
Vapor-phase synthesis of low-valent metal–organic frameworks from metal carbonyl synthons†
Carl Emil
Andersen
 a,
James Neill
McPherson
a,
Mónica
Giménez-Marqués
b,
Jingguo
Li
c,
Mariusz
Kubus
a,
Sho
Ito
d,
Christian R.
Göb
e,
Sascha
Ott
c,
René Wugt
Larsen
a,
Guillermo
Mínguez Espallargas
b and
Kasper S.
Pedersen
*a
a,
James Neill
McPherson
a,
Mónica
Giménez-Marqués
b,
Jingguo
Li
c,
Mariusz
Kubus
a,
Sho
Ito
d,
Christian R.
Göb
e,
Sascha
Ott
c,
René Wugt
Larsen
a,
Guillermo
Mínguez Espallargas
b and
Kasper S.
Pedersen
*a
aDepartment of Chemistry, Technical University of Denmark, Kemitorvet 207, Kgs. Lyngby 2800, Denmark. E-mail: kastp@kemi.dtu.dk
bInstituto de Ciencia Molecular (ICMol), Universidad de Valencia, Paterna 46980, Valencia, Spain
cDepartment of Chemistry–Ångström Laboratory, Uppsala University, Uppsala 75120, Sweden
dRigaku Corporation, 3-9-12 Matsubara-cho, Akishima, Tokyo 196-8666, Japan
eRigaku Europe SE, Hugenottenallee 167, Neu-Isenburg 63263, Germany
First published on 12th July 2023
Abstract
Incorporating low-valent metals as nodes in metal–organic frameworks (MOFs) is orthogonal to the commonly accepted strategy to robust and porous structures. The electronic structures of low-valent metals are chemically unique and have driven the successes of organometallic chemistry. The translation of these properties into MOFs is largely impeded by the lack of generalizable synthetic approaches. Metal carbonyls are bona fide low-valent and volatile synthons, which, by partial chemical substitution may tether ditopic ligands into MOF architectures. We herein demonstrate the formation of CO2 adsorbing MOFs from the direct vapor-phase substitution of CO by ditopic 4,4′-bipyridine at Mo(0) and W(0) centers, which allows facile coating of low-valent MOFs onto substrates such as indium tin oxide. We envisage this strategy could be generalized to a multitude of mono- and polynuclear metal carbonyls, paralleling the diversity of conventional MOFs.
Introduction
Metal–organic frameworks (MOFs) bridge the fields of coordination chemistry and materials science,1 and have received tremendous attention during the past two decades due to their extreme compositional and structural diversity and ensuing physical and chemical properties.2 MOFs containing zero-valent metal nodes have only recently been reported.3–6 In stark contrast to conventional MOFs—which are largely ‘held together’ through labile electrostatic interactions between cationic metal nodes and anionic linkers—more covalent interactions between the charge-neutral building blocks dominate the bonding within zero-valent MOFs. It is tempting to speculate that such fundamental differences in bonding will manifest in new chemical, physical, and mechanical properties. For example, zero- or low-valent metal ion-based frameworks appear well-positioned to address challenges such as multi-electron reduction catalysis. Indeed, polypyridyl low-valent metal carbonyl complexes are well-known molecular electrocatalysts for two (or more) electron reductions of carbon dioxide.7–16 There is already considerable interest in developing methods to incorporate these functional units into existing MOF scaffolds,17–21 while very recently they have also proven to be active photoredox catalysts.22The applications of MOFs, for example as electrocatalysts23 or sensors,24 often require their deposition on various substrates.25 MOF synthesis and deposition protocols largely rely on solution-phase processes to control homogeneity and crystallization. Solvents typically account for the vast majority of the masses used in MOF synthesis and are often incompatible with the delicate substrates used in, for example, micro-electronic devices.26,27 As such, the synthesis of MOF films from the gas phase, thereby alleviating the requirement for solvents, has seen a surge of interest. For example, chemical vapor deposition has been used in all-gas syntheses of thin films,28,29 and also to convert thin layers of metal oxides into MOF-films of controlled thickness,30–32 while atomic layer deposition has been used for layer-by-layer growth by repeated treatment with metal ion precursors and organic linkers.33,34 In many cases, solvents such as water,35 acetic acid,33 or dimethylformamide,36 have still been required for the deposition or crystallization of thin films. We have previously demonstrated that the dense fac-M(CO)3(pyz)3/2 frameworks (M = Cr, Mo or W, and pyz = pyrazine) can be accessed from the group 6 metal hexacarbonyl precursors.6 The volatility of these (and related) homoleptic carbonyl precursors—in stark contrast to the metal salt precursors typically used for conventional MOFs—presents an opportunity to construct zero-valent MOFs from vapor-phase reactions. For example, magnetic thin films have been deposited using V(CO)6,37 and molecular organometallic films from Mo(CO)6 have been reported more recently.38
Results and discussion
