
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRadiolabeling and in vivo evaluation of lanmodulin with biomedically relevant lanthanide isotopes†
Kirsten E.
Martin
a,
Joseph A.
Mattocks
 b,
Dariusz
Śmiłowicz
a,
Eduardo
Aluicio-Sarduy
cd,
Jennifer N.
Whetter
a,
Jonathan W.
Engle
cd,
Joseph A.
Cotruvo
Jr
*b and
Eszter
Boros
*a
b,
Dariusz
Śmiłowicz
a,
Eduardo
Aluicio-Sarduy
cd,
Jennifer N.
Whetter
a,
Jonathan W.
Engle
cd,
Joseph A.
Cotruvo
Jr
*b and
Eszter
Boros
*a
aDepartment of Chemistry, Stony Brook University, Stony Brook, New York 11794, USA. E-mail: eszter.boros@stonybrook.edu
bDepartment of Chemistry, The Pennsylvania State University, University Park, Pennsylvania 16802, USA. E-mail: juc96@psu.edu
cDepartment of Medical Physics, University of Wisconsin, Madison, Wisconsin 53705, USA
dDepartment of Radiology, University of Wisconsin, Madison, Wisconsin 53705, USA
First published on 13th April 2023
Abstract
Short-lived, radioactive lanthanides comprise an emerging class of radioisotopes attractive for biomedical imaging and therapy applications. To deliver such isotopes to target tissues, they must be appended to entities that target antigens overexpressed on the target cell's surface. However, the thermally sensitive nature of biomolecule-derived targeting vectors requires the incorporation of these isotopes without the use of denaturing temperatures or extreme pH conditions; chelating systems that can capture large radioisotopes under mild conditions are therefore highly desirable. Herein, we demonstrate the successful radiolabeling of the lanthanide-binding protein, lanmodulin (LanM), with medicinally relevant radioisotopes: 177Lu, 132/135La and 89Zr. Radiolabeling of the endogenous metal-binding sites of LanM, as well exogenous labeling of a protein-appended chelator, was successfully conducted at 25 °C and pH 7 with radiochemical yields ranging from 20–82%. The corresponding radiolabeled constructs possess good formulation stability in pH 7 MOPS buffer over 24 hours (>98%) in the presence of 2 equivalents of natLa carrier. In vivo experiments with [177Lu]-LanM, [132/135La]-LanM, and a prostate cancer targeting-vector linked conjugate, [132/135La]-LanM-PSMA, reveal that endogenously labeled constructs produce bone uptake in vivo. Exogenous, chelator-tag mediated radiolabeling to produce [89Zr]-DFO-LanM enables further study of the protein's in vivo behavior, demonstrating low bone and liver uptake, and renal clearance of the protein itself. While these results indicate that additional stabilization of LanM is required, this study establishes precedence for the radiochemical labeling of LanM with medically relevant lanthanide radioisotopes.
Introduction
Emerging radioactive lanthanide isotopes such as 177Lu (β−(max) = 501 keV, t1/2 = 6.64 d), 86Y (β+(max) = 4.2 MeV, t1/2 = 14.7 h), 161Tb (β−(max) = 593 keV, t1/2 = 6.90 d) and 132La (t1/2 = 4.59 h, 42.1% β+(max) = 3.67 MeV) offer promising avenues for the development of diagnostic and therapeutic radiopharmaceuticals.1 In order to selectively deliver these radionuclides to a target organ or tissue, they must be appended to a tissue/organ-specific targeting vector by way of a bifunctional chelator.2,3 Such chelators, once bound to the metal ion of interest, must exhibit high kinetic inertness to prevent isotope release prior to delivery. In many instances, and for most clinically used radiopharmaceuticals, this is achieved by use of macrocycle- or cryptand-based chelator systems that rigidify upon binding of the metal ion, forming exceptionally inert coordination complexes.4–7 However, the high thermodynamic stability of the corresponding chelate, combined with conformational changes required to form an in-cage chelate complex, often necessitates incubation at 80–100 °C for prolonged periods of time (1–2 hours).8–11 This poses no issue when the targeting vector is a small molecule or short peptide with good thermal stability. However, if biological targeting vectors such as monoclonal antibodies, antibody fragments, or affibodies are employed, the requirement for heating poses a significant challenge. This must be addressed by combining such targeting vectors with bifunctional chelators that form inert complexes at <40 °C with high radiochemical yield and purity – a challenge which remains pertinent, especially for lanthanide isotopes.12–14To date, few chelators have been employed to chelate large lanthanides under mild conditions, outlining a need for the development of such systems.15 Our group has previously investigated the chelation and targeted delivery of the 132La/135La pair.16 The 132La isotope is suitable for positron emission tomography (PET), while 135La (t1/2 = 18.9 h) decays by electron capture with the subsequent emission of 10.9 ± 3.2 high linear energy transfer (LET) Auger electrons per decay.17 These electrons travel less than a micron in tissue on average, suitable for targeted radiotherapy.18 Furthermore, due to the comparable size and chemical behavior to the Ac3+ ion, the La3+ ion and its isotopes are considered suitable surrogates to facilitate the development of 225Ac radiopharmaceuticals in light of the 225Ac isotope's scarcity and lack of imageable emissions.16,19 The 135La and 132La isotopes are produced concomitantly when natBa is irradiated with protons. Our previous work found that functionalized, tetra-azamacrocycle derived chelators required prolonged incubation at 80 °C to produce in cage formation, while the large cavity macrocycle macropa enabled room-temperature chelation but suffered from radiolytic degradation of the chelate-adjacent, peptide-linked thiourea bond in its functionalized state.16 Thus, alternative approaches, especially to 132/135La chelation of bioconjugates, remain elusive.
