
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNitrogen atom insertion into indenes to access isoquinolines†
Patrick
Finkelstein
 ,
Julia C.
Reisenbauer
,
Bence B.
Botlik
,
Ori
Green
,
Andri
Florin
and
Bill
Morandi
*
,
Julia C.
Reisenbauer
,
Bence B.
Botlik
,
Ori
Green
,
Andri
Florin
and
Bill
Morandi
*
Laboratorium für Organische Chemie, ETH Zürich, Vladimir-Prelog-Weg 3, HCI, 8093 Zürich, Switzerland. E-mail: bill.morandi@org.chem.ethz.ch
First published on 23rd February 2023
Abstract
We report a convenient protocol for a nitrogen atom insertion into indenes to afford isoquinolines. The reaction uses a combination of commercially available phenyliodine(III) diacetate (PIDA) and ammonium carbamate as the nitrogen source to furnish a wide range of isoquinolines. Various substitution patterns and commonly used functional groups are well tolerated. The operational simplicity renders this protocol broadly applicable and has been successfully extended towards the direct interconversion of cyclopentadienes into the corresponding pyridines. Furthermore, this strategy enables the facile synthesis of 15N labelled isoquinolines, using 15NH4Cl as a commercial 15N source.
Introduction
Isoquinoline is an important aromatic N-heterocycle scaffold with numerous applications in medicinal chemistry,1–6 materials science,7–9 and as a ligand in catalysis.10–13 Traditionally, this heterocycle has been synthesised through the assembly of pre-oxidised building blocks and amines,14–16 or oxidation of di- or tetrahydroisoquinoline,17,18 among other methods.19–21 A strategically different approach is the introduction of the key nitrogen atom at a later stage of a synthetic route, using a pre-decorated carbocyclic framework. A classical strategy to accomplish this task relies on the stepwise oxidative cleavage of an indene skeleton, usually through ozonolysis, followed by condensation with an amine (Scheme 1A).22–24 Recently, more direct methods have emerged, such as an electrochemical approach using gaseous ammonia as the nitrogen source,25 but which is only compatible with electron-rich indenes bearing additional aryl and alkyl substitutions on the double bond. Finally, an osmium nitride was shown to stoichiometrically react with 3-phenyl-1H-indene, which, after a subsequent step gave 1-phenylisoquinoline.26 However, the need for stoichiometric metal and the synthesis of the starting osmium nitride limits synthetic applications. These challenges highlight the demand for a synthetically useful and practical approach to directly transform indenes into isoquinolines.27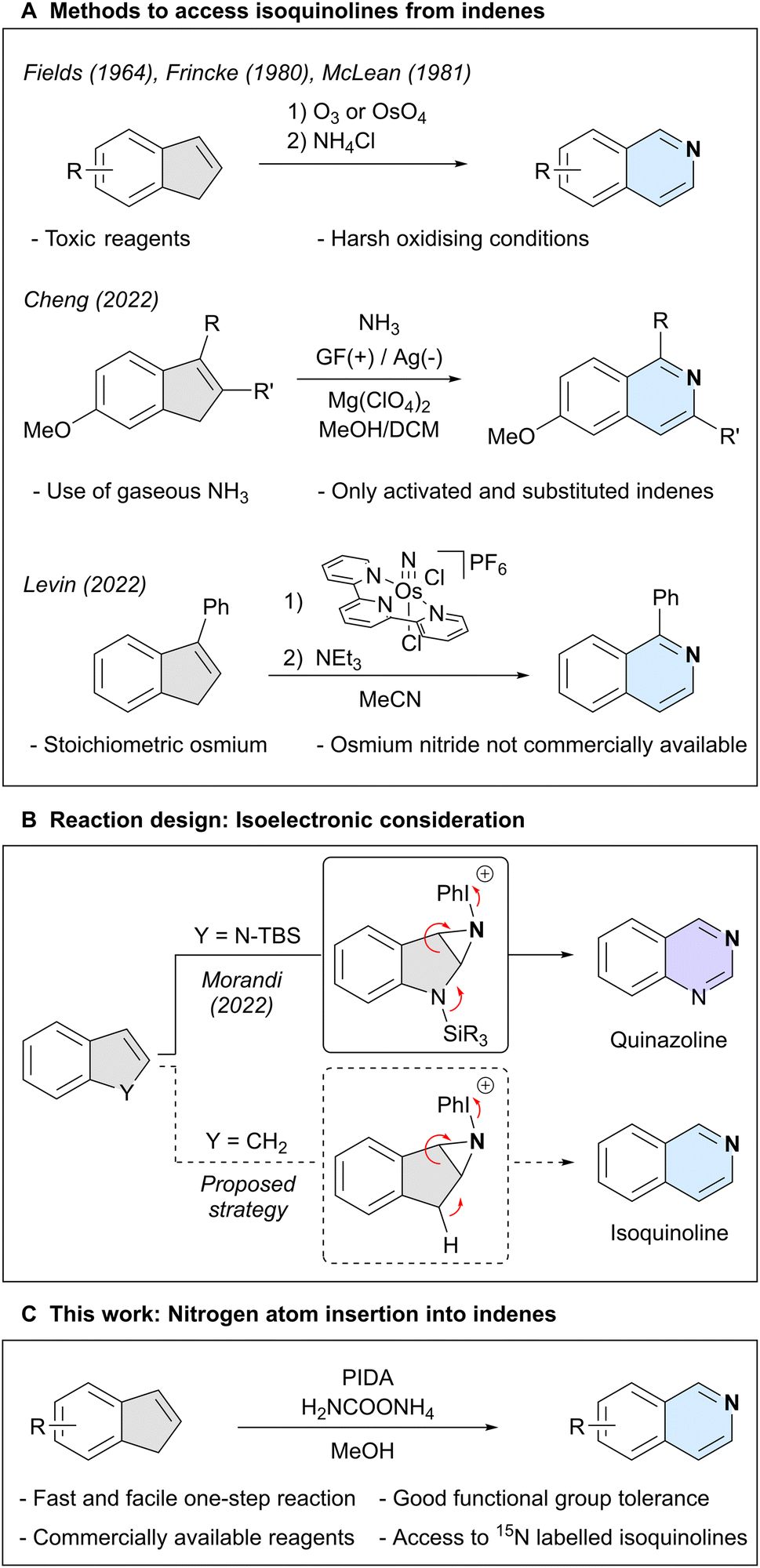 | ||
| Scheme 1 Context of this work. | ||
Our group has recently reported a nitrogen atom insertion method into silyl-protected indoles, affording either quinazolines or quinoxalines in a single step.28 In our proposed mechanism we postulated nitrogen lone pair participation in the fragmentation of an N-iodonium-aziridine intermediate. We surmised that indenes, which possess an acidic C–H moiety in place of the indole nitrogen atom, could possibly engage in a similar process through isoelectronic reactive intermediates (Scheme 1B). If successful, this would result in a simple and efficient conversion of indenes into isoquinolines.
Here, we report the direct nitrogen atom insertion into a broad range of indenes and cyclopentadienes, employing commercially available phenyliodine(III) diacetate (PIDA) as the oxidant and ammonium carbamate as the nitrogen source. We also report an extension of this protocol to enable the formation of isotopically labelled isoquinoline structures using commercially accessible 15NH4Cl (Scheme 1C).
