
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermoresponsive polymers with LCST transition: synthesis, characterization, and their impact on biomedical frontiers
Yichun
Yuan†
 a,
Konpal
Raheja†
a,
Nathalie B.
Milbrandt†
a,
Sophia
Beilharz†
a,
Steffy
Tene
a,
Solomon
Oshabaheebwa
b,
Umut A.
Gurkan
bc,
Anna Cristina S.
Samia
a and
Metin
Karayilan
*a
a,
Konpal
Raheja†
a,
Nathalie B.
Milbrandt†
a,
Sophia
Beilharz†
a,
Steffy
Tene
a,
Solomon
Oshabaheebwa
b,
Umut A.
Gurkan
bc,
Anna Cristina S.
Samia
a and
Metin
Karayilan
*a
aDepartment of Chemistry, Case Western Reserve University (CWRU), Cleveland, Ohio 44106, USA. E-mail: metin.karayilan@case.edu
bDepartment of Biomedical Engineering, CWRU, Cleveland, Ohio 44106, USA
cDepartment of Mechanical and Aerospace Engineering, Case Comprehensive Cancer Center, CWRU, Cleveland, Ohio 44106, USA
First published on 29th August 2023
Abstract
Advances in thermoresponsive materials have significantly impacted many biomedical fields. The unique behavior of reversible phase transition close to the physiological temperatures makes these types of materials a great candidate for a wide variety of biomedical applications including bioimaging, biosensing, injectables, smart surfaces, adhesives, biomanufacturing, and tissue engineering. Thermoresponsive behavior, mainly lower critical solution temperature (LCST) can be easily tuned by shifting the balance between hydrophobicity and hydrophilicity (e.g., by using comonomers or changing end groups) and modifying the molecular weight and architecture of the polymer. Hence, synthetic and characterization tools are critical in tailoring and precisely determining these properties. This review aims to show the full scope of the journey of thermoresponsive polymers from benchtop to potential applications. We especially intend to emphasize the effects of the structural heterogeneity of polymers on thermal transition and highlight the modern characterization techniques used to study thermoresponsive behavior. A better understanding of these structural effects and benchtop tools can help us design and implement more advanced materials for future applications in public health.
1. Introduction
Polymers can be engineered to exhibit responses to a diverse variety of external stimuli including changes in temperature,1–5 light,6–9 pH,10–12 magnetic fields,13–16 electric fields,17–19 ultrasonication,20,21 mechanical forces,22,23 and many others.24–28 Arguably, thermoresponsive behavior of polymers is studied most extensively in these stimuli-responsive materials space. The earliest works on thermoresponsive properties of poly(N-isopropylacrylamide) (PNIPAM) were documented in the late 1960s.29,30Thermoresponsive polymers can undergo reversible phase transition upon exposure to temperature change. Polymers that show a lower critical solution temperature (LCST) behavior in an aqueous solution are soluble below LCST due to extensive hydrogen bonding interactions between the polymer and surrounding water molecules. Above LCST, hydrogen bonding with water molecules is disturbed and the intra- and intermolecular hydrophobic and hydrogen bonding interactions become more dominant, as a result, the polymer becomes insoluble in an aqueous solution upon heating.31 When water molecules are repelled from the polymer chain at elevated temperatures, hydrogen bonds (between water and polymer chain) are broken and new hydrogen bonds are formed resulting in a change in enthalpy. In addition, entropy increases as water molecules are no longer constrained. Hence, based on Gibbs free energy equation (ΔG = ΔH − TΔS), phase transition from soluble to insoluble polymer chains becomes spontaneous as the temperature gets above the threshold value (LCST) due to so-called “hydrophobic effect”.32,33 PNIPAM chains, for example, are soluble in water below its LCST due to the interactions between amide groups on side chains and water molecules, forming solvated random coils. At elevated temperatures, side chains favorably interact with each other resulting in the transformation from soluble coils to insoluble globules. The temperature of the coil-to-globule transition is called the cloud point temperature (Tcp) at which a phase transition occurs from fully transparent to opaque solution (generally at 50% transmittance) due to the formation of stable polymer agglomerates with a size larger than visible light (Fig. 1).34 The LCST is defined as the minimum value of the Tcp in the temperature–concentration phase diagram.35 The LCST behavior of polymers in an aqueous solution can be tuned by chemically incorporating hydrophilic or hydrophobic character to the original polymer through copolymerization and end group transformation/functionalization, modifying the polymer architecture and molecular mass (e.g., weight average molecular weight, Mw or number average molecular weight, Mn), and adjusting ionic strength (e.g., salt type or concentration). The tunability of LCST makes the thermoresponsive polymers ideal for use in physiological temperatures, typically 35–40 °C range.36–39
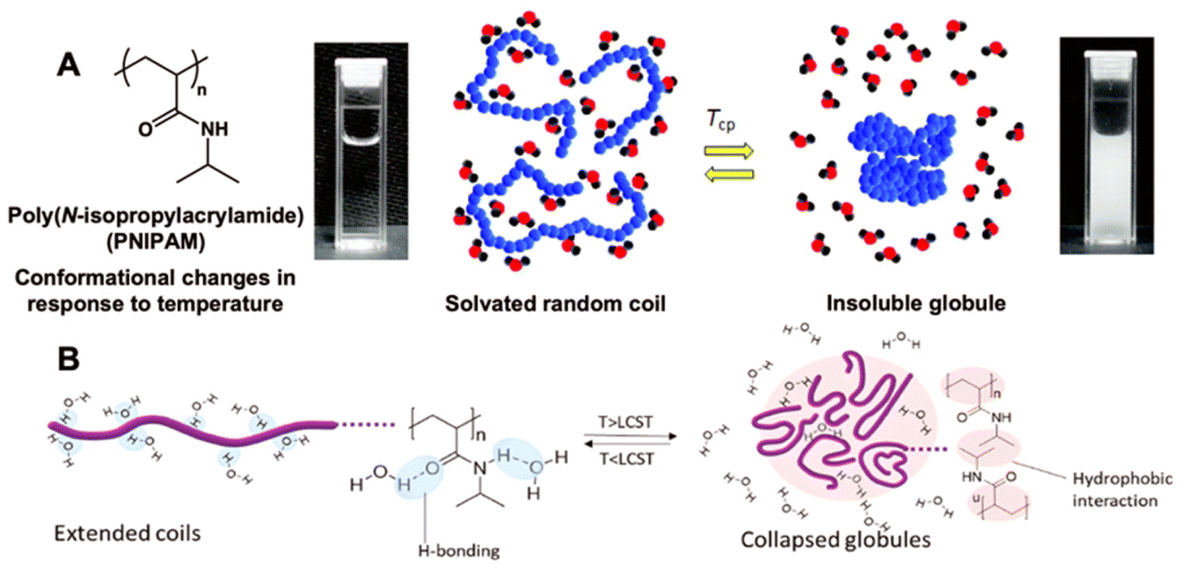 | ||
| Fig. 1 Schematic representation of (A) a polymer phase transition of PNIPAM in aqueous solution from a completely dissolved homogeneous state (left, solvated random coil) to a two-phase demixed system (right, insoluble globule) and (B) the coil-to-globule transition with polymer solvation through hydrogen bonding below the LCST and domination of the hydrophobic interactions above the LCST. Adapted with permission from ref. 34 and 40. Copyright 2022, RSC and 2020, Elsevier. | ||
These polymers have gained significant attention in recent years41–49 due to their potential applications in several biomedical fields such as bioimaging,50 drug delivery,51–56 injectables,57–63 smart surfaces,64–75 adhesives,76 and tissue engineering.77–80 However, the effects of the structural heterogeneity of polymers on thermal transition and modern characterization techniques have not been systematically discussed before. The precise determination of phase transition and LCSTs of these novel materials play a critical role in biomedical applications, where tolerance for error is generally infinitesimal. In this review, we aim to show the full scope of the journey of thermoresponsive polymers from their synthesis to biomedical frontiers. This review article (1) highlights the most common thermoresponsive polymers such as poly(N-alkyl acrylamide) and polyethylene glycol (PEG) derivatives; (2) provides a brief summary of controlled radical polymerization (CRP) techniques for synthesis and structural property control of thermoresponsive polymers; (3) discusses the effects of structural heterogeneity (e.g., architecture, molecular mass, grafting density) on LCST; (4) uncovers modern characterization tools for precisely determining thermoresponsive properties (e.g., LCST) and phase transition; and finally (5) touches on the state-of-the-art biomedical applications of thermoresponsive polymers with LCST transition.
2. Thermoresponsive polymers and their syntheses
2.1. Common types of thermoresponsive polymers
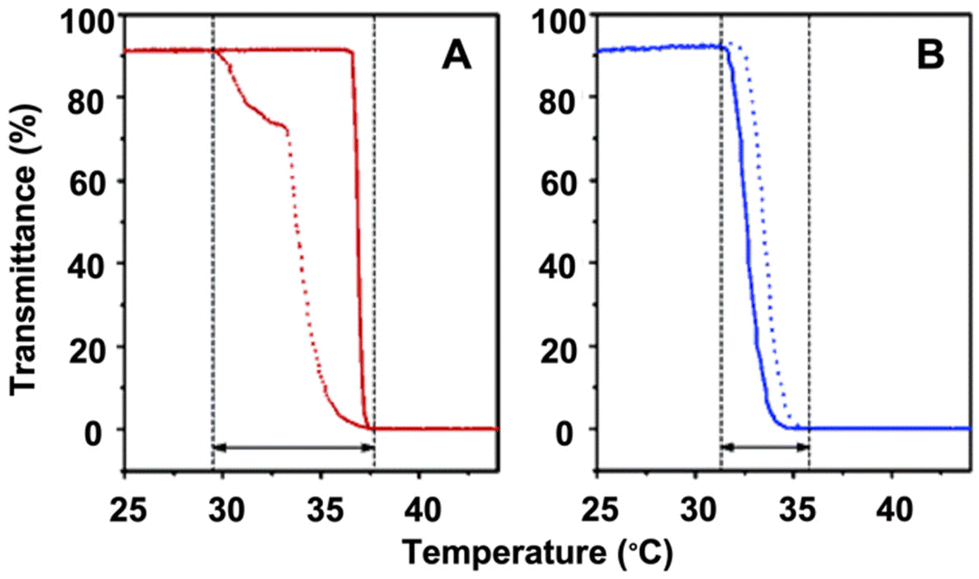 | ||
| Fig. 2 Plots of transmittance as a function of temperature measured for aqueous solutions of (A) a PNIPAM homopolymer (DPnca. 100; Mw/Mn = 1.12) (red) and (B) a copolymer P(MEO2MA-co-OEGMA) containing 5 mol% OEGMA per chain (DPnca. 100; Mw/Mn = 1.34) (blue). Transmittance drops to 0% when the size of polymer aggregates becomes larger than the visible light and the solution turns completely opaque. Adapted with permission from ref. 85. Copyright 2006, American Chemical Society. | ||
Numerous reports described the synthesis and use of (co)polymers of PNIPAM with other functional comonomers such as other acrylamides,86 (meth)acrylates,87,88 ethylene glycols,57 and pyrrolidines.89 In addition, PNIPAM-based crosslinked hydrogel systems51,80 and brushes72,90 were explored. Because of its tunable LCST, PNIPAM or its copolymers have broad application prospects such as medical diagnostics, drug delivery, biomedical devices, tissue engineering, and separations.91 Thermosensitivity of poly(N-substituted acrylamide) derivatives with the substituents on the nitrogen atom on the side chain has also been studied.34 Poly(N-alkylacrylamide)s (PAMs) such as poly(N-ethylacrylamide) (PNEAM), poly(N-cyclopropylacrylamide) (PNcPAM),92 and poly(N-n-propylacrylamide) (PNnPAM),93 and poly(N,N-dialkylacrylamide)s (PDAs) such as poly(N,N-ethylmethylacrylamide) (PEMAM),94 poly(N,N-diethylacrylamide) (PDEAM),95 poly(N-acryloylpyrrolidine) (PAPy),96 and poly(N-acryloylpiperidine) (PAPi) undergo thermal phase transition in water. It was reported that the thermoresponsive properties depend on the degree of hydrophobicity, bulkiness, and flexibility of the substituents on the amide groups.26,34,97–99Fig. 3 shows the chemical structures and reported LCST or Tcp values of PNIPAM copolymers and poly(N-substituted acrylamide) derivatives.
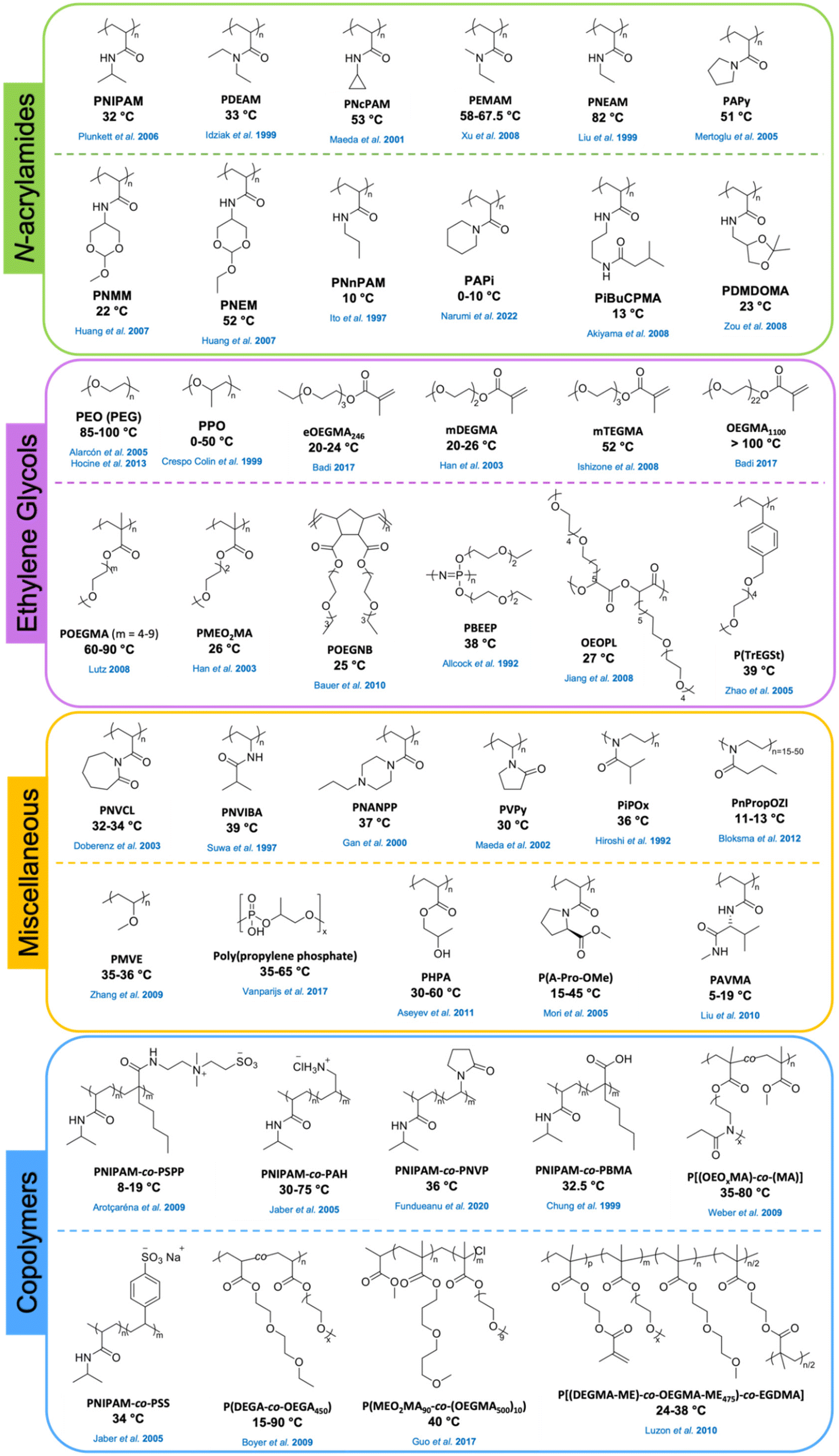 | ||
| Fig. 3 Chemical structures of common thermoresponsive homo- and co-polymers and their reported LCST or cloud point values. | ||
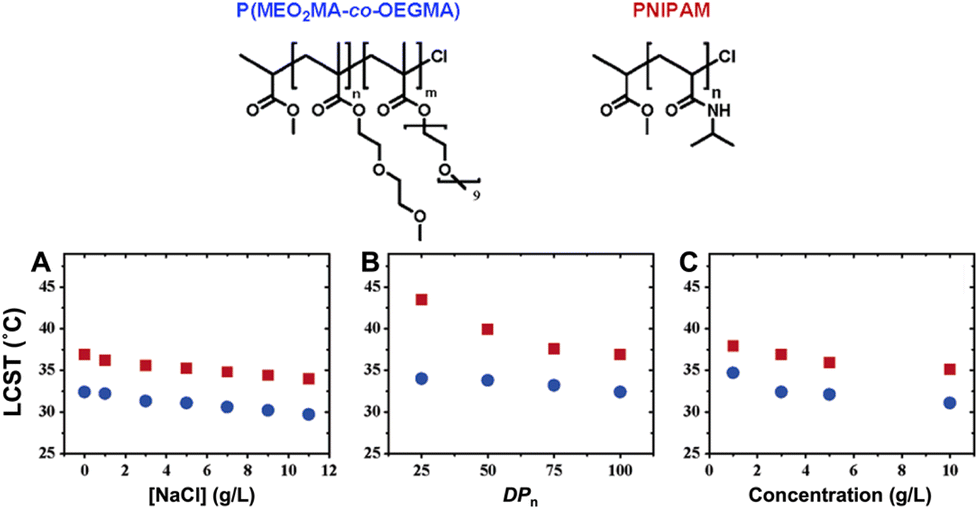 | ||
| Fig. 4 Molecular structures of ethylene glycol-based copolymer and PNIPAM (top). Plots of measured cloud points as a function of (A) NaCl concentration, (B) degree of polymerization (DPn), and (C) polymer concentration (bottom). In all figures, cloud points of P(MEO2MA-co-OEGMA475) and PNIPAM are represented by blue dots and red squares, respectively. Data in figures (A) and (C) were measured with a copolymer P(MEO2MA-co-OEGMA475) containing 5 mol% OEGMA475 per chain (DPnca. 100; Mw/Mn = 1.34) and a homopolymer of NIPAM (DPnca. 100; Mw/Mn = 1.12). Adapted with permission from ref. 85. Copyright 2006, American Chemical Society. | ||
Polymers containing carbon–carbon backbones (e.g., acrylic, methacrylic, styrenic) with short ethylene glycol side chains (e.g., mono-, di-, and tri-ethylene glycol) generally demonstrate lower LCSTs in aqueous solution than polymers with long ethylene glycol side chains (i.e., OEG).104,105 The hydrophobicity of the polymer increases as the length of the ethylene glycol units decreases due to the increase in the overall percentage of the hydrocarbon backbone. For instance, PMEO2MA with two EG units and PMEO3MA with three EG units exhibit LCST around 26 and 52 °C, respectively. On the other hand, POEGMAs with 4–10 EG units have transition temperatures in the range of 60–90 °C. To tune the LCST, short and long ethylene glycol (EG) (meth)acrylates can be copolymerized at different ratios of each monomer.106–109Fig. 5 shows representative interactions between P(MEO2MA-co-OEGMA475) copolymer and water molecules (top) and optical transparency of the aqueous solutions at 25 °C and 45 °C (bottom). Above LCST, P(MEO2MA-co-OEGMA475) copolymer becomes completely opaque due to the aggregation of large particles. Polymers can be decorated with ethylene glycol units on the side chain with various main chains composed of polymethacrylate (POEGMA),104,110 polyacrylate (POEGA),45 polystyrene (POEGSt),111 poly(vinyl ether) (POEGVE),112 polynorbornene (POEGNB),113 polyether (POEGE),114,115 polylactide (POEGLA),116,117 polymethylene (POEGM),118 polyphosphazene (PBEEP),119,120 and poly(amino acid)121,122 (Fig. 3). These thermoresponsive polymers can be tailored as linear or branched as well as three-dimensional structures.34,123 In addition, poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) and their copolymers with LCSTs varying from 20 °C to 85 °C contain EG units in their main chains and are commercially available under the names of Pluronics, Poloxamers, and Tetronics.124–126 Amphiphilic balance in OEG structure is the main reason for the thermoresponsive property of these types of polymers.127,128
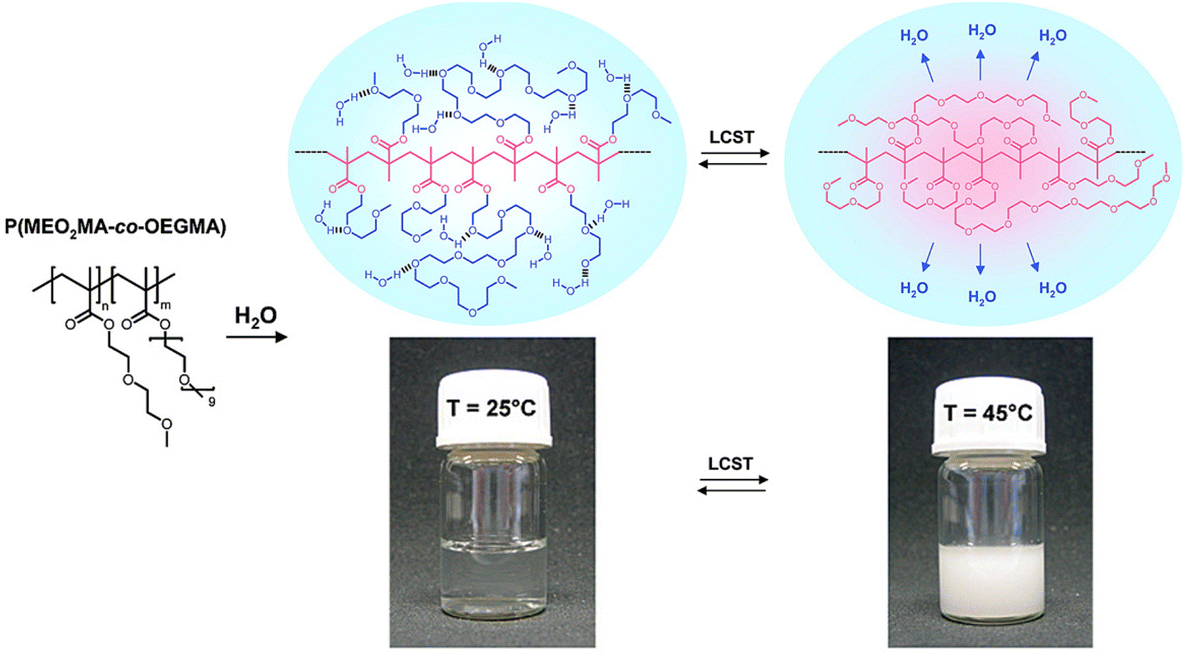 | ||
| Fig. 5 Proposed mechanism for the temperature-induced phase transition of poly(MEO2MA-co-OEGMA) copolymers in aqueous solution. Adapted with permission from ref. 106. Copyright 2007, American Chemical Society. | ||
The previous reports on thermoresponsive polymers are dominated by PNIPAM and POEGMA derivatives. However, a wide variety of other polymers demonstrated promising thermoresponsive behavior in aqueous solution.129–135 For example, poly(N-vinylcaprolactam) (PNVCL)136–138 and poly(oxazoline)s139–143 have been recognized as synthetic polymers exhibiting LCST behavior.26 Similar to PNIPAM, PNVCL is hydrophilic and soluble in water at room temperature, gradually becoming more hydrophobic and eventually insoluble in water when the temperature is raised from 25 °C to 35 °C.124 Moreover, there are several examples of thermoresponsive polymer–biopolymer conjugates to incorporate responsive behavior to the biopolymers such as cyclodextrin,144 enzymes,145 proteins,146 or oligonucleotides124 for target applications.