Lanmodulin (LanM) is a 12 kDa protein, produced by methylotrophic bacteria that preferentially acquires and utilizes the lighter, larger lanthanides for essential functions. LanM possesses three highly selective lanthanide-binding sites with average apparent dissociation constants (Kd) in the low picomolar range across the lanthanide series (at pH 7.2, ranging from 1.8 pM for La3+ to ∼5-fold higher for Ho3+ (the heaviest lanthanide for which the apparent Kd has been directly measured; the apparent Kd for Lu3+ is expected to be slightly weaker)).20–22 These apparent Kd values represent the two tightest, cooperative metal-binding sites (EF-hands 2 and 3); EF-hand 1 is ∼4-fold weaker in the case of La3+,20,23 and it is more susceptible to disruption by competing ligands.23–25 The protein itself is stable under extreme pH and thermally challenging conditions.20,26 LanM exhibits a large, cooperative, and highly selective conformational response to lanthanides that results in a shift from a disordered secondary structure to ∼60% alpha-helical character, a structure that retains the metal ions in their respective binding pockets with appropriate kinetic inertness.27 These properties have been leveraged for applications ranging from the sequestration and separation of rare earths from electronic waste24,26 to separation of trivalent actinides and lanthanides from di- or tetravalent contaminants on the macroscopic scale.28,29 Long-term cycling of LanM for the purification of 228Ac further demonstrated LanM's considerable resilience against radiolytic degradation.22 Additionally, through modification of metal-binding site, LanM can be tuned to possess enhanced selectivity for actinides over lanthanides (Cm vs. Eu).25
Based on the exceptional selectivity and resilience of LanM complexes with large lanthanides like La3+ against physiological competing chelators such as phosphate,20,22,26 achieved at biocompatible temperatures <40 °C, paired with stabilization of the metal-bound LanM species within a broad pH range, we hypothesized that the LanM protein could provide a suitable alternative to small molecular chelates with radiosensitive bifunctionalization handles for the radiolabeling and targeted delivery of biomedically relevant lanthanide isotopes. Herein, we demonstrate the endogenous and exogenous radiolabeling of LanM with 177Lu (endogenous), 132/135La (endogenous), and 89Zr (exogenous), as well as the synthesis of [132/135La]-LanM-PSMA, a LanM protein functionalized with a prostate cancer targeting peptide (Fig. 1). The corresponding radiolabeled constructs were probed for their in vitro, in vivo, and metabolic stabilities to determine the potential of LanM for the chelation and targeted delivery of the biomedical isotope 132/135La at tracer scale.