To actuate porosity in fac-M(CO)3(pyz)3/2-type framework architectures, glass ampoules were charged with M(CO)6 (M = Mo, W) and 4,4′-bipyridine (bipy, mp 114 °C) before they were vacuum sealed and heated (Scheme 1). Black, microcrystalline powders of fac-M(CO)3(bipy)3/2 (M = Mo or W) were isolated directly from the reactions, and excess bipy could be removed by soaking and washing the powders with acetonitrile. Rod-shaped microcrystals were stable under inert atmospheres but deteriorated over several weeks when exposed to air. This decomposition was further accelerated by sunlight. While the crystals in microcrystalline W powders were too small for conventional single-crystal X-ray diffraction analysis, their structure could be determined from continuous rotation 3D electron diffraction (3D-ED, Fig. 1 and Fig. S1, S3 and Tables S1, S3, ESI†), an emerging technique tailored directly for sub-micron crystals.39 The structure of W was solved in the monoclinic P2/c space group and constitutes three-dimensional, four-fold interpenetrated networks—vastly different to the two crystallographic phases previously reported for the fac-M(CO)3(pyz)3/2 frameworks.6 Two crystallographically independent fac-{W(CO)3(bipy)3/2} units were found in the asymmetric unit, which are assembled into tetrahedral {W(CO)3(bipy)}4 secondary building units (SBUs), with W centres at the four vertices (Fig. 1a). The bridging bipy ligands span four of the six edges of the tetrahedra, and each tetrahedron is linked to four other tetrahedra through bipy bridges. When viewed down the crystallographic c axis, the bipy bridges connect the upper vertices of a tetrahedron to the lower vertices of its neighbours, and vice versa, and as such, these form octagonal spirals winding along the c axis (Fig. 1b and Fig. S3, S4, ESI†), connecting eight W centres before superimposing on itself. The chirality of spirals which share a common bipy bridge are, necessarily, inverted (Fig. S5, ESI†). The pitch of each spiral is double the length of c, and so each spiral is ‘double threaded’—a second identical network (orange sticks in Fig. S3, ESI†) is wound between the thread of the octagonal spiral. The networks formed by these parallel threads are interpenetrated by another pair of parallel networks (green and pink sticks in Fig. S3, ESI†), offset by half a unit cell along b to predominantly occupy the large cylindrical voids inside the spirals. Three types of solvent accessible cavities make up the remainder of the structure—one inside the {W(CO)3(bipy)}4 tetrahedra and two types between the exterior walls of the tetrahedra of neighbouring, interpenetrated nets (Fig. S6, ESI†)—which combine to account for 26% (1063 Å3) of the unit cell volume. Larger crystals of Mo could be isolated directly from the analogous reaction of Mo(CO)6 and bipy and were suitable for single crystal X-ray diffraction structural analysis (Fig. S7, ESI†). Whilst Mo crystallizes in the lower symmetry, triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Tables S2 and S3, ESI†), its network can be described by the same topology as for W. Four independent Mo atoms form the vertices of the tetrahedral {Mo(CO)3(bipy)}4 SBUs, which are again linked by bridging bipy ligands to form the double-threaded octagonal spirals (down the a axis). Significant electron density (∼307 e− unit cell−1) was found in the pores of pristine Mo, which accounted for a similar volume (1041 Å3 or 26%) of the unit cell as in W. Three partially occupied (and positionally disordered) bipy guests were modelled in these voids, accounting for the majority of the residual electron density, with a formulation of 3.2 bipy guests per unit cell. Impurity phases of one-dimensional M(CO)3(bipy)2 (Mo*, W*, Fig. S8, ESI†) chains were also isolated, but are avoided through the reported, optimized synthetic procedures. The C, H, N and Mo or W elemental analysis results of evacuated Mo or W powders agreed with those expected for the empty-pore formulation, M(CO)3(bipy)3/2 (cf. ESI†).
(Tables S2 and S3, ESI†), its network can be described by the same topology as for W. Four independent Mo atoms form the vertices of the tetrahedral {Mo(CO)3(bipy)}4 SBUs, which are again linked by bridging bipy ligands to form the double-threaded octagonal spirals (down the a axis). Significant electron density (∼307 e− unit cell−1) was found in the pores of pristine Mo, which accounted for a similar volume (1041 Å3 or 26%) of the unit cell as in W. Three partially occupied (and positionally disordered) bipy guests were modelled in these voids, accounting for the majority of the residual electron density, with a formulation of 3.2 bipy guests per unit cell. Impurity phases of one-dimensional M(CO)3(bipy)2 (Mo*, W*, Fig. S8, ESI†) chains were also isolated, but are avoided through the reported, optimized synthetic procedures. The C, H, N and Mo or W elemental analysis results of evacuated Mo or W powders agreed with those expected for the empty-pore formulation, M(CO)3(bipy)3/2 (cf. ESI†).
