
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanistic studies of the palladium-catalyzed S,O-ligand promoted C–H olefination of aromatic compounds†
Kananat
Naksomboon
a,
Enrique
Gómez-Bengoa
 b,
Jaya
Mehara
c,
Jana
Roithová
c,
Edwin
Otten
d and
M. Ángeles
Fernández-Ibáñez
*a
b,
Jaya
Mehara
c,
Jana
Roithová
c,
Edwin
Otten
d and
M. Ángeles
Fernández-Ibáñez
*a
aVan't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: m.a.fernandezibanez@uva.nl
bDepartment of Organic Chemistry I, Universidad País Vasco, UPV/EHU, Apdo. 1072, 20080, San Sebastian, Spain
cInstitute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands
dStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 16th February 2023
Abstract
Pd-catalyzed C–H functionalization reactions of non-directed substrates have recently emerged as an attractive alternative to the use of directing groups. Key to the success of these transformations has been the discovery of new ligands capable of increasing both the reactivity of the inert C–H bond and the selectivity of the process. Among them, a new type of S,O-ligand has been shown to be highly efficient in promoting a variety of Pd-catalyzed C–H olefination reactions of non-directed arenes. Despite the success of this type of S,O-ligand, its role in the C–H functionalization processes is unknown. Herein, we describe a detailed mechanistic study focused on elucidating the role of the S,O-ligand in the Pd-catalyzed C–H olefination of non-directed arenes. For this purpose, several mechanistic tools, including isolation and characterization of reactive intermediates, NMR and kinetic studies, isotope effects and DFT calculations have been employed. The data from these experiments suggest that the C–H activation is the rate-determining step in both cases with and without the S,O-ligand. Furthermore, the results indicate that the S,O-ligand triggers the formation of more reactive Pd cationic species, which explains the observed acceleration of the reaction. Together, these studies shed light on the role of the S,O-ligand in promoting Pd-catalyzed C–H functionalization reactions.
Introduction
Over the past 20 years, transition-metal-catalyzed C–H functionalization reactions have become a powerful synthetic tool in organic synthesis due to their high atom efficiency compared with traditional methods.1 The majority of the reported methodologies rely on the use of directing groups to increase the reactivity and selectivity of the process.2 The C–H functionalization of non-directed substrates offers the possibility for orthogonal selectivity as well as broader substrate scope beyond those bearing directing groups. However, these processes suffer from low yields and low levels of site-selectivity even using high catalyst loading, an excess of the arene and harsh reaction conditions. It was not until recently that the discovery of new ligands has allowed to develop efficient transformations.3Mono-protected amino acids ligands (MPAA),4 pyridine-based ligands5 or the combinations of both6 as well as bis(carboxylate) anions7 have been identified as effective ligands for these transformations. Additionally, we have discovered a new type of S,O-ligand, namely thioether carboxylic acid, that enables palladium-catalyzed C–H olefination of simple arenes, thiophenes, anisole, and aniline derivatives (Scheme 1).8 The new catalytic system based on Pd/S,O-ligand is also compatible with the introduction of other functionalities8i,j and has inspired other research groups to incorporate the S,O-ligand into a heterogeneous catalyst.9
 | ||
| Scheme 1 Pd/S,O-ligand-catalyzed C–H olefination of non-directed arenes. | ||
A unique feature of the Pd/S,O-ligand catalytic system is its high catalytic activity, enabling the functionalization of substrates that are unreactive using other catalysts. Despite the potential applications of the Pd/S,O-ligand catalyst in C–H functionalization reactions, the mechanistic role of the S,O-ligand is unknown. A detailed understanding of the role of this type of ligands in the reaction mechanism will be essential for future developments.
Herein, we describe a detailed mechanistic investigation of the role of the S,O-ligand in Pd-catalyzed C–H functionalization reactions of aromatic compounds. We have isolated and characterized by X-ray diffraction analysis several complexes before and after C–H activation and evaluated their catalytic activities. NMR and kinetic studies reveal that the C–H activation is rate limiting. Moreover, these studies suggest that the S,O-ligand triggers the formation of Pd cationic species. Additionally, a cationic Pd-complex was detected by ESI-MS and its reactivity in C–H activation processes was studied. DFT calculations corroborate the feasibility of the cationic pathway in the C–H activation of benzene as well as reveal the higher reactivity of cationic complexes in these processes. These investigations provide insights into the role of the S,O-ligand in promoting Pd-catalyzed C–H functionalization reactions. We expect that our findings will not only serve to guide future ligand development in the growing field of non-directed C–H functionalization reactions but also in further applications of this type of ligands in metal-catalysis.
Results and discussion
Identification of active Pd complexes
Initially, we concentrated our efforts on the identification of Pd complexes bearing an S,O-ligand. To facilitate mechanistic studies, we performed our experiments using the 2-methyl-2-(phenylthio)propanoic acid ligand (L2) that shows similar performance in the C–H olefination of simple arenes as the 3-methyl-2-(phenylthio)butanoic acid (L1) previously used.8a With this purpose, we reacted Pd(OAc)2 with L2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in CH2Cl2 at room temperature overnight (Scheme 2a). From the 1H NMR spectrum of the crude reaction mixture, several complexes were detected. Fortunately, we were able to unequivocally characterize by X-ray analysis the major Pd complex 1-cis bearing two S,O-ligands in a cis geometry.8a,10 When the reaction was performed using 2 equiv. of ligand L2, Pd-complexes 1-cis and 1-trans were obtained in quantitative yield in a 2:1 ratio, respectively (Scheme 2b). In order to isolate the Pd complex with only one S,O-ligand attached, we performed the reaction in the presence of PPh3 using a 1:1:1 ratio of Pd:L2:PPh3 in CH2Cl2. To our delight, complex 2 bearing one S,O-ligand, one PPh3 and one acetate ligand was isolated in 84% yield and fully characterized by X-ray analysis (Scheme 2c).11
:1 ratio in CH2Cl2 at room temperature overnight (Scheme 2a). From the 1H NMR spectrum of the crude reaction mixture, several complexes were detected. Fortunately, we were able to unequivocally characterize by X-ray analysis the major Pd complex 1-cis bearing two S,O-ligands in a cis geometry.8a,10 When the reaction was performed using 2 equiv. of ligand L2, Pd-complexes 1-cis and 1-trans were obtained in quantitative yield in a 2:1 ratio, respectively (Scheme 2b). In order to isolate the Pd complex with only one S,O-ligand attached, we performed the reaction in the presence of PPh3 using a 1:1:1 ratio of Pd:L2:PPh3 in CH2Cl2. To our delight, complex 2 bearing one S,O-ligand, one PPh3 and one acetate ligand was isolated in 84% yield and fully characterized by X-ray analysis (Scheme 2c).11
 | ||
| Scheme 2 Synthesis of Pd/S,O-ligand complexes. | ||
With these complexes in hand, we performed several experiments to evaluate their catalytic activities (Table 1). We executed the reaction of benzene with ethyl acrylate under the optimal reaction conditions with and without ligand L2 (entries 1 and 2).8a The reaction without ligand provided, after 2 h, the olefinated product in 21% yield and in the presence of L2, the olefinated products 3 were obtained in 78% yield. When we performed the same reaction using 5 mol% of complex 1 (cis and trans) instead of Pd(OAc)2, the olefinated product was obtained in 53% yield (entry 3), indicating that complex 1 (cis and trans) might accelerate the C–H olefination reaction. To see the effect of PPh3 in the reaction, we tested the reaction using 5 mol% of a catalyst based on a 1:1:1 ratio of Pd:L2:PPh3. This reaction provided the desired products in 79% yield which is comparable to the result of the reaction without PPh3 (entries 2 and 4). To our delight, the reaction using 5 mol% of complex 2 gave products 3 in 77% yield (entry 5), suggesting that this complex is involved in the catalytic reaction in the presence of PPh3. Additionally, the kinetic profiles of the reactions using different Pd catalysts were also performed (Fig. 1). The reactions using a 1:1 ratio of Pd:L2 and complex 2 as catalyst provided comparable curves suggesting that PPh3 does not have a critical role in the reaction. The reaction using complex 1 showed an increase in the reaction rate compared with the reaction without ligand. However, the kinetic profile shows that the reaction is slower than when complex 2 or a 1:1 ratio of Pd:L2 is used.
