
Disparate reactivity of a chiral iron(II) tetracarbene complex with organic azides†
Jerred J.
Russell
,
Joseph F.
DeJesus
,
Brett A.
Smith
,
Phattananawee
Nalaoh
 ,
Konstantinos D.
Vogiatzis
* and
David M.
Jenkins
*
,
Konstantinos D.
Vogiatzis
* and
David M.
Jenkins
*
Department of Chemistry, The University of Tennessee, Knoxville, Tennessee 37996, USA. E-mail: kvogiatz@utk.edu; jenkins@ion.chem.utk.edu
First published on 12th June 2024
Abstract
A chiral tetra-NHC iron(II) complex and its disparate reactivity with multiple organic azides is reported. Both aryl and alkyl azides react with the iron(II) complex yielding three distinct products: an iron(IV) imide, an iron(IV) tetrazene, and a surprising and unprecedented double imide insertion complex.
Organic azides are key nitrene sources for many catalytic nitrogen transfer reactions, from amination to aziridination.1,2 During these catalytic cycles, organic azides undergo the release of dinitrogen to form a key intermediate, an imide complex.3,4 First row transition metals are often targeted for these catalytic reactions due to their high earth abundance (Fig. 1A),5 and of these metals, iron has been targeted specifically for catalysis of pharmaceutical intermediates due to its high biocompatibility.6
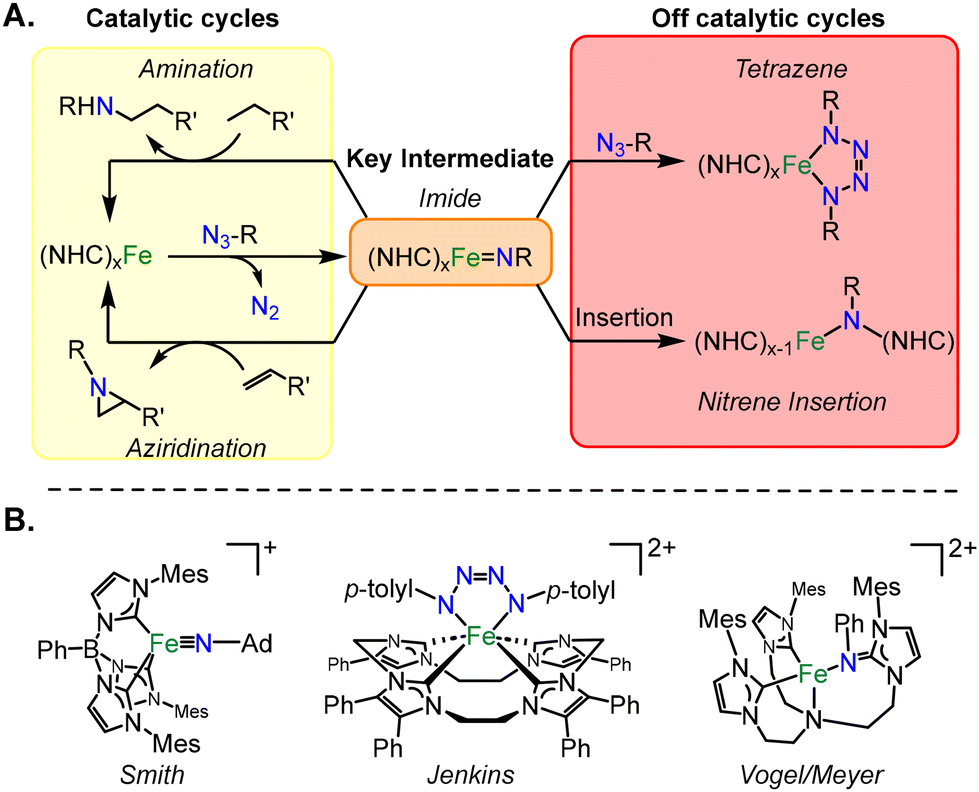 | ||
| Fig. 1 (A) Possible reactivities of (NHC)xFe complexes with organic azides. Notably, additional off-cycle reactions can occur from a metal imide. (B) Examples of the first imide, tetrazene, and imide insertion supported by NHC auxiliary ligands that were formed from organic azides. | ||