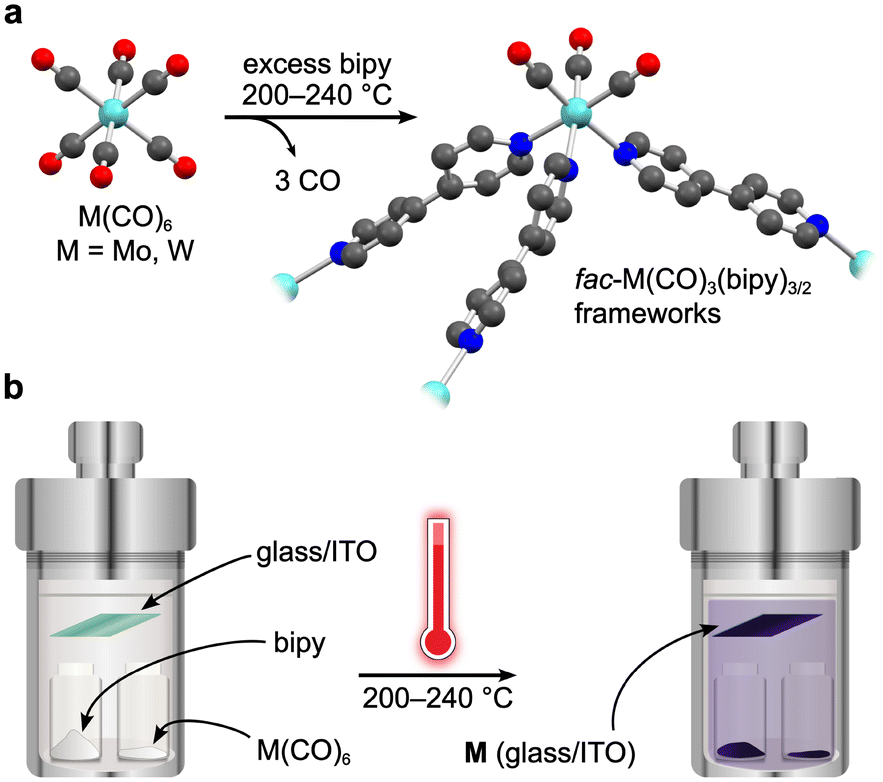 | ||
| Scheme 1 General synthetic route to fac-M(CO)3(bipy)3/2 (a), M = Mo, W) and a schematic presentation of the simplistic vapor-phase synthesis method (b). | ||
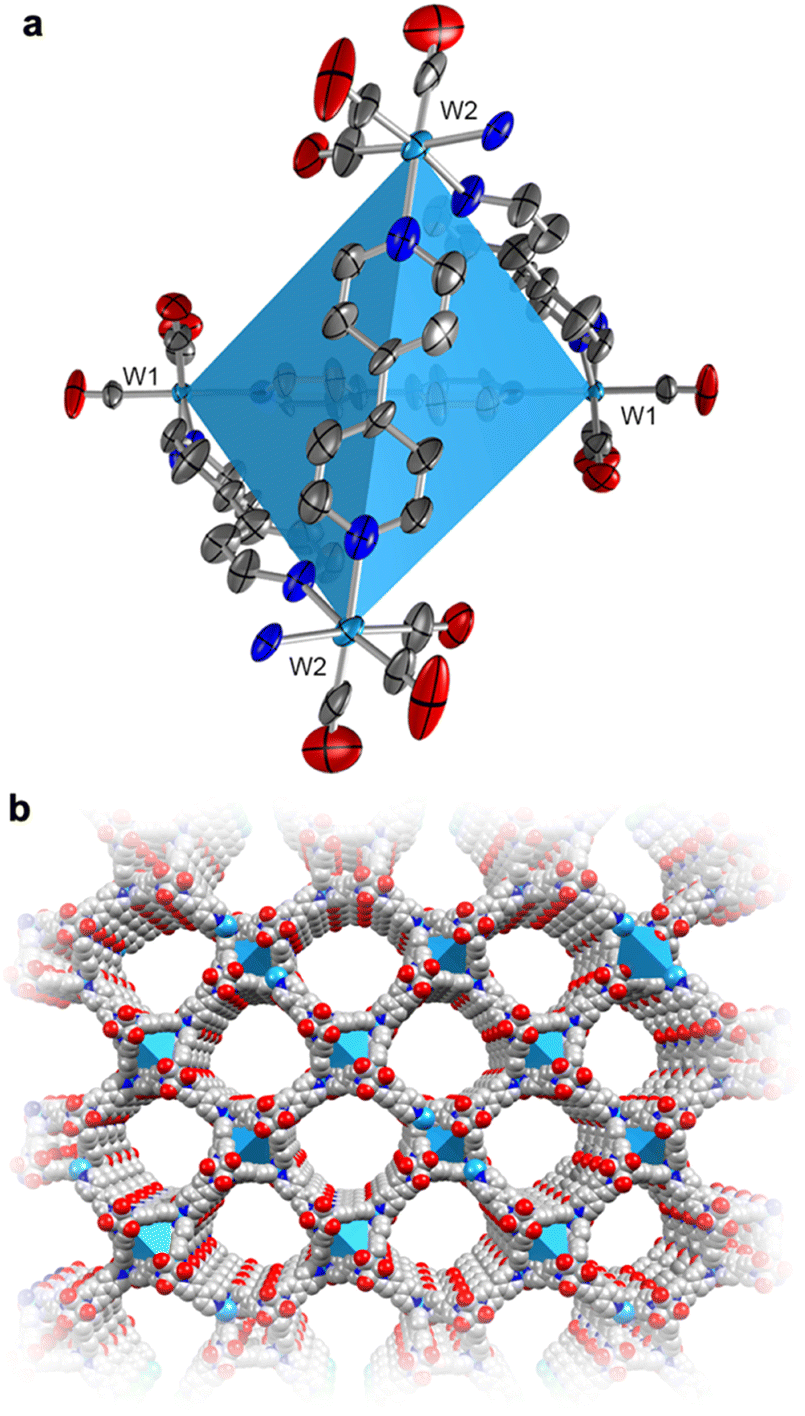 | ||
| Fig. 1 (a) Room-temperature 3D-ED structure of the tetrahedral SBU, {W(CO)3(bipy)}4, of W, showing the thermal displacement ellipsoids at 30% probability level. The coordination sphere is completed by half a bipy for each W center (with the N atom depicted). (b) The crystal structure of a single polymeric network of W as viewed down the crystallographic c axis (cf. Fig. S3, ESI†). H-atoms and the three identical, interpenetrated networks have been omitted for clarity. Color codes: W, light blue; O, red; N blue; C, grey. | ||
While Mo and W were heated to temperatures exceeding 200 °C during the synthesis, Mo and W decompose at ∼150 °C in a dry Ar stream (Fig. S9, ESI†). Soaking Mo in dichloromethane or tetrahydrofuran followed by filtration and drying at RT resulted in subtle changes to the powder X-ray diffractograms (Fig. S11, ESI†). These changes can be reversed by heating at 70 °C in a dynamic vacuum, leading to identical powder diffraction patterns, suggesting the removal of any solvent