|
|
||
|---|---|---|
| Entry | Catalyst | NMR yield (3-mono + 3-di) |
| a Yield was determined by 1H NMR analysis of the crude mixture using CH2Br2 as internal standard. | ||
| 1 | Pd(OAc)2 | 21% (21% + 0%) |
| 2 | Pd(OAc)2 + ligand L2 (1:1) |
78% (74% + 4%) |
| 3 | Complexes 1-cis and 1-trans | 53% (53% + 0%) |
| 4 | Pd(OAc)2 + ligand L2 + PPh3 (1:1:1) |
79% (78% + 1%) |
| 5 | Complex 2 | 77% (75% + 2%) |

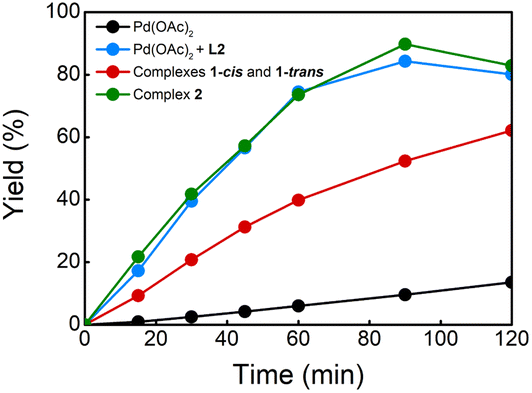 | ||
| Fig. 1 Kinetic profiles of C–H olefination of benzene. | ||
With the intention to identify a Pd complex after the C–H activation step, we carried out the reaction of complex 2 with benzene in a pressure tube at 100 °C. The reaction after 2 h provided a complex mixture where complex 4/4′ could not be identified. Therefore, we repeated the reaction and stopped after 10 min when the formation of Pd black was observed (Scheme 3a). From the 1H NMR spectrum of the reaction mixture, we tentatively assigned the peaks from 6.4–6.6 ppm to the formation of complexes 4 and/or 4′, which come after the C–H activation step, along with many other peaks that we could not identify. From the 31P NMR spectrum, mainly three equally intense peaks which belong to triphenylphosphine oxide, complexes 2 and 4 (or 4′) were observed. Unfortunately, from the reaction mixture, we were not able to isolate complex 4 (or 4′).
 | ||
| Scheme 3 Reaction of (a) Pd complex 2 with benzene and (b) Pd complex 4′ with ethyl acrylate. | ||
To confirm the formation of complex 4 (or 4′), we decided to synthesize 4′via a different synthetic route following a literature procedure.12 The structure of complex 4′ was confirmed by X-ray analysis that crystallizes in a non-centrosymmetric space group (see ESI†).13 Then, we compared the 1H and 31P NMR spectra of 4′ with the one obtained from the reaction of 2 with benzene. We found that both spectra match, confirming that 4′ was formed in the reaction of complex 2 with benzene. Next, the reaction of complex 4′ with an excess of ethyl acrylate furnished the olefinated product in 90% 1H NMR yield (Scheme 3b). From these results, we suggest that complexes 2 and 4′ are active catalysts in the reaction.
Identification of Pd complexes during the catalytic reaction
After having several Pd complexes characterized, we attempted to identify the complexes that were formed during the catalytic reaction. The reaction was performed in an NMR tube at 100 °C and was followed at different times by measuring the 1H and 31P NMR spectra at room temperature. We performed the reaction under standard conditions but using 50 mol% of the catalyst and deuterated acetic acid as a solvent. The comparison of 1H NMR spectra of ligand L2, Pd complexes 1, 2 and 4′ and the reaction at different times (0, 3, 5 and 10 min) are shown in Fig. 2. Before the reaction started (0 min), ligand L2 and complex 2 were clearly identified from their characteristic peaks of the methyl groups. After 3 and 5 min, we observed mainly complex 2 in the 1H NMR spectra. After 10 min, the reaction was completed and the signals of complex 2 were almost insignificant. After 3 min, we started to observe trace amounts of complex 1 and we did not detect any formation of complex 4′ during the reaction. The 1H NMR data were consistent with 31P NMR spectra as shown in Fig. 3. Mainly the peak at 27 ppm, which belongs to complex 2, was observed from 0 to 5 min and the intensity of this peak dramatically decreased after 10 min. The formation of complex 4′ (31P NMR at 26 ppm) was not detected during the reaction; however, other unidentified phosphorus species were formed. These results indicate that complex 2 is the resting state of the catalyst. | ||
| Fig. 2
1H NMR spectra of (a) ligand L2, (b) complexes 1-cis and 1-trans, (c) complex 2, (d) complex 4′ in AcOH-d4. C–H Olefination of benzene and ethyl acrylate using a 1:1:1 ratio of Pd:L2:PPh3 as catalyst in AcOH-d4 monitored by 1H NMR spectroscopy at (e) 0 min, (f) 3 min, (g) 5 min and (h) 10 min. ox = oxidant, EA = ethyl acrylate, BA = benzoic acid. | ||
 | ||
| Fig. 3
31P NMR spectra of (a) complex 2 and (b) complex 4′ in AcOH-d4. C–H Olefination of benzene and ethyl acrylate using a 1:1:1 ratio of Pd:L2:PPh3 as catalyst in AcOH-d4 monitored by 31P NMR spectroscopy at (c) 0 min, (d) 3 min, (e) 5 min and (f) 10 min. | ||
Next, we examined the reaction using benzene, Pd(OAc)2, ligand L2 and PPh3 in AcOD-d4 in the absence of ethyl acrylate in an NMR tube at 100 °C, recording the NMR data at the indicated time at room temperature. As shown in Fig. 4, at 0 min, ligand L2 and complex 2 were detected. After 2 and 4 min, complex 2 was the main complex observed, traces of complex 1 were detected and traces of complex 4′ were identified from the peaks around 6.5–7 ppm. 31P NMR data corroborated that complex 2 (27 ppm) was the main complex during the reaction and that traces of complex 4′ (26 ppm) were formed (Fig. 5).