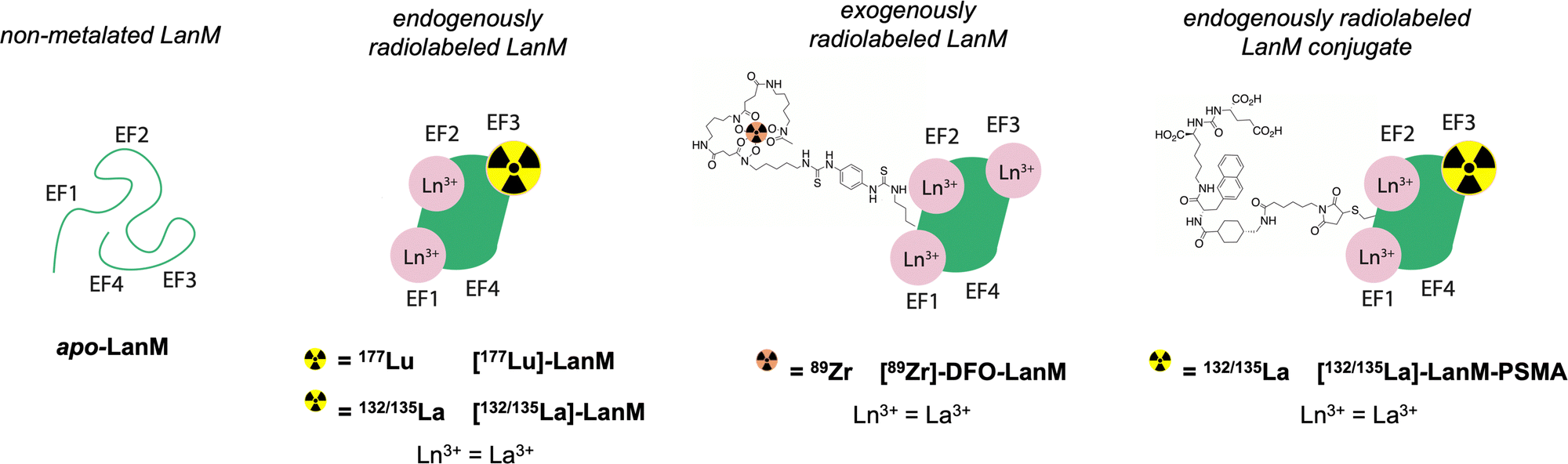 | ||
| Fig. 1 Schematic description of LanM variants subject of this work, including unfolded, non-metalated LanM, endogenously radiolabeled, folded LanM, exogenously radiolabeled, folded LanM and endogenously labelled LanM conjugated to a targeting peptide via modified, N-terminal cysteine-maleimide. Note: we are unable to localize incorporation of isotope to a specific site of EF1, EF2, and EF3 in the endogenously labeled LanM; schematic descriptions only include EF3 labelled for simplicity, in this and in subsequent figures. | ||
Experimental
Materials and methods
All chemicals were obtained from commercial suppliers in biochemical reagent grade or better and used without further purification. E. coli BL21(DE3) cells were obtained from New England Biolabs (NEB). NMR spectra (1H, 13C) were collected on a 700 MHz Advance III Bruker instrument at 25 °C and processed using TopSpin 4.0.7. Chemical shifts are reported as parts per million (ppm). Mass spectrometry of protein constructs was performed at the Proteomics and Mass Spectrometry Core Facility at Pennsylvania State University. The mass spectra were acquired on a Bruker Ultraflextreme MALDI TOF–TOF instrument using a factory-configured instrument method for the linear positive-ion detection over the 5000–20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m/z range. Laser power attenuation and pulsed ion extraction time were optimized to achieve the best signal. The instrument was calibrated with a five-protein mixture containing bovine insulin (MW 5733.6), bovine ubiquitin (MW 8564.8), horse heart cytochrome C (MW 12360.1), bovine ribonuclease A (MW 13682.3), and horse heart apomyoglobin (MW 16951.5). Mass spectra were smoothed (Savitzky–Golay, width 5 m/z, 1 cycle) and baseline subtracted (TopHat). Mass lists were generated using a centroid peak detection algorithm. High-resolution electrospray ionization (ESI) mass spectrometry of small molecules was carried out at the Stony Brook University Center for Advanced Study of Drug Action (CASDA) with a Bruker Impact II UHR QTOF MS system. Gel filtration chromatography was performed on an automated GE Healthcare Biosciences Akta Pure fast protein liquid chromatography system (FPLC). Preparative high performance liquid chromatography (HPLC) was carried out on a Phenomenex Luna C18 column (250 mm × 21.2 mm, 100 Å, AXIA packed) at a flow rate of 15 mL min−1 using a Shimadzu HPLC-20AR equipped with a binary gradient pump, UV-vis detector, and manual injector. UV absorption was recorded at 254 nm. Method A (Solvent A = H2O + 0.1% TFA and B = MeCN + 0.1% TFA): Gradient: 0–1 min: 5% B; 1–14 min: 5–50% B; 14–23 min: 50–95% B; 23–26 min: 95% B; 26–27 min: 95–5% B; 27–30 min: 5% B. Analytical HPLC was carried out on a Phenomenex Luna 5 μm C18 column (150 mm × 3 mm, 100 Å, AXIA packed) at a flow rate of 0.8 mL min−1 using a Shimadzu HPLC-20AR equipped with a binary gradient pump, UV-vis detector, autoinjector, and Laura radiodetector. Method B (Solvent A = H2O + 0.1% TFA and B = MeCN + 0.1% TFA): Gradient: 0–2 min: 5% B; 2–14 min: 5–95% B; 14–16 min: 95% B; 16–16.5 min: 95–5% B; 16.5–20 min 5% B. RadioHPLC analysis was carried out on the Shimadzu HPLC-20AR equipped with a binary gradient, pump, UV-Vis detector, autoinjector and a Laura radiodetector on a Phenomenex Yarra-2000 size exclusion column using an isocratic gradient (Method C: 3 μm, 145 Å, 150 × 7.8 mm; Solvent: 30 mM MOPS (3-(N-morpholino)propanesulfonic acid), 100 mM KCl, pH = 7.0; 0.75 mL min−1). Inductively coupled plasma optical emission spectroscopy (ICP-OES) was carried out using an Agilent 5110 inductively coupled plasma optical emission spectrometer. A 6-point standard curve with a check standard with respect to lutetium and lanthanum was used and fits were found to be at least R2 of 0.9999. Concentrations were then back calculated to the sample stock concentration. PET imaging was conducted using Siemens Inveon PET/CT Multimodality System and data reconstruction was done using ASIPro software and AMIDE. 