molecules without collapse of the Mo framework. Activation of Mo overnight at 100 °C under dynamic vacuum was also possible without the collapse of the framework, as confirmed by powder X-ray diffraction (Fig. S12, ESI†). Attenuated-total-reflectance Fourier-transform infrared (ATR-FTIR) spectroscopy confirmed that CO was retained at the metals during this process (Fig. S16, ESI†). The porosity of activated Mo was demonstrated through CO2 gas sorption experiments at 273 K. The uptake amounts to 0.7 mmol CO2 g−1, corresponding to ∼1.1 equivalent per {Mo(CO)3(bipy)}4 SBU, at p(CO2) = 1 bar (Fig. 2). Mo showed negligible N2 adsorption under similar conditions, a phenomenon often observed for materials with narrow pores.40
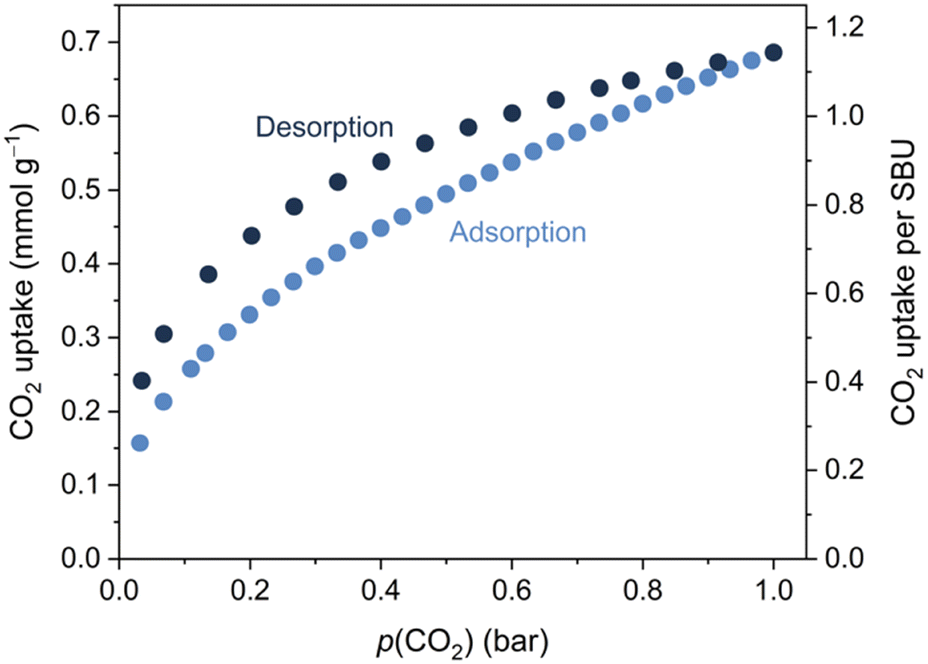 | ||
| Fig. 2 Low-pressure CO2 adsorption and desorption isotherms of bulk Mo obtained at T = 273 K. | ||
None of the previously reported fac-M(CO)3(pyz)3/2 networks showed permanent porosity: the 3D cubic phases were too densely packed to accommodate guests, while the 2D sheets collapsed on activation.6 The new topology that has emerged from the longer bipy linker has, despite the four-fold interpenetration, installed modest porosity in this new class of M(0)-tricarbonyl frameworks.