Results and discussion
We chose 3-phenyl-1H-indene 1a as our model substrate and started our study by using two equivalents of phenyliodine(III)-bis(trifluoroacetate) (PIFA) as the oxidant and four equivalents of ammonium carbamate as the nitrogen source. The combination of a hypervalent iodine compound and an ammonia source—proposed to generate an iodonitrene in situ—has been used successfully in several transformations in recent years.28–31 Our hypothesis was validated by observing the formation of the desired 1-phenylisoquinoline product 1b (Table 1, entry 1) in methanol at 0 °C after 10 minutes in 77% yield (as determined by 1H-NMR analysis of the crude reaction mixture, for more details see ESI†). However, further increasing the amount of oxidant and ammonia source did not improve the yield (Table 1, entry 2). We continued by investigating the remaining parameters of the reaction, starting with different ammonia sources and observed that these also led to the desired product, albeit in lower quantities (Table 1, entries 3–4). The reaction also proceeded in aprotic solvents such as MeCN (Table 1, entry 5), providing a potential alternative for substrates that are incompatible with protic conditions. Changing the oxidant had the largest net positive effect, with both bis(tert-butylcarbonyloxy)iodobenzene and PIDA increasing the NMR yield to over 90% (Table 1, entries 6–7). Finally, 2 equivalents of PIDA in combination with 4 equivalents of ammonium carbamate in MeOH at 0 °C proved to give the highest yield for our transformation. Other parameters such as the temperature (0 °C vs. r.t.) or concentration (0.033−0.20 M) only had minor effects on the overall outcome of the reaction (see ESI†). When scaling up the reaction to 1.0 mmol, we increased the oxidant loading to 2.5 equivalents to reach full conversion and thus were able to isolate the product 1b in 63% yield (Table 1, entry 8). In summary, the transformation of indene 1a into the corresponding isoquinoline 1b was rapidly achieved open-flask in a variety of conditions including multiple solvents, oxidants, and ammonia sources, highlighting the robustness of our protocol.|
|
||
|---|---|---|
| Entry | Deviation from above | Yieldb of 1b [%] |
| a Reaction conditions: indene (0.05 mmol), PIFA (0.10 mmol), ammonium carbamate (0.20 mmol), methanol-d4 (0.07 M), 0 °C, 10 min. b Yields in % obtained by 1H-NMR analysis of the crude reaction mixture using 1,1,2,2-tetrachloroethane as the internal standard. c Isolated yield. | ||
| 1 | None | 77 |
| 2 | PIFA (4.0 equiv.), ammonium carbamate (6.0 equiv.) | 72 |
| 3 | Ammonium formate instead of ammonium carbamate | 46 |
| 4 | Ammonium acetate instead of ammonium carbamate | 70 |
| 5 | MeCN instead of MeOH | 62 |
| 6 | Bis(tert-butylcarbonyloxy)iodobenzene instead of PIFA | 91 |
| 7 | PIDA instead of PIFA | 93 |
| 8 | 1-mmol scale, PIDA (2.5 equiv.) instead of PIFA | 77, (63)c |

With the optimised conditions in hand, we set out to examine the functional group tolerance and limitations of our method. For all reactions, we report the 1H-NMR yield on a 0.05-mmol scale and the isolated yield on a 1.00-mmol scale (Scheme 2). In the following sections, we discuss the substrate scope based on the NMR yields, to systematically compare substitution patterns and functional group effects.