177LuCl3 was obtained from the Department of Energy (DOE) isotope program, produced at the University of Missouri reactor. 132/135LaCl3 and 89Zr-oxalate were obtained from the Engle Lab at University of Wisconsin–Madison.
000 m/z range. Laser power attenuation and pulsed ion extraction time were optimized to achieve the best signal. The instrument was calibrated with a five-protein mixture containing bovine insulin (MW 5733.6), bovine ubiquitin (MW 8564.8), horse heart cytochrome C (MW 12360.1), bovine ribonuclease A (MW 13682.3), and horse heart apomyoglobin (MW 16951.5). Mass spectra were smoothed (Savitzky–Golay, width 5 m/z, 1 cycle) and baseline subtracted (TopHat). Mass lists were generated using a centroid peak detection algorithm. High-resolution electrospray ionization (ESI) mass spectrometry of small molecules was carried out at the Stony Brook University Center for Advanced Study of Drug Action (CASDA) with a Bruker Impact II UHR QTOF MS system. Gel filtration chromatography was performed on an automated GE Healthcare Biosciences Akta Pure fast protein liquid chromatography system (FPLC). Preparative high performance liquid chromatography (HPLC) was carried out on a Phenomenex Luna C18 column (250 mm × 21.2 mm, 100 Å, AXIA packed) at a flow rate of 15 mL min−1 using a Shimadzu HPLC-20AR equipped with a binary gradient pump, UV-vis detector, and manual injector. UV absorption was recorded at 254 nm. Method A (Solvent A = H2O + 0.1% TFA and B = MeCN + 0.1% TFA): Gradient: 0–1 min: 5% B; 1–14 min: 5–50% B; 14–23 min: 50–95% B; 23–26 min: 95% B; 26–27 min: 95–5% B; 27–30 min: 5% B. Analytical HPLC was carried out on a Phenomenex Luna 5 μm C18 column (150 mm × 3 mm, 100 Å, AXIA packed) at a flow rate of 0.8 mL min−1 using a Shimadzu HPLC-20AR equipped with a binary gradient pump, UV-vis detector, autoinjector, and Laura radiodetector. Method B (Solvent A = H2O + 0.1% TFA and B = MeCN + 0.1% TFA): Gradient: 0–2 min: 5% B; 2–14 min: 5–95% B; 14–16 min: 95% B; 16–16.5 min: 95–5% B; 16.5–20 min 5% B. RadioHPLC analysis was carried out on the Shimadzu HPLC-20AR equipped with a binary gradient, pump, UV-Vis detector, autoinjector and a Laura radiodetector on a Phenomenex Yarra-2000 size exclusion column using an isocratic gradient (Method C: 3 μm, 145 Å, 150 × 7.8 mm; Solvent: 30 mM MOPS (3-(N-morpholino)propanesulfonic acid), 100 mM KCl, pH = 7.0; 0.75 mL min−1). Inductively coupled plasma optical emission spectroscopy (ICP-OES) was carried out using an Agilent 5110 inductively coupled plasma optical emission spectrometer. A 6-point standard curve with a check standard with respect to lutetium and lanthanum was used and fits were found to be at least R2 of 0.9999. Concentrations were then back calculated to the sample stock concentration. PET imaging was conducted using Siemens Inveon PET/CT Multimodality System and data reconstruction was done using ASIPro software and AMIDE. 177LuCl3 was obtained from the Department of Energy (DOE) isotope program, produced at the University of Missouri reactor. 132/135LaCl3 and 89Zr-oxalate were obtained from the Engle Lab at University of Wisconsin–Madison.