2.2. Polymerization techniques and property control
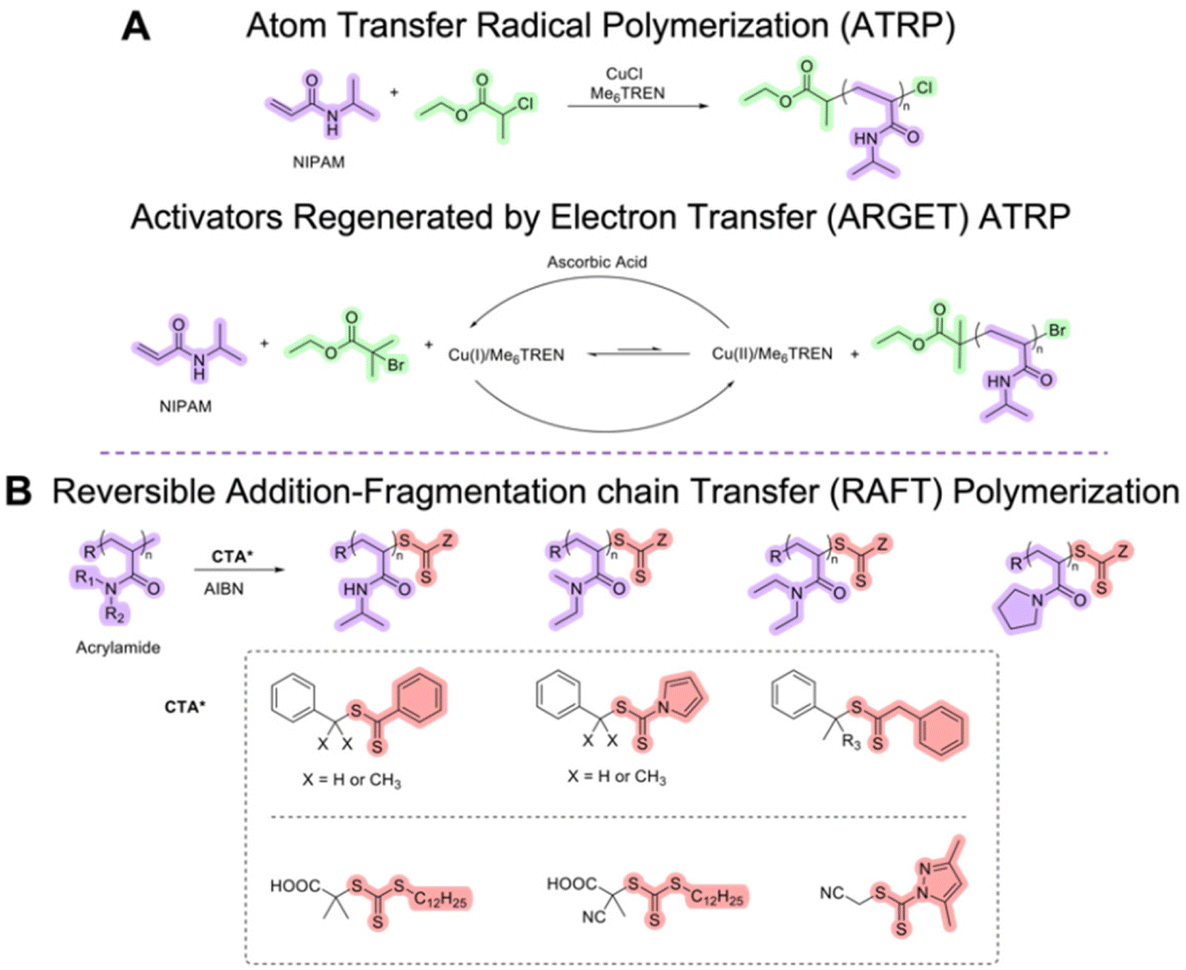 | ||
| Scheme 1 Synthetic schemes for ATRP and RAFT polymerization of acrylamide monomers. | ||
Acrylamides and (meth)acrylates can be successfully polymerized through ATRP and RAFT polymerization. Conventional ATRP can be used to polymerize most of the (meth)acrylates. However, the ATRP technique needs to be modified to be able to polymerize (meth)acrylamide-based monomers with high yield and narrow dispersity. As a typical methacrylate example, OEGMA was copolymerized with more hydrophobic MEO2MA or poly(propylene glycol methacrylate) (PPGMA) through conventional ATRP to decrease the LCST to below 37 °C.85,159 Conventional ATRP requires a metal catalyst (e.g., CuBr or CuCl), a ligand (e.g., linear amines or bipyridines), and an initiator (e.g., α-haloester).153 Several (meth)acrylamides (N,N-dimethylacrylamide, N-tert-butylacrylamide, and N-(2-hydroxypropyl)methacrylamide) were attempted to polymerize through conventional ATRP. When linear amines and bipyridines were employed as ligands in bulk or solution, very low monomer conversions were obtained. The low conversion can be attributed to the copper catalyst competitively complexing with poly(meth)acrylamides. The use of 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane (Me4Cyclam) as a ligand resulted in high yield in a short period of time, but the polymerization was not controlled potentially due to the slow deactivation and the loss of bromine end groups due to side reactions, yielding high dispersity.152 Masci et al. reported a successful ATRP of NIPAM using tris[2-(dimethylamino)ethyl]amine (Me6TREN) as a ligand, ethyl 2-chloropropionate as an initiator, CuCl as a catalyst in dimethylformamide (DMF)/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at room temperature (Scheme 1A). The reaction yielded a first-order kinetic plot with 92% conversion and dispersity of 1.19.160 Supplemental activator and reducing agent (SARA) ATRP in the presence of Cu0 and electrochemically mediated ATRP (eATRP) of NIPAM were also previously investigated. PEO-b-PNIPAM copolymers were successfully synthesized with narrow dispersity and the % conversion between 43–95% by SARA ATRP and eATRP.161 Moreover, activators regenerated by electron transfer (ARGET) ATRP162 can be utilized to synthesize thermoresponsive polymers from (meth)acrylic and (meth)acrylamide monomers (Scheme 1A). ARGET ATRP is a robust technique that is less sensitive to oxygen presence and requires CuII catalyst and a reducing agent such as tin(II) 2-ethylhexanoate (Sn(EH)2) or ascorbic acid. For the synthesis of PNIPAM, more active catalyst systems with a tetradentate ligand such as Me6TREN or tris(2-pyridylmethyl)amine (TPMA) which is 103–105 times more active than CuBr/bipyridine complexes are necessary.162 It should be noted that the potential toxicity of residual transition metal catalyst contamination is the key limiting factor for applications of thermoresponsive polymers synthesized via ATRP. The use of more active catalysts to lower the catalyst loading and more rigorous purification for the removal of metals after polymerization is critical for biomedical applications.163–165 Precisely controlled molecular architecture (topology, composition, functionality) of polymers, hybrid materials, and bioconjugates can also be achieved through ATRP.166–170
:1, v/v) at room temperature (Scheme 1A). The reaction yielded a first-order kinetic plot with 92% conversion and dispersity of 1.19.160 Supplemental activator and reducing agent (SARA) ATRP in the presence of Cu0 and electrochemically mediated ATRP (eATRP) of NIPAM were also previously investigated. PEO-b-PNIPAM copolymers were successfully synthesized with narrow dispersity and the % conversion between 43–95% by SARA ATRP and eATRP.161 Moreover, activators regenerated by electron transfer (ARGET) ATRP162 can be utilized to synthesize thermoresponsive polymers from (meth)acrylic and (meth)acrylamide monomers (Scheme 1A). ARGET ATRP is a robust technique that is less sensitive to oxygen presence and requires CuII catalyst and a reducing agent such as tin(II) 2-ethylhexanoate (Sn(EH)2) or ascorbic acid. For the synthesis of PNIPAM, more active catalyst systems with a tetradentate ligand such as Me6TREN or tris(2-pyridylmethyl)amine (TPMA) which is 103–105 times more active than CuBr/bipyridine complexes are necessary.162 It should be noted that the potential toxicity of residual transition metal catalyst contamination is the key limiting factor for applications of thermoresponsive polymers synthesized via ATRP. The use of more active catalysts to lower the catalyst loading and more rigorous purification for the removal of metals after polymerization is critical for biomedical applications.163–165 Precisely controlled molecular architecture (topology, composition, functionality) of polymers, hybrid materials, and bioconjugates can also be achieved through ATRP.166–170
RAFT polymerization has been widely used to synthesize thermoresponsive polymers from various types of monomers. RAFT polymerization can allow metal-free synthesis of these responsive polymers; however, cytotoxicity of the chain transfer agent (CTA) remaining at the end of the polymer chains was reported.171–173 The end group CTAs also need to be completely removed to prevent any complications.174,175 In early 2000, a series of PNIPAM homopolymers were synthesized by using benzyl and cumyl dithiobenzoate (Scheme 1B) in different organic solvents (benzene and 1,4-dioxane) at 60 °C.176,177 Convertine, McCormick, and co-workers successfully polymerized NIPAM through the RAFT process at room temperature. A commercial trithiocarbonate RAFT CTA, 2-(dodecylsulfanylthiocarbonylsulfanyl)-2-methylpropionic acid, along with an azo initiator, 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) was used to homopolymerize NIPAM in DMF at 25 °C. PNIPAM homopolymers with Mn of ca. 50000 g mol−1 and dispersity of ca. 1.10 were obtained. These homopolymers were then used as a macroCTA to yield block copolymers. Near-quantitative block copolymer formation with narrow dispersity was confirmed by size-exclusion chromatography (SEC).178 The same research group synthesized thermoresponsive N,N-dimethylacrylamide (DMA)/NIPAM di- and tri-block copolymers via aqueous RAFT polymerization at room temperature.179 Overall, trithiocarbonate RAFT agents (Scheme 1B) were reported as a more versatile CTA for more activated monomers such as acrylamides.180 RAFT polymerization of various N-alkylacrylamide and N,N-dialkylacrylamide derivatives was reviewed by Kakuchi et al. in 2022. Kanaoka and co-workers synthesized PNIPAM hydrogels via a polymerization-induced self-assembly process in which they employed PDMA as macroCTA and a divinyl cross-linker. Their research group also reported PNIPAM-based crosslinked hydrogel being formed in a star shape and observed greater dispersibility and uniform distribution of the star PNIPAM polymers at the crosslinking point.181 Other thermoresponsive star architectures synthesized through RAFT process have also been reported.182–185
Thermoresponsive polymers can also be made through photo-polymerization methods. Photo-induced electron/energy transfer (PET)-RAFT polymerization in the presence of a photo-redox catalyst which transmits the captured light energy to the CTA has been widely used.186 Photoiniferter (PI)-RAFT polymerization, on the other hand, involves direct photolytic cleavage of a CTA by the light, after which the procedure is changed to available RAFT mode. Corrigan, Boyer, and co-workers prepared PNIPAM homopolymers via PET-RAFT polymerization. 2-(n-Butyltrithiocarbonate) propionic acid as a CTA and 5,10,15,20-tetraphenyl-21H,23H-porphyrin zinc(II) (ZnTPP) as a photo-redox catalyst were used to polymerize NIPAM in methanol under green LED light (λmax = 520 nm) and inert atmosphere. Polymers with narrow dispersity (Đ < 1.18) and Mn ranging from 6.3–66.2 kg mol−1 were synthesized for cloud point comparison.187 Karpov and co-workers reported dimethyl sulfoxide (DMSO) as the best solvent for homopolymerization among other solvents (DMSO > tetrahydrofuran, THF > toluene) for photo-RAFT polymerization. Amphiphilic random and diblock alkoxy (C12–C14) OEGMA (with ca. EG units) and methoxy–OEGMA500 (1:1) bottlebrush copolymers have been synthesized by means of PI-RAFT polymerization in different solvents. Moreover, the authors performed an “on–off” experiment to confirm the easy switchability of the process and pseudo-living mechanism of polymerization.188
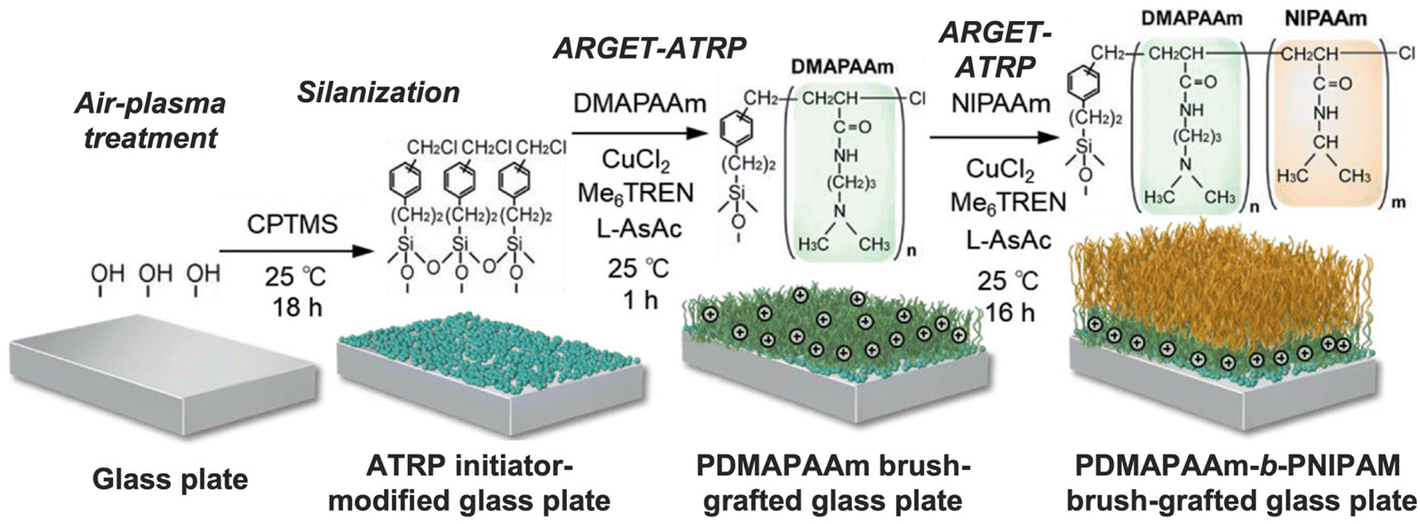 | ||
| Fig. 6 Preparation of a thermoresponsive block copolymer brush-modified glass plate through ARGET ATRP. Adapted with permission from ref. 90. Copyright 2020, John Wiley and Sons. | ||
3. Effects of structural heterogeneity on LCST
A hydrophilic–hydrophobic balance in the thermoresponsive polymer chains gives rise to changing their shape (i.e., random coil to globule) in response to temperature change. To finely tune the LCST of thermoresponsive polymers, several structural factors have been investigated including architectures, compositions, molecular mass, dispersity, and grafting length/density.3.1. Architectures
The responsive properties can be manipulated by changing the macromolecular architectures. Thermoresponsive polymers with different macromolecular architectures, such as block, cyclic, comb, bottlebrush, and star, have been synthesized using controlled/living polymerization techniques and the structural effects on thermal phase transition have been studied. Different polymer architectures used for thermoresponsive polymer design (A–E) and multi-arm star and hyperbranched copolymers with PEG side chains (F and G) are shown in Fig. 7.3,211 For PEG-based polymers, the differences in thermal behavior between block and gradient copolymers of 2-hydroxyethyl acrylate (HEA) and ethylene glycol methyl ether acrylate (mEGA) were reported by Steinhauer et al.212 The Tcp was shown to be tuned between 0 °C and 80 °C for the block copolymers and between 0 °C and 60 °C for the gradient copolymers. Sun et al. studied a reversible polymer architecture switching process between linear polymers and cross-linked nanogels. By investigating multifunctional block copolymers of acrylamide and ethylene glycol acrylate derivatives, they found that linear structures showed sharp thermal transitions while the crosslinked nanogels showed linear thermal transitions over a broad temperature range.213 For other polymer systems, the relationship between thermoresponsive behaviors and architectures is also demonstrated. Cheng et al. prepared a series of poly(3-ethyl-3-(hydroxymethyl)-oxetane) (PEHO)-star-PEO and PEHO-star-poly(2-(dimethylamino)ethyl methacrylate) (PDMAEMA) copolymers.211 The LCST transition of the first system is based on hydrophilic–hydrophobic balance and is highly dependent on the degree of branching of the PEHO core. EG-based polymers were synthesized in star–block architectures to produce thermoreversible hydrogels (thermogels or hydrogelators). MEO2MA and OEGMA475 were copolymerized using a 4-arm star PEG ATRP macroinitiator. These star–block copolymers with an inner PEG core and outer block thermoresponsive arms exhibited drastic changes in viscosity above LCST. The star copolymers showed a reversible thermogelation within the range of 35–42 °C over repeated heating/cooling cycles as opposed to the linear block copolymer solutions which only displayed a higher viscosity upon heating.214,215 Li et al. synthesized dendrimers containing PEG in the main chain, and OEG as a side chain.216 With an aim to examine how the length of the OEG side chain affects the thermoresponsive behavior, the research group of Deng synthesized a series of degradable copolymers with varying EG units from 1 to 3.217 Moreover, EG-based polymers exhibit a broad thermoresponsive behavior in dendronized form. For instance, OEG-based dendrons display hitherto unseen abrupt and rapid transitions in aqueous solution with LCSTs varying from 27 to 65 °C.216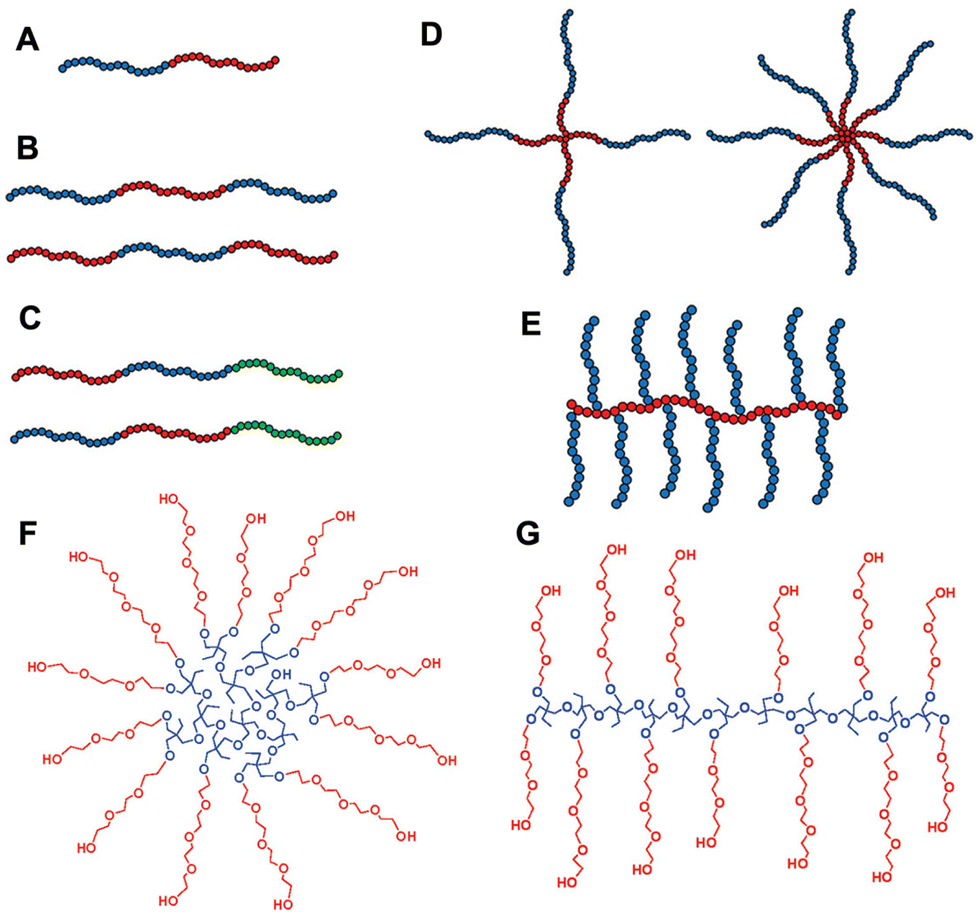 | ||
| Fig. 7 Polymer architectures used in the construction of thermoreversible polymers: (A) Di-block, (B) ABA tri-block, (C) ABC tri-block, (D) 4 and 8-arm star-shaped, (E) graft copolymers. (F and G) Multi-arm star and hyperbranched copolymers with poly(ethylene oxide) side chains. Adapted with permission from ref. 3 and 211. Copyright 2020, John Wiley and Sons and 2010, American Chemical Society. | ||
PNIPAM-based block copolymers and star polymers containing hydrophilic or hydrophobic segments exhibit various LCSTs (Fig. 3).218–222 A series of PNIPAM- and PDMA-based double hydrophilic multiblock copolymer architectures with hydrophobic dodecyl hydrocarbon end-groups were synthesized through RAFT polymerization. It was shown that incorporations of hydrophilic comonomer can increase the Tcp of the block copolymer systems.223 Lang et al. prepared ethyl- and dodecyl-terminated PNIPAM, PNnPAM, and PNcPAM to unveil the relationship between chain terminal groups and thermoresponsive behaviors.224 When below the critical temperature, polymers terminated with short hydrophobic groups would have large-scale, fractal assemblies while polymers with long hydrophobic terminal groups formed small micelles. As expected, Tcp of PNIPAM decreases when the polymer is decorated with hydrophobic end groups and the opposite effect can be observed when the end groups are hydrophilic.225 The star and cyclic architectures have been shown to affect thermoresponsive behavior, especially polymers with low molecular masses. Polarity of the core and end-groups and the number and length of the arms directly affect the thermoresponsive properties in the star architecture.226,227 PNIPAM-based star polymers showed lower Tcp (Fig. 8), and cyclic polymers showed higher Tcp compared with linear structures.34 The Tcp values of four-arm star PNIPAM were reported lower than those of linear counterparts.228 When working as a thermoresponsive shell of core–shell copolymers using tris(2-aminoethyl)amine (TREN) and a polyamido-amine hyperbranched polymer as the core, Tcp increased as the degree of branching increased.229 Winnik et al. reported the effects of cyclic architecture on the phase transitions of PNIPAM systems in aqueous solutions. Linear PNIPAMs with α-azido and ω-propargyl end-groups were synthesized through RAFT polymerization and subsequently, these end groups were clicked via aminolysis/Michael addition reactions to yield cyclic PNIPAM polymers.230 It was reported that polymer concentration and structural factors such as the absence of end groups and steric hindrance, affect the phase transition for cyclic PNIPAM systems.231 PNIPAM copolymers with different compositions and architectures (e.g., random, block, graft, or star) have been also shown to exhibit a sol–gel transition upon heating.232–236
 | ||
| Fig. 8 Structures and thermoresponsive properties of linear and four-arm star-shaped PNIPAM at different molecular weights. Reproduced with permission from ref. 34. Copyright 2022, RSC. | ||
3.2. Molecular mass and dispersity
Phase transition of thermoresponsive polymers from coil to globule in aqueous solutions is considered a function of molecular mass and Đ. This is especially relevant regarding synthetic polymers, whose molecular mass and Đ can be altered and controlled to manipulate the corresponding thermoresponsive behavior. The molecular mass dependency of the Tcp of PNIPAM was reported by Stöver et al. Polymers with Mw ranging from 2800 to 26500 g mol−1 with narrow dispersity (Đ = 1.1–1.2) were prepared by the ATRP technique. Turbidimetry and differential scanning calorimetry studies showed that the Tcp of the PNIPAM aqueous solutions decreased as the molecular mass increased. This inverse relationship between the Tcp and molecular mass has also been reported for other poly(N,N-dialkyl acrylamide)s (Fig. 9).94,237–239 Liu et al. synthesized a semirigid water-soluble thermoresponsive polymer and found that both the monomer bis(N-hydroxyisopropyl pyrrolidone) 2-vinylterephthalate (HIPPVTA) and PHIPPVTA exhibited thermoresponsive phase transition behaviors in aqueous solutions and that PHIPPVTA exhibited an increased Tcp with increasing molecular mass.240 PHIPPVTA cloud point is extremely dependent on Mw and Đ, while it also demonstrates a great solvent isotopic effect. The effect of molecular mass and Đ on thermal behaviors can also be seen in synthetic polymer mixtures. Corrigan, Boyer, and co-workers prepared a series of PNIPAM homopolymers with narrow dispersity via PET-RAFT polymerization. Their study showed that the cloud points are inversely proportional to the Mw. For binary blends of PNIPAM homopolymers with more highly skewed dispersity and large tailing, they determined that the thermoresponsive behavior changed from a single, sharp transition to a broad transition, and eventually to two distinct transitions.187 While this study is limited to PNIPAM samples, it illustrates the effect of the shape of dispersity on the thermal responsive behavior of polymer mixtures. PEGMA copolymer mixtures also demonstrated the effect of Mw and Đ on the thermoresponsive behaviors, supporting this idea that the thermoresponsiveness of polymer mixtures is dependent on Mw and Đ. Through synthesizing triblock copolymers with a PEGMA, n-butyl methacrylate (nBuMA), and DMAEMA block and with varying molecular mass and composition, Ward et al. demonstrated that composition influenced the pKa and that both molecular mass and composition influenced the cloud point, hydrodynamic diameter, and gelation of the polymers.241 These results suggest that the composition and molecular mass of triblock polymers can be manipulated with the promise of gel-formation abilities due to their thermoresponsive behavior. Overall, understanding the effect of molecular mass and Đ on polymers is crucial in developing polymeric thermoresponsive materials for drug delivery and tissue engineering.