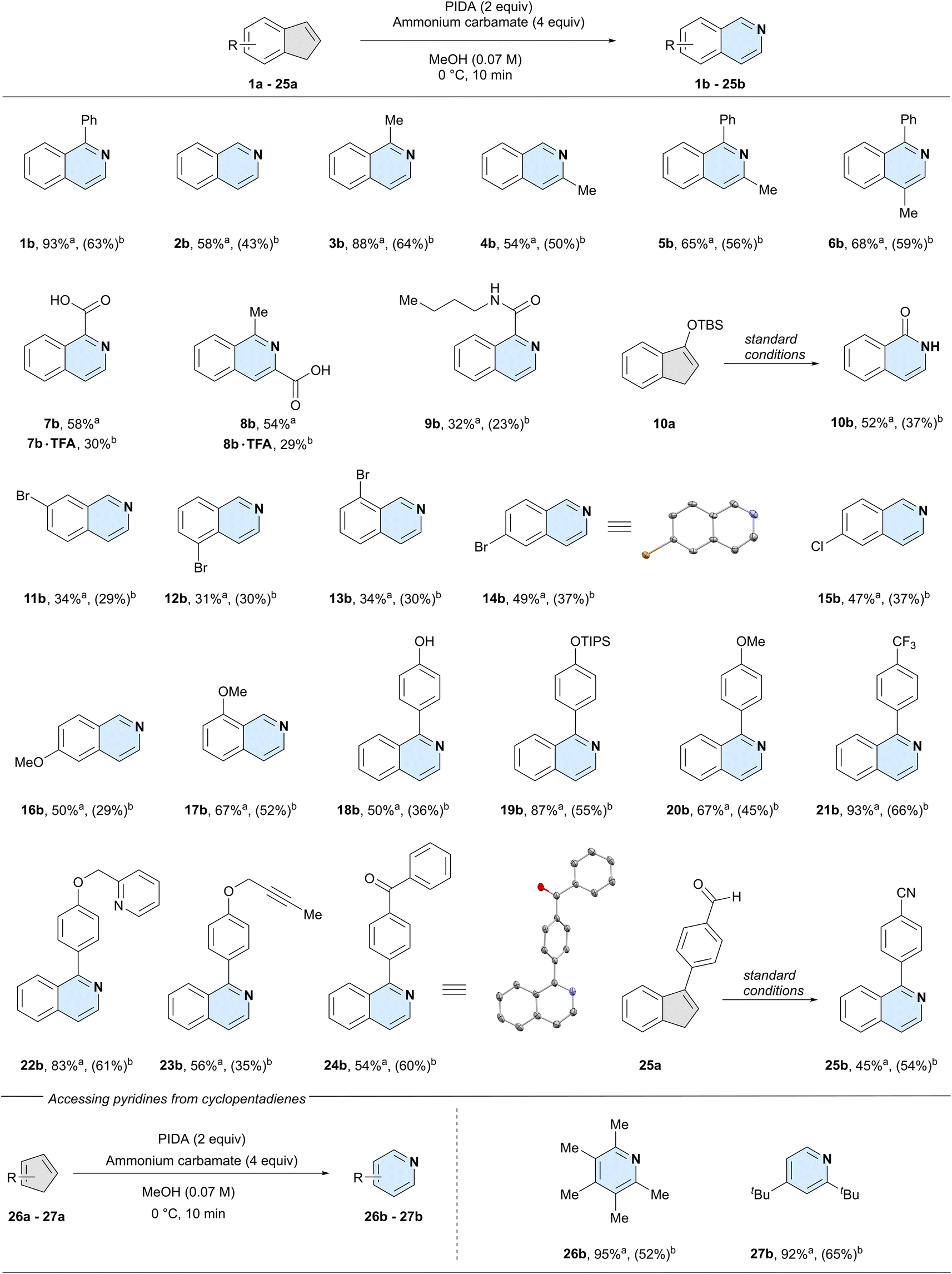 | ||
| Scheme 2 Substrate scope. For the scXRD structures, the ellipsoids are shown at 50% probability and hydrogen atoms are omitted for clarity.a 1H-NMR yield of 0.05-mmol scale crude reaction mixture using 1,1,2,2-tetrachloroethane as the internal standard.b Isolated yield of a 1.00-mmol scale reaction, conditions: indene (1.0 mmol), PIDA (2.5 mmol), ammonium carbamate (4.0 mmol), methanol (0.07 M), 0 °C for 20 min, then rt for 10 min. | ||
Unsubstituted indene 2a could successfully be converted into isoquinoline (2b), however, the lack of a phenyl ring at the 3-position led to a reduced, yet synthetically useful yield of 58%. This is an important feature, as recently reported one-step methods are not compatible with simple unsubstituted indene substrates.25,26 We next systematically investigated the effect of substitutions at the double bond of the five-membered ring. Like the model system, 3-phenyl-1H-indene (1a), 3-methyl-1H-indene (3a) gave the product 1-methylisoquinoline (3b) in high yield (88%), possibly hinting at the beneficial effect of a substitution at the 3-position. A methyl group at the 2-position of the indene (4a) gave 54% yield of 3-methylisoquinoline (4b), similar to the unsubstituted system. Next, we installed a phenyl at the 3-position and compared the results for a methyl group at either the 2- (5a), or 1-position (6a). Both reactions gave essentially identical yields (5b: 65% and 6b: 68%), albeit higher than the previous substrates with no phenyl substitution at the 3-position. Additionally, as showcased by accessing isoquinoline 5b from the tetrasubstituted indene precursor 5a, steric hindrance was well tolerated under the developed reaction conditions.
We next investigated the effect of polar groups at the 2- or 3-position on the reaction outcome. Indenes bearing a carboxylic acid on either 3- (7a) or 2-position (8a) gave the corresponding isoquinolines in comparable yields (7b: 58% and 8b: 54%). Likewise, an amide on the double bond of the indene starting material was also tolerated under the reaction conditions (9b). Intriguingly, silyl-protected alcohol 10a was converted into the lactam 10b, with loss of the silyl group.