Expression and purification of LanM variants
Wild type (WT) LanM was produced as described.30 Cys-LanM (WT-LanM with a Gly-Ser-Gly-Cys peptide added to the C-terminus) was purified as described.31 Briefly, plasmids containing genes encoding for each protein were obtained from TWIST Bioscience (pET-29b(+)-LanM). The proteins were grown in chemically competent E. coli BL21(DE3) cells (NEB), which were grown at 37 °C in Lysogeny broth (LB). Isopropyl β-D-1-thiogalactopyranoside (IPTG, 200 μM) was added at OD600nm = ∼0.6 to induce overexpression for 3 h at 37 °C. Cells were lysed by sonication and loaded to a gravity-flow 25 mL (2.5 × 5 cm) Q-Sepharose Fast Flow column and eluted using an 80 × 80 mL, 0.01–1 M NaCl gradient. Dithiothreitol (DTT, 10 mM) was included in these buffers for Cys-LanM purification to minimize thiol oxidation. LanM-containing fractions were identified by SDS-PAGE analysis and then purified using gel filtration chromatography (HiLoad 16/600 Superdex 75 pg). For Cys-LanM purifications, 5 mM DTT was included. LanM- or Cys-LanM-containing fractions were collected and concentrated to ∼2.0 mM. LanM was stored in 30 mM MOPS, 100 mM KCl, 5% glycerol, pH 7.0 and Cys-LanM was stored in the same buffer containing 5 mM DTT.
Chemical functionalization of LanM and Cys-LanM
000 ×g for 2 min to remove precipitate. The reaction product was confirmed in the remaining aqueous phase by mass spectrometry and subsequently purified using analytical-scale gel filtration chromatography (HiLoad 10/300 Superdex 75 pg, 1 mL loop, 0.75 mL min−1) using 30 mM MOPS, 100 mM KCl, 5% glycerol, pH 7.0 (no reductant). The protein conjugate eluted from the column over the course of 2 mL.
000 ×g for 2 min) to remove precipitate and concentrated to ∼400 μL using an Amicon Ultra 10 kDa molecular weight cup off (MWCO) centrifugal filter device. >50% incorporation of DFO-NCS was confirmed by mass spectrometry through formation of peaks at m/z values integer multiples of 752 above LanM base mass of 11686 Da (Fig. S5, ESI†).
Radiochemical labeling
:2 DPBS: Matrigel), on the right and left shoulders respectively. When tumors reached a suitable size (<200 mm3), mice were injected with the radiolabeled species of interest and biodistribution/metabolite analysis was conducted in accordance with biodistribution studies in naïve mice.
Results and discussion
Synthesis of LanM-PSMA and DFO-LanM conjugates
To enable targeting or exogenous labeling of LanM, we conducted chemical functionalization of LanM via the native N-terminus (and lysine sidechains) and Cys-LanM, a mutant incorporating a surface accessible cysteine at the C-terminus for facile bioconjugation.24 Previous work showed that the C-terminally Cys-modified LanM exhibits similar metal-binding properties to the wild-type protein.24To functionalize LanM with a disease-specific targeting vector, the protein was conjugated to a short, peptide-based targeting vector selective for the prostate specific membrane antigen (PSMA), a well-studied antigen overexpressed on the surface of prostate cancer cells.32–35 In addition to the well-established nature of the corresponding targeting vector's chemistry, a wealth of in vivo imaging and biodistribution data has been reported on PSMA-targeted constructs; therefore, previously published datasets serve as suitable means to evaluate the relative stability and performance of new PSMA-targeting entities. A maleimide-functionalized version of the small peptide targeting vector used in the clinically approved 177Lu-PSMA-617 was synthesized with manual solid phase peptide synthesis methods (Fig. S3, ESI†).36 With established maleimide–thiol coupling conditions, this maleimide linked-PSMA targeting vector was subsequently appended to Cys-LanM, to produce LanM-PSMA. Successful conjugation was affirmed by mass spectrometric analysis (Fig. S4, ESI†).