Since the formation of a metal imide from an organic azide is a redox process, the auxiliary ligands on iron are critical since they should not be able to be oxidized. For this reason, many researchers have often selected nitrogenous ligands for iron catalysis.7,8 Recently, N-heterocyclic carbenes (NHCs) have also been effective for these catalytic reactions,9,10 since they are highly effective at stabilizing high oxidation states on iron, notably, as high as iron(VII).11–13
Despite the ease of high valent iron imide formation with organic azides (example from Smith, Fig. 1B),11,14,15 off catalytic cycle reactions have been documented with iron NHC complexes (Fig. 1A). If a second equivalent of organic azide reacts with a metal imide in a 2 + 3 cycloaddition reaction, then a metallotetrazene is formed (Fig. 1B).16–18 Jenkins reported a rare example of an iron(IV) tetrazene which hinders catalytic aziridination.10 More remarkably, there is one report of an insertion of a nitrene into the metal–carbon bond on iron of an NHC, presumably that occurs via a metal imide intermediate (Fig. 1B).19 Yet, to date, all of these processes have not been documented on a single system.
In this manuscript, we describe the disparate reactivity of organic azides with a single chiral tetra NHC iron(II) complex. We isolated three unique species including an iron(IV) imide, an iron(IV) tetrazene, and a structurally novel bis-nitrene insertion product. All iron complexes were evaluated by multinuclear NMR, single crystal X-ray diffraction, and computational methods. The variance in reactivity can be justified by the energetics of the competing reactions to form a tetrazene or insertion product.
Aryl and alkyl azides with disparate steric bulk were reacted with the previously reported chiral iron(II) complex, ((S,S)-1,2-Cy,BMe2TCH)Fe (1) (Scheme 1).20 These organic azides were chosen because we have previously noted considerable differences in reactivity between aryl (more reactive) and alkyl (less reactive) azides on similar tetracarbene iron and chromium complexes for catalytic aziridination.9,21–23