 | ||
| Fig. 4 1H NMR spectra of (a) ligand L2, (b) complexes 1-cis and 1-trans, (c) complex 2 and (d) complex 4′ in AcOH-d4. The reaction of benzene with Pd(OAc)2, ligand L2 and PPh3 in AcOH-d4 monitored by 1H NMR spectroscopy at (e) 0 min, (f) 2 min and (g) 4 min. | ||
 | ||
| Fig. 5 31P NMR spectra of (a) complex 2 and (b) complex 4′ in AcOH-d4. The reaction of benzene with Pd(OAc)2, ligand L2 and PPh3 in AcOH-d4 monitored by 31P NMR spectroscopy at (c) 0 min, (d) 2 min and (e) 4 min. | ||
Kinetic investigations
Order of the reaction
The kinetic order of each reagent was determined in the reaction in the presence of ligand L1 by using the initial rate method.14Fig. 6 shows the plot of the logarithm of the reaction rate against the concentration of the reagent. A straight line with a slope of nearly one was measured for Pd-catalyst and benzene, revealing a first order in these reagents (Fig. 6a and b). Non-significant change in the reaction rate was observed using different concentrations of oxidant and olefin (Fig. 6c and d), indicating that the migratory insertion and oxidation of Pd(0) occur after the RDS. Nevertheless, the small negative fractional order in both cases can be explained by the formation of an off-cycle palladium complex bearing these reagents as a neutral ligand.15 | ||
| Fig. 6 Dependence of the observed rate constant on the concentration of (a) catalyst, (b) benzene, (c) PhCO3t-Bu, (d) ethyl acrylate and (e) NaOAc for the reaction of Pd(OAc)2, ligand L1, tert-butyl peroxybenzoate, ethyl acrylate and benzene in AcOH at 100 °C. | ||
Based on a previously reported observation of an inverse first-order dependence of the rate on [AcO−] in the C–H olefination of heteroarenes using a monodentate thioether ligand,16 we decided to determine the order of the reaction in NaOAc concentration and an inverse 0.7 order dependence was found (Fig. 6e).17 To explain the inverse order observed, we considered two different possibilities: (1) the association of acetate to form off-cycle [Pd(L1)(OAc)2]− species and (2) a reversible dissociation of acetate from [LPd(L1)(OAc)] to form cationic palladium species. To distinguish between these two possibilities, we added an excess of NaOAc to a 1 to 1 mixture of L2 and Pd(OAc)2 in AcOD-d4 to investigate whether the formation of the off-cycle [Pd(S,O-L)(OAc)2]− was the cause of the inverse order on acetate. After heating the mixture at 100 °C, we observed by 1H NMR that the relative intensity of peaks that belong to different Pd species changed but no major new Pd species were detected (see ESI†). Therefore, the observed inverse order with respect to NaOAc is not the result of the formation of off-cycle [Pd(S,O-L)(OAc)2]− species. Taking into account these results, it seems reasonable to propose that more reactive cationic palladium species are formed during the reaction and prior the RDS.16,18
Next, we investigated the order of each reagent in the reaction without ligand (Fig. 7). We observed a 0.3 order in the catalyst and the first order in the benzene concentration (Fig. 7a and b), suggesting a trimeric precatalyst and one benzene associated with the catalyst prior to the RDS.19 Again, near zero order in both oxidant and olefin were observed (Fig. 7c and d), indicating that olefin insertion and oxidation of Pd(0) occur after the RDS. Zero order in the acetate concentration is observed at lower concentrations of NaOAc and changed to the inverse 1.2 order dependence when the concentration of acetate was increased (Fig. 7e). We postulated that at higher concentrations of acetate the off-cycle [Pd(OAc)3]− is formed in the reaction.
 | ||
| Fig. 7 Dependence of the observed rate constant on the concentration of (a) catalyst, (b) benzene, (c) PhCO3t-Bu, (d) ethyl acrylate and (e) NaOAc for the reaction of Pd(OAc)2, tert-butyl peroxybenzoate, ethyl acrylate and benzene in AcOH at 100 °C. | ||
From these kinetic experiments, we suggest that the C–H activation is the RDS in both cases, with and without ligand and that the presence of the S,O-ligand triggers the formation of cationic species, which are expected to be more reactive.18 We also proposed that the reaction in the presence of the S,O-ligand occurs via catalytically active monomeric species which are the main species in solution.
KIE and H/D exchange experiments
The KIE values were examined by performing reactions with benzene and benzene-d6 in two different reaction flasks.20 The reaction in the presence of ligand L1 provided a kH/kD ratio of 4.0 and in the absence of ligand provided a kH/kD ratio of 5.4 (Scheme 4a). The observed primary KIE corroborates together with the kinetic and NMR studies that the C–H bond cleavage is the RDS in the reaction with and without the S,O-ligand. Additionally, the reversibility of the C–H activation step by performing H/D scrambling experiments using AcOD-d4 as a deuterium source was evaluated (see ESI†). We used mesitylene as a diagnostic arene to facilitate the interpretation of the 1H NMR spectra. The reaction with ligand L1 provided the olefinated product and deuterated mesitylene in 17% after 6 h. In the reaction without ligand, by increasing the amount of the catalyst and the reaction time, the formation of 25% deuterated mesitylene was observed. Based on these experimental results we suggest that the C–H activation is per se reversible in both cases, with and without ligand.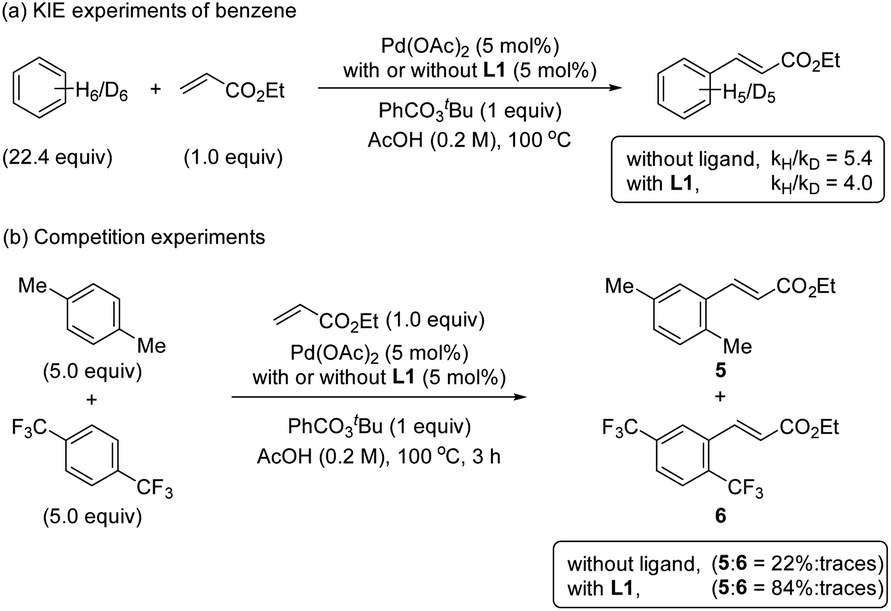 | ||
