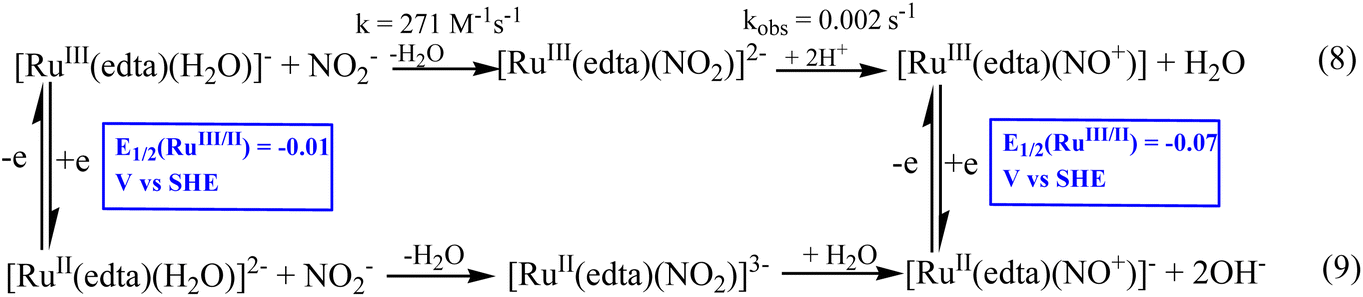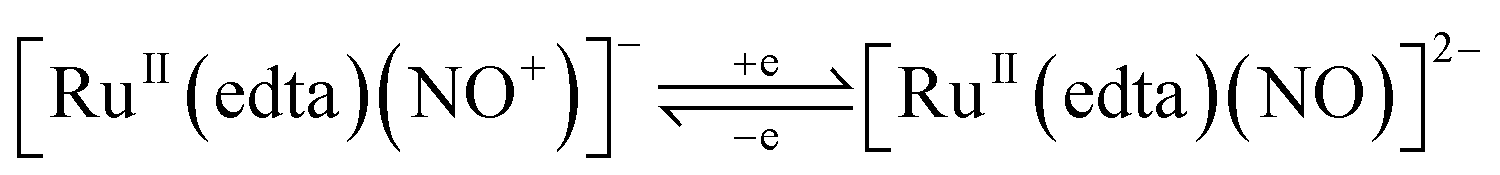DOI:
10.1039/D3QI00199G
(Review Article)
Inorg. Chem. Front., 2023,
10, 1958-1964
Prospect of Ru(edta) complexes in nitrogen cycle electrocatalysis: a mini review
Received
1st February 2023
, Accepted 27th February 2023
First published on 28th February 2023
Abstract
The nitrogen cycle is one of the most important biogeochemical cycles on Earth. This cycle mainly involves redox conversion of dinitrogen when it is converted into ammonia (nitrogen fixation pathway) and the cycle is completed with the conversion of ammonia to dinitrogen (involving nitrification and denitrification pathways). The application of Ru(edta) complexes (edta4− = ethylenediaminetetraacetate) in nitrogen cycle-related electrochemical transformation reactions has not been systematically reviewed to date. This review aims to report the research progression on the use of Ru(edta) complexes in catalyzing N-cycle electrochemical transformations. In this review, the role of Ru(edta) complexes in mediating electrochemical reactions pertaining to nitrogen fixation and denitrification in the nitrogen cycle has been discussed, providing in-depth mechanistic knowledge for understanding the varied roles of Ru(edta) complexes pertaining to the many N-cycle-related electrochemical transformations.
1. Introduction
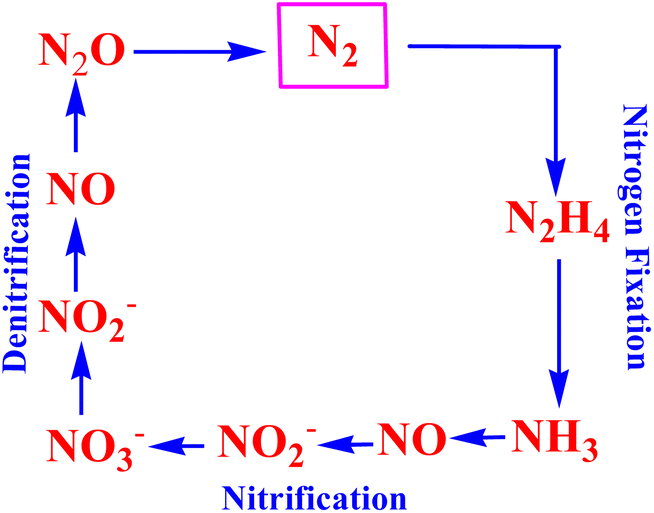
The nitrogen cycle (hereinafter referred to as the N-cycle) is a naturally occurring reaction cycle,1 wherein the inorganic compounds of nitrogen are controlled. It is one of the most significant and long lasting (ten thousand years) biogeochemical cycles on Earth. The nitrogen cycle relates a diverse range of sectors including agriculture and energy, and has ecological impacts on climate change and water bodies. Although the global nitrogen cycle in the current era includes the main forms of nitrogen and their circulation in different biogeochemical sources, viz. oceans, lakes, rivers, groundwater, the atmosphere, terrestrial biosphere, and geosphere,2,3 we depict herein a simplified nitrogen cycle as shown in Fig. 1, signifying the most essential pathways and the intermediates formed in the different interconversions of nitrogen. As shown typically in Fig. 1, dinitrogen (N2) is converted into ammonia (NH3) through the nitrogen fixation pathway, and the cycle is completed with the conversion of ammonia to dinitrogen involving the nitrification of ammonia to nitric oxide/nitrite/nitrate followed by their denitrification (Fig. 1). The intermediate compounds such as hydrazine (N2H4), hydroxylamine (NH3OH), nitric oxide (NO), nitrous and nitric acids (HNO2/HNO3), and nitrous oxide (N2O) produced during the nitrogen cycle are of value in the chemical industries.
