The data indicate that it is practically unnecessary in our method to perform any corrections because the purities after the separation procedures are already less than 10−6 for both Hf/W and Ta/W (Table S2†). We did not examine Os interference because no measurable effects were observed based on the signals monitored for 188Os (188Os/184W < 10−6).
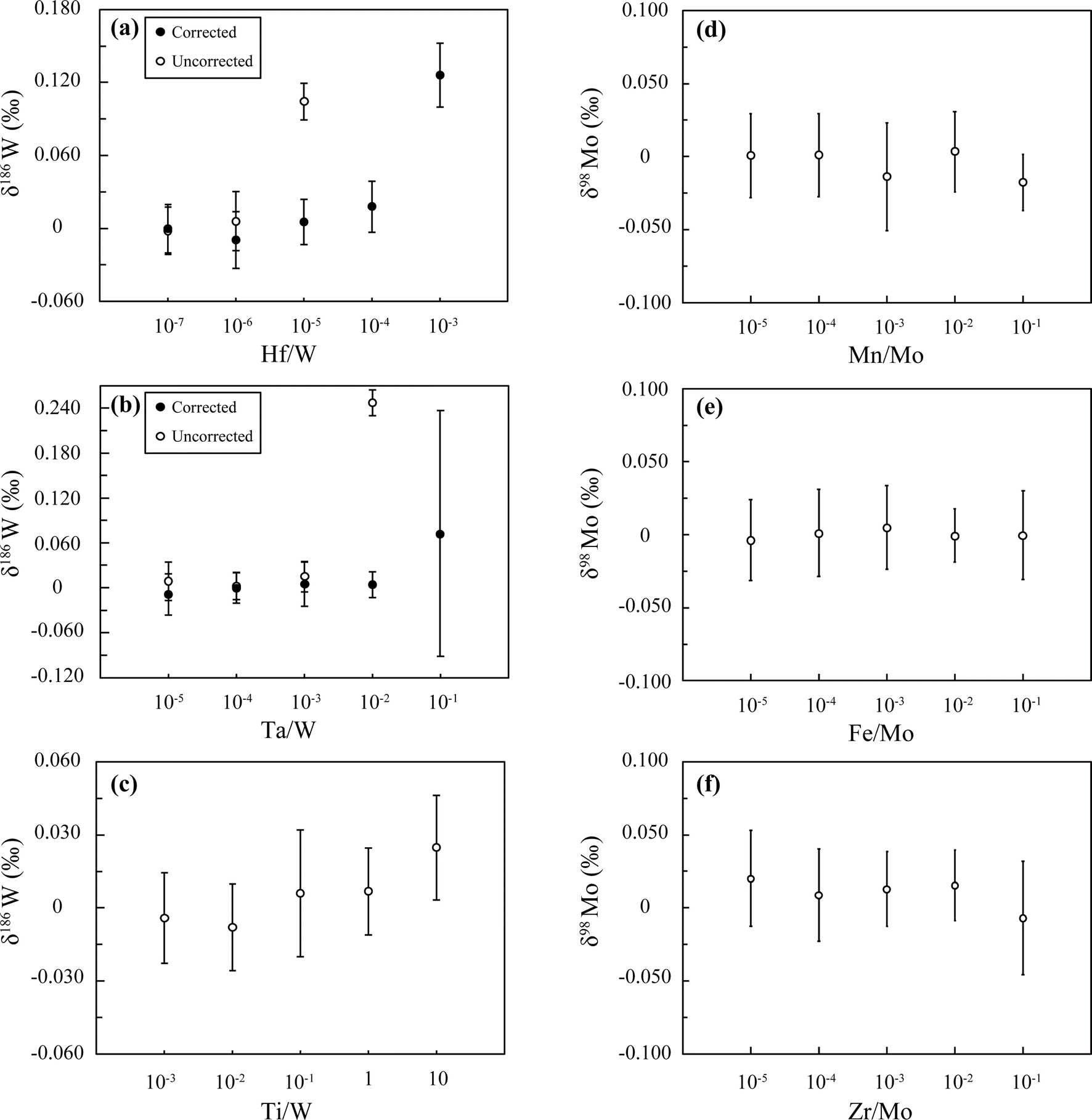 | ||
| Fig. 4 (a–c) δ186W and (d–f) δ98Mo values of NIST standard solutions doped with different elements in varying concentration ratios with W and Mo, respectively. White circles are uncorrected for interference; black circles are corrected for the interference of (a) Hf and (b) Ta on 180W. Error bars are 2 SD from repeated measurements of the same solutions (n = 3). | ||
On the other hand, we should also pay attention to the presence of Ti because this element also forms anionic complexes in HF-bearing media, as do W and Mo, which could preclude its complete removal from the W and Mo fractions. Although Ti has no significant spectral interference, it could practically cause inaccurate isotope measurements by suppressing ionization efficiencies or decreasing instrumental sensitivity over time.60,69 We found a slight increase of the δ186W values of the NIST solution when Ti was added above Ti/W > 10−1 (Fig. 4c), although this is much higher than that in our purified solutions (Ti/W or Ti/Mo ≈ 10−4) (Table S2†); therefore, our procedures seem to be effective enough to remove Ti from the samples analyzed here. Nevertheless, it still might be better to perform further tests if our procedures were to be applied to larger samples with high Ti contents, for example, >1 g of mantle-derived rocks.42,60
For the Mo-DS measurements, Mn, Fe, Zr, and Ru can cause isobaric (98Ru and 100Ru) or polyatomic interference by forming argides (40Ar57Fe, 40Ar58Fe and 40Ar55Mn) and hydrides (94Zr1H and 96Zr1H). We examined the effects of high-abundance elements such as Mn, Fe, and Zr (Fig. 4d–f) and confirmed no measurable effects on the precision and accuracy of our analyses of NIST solutions over a wide range of X/Mo ratios that were much higher than those in our purified solutions (Table S2†). The effect of Ru interference was not examined because Ru/Mo ratios in terrestrial samples are very low, and Ru can be quantitatively removed by separation and evaporation (Table S2†). Collectively, we found that any interference corrections for Mo are not necessary in practice for our purified solutions, as is the case for W.