We next compared different substituents on the six-membered ring. Substrates bearing a bromine atom at any of the four possible positions on the benzene ring were tolerated in the reaction (11a, 12a, 13a, and 14a). Similarly diminished yields were obtained when the bromide was either in the 4- (11b: 34%), 5- (12b: 31%), or 7-position (13b: 34%) of the starting indene, respectively. In contrast, 6-bromo-1H-indene 14a gave a yield closer to that of the unsubstituted system (14b: 49% vs.2b: 58%). We observed a similar yield when subjecting 6-chloro-1H-indene 15a to our reaction conditions, giving 15b in 47% yield. To investigate a potential electronic effect at the 6-position of the indene, we exchanged the halides for an electron-donating methoxy group. The reaction afforded 6-methoxyisoquinoline 16b in 50% yield, suggesting that electronic effects at this position do not influence the reaction's yield. In comparison, a methoxy group closer to the reaction centre resulted in an increase in yield to 67% of 8-methoxyisoquinoline 17b.
Having established that reactivity could be expected both with or without substitutions at any position on the indene core, we further explored the functional group tolerance of the reaction. A free phenol (18a) was well tolerated and gave phenol-containing isoquinoline 18b in 50% yield. Protecting the phenol with a tri-isopropyl silyl group (19a) resulted in an even higher yield of product 19b (87%). Other groups in para position on the phenyl group were well tolerated, such as a methoxy (20b: 67%) or a trifluoromethyl group (21b: 93%). Combining these results with that of our model system 1b, it seems that the reaction yield is not influenced by the electronics of the pendant aryl substituents.