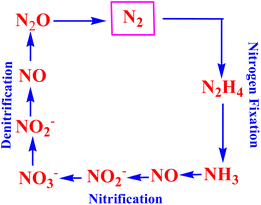 |
| | Fig. 1 Pictorial presentation of the nitrogen cycle typically representing the most significant reactions: nitrogen fixation, nitrification and denitrification. | |
It is noteworthy that all the aforesaid reactions as shown in Fig. 1 occur in nature, and are catalyzed by active sites of metalloenzymes. However, the syntheses of transition metal complexes and their application towards mimicking such enzymatic transformations shown in the N-cycle (Fig. 1) are intriguing areas of research and of enduring importance.4–15 In this context, an electrochemical approach to effect the transformations referred to as the N-cycle (Fig. 1) using transition metal complexes as ‘molecular electro-catalysts’ appears to be the most attractive for its veritable ‘eco-friendly’ aspects. Such metal complex catalysts bind to N-cycle molecules, thus providing a lower energy pathway (rendering a substantial drop in the over-potential required to initiate the electrochemical conversion of the substrate directly) for effecting multi-electron transfer redox transformations pertaining to the N-cycle. Furthermore, a properly designed electrocatalytic process does not produce any toxic or hazardous materials or introduce hazards of its own. On the other hand, the reaction selectivity of the electrode materials for the reactions in the N-cycle can be governed by varying the ligand structure of the metal complex. It is noteworthy that in 1965 the [RuII(NH3)5(N2)]2+ complex was reported to be the first stable metal complex of dinitrogen.16 In the same year, the activity of such complexes as models for nitrogenase with nitrogen on active sites was reported.17 Since then, ruthenium complexes have been of abiding importance in the literature for their application as molecular catalysts in various N-cycle electrochemical transformations, viz. nitrogen-to-ammonia conversion, nitrite reduction, conversion of ammonia, etc.18–23
While reports on the application of Ru(edta) complexes (edta4− = ethylenediaminetetraacetate) to mediate N-cycle electrochemical reactions are available in the literature, we for the first time systematically review the Ru(edta)-catalyzed N-cycle-related electrochemical transformations, highlighting the potential role of Ru(edta) complexes to function as efficient ‘molecular catalysts’ in a homogeneous solution involving an electrode as a heterogeneous outer-sphere electron donor or acceptor. The [RuIII(edta)(H2O)]− complex, due to its intrinsic lability, could easily bind to N-cycle molecules (via rapid substitution reactions), thus providing a lower energy pathway (rendering a substantial drop in the over-potential required to initiate the electrochemical transformation of N-cycle molecules directly). In this short review, we have included reports on the N-cycle-related electrochemical transformations catalyzed by Ru(edta) complexes and summarized our mechanistic understanding of such electrochemical conversions that occur resembling those in the active sites of enzymes involved in the nitrogen cycle.
2. Background chemistry
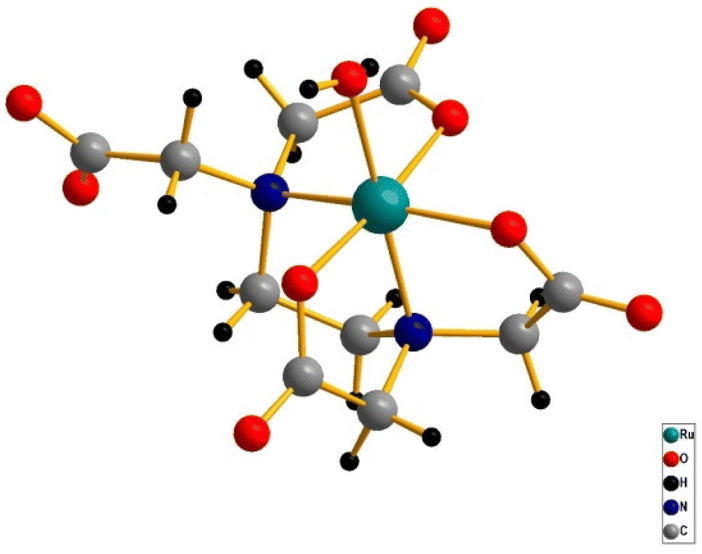
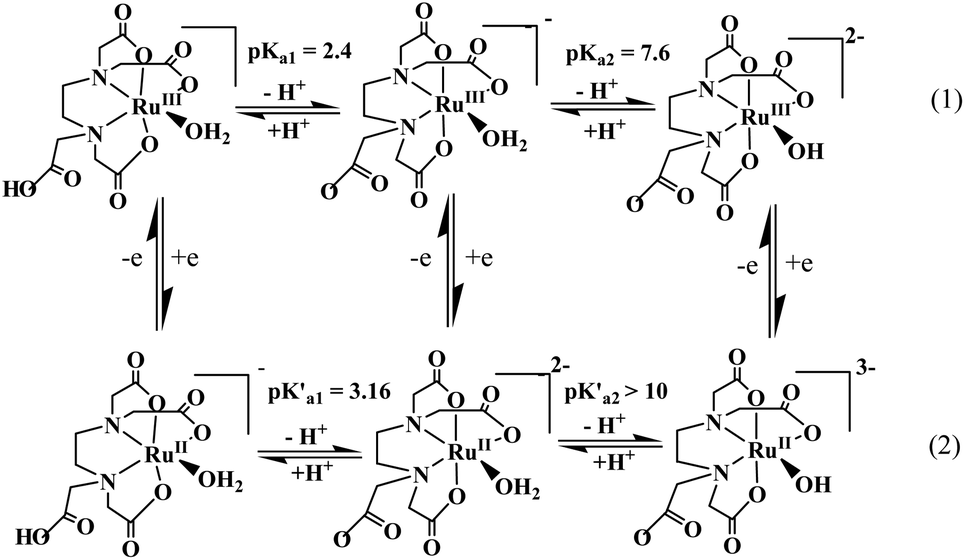
The ‘edta’ ligand forms a very stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal complex with ruthenium. It was established by crystallographic studies24 that ‘Hedta3−’ in the [RuIII(Hedta)(H2O)] complex acts as a pentadentate coordinating ligand (Fig. 2). The sixth coordination site of the ruthenium center is occupied by a water molecule; however, with the increase in the pH of the solution, deprotonation of the dangling carboxylic acid group (pKa1 = 2.4) and the coordinated water molecule (pKa2 = 7.6) occur successively as shown in Scheme 1.25
:1 metal complex with ruthenium. It was established by crystallographic studies24 that ‘Hedta3−’ in the [RuIII(Hedta)(H2O)] complex acts as a pentadentate coordinating ligand (Fig. 2). The sixth coordination site of the ruthenium center is occupied by a water molecule; however, with the increase in the pH of the solution, deprotonation of the dangling carboxylic acid group (pKa1 = 2.4) and the coordinated water molecule (pKa2 = 7.6) occur successively as shown in Scheme 1.25