3.4 Isotope measurements of geological reference materials
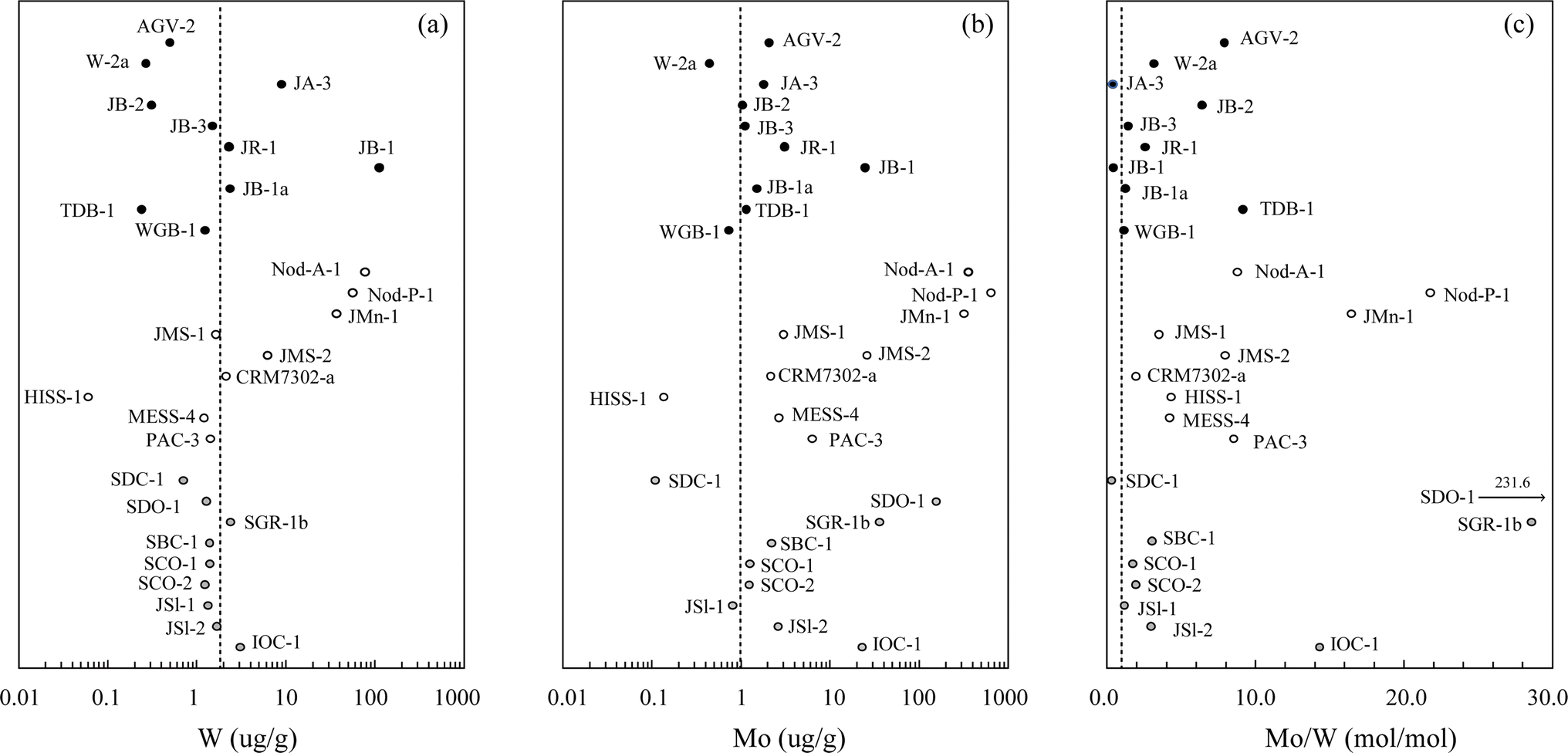 | ||
| Fig. 5 Concentrations of (a) W, (b) Mo, and (c) Mo/W in the series of geochemical reference materials analyzed in this study. Analytical uncertainties on individual data points are 2 SD (n = 3) for each sample and are smaller than the symbol size in most cases. Black circles, igneous rocks; white circles, sediments; gray circles, sedimentary and metasedimentary rocks. The dotted lines represent crustal values of 1.9 μg g−1 for W and 1.1 μg g−1 for Mo.52 | ||
| Sample | Providera | Description | Referenceb | W (μg g−1) | δ 186W (‰) | Mo (μg g−1) | δ 98Mo (‰) | Mo/W mol mol−1 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Average | RSDc (%) | Average | 2SD2 | Average | RSDc (%) | Average | 2SD2 | |||||
| a USGS, United States Geological Survey; GSJ, Geological Society of Japan; CANMET-CCRMP, Canadian Certified Reference Materials Project of Canada Centre for Mineral and Energy Technology; NMIJ, National Metrology Institute of Japan; NRC, National Research Council Canada. b The references in the table are shown in ESI. Recommended and certified values are provided by the certificates from USGS, and information or provisional values are provided by the certificates from NRC and CANMET-CCRMP. c The number of digestions in this study was 1 for most of the samples except for W-2a, JB-2, TDB-1, HISS, and SDC-1, where the solutions were separated for three individual column procedures and provided the error (n = 3). W-2a, JB-2, TDB-1, HISS, and SDC-1 were digested as three individual solutions, each of which was processed in an individual column procedure and provided the errors (n = 3). d Reported values of δ184/183W were converted to δ186/184W with multiplication by 2. | ||||||||||||
| Igneous rocks | ||||||||||||
| AGV-2 | USGS | Andesite | This study | 0.50 | 0.3 | −0.01 | 0.02 | 1.99 | 0.8 | 0.09 | 0.02 | 7.90 |
| Tsujisaka et al., 2019 | 0.42 | 4.5 | −0.02 | 0.04 | 1.86 | 6.5 | 0.11 | 0.02 | ||||
| Stubbs et al., 2022 | 0.50 | 0.2 | −0.03 | 0.02 | ||||||||
| Krabbe et al., 2017d | 0.49 | 0.9 | −0.01 | 0.08 | ||||||||
| Kurzweil et al., 2018 | 0.50 | 5.2 | 0.02 | 0.03 | ||||||||
| Roué et al., 2021 | 0.47 | 0.8 | 0.02 | 0.02 | ||||||||
| Zhang et al., 2022 | 0.49 | 0.02 | 0.02 | |||||||||
| Yang et al., 2022 | −0.01 | 0.05 | ||||||||||
| Breton and Quitté 2014 | 0.73 | 0.08 | ||||||||||
| Abraham et al., 2015 | 0.18 | 0.05 | ||||||||||
| Zhao et al., 2016 | 1.95 | 1.0 | 0.11 | 0.05 | ||||||||
| Willbold et al., 2016 | 1.96 | 1.3 | 0.10 | 0.01 | ||||||||
| W-2a | USGS | Diabase | This study | 0.27 | 1.1 | 0.08 | 0.03 | 0.44 | 1.2 | 0.20 | 0.03 | 3.15 |
| Stubbs et al., 2022 | 0.27 | 0.6 | 0.04 | 0.02 | ||||||||
| Roué et al., 2021 | 0.26 | 0.2 | 0.09 | 0.02 | ||||||||
| Kurzweil et al., 2018 | 0.26 | 1.2 | 0.08 | 0.02 | ||||||||
| Zhao et al., 2016 | 0.46 | 3.6 | 0.15 | 0.08 | ||||||||
| JA-3 | GSJ | Andesite | This study | 8.89 | <0.1 | 0.01 | 0.02 | 1.79 | <0.1 | 0.34 | 0.03 | 0.39 |
| Tsujisaka et al., 2019 | 7.62 | 0.7 | −0.03 | 0.02 | 1.48 | 2.9 | 0.34 | 0.04 | ||||
| Irisawa and Hirata, 2006d | −0.03 | 0.05 | ||||||||||
| Imai et al., 1995 | 8.07 | 19.0 | 1.89 | 41.3 | ||||||||
| JB-2 | GSJ | Basalt | This study | 0.31 | 4.1 | 0.09 | 0.02 | 1.04 | 5.0 | 0.32 | 0.02 | 6.39 |
| Stubbs et al., 2022 | 0.32 | 7.0 | 0.06 | 0.03 | ||||||||
| Mazza et al., 2019d | 0.30 | 0.11 | 0.02 | |||||||||
| Willbold et al., 2016 | 0.92 | 1.6 | 0.30 | 0.03 | ||||||||
| Zhao et al., 2016 | 1.00 | 11.0 | 0.27 | 0.09 | ||||||||
| Imai et al., 1995 | 0.26 | 1.08 | 43.5 | |||||||||
| JB-3 | GSJ | Basalt | This study | 1.50 | <0.1 | 0.02 | 0.02 | 1.11 | <0.1 | 0.07 | 0.02 | 1.42 |