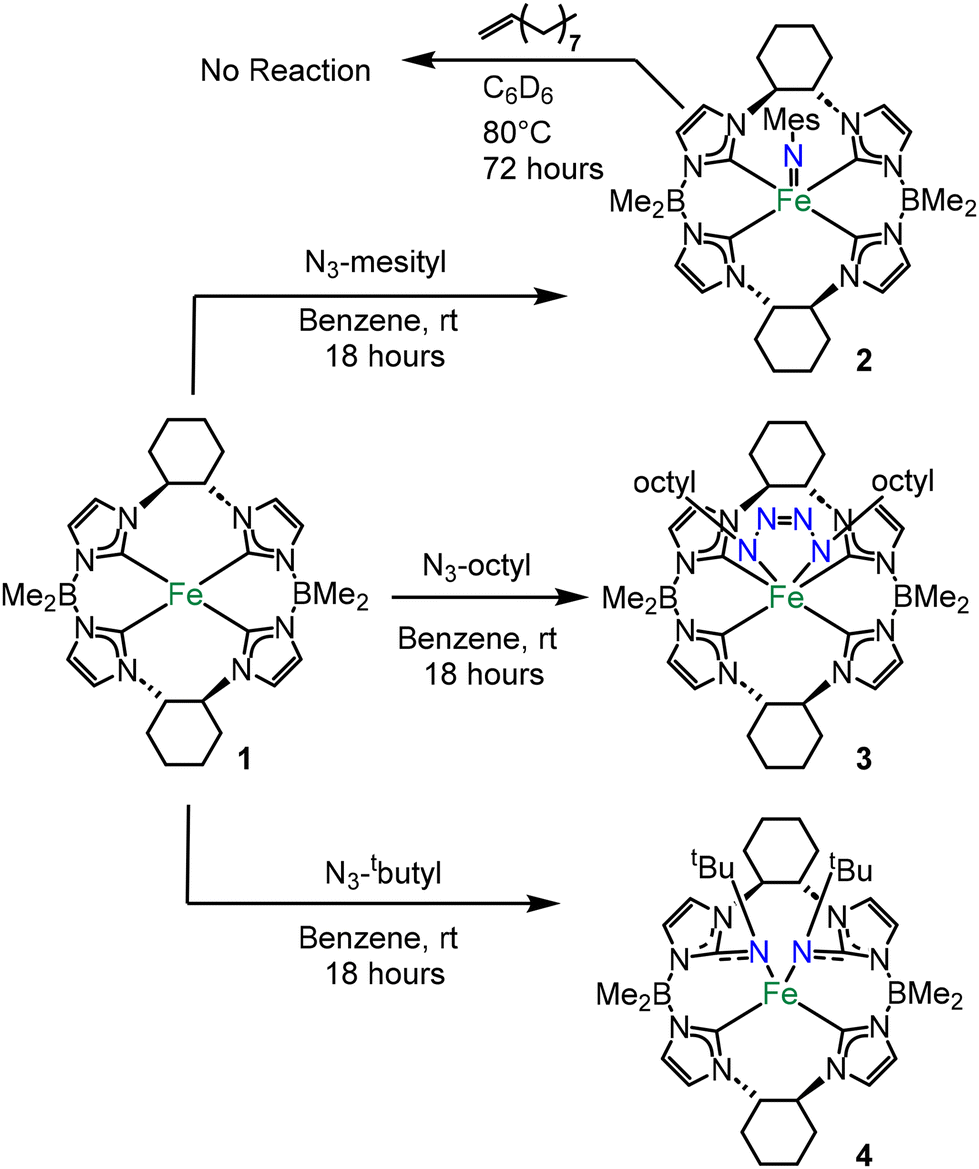 | ||
| Scheme 1 Reactions of 1 with both aryl and alkyl azides. | ||
Complex 1 was reacted with five equivalents of mesityl azide in benzene at room temperature which gave an immediate blue solution (Scheme 1). After removal of excess azide, a blue powder corresponding to ((S,S)-1,2-Cy,BMe2TCH)Fe(NMes) (2) was isolated in 52% yield. The 1H NMR spectrum of 2 revealed nineteen paramagnetically shifted resonances that are consistent with C2 symmetry in solution (Fig. S1†).20 Evans’ method data gave a measurement of 2.82 μB that is consistent with a S = 1 system (see ESI†) and similar to an isostructural iron(IV) imide that we reported previously.11
Single crystal X-ray diffraction for 2 confirmed a square pyramidal geometry with an iron imide bond (Fig. 2). Complex 2 features a highly elongated Fe–N bond distance of 1.758(4) Å. High spin iron(IV) imides typically have longer iron–imide bonds (1.70 Å or greater) whereas low-spin (S = 0) iron-imide complexes typically have bond lengths between 1.60–1.68 Å.11,24 For example, our previously reported five coordinate Fe(IV) imide with an S = 1 spin state had an iron–nitrogen bond distance of 1.730(1) Å.11 Likewise, Werncke, Munz, and coworkers synthesized an iron imidyl (NMes) complex with an iron–nitrogen bond length of 1.774(2) Å.24 The imide moiety on 2 is somewhat bent with a Fe–N1–C20 bond angle of 162.7(3)°, consistent with previous reports.11,25
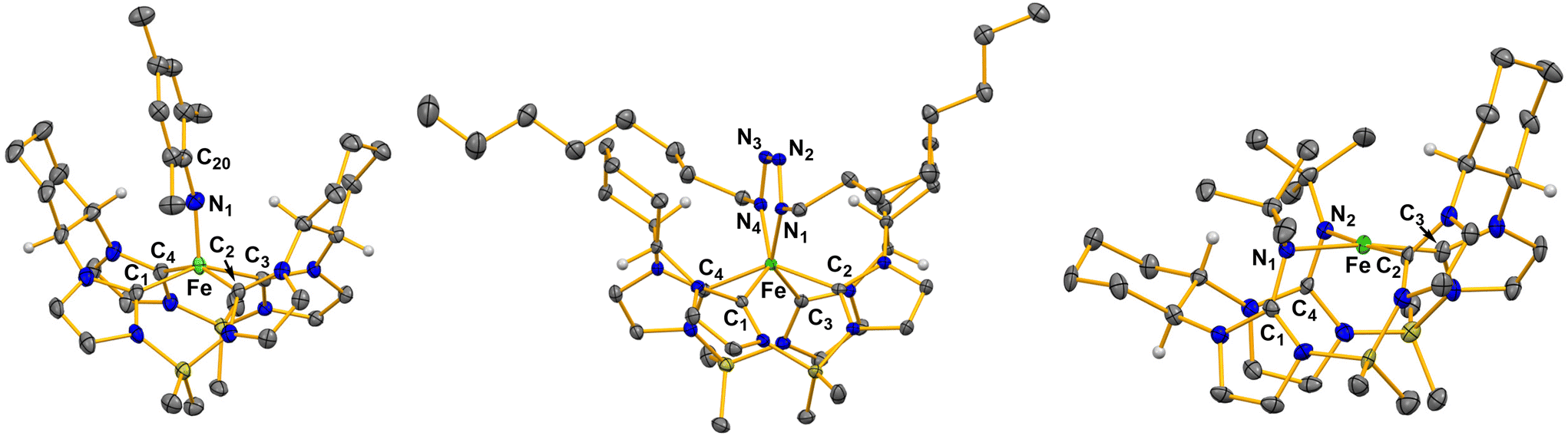 | ||