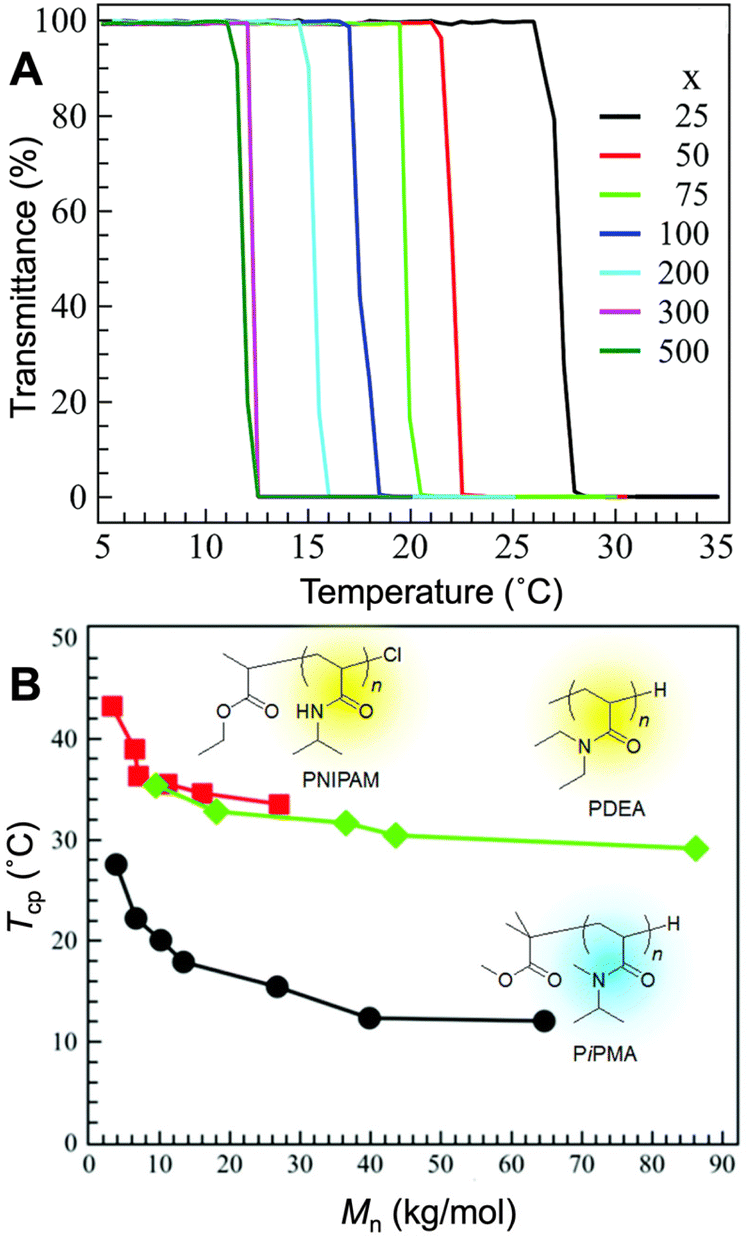 | ||
| Fig. 9 (A) The cloud point curves (transmittance vs. temperature) of poly(N,N-isopropylmethylacrylamide) (PiPMA) with different degrees of polymerization (x) in aqueous solution. (B) The molecular mass dependence of cloud point temperature of PNIPAM (■), poly(N,N-diethylacrylamide) (PDEA) (◆), and PiPMA (●). Adapted with permission from ref. 34, 225, 237 and 242. Copyright 2020 and 2022, RSC and 2019 and 2005, American Chemical Society. | ||
3.3. Grafting length/density
Grafting density has also proved to influence the thermoresponsive behaviors of polymers. PNIPAM is the most studied regarding grafting effects as it experiences a conformational change above its LCST and the physical properties of PNIPAM are readily altered by changing the temperatures even when grafted to surfaces. Both graft length and density were discovered to impact thermoresponsive behavior and self-assembly morphology. Jiang et al. determined that both graft length and density experienced a positive correlation with LCST, mean diameter, and size distribution, with graft length having a more significant effect than density, and that graft length and density had a more significant effect on diameter than self-assemble morphology.243 The grafted polymers with dense side chains and higher molecular masses had higher LCSTs with the aggregates having bigger diameters and stronger steric repulsive interactions. Thus, the desired thermoresponsive behavior and self-assembly morphology can be achieved by altering the grafting length and grafting density to the proper length. When looking at the effect of grafting on thermoresponsive polymers, the effect of molecular mass should also be considered because the magnitude of the conformational changes is usually dependent on size. Yim et al. studied the temperature-dependent conformational changes of PNIPAM chains grafted over a range of grafting lengths and densities. They found that the conformational change of the grafted PNIPAM brushes varied greatly with molecular mass and grafting density, with the maximum conformational change observed for the brushes with high molecular mass and intermediate grafting density.244 This is due to the competition between the stretching effect of laterally interacting tethered chains. Moreover, molecular mass and grafting density were also found to affect the collapse of these end-grafted PNIPAM polymers above the LCST. Plunkett et al. observed thermally induced chain collapse at a high molecular mass and high grafting density.245 It was concluded that PNIPAM behaves as an ideal polymer (i.e., the same distribution was observed with a different effective bond length) at and above the LCST but not below the LCST. Overall, the ability to tune the thermal properties of PNIPAM films depends strongly on the grafting parameters.Other external components along with temperature influence the thermoresponsive behavior of polymers, the main being pressure, salts, and buffers since hydrogen bonding ability or other electrostatic interactions in solution can be affected. The pressure was found to induce structural changes, transitioning from coil to globule.246 The effect of salt types and concentrations on the thermal transitions of non-ionic POEGMA-based copolymers synthesized via ATRP was investigated. The LCST of the copolymer increased or decreased depending on the salt type.247 Dendritic polymers were found to be more sensitive to the addition of salt, experiencing a nonlinear decrease in LCSTs with rising salt concentrations.248 The specific type of ion affects the LCST as thermoresponsive polymers experience decreasing LCST with increasing kosmotropic (water “structure maker”) anion concentration and abnormal salting out properties at low chaotropic (water “structure breaker”) concentrations.249 The type of ion has also been found to affect the structure of polymer brushes, with kosmotropes resulting in relatively collapsed structures whereas chaotropes resulting in relatively swollen structures.250 The presence of salt in buffer solutions was determined to be non-additive, and the influence of salt on polymer aggregation was dependent on salt ion type, concentration, and polymer main chain structure.251
4. Characterization of thermoresponsive behavior
General characterization techniques to confirm successful syntheses of thermoresponsive polymers usually include Nuclear Magnetic Resonance (NMR) spectroscopy, Size-Exclusion Chromatography (SEC), and UV-Vis and Fourier transform infrared (FTIR) spectroscopy techniques. To better understand the LCST behavior, other advanced characterization techniques will be introduced in this section. We categorized these techniques into two parts: solution characterization and bulk characterization. An overall summary of these characterization techniques is given in Table 1.| Characterization technique | Results collected | Polymers tested | Ref. |
|---|---|---|---|
| Solution characterization | |||
| Rheological studies | Phase transition behavior, viscoelasticity, and time–temperature superposition | PNVCL, PNIPAM, PDMAEMA, POEGMA | 52, 229, 241 and 252 |
| Optical studies | Cloud point measurement: temperature of phase inversions in polymer solutions | POEGMA, PNIPAM, EG–PLLA, EG–PDLA, P(OEGA)-co-(DEGA), PDMA-b-P(MEA-co-PEGA-co-PDSEA-co-Flu) | 97, 213 and 253–256 |
| Light scattering measurements | Size distribution profile of polymers in solution | P(NVCL/VP), poly(OEGMA-co-DEGMA), P(OEOMA-co-HMStBA), P(DEGMA-co-HPMA-co-PEGMA), PDMA-b-P(NIPAM-co-ACPBA), P(DEGMA-co-HPMA), PNIPAM, P(DMA-b-(NIPAM-co-DMAEA-co-BA)), PDEA | 182, 252 and 257–264 |
| Small-angle scattering (SANS/SAXS) | Shape and organization of large molecules, conformational polydispersity | PNIPAM, P(DEGMA) | 254 and 265–267 |
| Diffusion coefficients measurements | Hydrodynamic radius, formation of aggregated species | Hyperbranched poly(ethylene imine), OEG | 229, 268 and 269 |
| Bulk characterization | |||
| Contact angle measurements | The contact angle between polymer surfaces and a water droplet | PMEO2MA, P(MEO2MA-co-OEGMA), PNIPAM, PNtBAM, poly(NIPAM-co-NtBA) | 270–272 |
| Ellipsometry | The thickness of polymer films | PMEO2MA, P(MEO2MA-co-OEGMA), PNIPAM, P(MEO2MA-stat-OEGMA), PNIPAM-g-PEG | 270, 271, 273 and 274 |
| Thermal analysis (DSC, TGA) | Identification of physical properties and thermal transitions of polymers | P(NVCL/VP), PNIPAM-based polymers, pMPEGMAs, pAOEGMAs, PEGMA-b-PBuMA-b-PDMAEMA, poly(2-isopropyl-2-oxazoline) | 229, 241, 252 and 275–283 |
| Electron microscopy (SEM, TEM) | Particle counting, size determination, topography, and atomic composition | P(VP-stat-NVCL), PNIPAM, P2VP–PEO | 229, 252 and 284 |
| X-ray photoelectron spectroscopy (XPS) | Composition of surface material elements and their relative abundance, chemical state of the polyvalent ions | PNIPAM, OEGA-based polymers, poly(2-isopropyl-2-oxazoline) | 201, 256, 285 and 286 |
| Atomic force microscopy (AFM) | Adhesion strength, magnetic forces, mechanical properties, topography | Poly(NIPAM-co-acrylamidebenzophenone (AcBzPh)) poly(NIPAM-co-AcBzPh-co-N-tert-butylacrylamide), PNIPAM, poly(2-isopropyl-2-oxazoline) | 76, 101, 281, 285 and 286 |
| X-ray powder diffraction (XRD) | Degree of crystallinity and phase ratio | PNIPAM, β-cyclodextrin thermoresponsive diblock copolymer | 283 and 287 |
4.1. Solution characterization
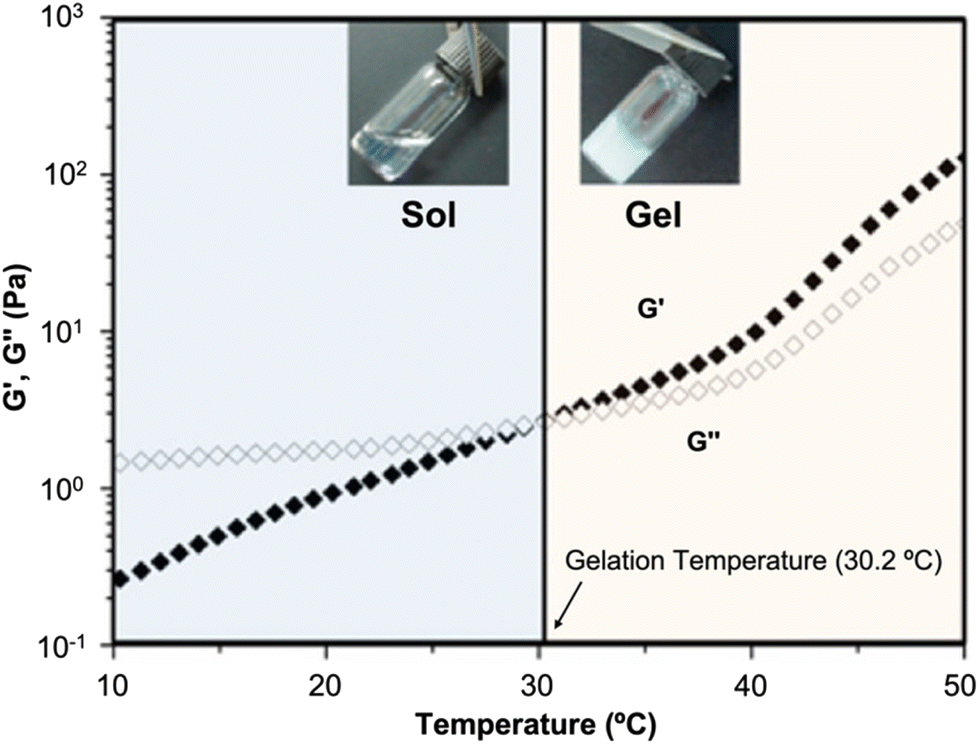 | ||
| Fig. 10 Thermogelling behavior at LCST. Dynamic rheological analysis of a thermoresponsive copolymer in aqueous solution as a function of temperature. Reprinted with permission from ref. 289. Copyright 2012, American Chemical Society. | ||
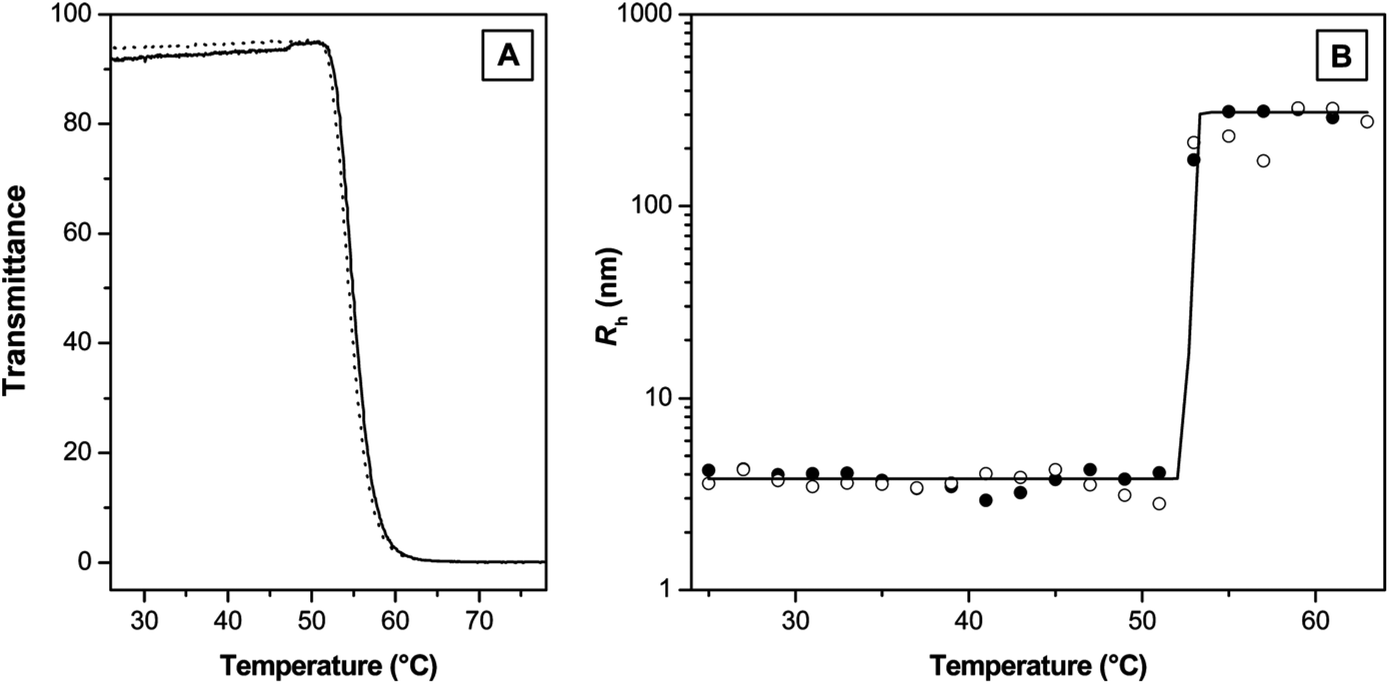 | ||
| Fig. 11 (A) Transmittance as a function of temperature measured by turbidimetry (solid line, heating; dotted line, cooling) and (B) hydrodynamic radius (RH) as a function of temperature recorded by dynamic light scattering for an aqueous solution of a copolymer poly(MEO2MA-co-OEGMA) (full symbols, heating; empty symbols, cooling; full line, a Boltzmann fit). Reprinted with permission from ref. 106. Copyright 2007, American Chemical Society. | ||
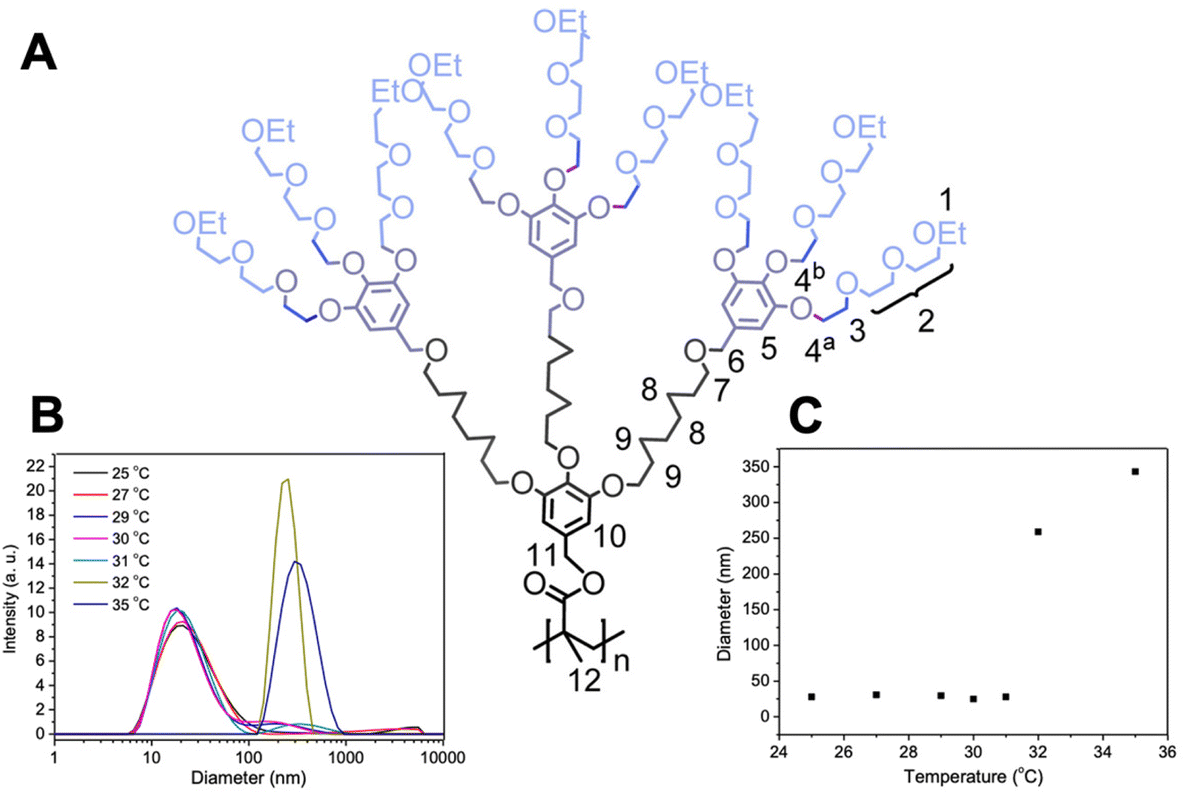 | ||
| Fig. 12 (A) Chemical structure of a thermoresponsive dendronized polymer. Dynamic light scattering measurements at different temperatures: (B) particle size distribution at different temperatures and (C) hydrodynamic diameter vs. temperature. Adapted with permission from ref. 269. Copyright 2016, American Chemical Society. | ||
In SANS experiments, neutrons penetrate into samples and scatter from the nuclei as a function of angle. The SANS technique can be used to analyze individual polymer chain conformation in a dilute aqueous solution of thermoresponsive polymers. SANS is a more suitable technique for low electron density polymers than SAXS since the difference in neutron scattering lengths between two different nuclei (e.g., hydrogen and deuterium) is large and detectable and SAXS is only sensitive to electron density rather than the nuclei.294 The growth and morphology of microgel P(MEO2MA-co-OEGMA500) particles were previously investigated by using SANS.295 Nishi et al. performed SANS measurements to understand the tacticity effect on phase separation for PNIPAM aqueous solution. The results revealed that hydrophobicity of PNIPAM containing meso- and racemo-isomers increased with increasing the meso-content.296 SANS and DLS were used to study the aggregate behavior of thermoresponsive triblock PSt–PNIPAM–PSt copolymer in water as a function of temperature. The SANS and DLS measurements showed that the triblock copolymer underwent first shrinking close to the LCST and further clustering above the LCST.297 Papadakis and their team used thermoresponsive poly(2-oxazoline) gradient copolymers and showed the collapse of small aggregates right above the cloud point and the formation of larger aggregates at higher temperatures by utilizing time-resolved SANS experiments.298,299 SANS was also used to investigate changes in the nanostructure of double network thermoresponsive hydrogels induced by temperature change. The double network hydrogels were prepared from densely crosslinked PNIPAM first network and loosely crosslinked hydrophilic polyacrylamide second network. The SANS intensities from the double network were reported lower than those from PNIPAM single network. Lower scattering was attributed to increased water content in the double network due to the presence of the hydrophilic second network.300 Overall, small-angle scattering techniques have become a powerful tool for the structural analysis of thermoresponsive polymers in physiological conditions. However, these techniques present several challenges including high-quality sample preparation and data interpretation and more importantly the cost of the analysis. Although the applicability of these techniques spans across disciplines, the availability of the instruments may be limited to the national labs or shared instrument facilities.