| Irisawa and Hirata, 2006d | 0.02 | 0.09 | ||||||||||
| Imai et al., 1995 | 0.9–1.38 | 1.09 | ||||||||||
| JR-1 | GSJ | Rhyolite | This study | 2.29 | <0.1 | 0.08 | 0.04 | 3.06 | <0.1 | 0.16 | 0.06 | 2.56 |
| Irisawa and Hirata, 2006d | 0.04 | 0.07 | ||||||||||
| Imai et al., 1995 | 1.59 | 52.8 | 3.25 | 17.8 | ||||||||
| JB-1 | GSJ | Basalt | This study | 112 | <0.1 | −0.11 | 0.03 | 24.7 | <0.1 | 0.23 | 0.02 | 0.42 |
| Imai et al., 1995 | 17.1 | 20.5 | 27.4 | 37.2 | ||||||||
| JB-1a | GSJ | Basalt | This study | 2.36 | <0.1 | 0.04 | 0.03 | 1.51 | <0.1 | 0.11 | 0.02 | 1.22 |
| Imai et al., 1995 | 1.83 | 39.3 | 1.57 | 19.1 | ||||||||
| TDB-1 | CANMET-CCRMP | Diabase | This study | 0.24 | 0.5 | 0.09 | 0.03 | 1.14 | <0.1 | 0.14 | 0.03 | 9.15 |
| Information or provisional values | 0.6 | 0.9–2.3 | ||||||||||
| WGB-1 | CANMET-CCRMP | Gabbro | This study | 1.24 | <0.1 | 0.12 | 0.02 | 0.73 | 3.8 | 0.41 | 0.03 | 1.12 |
| Zhao et al., 2016 | 0.67 | 3.3 | 0.45 | 0.11 | ||||||||
| Information or provisional values | 1–3.5 | 0.7–1.7 | ||||||||||
|
||||||||||||
| Sediments | ||||||||||||
| Nod-A-1 | USGS | Mn nodule | This study | 77.7 | <0.1 | 0.00 | 0.03 | 356 | <0.1 | −0.39 | 0.03 | 8.78 |
| Tsujisaka et al., 2019 | 62.7 | 2.4 | 0.04 | 0.02 | 330 | 0.8 | −0.33 | 0.04 | ||||
| Stubbs et al., 2022 | 76.3 | <0.1 | −0.01 | 0.02 | ||||||||
| Kurzweil et al., 2018 | 80.0 | 2.2 | 0.03 | 0.01 | ||||||||
| Zhang et al., 2022 | 80.0 | 0.03 | 0.02 | |||||||||
| Yang et al., 2022 | 0.00 | 0.04 | ||||||||||
| Abraham et al., 2015 | 0.07 | 0.05 | ||||||||||
| Barling et al., 2001 | −0.95 | 0.15 | ||||||||||
| Asael et al., 2013 | −0.42 | 0.04 | ||||||||||
| Goto et al., 2015 | −0.41 | 0.05 | ||||||||||
| Li et al., 2016 | 484 | 0.4 | −0.48 | 0.05 | ||||||||
| Zhao et al., 2016 | 589 | 0.5 | −0.58 | 0.05 | ||||||||
| Gaspers et al., 2020 | 364 | 1.6 | −0.43 | 0.01 | ||||||||
| Nod-P-1 | USGS | Mn nodule | This study | 56.0 | <0.1 | 0.14 | 0.01 | 637 | <0.1 | −0.63 | <0.01 | 21.8 |
| Tsujisaka et al., 2019 | 52.3 | 0.1 | 0.15 | <0.01 | 680 | 0.3 | −0.60 | <0.01 | ||||
| Stubbs et al., 2022 | 57.4 | <0.1 | 0.11 | 0.02 | ||||||||
| Kurzweil et al., 2018 | 55.3 | 2.7 | 0.15 | 0.01 | ||||||||
| Zhang et al., 2022 | 55.4 | 0.14 | 0.02 | |||||||||
| Yang et al., 2022 | 0.11 | 0.04 | ||||||||||
| Abraham et al., 2015 | 0.31 | 0.04 | ||||||||||
| Barling et al., 2001 | −0.63 | 0.15 | ||||||||||
| Asael et al., 2013 | −0.63 | 0.15 | ||||||||||
| Goto et al., 2015 | −0.61 | 0.04 | ||||||||||
| Li et al., 2016 | 777 | 0.7 | −0.66 | 0.05 | ||||||||
| Zhao et al., 2016 | 637 | 0.4 | −0.63 | 0.01 | ||||||||
| JMn-1 | GSJ | Mn nodule | This study | 37.0 | 0.5 | 0.11 | 0.01 | 318 | <0.1 | −0.64 | 0.04 | 16.5 |
| Tsujisaka et al., 2019 | 25.9 | 2.3 | 0.05 | <0.01 | 265 | 0.3 | −0.55 | <0.01 | ||||
| Irisawa and Hirata, 2006d | 0.08 | 0.07 | ||||||||||
| JMS-1 | GSJ | Marine sediment | This study | 1.64 | <0.1 | 0.00 | 0.03 | 2.99 | <0.1 | 0.95 | 0.02 | 3.50 |
| Tsujisaka et al., 2019 | 1.17 | 4.0 | −0.06 | 0.01 | 2.42 | 3.1 | 0.94 | 0.01 | ||||
| Gaspers et al., 2020 | 2.70 | 0.2 | 0.98 | 0.05 | ||||||||
| JMS-2 | GSJ | Marine sediment | This study | 6.24 | <0.1 | 0.11 | 0.01 | 25.9 | <0.1 | −0.50 | 0.03 | 7.96 |
| Tsujisaka et al., 2019 | 4.20 | 1.2 | 0.15 | 0.02 | 19.6 | 4.0 | −0.47 | 0.01 | ||||
| CRM7302-a | NMIJ | Marine sediment | This study | 2.13 | <0.1 | 0.00 | 0.01 | 2.15 | <0.1 | 0.51 | 0.02 | 1.94 |
| Tsujisaka et al., 2019 | 2.47 | 2.7 | 0.04 | 0.04 | 1.98 | 2.5 | 0.64 | 0.06 | ||||
| HISS-1 | NRC | Marine sediment | This study | 0.06 | 6.2 | 0.07 | 0.04 | 0.14 | 2.8 | 0.63 | 0.03 | 4.31 |
| Tsujisaka et al., 2019 | 0.06 | 1.2 | 0.15 | 0.04 | 0.14 | 3.3 | 0.74 | 0.06 | ||||
| MESS-4 | NRC | Marine sediment | This study | 1.21 | <0.1 | 0.09 | 0.03 | 2.65 | <0.1 | 0.43 | 0.03 | 4.21 |