| Fig. 2 X-ray crystal structures of ((S,S)-1,2-Cy,BMe2TCH)Fe(NMes) (2), ((S,S)-1,2-Cy,BMe2TCH)Fe((n-octyl)N4(n-octyl)) (3), and ((S,S)-1,2-Cy,BMe2TCH)**Fe(NtButyl)2 (4). Green, blue, grey, olive, and white ellipsoids (50% probability) represent Fe, N, C, B, and H atoms, respectively. Solvent molecules and H-atoms on non-stereogenic carbons are omitted for clarity. | ||
With the successful isolation of the iron imide, we attempted a nitrene transfer reaction with an alkene to form an aziridine (Scheme 1). Three equivalents of 1-decene was added to 2 in C6D6 at 80 °C for 72 hours, but no transfer was observed (see ESI†), which is consistent with the vast majority of isolated iron imides excluding a key example by Betley.26
In contrast to the reaction with mesityl azide, five equivalents of octyl azide reacted with 1 producing a dark red solution (Scheme 1). After work up, ((S,S)-1,2-Cy,BMe2TCH)Fe((n-octyl)N4(n-octyl)) (3) was isolated as a burgundy-coloured powder in 76% yield. The 1H NMR spectrum of 3 showed a diamagnetic species with apparent C2 symmetry (Fig. S8†). The C2 symmetry was confirmed when analysing the 13C NMR carbon resonances at 171.26 and 161.10 ppm which correspond to two distinct carbene peaks (Fig. S9†).20 However, the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by integration of the octyl protons relative to the macrocycle protons in the 1H NMR suggested a tetrazene moiety.17 Single crystal X-ray diffraction for 3 verified the formation of an iron(IV) tetrazene (Fig. 2). Complex 3 has a rare trigonal prismatic geometry, which we have noted is consistent with our previous reported tetrazene complexes.17,27 The formation of a tetrazene from octyl azide can easily be rationalized by differences in steric bulk between the organic azides, where the mesityl azide is too large for an attack on the imide bond the octyl is small enough for a second equivalent to react easily.