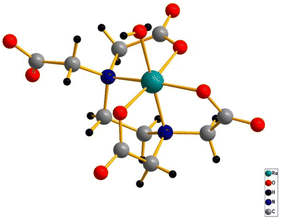 |
| | Fig. 2 3D crystal structure of the [RuIII(Hedta)(H2O)] complex. | |
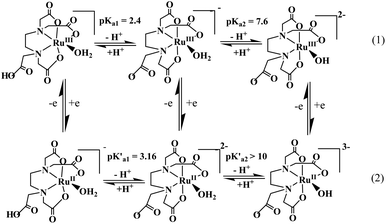 |
| | Scheme 1 Electrode reactions and proton dissociation equilibria of [RuIII/II(Hedta)(H2O)]0/− complexes. | |
The one-electron reduction of [RuIII(Hedta)(H2O)] to [RuII(Hedta)(H2O)]− was found to be reversible in the pH range 3–5, and the E1/2 values corresponding to the RuIII/RuII redox couple became more negative with increasing pH, and tended to reach a limiting value at a pH higher than 5.0. Such a trend of the E1/2 values is explicable in terms of the proton dissociation of the uncoordinated –COOH group of the complex as shown in Scheme 1. At higher pH (>6), the lack of reversibility is caused by the considerable difference in the proton-dissociation equilibrium (Scheme 1) values of [RuIII(edta)(H2O)]− (pKa2 = 7.6) and [RuII(edta)(H2O)]2− (pK′a2 > 10) complexes.26 The intriguing electrochemical properties of Ru(edta) complexes were brought together and thoroughly evaluated in a recent review.27 The electrochemical data (including a wide range of metal-centered redox potentials) described in the review article for a number of mixed-ligand and mixed-chelate complexes of Ru(edta) ascertain their redox-mediating ability, and are of significance in designing new catalytic systems appreciating the potential range required for the particular redox reaction.27
During the late seventies to the late nineties, research interest in [RuIII(edta)(H2O)]− was limited mainly due to its reactivity towards aqua-substitution reactions.28 It was shown that the [RuIII(edta)(H2O)]− complex by virtue of its remarkable lability towards the aqua-substitution reaction could bind DNA and DNA constituents and cellular thiols and nitric oxide (NO) in a facile and straightforward manner under physiological conditions exhibiting pharmaceutical activity.29 However, later studies demonstrated the ability of the Ru(edta) complexes to mediate the chemical and electrochemical transformations of small molecules, viz. CO2, O2/H2O2, and NO/NO2−, and biologically important thiols (RSH) resembling metalloenzymes. The results of all the aforementioned studies have recently been systematically reviewed.30–33
3. Application of Ru(edta) complexes in N-cycle electrocatalysis
3.1 Ru(edta) complex mediated nitrogen fixation
While reports on the synthesis and physico-chemical characterization of both the terminal [RuII(edta)N2]2− and bridging [{RuII(edta)}2(N2)]4− complexes are available in the literature,34 they are comparatively less stable and much more labile than their corresponding pentaammine analogues, [Ru(NH3)5N2]2+ and [{Ru(NH3)5}2N2]2+.16 Although no report on the use of the aforesaid dinitrogen complexes of Ru(edta) as a nitrogenase model17 in the electrochemical reduction of dinitrogen (N2) to ammonia (NH3) is available in the literature, the intermediacy of [Ru(Hedta)(N2)] in the photocatalytic N2 fixation over the surface of visible light-irradiated CdS/Pt/RuO2 semiconductor particles under a nitrogen atmosphere (1 atm) was reported by Taquikhan and co-workers in the late eighties.35 Their studies revealed that upon light excitation, an electron from the conduction band of the semiconductor is relayed through the surface-bound metal-complex to effect the reduction of the coordinated nitrogen (N2) in [Ru(Hedta)(N2)].35 The results of the aforesaid studies will encourage further studies towards the use of the [Ru(Hedta)(N2)] complex anchored to the electrode surface for achieving the reduction of coordinated N2 to NH3 electrochemically.
However, in 1994, Ramachandraiah explored whether the [RuIII(Hedta)(H2O)] complex could effectively catalyze the reduction of hydrazine to ammonia electrochemically.36 Hydrazine, a penultimate compound, is formed prior to the formation of ammonia in the nitrogen fixation pathway (eqn (3)).