| Information value | 1.3 | 2.41–2.65 | ||||||||||
| PACS-3 | NRC | Marine sediment | This study | 1.42 | <0.1 | 0.03 | 0.01 | 6.31 | <0.1 | 1.35 | 0.02 | 8.53 |
| Information value | 5.9 | |||||||||||
|
||||||||||||
| Sedimentary and metasedimentary rocks | ||||||||||||
| SDC-1 | USGS | Mica shist | This study | 0.71 | 0.3 | 0.04 | 0.03 | 0.11 | 0.7 | −0.10 | 0.04 | 0.30 |
| Kurzweil et al., 2018 | 0.69 | 0.7 | 0.05 | 0.02 | ||||||||
| Zhang et al., 2022 | 0.75 | 0.05 | 0.02 | |||||||||
| Abraham et al., 2015 | 0.36 | 0.05 | ||||||||||
| SDO-1 | USGS | Shale | This study | 1.28 | <0.1 | 0.11 | 0.02 | 155 | <0.1 | 1.06 | 0.03 | 232 |
| Abraham et al., 2015 | 0.26 | 0.06 | ||||||||||
| Kendall et al., 2023 | 1.02 | 0.04 | ||||||||||
| Goldberg et al., 2013 | 1.05 | 0.14 | ||||||||||
| Kane et al., 1990 | 134 | 2.1 | ||||||||||
| SGR-1b | USGS | Shale | This study | 2.69 | <0.1 | 0.11 | 0.01 | 35.8 | <0.1 | 0.65 | 0.02 | 28.6 |
| Zhao et al., 2016 | 35.5 | 2.3 | 0.69 | 0.11 | ||||||||
| Li et al., 2016 | 44.7 | 0.2 | 0.63 | 0.02 | ||||||||
| Gaspers et al., 2020 | 36.6 | 1.6 | 0.69 | 0.06 | ||||||||
| Kendall et al., 2023 | 0.58 | 0.02 | ||||||||||
| Recommended values | 2.60 | 2.3 | 35.1 | 2.6 | ||||||||
| SBC-1 | USGS | Shale | This study | 1.39 | <0.1 | 0.06 | 0.04 | 2.21 | <0.1 | 0.69 | 0.01 | 3.03 |
| Krabbe et al., 2017d | 1.60 | 0.1 | 0.06 | 0.02 | ||||||||
| Kendall et al., 2023 | 0.64 | 0.02 | ||||||||||
| Gaspers et al., 2020 | 2.33 | 5.4 | 0.61 | 0.08 | ||||||||
| SCO-1 | USGS | Shale | This study | 1.40 | <0.1 | 0.04 | <0.01 | 1.26 | <0.1 | −0.15 | 0.04 | 1.72 |
| Roué et al., 2021 | 1.48 | 2.4 | 0.06 | 0.02 | ||||||||
| Zhao et al., 2016 | 1.20 | 1.3 | −0.16 | 0.03 | ||||||||
| Li et al., 2016 | 1.40 | 14.3 | −0.24 | 0.06 | ||||||||
| SCO-2 | UGSG | Shale | This study | 1.23 | 0.1 | 0.06 | 0.02 | 1.24 | <0.1 | 0.38 | 0.03 | 1.92 |
| Kurzweil et al., 2018 | 1.48 | 2.4 | 0.06 | 0.03 | ||||||||
| Certified values | 1.20 | 4.2 | ||||||||||
| JSl-1 | GSJ | Slate | This study | 1.34 | <0.1 | 0.02 | 0.02 | 0.80 | <0.1 | 0.33 | 0.03 | 1.15 |
| Tsujisaka et al., 2019 | 0.76 | 0.3 | −0.03 | 0.03 | 0.80 | 5.8 | 0.22 | 0.08 | ||||
| Gaspers et al., 2020 | 0.73 | 0.7 | 0.29 | 0.03 | ||||||||
| Imai et al., 1996 | 0.58–4.7 | 0.82 | 29.2 | |||||||||
| JSl-2 | GSJ | Slate | This study | 1.68 | <0.1 | 0.01 | 0.01 | 2.59 | <0.1 | 0.10 | 0.02 | 2.96 |
| Tsujisaka et al., 2019 | 1.02 | 8.3 | −0.04 | 0.02 | 1.41 | 19.7 | −0.03 | 0.07 | ||||
| Imai et al., 1996 | 0.61–2.7 | |||||||||||
| IOC-1 | CANMET-CCRMP | Iron ore | This study | 3.06 | <0.1 | 0.20 | 0.01 | 22.9 | <0.1 | 0.24 | 0.04 | 14.3 |
| Sindol et al., 2022 | 3.09 | 5.2 | 23.0 | 1.7 | ||||||||
 | ||
| Fig. 6 (a) δ186W and (b) δ98Mo values of geochemical reference materials of the igneous rock series analyzed in this study. Black circles are data obtained in this study; white circles are data from the literature. Error bars represent the 2SD of triplicate measurements of individual samples. δ98Mo is normalized relative to NIST 3134 (=0‰) + 0.25‰. | ||
We also measured JB-3, JR-1, JB-1, JB-1a, TDB-1, and WGB-1 (Fig. 6 and Table 4). Because our δ186W and/or δ98Mo values are mostly the first reports, their accuracy as reference materials should be established by future additional reports. JB-3 and JR-1 are a basalt and rhyolite, respectively, for which δ186W values were reported by Irisawa and Hirata (2006),62 and WGB-1 is a gabbro for which the δ98Mo value was reported by Zhao et al. (2016).43 Our δ186W for JB-3 and JR-1 and δ98Mo for WGB-1 agreed well with these reports. The results indicate that JB-3 and JR-1, and WGB-1 could be homogeneous references for δ186W and δ98Mo, respectively, at this stage.