:1 ratio by integration of the octyl protons relative to the macrocycle protons in the 1H NMR suggested a tetrazene moiety.17 Single crystal X-ray diffraction for 3 verified the formation of an iron(IV) tetrazene (Fig. 2). Complex 3 has a rare trigonal prismatic geometry, which we have noted is consistent with our previous reported tetrazene complexes.17,27 The formation of a tetrazene from octyl azide can easily be rationalized by differences in steric bulk between the organic azides, where the mesityl azide is too large for an attack on the imide bond the octyl is small enough for a second equivalent to react easily.
While the previous two reactions with 1 can be justified based on the size of the organic azide, the reaction of 1 with tert-butyl azide was unexpected. A reaction of five equivalents of tert-butyl azide with 1 yielded an amber solution that was similar in colour to 3. ((S,S)-1,2-Cy,BMe2TCH)**Fe(NtButyl)2 (4) was isolated in 63% yield after work up. The 1H NMR spectrum of 4 showed a paramagnetic species with 38 unique resonances (Fig. S13†), suggesting that the complex is asymmetric (C1).
Single crystal X-ray diffraction for 4 revealed a double insertion of two nitrene moieties into two NHCs (Fig. 2). The nitrogens bound to the iron are canted out of the plane of the tetra-NHCs leading to a square planar complex. The Fe–N1 and Fe–N2 bond distances are 2.029(3) Å and 1.995(3) Å, respectively. Evans’ method measurements on 4 gave a value of 3.14 μB, consistent with a square planar S = 1 iron(II) complex. Notably, reaction of 1 with p-tolyl azide also resulted in a double insertion product, but we could not isolate more than a few crystals (see ESI†).
Nitrene insertion into metal–NHC bonds from an organic azide source is a very rare phenomenon that has only been described in two cases. First, on a tripodal NHC cobalt complex by Meyer and, second, with its isostructural iron complex.19,28 For the iron complex, the insertion has resulted in a two electron reduction at the metal centre, with a reported iron nitrogen bond distance of 2.017(3) Å.
A mechanistic study based on density functional theory (DFT) calculations can assist in explaining the divergent reactivity between the three azides with 1. Each azide reacts with 1 to form an iron imide species with relatively low energy reaction barriers (TS1) which is consistent with their reactivities at room temperature (Fig. 3). Notably, the alkyl azides give calculated diamagnetic (S = 0) iron(IV) imide complexes at −35.6 kcal mol−1 (tBu) and −38.3 kcal mol−1 (octyl), which is consistent with our previous results (Fig. 3A, blue and red lines).11 Mesityl azide leads to a paramagnetic (S = 1) iron(IV) imide complex that is much lower in energy at −55.2 kcal mol−1 (Fig. 3A, black line).