| Scheme 4 Mechanistic studies (a) KIE experiments of benzene and (b) competition experiments. | ||
Competition experiments
To get more insights into the mechanism of the C–H activation step, we performed one-pot intermolecular competition experiments between an electron-rich arene, p-xylene, and an electron-poor arene, 1,4-bis(trifluoromethyl)benzene, with and without the ligand (Scheme 4b). We found that in both cases electron-rich arene reacted preferentially. These results together with the C–H bond cleavage being the RDS are consistent with an asynchronous concerted metalation deprotonation (CMD)21,22 or a base-assisted internal electrophilic-type substitution (BIES) mechanism.23,24Detection of reactive palladium complexes by ESI-MS and their reactivities in C–H activation processes
With the aim to identify cationic reactive intermediates in solution, we analyzed the system by electrospray ionization mass spectrometry (ESI-MS).25 Electrospray ionization of a solution of Pd(OAc)2 and S,O-ligand L2 in benzene led to the detection of larger palladium clusters with a combination of L2 and AcO− ligands (Fig. S14 in the ESI†). The addition of PPh3 to the mixture resulted in the disappearance of the clusters and in the dominant formation of monomeric [(PPh3)2Pd(L2)]+ complex (m/z 825, Fig. 8a).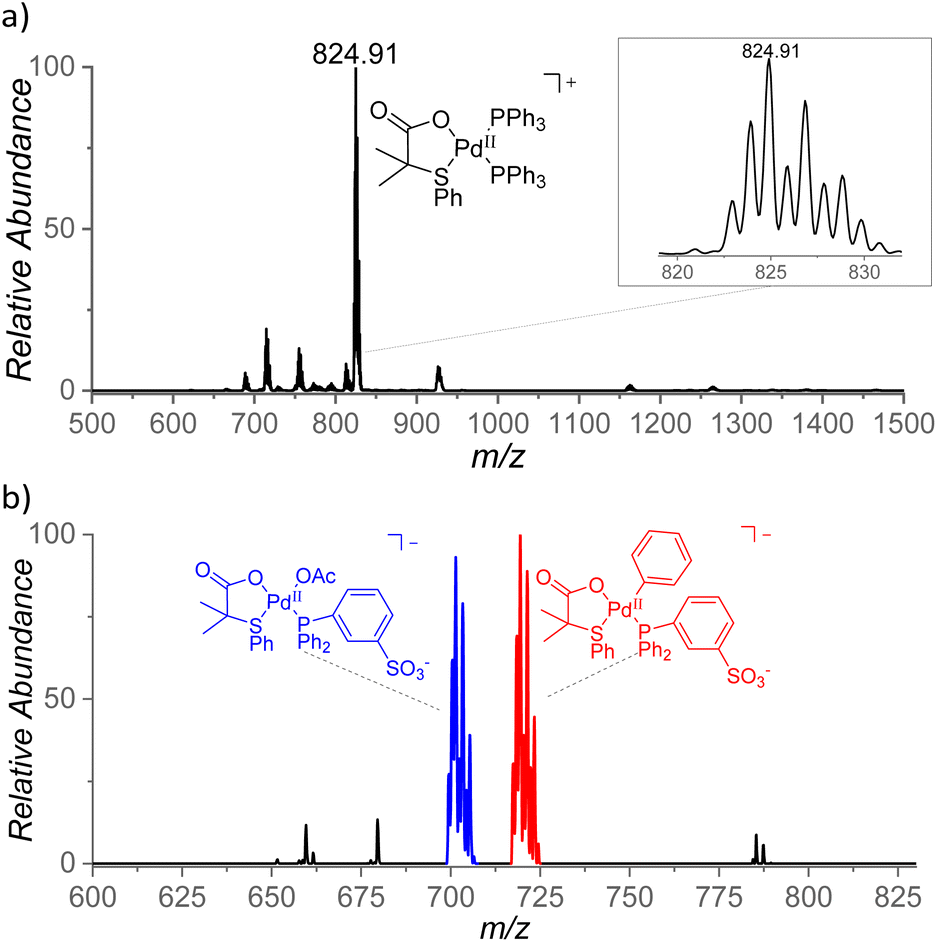 | ||
| Fig. 8 (a) ESI mass spectrum of a mixture of Pd(OAc)2, L2, and Ph3P in benzene and (b) ESI mass spectrum of Na[(PPh2PhSO3−)Pd(L2)(OAc)] in benzene, heated for 5 min at 100 °C. | ||
The key intermediates [(PPh3)Pd(L2)(Ph)] are neutral and thus not detectable by ESI-MS; therefore, we performed the experiments also with the negatively charged ligand PPh2PhSO3− (PhSO3− = 3-phenylsulfonate). The addition of the remote charge to the phosphine ligand allowed us to detect [(PPh2PhSO3−)Pd(L2)(OAc)]− and [(PPh2PhSO3−)Pd(L2)2]− ions (m/z 701 and m/z 836, respectively, Fig. S15†). The key intermediate [(PPh2PhSO3−)Pd(L2)(Ph)]− was detected only after heating of the [(PPh2PhSO3−)Pd(L2)(OAc)]Na complex in benzene for 5 min at 100 °C (m/z 719, Fig. 8b) which points to a substantial energy barrier for the C–H activation reaction. The assignment of [(PPh2PhSO3−)Pd(L2)(Ph)]− was confirmed by repeating the experiment in deuterated benzene and by high-resolution measurements (Fig. S16 and S17†). The [(PPh2PhSO3−)Pd(L2)(Ph)]− can have two isomers (4/4′). We attempted to identify these isomers by ion mobility separation. The ion mobility profile is related to the shape of the ions and therefore it is expected to be different for the 4 and 4′ isomers. However, the ion mobility experiment indicated only one major isomer of the detected [(PPh2PhSO3−)Pd(L2)(X)]− complexes with X = Cl, OAc, and Ph. (Fig. S18†).
The role of the S,O-ligand in the C–H activation process was further studied by intramolecular reactivity of the [(PPh3)2Pd(L2)]+ complex. This complex eliminates the neutral L2 ligand upon collisional activation (196 Da) suggesting that L2 engaged in C–H activation of a phenyl ring of the PPh3 ligand (Fig. 9a). This C–H activation process was confirmed by an experiment with [(PPh3-d15)2Pd(L2)]+ that indeed eliminated monodeuterated S,O-ligand L2D (197 Da) (Fig. 9b). The kinetic isotope effect of this process can be determined from the ratio of the L2-H:L2-D eliminations from mixed complex [(PPh3)(PPh3-d15)Pd(L2)]+ (m/z = 840) and it corresponds to KIE = ∼2. The energy demand of this process is 41 kcal mol−1 (∼1.8 eV, Fig. 9d).
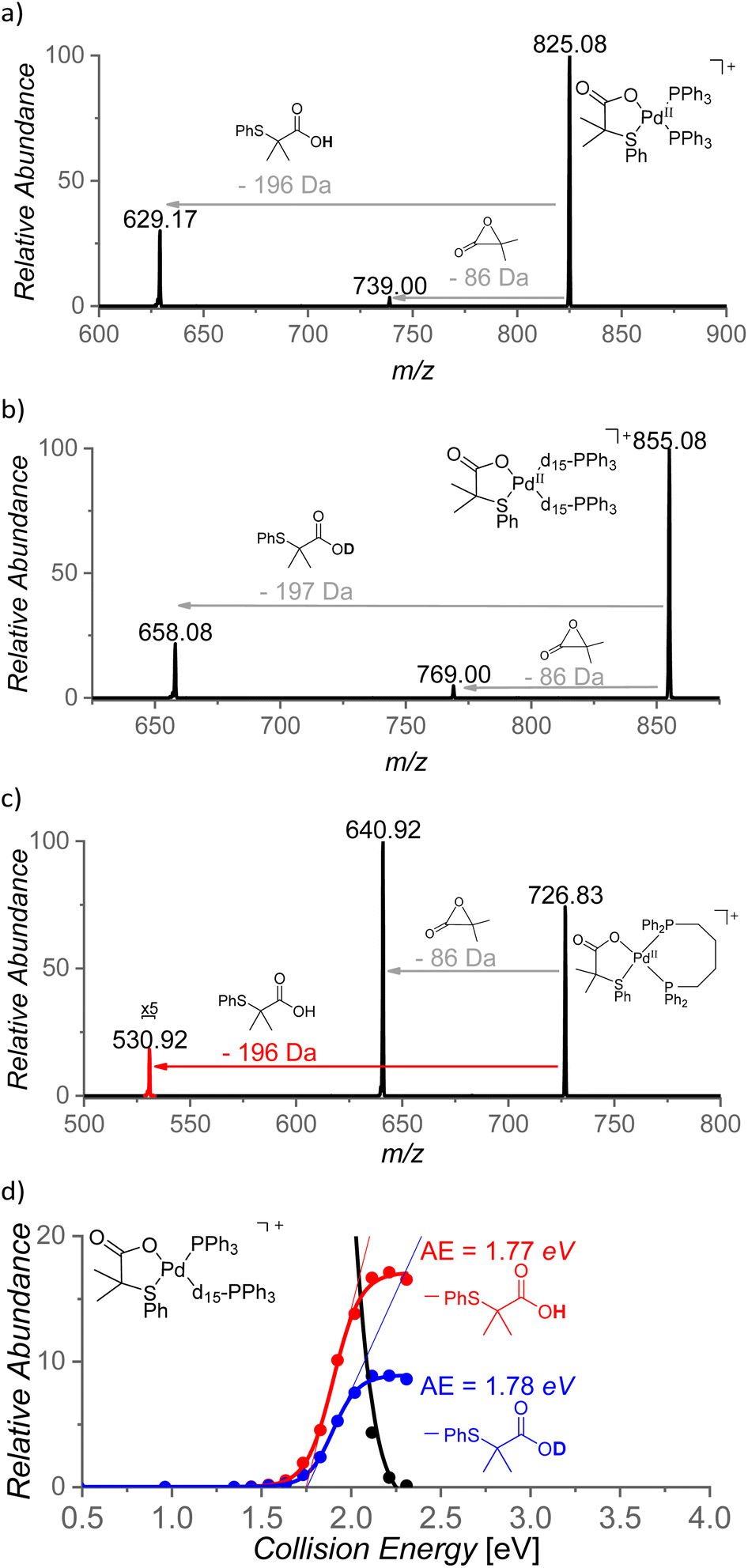 | ||
| Fig. 9 CID mass spectra of (a) [(PPh3)2Pd(L2)]+ (m/z = 825), (b) [(P(C6D5)3)2Pd(L2)]+ (m/z = 855), (c) [(dppb)Pd(L2)]+ (m/z = 727) and (d) energy resolved CID of mixed phosphine complex [(PPh3)(d15-PPh3)Pd(L2)]+ (m/z = 840). | ||
The steric requirements for the C–H activation process can be explored by changing the bite angles between the ligands in [(PPh3)2Pd(L2)]+. Hence, the PPh3 ligands were exchanged by diphosphine ligands: dppm [1,1-bis(diphenylphosphino)methane], dppe [1,1-bis(diphenylphosphino)ethane], dppp [1,1-bis(diphenylphosphino)propane] and dppb [1,1-bis(diphenylphosphino)butane] (Fig. 9c). Complexes [(dppm)Pd(L2)]+ and [(dppe)Pd(L2)]+ do not show any C–H activation process and instead reveal a degradation of the S,O-ligand (a loss of 86 Da). On contrary, the complexes with the larger and more flexible ligands dppp and dppb show the C–H activation reaction (Fig. 9c and S21(iii)†). Hence, the C–H activation reaction requires proximity of the phenyl ring and the S,O-ligand which points towards a tight transition structure for this process.