We further investigated other common functional groups, including a benzylic pyridine (22a) which was converted into the corresponding product 22b in 83% yield, an internal alkyne (23a), which gave the product 23b in 56% yield, and an indene bearing a ketone (24a) which afforded the desired product 24b in 54% yield. As aldehydes are prone to oxidise to nitriles in the presence of ammonia and an oxidant,32 we wondered whether we could achieve both the oxidation and the nitrogen insertion simultaneously. Indeed, aldehyde 25a was converted to the nitrile product 25b in 45% yield. Finally, we decided to expand our scope beyond indenes by testing two cyclopentadienes. We chose 1,2,3,4,5-pentamethyl cyclopentadiene and 1,3-di-tert-butylcyclopentadiene as our two substrates, due to their higher stability compared to the unsubstituted parent structure. To our delight, both 2,3,4,5,6-pentamethyl pyridine 26b and 2,4-di-tert-butylpyridine 27b were formed in high yields (26b: 95%, 27b: 92%), clearly demonstrating the possibility to synthesise densely functionalised pyridine products from cyclopentadienes. Overall, commonly used functional groups were well tolerated in the reaction and synthetically useful yields were observed for non-, mono-, and di-substituted indene cores.
Apart from easily accessing isoquinolines, an intriguing application of our method would be the incorporation of the heavier 15N isotope, which has a natural abundance of only around 0.3%. 15N labelled molecules have been used in many different settings for their nuclear magnetic resonance (NMR)33 and mass-related properties, such as in proteomics,34 as sensitive protonation probes,35,36 reaction-progress and complexation monitoring of COFs,37,38 and more generally for studies on N-heterocycles.39–4115N NMR spectroscopy has some significant advantages compared to the method using the lighter isotope,42 but suffers from high costs for the typically laborious syntheses of 15N labelled precursors. Thus, an inexpensive protocol to incorporate the valuable 15N-label at a later stage in the synthesis could be attractive to access relevant labelled organic or organometallic structures.
We thus set out to evaluate whether our protocol could insert 15N into indenes to form isotopically labelled isoquinolines. Due to the lack of commercially available 15N ammonium carbamate, we changed our nitrogen source to NH4Cl and envisaged that adding a base could unlock the desired reactivity.29,43 Surprisingly, when using four equivalents of NH4Cl as the ammonia source without any base, we could already see conversion of 1a to the product 1b, albeit in low quantities (Table 2, entry 1). By adding a base and changing the equivalents of the reagents, the reaction was optimised to afford the product 1b in 80% yield on a 0.05-mmol scale and 49% NMR yield on a 1.0-mmol scale (Table 2, entries 2–7).
|
|
||
|---|---|---|
| Entry | Deviation from above | Yieldb of 1b [%] |
| a Reaction conditions: indene (0.05 mmol), PIDA (0.10 mmol), ammonium chloride (0.20 mmol), methanol-d4 (0.07 M), 0 °C, 10 min. b Yields in % obtained by 1H-NMR analysis of the crude reaction using 1,1,2,2-tetrachloroethane as the internal standard. | ||
| 1 | None | 7 |
| 2 | With NaOAc (4.0 equiv.) | 27 |
| 3 | With K3PO4 (4.0 equiv.) | 48 |
| 4 | With K2CO3 (4.0 equiv.) | 69 |
| 5 | With K2CO3 (2.0 equiv.) | 71 |
| 6 | NH4Cl (2.0 equiv.), K2CO3 (2.0 equiv.) | 80 |
| 7 | 1-mmol scale, NH4Cl (2.0 equiv.), K2CO3 (2.0 equiv.), PIDA (2.5 equiv.) | 49 |

We next used the isotopically labelled salt, 15NH4Cl, to convert 3-phenyl-1H-indene (1a) into 1-phenylisoquinoline-15N (15N-1b) in 51% isolated yield (Scheme 3A). We confirmed the incorporation of the heavier nitrogen isotope by high-resolution mass spectrometry (HRMS) and NMR analysis. The usual characteristic doublet in the 1H NMR spectrum of the H at the 3-position of 1b was split into a doublet of doublets (dd, J = 10.9, 5.7 Hz), as was the signal corresponding to the H in the 4-position (ddd, J = 5.7, 2.0, 0.9 Hz). We recorded a 15N NMR spectrum, noting a doublet of doublets at δ 306.7 (dd, J = 10.8, 1.9 Hz). This splitting is caused by the coupling of the nitrogen with the adjacent hydrogens at the 3- and 4-position of the isoquinoline, which was also confirmed by 2D 1H–15N-HMBC experiments (see ESI†).