The volatility of the starting materials prompted the exploitation of a fully vapor-phase approach to Mo and W. Heating a Teflon-lined autoclave containing the M(CO)6 and bipy precursors in separate vials below silica glass and indium tin oxide (ITO) (Scheme 1) substrates led to the formation of coatings, which were confirmed to consist of phase-pure W and Mo by powder X-ray diffraction analysis (Fig. 3 and Fig. S13, ESI,† respectively). Similar to the bulk products described above, the Mo and W coatings are moderately stable in air but decompose more rapidly when exposed to sunlight. No signs of decomposition were observed when Mo and W coatings were kept under inert atmospheres. The powder X-ray diffractograms of Mo and W coatings on glass or ITO were in excellent agreement with the experimental and simulated patterns of bulk Mo and W superimposed onto the backgrounds matching the diffractograms of the clean coatings.
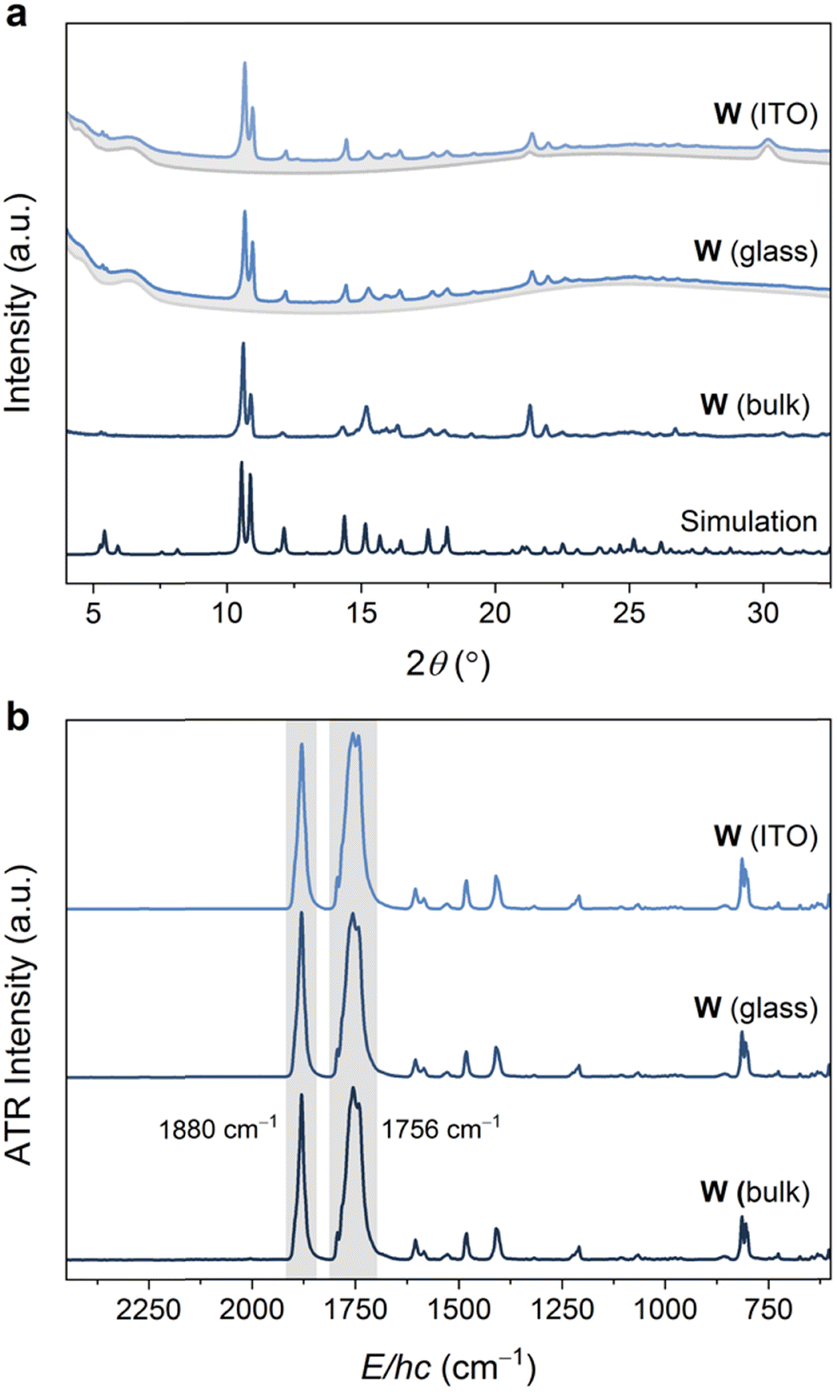 | ||
| Fig. 3 (a) Powder X-ray diffractograms (Cu Kα, λ = 1.5406 Å) of W coated on ITO and glass (blue traces, with grey traces representing uncoated substrate), compared with an experimental diffractogram of bulk W, as well as a simulated diffractogram. (b) ATR-FTIR spectra of W coated on ITO and glass, compared with the experimental spectrum collected on bulk W. See Fig. S13 and S17 (ESI†) for analogous Mo data. | ||
The ATR-FTIR spectra of the W or Mo coatings were identical to those collected for the bulk MOFs (Fig. 3b and Fig. S17, ESI†). In particular, the spectra are dominated by two intense bands assigned to IR-active E-asymmetric (∼1765 cm−1 for Mo, 1756 cm−1 for W) and A1-symmetric (∼1893 cm−1 for Mo, ∼1879 cm−1 for W) CO stretching modes for the pseudo-C3v symmetric fac-M(CO)3(bipy)3/2 nodes. These energies are significantly red-shifted from that of free CO (2143 cm−1) and the M(CO)6 precursors (2027–2029 cm−1),41 reflecting the weak π-acidity of bipy. While the A1 fundamentals were observed at the same energies (within ±4 cm−1) as those previously reported for fac-M(CO)3(pyz)3/2, the E fundamentals for the bipy networks, Mo or W, were observed at even lower energies (by ∼45 cm−1 for Mo, ∼55 cm−1 for W) than those in fac-M(CO)3(pyz)3/2. The overall larger redshifts of the CO stretching fundamentals may reflect the stronger basicity of bipy compared to pyz.42 Scanning electron microscopy (SEM) images were obtained for Mo and W-coated glass and ITO. The images (Fig. 4 and Fig. S18–S22, ESI†) reveal rod-shaped μm-sized crystallites coating the substrates. The macroscopic topology of Mo (ITO) could be modified by changing the vapor-phase reaction conditions. Smaller crystallites could be produced by rapid heating and shorter reaction time (Fig. 4 and Fig. S18, S19, ESI†) without compromising crystallinity (Fig. S15, ESI†).