The size of thermoresponsive polymers can be further determined as the diffusion coefficient can be expressed as a function of viscosity and temperature in regard to the Stokes–Einstein equation: D = kT/6πηRH (where k is the Boltzmann constant, T is temperature, η is the viscosity of the solution, and RH is the radius of the solvated species).229 As temperature increases, polymers will have coil–globule structural changes and the size of the aggregates will change. This relationship is illustrated through the DOSY experiment in which peaks in NMR spectra of different compounds are separated based on varying diffusion coefficients and therefore differences in viscosity and temperature. DOSY can also measure molecular translational diffusion coefficients and particle size distributions, and because aggregates result in different diffusion coefficients, DOSY can determine how many aggregates with different sizes are formed. Additionally, DOSY can determine the number of aggregated species that are formed in connection with thermoresponsive globular structure formation in the solution.
The diffusion coefficients determined by DOSY can be used to derive the hydrodynamic radius of the polymers for determining size distribution and aggregation. In an aggregation study utilizing DOSY conducted by Zhang et al., DOSY spectra of dendronized polymers displayed similar diffusion coefficients below the LCST (25 °C) but different diffusion coefficients above the LCST (32 °C).269 It was concluded that above the LCST, OEG chains likely aggregate from the same and adjacent dendron units, resulting in a larger molecule, as supported by smaller diffusion coefficients. It is possible that the OEG chains also nonuniformly aggregate from peripheral dendrons, hence differences in diffusion coefficients and molecular size are observed. Other 1D and 2D NMR techniques can also be utilized to monitor the aggregation process and phase transition of thermoresponsive polymers.303,304
4.2. Bulk characterization
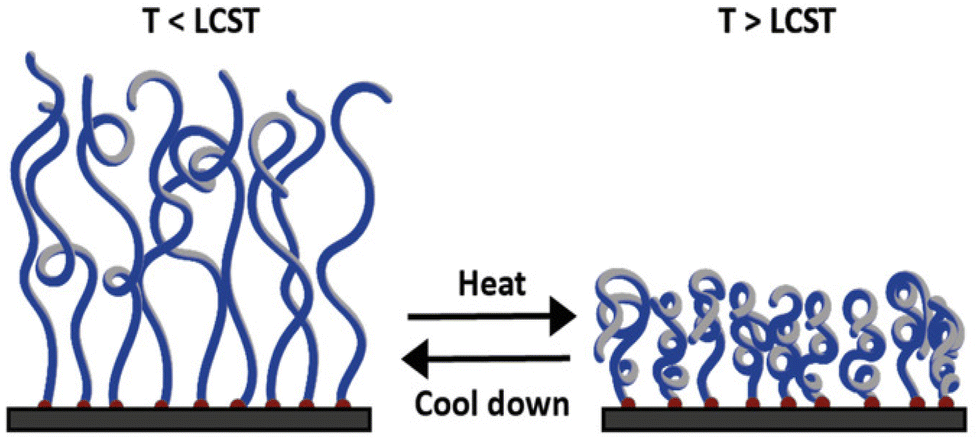 | ||
| Fig. 13 Schematic illustration of the conformational change of a polymer brush below the LCST at extended state (left) and above the LCST at collapsed state (right). Adapted with permission from ref. 201. Copyright 2022, John Wiley and Sons. | ||
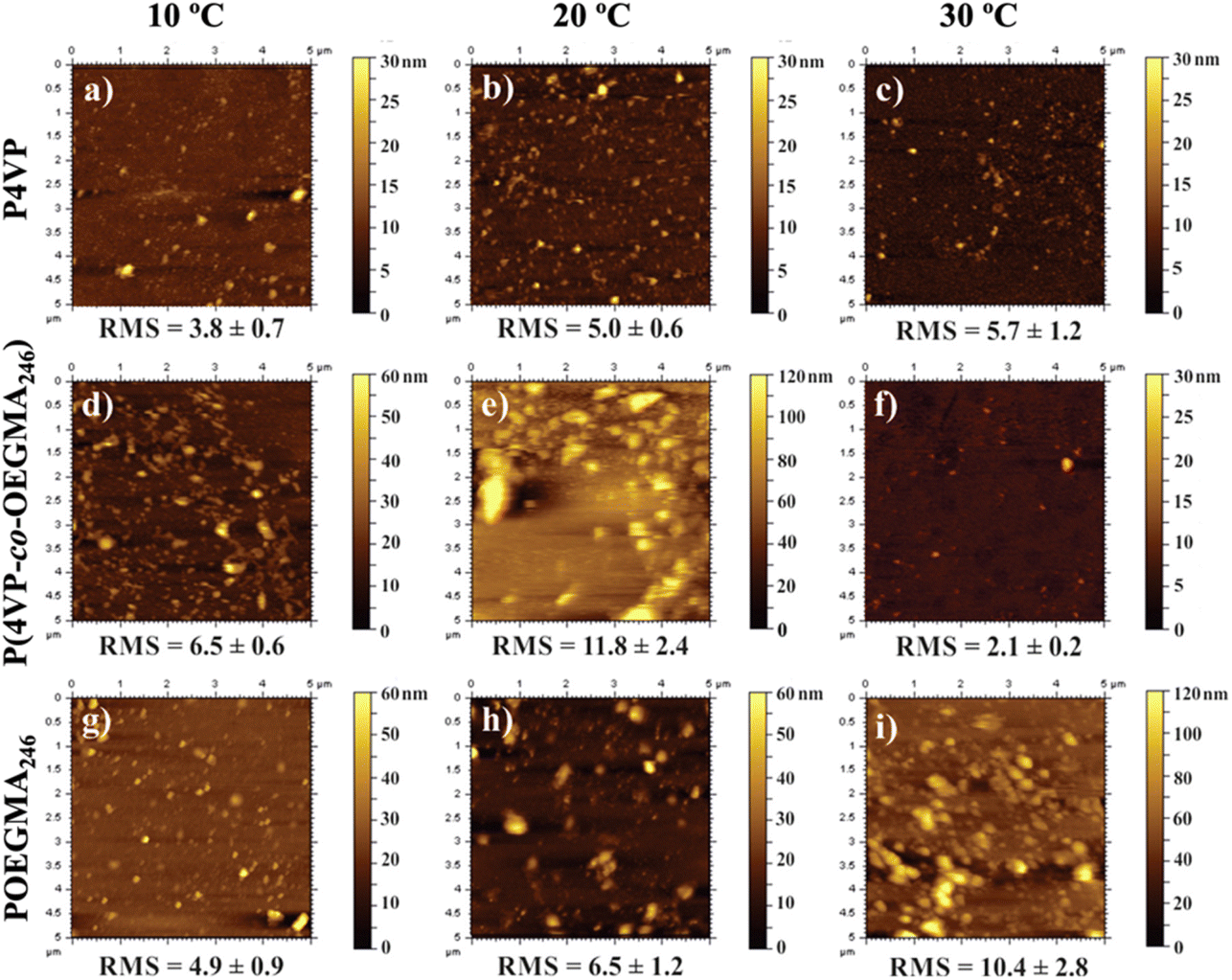 | ||
| Fig. 14 AFM topography of P4VP (a–c), P(4VP-co-OEGMA246) (d–f), and P(OEGMA246) (g–i) grafted brushes at different temperatures. Adapted with permission from ref. 307. Copyright 2017, American Chemical Society. | ||
AFM is also used to determine the topological effects on thermoresponsive behavior. Changes in structures, number of arms, and crosslinking can alter the thermoresponsiveness of polymers as it affects phase transitions. Phase transitions from a rigid to a flowing state or vice versa can alter how cells interact with each other and on surfaces. More specifically, Oleszko et al. utilized AFM to determine how poly(2-isopropyl-2-oxazoline) (PIPOx) surface crystallinity on a thermoresponsive influence on cell adhesion and detachment.285 AFM analysis detected thermoresponsive-induced changes in physicochemical properties, such as the formation of PIPOx crystallites, that promote cell adhesion and cell detachment. Regarding AFM's ability to detect topography, Tao et al. observed the thermoresponsive properties of dendritic macromonomers and dendronized polymers through the examination of topology with AFM.101 AFM measurements verified a mixture of bulk polymers. Additionally, it was found that the fan-shaped topology of the dendritic macromonomers changes to cylindrical for dendronized polymers, having significant effects on the thermoresponsive behavior. Similarly, Arias et al. observed the chiro-thermoresponsive (i.e., thermoresponsive behavior through conformational changes) properties of helical poly(phenylacetylene) bearing elastin-based side chain polymers.281 The AFM revealed a spherical morphology of the polymers, an unexpected thermoresponsive conformational change on the synthesized helical side chains of the elastin polymers. This is suggesting that the helical structure is of great importance in thermoresponsive behavior.
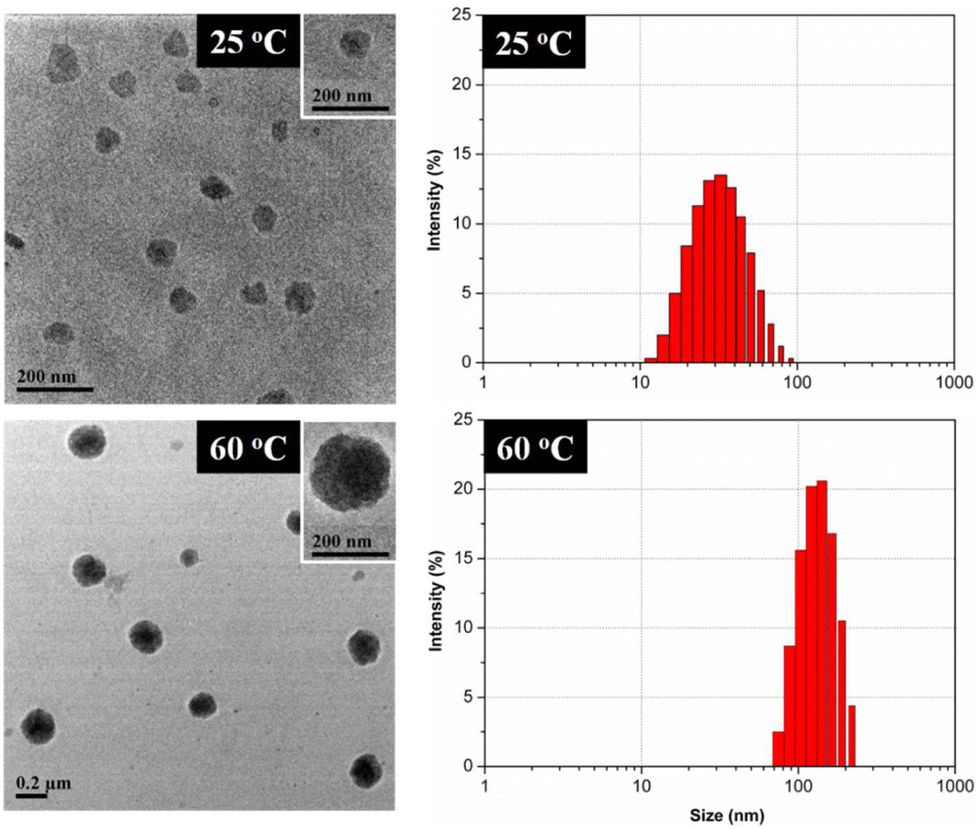 | ||
| Fig. 15 TEM micrographs (left) and DLS particle size distribution by intensity (right) of micelles in copolymer solution. Reprinted with permission from ref. 289. Copyright 2012, American Chemical Society. | ||
XPS is not limited to biomedical applications, but also to the biocompatibility of polymers, as surface adhesion is also of importance in cell cultures. By determining the composition of the surface layers, conclusions can be made on cell adhesion and attachment. Cell adhesion is fundamental in cell communication, regulation, maintenance, and growth, whereas cell detachment is essential in culturing cells. Thermoresponsive polymers can be used as a non-invasive control for adhesion and detachment by altering the temperature. Two studies examined the characteristics of thermoresponsive polymer layers used in the development of cell cultures. Oleszko et al. focused on thermoresponsive PIPOx layers, as these layers are used as biomaterials for human dermal fibroblast culture and detachment.285 The surface composition was determined through XPS, revealing the contents of carbon and nitrogen atoms. Therefore, the high crystallite content of PIPOx promoted the adhesion of the fibroblasts. PNIPAM has also been regarded for cell culture. Its chemical composition was analyzed through XPS in a study by Healy and co-workers, with the goal to make cell sheet regeneration more feasible and accessible.286 It was concluded that thermoresponsive film based on PNIPAM supports cell adhesion and temperature-controlled cell sheet detachment. The results demonstrated that thermoresponsive films successfully promoted cell adhesion and temperature-controlled cell detachment. This is beneficial as thermoresponsive surfaces can detach confluent cell sheets in a less invasive manner, allowing the detached sheets to be transplanted for further in vitro and in vivo adhesion and applications.
5. Biomedical applications of thermoresponsive polymers
Advancements in thermoresponsive materials have had a profound influence on numerous biomedical domains. The distinctive characteristic of reversible phase transition, occurring near physiological temperatures and allowing thermogelation for local treatment or use in wound healing, positions these materials as highly promising for an extensive range of biomedical applications. Such applications encompass bioimaging, biosensing, drug delivery systems, injectables, smart surfaces, bioadhesion, biomanufacturing, and tissue engineering.5.1. Bioimaging and biosensing
Thermoresponsive polymers can be functionalized with different types of contrast agents or biomolecular probes for imaging and sensing applications. These materials can be chemically tuned to enable temperature-dependent phase transitions to respond to disease biomarkers, such as in cancer diagnostics, and in general assist in the monitoring of disease progression and drug delivery efficiency.308–312 The unique properties of thermoresponsive polymers to undergo sol–gel transition above a certain temperature makes them promising materials for local delivery and long-term monitoring of therapeutic and diagnostic (theranostic) agents. For example, Kim et al. developed a poly(organophosphazene)-based hydrogel system that contained magnetic nanoparticles for long-term magnetic resonance (MR) imaging and cancer drug delivery application. Using their thermosensitive hydrogel system they were able to demonstrate the in vivo efficacy of long-term MR theranostics over a period of three weeks using their tumor-bearing mice model.308 In another study conducted by Wang et al., they used a PNIPAM-coated multifunctional nanoparticle system for combined fluorescence and MR imaging to track transfused cells in vivo. This new modality of cell tracking can be used for monitoring cell therapeutics such as in stem cell transfusion to cure leukaemia and other diseases.309In addition, thermoresponsive polymers have been exploited for applications in biosensing and can be utilized to detect various biological signals, such as changes in pH, temperature, and the presence of specific biomarkers or pathogens. Antunez et al. synthesized a thermoreversible biosensor for detecting heat-labile enterotoxin from the pathogen Escherichia coli which is known to cause diarrhea. The biosensing platform is composed of a porous silicon interferometric transducer and a thermoresponsive multivalent glycopolymer receptor that allows the reuse of the biosensor and subsequent detection cycles.310 In another study, Elshaarani et al. developed a glucose sensor based on 3-acrylamidophenylboronic acid and PNIPAM. Moreover, this sensor can detect different concentrations of glucose as glucose accumulates on the hydrophilic PNIPAM chains through hydrogen bonding, and as the temperature increases above 25 °C the hydrogel shrinks due to the thermoresponsive nature of the PNIPAM chain. This phenomenon in turn limits the accessibility of the phenylboronic acid component for glucose complexation as its conformation changes from coil to globule shape.311
Fluorescent dyes can also be incorporated into the polymers for development of biosensors. Mao et al. utilized OEGylated cyclodextrin-based thermoresponsive polymers as switchable inclusion complexes for fluorescent dyes. The OEG units work cooperatively towards the formation of inclusion complexes with fluorescent dyes that leads to significant changes in the monitored optical response of the system. The encapsulated dyes can be released upon heating, resulting in a noticeable decrease in fluorescence via quenching. This makes cyclodextrin polymers suitable for use as the foundation for temperature-responsive fluorescent sensors.312 Overall, the temperature sensitivity of thermoresponsive polymers makes them useful tools in biosensing, allowing for the detection of various biological signals in real-time and providing new methods for monitoring biological processes.