| | | NH2 − NH2 + 2H+ + 2e− → 2NH3 | (3) |
The catalytic performance of the [RuIII(Hedta)(H2O)] complex towards the electrochemical reduction of hydrazine was studied, and the results of the detailed electrochemical studies revealed that the [RuIII(Hedta)(H2O)] complex could reduce hydrazine to ammonia (at 0.19 V vs. SHE) at the surface of a mercury electrode with 100% coulombic efficiency, and the turn-over rates (in terms of NH3 formation via the reduction of hydrazine) were 18.4 and 9.5 (mol.h−1) at pH 2.8 and 1.9, respectively.36
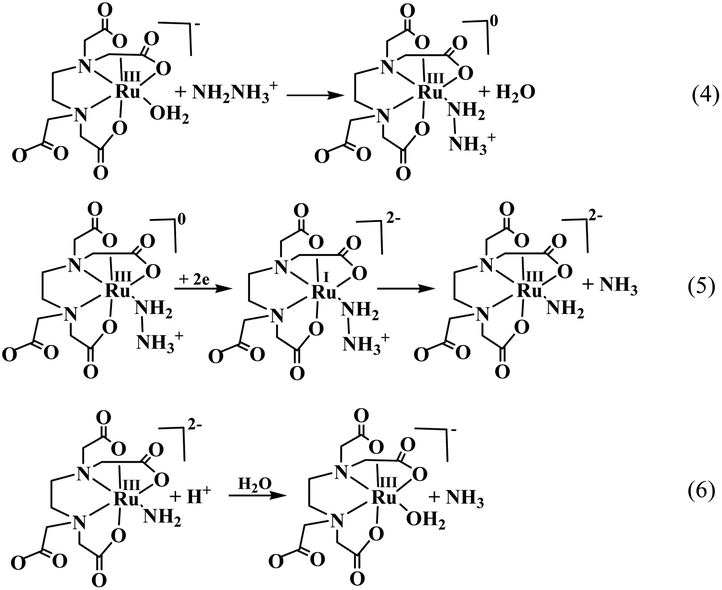
The overall catalytic process shown in Scheme 2 involves the rapid binding of the substrate hydrazine (NH2NH3+) to the metal center of the catalyst complex (eqn (4)), resulting in the formation of a catalytically active [RuIII(Hedta)(NH2NH3)]+ intermediate species, which then undergoes a two-electron electrochemical reduction to produce a highly unstable [RuI(Hedta)(NH2NH3)]− intermediate species (eqn (5)).36 The [RuI(Hedta)(NH2NH3)]− intermediate rapidly undergoes N–N bond cleavage involving an intra-molecular electron transfer pathway to yield [RuIII(Hedta)(NH2)]− with the concomitant formation of NH3 (eqn (5)) in the reaction system. Hydrolysis of the [RuIII(edta)(NH2)]− complex at lower pH (<3) generated another molecule of NH3 (eqn (6)) along with the [RuIII(Hedta)(H2O)] catalyst complex in the reaction system.36 In a follow-up study,37 the same group reported that the [RuIII(Hedta)(H2O)] complex could catalyze the electrochemical transformation of phenylhydrazine to ammonia and aniline at 0.065 V (vs. SHE) at a turnover rate of 5.98 (mol.h−1) at pH 2.82.37 Formation of the [RuIII(Hedta)(NH2NHPh)] complex through aqua-substitution of [RuIII(Hedta)(H2O)] followed by two-electron electrochemical reduction of coordinated phenylhydrazine in [RuIII(Hedta)(NH2NHPh)] yielding ammonia and aniline was suggested.37
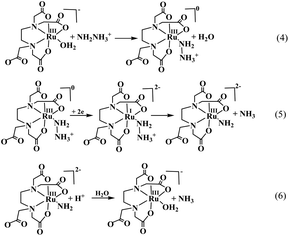 |
| | Scheme 2 Electrochemical reduction of hydrazine to ammonia catalyzed by the [RuIII(Hedta)(H2O)] complex. | |
3.2 Ru(edta) complex-mediated denitrification
The reduction of nitrite (NO2−) to ammonia (NH3) is a multi-electron transfer process (eqn (7)) catalyzed by nitrite reductase enzymes.38| | | NO2− + 7H+ + 6e− → NH3 + 2H2O | (7) |
During the late eighties, Meyer et al.39,40 explored whether the [Ru(edta)(H2O)]− complex could catalyze the electrochemical transformation of nitrite (NO2−) to various N-based products (N2O, N2, NH2OH and NH3) involved in the N-cycle transformation. The electrochemical reduction process was investigated thoroughly by performing extensive controlled potential electrolysis (using a mercury pool as the working electrode) at different pH values and applied potentials under turnover conditions of excess nitrite.40 The selectivity and product yield of each product in the mixture of N-based products, N2O, N2, NH2OH and NH3, formed in the aforesaid nitrite reduction depend on the applied potential and pH of the reaction system.40 Furthermore, the results of studies at varied pH values exhibited 97% conversion of total nitrite, which could be reduced at pH 2.1 (at −0.31 V vs. SHE); however, at pH 5.0 (at −0.51 V vs. SHE), a lower conversion (86%) was noticed.40 It is noteworthy that at higher pH, the catalytic efficiency of the system diminished due to the degradation of the Ru(edta) catalyst complexes.40 The [RuII(edta)(NO+)]− species is shown to be the key intermediate species in the overall electrocatalytic process.40
The results of kinetic and mechanistic studies revealed that the [RuIII(edta)(H2O)]− complex reacts rapidly with NO2− to form the [RuIII(edta)(NO2)]2− complex (k = 271 M−1s−1 at 25 °C).41 The [RuIII(edta)(NO2)]2− complex is unstable at lower values of pH (<5) and undergoes decomposition (eqn (8) in Scheme 3), yielding [RuIII(edta)(NO+)]0 species in the reaction system.41 [RuIII(edta)(NO+)]0 could exhibit a metal-based electron-transfer reaction at the electrode to produce [RuII(edta)(NO+)]− as shown in Scheme 3. The E1/2 value reported for the [RuIII(edta)(NO+)]0/[RuII(edta)(NO+)]− redox couple is −0.07 V (vs. SHE).42 However, a report on the formation of [RuII(edta)(NO2)]3−via an alternative route involving aqua-substitution of [RuII(edta)(H2O)]2− with NO2− followed by its conversion to [RuII(edta)(NO+)]− (eqn (9) in Scheme 3) is also available in the literature.43
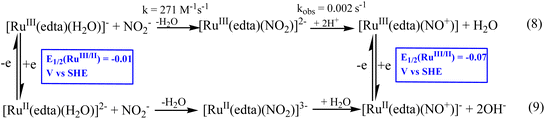 |
| | Scheme 3 Formation of [RuII(edta)(NO+)]−via NO2− reduction. | |