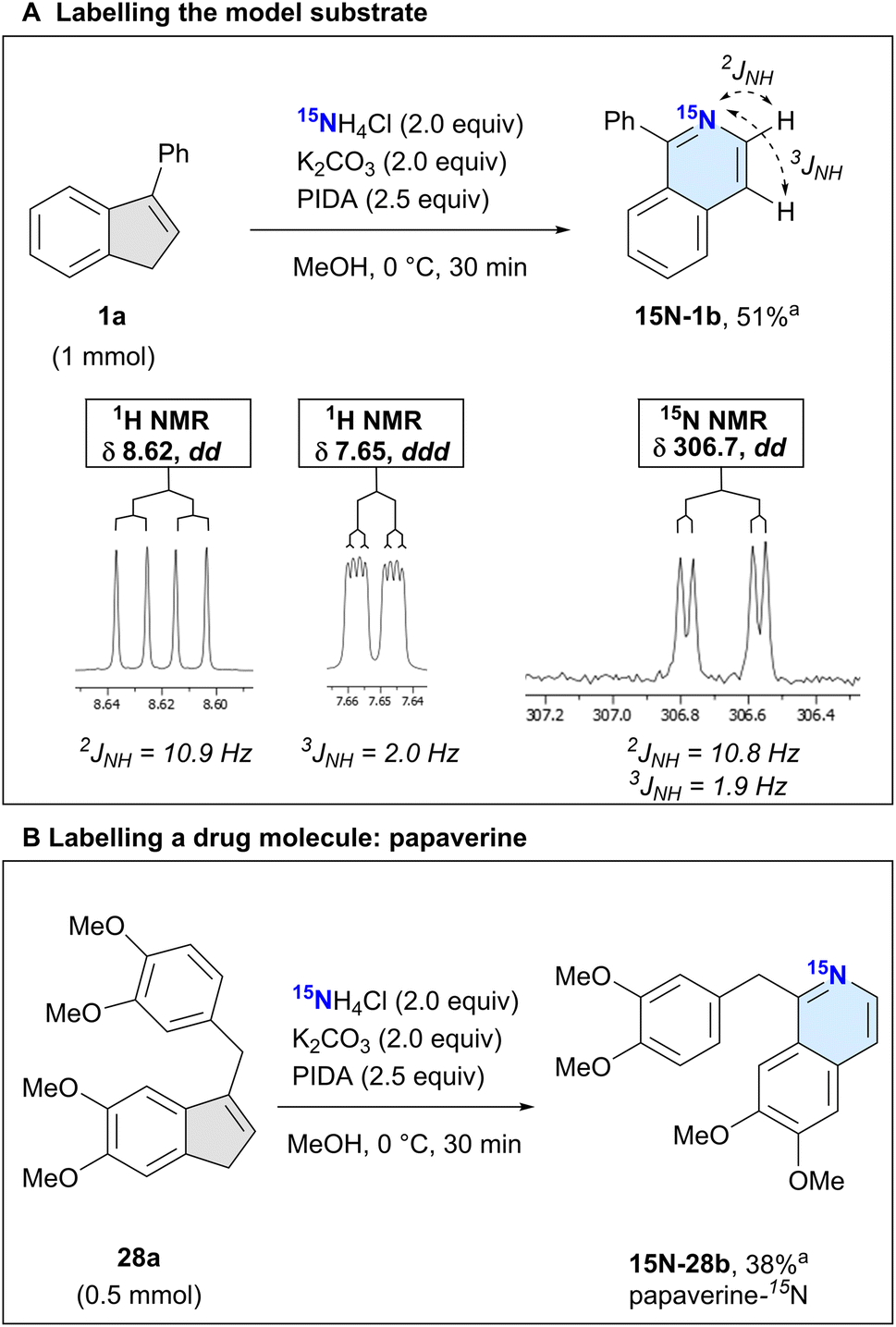 | ||
| Scheme 3 Isotopically labelled structures. aIsolated yields. | ||
After this successful initial example, we next sought to demonstrate the utility of our labelling method by incorporating 15N into the isoquinoline-based drug papaverine, which is used to treat visceral spasms and vasospasms.44 Having access to an isotopically labelled drug can greatly help identify metabolic pathways.45,46 Subjecting 28a to our reaction conditions, we obtained the product papaverine-15N (15N-28b) in 38% isolated yield (Scheme 3B). As with 15N-1b, we confirmed the nitrogen isotope incorporation by HRMS and 1H/15N NMR analyses.