5.2. Therapeutics
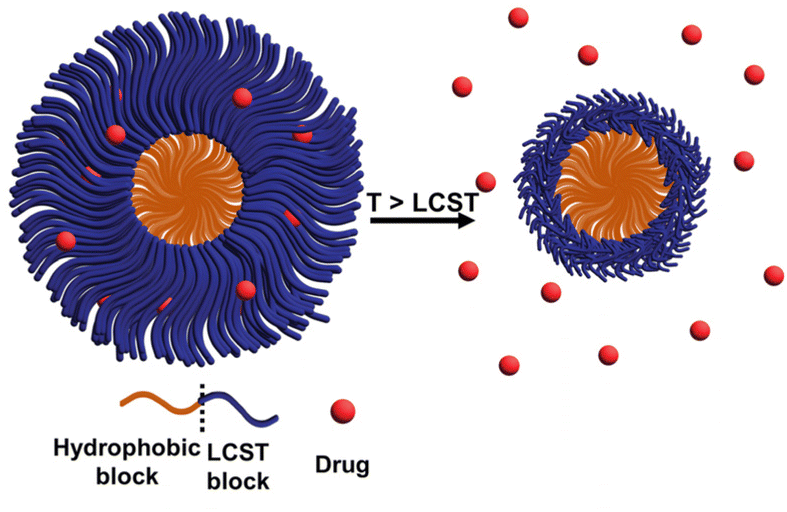 | ||
| Fig. 16 Schematic representation of a thermoresponsive diblock copolymer drug delivery system, which contains a hydrophobic block that serves as a scaffold and an LCST block that responds to temperature changes in the environment. At higher temperatures, the LCST block shrinks and the drug is released from the system. | ||
In the past, many PNIPAM-based delivery systems have been engineered. Nayak et al. successfully synthesized a structurally hollow PNIPAM gel from core–shell nanoparticles. These hollow structures show higher equilibrium swelling volumes below LCST and a greater deswelling ratio above LCST.318 Aside from thermal control, the drug delivery systems must be stable at physiological pH, but to enable degradation for clearance should hydrolyze in a slightly acidic environment. To accelerate the hydrolysis reaction the polymer side changes were modified with additional hydroxyl groups.320 Thermoresponsive delivery systems are highly desirable for cancer treatment as they show strong enhanced permeation and retention effect in targeting cytotoxic drugs to solid tumor tissues.280,321 An example of a cancer drug that exhibits adverse side effects is doxorubicin. Using a star–block poly(ε-caprolactone)-block-PME2OMA-co-POEGMA that self-assembles into a micelle, Li et al. were able to show low cytotoxic delivery of doxorubicin that also was effective in inhibiting HepG2 cell proliferation.54 Another promising delivery system for doxorubicin is thermogelling glycol chitin, which displayed easy encapsulation and sustained drug release over a 13-day period.322 Paclitex is a natural anticancer drug containing Cremophor EL (polyoxyethylated castor oil) that also has many side effects including bronchospasm, hypotension, and hypersensitivity. Paclitex also faces multi-drug resistance from cancer cells and has poor water solubility. These challenges restrict the drug's dosage and infusion period. Song et al. demonstrated that by using a thermoresponsive polymer system consisting of a β-cyclodextrin as a core to a four-armed PNIPAM to encapsulate Paclitex, the cellular uptake of the drug can be enhanced, and multidrug resistance can be overcome making the polymer an ideal candidate for cancer therapeutics.144 A very few of these thermoresponsive micellar drug delivery systems have been tested for the later stages of clinical trials. One PEG–poly(D,L-lactide) system under the name of Genexol-PM has received approval for breast and lung cancer treatments.323 There are other several PEG-based micellar systems currently under Phase I–III clinical trials.14
In addition to cancer therapy, ocular diseases are promising targets for thermoresponsive therapeutic systems.324 Eye diseases are generally treated by the ocular installation of drugs as it is a non-invasive route and allows for easy administration. However, only about 5% of the administered drug infiltrates the cornea to the intraocular tissue through conventional eye drop formulations as such improving the bioavailability and targeted delivery of ocular drugs remains a challenge. By taking advantage of the interactions of cyclodextrins with hydrophobically modified polymers, Iohara et al. prepared a thermoresponsive hydrogel system for ocular applications. By adding α-cyclodextrin to hydrophobically modified hydroxypropyl methylcellulose, the resulting cellulose/cyclodextrin gel could undergo a reversible sol–gel transition under physiological temperature, and they demonstrated the effective delivery of diclofenac sodium in the eyes of a rabbit animal model. Moreover, the cyclodextrin improved the drug's bioavailability and bioabsorption.52
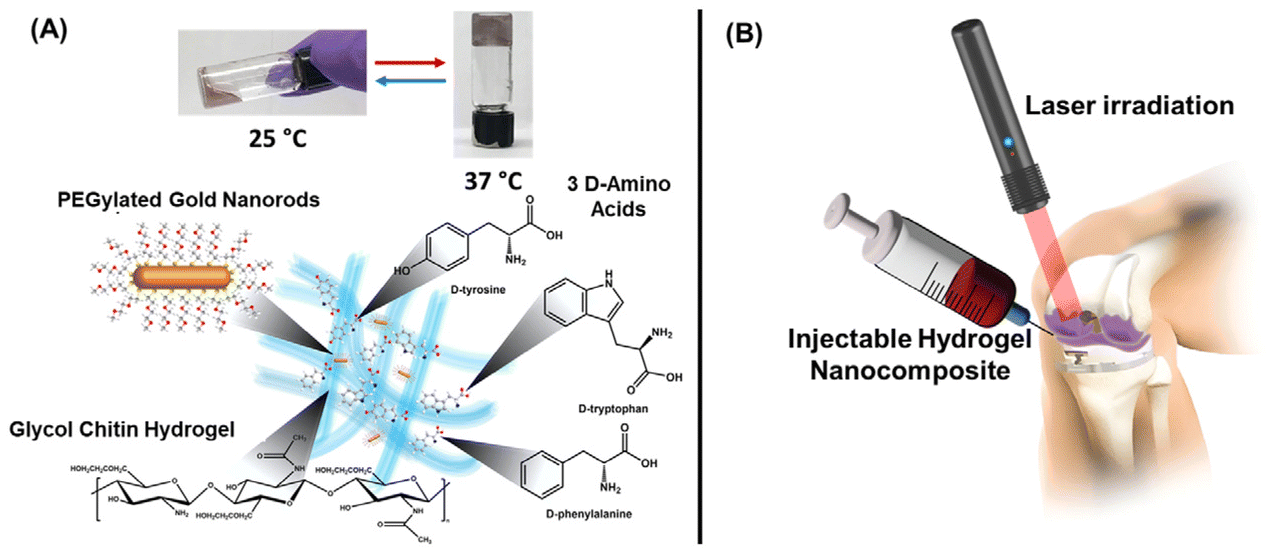 | ||
| Fig. 17 Schematic representations of the components (A) and injectable treatment approach (B) of a thermoreversible hydrogel system for the treatment of joint infections. Adapted with permission from ref. 326 and 327. Copyright 2020 and 2023, American Chemical Society. | ||
In the case of dramatic eye injuries or diseases, rapid treatment is crucial to avoid vision deterioration. PNIPAM and its derivatives can be used as a bioadhesive for treating ocular complications.329–331 As a substitute for sutures or cyanoacrylate-based adhesive materials (i.e., biomedical grade super glue), Bayat et al. developed a temperature-sensitive injectable hydrogel consisting of physically crosslinked PNIPAM copolymerized with n-butyl acrylate that functions as a temporary sealant. When tested on a rabbit model with open globe injury, the hydrogel was easily applied using custom-made temperature-controlled syringes and effectively maintained intraocular pressure without causing neurotoxicity, retinal tissue degradation, or significant chronic inflammatory response. The hydrogel can be removed by exposing it to cold water once it has set. This hydrogel can be beneficial in areas with limited resources, where it can temporarily close wounds to prevent further tissue damage or vision loss prior to surgery.330 Although the mechanism was not explained, PEG-based in situ forming hydrogels (ReSure® sealant) were used to prevent incision leakage after cataract surgery and approved by Food and Drug Administration (FDA) and a Phase 3 clinical trial was also completed.332Fig. 18 shows PEG-PNIPAM-based thermoresponsive injectable polymers for various biomedical applications.57
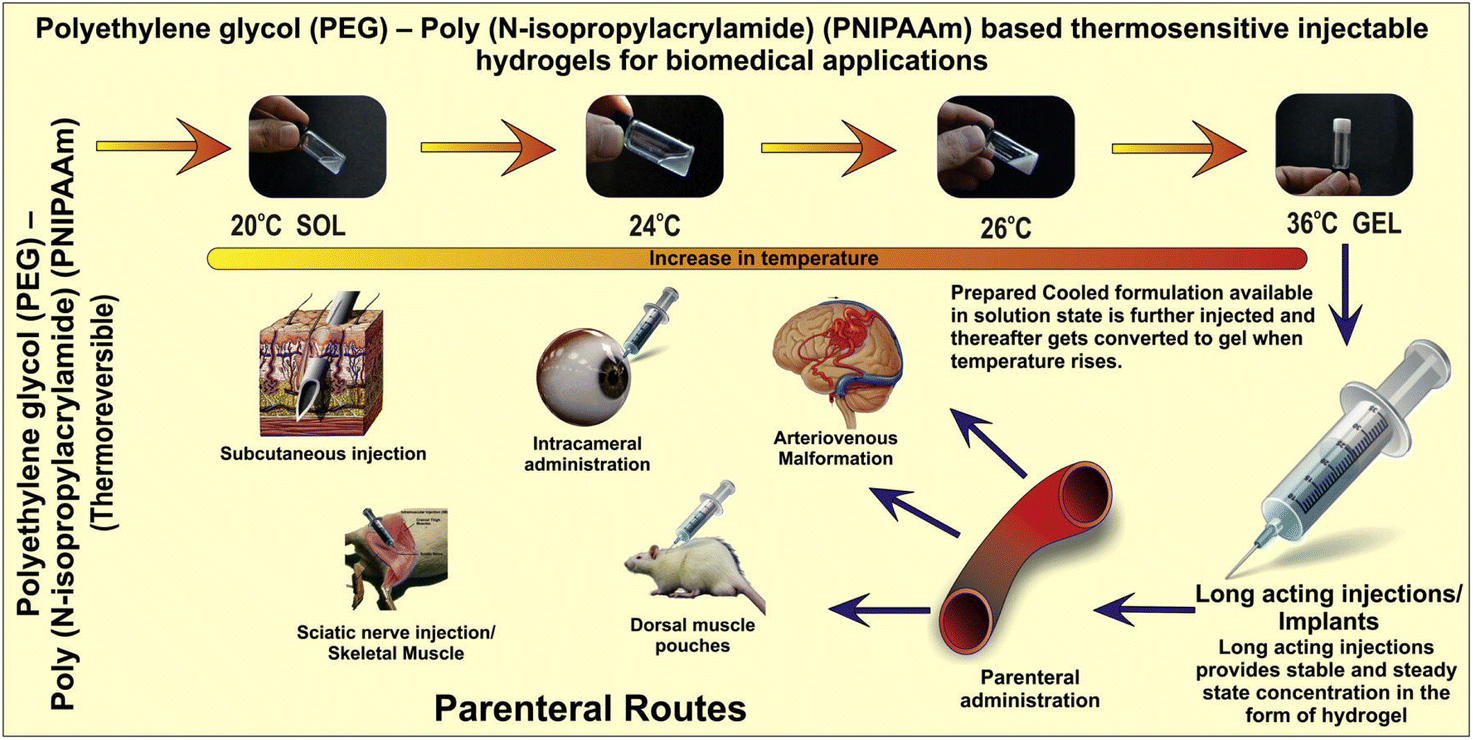 | ||
| Fig. 18 PEG–PNIPAM-based thermoresponsive injectable polymers for biomedical applications. Adapted with permission from ref. 57. Copyright 2014, Elsevier. | ||
Although limited, injectable thermoresponsive hydrogels have been used in clinical trials for cancer treatment. Triblock copolymers of poly(lactic-co-glycolic acid) (PLGA)–PEG–PLGA with a sol–gel transition at 37 °C commercialized under the names of ReGel® and OncoGel® were used to solubilize poorly soluble drugs and apply for local tumor management. This thermoresponsive system increased the drug loading (e.g., paclitaxel) by more than 400-fold with high drug release results and degradation over 4–6 weeks.333–335 However, these clinical trials were terminated due to lacking impact on tumor regression.336 Additionally, PEG-based polymers and related technologies are the only synthetic polymeric biomaterials with a thermoresponsive transition around human body temperature approved by FDA.337
5.3. Smart surfaces
Thermoresponsive polymers have been quite often used as a coating material to design stimuli-responsive smart surfaces by generally chemical or sometimes physical tethering. Protocols for the production of thermoresponsive surfaces are complex, costly, and can often not yield the desired product (no control over the geometry of the surface or layer thickness). Sponchioni et al. developed a synthetic technique and fabricated thermoresponsive polymers enabling the coating of tissue culture polystyrene with styrene as an anchor and arginine–glycine–aspartic acid (RGD) for the collection of cell sheets in serum-free conditions. This technique required no particular equipment and allowed copolymer adsorption regardless of geometry and size.64 Another approach to increasing cell adhesion is the copolymerization of dopamine methacrylamide with PNIPAM. As opposed to pure PNIPAM microgels, the resulting P(NIPAM-co-dopamine methacrylamide) microgel exhibited enhanced adhesion even under harsh conditions (i.e., mechanical stress).76 It is important to note that the nature and concentration of the ion strongly influence the thermoresponsive properties of polymeric gels and brushes.250Polymeric hydrogels are often modified with amino acid sequences like RGD to increase their cell adhesion properties.74,338 Marrow stromal cells were used to test the adhesive properties of these RGD-modified hydrogels. After 28 days, adhesion of the cells was observed as well as successful proliferation.338 Additionally, the copolymerization of insulin and RGDS (arginine–glycine–aspartic acid–serine) demonstrated site-selective cell adhesion and growth along with the temperature-dependent spatiotemporal detachment of sparsely adherent and confluent cells. From a peptide-based approach, RGDS and insulin were immobilized onto thermoresponsive surfaces for site-selective cell adhesion and growth, which allowed for site-specific harvest. Moreover, selective recovery of either contiguous monolayer or mesh-like designed monolayer tissue constructs on identical surfaces was observed.74
In addition, thermoresponsive polymers have drawn much attention in the design of smart surfaces that can be used for bioanalysis and bioseparation because of their switchable wetting properties that can be readily tuned through temperature changes in the environment. In chromatographic applications, thermoresponsive polymers can be useful in the separation of proteins and cells that can be deactivated by organic solvents that work with traditional reversed-phase chromatography column packing materials.189,339–343 Kanazawa et al. developed an early type of thermoresponsive chromatography stationary phase made by modifying silica bead surfaces with PNIPAM via a coupling reaction (Fig. 19). The PNIPAM-modified silica beads were packed into a stainless-steel column and used to separate different steroids. At 5 °C all the steroids were eluted at once, but with increasing temperature the retention times were prolonged and effective separation was achieved at 35 °C and above. The effective separation could be attributed to the enhanced hydrophobic interactions between the PNIPAM-modified silica beads and the steroids at elevated temperatures.344
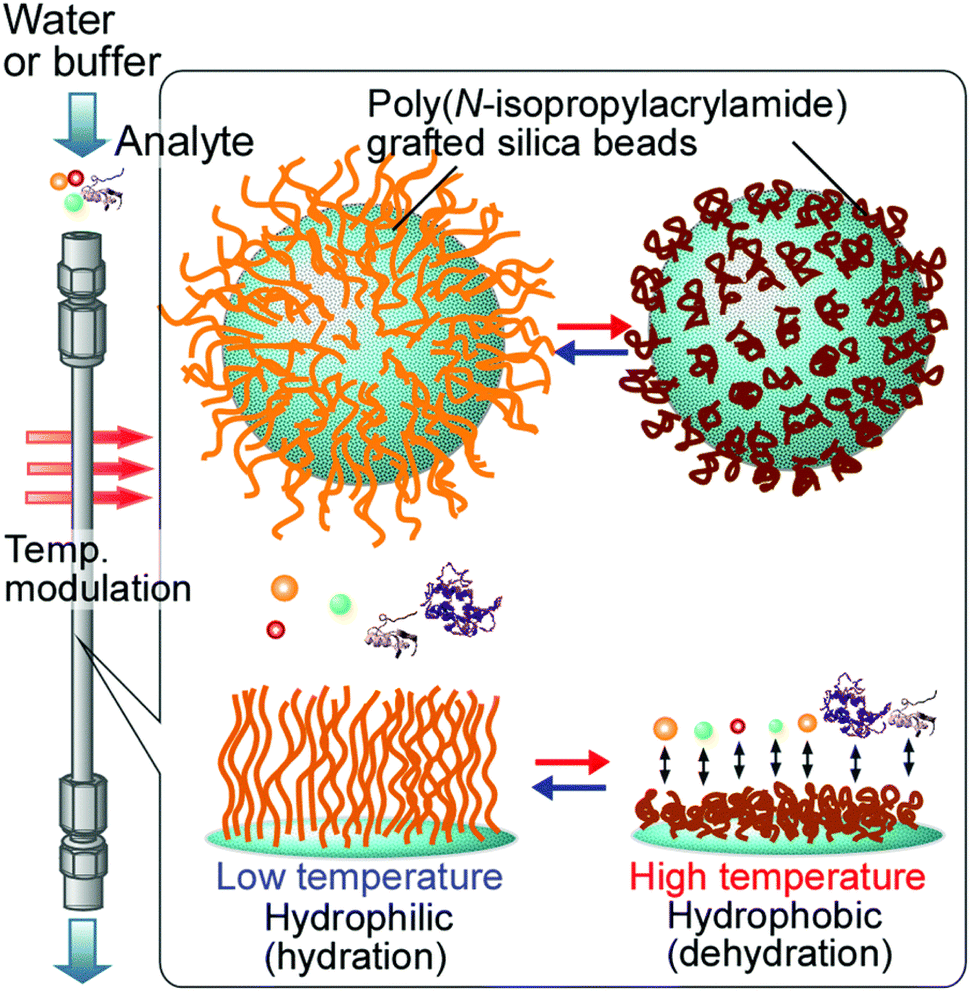 | ||
| Fig. 19 Concept of thermoresponsive bioanalysis and bioseparation using surfaces modified with thermoresponsive polymers. Reprinted with permission from ref. 342. Copyright 2016, RSC. | ||
In biomanufacturing, microfluidic systems offer low-cost solutions for scaling up cell-based therapies to reduce health disparities. However, a major challenge is the efficient recovery of cells that are bound to microfluidic channels while maintaining their viability. Thermoresponsive polymers provide a promising approach for overcoming this limitation. By functionalizing the polymers with specific antibodies, they can be used to bind a targeted cell type for isolation, on-chip transduction, and expansion. Importantly, the temperature-triggered response of these polymers allows for the controlled release of modified cells without affecting their viability. Moreover, recent research has shown that not all CD34+ stem cells are relevant for hematopoiesis, and instead, a small sub-population of CD34+ cells co-expressing CD90+ cells is responsible for repopulation after hematopoietic stem cell transplantation.345 However, it is not currently possible to target cells that co-express multiple markers using immunomagnetic bead-based purification. To overcome this drawback, thermoresponsive polymers offer a viable platform for double positive selection, where isolated CD34+ cells can be further processed to select only cells co-expressing CD90 markers, potentially improving treatment outcomes. Gurkan et al. demonstrated the use of PNIPAM-coated microfluidic channels for isolation of CD4+ and CD34+ hematopoietic stem cells from unprocessed human whole blood with high specificity (90%), viability (95%), and release efficiency (60%).346,347 Although selective capture of cells from bodily fluids in microchannels has been previously demonstrated by other groups, it remains a significant challenge to release the desired cells due to their strong adherence to the microchannel surface. By using thermoresponsive polymers, the same research lab was able to capture the CD4+ lymphocytes at physiological temperature and subsequently release them below the LCST (∼32 °C) of the polymer.346 By synthesizing a novel UV cross-linkable copolymer of 4-(N-cinnamoylcarbamide)methylstyrene and NIPAM, Recum et al. were able to develop a thermoreversible cell culture coating and grew retinal pigment epithelium cells to confluence. Then by decreasing the temperature from 37 °C to 20 °C the cells readily detach. This technology that gives rise to cell sheets without scaffolds will be useful for tissue engineering applications as it can provide cells without cell damage and serve as implanted scaffolds.348
In the case of cell transplantation to treat intractable diseases, it is crucial to develop cell separation methods whereby thermoresponsive polymers have also great potential. Recently, Nagase et al. synthesized a thermoresponsive cationic copolymer brush (PDMAPAM-b-PNIPAM) that could successfully separate targeted umbilical cord-derived mesenchymal stem cells from fibroblasts and macrophages. At physiological temperature, all cell types adhere to the cationic polymer brush, however, when the temperature is reduced to 20 °C the mesenchymal stem cells detached from the polymer brush while the fibroblasts and macrophages remain adhered. As such, the prepared thermoresponsive cationic polymer brush could be used as an effective cell separation tool for the purification of mesenchymal stem cells.90 By leveraging the unique properties of thermoresponsive polymers, researchers can potentially overcome some of the challenges associated with traditional cell separation techniques and develop more efficient and cost-effective methods for cell-based therapies.
5.4. Tissue engineering
Due to the shortage of organ and tissue donors, much attention has been devoted to finding replacements for use in regenerative medicine and advanced tissue engineering. With cell sheet engineering, tissue-specific cells can be used for the fabrication of physically connective, functionally synchronous three-dimensional tissues.64 In the case of cell-based therapies, thermoresponsive polymers serve as a good platform for scaffold-free cell growth as they permit little to no immune rejection, and display greater regenerating properties compared to directly injected suspended cells. This type of biomaterials has found applications in orthopaedic tissue engineering, wound healing, oral disease treatments, and neural and corneal tissue therapy (Fig. 20). Moreover, the ability of thermoresponsive polymers to swell in water allows for their tissue-like elastic properties and high permeability to biological fluids.349,350 While designing a smart polymer material, it is important that candidate polymers are highly porous to provide cell diffusion and promotion of the developing tissues’ vascularization, have easily tunable functions and properties, and be completely biodegradable. Kang et al. synthesized highly porous gelatin-based scaffolds that successfully provided a way to fabricate biodegradable scaffolds with interconnective pores for mass diffusion control.349 Moreover, their gelatin-based scaffolds were found to be easily integrated into human tissues.351 These scaffolds can also be produced by 3D bioprinting of thermoresponsive hydrogels which has been previously reviewed.352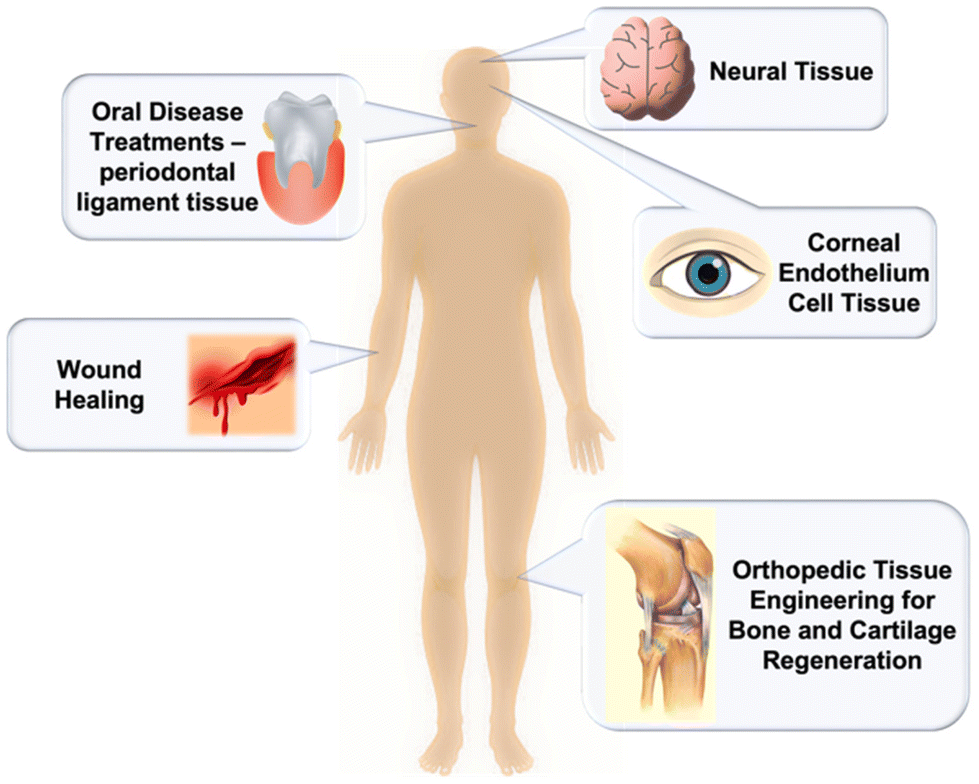 | ||
| Fig. 20 Illustration of the different types of applications of thermoresponsive polymers in tissue engineering. | ||
Macroporous constructs have become favorable materials for bone-tissue engineering as they serve as ideal scaffolds for tissue growth while allowing the use of different types of biomaterials including those that are metallic, ceramic, or synthetic polymer in nature.338,353 Fu et al. synthesized a biodegradable triblock copolymer PEG–poly(ε-caprolactone)–PEG (PEG–PCL–PEG) with nanohydroxyapatite, which showed potential use in cell proliferation for bone regeneration applications.63 In a similar study, Shin et al. were able to design a novel oligo(poly(ethylene glycol) fumarate) (OPF) hydrogel and they assessed the in vivo bone and soft tissue behavior in a rabbit animal model. The group was able to show the formation of a fibrous capsule around the cranial implant with a thickness that remained consistent during implantation thereby demonstrating that the OPF hydrogel invokes a tissue response. Moreover, histology revealed that OPF made from higher molecular mass PEG exhibited enhanced fragmentation and tissue infiltration by inflammatory cells making this type of material promising as a biodegradable scaffold for bone tissue engineering.350
Additionally, thermoresponsive polymers have a variety of applications in cutaneous wound healing. Injury to the skin can be treated through a multi-phase healing process involving tissue granulation and re-epithelialization. Failure in the wound-healing process can lead to chronic unhealed wounds or abnormal scarring. In a proof-of-concept experiment, Zhong et al. demonstrated that a citrate-based poly(poly(ethylene glycol)citrate-co-NIPAM) copolymer scaffold embedded with iKera cells provided more effective cutaneous wound healing and re-epithelialization than either the copolymer or iKera cells alone.354 On a similar note, Pawar et al. prepared polymer blends of PNIPAM, egg albumen, and poly(ε-caprolactone) with the antibiotic gatifloxacin hydrochloride (Gati) to prepare nanofibers for wound healing applications. The in vivo studies showed faster tissue regeneration using the nanofibers which supports the suitability of Gati-loaded nanofibers for wound healing applications.355 In the case of periodontal diseases, these polymers can be used to create scaffolds that mimic the natural environment of the periodontium and promote the growth of new tissue. By incorporating thermoresponsive polymers into such scaffolds, researchers can potentially create a treatment that is minimally invasive and maximally effective in treating periodontal diseases.356–359 For example, Ji et al. developed a chitosan-based thermosensitive hydrogel made from quaternized chitosan with α,β-glycerophosphate that they could use to promote the development of alkaline phosphatase activity in human periodontal ligament cells.356
Tissue engineering involving the neural system has generally been more challenging because of the presence of the blood–brain barrier that separates the nervous system from systemic circulation and the limited self-regenerating properties of the nervous system. However, injectable hydrogels, such as 0.5%:3% and 0.75%:3% chitosan/β-glycerophosphate hydrogels developed by Bhuiyan et al. have been shown to have suitable properties for neural tissue engineering and supported cell growth and proliferation in vitro. This type of hydrogel has been demonstrated to be suitable for porosity, osmolality, and swelling behavior for neural tissue engineering and also has desirable biodegradation pathways.79 Aside from neural repair applications, thermoresponsive polymers have also been demonstrated to show potential for regenerative medicine applications in the eye.360–363 Khalili et al. were able to generate corneal endothelium-like cell sheets using alternative cells that were embedded in elastin-mimetic dendrimers.363 Overall, the application of thermoresponsive polymers in tissue engineering holds great promise for developing new approaches that could be useful for regenerative medicine and pharmaceutical research.