It was shown that the [RuII(edta)(NO+)]− complex formed during the electrochemical nitrite reduction process40 could undergo one-electron reduction at −0.11 V (vs. SHE) to produce an unstable intermediate species, [RuII(edta)(NO)]2− (eqn (10)),40,44 which underwent further reduction (eqn (11) and (12)) as noticed in cyclic voltammetric studies at pH 2.1.40
| | 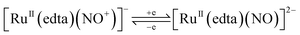 | (10) |
| | | [RuII(edta)(NO)]2− + e → [RuI(edta)(NO)]3− | (11) |
| | | [RuII(edta)(NO)]2− + e + H+ → [RuI(edta)(NHO)]2− | (12) |
The [RuI(edta)(NO)]3− and [RuI(edta)(NHO)]2− intermediate species (admittedly speculative) were very active and they underwent the following reactions (eqn (13)–(18)) as outlined below, leading to the formation of various N-based products, viz. N2O, N2, NH2OH, and NH3 at pH 2.1 and an applied potential of −0.31 V (vs. SHE).40
| | | 2[Ru(edta)(NHO)]2− + H2O → 2[RuII(edta)(H2O)]2− + N2O | (13) |
| | | 2[Ru(edta)(NHO)]2− + 2H+ + 2e → 2[RuII(edta)(H2O)]2− + N2 | (14) |
| | | [Ru(edta)(NHO)]2− + 2H+ + 2e → [RuII(edta)(NH2OH)]2− | (15) |
| | | [RuII(edta)(NH2OH)]2− + H+ → [RuII(edta)(NH3OH)]− | (16) |
| | | [RuII(edta)(NH3OH)]− + H+ + 2e → [RuII(edta)(NH3)]2− + H2O | (17) |
While the formation of N2O involved initial disproportionation of [Ru(edta)(NHO)]2−, followed by N–N coupling (eqn (13)), N2 production was accounted for the further reduction of [Ru(edta)(NHO)]2− including N–N coupling (eqn (14)).40 More highly reduced products, NH3OH+ and NH4+, were formed via two-electron reduction and the formation of NH2OH as outlined in eqn (15)–(18). It was reported that NH3OH+ appeared to be a precursor of NH4+ which is formed as a transient species that prevents the binding and further reduction to NH3/NH4+.40 It is noteworthy that in the aforesaid electrocatalytic denitrification process,40 N2O (an environmental pollutant) formed via N–N coupling was found to be a major N-based product (47%) at lower pH (2.1) and an applied potential of (−0.31 V vs. SHE) in comparison with the formation of NH3 (14%).40 However, at higher pH (5.0) and a larger potential (−0.51 V vs. SHE), reduction in the formation of N2O (26%) was noticed along with enhancement of the formation of NH3 (29%). The percentages of N2 (involving N–N coupling) in the N-based products formed both at pH 2.1 and at pH 5.0 are more or less the same (15–17%).40 It appears that both pH and applied potential play a significant role in governing the N–N coupling step on the way to N2O (eqn (13)) formation vis-à-vis further reduction of nitrosyl intermediates to produce NH3 in the reacting solution.40 While application of Ru(edta) complexes pertaining to NO utilization is well recognized,32 it would be of interest to further investigate whether Ru(edta) complexes could drive the NO dismutation reaction resulting in the formation of N2 and O2 in the N cycle.45 Moreover, the [RuII(edta)(NO+)]− complex anchored on poly(amidoamine)dendrimers (PAMAM) through peptide bonds formed in the reaction of the uncoordinated carboxylic acid arm of the metal complex reveals similar electrochemical properties to those exhibited by the unbound complex.46 Further research is necessitated in order to examine whether the tethered [(RuII(edta)NO)]2− species could participate in the N–N coupling reaction (that leads to the formation of N2O) or not, and improve our understanding of the mechanism of N2O production.
3.3 Perspectives on Ru(edta) mediated reversal of nitrogen fixation
While in the electrocatalytic denitrification process (Fig. 1) a substantial amount of N2O is produced which has a detrimental environmental impact, recent results suggest that nitrification also affords N2O.47 In this context, oxidation of NH3 along with N2O production and degradation in the nitrogen cycle is a demanding area of research.48 Recent studies explored whether ruthenium complexes could be used as catalysts or electrocatalysts for reversing the nitrogen fixation reaction through oxidation of NH3 to N2 under ambient conditions.21,22
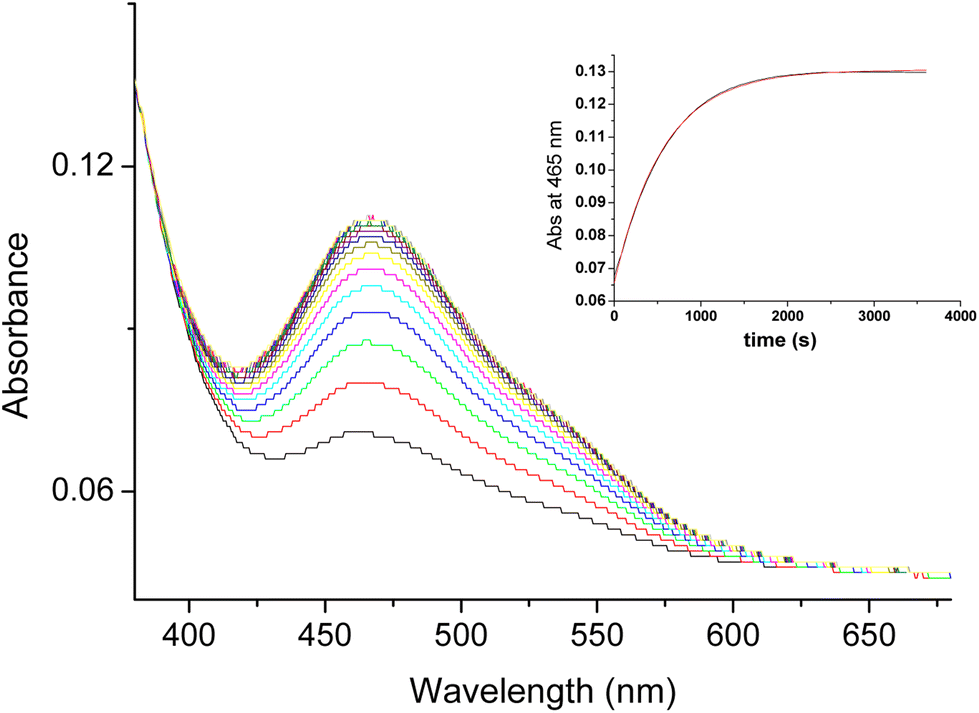
While studying electron transfer reactions of [RuIII(edta)(pz)]− with different electron donors, viz. ascorbic acid, catechol, sulfite, thiols, and bisulfide,49 we noticed that the addition of hydrazine to the pale-yellow solution of [RuIII(edta)(pz)]− resulted in spectral changes (shown in Fig. 3) consistent with the formation of the [RuII(edta)(pz)]2− complex (λmax at 465 nm), confirming the ability of hydrazine (N2H4) to reduce [RuIII(edta)(pz)]− to [RuII(edta)(pz)]2− (eqn (18)).