Conclusions
In conclusion, we report a facile method to insert a nitrogen atom into indenes and cyclopentadienes, granting access to a wide variety of differently substituted and functionalised isoquinolines and two bulky pyridines. We could further expand our method to use 15NH4Cl as the nitrogen source, allowing for the convenient synthesis of 15N labelled isoquinolines.Data availability
All experimental and characterization data, as well as NMR spectra are available in the ESI.† Crystallographic data for compounds 14b and 24b have been deposited in the Cambridge Crystallographic Data Centre under accession number CCDC 2221959 (14b) and 2221960 (24b).Author contributions
P. F. conceived the project. P. F., J. C. R., B. B. B., O. G., and A. F. performed the experimental studies. B. M. supervised the research. P. F. wrote the original draft of the manuscript which was edited by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by ETH Zürich, the Swiss National Science Foundation (SNSF 184658), and the European Research Council under the European Union's Horizon 2020 Research and Innovation Program (Shuttle Cat, project ID: 757608). J. C. R. acknowledges a fellowship from the Stipendienfonds der Schweizerischen Chemischen Industrie (SSCI). O. G. acknowledges a fellowship from the International Human Frontier Science Program Organization (grant LT000861/2020-L). We thank the NMR, MS (MoBiAS), and X-ray (SMoCC) service departments at ETH Zürich for technical assistance and the Morandi group for critical proofreading of the manuscript.Notes and references
- H. Hidaka, M. Inagaki, S. Kawamoto and Y. Sasaki, Biochemistry, 1984, 23, 5036–5041 CrossRef CAS PubMed.
- M. S. C. Pedras, A. Abdoli and V. K. Sarma-Mamillapalle, Molecules, 2017, 22, 1345 CrossRef PubMed.
- M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS PubMed.
- J. W. Wilson, N. D. Dawson, W. Brooks and G. E. Ullyot, J. Am. Chem. Soc., 1949, 71, 937–938 CrossRef CAS PubMed.
- S. Theeramunkong, A. Thiengsusuk, O. Vajragupta and P. Muhamad, Med. Chem. Res., 2021, 30, 109–119 CrossRef CAS.
- M. Croisy-Delcey, A. Croisy, D. Carrez, C. Huel, A. Chiaroni, P. Ducrot, E. Bisagni, L. Jin and G. Leclercq, Bioorg. Med. Chem., 2000, 8, 2629–2641 CrossRef CAS PubMed.
- P. S. Hariharan, E. M. Mothi, D. Moon and S. P. Anthony, ACS Appl. Mater. Interfaces, 2016, 8, 33034–33042 CrossRef CAS PubMed.
- Y. Chen, C. Dai, X. Xu, Y. Zhou, Y. Lei, M. Liu, W. Gao, X. Huang and H. Wu, J. Phys. Chem. C, 2021, 125, 24180–24188 CrossRef CAS.
- K.-H. Fang, L.-L. Wu, Y.-T. Huang, C.-H. Yang and I.-W. Sun, Inorg. Chim. Acta, 2006, 359, 441–450 CrossRef CAS.
- B. A. Sweetman, H. Müller-Bunz and P. J. Guiry, Tetrahedron Lett., 2005, 46, 4643–4646 CrossRef CAS.
- G. Cheng, P. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 8183–8186 CrossRef CAS PubMed.
- C. W. Lim, O. Tissot, A. Mattison, M. W. Hooper, J. M. Brown, A. R. Cowley, D. I. Hulmes and A. J. Blacker, Org. Process Res. Dev., 2003, 7, 379–384 CrossRef CAS.
- P. Wang, G. Liang, M. R. Reddy, M. Long, K. Driskill, C. Lyons, B. Donnadieu, J. C. Bollinger, C. E. Webster and X. Zhao, J. Am. Chem. Soc., 2018, 140, 9219–9229 CrossRef CAS PubMed.
- L. Zheng, J. Ju, Y. Bin and R. Hua, J. Org. Chem., 2012, 77, 5794–5800 CrossRef CAS PubMed.
- S.-G. Lim, J. H. Lee, C. W. Moon, J.-B. Hong and C.-H. Jun, Org. Lett., 2003, 5, 2759–2761 CrossRef CAS PubMed.
- Z. Xiang, T. Luo, K. Lu, J. Cui, X. Shi, R. Fathi, J. Chen and Z. Yang, Org. Lett., 2004, 6, 3155–3158 CrossRef CAS PubMed.
- M. A. Esteruelas, V. Lezáun, A. Martínez, M. Oliván and E. Oñate, Organometallics, 2017, 36, 2996–3004 CrossRef CAS.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS PubMed.
- K. R. Roesch and R. C. Larock, Org. Lett., 1999, 1, 553–556 CrossRef CAS.
- R. He, Z.-T. Huang, Q.-Y. Zheng and C. Wang, Tetrahedron Lett., 2014, 55, 5705–5713 CrossRef CAS.
- K. Narasimhan and P. Raja Kumar, Heterocycles, 1984, 22, 1369 CrossRef CAS.
- R. B. Miller and J. M. Frincke, J. Org. Chem., 1980, 45, 5312–5315 CrossRef CAS.
- M. I. Fremery and E. K. Fields, J. Org. Chem., 1964, 29, 2240–2243 CrossRef CAS.
- D. S. Dime and S. McLean, J. Org. Chem., 1981, 46, 4999–5000 CrossRef CAS.