6. Conclusions and future perspectives
Polymers have the ability to respond to various external stimuli such as temperature, light, and pH. Among these stimuli, thermoresponsive behavior of polymers is studied extensively. The LCST behavior of thermoresponsive polymers can be tailored through various synthetic tools including copolymerization, end-group functionalization, and modifying the architecture of the polymer. Thermoresponsive polymers with narrow molecular mass distribution, controlled molecular mass, high end-group fidelity, and various compositions and architectures can be synthesized through controlled radical polymerization techniques such as ATRP and RAFT polymerization which are ideal synthetic tools for better structural design, control, and tunability in solution and from the surface. The structural heterogeneity in polymers has a significant effect on the thermoresponsive properties. Thus, these thermal transitions should be precisely determined by using modern characterization tools such as NMR and UV-Vis spectroscopy, AFM, rheometer, and electron microscopy. The tunability of the LCST makes these polymers ideal for use in physiological temperatures. The study of thermoresponsive polymers has a broad range of current and potential future applications in drug delivery for cancer treatment, tissue engineering for regenerative medicine, injectable polymers for therapeutics, smart surface for cell separation, and several other areas. Although the earliest works on temperature-induced phase transition were reported in the late 1960s, thermoresponsive materials in the biomedical field have still been emerging and more advanced materials for use in patients can be designed by leveraging the synthetic and characterization tools highlighted in this review article.Abbreviations
| AcBzPh | Acrylamidebenzophenone |
| ACPBA | Acetamidophenylboronic acid |
| AFM | Atomic force microscopy |
| AgNPs | Silver nanoparticles |
| ARGET | Activators regenerated by electron transfer |
| ATRP | Atom transfer radical polymerization |
| BuMA | Butyl methacrylate |
| CRP | Controlled radical polymerization |
| CTA | Chain transfer agent |
| DEGA | Di(ethylene glycol) monomethyl ether acrylate |
| DLS | Dynamic light scattering |
| DMA | Dimethyl acrylamide |
| DMAPAM | N,N-Dimethylaminopropyl acrylamide |
| DOSY | Diffusion-ordered spectroscopy |
| DSC | Differential scanning calorimetry |
| eATRP | Electrochemically-mediated atom transfer radical polymerization |
| EG | Ethylene glycol |
| eOEGMA | Ethoxy oligoethylene glycol methacrylate |
| FDA | Food and Drug Administration |
| Flu | Fluorescein |
| FTIR | Fourier transform infrared |
| HEA | 2-Hydroxyethyl acrylate |
| HIPPVTA | Bis(N-hydroxyisopropyl pyrrolidone)-2-vinylterephthalate |
| HMStBA | Pendant disulfide functionalized t-butyl acrylate |
| HPEI | Hyperbranched poly(ethylene imine) |
| HPMA | N-(2-Hydroxypropyl) methacrylamide |
| LCST | Lower critical solution temperature |
| mDEGMA | Diethylene glycol methyl ether methacrylate |
| Me4Cyclam | 1,4,8,11-Tetramethyl-1,4,8,11-tetraazacyclotetradecane |
| Me6TREN | Tris[2-(dimethylamino)ethyl]amine |
| MEA | Monoethanolamine |
| mEGA | Ethylene glycol methyl ether acrylate |
| ME2OMA | 2-(2-Methoxyethoxy)ethyl methacrylate |
| M n | Number-average molecular weight |
| MPEGMA | Methyl polyethylene glycol methacrylate |
| MR | Magnetic resonance |
| mTEGMA | Tetraethylene glycol methyl ether methacrylate |
| M w | Weight-average molecular weight |
| NEM | N-Ethylmaleimide |
| NMM | N-Methylmorpholine |
| NMP | Nitroxide-mediated polymerization |
| NMR | Nuclear magnetic resonance |
| OEG | Oligo(ethylene glycol) |
| OEGA | Oligo(ethylene glycol) acrylate |
| OEGMA | Oligo(ethylene glycol)methacrylate) |
| OPF | Oligo(poly(ethylene glycol) fumarate) |
| PAMs | Poly(N-alkylacrylamide)s |
| PAPi | Poly(N-acryloylpiperidine) |
| PAPy | Poly(N-acryloylpyrrolidine) |
| PBEEP | Poly(benzophenenone ethylene ethyl phosphate) |
| PCL | Poly(ε-caprolactone) |
| PDEA | Poly(N,N-diethylacrylamide) |
| PDEGMA | Poly(diethylene glycol methyl ether methacrylate) |
| PDAs | Poly(N,N-dialkylacrylamide)s |
| PDEA | Poly(N,N-diethylacrylamide) |
| PDFEA | Poly[N-(2,2-difluoroethyl)acrylamide] |
| PDLA | Polymer D-lactic acid |
| PDMAEMA | Poly(2-(N,N-dimethylamino) ethyl methacrylate |
| PDMDOMA | Poly[N-(2,2-dimethyl-1,3-dioxolane)-methyl)]acrylamide |
| PDSEA | Pyridyldisulfide ethylacrylamide |
| PEG | Polyethylene glycol |
| PEGA | Poly(ethylene glycol acrylate) |
| PEGMA | Poly(ethylene glycol) methyl ether methacrylate |
| PEHO | Poly-(hydroxymethyl)oxetane |
| PEMA | Poly(N,N-ethylmethylacrylamide) |
| PEO | Poly(ethylene oxide) |
| PET | Photo-induced electron/energy transfer |
| PEtOx | Poly(2-ethyl-2-oxazoline) |
| PiPMA | Poly(isopropyl methacrylate) |
| PIPOx | Poly(2-isopropyl-2-oxazoline) |
| PI-RAFT | Photoiniferter reversible addition–fragmentation chain transfer |
| PLGA | Poly(lactic-co-glycolic acid) |
| PLLA | Poly(L-lactide) |
| PMEO2MA | Poly(2-(2-methoxyethoxy)ethyl methacrylate) |
| PMVE | Poly(methyl vinyl ether) |
| PNANPP | Poly(N-acryloyl-N′-propylpiperazine) |
| PNcPAM | Poly(N-cyclopropylacrylamide) |
| PNEAM | Poly(N-ethylacrylamide) |
| PNIPAM | Poly(N-isopropylacrylamide) |
| PNnPAM | Poly(N-n-propylacrylamide) |
| PNtBAM | Poly(N-tert-butylacrylamide) |
| PNVCL | Poly(N-vinylcaprolactam) |
| PNVIBA | Poly(N-vinylisobutyramide) |
| POEGMA | Poly(oligo(ethylene glycol)methacrylate) |
| POEGNB | Poly(oligoethylene glycol) norbornene |
| POEGSt | Poly(oligoethylene glycol) styrene |
| POEGVE | Poly(oligoethylene glycol) vinyl ether |
| poly(NMEP) | Poly(N-(2-methacryloyloxyethyl)pyrrolidone |
| PPGMA | Poly(propylene glycol methacrylate) |
| PPO | Poly(propylene oxide) |
| PPrOx | Poly(2-propyl-2-oxazoline) |
| Pro | Proline |
| PSS | Polystyrene sulfonate |
| PVP/PVPy | Polyvinylpyrrolidone |
| RAFT | Reversible addition–fragmentation chain transfer |
| RGD | Arginine–glycine–aspartic acid |
| RGDS | Arginine–glycine–aspartic acid–serine |
| ROMP | Ring-opening metathesis polymerization |
| SARA | Supplemental activator and reducing agent |
| SANS | Small-angle neutron scattering |
| SAXS | Small-angle X-ray scattering |
| SEC | Size-exclusion chromatography |
| SEM | Scanning electron microscopy |
| Sn(EH)2 | Tin(II) 2-ethylhexanoate |
| T cp | Cloud point temperature |
| TEM | Transmission electron microscopy |
| TGA | Thermogravimetric analysis |
| TPMA | Tris(2-pyridylmethyl)amine |
| TREN | Tris(2-aminoethyl)amine |
| XPS | X-ray photoelectron spectroscopy |
| XRD | X-ray powder diffraction |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by Case Western Reserve University (CWRU) College of Arts and Science (CAS) and Expanding Horizons Initiative at CWRU. YY thanks CWRU Flora Stone Mather Center for Women for financial support.References
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2009, 5, 2513–2533 RSC.
- Y.-J. Kim and Y. T. Matsunaga, J. Mater. Chem. B, 2017, 5, 4307–4321 RSC.
- M. T. Cook, P. Haddow, S. B. Kirton and W. J. McAuley, Adv. Funct. Mater., 2021, 31, 2008123 CrossRef CAS.
- F. Doberenz, K. Zeng, C. Willems, K. Zhang and T. Groth, J. Mater. Chem. B, 2020, 8, 607–628 RSC.
- P. Zarrintaj, M. Jouyandeh, M. R. Ganjali, B. S. Hadavand, M. Mozafari, S. S. Sheiko, M. Vatankhah-Varnoosfaderani, T. J. Gutiérrez and M. R. Saeb, Eur. Polym. J., 2019, 117, 402–423 CrossRef CAS.
- O. Bertrand and J.-F. Gohy, Polym. Chem., 2017, 8, 52–73 RSC.
- Y. Takashima, S. Hatanaka, M. Otsubo, M. Nakahata, T. Kakuta, A. Hashidzume, H. Yamaguchi and A. Harada, Nat. Commun., 2012, 3, 1270 CrossRef PubMed.
- F. D. Jochum, L. zur Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854–7862 CrossRef CAS.
- J. Jiang, X. Tong and Y. Zhao, J. Am. Chem. Soc., 2005, 127, 8290–8291 CrossRef CAS PubMed.
- G. Kocak, C. Tuncer and V. Bütün, Polym. Chem., 2017, 8, 144–176 RSC.
- S. Dai, P. Ravi and K. C. Tam, Soft Matter, 2008, 4, 435–449 RSC.
- A. Nese, N. V. Lebedeva, G. Sherwood, S. Averick, Y. Li, H. Gao, L. Peteanu, S. S. Sheiko and K. Matyjaszewski, Macromolecules, 2011, 44, 5905–5910 CrossRef CAS.
- M. Kohri, K. Yanagimoto, K. Kohaku, S. Shiomoto, M. Kobayashi, A. Imai, F. Shiba, T. Taniguchi and K. Kishikawa, Macromolecules, 2018, 51, 6740–6745 CrossRef CAS.
- K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS PubMed.
- M. Czaun, L. Hevesi, M. Takafuji and H. Ihara, Chem. Commun., 2008, 2124–2126, 10.1039/b717721f.
- P. M. Xulu, G. Filipcsei and M. Zrínyi, Macromolecules, 2000, 33, 1716–1719 CrossRef CAS.
- T. Tanaka, I. Nishio, S.-T. Sun and S. Ueno-Nishio, Science, 1982, 218, 467–469 CrossRef CAS PubMed.
- T. Shiga, Neutron Spin Echo Spectroscopy Viscoelasticity Rheology, Springer Berlin Heidelberg, Berlin, Heidelberg, 1997, pp. 131–163. DOI:10.1007/3-540-68449-2_2.
- S. Ramanathan and L. H. Block, J. Controlled Release, 2001, 70, 109–123 CrossRef CAS PubMed.
- J. Kost, K. Leong and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 7663–7666 CrossRef CAS PubMed.
- A. Marin, M. Muniruzzaman and N. Rapoport, J. Controlled Release, 2001, 75, 69–81 CrossRef CAS PubMed.
- J. Li, C. Nagamani and J. S. Moore, Acc. Chem. Res., 2015, 48, 2181–2190 CrossRef CAS PubMed.
- Y. C. Simon and S. L. Craig, Mechanochemistry in Materials, Royal Society of Chemistry, 2017 Search PubMed.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- M. Wei, Y. Gao, X. Li and M. J. Serpe, Polym. Chem., 2017, 8, 127–143 RSC.
- A. P. Vogt and B. S. Sumerlin, Soft Matter, 2009, 5, 2347–2351 RSC.
- J. S. Scarpa, D. D. Mueller and I. M. Klotz, J. Am. Chem. Soc., 1967, 89, 6024–6030 CrossRef CAS.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Chem., 1968, 2, 1441–1455 CAS.
- S. Fujishige, K. Kubota and I. Ando, J. Phys. Chem., 1989, 93, 3311–3313 CrossRef CAS.
- M. Lemanowicz, A. Gierczycki, W. Kuźnik, R. Sancewicz and P. Imiela, Miner. Eng., 2014, 69, 170–176 CrossRef CAS.
- J. C. Foster, I. Akar, M. C. Grocott, A. K. Pearce, R. T. Mathers and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 1700–1707 CrossRef CAS PubMed.
- A. Narumi, S.-i. Sato, X. Shen and T. Kakuchi, Polym. Chem., 2022, 13, 1293–1319 RSC.
- A. Bordat, T. Boissenot, J. Nicolas and N. Tsapis, Adv. Drug Delivery Rev., 2019, 138, 167–192 CrossRef CAS PubMed.
- M. Shibayama and T. Tanaka, in Responsive Gels: Volume Transitions I, ed. K. Dušek, Springer Berlin Heidelberg, Berlin, Heidelberg, 1993, pp. 1–62. DOI:10.1007/3-540-56791-7_1.
- G. Chen and A. S. Hoffman, Nature, 1995, 373, 49–52 CrossRef CAS PubMed.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Macromolecules, 1993, 26, 2496–2500 CrossRef CAS.
- X. Qiu, T. Koga, F. Tanaka and F. M. Winnik, Sci. China: Chem., 2013, 56, 56–64 CrossRef CAS.
- G. Pasparakis and C. Tsitsilianis, Polymer, 2020, 211, 123146 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- R. Liu, M. Fraylich and B. R. Saunders, Colloid Polym. Sci., 2009, 287, 627–643 CrossRef CAS.
- Z. Hu, T. Cai and C. Chi, Soft Matter, 2010, 6, 2115–2123 RSC.
- V. Aseyev, H. Tenhu and F. M. Winnik, in Self Organized Nanostructures of Amphiphilic Block Copolymers II, ed. A. H. E. Müller and O. Borisov, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, pp. 29–89. DOI:10.1007/12_2010_57.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS PubMed.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- R. Hoogenboom, in Smart Polymers and their Applications, ed. M. R. Aguilar and J. San Román, Woodhead Publishing, 2nd edn, 2019, pp. 13–44. DOI:10.1016/B978-0-08-102416-4.00002-8.
- C. Zhao, Z. Ma and X. X. Zhu, Prog. Polym. Sci., 2019, 90, 269–291 CrossRef CAS.
- X. Chen, J. Zhang, K. Wu, X. Wu, J. Tang, S. Cui, D. Cao, R. Liu, C. Peng, L. Yu and J. Ding, Small Methods, 2020, 4, 2000310 CrossRef CAS.
- P. Gheysoori, A. Paydayesh, M. Jafari and H. Peidayesh, Eur. Polym. J., 2023, 186, 111846 CrossRef CAS.
- D. Iohara, M. Okubo, M. Anraku, S. Uramatsu, T. Shimamoto, K. Uekama and F. Hirayama, Mol. Pharmaceutics, 2017, 14, 2740–2748 CrossRef CAS PubMed.
- S. Kim, K. Lee and C. Cha, J. Biomater. Sci., Polym. Ed., 2016, 27, 1698–1711 CrossRef CAS PubMed.
- J. Li, J. Leng, Y. Qu, L. Deng and J. Ren, Mater. Lett., 2014, 131, 5–8 CrossRef CAS.
- A. K. Bajpai, S. K. Shukla, S. Bhanu and S. Kankane, Prog. Polym. Sci., 2008, 33, 1088–1118 CrossRef CAS.
- G.-H. Hsiue, R.-W. Chang, C.-H. Wang and S.-H. Lee, Biomaterials, 2003, 24, 2423–2430 CrossRef CAS PubMed.
- A. Alexander, Ajazuddin, J. Khan, S. Saraf and S. Saraf, Eur. J. Pharm. Biopharm., 2014, 88, 575–585 CrossRef CAS PubMed.
- A. Alexander, Ajazuddin, J. Khan, S. Saraf and S. Saraf, J. Controlled Release, 2013, 172, 715–729 CrossRef CAS PubMed.
- M. Patenaude and T. Hoare, ACS Macro Lett., 2012, 1, 409–413 CrossRef CAS PubMed.
- Z. W. Cui, B. H. Lee, C. Pauken and B. L. Vernon, J. Biomed. Mater. Res., Part A, 2011, 98A, 159–166 CrossRef CAS PubMed.
- W. X. Wang, H. Liang, R. C. Al Ghanami, L. Hamilton, M. Fraylich, K. M. Shakesheff, B. Saunders and C. Alexander, Adv. Mater., 2009, 21, 1809–1813 CrossRef CAS.
- E. Henderson, B. H. Lee, Z. W. Cui, R. McLemore, T. A. Brandon and B. L. Vernon, J. Biomed. Mater. Res., Part A, 2009, 90A, 1186–1197 CrossRef CAS PubMed.
- S. Fu, G. Guo, C. Gong, S. Zeng, H. Liang, F. Luo, X. Zhang, X. Zhao, Y. Wei and Z. Qian, J. Phys. Chem. B, 2009, 113, 16518–16525 CrossRef CAS PubMed.
- M. Sponchioni, N. Manfredini, A. Zanoni, E. Scibona, M. Morbidelli and D. Moscatelli, ACS Biomater. Sci. Eng., 2020, 6, 5337–5345 CrossRef CAS PubMed.
- K. Nagase, M. Yamato, H. Kanazawa and T. Okano, Biomaterials, 2018, 153, 27–48 CrossRef CAS PubMed.
- T. Koriyama, Y. Takayama, C. Hisatsune, T. A. Asoh and A. Kikuchi, J. Biomater. Sci., Polym. Ed., 2017, 28, 900–912 CrossRef CAS PubMed.
- J. Kobayashi, M. Yamato and T. Okano, J. Biomater. Sci., Polym. Ed., 2017, 28, 939–957 CrossRef CAS PubMed.
- H. Takahashi, M. Nakayama, K. Itoga, M. Yamato and T. Okano, Biomacromolecules, 2011, 12, 1414–1418 CrossRef CAS PubMed.
- Q. Yu, Y. Zhang, H. Chen, F. Zhou, Z. Wu, H. Huang and J. L. Brash, Langmuir, 2010, 26, 8582–8588 CrossRef CAS PubMed.
- K. Uhlig, E. Wischerhoff, J. F. Lutz, A. Laschewsky, M. S. Jaeger, A. Lankenau and C. Duschl, Soft Matter, 2010, 6, 4262–4267 RSC.
- T. Chen, R. Ferris, J. Zhang, R. Ducker and S. Zauscher, Prog. Polym. Sci., 2010, 35, 94–112 CrossRef CAS.
- A. Mizutani, A. Kikuchi, M. Yamato, H. Kanazawa and T. Okano, Biomaterials, 2008, 29, 2073–2081 CrossRef CAS PubMed.
- P. M. Mendes, Chem. Soc. Rev., 2008, 37, 2512–2529 RSC.
- H. Hatakeyama, A. Kikuchi, M. Yamato and T. Okano, Biomaterials, 2007, 28, 3632–3643 CrossRef CAS PubMed.
- N. Nath and A. Chilkoti, Adv. Mater., 2002, 14, 1243–1247 CrossRef CAS.
- S. Forg, A. Karbacher, Z. Ye, X. Guo and R. von Klitzing, Langmuir, 2022, 38, 5275–5285 CrossRef CAS PubMed.
- K. J. Hogan and A. G. Mikos, Polymer, 2020, 211, 123063 CrossRef CAS.
- M. Sponchioni, U. Capasso Palmiero and D. Moscatelli, Mater. Sci. Eng., C, 2019, 102, 589–605 CrossRef CAS PubMed.
- M. H. Bhuiyan, A. N. Clarkson and M. A. Ali, Colloids Surf., B, 2023, 224, 113193 CrossRef CAS PubMed.
- S. Ashraf, H.-K. Park, H. Park and S.-H. Lee, Macromol. Res., 2016, 24, 297–304 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS PubMed.
- X. Wang, X. Qiu and C. Wu, Macromolecules, 1998, 31, 2972–2976 CrossRef CAS.
- F. Tanimoto, Y. Kitamura, T. Ono and H. Yoshizawa, ACS Appl. Mater. Interfaces, 2010, 2, 606–610 CrossRef CAS PubMed.
- S. Tang, M. Floy, R. Bhandari, M. Sunkara, A. J. Morris, T. D. Dziubla and J. Z. Hilt, ACS Omega, 2017, 2, 8723–8729 CrossRef CAS PubMed.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- B. Yıldız, B. Işık and M. Kış, Polymer, 2001, 42, 2521–2529 CrossRef.
- G. Fundueanu, M. Constantin, F. Bortolotti, P. Ascenzi, R. Cortesi and E. Menegatti, Macromol. Biosci., 2005, 5, 955–964 CrossRef CAS PubMed.
- Y. Huang, P. Yong, Y. Chen, Y. Gao, W. Xu, Y. Lv, L. Yang, R. L. Reis, R. P. Pirraco and J. Chen, RSC Adv., 2017, 7, 28711–28722 RSC.
- S. Senel, B. Isik-Yuruksoy, H. Cicek and A. Tuncel, J. Appl. Polym. Sci., 1997, 64, 1775–1784 CrossRef CAS.
- K. Nagase, A. Ota, T. Hirotani, S. Yamada, A. M. Akimoto and H. Kanazawa, Macromol. Rapid Commun., 2020, 41, 2000308 CrossRef CAS PubMed.
- Y. Nakayama, R. Okahashi, R. Iwai and K. Uchida, Langmuir, 2007, 23, 8206–8211 CrossRef CAS PubMed.