DFT calculations
Density Functional Theory calculations [M06/def2tzvpp (IEF-PCM, acetic acid) with Gaussian software] were also carried out to get more insight into the species involved in the mechanism,26 and more specifically, about the role played by the S,O ligand L2 in the reactivity enhancement. We considered that the observed reactivity is the result of the sum of different factors. First, we evaluated the possibility that the ligand is helping the formation of catalytically relevant monomeric palladium species. Indeed, palladium acetate is known to exist as a trimer, Pd3(OAc)6, which persists even in solution.27 In our hands, the dissociation of the trimer into the monomeric acetate is greatly disfavored, uphill by 12.5 kcal mol−1 (per palladium unit, Fig. 10). To our delight, the exchange of an acetate by ligand L2 stabilizes the monomeric complex by 13.4 kcal mol−1 (II). Following this trend, a second equivalent of L2 completely reverts the trimer/monomer equilibrium, ensuring the efficient dissociation of Pd3(OAc)6 with formation of complex 1-cis (III), as found experimentally (Scheme 2). | ||
| Fig. 10 Computed relative stability of trimeric and monomeric palladium acetate with L2 containing complexes. | ||
We also wanted to prove theoretically the higher reactivity of the cationic complexes that are formed during the reaction, as suggested by the experimental results. In this regard, a crucial step in non-directed C–H activations is the coordination of the aromatic substrate to the palladium center, which is inherently difficult. Indeed, the calculations show that benzene hardly coordinates to neutral Pd(OAc)2 (IV, ΔGeq = +8.9 kcal mol−1, Fig. 11a). Obviously, the situation would be quite different in a cationic complex like V (Fig. 11b), which is coordinatively unsaturated. But even in this equilibrium, the formation of the benzene coordinated complex VI is just isoenergetic with V. Noteworthy, complex VII, which contains our ligand L2 (instead of acetate) shows a much larger affinity towards the benzene coordination, forming VIII, which is ca. 3 kcal mol−1 lower in energy than VII.
 | ||
| Fig. 11 Relative affinity towards benzene coordination of neutral and cationic complexes. | ||
Considering the intermediacy of complex VIII as an active species in the potential energy surface, we next evaluated different alternatives for a plausible C–H bond cleavage (Fig. 12).28 In line to what has been proposed in other catalytic systems, we computed the involvement of the internal S,O-ligand in 4- or 6-membered transition states, and an external acetate anion present in the reaction medium.29 As expected, the participation of the O atom of the S,O-ligand in a 4-membered transition state (TS-A) was computed to demand a high activation energy (ΔG‡ = 33.6 kcal mol−1), due to the strained disposition of the Pd–O–H–C atoms in the structure. This type of TS has been seldom proposed in some CMD mechanisms but it has never been confirmed computationally.30 A much more favored situation was expected in TS-B, which is a conventional 6-membered CMD transition state.31 However, in this mechanism, the coordination pattern of the S,O-ligand must completely switch from VIII to IX, via decoordination of the S atom and formation of an acetate-type coordination to palladium. This change in coordination involves an increase of 13.5 kcal mol−1 in the energy of IX. Furthermore, the cationic character of the palladium center weakens the C–H bonds of benzene, but at the same time it decreases the basicity of the acetate fragment, rendering a final activation energy of 35.3 kcal mol−1 for TS-B. This value is again too high to be feasible at the experimental conditions. Furthermore, TS-B would make difficult to rationalize the distinguishing reactivity of our S,O-ligand compared to a standard acetate, since the S atom does not directly participate in the coordination during the transition state. We note, that both TS-A and TS-B activation energies agree well with large C–H activation energies observed in the gas-phase unimolecular reactions (vide supra).
 | ||
| Fig. 12 Computed transition states for the C–H activation promoted by the internal S,O ligand (TS-A and TS-B) or an external acetate (TS-C). Energies are in kcal mol−1 referred to VIII as G = 0. | ||
Alternatively, bimolecular mechanism through the activation of the C–H bond by an external acetate was found to be strikingly favored, as indicated by the very low activation energy of the transition state TS-C. The incoming acetate unit can initially form an ion pair as in VIII-OAc, stabilizing the complex in 7.5 kcal mol−1. From there, the activation energy of the C–H bond cleavage amounts to only 8.2 kcal mol−1, one of the lowest values computed for a C–H bond cleavage. The participation of an external base has been hypothesized and computed by different authors,32 but it is not in general the preferred option. In neutral systems, it presents the drawback that the deprotonation renders an anionic palladium complex. Therefore, we believe that in our case, the cationic palladium system VIII is perfectly suited for the participation of an external base, since after deprotonation, a more stable neutral complex like 4 would be formed (Scheme 5). Furthermore, the reactive cationic palladium promotes the cleavage of aromatic C–H bonds by increasing their acidity, while the incoming acetate maintains its full basic strength.