One problem specific to both W and Mo isotopes is the higher risk of contamination in sampling procedures because (i) sampling of original hard rocks is often conducted using tungsten carbide,12,73 and (ii) large amounts of sample powder for reference materials are often processed in large batches using hardened steel equipment.3,23 This risk of contamination is particularly higher for igneous rocks because of their low concentrations of W and Mo (Fig. 5a and b). Then, we examined this effect by comparing JB-1 and JB-1a, for which both powders were prepared from the same stock chip but JB-1 shows larger W and Mo concentrations due to contamination during sample processing.78–80 We found clear discrepancies between JB-1 and JB-1a for both δ186W and δ98Mo (Fig. 6a and b) and our W concentration for JB-1 was much higher than the reported values (Table 4). The data indicate that (i) JB-1 is clearly contaminated in terms of both δ186W and δ98Mo, (ii) this contamination may not be homogeneous within the sample powder, as is known for the nugget effect,81,82 and (iii) the geological context of their isotope data can only be interpreted with appropriate sampling procedures without contamination.
Nevertheless, with the exception of JB-1, the small but resolvable variations observed in both δ186W and δ98Mo seem to show weak correlations with MgO contents (Fig. S4†), as was reported for δ186W in USGS reference rocks.3 Recent studies have suggested that both W and Mo isotopes show similarly lighter compositions in chemically more evolved rocks than in more mafic rocks, which could be caused by the isotope fractionation during magmatic differentiation.10,23,83 Although geochemical discussion should be conducted using a suite of well-characterized, co-genetic, and properly prepared samples, our measurements of the igneous rock series here might capture the signatures of δ186W and δ98Mo simultaneously, both of which could be produced by high-temperature igneous processes.
 | ||
| Fig. 7 (a) δ186W and (b) δ98Mo values of geochemical reference materials of the sediment series analyzed in this study. Black circles are data obtained in this study; white circles are data from the literature. Error bars represent the 2SD of triplicate measurements of individual samples. δ98/95Mo is normalized relative to NIST 3134 (=0‰) + 0.25‰. | ||
This data set allows us to evaluate the validity of the sediments analyzed here as reference materials. For δ98Mo (Fig. 7b), sediments such as Nod-P-1 and JMS-1 are suitable for interlaboratory calibration because of the remarkable agreement among many independent groups. Most of the other sediments, despite the few available δ98Mo reports, also show good agreement and could be established as references covering a wide range of δ98Mo values by further analyses. In contrast, the δ186W values for each sample seem to exhibit somewhat larger spreads, even within analytical uncertainty (Fig. 7a). This data spread is apparently emphasized by the smaller range of variation of δ186W values (0.00–0.18‰) relative to that of δ98Mo values (−0.64 to +1.35‰), but several groups have also discussed that δ186W values obtained via DS calculation may be systematically lower or higher due to potential 183W deficits in a series of procedures.17,45,64 At this stage, sediment materials may not necessarily be suitable for the method validation of W isotope measurements including the small effects such as the potential 183W deficit in the DS method.
To avoid large data spreads in both concentrations and isotope ratios, sediment materials may require more specific attention. First, the large variations observed in W and Mo concentrations (Fig. 5a and b) practically mean that different sampling scales could be required for isotope analysis. Because sample heterogeneities and blank contributions are closely dependent on the sampling scale, this effect should be avoided to obtain their representative data.81 Second, large variations in Mo/W ratios (Fig. 5c) lead to different proportions of the two DSs, leading to variable contributions of the spike impurities, i.e., a small amount of W in the Mo DS or vice versa. We confirmed that this effect is negligible in the range of Mo/W ratios here (Mo/W ≈ 0.30–232), but care should still be taken to ensure simultaneous DS spiking of these two elements because the case is completely dependent on the purity of the isotope spikes used. Third, some mineral components in the sediments strongly absorb moisture from the air, which can cause weighing errors and therefore deviations from the optimal spiking. These effects could be particular pitfalls in dealing with sediment samples as reference materials in general and also be possible reasons for the observed discrepancies among the data compiled here.
From a geochemical standpoint, the large variation observed in δ98Mo values (Fig. 7b) seems to correlate with typical observations for marine sediments under different redox conditions.21 Oxic sediments such as Mn nodules (Nod-A-1, Nod-P-1, and JMn-1) and deep-sea pelagic sediments (JMS-2) show large Mo isotope fractionations from homogeneous modern seawater (δ98MoSW: 2.34 ± 0.10‰),85,86 whereas other organic-rich sediments (JMS-1, CRM7302-a, HISS, MESS-4, and PACS-3) show a wide variation of δ98Mo values as a consequence of capturing seawater depending on local/global conditions.87,88 In this context, the δ186W values of oxic sediments (Nod-A-1, Nod-P-1, JMn-1, and JMS-2) similarly show large isotope fractionations relative to modern seawater (δ186WSW: 0.55 ± 0.12‰) (Fig. 7a),14,16 as is the case of Mo. In contrast, the δ186W values of other organic-rich sediments (JMS-1, CRM7302-a, HISS, MESS-4, and PACS-3) are scattered around the crustal value (δ186W = 0.01 ± 0.03‰, estimated from loess),12 thus still showing a larger offset from seawater. This could be because, unlike for MoO42−, euxinic conditions can facilitate the solubility of WO42− and do not provide an effective burial pathway for W from seawater to the sediments, leading to the dominant δ186W contribution being from clastic materials.11,89 This difference between W and Mo isotopes at the solid/water interface is a unique contrast to high-temperature igneous processes and could reflect the redox conditions in water masses.
 | ||
| Fig. 8 (a) δ186W and (b) δ98Mo values of geochemical reference materials of sedimentary and metasedimentary rock series analyzed in this study. Black circles are data obtained in this study; white symbols are data from the literature. Error bars represent the 2SD of triplicate measurements of individual samples. δ98/95Mo is normalized relative to NIST 3134 (=0‰) + 0.25‰. | ||
Only limited reports of δ186W and δ98Mo values are available for other sedimentary and metasedimentary rocks (Fig. 8); thus, our data can contribute to evaluating the validity of these samples as reference materials. Our data for SBC-1, SCO-1, and SCO-2 all matched with the literature, indicating their homogeneity. In contrast, our data for JSl-1 and JSl-2 showed discrepancies with those of Tsujisaka et al. (2019).44 These discrepancies are likely caused by sample heterogeneity, because large spreads were also observed among previous reports with respect to the W and Mo concentrations90 (Table 4). Our data for IOC-1 are the first reports for both δ186W and δ98Mo. Further analyses will be necessary to establish the accuracies for these samples for which there is a lack of data or lack of agreement with the literature as reference materials.