- Y. Maeda, T. Nakamura and I. Ikeda, Macromolecules, 2001, 34, 1391–1399 CrossRef CAS.
- D. Ito and K. Kubota, Macromolecules, 1997, 30, 7828–7834 CrossRef CAS.
- J. Xu, X. Jiang and S. Liu, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 60–69 CrossRef CAS.
- I. Idziak, D. Avoce, D. Lessard, D. Gravel and X. X. Zhu, Macromolecules, 1999, 32, 1260–1263 CrossRef CAS.
- M. Mertoglu, S. Garnier, A. Laschewsky, K. Skrabania and J. Storsberg, Polymer, 2005, 46, 7726–7740 CrossRef CAS.
- X. Huang, F. Du, R. Ju and Z. Li, Macromol. Rapid Commun., 2007, 28, 597–603 CrossRef CAS.
- Y. Akiyama, Y. Shinohara, Y. Hasegawa, A. Kikuchi and T. Okano, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5471–5482 CrossRef CAS.
- Y. Zou, D. E. Brooks and J. N. Kizhakkedathu, Macromolecules, 2008, 41, 5393–5405 CrossRef CAS.
- N. Badi, Prog. Polym. Sci., 2017, 66, 54–79 CrossRef CAS.
- X. Tao, K. Liu, W. Li and A. Zhang, Polymer, 2014, 55, 3672–3679 CrossRef CAS.
- L. Liu, W. Li, K. Liu, J. Yan, G. Hu and A. Zhang, Macromolecules, 2011, 44, 8614–8621 CrossRef CAS.
- J. F. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- S. Han, M. Hagiwara and T. Ishizone, Macromolecules, 2003, 36, 8312–8319 CrossRef CAS.
- T. Ishizone, A. Seki, M. Hagiwara, S. Han, H. Yokoyama, A. Oyane, A. Deffieux and S. Carlotti, Macromolecules, 2008, 41, 2963–2967 CrossRef CAS.
- J.-F. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- T. Cai, M. Marquez and Z. Hu, Langmuir, 2007, 23, 8663–8666 CrossRef CAS PubMed.
- C. Boyer, M. R. Whittaker, M. Luzon and T. P. Davis, Macromolecules, 2009, 42, 6917–6926 CrossRef CAS.
- Y. Guo, X. Dong, W. Ruan, Y. Shang and H. Liu, Colloid Polym. Sci., 2017, 295, 327–340 CrossRef CAS.
- J. F. Lutz and A. Hoth, Macromolecules, 2006, 39, 893–896 CrossRef CAS.
- B. Zhao, D. Li, F. Hua and D. R. Green, Macromolecules, 2005, 38, 9509–9517 CrossRef CAS.
- S. Aoshima, H. Oda and E. Kobayashi, J. Polym. Sci., Part A: Polym. Chem., 1992, 30, 2407–2413 CrossRef CAS.
- T. Bauer and C. Slugovc, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2098–2108 CrossRef CAS.
- T. Isono, K. Miyachi, Y. Satoh, S.-i. Sato, T. Kakuchi and T. Satoh, Polym. Chem., 2017, 8, 5698–5707 RSC.
- T. He, Y. Wang, L. Xu, X. Fu, A. Narumi, S.-i. Sato, X. Shen and T. Kakuchi, Polym. Chem., 2021, 12, 2580–2591 RSC.
- X. Jiang, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 318–324 CrossRef CAS.
- X. Jiang, E. B. Vogel, M. R. Smith and G. L. Baker, Macromolecules, 2008, 41, 1937–1944 CrossRef CAS.
- H. Shimomoto, K. Shimizu, C. Takeda, M. Kikuchi, T. Kudo, H. Mukai, T. Itoh, E. Ihara, N. Hoshikawa, A. Koiwai and N. Hasegawa, Polym. Chem., 2015, 6, 8124–8131 RSC.
- H. R. Allcock and G. K. Dudley, Macromolecules, 1996, 29, 1313–1319 CrossRef CAS.
- H. R. Allcock, S. R. Pucher, M. L. Turner and R. J. Fitzpatrick, Macromolecules, 1992, 25, 5573–5577 CrossRef CAS.
- C. Geng, S. Wang and H. Wang, Polymers, 2021, 13, 1813 CrossRef CAS PubMed.
- X. Fu, Y. Shen, W. Fu and Z. Li, Macromolecules, 2013, 46, 3753–3760 CrossRef CAS.
- Y. Haba, A. Harada, T. Takagishi and K. Kono, J. Am. Chem. Soc., 2004, 126, 12760–12761 CrossRef CAS PubMed.
- C. de las Heras Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- S. Hocine and M.-H. Li, Soft Matter, 2013, 9, 5839–5861 RSC.
- A. Crespo Colin, S. M. Cancho, R. G. Rubio and A. Compostizo, Phys. Chem. Chem. Phys., 1999, 1, 319–322 RSC.
- P. Alexandridis and T. Alan Hatton, Colloids Surf., A, 1995, 96, 1–46 CrossRef CAS.
- P. Alexandridis, J. F. Holzwarth and T. A. Hatton, Macromolecules, 1994, 27, 2414–2425 CrossRef CAS.
- K. Suwa, Y. Wada, Y. Kikunaga, K. Morishita, A. Kishida and M. Akashi, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1763–1768 CrossRef CAS.
- L. H. Gan, Y. Y. Gan and G. R. Deen, Macromolecules, 2000, 33, 7893–7897 CrossRef CAS.
- Y. Maeda, T. Nakamura and I. Ikeda, Macromolecules, 2002, 35, 217–222 CrossRef CAS.
- N. Vanparijs, L. Nuhn and B. G. De Geest, Chem. Soc. Rev., 2017, 46, 1193–1239 RSC.
- W. Z. Zhang, X. D. Chen, W. Luo, J. Yang, M. Q. Zhang and F. M. Zhu, Macromolecules, 2009, 42, 1720–1725 CrossRef CAS.
- H. Mori, H. Iwaya, A. Nagai and T. Endo, Chem. Commun., 2005, 4872–4874, 10.1039/b509212d.
- Z. L. Liu, J. W. Hu, J. P. Sun, G. P. He, Y. H. Li and G. W. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3573–3586 CrossRef CAS.
- N. A. Cortez-Lemus and A. Licea-Claverie, Prog. Polym. Sci., 2016, 53, 1–51 CrossRef CAS.
- A. C. W. Lau and C. Wu, Macromolecules, 1999, 32, 581–584 CrossRef CAS.
- L. Marsili, M. Dal Bo, G. Eisele, I. Donati, F. Berti and G. Toffoli, Polymers, 2021, 13, 2639 CrossRef CAS PubMed.
- C. Weber, C. R. Becer, R. Hoogenboom and U. S. Schubertt, Macromolecules, 2009, 42, 2965–2971 CrossRef CAS.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686–714 CrossRef CAS.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- U. Hiroshi and K. Shiro, Chem. Lett., 1992, 21, 1643–1646 CrossRef.
- M. M. Bloksma, R. M. Paulus, H. P. C. van Kuringen, F. van der Woerdt, H. M. L. Lambermont-Thijs, U. S. Schubert and R. Hoogenboom, Macromol. Rapid Commun., 2012, 33, 92–96 CrossRef CAS PubMed.
- X. Song, Y. Wen, J.-l. Zhu, F. Zhao, Z.-X. Zhang and J. Li, Biomacromolecules, 2016, 17, 3957–3963 CrossRef CAS PubMed.
- H. Liu, J. Zhang, X. Luo, N. Kong, L. Cui and J. Liu, Eur. Polym. J., 2013, 49, 2949–2960 CrossRef CAS.
- P. S. Stayton, T. Shimoboji, C. Long, A. Chilkoti, G. Ghen, J. M. Harris and A. S. Hoffman, Nature, 1995, 378, 472–474 CrossRef CAS PubMed.
- C. Li, C. Wang, Z. Ji, N. Jiang, W. Lin and D. Li, Eur. Polym. J., 2019, 113, 404–410 CrossRef CAS.
- X. Savelyeva and M. Marić, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2011–2024 CrossRef CAS.
- O. W. Webster, W. R. Hertler, D. Y. Sogah, W. B. Farnham and T. V. RajanBabu, J. Am. Chem. Soc., 1983, 105, 5706–5708 CrossRef CAS.
- K. Fuchise, Y. Chen, T. Satoh and T. Kakuchi, Polym. Chem., 2013, 4, 4278–4291 RSC.
- P. Zhang, P. She, J. He, Z. Xiang, Z. Li, Y. Cao and X. Zhang, React. Funct. Polym., 2019, 142, 128–133 CrossRef CAS.
- M. Teodorescu and K. Matyjaszewski, Macromolecules, 1999, 32, 4826–4831 CrossRef CAS.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- G. Moad, Polym. Chem., 2017, 8, 177–219 RSC.
- A. E. Smith, X. Xu and C. L. McCormick, Prog. Polym. Sci., 2010, 35, 45–93 CrossRef CAS.
- W. Wang, H. Liang, R. Cheikh Al Ghanami, L. Hamilton, M. Fraylich, K. M. Shakesheff, B. Saunders and C. Alexander, Adv. Mater., 2009, 21, 1809–1813 CrossRef CAS.
- G. Masci, L. Giacomelli and V. Crescenzi, Macromol. Rapid Commun., 2004, 25, 559–564 CrossRef CAS.
- P. Chmielarz, P. Krys, S. Park and K. Matyjaszewski, Polymer, 2015, 71, 143–147 CrossRef CAS.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 CrossRef CAS PubMed.
- J. R. Adams and S. K. Mallapragada, Macromol. Chem. Phys., 2013, 214, 1321–1325 CrossRef CAS.
- T. G. Ribelli, M. Fantin, J.-C. Daran, K. F. Augustine, R. Poli and K. Matyjaszewski, J. Am. Chem. Soc., 2018, 140, 1525–1534 CrossRef CAS PubMed.
- S. d. Á. Gonçalves and R. P. Vieira, React. Funct. Polym., 2020, 147, 104453 CrossRef.
- Y.-Y. Liu, Y.-B. Zhong, J.-K. Nan and W. Tian, Macromolecules, 2010, 43, 10221–10230 CrossRef CAS.
- Z. Gao, J. Liang, X. Tao, Y. Cui, T. Satoh, T. Kakuchi and Q. Duan, Macromol. Res., 2012, 20, 508–514 CrossRef CAS.
- Z.-X. Zhang, X. Liu, F. J. Xu, X. J. Loh, E.-T. Kang, K.-G. Neoh and J. Li, Macromolecules, 2008, 41, 5967–5970 CrossRef CAS.
- P. Pan, M. Fujita, W.-Y. Ooi, K. Sudesh, T. Takarada, A. Goto and M. Maeda, Langmuir, 2012, 28, 14347–14356 CrossRef CAS PubMed.
- M. Karayilan, K. C. McCleary-Petersen, M. O. B. Hamilton, L. Fu, K. Matyjaszewski, R. S. Glass, D. L. Lichtenberger and J. Pyun, Macromol. Rapid Commun., 2020, 41, 1900424 CrossRef CAS PubMed.
- C.-W. Chang, E. Bays, L. Tao, S. N. S. Alconcel and H. D. Maynard, Chem. Commun., 2009, 3580–3582, 10.1039/B904456F.
- D. Pissuwan, C. Boyer, K. Gunasekaran, T. P. Davis and V. Bulmus, Biomacromolecules, 2010, 11, 412–420 CrossRef CAS PubMed.
- B. D. Fairbanks, P. A. Gunatillake and L. Meagher, Adv. Drug Delivery Rev., 2015, 91, 141–152 CrossRef CAS PubMed.
- J. M. Spruell, B. A. Levy, A. Sutherland, W. R. Dichtel, J. Y. Cheng, J. F. Stoddart and A. Nelson, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 346–356 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- F. Ganachaud, M. J. Monteiro, R. G. Gilbert, M.-A. Dourges, S. H. Thang and E. Rizzardo, Macromolecules, 2000, 33, 6738–6745 CrossRef CAS.
- C. Schilli, M. G. Lanzendörfer and A. H. E. Müller, Macromolecules, 2002, 35, 6819–6827 CrossRef CAS.
- A. J. Convertine, N. Ayres, C. W. Scales, A. B. Lowe and C. L. McCormick, Biomacromolecules, 2004, 5, 1177–1180 CrossRef CAS PubMed.
- A. J. Convertine, B. S. Lokitz, Y. Vasileva, L. J. Myrick, C. W. Scales, A. B. Lowe and C. L. McCormick, Macromolecules, 2006, 39, 1724–1730 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- M. Morimura, S. Ida, M. Oyama, H. Takeshita and S. Kanaoka, Macromolecules, 2021, 54, 1732–1741 CrossRef CAS.
- R. Plummer, D. J. T. Hill and A. K. Whittaker, Macromolecules, 2006, 39, 8379–8388 CrossRef CAS.
- J. Xu, S. Luo, W. Shi and S. Liu, Langmuir, 2006, 22, 989–997 CrossRef CAS PubMed.
- C. Herfurth, A. Laschewsky, L. Noirez, B. von Lospichl and M. Gradzielski, Polymer, 2016, 107, 422–433 CrossRef CAS.
- M. Cao, H. Nie, Y. Hou, G. Han and W. Zhang, Polym. Chem., 2019, 10, 403–411 RSC.
- M. L. Allegrezza and D. Konkolewicz, ACS Macro Lett., 2021, 10, 433–446 CrossRef CAS PubMed.
- S. Xu, F. J. Trujillo, J. Xu, C. Boyer and N. Corrigan, Macromol. Rapid Commun., 2021, 42, 2100212 CrossRef CAS PubMed.
- A. Sivokhin, D. Orekhov, O. Kazantsev, O. Sivokhina, S. Orekhov, D. Kamorin, K. Otopkova, M. Smirnov and R. Karpov, Polymers, 2022, 14, 137 CrossRef CAS PubMed.
- K. Nagase, T. Onuma, M. Yamato, N. Takeda and T. Okano, Macromol. Rapid Commun., 2015, 36, 1965–1970 CrossRef CAS PubMed.
- F. Liu, C.-H. Du, B.-K. Zhu and Y.-Y. Xu, Polymer, 2007, 48, 2910–2918 CrossRef CAS.
- Z. Zhang, A. J. Morse, S. P. Armes, A. L. Lewis, M. Geoghegan and G. J. Leggett, Langmuir, 2011, 27, 2514–2521 CrossRef CAS PubMed.
- A. Olivier, F. Meyer, J.-M. Raquez, P. Damman and P. Dubois, Prog. Polym. Sci., 2012, 37, 157–181 CrossRef CAS.
- G. Conzatti, S. Cavalie, C. Combes, J. Torrisani, N. Carrere and A. Tourrette, Colloids Surf., B, 2017, 151, 143–155 CrossRef CAS PubMed.
- B. Zdyrko and I. Luzinov, Macromol. Rapid Commun., 2011, 32, 859–869 CrossRef CAS PubMed.
- L. Wu, U. Glebe and A. Böker, Polym. Chem., 2015, 6, 5143–5184 RSC.
- R. Barbey, L. Lavanant, D. Paripovic, N. Schüwer, C. Sugnaux, S. Tugulu and H.-A. Klok, Chem. Rev., 2009, 109, 5437–5527 CrossRef CAS PubMed.
- P. Shivapooja, L. K. Ista, H. E. Canavan and G. P. Lopez, Biointerphases, 2012, 7, 32 CrossRef CAS PubMed.
- B. Lego, M. François, W. G. Skene and S. Giasson, Langmuir, 2009, 25, 5313–5321 CrossRef CAS PubMed.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14–22 RSC.
- M. Krishnamoorthy, S. Hakobyan, M. Ramstedt and J. E. Gautrot, Chem. Rev., 2014, 114, 10976–11026 CrossRef CAS PubMed.
- L. W. Teunissen, A. R. Kuzmyn, F. S. Ruggeri, M. M. J. Smulders and H. Zuilhof, Adv. Mater. Interfaces, 2022, 9, 2101717 CrossRef CAS.
- P.-F. Guo, H.-Y. Gong, H.-W. Zheng, M.-L. Chen, J.-H. Wang and L. Ye, Colloids Surf., B, 2020, 196, 111282 CrossRef CAS PubMed.
- A. Khabibullin, E. Mastan, K. Matyjaszewski and S. Zhu, in Controlled Radical Polymerization at and from Solid Surfaces, ed. P. Vana, Springer International Publishing, Cham, 2016, pp. 29–76. DOI:10.1007/12_2015_311.
- N. Matsuzaka, H. Takahashi, M. Nakayama, A. Kikuchi and T. Okano, J. Biomater. Sci., Polym. Ed., 2012, 23, 1301–1314 CrossRef CAS PubMed.
- A. B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32, 283–351 CrossRef CAS.
- R. D. Roeder, O. Garcia-Valdez, R. A. Whitney, P. Champagne and M. F. Cunningham, Polym. Chem., 2016, 7, 6383–6390 RSC.
- M. Li, M. Fromel, D. Ranaweera, S. Rocha, C. Boyer and C. W. Pester, ACS Macro Lett., 2019, 8, 374–380 CrossRef CAS PubMed.
- A. R. Kuzmyn, A. T. Nguyen, L. W. Teunissen, H. Zuilhof and J. Baggerman, Langmuir, 2020, 36, 4439–4446 CrossRef CAS PubMed.
- A. R. Kuzmyn, L. W. Teunissen, P. Fritz, B. van Lagen, M. M. J. Smulders and H. Zuilhof, Adv. Mater. Interfaces, 2022, 9, 2101784 CrossRef CAS.
- J. E. Krause, N. D. Brault, Y. Li, H. Xue, Y. Zhou and S. Jiang, Macromolecules, 2011, 44, 9213–9220 CrossRef CAS.
- H. Cheng, S. Xie, Y. Zhou, W. Huang, D. Yan, J. Yang and B. Ji, J. Phys. Chem. B, 2010, 114, 6291–6299 CrossRef CAS PubMed.
- W. Steinhauer, R. Hoogenboom, H. Keul and M. Moeller, Macromolecules, 2013, 46, 1447–1460 CrossRef CAS.
- W. Sun, Z. An and P. Wu, Macromol. Rapid Commun., 2017, 38, 1600808 CrossRef PubMed.
- N. Fechler, N. Badi, K. Schade, S. Pfeifer and J.-F. Lutz, Macromolecules, 2009, 42, 33–36 CrossRef CAS.
- N. Badi and J.-F. Lutz, J. Controlled Release, 2009, 140, 224–229 CrossRef CAS PubMed.
- W. Li, A. Zhang, Y. Chen, K. Feldman, H. Wu and A. D. Schlüter, Chem. Commun., 2008, 5948–5950, 10.1039/B814192D.
- L. Feng, Y. Liu, J. Hao, X. Li, C. Xiong and X. Deng, Macromol. Chem. Phys., 2011, 212, 2626–2632 CrossRef CAS.
- G.-F. Luo, W.-H. Chen and X.-Z. Zhang, ACS Macro Lett., 2020, 9, 872–881 CrossRef CAS PubMed.
- J. E. Chung, M. Yokoyama, M. Yamato, T. Aoyagi, Y. Sakurai and T. Okano, J. Controlled Release, 1999, 62, 115–127 CrossRef CAS PubMed.
- M. Arotçaréna, B. Heise, S. Ishaya and A. Laschewsky, J. Am. Chem. Soc., 2002, 124, 3787–3793 CrossRef PubMed.
- J. A. Jaber and J. B. Schlenoff, Macromolecules, 2005, 38, 1300–1306 CrossRef CAS.
- G. Fundueanu, M. Constantin, S. Bucatariu and P. Ascenzi, eXPRESS Polym. Lett., 2020, 14, 63–76 CrossRef CAS.
- M. L. Ohnsorg, J. M. Ting, S. D. Jones, S. Jung, F. S. Bates and T. M. Reineke, Polym. Chem., 2019, 10, 3469–3479 RSC.
- X. Lang, A. D. Patrick, B. Hammouda and M. J. A. Hore, Polymer, 2018, 145, 137–147 CrossRef CAS.
- Y. Xia, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2006, 39, 2275–2283 CrossRef CAS.
- N. Xue, X.-P. Qiu, Y. Chen, T. Satoh, T. Kakuchi and F. M. Winnik, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 2059–2068 CrossRef CAS.
- K. Zhu, R. Pamies, N. Al-Manasir, J. Ginés Hernández Cifre, J. García de la Torre, B. Nyström and A.-L. Kjøniksen, ChemPhysChem, 2020, 21, 1258–1271 CrossRef CAS PubMed.
- Y. Chen, N. Xiao, M. Fukuoka, K. Yoshida, Q. Duan, T. Satoh and T. Kakuchi, Polym. Chem., 2015, 6, 3608–3616 RSC.
- H. H. Nguyen, B. Payré, J. Fitremann, N. Lauth-de Viguerie and J.-D. Marty, Langmuir, 2015, 31, 4761–4768 CrossRef CAS PubMed.
- X.-P. Qiu, F. Tanaka and F. M. Winnik, Macromolecules, 2007, 40, 7069–7071 CrossRef CAS.
- J. Xu, J. Ye and S. Liu, Macromolecules, 2007, 40, 9103–9110 CrossRef CAS.
- C. K. Han and Y. H. Bae, Polymer, 1998, 39, 2809–2814 CrossRef CAS.
- A. Durand and D. Hourdet, Polymer, 1999, 40, 4941–4951 CrossRef CAS.
- H.-H. Lin and Y.-L. Cheng, Macromolecules, 2001, 34, 3710–3715 CrossRef CAS.
- C. Li, J. Madsen, S. P. Armes and A. L. Lewis, Angew. Chem., Int. Ed., 2006, 45, 3510–3513 CrossRef CAS PubMed.
- S. E. Kirkland, R. M. Hensarling, S. D. McConaughy, Y. Guo, W. L. Jarrett and C. L. McCormick, Biomacromolecules, 2008, 9, 481–486 CrossRef CAS PubMed.
- J. Li, S. Mizutani, S.-i. Sato, A. Narumi, O. Haba, S. Kawaguchi, M. Kikuchi, T. Kakuchi and X. Shen, Polym. Chem., 2020, 11, 2346–2359 RSC.
- R. Watanabe, K. Takaseki, M. Katsumata, D. Matsushita, D. Ida and M. Osa, Polym. J., 2016, 48, 621–628 CrossRef CAS.
- J. Li, S. Mizutani, S.-i. Sato, A. Narumi, O. Haba, S. Kawaguchi, T. Kakuchi and X. Shen, Polymer, 2020, 202, 122678 CrossRef CAS.
- P. Liu, H. Xie, H. Tang, G. Zhong and H. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3664–3673 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, Soft Matter, 2012, 8, 2737–2745 RSC.
- J. Li, S. Kikuchi, S.-i. Sato, Y. Chen, L. Xu, B. Song, Q. Duan, Y. Wang, T. Kakuchi and X. Shen, Macromolecules, 2019, 52, 7207–7217 CrossRef CAS.