 | ||
| Fig. 4 SEM images of Mo (ITO) fabricated with a reaction time of 5 h (a) and 2 h (b) (cf. Fig. S18–S22, ESI†). | ||
The electrochemical properties of Mo (ITO) were investigated by cyclic voltammetry (see ESI†). A pseudo-reversible redox couple was observed at E1/2 ≈ −0.4 V vs. Fc+/Fc (ΔEp = 235 mV, ip,c/ip,a = 87%) (Fig. 5 and Fig. S23, ESI†). Kitzmann et al. have recently assigned a reversible couple at E1/2 = −0.20 V (vs. Fc+/Fc) to the one-electron oxidation of the structurally related fac-Mo(CO)3(tpe) (tpe = 1,1,1-tris(pyrid-2-yl)ethane) complex in DMF.22 Also shown in Fig. 5 and Fig. S23 (ESI†) are the CVs of the analogous molecular complex. The CVs for fac-Mo(CO)3(py)3 (py = pyridine) were obtained under identical conditions to Mo (ITO), but with an untreated ITO glass coated slide working electrode (Fig. 5 and Fig. S23, ESI†). While the reversibility of the redox couple was poorer for fac-Mo(CO)3(py)3 (ΔEp = 370 mV, ip,c/ip,a = 40%), we are confident in assigning the couple observed at −0.4 V to the same cycling between Mo(I) and Mo(0) in both Mo (ITO) and fac-Mo(CO)3(py)3. When scanning through more reducing potentials (below −1 V vs. Fc+/Fc), large currents were observed for the Mo (ITO) electrode (Fig. S23, ESI†). These currents appear to be related to decomposition of the framework, as subsequent cycles produce dramatically different CVs. The instability of the Mo coating under strongly reducing conditions is unsurprising, as reduction of the molecular fac-Mo(CO)3(py)3 analogue is known to lead to chemically unstable products.43
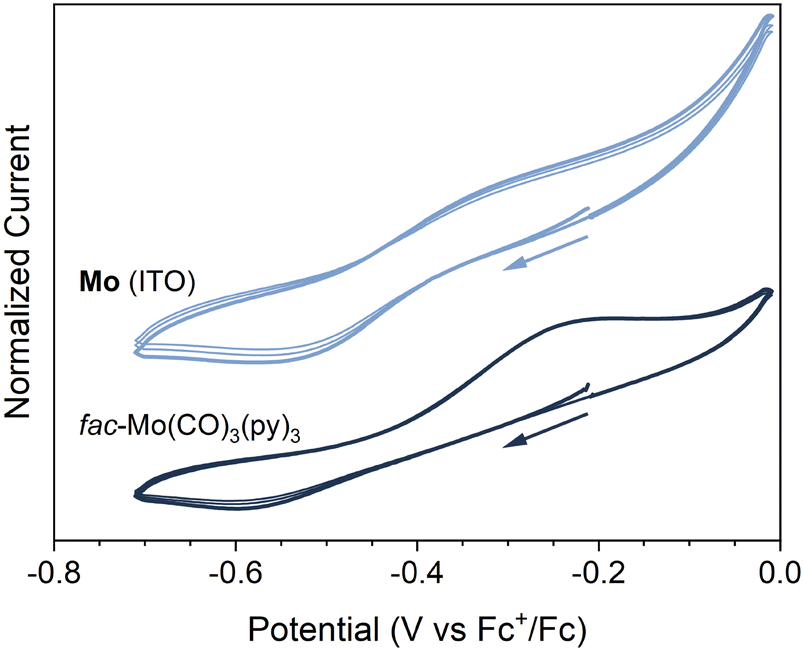 | ||
| Fig. 5 Smoothed and normalized cyclic voltammograms (three cycles, ν = 100 mV s−1, 0.1 M LiClO4 in DMF) of an Mo (ITO) working electrode (light blue) and a DMF solution of fac-Mo(CO)3(py)3 at a bare ITO working electrode (dark blue). | ||
Conclusions
In summary, we have presented a route to robust, porous, and redox-active low-valent MOFs, using chemical substitution at homoleptic hexacarbonyl precursors. The use of high-volatility starting materials enabled deposition of phase-pure, crystalline MOF coatings on glass or ITO substrates. The Mo (ITO) materials serve as electrodes which retain the ability to cycle between Mo(I) and Mo(0). This strategy to coat conductive substrates with electroactive MOFs may lead to novel hybrid electrodes for, e.g., electrocatalysis and sensing. Reversible CO2 uptake was shown for these porous compounds, the nodes of which resemble those found in (Mo/W)(CO)4(diimine)7–10 and (Mn/Re)X(CO)3(diimine)13–15 electrocatalysts. These perspectives are further broadened by the synthetic simplicity and the plethora of mono- and polynuclear metal carbonyls as potential synthons for low-valent MOFs.Author contributions