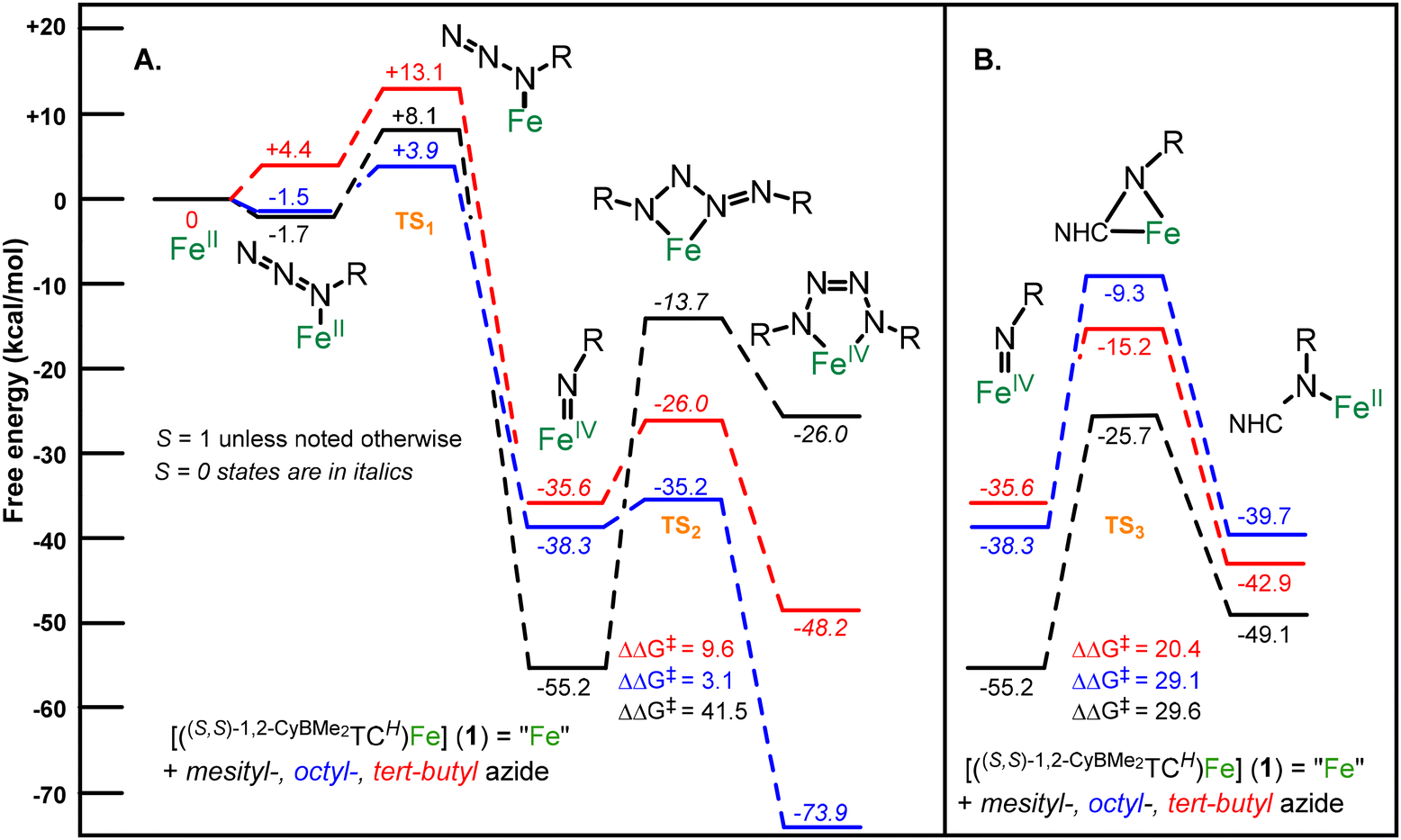 | ||
| Fig. 3 (A.) DFT-computed free-energy pathway for formation of iron imide from reaction between an organic azide and 1 followed by the further reactivity with an additional equivalent of azide to form an iron tetrazene. All species are in S = 1 (triplet) spin state unless designated by italics in which case they are in S = 0 (singlet) spin state. Structures shown at each interval represent the lowest energy spin state for each intermediate and product. TS designates a transition state. (B.) Depicts DFT-computed free-energy pathway for formation of insertion products starting at iron imides. Free energies (ΔG) are given in kcal mol−1. | ||
Three scenarios are possible upon formation of the imide complex: the imide complex reacts with additional azide to form a tetrazene, the imide inserts into the NHC, or the imide complex is thermodynamically stable. For the mesityl azide case, the imide complex is the most stable thermodynamic product (Fig. 3A and B, black lines). Both reactions are uphill in energy and, likewise, the kinetic barrier to further reaction is so high (ΔΔG‡ = 41.5 for tetrazene formation and 29.6 kcal mol−1 for imide insertion) as to be dubious at room temperature.