| | | 2[RuIII(edta)(pz)]− + N2H4 → 2[RuII(edta)(pz)]2− + N2 + 4H+ | (18) |
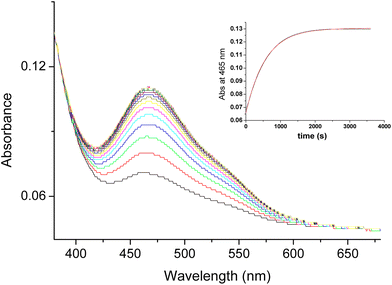 |
| | Fig. 3 Spectral changes that occurred for the reaction of [RuIII(edta)(pz)]− with hydrazine (N2H4). | |
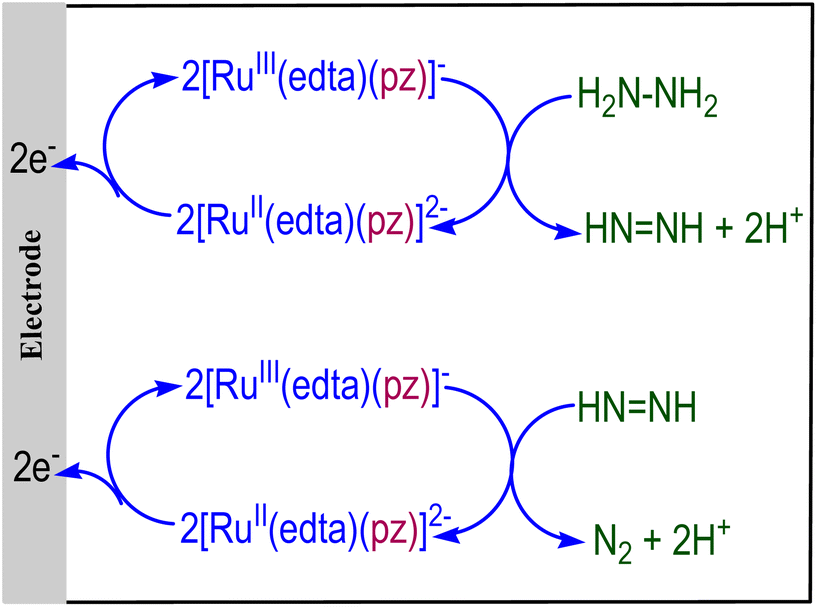
The above observation pertaining to the oxidation of hydrazine by [RuIII(edta)(pz)]− leads to the idea of making the system electrocatalytic by re-oxidizing [RuII(edta)(pz)]2− formed during hydrazine oxidation (eqn (18)) at the electrode, as pictorially demonstrated in Fig. 4. Although the [RuIII(edta)(pz)]− catalysed electrochemical oxidation of N2H4 to N2 is speculatively shown in Fig. 4, it may provide the basis for further research towards a successful demonstration. Four-electron oxidation of N2H4 to produce N2 is important per se in hydrazine-based fuel cells.50
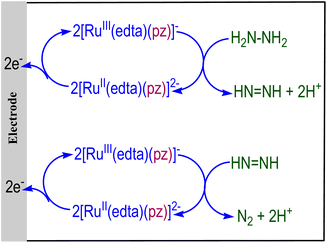 |
| | Fig. 4 Electrochemical oxidation of N2H4 to N2 catalyzed by [RuIII(edta)(pz)]−. | |
4. Summary and prospects
In this short review, we have presented an account of Ru(edta)-mediated electrochemical transformation reactions involving inorganic compounds of nitrogen cycles. We have shown the ability of Ru(edta) in catalyzing the electrochemical reduction of hydrazine to ammonia, and shown the role of the nitrosyl complex of Ru(edta) in effecting the reduction of nitrite to various N-based products formed in N-cycle denitrification. The mechanistic details discussed in this review may be of use in outlining reaction conditions for optimizing the product yield and selectivity in the denitrification reaction. Carrying out N-cycle electrocatalysis with an electrode surface modified by polymer-bound Ru(edta) complexes could be promising to achieve selectivity in N-cycle nitrification and denitrification. Considering the comparatively simple and straightforward synthesis of Ru(edta) complexes, and their remarkable coordinating ability towards binding substrate molecules (thus lowering the activation energy barrier of the electrochemical transformation of such substrate molecules), they may be advantageous to the researcher for developing improved catalytic systems for electrochemical transformations of inorganic molecules of N-cycle reactions with higher efficiency and selectivity. In this regard, research exploring the efficacy of Ru(edta) complexes in nitrate reduction and ammonia oxidation would be of interest in further understanding enzymatic transformation in the nitrogen cycle.51
Author contributions
Olga Impert: writing original draft. Debabrata Chatterjee: review & editing. Rudi van Eldik: review & editing.
Conflicts of interest
There is no conflict to declare.
Acknowledgements
DC and RvE thank their co-workers and collaborators who have contributed to this study.