In addition to LanM-PSMA, we also proposed the synthesis of a LanM conjugate to selectively incorporate a radionuclide via exogenous radiolabeling of fully metalated LanM. An exogenously tagged construct can provide information on the protein's native pharmacokinetic behavior without concern of potential metal dissociation from the endogenous metal-binding sites. To this end, we employed desferrioxamine (DFO), a chelator with poor affinity for large, trivalent lanthanides but high affinity for the Zr4+ ion.37 Antibody-linked DFO is commonly employed for the chelation of the 89Zr PET isotope (β+(max) = 897 keV, t1/2 = 78 hours).38 In a similar manner to well-established antibody conjugation, DFO-NCS was appended to LanM via available and reactive lysine side chains (and/or the N-terminus) and formed the corresponding thiourea bond (Fig. S5, ESI†).39 Formation of the desired conjugate was confirmed using mass spectrometric analysis, revealing the formation of multiple LanM-DFO conjugates (LanM(DFO)n, n = 1–3).
Endogenous radiochemical labeling of Ln binding sites
To investigate the feasibility of radiolabeling LanM within its metal-binding sites, we probed radiochemical labeling with the largest and smallest lanthanides, La3+ and Lu3+, both of which possess readily available radioisotopes, 132/135La and 177Lu, respectively.Radiolabeling of LanM (10 nmol) was achieved with 177Lu at room temperature in 90 minutes using a low-specific-activity radiolabeling approach by addition of natLu. Initial attempts to radiolabel LanM at tracer concentration (<5 pmol) led to no significant incorporation of the isotope. We posited that at tracer levels, the binding of (statistically) no more than one metal ion per LanM protein did not allow for a cooperative conformational change, which has been documented to stabilize the metal–protein complex.20,23 Therefore, we proceeded with a cold-carrier-mediated radiolabeling approach.
To probe the optimal quantity of non-radioactive metal necessary to maximize radiolabeling yields, apo-LanM was incubated with 1–4 equivalents of natLu (cold carrier) relative to protein quantity in presence of 177Lu (0.1 mCi). Two equivalents of carrier natLu produced the most labeled protein (Fig. S7, ESI†) according to radioHPLC size exclusion chromatography (SEC) analysis (Fig. 2A). After radiolabeling, unbound metal was separated from [177Lu]-LanM using size exclusion chromatography to produce the purified, endogenously labeled construct in 75% isolated radiochemical yield (RCY). We also assessed the achievable radiochemical yield using natLa as the non-radioactive carrier, which also resulted in formation of the desired product, albeit with only 20% radiochemical labeling yield, possibly due to preferential incorporation of the larger La3+ ion over trace 177Lu3+. Formulation stability measurements over 24 hours indicated that natLa carrier resulted in good retention of radioactivity within the protein, whereas natLu resulted in release of the radioactive metal ion over the same time course (Fig. 2B and C). This result may be explained by Lu3+ being the lanthanide that is least efficient in inducing LanM's conformational change, and/or that formation of Lu hydroxides occurs over long periods of time because of the presence of two solvent molecules in the metal-binding sites.28
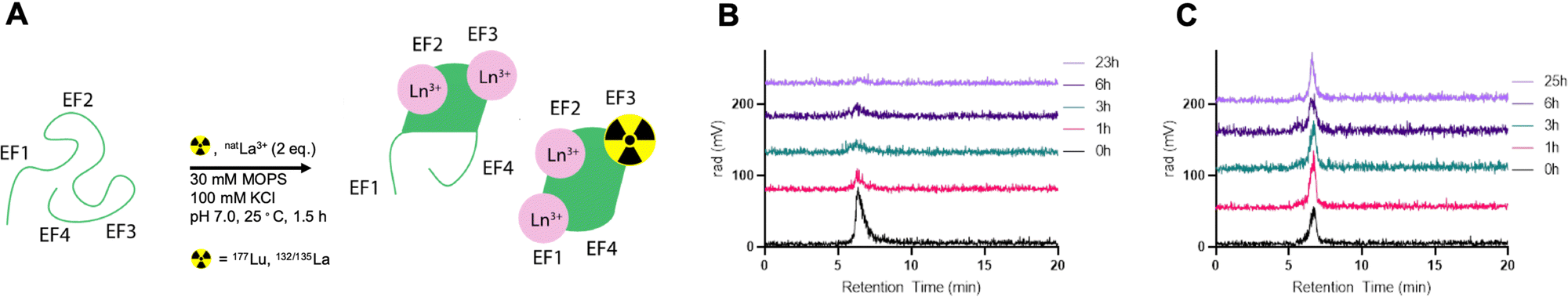 | ||
| Fig. 2 (A) Schematic description of carrier-added radiochemical labeling procedure for the introduction of 177Lu or 132/135La to the endogenous metal ion binding sites of LanM. (B) Formulation stability analysis using SEC-radioHPLC of [177Lu]-LanM using natLu as non-radioactive carrier shows the degradation of the LanM-associated peak over the course of the experiment while (C) shows retention of the 177Lu isotope within the endogenous LanM metal ion binding site with natLa as the non-radioactive carrier. | ||
Accordingly, radiolabeling of apo-LanM with 132/135La was conducted using 2 equivalents of natLa carrier and the crude reaction mixture was purified using size exclusion chromatography. The purified [132/135La]-LanM was obtained in 28% radiochemical yield. Both [177Lu]-LanM and [132/135La]-LanM with natLa as non-radioactive carrier exhibited excellent formulation stability in pH 7.4 MOPS buffer (compatible with in vivo injection) for 24 hours.