Most of the (meta)sedimentary rocks analyzed here (Fig. 8) are organic-rich and showed large variations of δ98Mo values with constant δ186W values around the crustal value (δ186W = 0.01 ± 0.03‰, estimated from loess).12 This observation is consistent with the typical trend observed for the sediment series (Fig. 7), indicating that the δ186W and δ98Mo of (meta)sedimentary rocks likely reflect the redox conditions of the water masses in the past. Furthermore, these constant δ186W values (Fig. 8a), as a consequence, emphasize the relatively higher δ186W of SGR-1b (δ186W = 0.11 ± 0.01‰), SDO-1 (δ186W = 0.11 ± 0.02‰), and IOC-1 (δ186W = 0.20 ± 0.01‰) compared to other sediments derived from clastic contributions. We consider that such deviations from the crustal values could commonly reflect larger contributions of authigenic signatures from ambient seawater, because the two black shales contain significant amounts of organic carbons (24.8% for SGR-1b91,92 and 9.67% for SDO-1 (ref. 93)), and IOC-1 is an iron ore which is likely precipitated from seawater.94 These results may indicate that capturing authigenic δ186W signatures may require sample selection that could be different from those for Mo isotopes depending on redox conditions.
4 Conclusions
We have established the simultaneous application of the DS method to isotope measurements of W and Mo for a single sample aliquot. This was achieved by optimizing two-stage anion-exchange procedures, which enables the efficient removal of critical interference for their DS method and the collection of sharply separated W and Mo fractions. The obtained recovery is quantitative for both W and Mo, and the purity of both elements is sufficiently high to achieve high-precision measurements that are comparable to previous DS studies of individual elements. The application of this method to 27 geological reference materials, including igneous rocks, sediments, and (meta)sedimentary rocks, provided comprehensive data sets that confirmed the accuracy of our measurements and expanded the reference materials available for interlaboratory comparisons of W and Mo isotope measurements. The data sets also highlight (i) pitfalls in sample preparation that should be considered when making appropriate geological interpretations, (ii) the suitability of analyzed samples as reference materials in terms of W and Mo isotopes, and (iii) several δ186W and δ98Mo variations possibly associated with the geochemistry of the sample formation processes and/or environments. Our simple, efficient, and robust method, along with the reference data sets, will serve as a basis to clarify how W isotopes behave in the Earth system relative to Mo isotopes and facilitate the development of the stable isotope geochemistry of W and Mo, two elements of increasing importance in Earth sciences.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Akira Ishikawa (Tokyo Tech), Dr Qing Chang (JAMSTEC), Dr Hatsuki Enomoto and Dr Yuki Hibiya (the Univ. of Tokyo) for their assistance in obtaining geological reference materials. This work was conducted based on the knowledge from several synchrotron experiments with approvals from JASRI (2012A1767 and 2017A1093) and KEK (2013G562 and 2021G123). This study was financially supported by Promotion of Science (JSPS) KAKENHI grants (no. JP17H06455 and 20H04316).References
- K. Abraham, J. Barling, C. Siebert, N. Belshaw, L. Gall and A. N. Halliday, J. Anal. At. Spectrom., 2015, 30, 2334–2342 RSC
.
- T. Breton and G. Quitté, J. Anal. At. Spectrom., 2014, 29, 2284–2293 RSC
- N. Krabbe, T. S. Kruijer and T. Kleine, Chem. Geol., 2017, 450, 135–144 CrossRef CAS
- F. Kurzweil, C. Münker, J. Tusch and R. Schoenberg, Chem. Geol., 2018, 476, 407–417 CrossRef CAS
- T. Kleine and R. J. Walker, Annu. Rev. Earth Planet. Sci., 2017, 45, 389–417 CrossRef CAS PubMed
- M. Willbold, T. Elliott and S. Moorbath, Nature, 2011, 477, 195–198 CrossRef CAS PubMed
- F. Kurzweil, C. Münker, M. Grupp, N. Braukmüller, L. Fechtner, M. Christian, S. V. Hohl and R. Schoenberg, Geochim. Cosmochim. Acta, 2019, 251, 176–191 CrossRef CAS
- F. Kurzweil, C. Archer, M. Wille, R. Schoenberg, C. Munker and O. Dellwig, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023544118 CrossRef CAS PubMed
- F. Kurzweil, O. Dellwig, M. Wille, R. Schoenberg, H. W. Arz and C. Münker, Earth Planet. Sci. Lett., 2022, 578, 117303 CrossRef CAS
- S. E. Mazza, A. Stracke, J. B. Gill, J.-I. Kimura and T. Kleine, Earth Planet. Sci. Lett., 2020, 530, 115492 CrossRef
- L. Roué, F. Kurzweil, M. Wille, A. Wegwerth, O. Dellwig, C. Münker and R. Schoenberg, Geochim. Cosmochim. Acta, 2021, 309, 366–387 CrossRef