C. E. A. carried out the synthesis of all compounds, performed the SEM- and powder X-ray diffraction analysis. M. K. collected single crystal X-ray diffraction data, and the data were solved and refined by M. K. and J. N. M., S. I. and C. R. G. carried out the 3D-ED analysis and refinement. R. W. L. performed the IR-analysis and subsequent data treatment. M. G.-M. and G. M. E. carried out the gas sorption experiments. J. L. and S. O. performed the electrochemical characterizations. K. S. P. conceived and supervised the project. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. S. P. thanks the Carlsberg Foundation for a Carlsberg Foundation Young Researcher Fellowship (CF21-0416), and the Independent Research Fund Denmark for a DFF-Research Project 1 grant (0135-00291B) and a Sapere Aude: DFF-Starting Grant (0165-00073B). The work has been supported by grants PID2020-117177GB-I00, PID2020-118564GA-I00 and CEX2019-000919-M, funded by MCIN/AEI/10.13039/501100011033. J. L. and S. O. acknowledge financial support from the Olle Engkvists Foundation (212-0147).Notes and references
- C. S. Diercks, M. J. Kalmutzki, N. J. Diercks and O. M. Yaghi, ACS Cent. Sci., 2018, 4, 1457–1464 CrossRef CAS PubMed.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS PubMed.
- D. W. Agnew, I. M. Dimucci, A. Arroyave, M. Gembicky, C. E. Moore, S. N. MacMillan, A. L. Rheingold, K. M. Lancaster and J. S. Figueroa, J. Am. Chem. Soc., 2017, 139, 17257–17260 CrossRef CAS.
- R. E. Sikma and S. M. Cohen, Angew. Chem., Int. Ed., 2022, 61, e202115454 CrossRef CAS.
- R. E. Sikma, K. P. Balto, J. S. Figueroa and S. M. Cohen, Angew. Chem., Int. Ed., 2022, 61, e202206353 CrossRef CAS.
- L. Voigt, R. Wugt Larsen, M. Kubus and K. S. Pedersen, Chem. Commun., 2021, 57, 3861–3864 RSC.
- F. Franco, C. Cometto, F. Sordello, C. Minero, L. Nencini, J. Fiedler, R. Gobetto and C. Nervi, ChemElectroChem, 2015, 2, 1372–1379 CrossRef CAS.
- N. W. Kinzel, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 11628–11686 CrossRef CAS.
- B. Rajeshwaree, A. Ali, A. Q. Mir, J. Grover, G. K. Lahiri, A. Dutta and D. Maiti, Catal. Sci. Technol., 2022, 12, 390–408 RSC.
- M. L. Clark, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Chem. Sci., 2014, 5, 1894–1900 RSC.
- K. A. Grice and C. Saucedo, Inorg. Chem., 2016, 55, 6240–6246 CrossRef CAS PubMed.
- D. Sieh, D. C. Lacy, J. C. Peters and C. P. Kubiak, Chem. – Eur. J., 2015, 21, 8497–8503 CrossRef CAS PubMed.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS.
- A. Zhanaidarova, S. C. Jones, E. Despagnet-Ayoub, B. R. Pimentel and C. P. Kubiak, J. Am. Chem. Soc., 2019, 141, 17270–17277 CrossRef CAS PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1984, 328–330 RSC.
- K. Kamogawa, Y. Shimoda, K. Miyata, K. Onda, Y. Yamazaki, Y. Tamaki and O. Ishitani, Chem. Sci., 2021, 12, 9682–9693 RSC.
- P. M. Stanley, J. Haimerl, N. B. Shustova, R. A. Fischer and J. Warnan, Nat. Chem., 2022, 14, 1342–1356 CrossRef CAS.
- K. M. Choi, D. Kim, B. Rungtaweevoranit, C. A. Trickett, J. T. D. Barmanbek, A. S. Alshammari, P. Yang and O. M. Yaghi, J. Am. Chem. Soc., 2017, 139, 356–362 CrossRef CAS PubMed.