 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Proposed catalytic cycle in the presence of S,O-ligand
By combining all the experimental data, we propose the mechanism outlined in Scheme 5 for the C–H olefination of arenes in the presence of the S,O-ligand. We draw the intermediates with PPh3 as ligand since we isolated and identified many of these complexes in the catalytic reaction and similar mechanism is expected to occur with and without PPh3. Nevertheless, in the case of performing the reaction without PPh3, complexes bearing an innocent ligand or polymeric species might be formed. First, Pd(OAc)2 reacts with ligand L2 and PPh3 forming complex 2, which we isolated and demonstrated that is catalytically active. We observed by NMR that complex 2 is the catalyst resting state, indicating that C–H activation is the RDS. This is in agreement with the order of the reaction and the large KIE values observed. After the C–H activation step, complex 4′ is formed. We propose that the C–H activation takes place via cationic monomeric palladium species 7, which are formed from complex 2 after losing an AcO− ligand. The formation of cationic species is supported by the inverse order in AcO−. Additionally, a monomeric cationic [(PPh3)2Pd(L2)]+ complex, which is related with the structure of palladium species 7, was detected by ESI-MS and its reactivity in intramolecular C–H activation processes proved. DFT calculations corroborate the higher reactivity of cationic complexes in the C–H activation of benzene. After the C–H activation step, complex 4 reacts with ethyl acrylate providing complex 9 which then undergoes β-H elimination to provide the olefinated compound and Pd(0) species. The oxidation of these species by an oxidant completes the catalytic cycle. From the comparison of the experiments performed with and without ligand, we propose that the S,O-ligand triggers the formation of cationic palladium species that are more reactive than the neutral species in the C–H activation step, which is in both cases the RDS.Conclusions
This article describes a detailed mechanistic study on the role of the S,O-ligand in the Pd-catalyzed C–H olefination reactions of non-directed arenes. Several monomeric Pd/S,O-ligand complexes before and after the C–H activation step have been isolated and fully characterized and their reactivities have been evaluated. NMR and kinetics studies, and KIE values indicate that the C–H activation step is RDS in both cases with and without S,O-ligand. Moreover, these studies suggest that the S,O-ligand triggers the formation of Pd cationic species. Additionally, a cationic Pd-complex has been detected by ESI-MS and its reactivity in C–H activation processes studied. DFT calculations corroborate the higher reactivity of cationic complexes in C–H activation processes.Together, these studies shed light on the features of the Pd/S,O-ligand catalyst that make it so effective in promoting Pd-catalyzed C–H functionalization reactions. We expect that the insights presented herein will provide the foundations for the rational design of novel and more active catalyst for C–H activation processes.
Data availability
The ESI† is available and contains experimental procedures, compounds characterization and computational studies.Author contributions
K. N. performed all the reactions, NMR and kinetic experiments. E. G. B. performed the DFT calculations. E. O. resolved the crystal structures. J. M and J. R. performed the MS experiments. M. A. F.-I. supervised the project. K. N and M. A. F.-I. wrote the manuscript with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from NWO through a VIDI grant (723.013.006) and from MCIN-PID2019-110008GB-I00. E. G.-B. thanks SGIker (UPV/EHU) for providing human and computational resources.Notes and references
- For general review of C–H activation reaction, see: (a) K. Godula and D. Sames, Science, 2006, 312, 67 CrossRef CAS PubMed; (b) R. G. Bergman, Nature, 2007, 446, 391 CrossRef CAS PubMed; (c) X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS; (d) J.-Q. Yu, Z. Shi, C–H Activation, Springer Berlin, Heidelberg, 2010 CrossRef; (e) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2 CrossRef CAS; (f) P. H. Dixneuf and H. Doucet, C–H Bond Activation and Catalytic Functionalization I, Springer Cham, Switzerland, 2016 Search PubMed; (g) W. Ali, G. Prakash and D. Maiti, Chem. Sci., 2021, 12, 2735 RSC.
- For selected reviews on directing group assisted C–H activation, see: (a) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC; (b) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS; (c) Y. Xia and G. Dong, Nat. Rev. Chem., 2020, 4, 600 CrossRef CAS; (d) K. Murali, L. A. Machado, R. L. Carvalho, L. F. Pedrosa, R. Mukherjee, E. N. Da Silva Júnior and D. Maiti, Chem.–Eur. J., 2021, 27, 12453 CrossRef CAS PubMed.
- For general reviews on non-directed C–H Activation, see: (a) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 ( Angew. Chem. , 2012 , 124 , 10382 ) CrossRef CAS PubMed; (b) A. Dey, S. Maity and D. Maiti, Chem. Commun., 2016, 52, 12398 RSC; (c) P. Wedi and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 13016 ( Angew. Chem. , 2018 , 130 , 13198 ) CrossRef CAS PubMed; (d) S. Kancherla, K. B. Jorgensen and M. Á. Fernández-Ibáñez, Synth, 2019, 51, 643 CrossRef CAS , for examples of Pd-catalyzed non-directed C–H Olefination, see; (e) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. W. Cheng, M. A. Poss, M. E. Farmer, K. S. Yeung and J. Q. Yu, Nature, 2017, 551, 489 CrossRef CAS PubMed; (f) H. Chen, P. Wedi, T. Meyer, G. Tavakoli and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 2497 ( Angew. Chem. , 2018 , 130 , 2523 ) CrossRef CAS PubMed ; for examples of non-directed C–H cyanation, see; (g) L. Y. Liu, K. S. Yeung and J. Q. Yu, Chem.–Eur. J., 2019, 25, 2199 CrossRef CAS; (h) D. Zhao, P. Xu and T. Ritter, Chem, 2019, 5, 97 CrossRef CAS; (i) H. Chen, A. Mondal, P. Wedi and M. Van Gemmeren, ACS Catal., 2019, 9, 1979 CrossRef CAS; (j) H. T. Kim, E. Kang, M. Kim and J. M. Joo, Org. Lett., 2021, 23, 3657 CrossRef CAS; (k) A. Saha, S. Guin, W. Ali, T. Bhattacharya, S. Sasmal, N. Goswami, G. Prakash, S. K. Sinha, H. B. Chandrashekar, S. Panda, S. S. Anjana and D. Maiti, J. Am. Chem. Soc., 2022, 144, 1929 CrossRef CAS PubMed; (l) S. Panja, S. Ahsan, T. Pal, S. Kolb, W. Ali, S. Sharma, C. Das, J. Grover, A. Dutta, D. B. Werz, A. Paul and D. Maiti, Chem. Sci., 2022, 13, 9432 RSC.
- For selected examples of C–H functionalization reactions of directed arenes using MPAA as ligand, see: (a) B. F. Shi, N. Maugel, Y. H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS PubMed; (b) P. Novák, A. Correa, J. Gallardo-Donaire and R. Martin, Angew. Chem., Int. Ed., 2011, 50, 12236 CrossRef PubMed; (c) X.-F. Cheng, Y. Li, Y.-M. Su, F. Yin, J.-Y. Wang, J. Sheng, H. U. Vora, X.-S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 1236 CrossRef CAS PubMed; (d) G. Li, D. Leow, L. Wan and J.-Q. Yu, Angew. Chem., Int. Ed., 2013, 52, 1245 CrossRef CAS PubMed; (e) L. Chu, K.-J. Xiao and J.-Q. Yu, Science, 2014, 346, 451 CrossRef CAS PubMed; (f) C. Zhu, Y. Zhang, J. Kan, H. Zhao and W. Su, Org. Lett., 2015, 17, 3418 CrossRef CAS PubMed , for selected examples of C–H functionalization reactions of non-directed arenes using MPAA as ligand, see; (g) X. Cong, H. Tang, C. Wu and X. Zeng, Organometallics, 2013, 32, 6565 CrossRef CAS; (h) Q. Huang, X. Zhang, L. Qiu, J. Wu, H. Xiao, X. Zhang and S. Lin, Adv. Synth. Catal., 2015, 357, 3753 CrossRef CAS; (i) Z. Zhuang, A. N. Herron, S. Liu and J.-Q. Yu, J. Am. Chem. Soc., 2021, 143, 687 CrossRef CAS PubMed.