- R. Yang, T. Li, D. Stubbs, T. Chen, S. Liu, D. B. Kemp, W. Li, S. Yang, J. Chen, T. Elliott, O. Dellwig, J. Chen and G. Li, Geochim. Cosmochim. Acta, 2022, 322, 227–243 CrossRef CAS
- R. Yang, D. Stubbs, T. Elliott, T. Li, T. Chen, A. Paytan, D. B. Kemp, H. Ling, J. Chen, J. R. Hein, C. D. Coath and G. Li, Geology, 2023, 51, 728–732 CrossRef CAS
- T. Kashiwabara, S. Kubo, M. Tanaka, R. Senda, T. Iizuka, M. Tanimizu and Y. Takahashi, Geochim. Cosmochim. Acta, 2017, 204, 52–67 CrossRef CAS
- E. A. Schauble, Rev. Mineral. Geochem., 2004, 55, 65–111 CrossRef CAS
- Y. Fujiwara, M. Tsujisaka, S. Takano and Y. Sohrin, Chem. Geol., 2020, 555, 119835 CrossRef CAS
- D. Stubbs, R. Yang, C. D. Coath, T. John and T. Elliott, Geochim. Cosmochim. Acta, 2022, 334, 135–154 CrossRef CAS
- A. D. Anbar, Rev. Mineral. Geochem., 2004, 55, 429–454 CrossRef
- A. D. Anbar and O. Rouxel, Annu. Rev. Earth Planet. Sci., 2007, 35, 717–746 CrossRef CAS
- N. Breilatt, C. Guerrot, E. Marcoux and P. Négrel, J. Geochem. Explor., 2016, 161, 1–15 CrossRef
- B. Kendall, T. W. Dahl and A. D. Anbar, Rev. Mineral. Geochem., 2017, 82, 683–732 CrossRef CAS
- B. Kendall, S. Wang, P. Lillis, L. Xing, W. Zheng and C. Zhu, Chem. Geol., 2023, 617, 121244 CrossRef CAS
- M. Willbold and T. Elliott, Chem. Geol., 2017, 449, 253–268 CrossRef CAS
- K. Matsuoka, T. Tatsuyama, S. Takano and Y. Sohrin, Front. Mar. Sci., 2023, 10, 682 Search PubMed
- T. Kashiwabara, Y. Takahashi, M. Tanimizu and A. Usui, Geochim. Cosmochim. Acta, 2011, 75, 5762–5784 CrossRef CAS
- Y. Sohrin, K. Isshiki, E. Nakayama, S. Kihara and M. Matsui, Anal. Chim. Acta, 1989, 218, 25–35 CrossRef CAS
- Y. Sohrin, M. Matsui and E. Nakayama, Geochim. Cosmochim. Acta, 1999, 63, 3457–3466 CrossRef CAS
- O. Dellwig, A. Wegwerth, B. Schnetger, H. Schulz and H. W. Arz, Earth-Sci. Rev., 2019, 193, 1–23 CrossRef CAS
- T. J. Mohajerin, G. R. Helz and K. H. Johannesson, Geochim. Cosmochim. Acta, 2016, 177, 105–119 CrossRef CAS
- A. Kletzin and M. W. Adams, FEMS Microbiol. Rev., 1996, 18, 5–63 CrossRef CAS PubMed
- R. Hille, Trends Biochem. Sci., 2002, 27, 360–367 CrossRef CAS PubMed
- T. Kashiwabara, Y. Takahashi, T. Uruga, H. Tanida, Y. Terada, Y. Niwa and M. Nomura, Chem. Lett., 2010, 39, 870–871 CrossRef CAS
- T. Kashiwabara, Y. Takahashi, M. A. Marcus, T. Uruga, H. Tanida, Y. Terada and A. Usui, Geochim. Cosmochim. Acta, 2013, 106, 364–378 CrossRef CAS
- D. Malinovsky, I. Rodushkin, D. C. Baxter, J. Ingri and B. Öhlander, Int. J. Mass Spectrom., 2005, 245, 94–107 CrossRef CAS
- T. Goldberg, G. Gordon, G. Izon, C. Archer, C. R. Pearce, J. McManus, A. D. Anbar and M. Rehkamper, J. Anal. At. Spectrom., 2013, 28, 724–735 RSC
- V. Migeon, B. Bourdon, E. Pili and C. Fitoussi, J. Anal. At. Spectrom., 2015, 30, 1988–1996 RSC
- J. Li, X. R. Liang, L. F. Zhong, X. C. Wang, Z. Y. Ren, S. L. Sun, Z. F. Zhang and J. F. Xu, Geostand. Geoanal. Res., 2014, 38, 345–354 CrossRef CAS
- J. Li, X. k. Zhu, S. h. Tang and K. Zhang, Geostand. Geoanal. Res., 2016, 40, 405–415 CrossRef CAS
- J. Liu, H. Wen, Y. Zhang, H. Fan and C. Zhu, J. Anal. At. Spectrom., 2016, 31, 1287–1297 RSC
- T. F. Nägler, A. D. Anbar, C. Archer, T. Goldberg, G. W. Gordon, N. D. Greber, C. Siebert, Y. Sohrin and D. Vance, Geostand. Geoanal. Res., 2014, 38, 149–151 CrossRef
- C. Siebert, T. F. Nagler and J. D. Kramers, Geochem., Geophys., Geosyst., 2001, 2, 7 CrossRef
- M. Willbold, K. Hibbert, Y.-J. Lai, H. Freymuth, R. C. Hin, C. Coath, F. Vils and T. Elliott, Geostand. Geoanal. Res., 2016, 40, 389–403 CAS
- P. P. Zhao, J. Li, L. Zhang, Z. B. Wang, D. X. Kong, J. L. Ma, G. J. Wei and J. F. Xu, Geostand. Geoanal. Res., 2016, 40, 217–226 CrossRef CAS
- M. Tsujisaka, S. Takano, M. Murayama and Y. Sohrin, Anal. Chim. Acta, 2019, 1091, 146–159 CrossRef CAS PubMed
- T. Zhang, J. Liu, Q. Zhang, Y. Zhang and L. Qin, Geostand. Geoanal. Res., 2022, 47, 169–183 CrossRef
- E. Bura-Nakić, M. B. Andersen, C. Archer, G. F. de Souza, M. Marguš and D. Vance, Geochim. Cosmochim. Acta, 2018, 222, 212–229 CrossRef
- J.-M. Brazier, A.-D. Schmitt, S. Gangloff, E. Pelt, M. I. Gocke and G. L. B. Wiesenberg, Chem. Geol., 2020, 545, 119641 CrossRef CAS
- A. J. Dickson, E. Idiz, D. Porcelli and S. H. J. M. van den Boorn, Geochim. Cosmochim. Acta, 2020, 287, 205–220 CrossRef CAS
- T. S. Kruijer, C. Burkhardt, L. E. Borg and T. Kleine, Earth Planet. Sci. Lett., 2022, 584, 117440 CrossRef CAS
- J. Bai, C. Wu, H. Wu, Z. Wang, L. Zhang, S. Zhong, J. Ma and G. Wei, Earth Planet. Sci. Lett., 2024, 629, 118597 CrossRef CAS