- S. Liu and X. Cheng, Nat. Commun., 2022, 13, 425 CrossRef CAS PubMed.
- P. Q. Kelly, A. S. Filatov and M. D. Levin, Angew. Chem., Int. Ed., 2022, 61, e202213041 CAS.
- During the final preparation of this manuscript, an independently developed strategy to access isoquinolines from indenes was reported: J. Wang, H. Lu, Y. He, C. Jing and H. Wei, J. Am. Chem. Soc., 2022, 144, 22433–22439 CrossRef CAS PubMed.
- J. C. Reisenbauer, O. Green, A. Franchino, P. Finkelstein and B. Morandi, Science, 2022, 377, 1104–1109 CrossRef CAS PubMed.
- T. Glachet, H. Marzag, N. Saraiva Rosa, J. F. P. Colell, G. Zhang, W. S. Warren, X. Franck, T. Theis and V. Reboul, J. Am. Chem. Soc., 2019, 141, 13689–13696 CrossRef CAS PubMed.
- A. Tota, M. Colella, C. Carlucci, A. Aramini, G. Clarkson, L. Degennaro, J. A. Bull and R. Luisi, Adv. Synth. Catal., 2021, 363, 194–199 CrossRef CAS.
- M. Zenzola, R. Doran, L. Degennaro, R. Luisi and J. A. Bull, Angew. Chem., Int. Ed., 2016, 55, 7203–7207 CrossRef CAS PubMed.
- C. Zhu, L. Ji and Y. Wei, Synthesis, 2010, 18, 3121–3125 Search PubMed.
- N. V. Chukanov, R. V. Shchepin, S. M. Joshi, M. S. H. Kabir, O. G. Salnikov, A. Svyatova, I. V. Koptyug, J. G. Gelovani and E. Y. Chekmenev, Chem.–Eur., 2021, 27, 9727–9736 CrossRef CAS PubMed.
- T. Geiger, J. Cox, P. Ostasiewicz, J. R. Wisniewski and M. Mann, Nat. Methods, 2010, 7, 383–385 CrossRef CAS PubMed.
- W. Jiang, L. Lumata, W. Chen, S. Zhang, Z. Kovacs, A. D. Sherry and C. Khemtong, Sci. Rep., 2015, 5, 9104 CrossRef PubMed.
- I. G. Shenderovich, G. Buntkowsky, A. Schreiber, E. Gedat, S. Sharif, J. Albrecht, N. S. Golubev, G. H. Findenegg and H.-H. Limbach, J. Phys. Chem. B, 2003, 107, 11924–11939 CrossRef CAS.
- Q. Li, W. Zhang, O. Š. Miljanić, C.-H. Sue, Y.-L. Zhao, L. Liu, C. B. Knobler, J. F. Stoddart and O. M. Yaghi, Science, 2009, 325, 855–859 CrossRef CAS PubMed.
- S. J. Lyle, T. M. Osborn Popp, P. J. Waller, X. Pei, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2019, 141, 11253–11258 CrossRef CAS PubMed.
- S. L. Deev, I. A. Khalymbadzha, T. S. Shestakova, V. N. Charushin and O. N. Chupakhin, RSC Adv., 2019, 9, 26856–26879 RSC.
- M. S. Solum, K. L. Altmann, M. Strohmeier, D. A. Berges, Y. Zhang, J. C. Facelli, R. J. Pugmire and D. M. Grant, J. Am. Chem. Soc., 1997, 119, 9804–9809 CrossRef CAS.
- R. Marek, O. Humpa, J. Dostál, J. Slavík and V. Sklenář, Magn. Reson. Chem., 1999, 37, 195–202 CrossRef CAS.
- G. Bodenhausen and D. J. Ruben, Chem. Phys. Lett., 1980, 69, 185–189 CrossRef CAS.
- J.-F. Lohier, T. Glachet, H. Marzag, A.-C. Gaumont and V. Reboul, Chem. Commun., 2017, 53, 2064–2067 RSC.
- G. H. Whipple, Angiology, 1977, 28, 737–749 CrossRef CAS PubMed.
- G. R. Waller, R. Ryhage and S. Meyerson, Anal. Biochem., 1966, 16, 277–286 CrossRef CAS PubMed.
- A. Le, A. N. Lane, M. Hamaker, S. Bose, A. Gouw, J. Barbi, T. Tsukamoto, C. J. Rojas, B. S. Slusher, H. Zhang, L. J. Zimmerman, D. C. Liebler, R. J. C. Slebos, P. K. Lorkiewicz, R. M. Higashi, T. W. M. Fan and C. V. Dang, Cell Metab., 2012, 15, 110–121 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2221959 and 2221960. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06952k |
| This journal is © The Royal Society of Chemistry 2023 |