Exogenous radiochemical labeling
The exogenous radiochemical labeling of DFO-LanM (for synthesis and characterization vide supra) was conducted using DFO-LanM with fully occupied LanM metal binding sites using 3 equivalents of natLa. Radiolabeling with 89Zr was carried out without the addition of additional non-radioactive carrier to form [89Zr]-DFO-LanM at room temperature and pH 7, followed by separation and isolation of the product with an 83% radiochemical labeling yield using size exclusion chromatography (Fig. 3).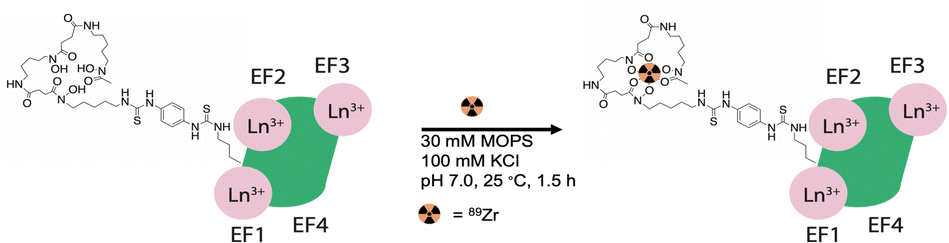 | ||
| Fig. 3 Schematic description of no carrier-added radiochemical labeling procedure for the introduction of 89Zr to the exogenous, DFO metal ion binding site of DFO-LanM. | ||
In vivo biodistribution in naïve mice
One of the most important evaluation criteria of new radiochemical labeling methods involves the determination of in vivo pharmacokinetics and stability of the corresponding labeled constructs. To this end, we administered the radiolabeled [132/135La]-LanM, [177Lu]-LanM and 89Zr-DFO-LanM intravenously to naïve, balb/C mice, followed by biodistribution and urine metabolite analysis 2 hours post injection. Additional cohorts received [177Lu]LuCl3, [132/135La]La-citrate or 89Zr-oxalate to determine the corresponding metal ion's native biodistribution. As expected and in accordance with previous studies, the administration of [177Lu]LuCl3 results in significant bone (8.30 ± 4.47% ID/g, Fig. 4A) and liver uptake (19.27 ± 0.69% ID/g, Fig. 4A). 