- B. Jiang, L. Zhang, J. Yan, Q. Huang, B. Liao and H. Pang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2442–2453 CrossRef CAS.
- H. Yim, M. S. Kent, S. Mendez, G. P. Lopez, S. Satija and Y. Seo, Macromolecules, 2006, 39, 3420–3426 CrossRef CAS.
- K. N. Plunkett, X. Zhu, J. S. Moore and D. E. Leckband, Langmuir, 2006, 22, 4259–4266 CrossRef CAS PubMed.
- S. Kunugi, K. Takano, N. Tanaka, K. Suwa and M. Akashi, Macromolecules, 1997, 30, 4499–4501 CrossRef CAS.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS PubMed.
- X. Liu, F. Cheng, H. Liu and Y. Chen, Soft Matter, 2008, 4, 1991–1994 RSC.
- X.-Y. Liu, X.-R. Mu, Y. Liu, H.-J. Liu, Y. Chen, F. Cheng and S.-C. Jiang, Langmuir, 2012, 28, 4867–4876 CrossRef CAS PubMed.
- T. J. Murdoch, B. A. Humphreys, E. C. Johnson, G. B. Webber and E. J. Wanless, J. Colloid Interface Sci., 2018, 526, 429–450 CrossRef CAS PubMed.
- Ł. Otulakowski, M. Kasprów, A. Strzelecka, A. Dworak and B. Trzebicka, Polymers, 2021, 13, 90 CrossRef PubMed.
- X. Zhao, O. Coutelier, H. H. Nguyen, C. Delmas, M. Destarac and J.-D. Marty, Polym. Chem., 2015, 6, 5233–5243 RSC.
- C. Boutris, E. G. Chatzi and C. Kiparissides, Polymer, 1997, 38, 2567–2570 CrossRef CAS.
- X. Chang, H. Mao, G. Shan, Y. Bao and P. Pan, ACS Macro Lett., 2019, 8, 357–362 CrossRef CAS PubMed.
- L. Liu, W. Li, J. Yan and A. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1706–1713 CrossRef CAS.
- A. Li, J. Liu, G. Liu, J. Zhang and S. Feng, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 87–95 CrossRef CAS.
- M. Luzon, C. Boyer, C. Peinado, T. Corrales, M. Whittaker, L. Tao and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2783–2792 CrossRef CAS.
- K. Rahimian-Bajgiran, N. Chan, Q. Zhang, S. M. Noh, H.-i. Lee and J. K. Oh, Chem. Commun., 2013, 49, 807–809 RSC.
- D. Lipowska-Kur, R. Szweda, B. Trzebicka and A. Dworak, Eur. Polym. J., 2018, 109, 391–401 CrossRef CAS.
- N. P. Truong, J. F. Quinn, A. Anastasaki, M. Rolland, M. N. Vu, D. M. Haddleton, M. R. Whittaker and T. P. Davis, Polym. Chem., 2017, 8, 1353–1363 RSC.
- W. L. A. Brooks, G. Vancoillie, C. P. Kabb, R. Hoogenboom and B. S. Sumerlin, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2309–2317 CrossRef CAS.
- N. P. Truong, M. R. Whittaker, A. Anastasaki, D. M. Haddleton, J. F. Quinn and T. P. Davis, Polym. Chem., 2016, 7, 430–440 RSC.
- N. T. D. Tran, Z. Jia, N. P. Truong, M. A. Cooper and M. J. Monteiro, Biomacromolecules, 2013, 14, 3463–3471 CrossRef CAS PubMed.
- K. Kolouchová, V. Lobaz, H. Beneš, V. R. de la Rosa, D. Babuka, P. Švec, P. Černoch, M. Hrubý, R. Hoogenboom, P. Štěpánek and O. Groborz, Polym. Chem., 2021, 12, 5077–5084 RSC.
- E. Read, B. Lonetti, S. Gineste, A. T. Sutton, E. Di Cola, P. Castignolles, M. Gaborieau, A. F. Mingotaud, M. Destarac and J. D. Marty, J. Colloid Interface Sci., 2021, 590, 268–276 CrossRef CAS PubMed.
- J. Lyngsø, N. Al-Manasir, M. A. Behrens, K. Zhu, A.-L. Kjøniksen, B. Nyström and J. S. Pedersen, Macromolecules, 2015, 48, 2235–2243 CrossRef.
- J. Korpanty, L. R. Parent, N. Hampu, S. Weigand and N. C. Gianneschi, Nat. Commun., 2021, 12, 6568 CrossRef CAS PubMed.
- B. Wang, T. Xiao, X.-B. Fu, T.-T. Jiang, Y. Chen and Y.-F. Yao, Macromolecules, 2017, 50, 9647–9655 CrossRef CAS.
- C. Zhang, H. Peng, W. Li, L. Liu, S. Puttick, J. Reid, S. Bernardi, D. J. Searles, A. Zhang and A. K. Whittaker, Macromolecules, 2016, 49, 900–908 CrossRef CAS.
- X. Laloyaux, B. Mathy, B. Nysten and A. M. Jonas, Langmuir, 2010, 26, 838–847 CrossRef CAS PubMed.
- L. Li, Y. Zhu, B. Li and C. Gao, Langmuir, 2008, 24, 13632–13639 CrossRef CAS PubMed.
- V. P. Gilcreest, W. M. Carroll, Y. A. Rochev, I. Blute, K. A. Dawson and A. V. Gorelov, Langmuir, 2004, 20, 10138–10145 CrossRef CAS PubMed.
- H. Robertson, E. C. Johnson, I. J. Gresham, S. W. Prescott, A. Nelson, E. J. Wanless and G. B. Webber, J. Colloid Interface Sci., 2021, 586, 292–304 CrossRef CAS PubMed.
- D. Schmaljohann, M. Nitschke, R. Schulze, A. Eing, C. Werner and K.-J. Eichhorn, Langmuir, 2005, 21, 2317–2322 CrossRef CAS PubMed.
- A. Sivokhin, D. Orekhov, O. Kazantsev, O. Sivokhina, S. Orekhov, D. Kamorin, K. Otopkova, M. Smirnov and R. Karpov, Polymers, 2022, 14, 137 CrossRef CAS PubMed.
- L. Ma, G. Wang, S. Sun and P. Wu, Phys. Chem. Chem. Phys., 2017, 19, 22263–22271 RSC.
- F. Käfer, M. Pretscher and S. Agarwal, Macromol. Rapid Commun., 2018, 39, 1800640 CrossRef PubMed.
- F. Käfer, A. Lerch and S. Agarwal, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 274–279 CrossRef.
- A. Espinha, M. C. Serrano, Á. Blanco and C. López, Adv. Opt. Mater., 2014, 2, 516–521 CrossRef CAS.
- J. Akimoto, Y. Ito, T. Okano and M. Nakayama, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1695–1704 CrossRef CAS.
- S. Arias, F. Freire, M. Calderón and J. Bergueiro, Angew. Chem., Int. Ed., 2017, 56, 11420–11425 CrossRef CAS PubMed.
- L. Xiang, X. Liu, H. Zhang, N. Zhao and K. Zhang, Polym. Chem., 2020, 11, 6157–6162 RSC.
- Kiran, R. Koyilapu, R. Tiwari, S. Krishnamoorthi and K. Kumar, Global Chall., 2020, 4, 1900089 CrossRef CAS PubMed.
- A. Rank, S. Hauschild, S. Förster and R. Schubert, Langmuir, 2009, 25, 1337–1344 CrossRef CAS PubMed.
- N. Oleszko, W. Wałach, A. Utrata-Wesołek, A. Kowalczuk, B. Trzebicka, A. Klama-Baryła, D. Hoff-Lenczewska, M. Kawecki, M. Lesiak, A. L. Sieroń and A. Dworak, Biomacromolecules, 2015, 16, 2805–2813 CrossRef CAS PubMed.
- D. Healy, M. E. Nash, A. Gorelov, K. Thompson, P. Dockery, S. Beloshapkin and Y. Rochev, Macromol. Biosci., 2017, 17, 1600175 CrossRef PubMed.
- S. A. Jadhav, I. Miletto, V. Brunella, G. Berlier and D. Scalarone, Polym. Adv. Technol., 2015, 26, 1070–1075 CrossRef CAS.
- M. A. da Silva, P. Haddow, S. B. Kirton, W. J. McAuley, L. Porcar, C. A. Dreiss and M. T. Cook, Adv. Funct. Mater., 2022, 32, 2109010 CrossRef CAS.
- Z. Li, Z. Zhang, K. L. Liu, X. Ni and J. Li, Biomacromolecules, 2012, 13, 3977–3989 CrossRef CAS PubMed.
- J. Bassi da Silva, P. Haddow, M. L. Bruschi and M. T. Cook, J. Mol. Liq., 2022, 346, 117906 CrossRef CAS.
- Z. Osváth and B. Iván, Macromol. Chem. Phys., 2017, 218, 1600470 CrossRef.
- Q. Li, A. P. Constantinou and T. K. Georgiou, J. Polym. Sci., 2021, 59, 230–239 CrossRef CAS.
- C. M. Jeffries, J. Ilavsky, A. Martel, S. Hinrichs, A. Meyer, J. S. Pedersen, A. V. Sokolova and D. I. Svergun, Nat. Rev. Methods Primers, 2021, 1, 70 CrossRef CAS.
- P. S. Singh, in Membrane Characterization, ed. N. Hilal, A. F. Ismail, T. Matsuura and D. Oatley-Radcliffe, Elsevier, 2017, pp. 95–111. DOI:10.1016/B978-0-444-63776-5.00006-1.
- S. Wellert, A. Radulescu, A. Carl, R. von Klitzing and K. Gawlitza, Macromolecules, 2015, 48, 4901–4909 CrossRef CAS.
- K. Nishi, T. Hiroi, K. Hashimoto, K. Fujii, Y.-S. Han, T.-H. Kim, Y. Katsumoto and M. Shibayama, Macromolecules, 2013, 46, 6225–6232 CrossRef CAS.
- A. Papagiannopoulos, J. Zhao, G. Zhang, S. Pispas and A. Radulescu, Eur. Polym. J., 2014, 56, 59–68 CrossRef CAS.
- S. Salzinger, S. Huber, S. Jaksch, P. Busch, R. Jordan and C. M. Papadakis, Colloid Polym. Sci., 2012, 290, 385–400 CrossRef CAS.
- S. Jaksch, A. Schulz, K. Kyriakos, J. Zhang, I. Grillo, V. Pipich, R. Jordan and C. M. Papadakis, Colloid Polym. Sci., 2014, 292, 2413–2425 CrossRef CAS.
- I. Krakovský, L. Hanyková, G. Paladini and L. Almásy, Eur. Polym. J., 2020, 137, 109929 CrossRef.
- Q. Zhang, J.-D. Hong and R. Hoogenboom, Polym. Chem., 2013, 4, 4322–4325 RSC.
- M. Sahn, T. Yildirim, M. Dirauf, C. Weber, P. Sungur, S. Hoeppener and U. S. Schubert, Macromolecules, 2016, 49, 7257–7267 CrossRef CAS.
- B. Yu, A. B. Lowe and K. Ishihara, Biomacromolecules, 2009, 10, 950–958 CrossRef CAS PubMed.
- S. Jiang, Y. Yao, Q. Chen and Y. Chen, Macromolecules, 2013, 46, 9688–9697 CrossRef CAS.
- C. Xue, N. Yonet-Tanyeri, N. Brouette, M. Sferrazza, P. V. Braun and D. E. Leckband, Langmuir, 2011, 27, 8810–8818 CrossRef CAS PubMed.
- Y. Zou, N. A. A. Rossi, J. N. Kizhakkedathu and D. E. Brooks, Macromolecules, 2009, 42, 4817–4828 CrossRef CAS.
- Y. Stetsyshyn, J. Raczkowska, O. Lishchynskyi, A. Bernasik, A. Kostruba, K. Harhay, H. Ohar, M. M. Marzec and A. Budkowski, ACS Appl. Mater. Interfaces, 2017, 9, 12035–12045 CrossRef CAS PubMed.
- J. I. Kim, B. S. Lee, C. Chun, J.-K. Cho, S.-Y. Kim and S.-C. Song, Biomaterials, 2012, 33, 2251–2259 CrossRef CAS PubMed.
- P. Wang, J. He, P.-N. Wang and J.-Y. Chen, Photomed. Laser Surg., 2010, 28, 201–205 CrossRef CAS PubMed.
- E. E. Antunez, C. S. Mahon, Z. Tong, N. H. Voelcker and M. Müllner, Biomacromolecules, 2021, 22, 441–453 CrossRef CAS PubMed.
- T. Elshaarani, H. Yu, L. Wang, R. S. Ullah, S. Fahad, K. Ur Rahman, A. Khan, A. Nazir, M. Usman, R. U. Khan, F. Haq, R. Liang, X. Chen and M. Haroon, J. Mater. Sci., 2019, 54, 10009–10023 CrossRef CAS.
- Q. Mao, K. Liu, W. Li, J. Yan and A. Zhang, Polym. Chem., 2015, 6, 1300–1308 RSC.
- A. D'Emanuele and R. Dinarvand, Int. J. Pharm., 1995, 118, 237–242 CrossRef.
- S. K. Li and A. D'Emanuele, J. Controlled Release, 2001, 75, 55–67 CrossRef CAS PubMed.
- X.-Z. Zhang, R.-X. Zhuo, J.-Z. Cui and J.-T. Zhang, Int. J. Pharm., 2002, 235, 43–50 CrossRef CAS PubMed.
- S. Ilić-Stojanović, L. Nikolić, V. Nikolić, I. Ristić, S. Cakić and S. D. Petrović, Gels, 2023, 9, 70 CrossRef PubMed.
- R. Dinarvand and A. D'Emanuele, J. Controlled Release, 1995, 36, 221–227 CrossRef CAS.
- S. Nayak, D. Gan, M. J. Serpe and L. A. Lyon, Small, 2005, 1, 416–421 CrossRef CAS PubMed.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Biomacromolecules, 2009, 10, 1331–1336 CrossRef CAS PubMed.
- Q. Zhang, Z. Hou, B. Louage, D. Zhou, N. Vanparijs, B. G. De Geest and R. Hoogenboom, Angew. Chem., Int. Ed., 2015, 54, 10879–10883 CrossRef CAS PubMed.
- M. Hruby, C. Konak, J. Kucka, M. Vetrik, S. K. Filippov, D. Vetvicka, H. Mackova, G. Karlsson, K. Edwards, B. Rihova and K. Ulbrich, Macromol. Biosci., 2009, 9, 1016–1027 CrossRef CAS PubMed.
- Z. Li, S. Cho, I. C. Kwon, M. M. Janát-Amsbury and K. M. Huh, Carbohydr. Polym., 2013, 92, 2267–2275 CrossRef CAS PubMed.
- Retrieved from https://clinicaltrials.gov/, search term: Genexol-PM, accessed in July, 2023.
- M. Karayilan, L. Clamen and M. L. Becker, Biomacromolecules, 2021, 22, 223–261 CrossRef CAS PubMed.
- E. C. Abenojar, S. Wickramasinghe, M. Ju, S. Uppaluri, A. Klika, J. George, W. Barsoum, S. J. Frangiamore, C. A. Higuera-Rueda and A. C. S. Samia, ACS Infect. Dis., 2018, 4, 1246–1256 CrossRef CAS PubMed.
- S. Wickramasinghe, M. Ju, N. B. Milbrandt, Y. H. Tsai, M. Navarreto-Lugo, A. Visperas, A. Klika, W. Barsoum, C. A. Higuera-Rueda and A. C. S. Samia, ACS Appl. Nano Mater., 2020, 3, 5862–5873 CrossRef CAS.
- N. B. Milbrandt, Y. H. Tsai, K. Cui, C. S. Ngompe Massado, H. Jung, A. Visperas, A. Klika, N. Piuzzi, C. A. Higuera-Rueda and A. C. S. Samia, ACS Appl. Bio Mater., 2023, 6, 1231–1241 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- L. H. Lima, Y. Morales and T. Cabral, J. Ophthalmol., 2016, 2016, 5356371 Search PubMed.
- N. Bayat, Y. Zhang, P. Falabella, R. Menefee, J. J. Whalen, M. S. Humayun and M. E. Thompson, Sci. Transl. Med., 2017, 9, eaan3879 CrossRef PubMed.
- G.-H. Hsiue, S.-h. Hsu, C.-C. Yang, S.-H. Lee and I. K. Yang, Biomaterials, 2002, 23, 457–462 CrossRef CAS PubMed.
- F. S. Mah, J. Ocul. Pharmacol. Ther., 2016, 32, 396–399 CrossRef CAS PubMed.
- H. Cho, J. Gao and G. S. Kwon, J. Controlled Release, 2016, 240, 191–201 CrossRef CAS PubMed.
- N. L. Elstad and K. D. Fowers, Adv. Drug Delivery Rev., 2009, 61, 785–794 CrossRef CAS PubMed.
- S. Tanga, M. Aucamp and P. Ramburrun, Gels, 2023, 9, 418 CrossRef CAS PubMed.
- A. P. Constantinou and T. K. Georgiou, Polym. Int., 2021, 70, 1433–1448 CrossRef CAS.
- M. M. Allyn, R. H. Luo, E. B. Hellwarth and K. E. Swindle-Reilly, Front. Med., 2022, 8, 1–25 Search PubMed.
- E. Behravesh and A. G. Mikos, J. Biomed. Mater. Res., Part A, 2003, 66A, 698–706 CrossRef CAS PubMed.
- K. Nagase, J. Kobayashi, A. Kikuchi, Y. Akiyama, H. Kanazawa and T. Okano, Langmuir, 2007, 23, 9409–9415 CrossRef CAS PubMed.
- K. Nagase, J. Kobayashi and T. Okano, J. R. Soc., Interface, 2009, 6, S293–S309 CrossRef CAS PubMed.
- K. Nagase, M. Watanabe, A. Kikuchi, M. Yamato and T. Okano, Macromol. Biosci., 2011, 11, 400–409 CrossRef CAS PubMed.
- K. Nagase and T. Okano, J. Mater. Chem. B, 2016, 4, 6381–6397 RSC.
- K. Nagase and T. Okano, Polymer and Biopolymer Brushes, 2017, pp. 361–375. DOI:10.1002/9781119455042.ch12.
- H. Kanazawa, K. Yamamoto, Y. Matsushima, N. Takai, A. Kikuchi, Y. Sakurai and T. Okano, Anal. Chem., 1996, 68, 100–105 CrossRef CAS PubMed.
- S. Radtke, J. E. Adair, M. A. Giese, Y. Y. Chan, Z. K. Norgaard, M. Enstrom, K. G. Haworth, L. E. Schefter and H. P. Kiem, Sci. Transl. Med., 2017, 9, 1–22 Search PubMed.
- U. A. Gurkan, T. Anand, H. Tas, D. Elkan, A. Akay, H. O. Keles and U. Demirci, Lab Chip, 2011, 11, 3979–3989 RSC.
- U. A. Gurkan, S. Tasoglu, D. Akkaynak, O. Avci, S. Unluisler, S. Canikyan, N. MacCallum and U. Demirci, Adv. Healthcare Mater., 2012, 1, 661–668 CrossRef CAS PubMed.
- H. A. von Recum, S. W. Kim, A. Kikuchi, M. Okuhara, Y. Sakurai and T. Okano, J. Biomed. Mater. Res., 1998, 40, 631–639 CrossRef CAS PubMed.
- H.-W. Kang, Y. Tabata and Y. Ikada, Biomaterials, 1999, 20, 1339–1344 CrossRef CAS PubMed.
- H. Shin, P. Quinten Ruhé, A. G. Mikos and J. A. Jansen, Biomaterials, 2003, 24, 3201–3211 CrossRef CAS PubMed.
- T. Sun and G. Qing, Adv. Mater., 2011, 23, H57–H77 CrossRef CAS PubMed.
- R. Suntornnond, J. An and C. K. Chua, Macromol. Mater. Eng., 2017, 302, 1600266 CrossRef.
- I. K. Kwon and T. Matsuda, Biomaterials, 2006, 27, 986–995 CrossRef CAS PubMed.
- J. Zhong, H. Wang, K. Yang, H. Wang, C. Duan, N. Ni, L. An, Y. Luo, P. Zhao, Y. Gou, S. Sheng, D. Shi, C. Chen, W. Wagstaff, B. Hendren-Santiago, R. C. Haydon, H. H. Luu, R. R. Reid, S. H. Ho, G. A. Ameer, L. Shen, T.-C. He and J. Fan, Bioact. Mater., 2022, 9, 523–540 CrossRef CAS PubMed.
- M. D. Pawar, G. V. N. Rathna, S. Agrawal and B. S. Kuchekar, Mater. Sci. Eng., C, 2015, 48, 126–137 CrossRef CAS PubMed.
- Q. X. Ji, J. Deng, X. M. Xing, C. Q. Yuan, X. B. Yu, Q. C. Xu and J. Yue, Carbohydr. Polym., 2010, 82, 1153–1160 CrossRef CAS.
- I. Ishikawa, T. Iwata, K. Washio, T. Okano, T. Nagasawa, K. Iwasaki and T. Ando, Periodontol. 2000, 2009, 51, 220–238 CrossRef PubMed.
- T. Iwata, M. Yamato, H. Tsuchioka, R. Takagi, S. Mukobata, K. Washio, T. Okano and I. Ishikawa, Biomaterials, 2009, 30, 2716–2723 CrossRef CAS PubMed.
- K. Washio, T. Iwata, M. Mizutani, T. Ando, M. Yamato, T. Okano and I. Ishikawa, Cell Tissue Res., 2010, 341, 397–404 CrossRef PubMed.
- K. Nishida, M. Yamato, Y. Hayashida, K. Watanabe, N. Maeda, H. Watanabe, K. Yamamoto, S. Nagai, A. Kikuchi, Y. Tano and T. Okano, Transplantation, 2004, 77, 379–385 CrossRef PubMed.
- K. Nishida, M. Yamato, Y. Hayashida, K. Watanabe, K. Yamamoto, E. Adachi, S. Nagai, A. Kikuchi, N. Maeda, H. Watanabe, T. Okano and Y. Tano, N. Engl. J. Med., 2004, 351, 1187–1196 CrossRef CAS PubMed.
- T. Ide, K. Nishida, M. Yamato, T. Sumide, M. Utsumi, T. Nozaki, A. Kikuchi, T. Okano and Y. Tano, Biomaterials, 2006, 27, 607–614 CrossRef CAS PubMed.
- M. Khalili, A. Zarebkohan, H. Dianat-Moghadam, M. Panahi, H. Andre and E. Alizadeh, Chem. Eng. J., 2022, 429, 132523 CrossRef CAS.
Footnote |
| † These authors contributed to writing the original draft equally. |
| This journal is © The Royal Society of Chemistry 2023 |