References
-
H. Bothe, S. Ferguson and W. E. Newton, Ed. The Biology of the Nitrogen Cycle, Elsevier, Amsterdam, 2006 Search PubMed.
-
N. Lehnert, G. Coruzzi, E. Hegg, L. Seefeldt and L. Stein, NSF Workshop Report: Feeding the World in the 21st Century: Grand Challenges in the Nitrogen Cycle, National Science Foundation, Arlington, VA, 2016 Search PubMed.
- X. Zhang, B. B. Ward and D. M. Sigman, Global nitrogen cycle: Critical enzymes organisms and processes for nitrogen budget and dynamics, Chem. Rev., 2020, 120, 5308–5351 CrossRef CAS PubMed.
- J. Chatt, J. R. Dilworth and R. L. Richards, Recent advances in the chemistry of nitrogen fixation, Chem. Rev., 1978, 78, 589–625 CrossRef CAS.
- B. A. MacKay and M. D. Fryzuk, Dinitrogen coordination chemistry: On the biomimetic borderlands, Chem. Rev., 2004, 104, 385–402 CrossRef CAS PubMed.
- V. Rosca, M. Duca, M. T. de Groot and M.T. M. Koper, Nitrogen cycle electrocatalysis, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS PubMed.
- S. Hinrichsen, H. Broda, C. Gradert, L. Söncksen and F. Tuczek, Recent developments in synthetic nitrogen fixation, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2012, 108, 17–47 RSC.
- Y. Nishibayashi, Recent progress in transition-metal catalyzed reduction of molecular dinitrogen under ambient reaction conditions, Inorg. Chem., 2015, 54, 9234–9247 CrossRef CAS PubMed.
- H. Tanaka, Y. Nishibayashi and K. Yoshizawa, Interplay between theory and experiment for ammonia synthesis catalyzed by transition metal complexes, Acc. Chem. Res., 2016, 49, 987–995 CrossRef CAS PubMed.
- K. M. Lancaster, J. D. Caranto, S. H. Majer and M. A. Smith, Joule, alternative bioenergy: Updates to and challenges in nitrification, Metalloenzymology, 2018, 2, 421–441 CAS.
- X. Cui, C. Tang and Q. Zhang, A review of electrocatalytic reduction of dinitrogen to ammonia under ambient conditions, Adv. Energy Mater., 2018, 8, 1800369 CrossRef.
- B. H. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Challenges and prospects in the catalysis of electroreduction of nitrogen to ammonia, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- M. J. Chalkley, M. W. Drover and J. C. Peters, Catalytic N2-to-NH3 (or -N2H4) conversion by well-defined molecular coordination complexes, Chem. Rev., 2020, 120, 5582–5636 CrossRef CAS PubMed.
- D. Hao, Y. Liu, S. Gao, H. Arandiyan, X. Bai, Q. Kong, W. Wei, P. Kang and S. B-J. Ni, Emerging artificial nitrogen cycle processes through novel electrochemical and photochemical synthesis, Mater. Today, 2021, 46, 212–233 CrossRef CAS.
- A. F. Ibrahim, P. Garrido-Barros and J. C. Peters, Electrocatalytic nitrogen reduction on a molybdenum complex bearing a PNP pincer ligand, ACS Catal., 2023, 13, 72–78 CrossRef CAS.
- A. D. Allen and C. V. Senoff, Nitrogenopentammineruthenium(II) complexes, J. Chem. Soc., Chem. Commun., 1965, 621–622 RSC.
- J. Chatt, R. L. Richards, J. R. Sanders and J. E. Fergusson, Nitrogen complexes as models for nitrogenase with nitrogen on the active site, Nature, 1969, 221, 551–552 CrossRef CAS.
- J. J. Warren, T. A. Tronic and J. M. Mayer, Thermochemistry of proton-coupled electron transfer reagents and its implications, Chem. Rev., 2010, 110, 6961–7001 CrossRef CAS PubMed.
- J. Fajardo Jr. and J. C. Peters, Catalytic nitrogen-to-ammonia conversion by osmium and ruthenium complexes, J. Am. Chem. Soc., 2017, 139, 16105–16108 CrossRef PubMed.
- Y. Arikawa, Y. Otsubo, H. Fujino, S. Horiuchi, E. Sakuda and K. Umakoshi, Nitrite reduction cycle on a dinuclear ruthenium complex producing ammonia, J. Am.
Chem. Soc., 2018, 140, 842–847 CrossRef CAS PubMed.
- K. Nakajima, H. Toda, K. Sakata and Y. Nishibayashi, Ruthenium-catalysed oxidative conversion of ammonia into dinitrogen, Nat. Chem., 2019, 11, 702–709 CrossRef CAS PubMed.
- F. Habibzadeh, S. L. Miller, T. W. Hamann and M. R. Smith, Homogeneous electrocatalytic oxidation of ammonia to N2 under mild conditions, Proc. Natl. Acad. Sci., 2019, 116, 2849–2853 CrossRef CAS PubMed.
- S. Kim, J. Kim, H. Zhong, G. B. Panetti and P. J. Chirik, Catalytic N–H bond formation promoted by a ruthenium hydride complex bearing a redox-active pyrimidine–imine ligand, J. Am. Chem. Soc., 2022, 144, 20661–20671 CrossRef CAS PubMed.
- M. M. Taqui Khan, H. C. Bajaj, Z. Shirin and K. Venkatasubraminan, Crystal and molecular structure of aquoethylenediaminetetra-acetatoruthenium(III) and its extraordinary lability towards substitution, Indian J. Chem., 1992, 31, 303–308 Search PubMed.
- T. Matsubara and C. Creutz, Properties and reactivities of ethylenediaminetetraacetate complexes of ruthenium(III) and -(II), Inorg. Chem., 1979, 18, 1956–1966 CrossRef CAS.
- N. Oyama and F. A. Anson, Attachment of the EDTA complex of ruthenium(III) to the surface of graphite electrodes: Electrochemistry and ligand substitution chemistry with the attached complex, J. Electroanal. Chem., 1978, 88, 289–297 CrossRef CAS.
- D. Chatterjee, M. Oszajca, A. Katafias and R. van Eldik, Coord. Chem. Rev., 2021, 436, 213773 CrossRef CAS.