- J.-H. Bai, J.-L. Ma, G.-J. Wei, L. Zhang and S.-X. Zhong, J. Anal. At. Spectrom., 2022, 37, 1618–1628 RSC
-
R. L. Rudnick and S. Gao, Composition of the continental crust, in The Crust (Treatise on Geochemistry), ed. R. L. Rudnick, Elsevier-Pergamon, Oxford, 2005, vol. 3, pp. 1–64 Search PubMed
- N. Gaspers, T. Magna and L. Ackerman, Geostand. Geoanal. Res., 2020, 44, 363–374 CrossRef CAS
- M. H. Dodson, J. Sci. Instrum., 1963, 40, 289–295 CrossRef
- K. Dideriksen, J. A. Baker and S. L. S. Stipp, Geochim. Cosmochim. Acta, 2006, 70, 118–132 CrossRef CAS
- J. F. Rudge, B. C. Reynolds and B. Bourdon, Chem. Geol., 2009, 265, 420–431 CrossRef CAS
- M. Klaver and C. D. Coath, Geostand. Geoanal. Res., 2019, 43, 5–22 CrossRef CAS
- J. Volkening, M. Koppe and K. G. Heumann, Int. J. Mass Spectrom. Ion Processes, 1991, 107, 361–368 CrossRef
- Y. V. Sahoo, S. Nakai and A. Ali, Analyst, 2006, 131, 434–439 RSC
- B. J. Peters, A. Mundl-Petermeier, M. F. Horan, R. W. Carlson and R. J. Walker, Geostand. Geoanal. Res., 2019, 43, 245–259 CrossRef CAS
- J. Tusch, P. Sprung, J. van de Löcht, J. E. Hoffmann, A. J. Boyd, M. T. Rosing and C. Münker, Geochim. Cosmochim. Acta, 2019, 257, 284–310 CrossRef CAS
- K. Irisawa and T. Hirata, J. Anal. At. Spectrom., 2006, 21, 1387 RSC
- Y. Nagai and T. Yokoyama, Anal. Chem., 2014, 86, 4856–4863 CrossRef CAS PubMed
- G. Budde, G. J. Archer, F. L. H. Tissot, S. Tappe and T. Kleine, J. Anal. At. Spectrom., 2022, 37, 2005–2021 RSC
- N. D. Greber, C. Siebert, T. F. Nägler and T. Pettke, Geostand. Geoanal. Res., 2012, 36, 291–300 CrossRef CAS
- W. A. Brand, T. B. Coplen, J. Vogl, M. Rosner and T. Prohaska, Pure Appl. Chem., 2014, 86, 425–467 CAS
- G. Quitté, J.-L. Birck, F. Capmas and C. J. Allègre, Geostand. Geoanal. Res., 2002, 26, 149–160 CrossRef
- J. I. Kim, H. Lagally and H.-J. Born, Anal. Chim. Acta, 1973, 64, 29–43 CrossRef CAS
- C. Munker, S. Weyer, E. Scherer and K. Mezger, Geochem., Geophys., Geosyst., 2001, 2, 12 CrossRef
- K. A. Kraus, F. Nelson and G. E. Moore, J. Am. Chem. Soc., 1955, 77, 3972–3977 CrossRef CAS
- M. Touboul and R. J. Walker, Int. J. Mass Spectrom., 2012, 309, 109–117 CrossRef CAS
- Q.-F. Mei, J.-H. Yang and Y.-H. Yang, J. Anal. At. Spectrom., 2018, 33, 569–577 RSC
- A. Takamasa, K. Suzuki, Y. Fukami, T. Iizuka, M. L. G. Tejada, W. Fujisaki, Y. Orihashi and T. Matsumoto, Geochem. J., 2020, 54, 117–127 CrossRef CAS
- L. Qin, N. Dauphas, P. E. Janney and M. Wadhwa, Anal. Chem., 2007, 79, 3148–3154 CrossRef CAS PubMed
- D. L. Cook and M. Schönbächler, J. Anal. At. Spectrom., 2016, 31, 1400–1405 RSC
- T. Schulz, C. Münker and S. T. M. Peters, Earth Planet. Sci. Lett., 2013, 362, 246–257 CrossRef CAS
- T. Kleine, K. Mezger, C. Münker, H. Palme and A. Bischoff, Geochim. Cosmochim. Acta, 2004, 68, 2935–2946 CrossRef CAS
- A. Ando, N. Mita and S. Terashima, Geostand. Newsl., 1987, 11, 159–166 CrossRef CAS
- N. Imai, S. Terashima, S. Itoh and A. Ando, Geochem. J., 1995, 29, 91–95 CrossRef CAS
- S. Terashima, Geostand. Newsl., 1996, 21, 93–96 CrossRef
- T. Meisel and J. Moser, Chem. Geol., 2004, 208, 319–338 CrossRef CAS
- A. Ishikawa, R. Senda, K. Suzuki, C. W. Dale and T. Meisel, Chem. Geol., 2014, 384, 27–46 CrossRef CAS
- W. Fang, L.-Q. Dai, Y.-F. Zheng and Z.-F. Zhao, Geochim. Cosmochim. Acta, 2022, 334, 273–292 CrossRef CAS
- J. Barling, G. L. Arnold and A. D. Anbar, Earth Planet. Sci. Lett., 2001, 193, 447–457 CrossRef CAS
- C. Siebert, T. F. Nägler, F. von Blanckenburg and J. D. Kramers, Earth Planet. Sci. Lett., 2003, 211, 159–171 CrossRef CAS
- Y. Nakagawa, S. Takano, M. L. Firdaus, K. Norisuye, T. Hirata, D. Vance and Y. Sohrin, Geochem. J., 2012, 46, 131–141 CrossRef CAS
- Y. Duan, A. D. Anbar, G. L. Arnold, T. W. Lyons, G. W. Gordon and B. Kendall, Geochim. Cosmochim. Acta, 2010, 74, 6655–6668 CrossRef CAS
- M. Wille, O. Nebel, M. J. Van Kranendonk, R. Schoenberg, I. C. Kleinhanns and M. J. Ellwood, Chem. Geol., 2013, 340, 68–76 CrossRef CAS
- O. Dellwig, A. Wegwerth, B. Schnetger, H. Schulz and H. W. Arz, Earth-Sci. Rev., 2019, 193, 1–23 CrossRef CAS
- N. Imai, S. Terashima, S. Itoh and A. Ando, Geostand. Newsl., 1996, 20, 165–216 CrossRef CAS
- J. Li and L. Yin, Geostand. Geoanal. Res., 2019, 43, 497–507 CrossRef CAS
- M. Wang, Z. Chu, T. C. Meisel and J. Guo, Geostand. Geoanal. Res., 2022, 46, 333–349 CrossRef CAS
- J. S. Kane, B. Arbogast and J. Leventhal, Geostand. Newsl., 1990, 14, 169–196 CrossRef
- G. P. Sindol, M. G. Babechuk, J. Conliffe, J. F. Slack, C. Rosca and R. Schoenberg, Precambrian Res., 2022, 379, 106750 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ja00059e |
| This journal is © The Royal Society of Chemistry 2024 |