- For selected examples of C–H functionalization reactions of directed arenes using pyridine-based ligands, see: (a) C. Wang, S. Rakshit and F. Glorius, J. Am. Chem. Soc., 2010, 132, 14006 CrossRef CAS PubMed; (b) H. Xu, M. Shang, H.-X. Dai and J.-Q. Yu, Org. Lett., 2015, 17, 3830 CrossRef CAS PubMed; (c) D. Zhu, G. Yang, J. He, L. Chu, G. Chen, W. Gong, K. Chen, M. D. Eastgate and J.-Q. Yu, Angew. Chem., Int. Ed., 2015, 54, 2497 CrossRef CAS; (d) M.-Z. Lu, X.-R. Chen, H. Xu, H.-X. Dai and J.-Q. Yu, Chem. Sci., 2018, 9, 1311 RSC , for selected examples of C–H functionalization reactions of non-directed arenes using pyridine-based ligands, see; (e) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390 CrossRef CAS PubMed; (f) E. M. Ferreira and B. M. Stoltz, J. Am. Chem. Soc., 2003, 125, 9578 CrossRef CAS; (g) J. M. Murphy, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 15434 CrossRef CAS PubMed; (h) Y. Izawa and S. S. Stahl, Adv. Synth. Catal., 2010, 352, 3223 CrossRef CAS PubMed; (i) M. Ye, G.-L. Gao and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 6964 CrossRef CAS PubMed; (j) M. H. Emmert, A. K. Cook, Y. J. Xie and M. S. Sanford, Angew. Chem., Int. Ed., 2011, 50, 9409 CrossRef CAS PubMed; (k) A. K. Cook, M. H. Emmert and M. S. Sanford, Org. Lett., 2013, 15, 5428 CrossRef CAS PubMed; (l) J. B. Gary, A. K. Cook and M. S. Sanford, ACS Catal., 2013, 3, 700 CrossRef CAS; (m) C. Cheng and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 592 CrossRef CAS PubMed; (n) C. Valderas, K. Naksomboon and M. Á. Fernández-Ibáñez, ChemCatChem, 2016, 8, 3213 CrossRef CAS; (o) P. Wang, P. Verma, G. Xia, J. Shi, J. X. Qiao, S. Tao, P. T. Cheng, M. A. Poss, M. E. Farmer and K.-S. Yeung, Nature, 2017, 551, 489 CrossRef CAS PubMed; (p) Y. Álvarez-Casao, C. A. van Slagmaat, G. K. Verzijl, L. Lefort, P. L. Alsters and M. Á. Fernández-Ibáñez, ChemCatChem, 2018, 10, 2620 CrossRef; (q) C. Karmel, Z. Chen and J. F. Hartwig, J. Am. Chem. Soc., 2019, 141, 7063 CrossRef CAS PubMed; (r) L.-Y. Liu, J. X. Qiao, K.-S. Yeung, W. R. Ewing and J.-Q. Yu, Angew. Chem., Int. Ed., 2020, 59, 13831 CrossRef CAS PubMed.
- For selected examples of C–H functionalization reactions using pyridine derivative and MPAA as ligands, see: (a) H. Chen, P. Wedi, T. Meyer, G. Tavakoli and M. van Gemmeren, Angew. Chem., Int. Ed., 2018, 57, 2497 CrossRef CAS PubMed; (b) D. Zhao, P. Xu and T. Ritter, Chem, 2019, 5, 97 CrossRef CAS; (c) A. Mondal, H. Chen, L. Flämig, P. Wedi and M. van Gemmeren, J. Am. Chem. Soc., 2019, 141, 18662 CrossRef CAS PubMed; (d) H. Chen, A. Mondal, P. Wedi and M. van Gemmeren, ACS Catal., 2019, 9, 1979 CrossRef CAS; (e) H. Chen, M. Farizyan, F. Ghiringhelli and M. van Gemmeren, Angew. Chem., Int. Ed., 2020, 59, 12213 CrossRef CAS PubMed; (f) A. Mondal and M. van Gemmeren, Angew. Chem., Int. Ed., 2021, 60, 742 CrossRef CAS PubMed.
- (a) J. Dhankhar, E. González-Fernández, C.-C. Dong, T. K. Mu-khopadhyay, A. Linden and I. Čorić, J. Am. Chem. Soc., 2020, 142, 19040 CrossRef CAS PubMed; (b) J. Dhankhar, M. D. Hofer, A. Linden and I. Čorić, Angew. Chem., Int. Ed., 2022, 61, e202205470 CrossRef CAS PubMed.
- (a) K. Naksomboon, C. Valderas, M. Gómez-Martínez, Y. Álvarez-Casao and Y. M. Á. Fernández-Ibáñez, ACS Catal., 2017, 7, 6342 CrossRef CAS PubMed; (b) K. Naksomboon, Y. Álvarez-Casao, M. Uiterweerd, N. Westerveld, B. Maciá and M. Á. Fernández-Ibáñez, Tetrahedron Lett., 2018, 59, 379 CrossRef CAS; (c) Y. Álvarez-Casao and M. Á. Fernández-Ibáñez, Eur. J. Org. Chem., 2019, 1842 CrossRef; (d) K. Naksomboon, J. Poater, F. M. Bickelhaupt and M. Á. Fernández-Ibáñez, J. Am. Chem. Soc., 2019, 141, 6719 CrossRef CAS PubMed; (e) W.-L. Jia, N. Westerveld, K. M. Wong, T. Morsch, M. Hakkennes, K. Naksomboon and M. Á. Fernández-Ibáñez, Org. Lett., 2019, 21, 9339 CrossRef CAS PubMed; (f) B. Yin, M. Fu, L. Wang, J. Liu and Q. Zhu, Chem. Commun., 2020, 56, 3293 RSC; (g) V. Sukowski, W.-L. Jia, R. van Diest, M. van Borselen and M. Á. Fernández-Ibáñez, Eur. J. Org. Chem., 2021, 4132 CrossRef CAS; (h) W.-L. Jia, S. V. Ces and M. Á. Fernández-Ibáñez, J. Org. Chem., 2021, 86, 6259 CrossRef CAS PubMed; (i) K.-Z. Deng, W.-L. Jia and M. Á. Fernández-Ibáñez, Chem.–Eur. J., 2022, 28, e20210410 Search PubMed; (j) V. Sukowski, M. van Borselen, S. Mathew and M. Á. Fernández-Ibáñez, Angew. Chem., Int. Ed., 2022, 61, e20220175 CrossRef PubMed.
- (a) N. Van Velthoven, Y. Wang, H. Van Hees, M. Henrion, A. L. Bugaev, G. Gracy, K. Amro, A. V. Soldatov, J. G. Alauzun, P. H. Mutin and D. E. De Vos, ACS Appl. Mater. Interfaces, 2020, 12, 47457 CrossRef CAS PubMed; (b) N. Van Velthoven, M. Henrion, J. Dallenes, A. Krajnc, A. L. Bugaev, P. Liu, S. Bals, A. V. Soldatov, G. Mali and D. E. De Vos, ACS Catal., 2020, 10, 5077 CrossRef CAS.
- CCDC 1567101 contains the supplementary crystallographic data for Pd complex 1-cis.
- CCDC 1969711 contains the supplementary crystallographic data for Pd complex 2.
- (a) V. V. Grushin and H. Alper, Organometallics, 1993, 12, 1890 CrossRef CAS; (b) L. A. Perego, L. Grimaud and F. Bellina, Adv. Synth. Catal., 2016, 358, 597 CrossRef CAS.
- CCDC 1969712 and CCDC 1969713 contains the supplementary crystallographic data for Pd complex 4′.
- J. H. Espenson, Chemical kinetics and reaction mechanisms, McGraw Hill, 2nd edn, 1987 Search PubMed.
- (a) A. K. Cook and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 3109 CrossRef CAS PubMed; (b) D. G. Blackmond, J. Am. Chem. Soc., 2015, 137, 10852 CrossRef CAS PubMed; (c) P. Wedi, M. Farizyan, K. Bergander, C. Mück-Lichtenfeld and M. van Gemmeren, Angew. Chem., Int. Ed., 2021, 60, 15641 CrossRef CAS PubMed.