- D. Chatterjee, Properties and reactivities of polyaminopolycarboxylate (pac) complexes of ruthenium, Coord. Chem. Rev., 1998, 168, 273–293 CrossRef CAS.
- D. Chatterjee, A. Mitra and G. S. De, Ruthenium polyaminocarboxylate complexes prospects for their use as metallopharmaceuticals, Platinum Met. Rev., 2006, 50, 1–12 CrossRef.
- D. Chatterjee and R. van Eldik, Prospect of RuIII(edta) in catalysis of bicarbonate reduction, Curr. Catal., 2020, 9, 23–31 CrossRef CAS.
- D. Chatterjee, Chemistry of Ru(edta) complexes relevant to oxidoreductase mimicking: A personal perspective, New J. Chem., 2020, 44, 18972–18979 RSC.
- D. Chatterjee, M. Chrzanowska, A. Katafias and R. van Eldik, J. Inorg. Biochem., 2021, 225, 111595 CrossRef CAS PubMed.
- D. Chatterjee and R. van Eldik, Application of Ru(edta) complexes in biomimetic activation of small molecules. Kinetic and mechanistic impact, Adv. Inorg. Chem., 2022, 81, 389–431 CrossRef.
- A. A. Diamantis and J. V. Dubrwaski, Preparation and structure of ethylenediaminetetraacetate complexes of ruthenium(II) with dinitrogen, carbon monoxide, and other π-acceptor ligands, Inorg. Chem., 1981, 20, 1142–1150 CrossRef CAS.
- M. M. Taqui Khan, R. C. Bhardwaj and C. Bhardwaj, Catalytic fixation of nitrogen by the photocatalytic CdS/Pt/RuO2 particulate system in the presence of aqueous [Ru(Hedta)N2]− complex, Angew. Chem., Int. Ed. Engl., 1988, 27, 923–925 CrossRef.
- G. Ramachandraiah, Spectrophotometric, kinetic and electrochemical investigations of new monomeric hydrazinium adducts with ethylenediaminetetraacetatoruthenium(III) complexes: Catalytic reduction of hydrazine to ammonia in aqueous acidic solution, J. Am. Chem. Soc., 1994, 116, 6733–6738 CrossRef CAS.
- R. Prakash, B. Tyagi, D. Chatterjee and G. Ramachandraiah, Interaction of phenylhydrazine with RuIII–EDTA complexes: Reduction of phenylhydrazine to ammonia and aniline in aqueous acidic conditions, Polyhedron, 1997, 16, 1235–1240 CrossRef CAS.
- M. Losada, Metalloenzymes of the nitrate-reducing system, J. Mol. Catal., 1976, 1, 245–264 CrossRef CAS.
- M. R. Rhodes, M. H. Barley and T. J. Meyer, Electrocatalytic reduction of nitrite using simple. coordination complexes of iron and ruthenium, Inorg. Chem., 1988, 27, 4772–4774 CrossRef CAS.
- M. R. Rhodes, M. H. Barley and T. J. Meyer, Electrocatalytic reduction of nitrite ion by edta complexes of iron(II) and ruthenium(II), Inorg. Chem., 1991, 30, 629–635 CrossRef CAS.
- D. Chatterjee, S. Shome, N. Jaiswal and P. Banerjee, Nitrite
reduction mediated by the complex RuIII(EDTA), Dalton Trans., 2014, 43, 13596–13600 RSC.
- M. M. Taqui Khan, K. Venkatasubramanian, Z. Shirin and M. M. Bhadbhade, (Ethylenediaminetetraacetato)nitrosylruthenium, an efficient oxygen-atom transfer agent, J. Chem. Soc., Dalton Trans., 1995, 1031–1035 Search PubMed.
- Y. Chen, F.-T. Lin and R. E. Shepherd,
15N NMR and electrochemical studies of [RuII(hedta)]− complexes of NO, NO+, NO2−, and NO−, Inorg. Chem., 1999, 38, 973–983 CrossRef CAS PubMed.
- P. G. Zanichelli, A. M. Miotto, H. F. G. Estrela, F. R. Soares, D. M. Grassi-Kassisse, R. C. Spadari-Bratfisch, E. E. Castellano, F. Roncarroli, A. R. Parise, J. A. Olabe, A. R. M. S. de Brito and D. W. Franco, The [Ru(Hedta)(NO)]0,–1 system: structure, chemical activity, and biological assays, J. Inorg. Biochem., 2004, 98, 1921–1932 CrossRef CAS PubMed.
- C. Ferousi, S. H. Majer, I. M. DiMucci and K. M. Lancaster, Biological and bioinspired inorganic N–N Bond-forming reactions, Chem. Rev., 2020, 120, 5252–5307 CrossRef CAS PubMed.
- P. C. G. Benini, B. R. McGarvey and D. W. Franco, Functionalization of PAMAM dendrimers with [RuIII(edta)(H2O)]−, Nitric Oxide, 2008, 19, 245–251 CrossRef CAS PubMed.
- P. Li, Y. Peng, S. Wang and Y. Liu, N2O emission from partial nitrification and full nitrification in domestic wastewater treatment process, Water, 2022, 14, 3195 CrossRef CAS.
- N. Lehnert, H. T. Dong, J. B. Harland, A. P. Hunt and C. J. White, Reversing nitrogen fixation, Nat. Rev. Chem., 2018, 2, 278–289 CrossRef CAS.
- D. Chatterjee and R. van Eldik, Electron transfer reactions of RuIII(edta) containing N-heterocyclic ligand pyrazine: Kinetic and mechanistic studies, Macrohetercycles, 2020, 13, 193–200 CrossRef CAS.
- N. V. Reesa and R. G. Compton, Carbon-free energy: A review of ammonia- and hydrazine-based electrochemical fuel cells, Energy Environ. Sci., 2011, 4, 1255–1260 RSC.
- N. Lehnert, B. W. Musselman and L. C. Seefeldt, Grand challenges in the nitrogen cycle, Chem. Soc. Rev., 2021, 50, 3640–3646 RSC.
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Olga
Impert
*ab,
Olga
Impert