- M. B. Majewski, A. W. Peters, M. R. Wasielewski, J. T. Hupp and O. K. Farha, ACS Energy Lett., 2018, 3, 598–611 CrossRef CAS.
- T. Kajiwara, M. Fujii, M. Tsujimoto, K. Kobayashi, M. Higuchi, K. Tanaka and S. Kitagawa, Angew. Chem., Int. Ed., 2016, 55, 2697–2700 CrossRef CAS PubMed.
- A. J. Blake, N. R. Champness, T. L. Easun, D. R. Allan, H. Nowell, M. W. George, J. Jia and X. Z. Sun, Nat. Chem., 2010, 2, 688–694 CrossRef CAS PubMed.
- W. R. Kitzmann, M.-S. Bertrams, P. Boden, A. C. Fischer, R. Klauer, J. Sutter, R. Naumann, C. Förster, G. Niedner-Schatteburg, N. H. Bings, J. Hunger, C. Kerzig and K. Heinze, J. Am. Chem. Soc., 2023 DOI:10.1021/jacs.3c03832.
- Y. H. Xiao, Z. G. Gu and J. Zhang, Nanoscale, 2020, 12, 12712–12730 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS.
- J. Meng, X. Liu, C. Niu, Q. Pang, J. Li, F. Liu, Z. Liu and L. Mai, Chem. Soc. Rev., 2020, 49, 3142–3186 RSC.
- A. J. Cruz, I. Stassen, M. Krishtab, K. Marcoen, T. Stassin, S. Rodríguez-Hermida, J. Teyssandier, S. Pletincx, R. Verbeke, V. Rubio-Giménez, S. Tatay, C. Martí-Gastaldo, J. Meersschaut, P. M. Vereecken, S. de Feyter, T. Hauffman and R. Ameloot, Chem. Mater., 2019, 31, 9462–9471 CrossRef CAS.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- M. Choe, J. Y. Koo, I. Park, H. Ohtsu, J. H. Shim, H. C. Choi and S. S. Park, J. Am. Chem. Soc., 2022, 144, 16726–16731 CrossRef CAS.
- F. J. Claire, M. A. Solomos, J. Kim, G. Wang, M. A. Siegler, M. F. Crommie and T. J. Kempa, Nat. Commun., 2020, 11, 5524 CrossRef CAS PubMed.
- I. Stassen, M. Styles, G. Grenci, H. van Gorp, W. Vanderlinden, S. de Feyter, P. Falcaro, D. de Vos, P. Vereecken and R. Ameloot, Nat. Mater., 2016, 15, 304–310 CrossRef CAS.
- T. Stassin, I. Stassen, J. Marreiros, A. J. Cruz, R. Verbeke, M. Tu, H. Reinsch, M. Dickmann, W. Egger, I. F. J. Vankelecom, D. E. de Vos and R. Ameloot, Chem. Mater., 2020, 32, 1784–1793 CrossRef CAS.
- T. Stassin, S. Rodríguez-Hermida, B. Schrode, A. J. Cruz, F. Carraro, D. Kravchenko, V. Creemers, I. Stassen, T. Hauffman, D. de Vos, P. Falcaro, R. Resel and R. Ameloot, Chem. Commun., 2019, 55, 10056–10059 RSC.
- K. B. Lausund and O. Nilsen, Nat. Commun., 2016, 7, 13578 CrossRef CAS PubMed.
- E. Ahvenniemi and M. Karppinen, Chem. Commun., 2016, 52, 1139–1142 RSC.
- J. K. Huang, N. Saito, Y. Cai, Y. Wan, C. C. Cheng, M. Li, J. Shi, K. Tamada, V. C. Tung, S. Li and L. J. Li, ACS Mater. Lett., 2020, 2, 485–491 CrossRef CAS.
- L. D. Salmi, M. J. Heikkilä, E. Puukilainen, T. Sajavaara, D. Grosso and M. Ritala, Microporous Mesoporous Mater., 2013, 182, 147–154 CrossRef CAS.
- K. I. Pokhodnya, A. J. Epstein and J. S. Miller, Adv. Mater., 2000, 12, 410–413 CrossRef CAS.
- M. Choe, S. Kim and H. C. Choi, RSC Adv., 2020, 10, 9536–9538 RSC.
- T. Yang, T. Willhammar, H. Xu, X. Zou and Z. Huang, Nat. Protoc., 2022, 17, 2389–2413 CrossRef CAS PubMed.
- O. Cheung and N. Hedin, RSC Adv., 2014, 4, 14480–14494 RSC.
- C. D. L. Cruz and N. Sheppard, J. Mol. Struct., 1990, 224, 141–161 CrossRef.
- J. K. Clegg, L. F. Lindoy, J. C. McMutrie and D. Schilter, Dalton Trans., 2006, 3114–3121 RSC.
- R. E. Dessy, R. B. King and M. Waldrop, J. Am. Chem. Soc., 1966, 88, 5112–5117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, additional physical characterization details and data. CCDC 2233877–2233880. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3tc02088f |
| This journal is © The Royal Society of Chemistry 2023 |