The octyl azide case demonstrates a key divergence between the two forward reaction pathways from an imide intermediate. The addition of a second equivalent of octyl azide must undergo only a 3.1 kcal mol−1 barrier (TS2) to form a diamagnetic tetrazene that is −73.9 kcal mol−1 lower in energy than the starting iron complex (Fig. 3A, blue line). While the competitive insertion reaction is slightly thermodynamically favourable, the kinetic barrier for the transition state TS3 is +29.1 kcal mol−1, which precludes this reaction at room temperature. This large reaction barrier is primarily a consequence of the steric repulsion between the long alkyl chains and the cyclohexane groups of the ligand. Finally, like the mesityl imide case, the calculated tetrazene product is in good agreement with the experimentally measured 3 and consistent with our previously reported tetrazene complexes (see ESI† for details).17
The calculated results for the tert-butyl azide reaction with 1 are more ambiguous. The formation of the imide complex is thermodynamically favourable, but at a somewhat higher energy barrier (+13.1 kcal mol−1versus1). Both the tetrazene formation and insertion into the NHC are similar in energy thermodynamically, although the formation of tetrazene has a lower activation energy (Fig. 3, red lines), which suggests tetrazene formation should take precedence. However, since no mechanism has been previously reported for an imide insertion into an NHC, there could be alternative pathways that have a lower kinetic barrier. A calculation of the thermodynamic free energy of the double insertion product 4 is −88.8 kcal mol−1 (see ESI†), suggesting that insertion product is far more stable than the tetrazene product at −48.2 kcal mol−1 (Fig. 3). Finally, the calculated structure for 4 reproduces the experimental X-ray crystal structure and experimentally determined spin state (S = 1).
In conclusion, a chiral iron(II) tetracarbene complex demonstrates highly disparate reactivity with a variety of organic azides, but exhibits no catalytic nitrene transfer activity. Mesityl azide reacted to form a stable S = 1 iron(IV) imide complex that was comparable to previous isostructural species. Alternatively, a reaction with octyl azide produced an diamagnetic iron(IV) tetrazene. Finally, we isolated the first bis-nitrene insertion into an NHC complex. Single crystal X-ray diffraction and magnetic data are consistent with a reduction to iron(II). Computational results assist in explaining the divergent reactivity between these azides.
Author contributions
J. J. R. performed final complex syntheses and collected spectroscopic data. J. F. D. performed initial complex syntheses and initial single crystal X-ray diffraction analysis. B. A. S. and K. D. V. performed theoretical calculations. P. N. performed final single crystal X-ray diffraction analysis. K. D. V. and D. M. J. designed and supervised the project. Manuscript was written and prepared by J. J. R., K. D. V., and D. M. J.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the National Science Foundation (NSF CHE-2154697) and NIH (R15GM117494-02) for support. We also thank the Infrastructure for Scientific Applications and Advanced Computing (ISAAC) for computational resources and the University of Tennessee for additional support. Any opinions, findings, and conclusions expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.References
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed
.