- B. J. Gorsline, L. Wang, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2017, 139, 9605 CrossRef CAS PubMed.
- To rule out that the negative order observed on [OAc]− is the result of the competition between the acetate and the S,O-ligand, we have determined the order on [OAc]− using complex 2, where there is no competition between the OAc and the S,O-ligand, We observed again an inverse 0.6 order dependence on [OAc]−, indicating that the negative order is not the result of the competition between the acetate and the S,O-ligand.
- (a) M. D. K. Boele, G. P. F. van Strijdonck, A. H. M. de Vries, P. C. J. Kamer, J. G. de Vries and P. W. N. M. van Leeuwen, J. Am. Chem. Soc., 2002, 124, 1586 CrossRef CAS PubMed; (b) T. Nishikata and B. H. Lipshutz, Org. Lett., 2010, 12, 1972 CrossRef CAS PubMed; (c) T. Nishikata, A. R. Abela and B. H. Lipshutz, Angew. Chem., Int. Ed., 2010, 49, 781 CrossRef CAS PubMed.
- J. Vana, J. Lang, M. Soltesova, J. Hanusek, A. Ruzicka, M. Sedlak and J. Roithova, Dalton Trans., 2017, 46, 16269 RSC.
- (a) M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857 CrossRef PubMed; (b) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- For selected examples of C–H activation reaction that proceed through a CMD/AMLA mechanism, see: (a) M. Lafrance, C. N. Rowley, T. K. Woo and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 8754 CrossRef CAS PubMed; (b) D. García-Cuadrado, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 1066 CrossRef PubMed; (c) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS PubMed; (d) D. García-Cuadrado, P. de Mendoza, A. A. C. Braga, F. Maseras and A. M. Echavarren, J. Am. Chem. Soc., 2007, 129, 6880 CrossRef PubMed; (e) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848 CrossRef CAS PubMed; (f) L. Ackermann, R. Vicente and A. Althammer, Org. Lett., 2008, 10, 2299 CrossRef CAS PubMed; (g) H.-Y. Sun, S. I. Gorelsky, D. R. Stuart, L.-C. Campeau and K. Fagnou, J. Org. Chem., 2010, 75, 8180 CrossRef CAS PubMed; (h) A. Gray, A. Tsybizovaa and J. Roithova, Chem. Sci., 2015, 6, 5544 RSC; (i) P. Wedi, M. Farizyan, K. Bergander, C. Mück-Lichtenfeld and M. van Gemmeren, Angew. Chem., Int. Ed., 2021, 60, 15641 CrossRef CAS PubMed , for a selected example of C–H activation reaction that proceed through a IES mechanism, see; (j) J. Oxgaard, W. J. Tenn, R. J. Nielsen, R. A. Periana and W. A. Goddard, Organometallics, 2007, 26, 1565 CrossRef CAS , for a selected example of C–H activation reaction that proceed through an eCMD mechanism, see; (k) L. Wang and B. P. Carrow, ACS Catal., 2019, 9, 6821 CrossRef CAS PubMed.
- Macgregor and Davies observed that electronic subsituent effects depend on whether the reaction is under kinetic or thermodynamic control. Under kinetic control, electron-rich arenes react faster and under thermodynamic control the products with electron withdrawing substituents are preferred. In our competition experiments, we found the same electronic substituent effect independently of the conditions used, R. A. Alharis, C. L. McMullin, D. L. Davies, K. Singh and S. A. Macgregor, J. Am. Chem. Soc., 2019, 141, 8896 CrossRef CAS PubMed.
- For selected examples of C–H activation reaction that proceed through a BIES mechanism, see: (a) W. Ma, R. Mei, G. Tenti and L. Ackermann, Chem.–Eur. J., 2014, 20, 15248 CrossRef CAS PubMed; (b) W. Liu, S. C. Richter, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7747 CrossRef CAS PubMed; (c) D. Zell, M. Bursch, V. Müller, S. Grimme and L. Ackermann, Angew. Chem., Int. Ed., 2017, 56, 10378 CrossRef CAS PubMed; (d) K. Raghuvanshi, D. Zell and L. Ackermann, Org. Lett., 2017, 19, 1278 CrossRef CAS PubMed; (e) Q. Bu, T. Rogge, V. Kotek and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 765 CrossRef CAS PubMed; (f) Y.-F. Liang, L. Yang, T. Rogge and L. Ackermann, Chem.–Eur. J., 2018, 24, 16548 CrossRef CAS PubMed; (g) W. Ma, Z. Weng, T. Rogge, L. Gu, J. Lin, A. Peng, X. Luo, X. Gou and L. Ackermann, Adv. Synth. Catal., 2018, 360, 704 CrossRef CAS; (h) M. R. Sk, S. S. Bera and M. S. Maji, Adv. Synth. Catal., 2019, 361, 585 CrossRef CAS.
- For examples of electrophilic aromatic substitution reaction that provide small KIE, see and references therein: (a) H. Zollinger, in Advances in Physical Organic Chemistry, Academic Press, 1964, vol. 2, pp. 163–200, for a selected example of C−H activation reaction that proceed through an electrophilic aromatic metalation mechanism with a small KIE, see Search PubMed; (b) C.-H. Park, V. Ryabova, I. V. Seregin, A. W. Sromek and V. Gevorgyan, Org. Lett., 2004, 6, 1159 CrossRef CAS PubMed , for selected examples of C−H activation reaction that proceed through an electrophilic aromatic metalation mechanism with large KIE, see; (c) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137 CrossRef CAS PubMed , and ref. 17a.
- J. Mehara and J. Roithova, Chem. Sci., 2020, 11, 11960 RSC.
- The DFT calculations were carried out with the Gaussian16 software, using M06-IEFPCM/def2tzvpp energies on B3LYP/6-31G(d, p)(Pd,SDD) optimized structures. For more details, see ESI.†.
- (a) C. S. Buettner, D. Willcox, B. G. N. Chapell and M. J. Gaunt, Chem. Sci., 2019, 10, 83 RSC; (b) A. C. Skapski and M. L. Smart, J. Chem. Soc., Chem. Commun., 1970, 658 RSC; (c) S. A. Kurzeev, G. M. Kazankov and A. D. Ryabov, Inorg. Chim. Acta, 2002, 340, 192 CrossRef CAS.
- (a) B. E. Haies, J. F. Berry, J.-Q. Yu and D. G. Musaev, ACS Catal., 2016, 6, 829 CrossRef; (b) Y.-F. Yang, X. Hong, J.-Q. Yu and K. N. Houk, Acc. Chem. Res., 2017, 60, 2853 CrossRef PubMed; (c) J. J. Gair, B. E. Haines, A. S. Filatov, D. G. Musaev and J. C. Lewis, Chem. Sci., 2017, 8, 5746 RSC.
- (a) K. M. Engle, Pure Appl. Chem., 2016, 88, 119 CrossRef CAS; (b) Q. Shao, K. Wu, Z. Zhuang, S. Qian and J.-Q. Yu, Acc. Chem. Res., 2020, 53, 833 CrossRef CAS PubMed.
- M. Gómez, J. Granell and M. Martínez, Organometallics, 1997, 16, 2539 CrossRef.
- D. L. Davies, S. M. A. Donald and S. A. MacGregor, J. Am. Chem. Soc., 2005, 127, 13754 CrossRef CAS PubMed.
- (a) D. G. Musaev, A. Kaledin, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 1690 CrossRef CAS PubMed; (b) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1567101 and 1969711–1969713. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc06840k |
| This journal is © The Royal Society of Chemistry 2023 |