- C. Dank and L. Ielo, Org. Biomol. Chem., 2023, 21, 4553–4573 RSC
- D. M. Jenkins, Synlett, 2012, 1267–1270 CrossRef CAS
- Y. Liu, K.-P. Shing, V. K.-Y. Lo and C.-M. Che, ACS Catal., 2023, 13, 1103–1124 CrossRef CAS
- J. E. Zweig, D. E. Kim and T. R. Newhouse, Chem. Rev., 2017, 117, 11680–11752 CrossRef CAS PubMed
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed
- E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Chem. Sci., 2014, 5, 1526–1532 RSC
- P. Liu, E. L.-M. Wong, A. W.-H. Yuen and C.-M. Che, Org. Lett., 2008, 10, 3275–3278 CrossRef CAS PubMed
- S. A. Cramer and D. M. Jenkins, J. Am. Chem. Soc., 2011, 133, 19342–19345 CrossRef CAS PubMed
- S. B. Isbill, P. P. Chandrachud, J. L. Kern, D. M. Jenkins and S. Roy, ACS Catal., 2019, 9, 6223–6233 CrossRef CAS PubMed
- M. R. Anneser, G. R. Elpitiya, J. Townsend, E. J. Johnson, X. B. Powers, J. F. DeJesus, K. D. Vogiatzis and D. M. Jenkins, Angew. Chem., Int. Ed., 2019, 58, 8115–8118 CrossRef CAS PubMed
- M. Keilwerth, W. Mao, S. A. V. Jannuzzi, L. Grunwald, F. W. Heinemann, A. Scheurer, J. Sutter, S. DeBeer, D. Munz and K. Meyer, J. Am. Chem. Soc., 2023, 145, 873–887 CrossRef CAS PubMed
- M. Keilwerth, W. Mao, M. Malischewski, S. A. V. Jannuzzi, K. Breitwieser, F. W. Heinemann, A. Scheurer, S. DeBeer, D. Munz, E. Bill and K. Meyer, Nat. Chem., 2024, 16, 514–520 CrossRef CAS PubMed
- I. Nieto, F. Ding, R. P. Bontchev, H. Wang and J. M. Smith, J. Am. Chem. Soc., 2008, 130, 2716–2717 CrossRef CAS PubMed
- L. Wang, L. Hu, H. Zhang, H. Chen and L. Deng, J. Am. Chem. Soc., 2015, 137, 14196–14207 CrossRef CAS PubMed
- R. E. Cowley, E. Bill, F. Neese, W. W. Brennessel and P. L. Holland, Inorg. Chem., 2009, 48, 4828–4836 CrossRef CAS PubMed
- S. A. Cramer, R. Hernández Sánchez, D. F. Brakhage and D. M. Jenkins, Chem. Commun., 2014, 50, 13967–13970 RSC
- B. M. Hakey, J. M. Darmon, N. G. Akhmedov, J. L. Petersen and C. Milsmann, Inorg. Chem., 2019, 58, 11028–11042 CrossRef CAS PubMed
-
C. S. Vogel, High-and Low-Valent Tris-N-Heterocyclic Carbene Iron Complexes: A Study of Molecular and Electronic Structure, Springer Science & Business Media, 2012 Search PubMed
- J. F. DeJesus and D. M. Jenkins, Chem. – Eur. J., 2020, 26, 1429–1435 CrossRef CAS PubMed
- C. L. Keller, J. L. Kern, B. D. Terry, S. Roy and D. M. Jenkins, Chem. Commun., 2018, 54, 1429–1432 RSC
- P. P. Chandrachud, H. M. Bass and D. M. Jenkins, Organometallics, 2016, 35, 1652–1657 CrossRef CAS
- K. M. Blatchford, C. J. Mize, S. Roy and D. M. Jenkins, Dalton Trans., 2022, 51, 6153–6156 RSC
- S. Reith, S. Demeshko, B. Battistella, A. Reckziegel, C. Schneider, A. Stoy, C. Lichtenberg, F. Meyer, D. Munz and C. G. Werncke, Chem. Sci., 2022, 13, 7907–7913 RSC
- B. P. Jacobs, P. T. Wolczanski, Q. Jiang, T. R. Cundari and S. N. MacMillan, J. Am. Chem. Soc., 2017, 139, 12145–12148 CrossRef CAS PubMed
- E. R. King, E. T. Hennessy and T. A. Betley, J. Am. Chem. Soc., 2011, 133, 4917–4923 CrossRef CAS PubMed
- G. R. Elpitiya, B. J. Malbrecht and D. M. Jenkins, Inorg. Chem., 2017, 56, 14101–14110 CrossRef CAS PubMed
- X. Hu and K. Meyer, J. Am. Chem. Soc., 2004, 126, 16322–16323 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, including selected NMRs, IRs, X-ray analysis, and computational details (PDF). CCDC 2332148–2332151. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt01422g |
| This journal is © The Royal Society of Chemistry 2024 |