177Lu-LanM demonstrates similar bone uptake (8.89 ± 3.15% ID g−1), indicative of likely dissociation of 177Lu from the metal binding sites within LanM during in vivo circulation. In contrast, in vivo administration of [132/135La]-LanM results in enhanced renal clearance and significant reduction of bone uptake (3.53 ± 0.49% ID g−1) when compared to results obtained with the free ion control [132/135La]La-citrate (9.07 ± 1.89% ID g−1). These results indicate a better retention of La3+, over the smaller Lu3+ (1.21 vs. 0.98 Å, CN = 8) in accordance with the protein's native selectivity and higher binding affinity for the former (Fig. 4B) and are also consistent with the greater stability of LanM complexes with larger Ln3+ ions against competitors, such as citrate, which is present in serum. Urine metabolite analysis using SEC-HPLC detected intact, radiometalated protein for both studies (Fig. 4D and E).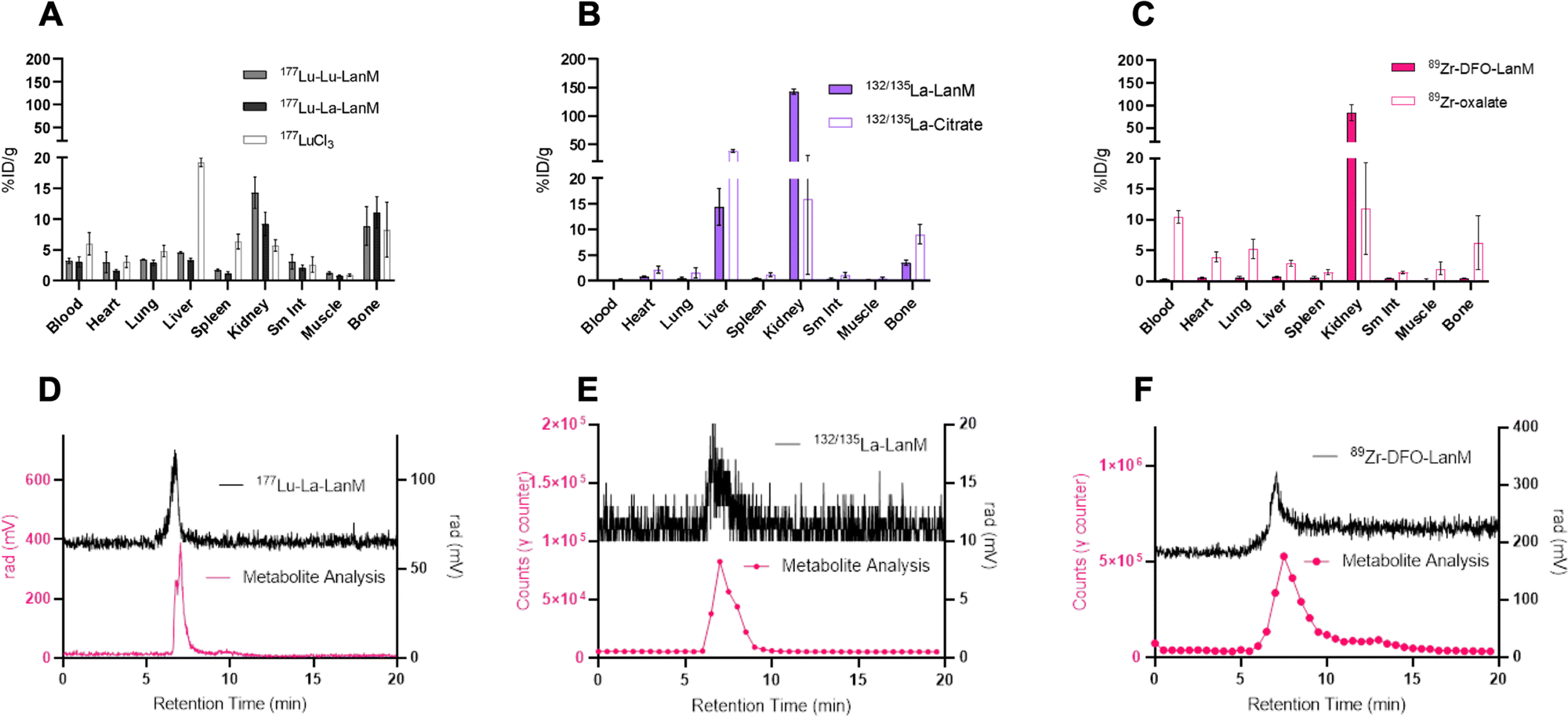 | ||
| Fig. 4 (A) Two-hour post-injection biodistribution analysis of [177Lu]-LanM using natLu (38 μCi nmol−1, 68–77 μCi administered per mouse, n = 3) or natLa (35 μCi nmol−1, 56–75 μCi administered per mouse, n = 4) as carrier in direct comparison with the distribution of 177LuCl3 as the free ion surrogate (n = 3). (B) Two-hour post-injection biodistribution analysis of [132/135La]-LanM (4 μCi nmol−1, 10–14 μCi administered per mouse, n = 3) with natLa as carrier in direct comparison with the distribution of 132/135La-citrate as the free ion surrogate (n = 3). (C) Two-hour post-injection biodistribution analysis of [89Zr]-DFO-LanM (40 μCi nmol−1, 25–40 μCi administered per mouse, n = 5) in direct comparison with the distribution of 89Zr-oxalate as the free ion surrogate (n = 3). (D) SEC-HPLC and urine metabolite analysis of [177Lu]-LanM using natLa as non-radioactive carrier (E) SEC-HPLC and urine metabolite analysis of [132/135La]-LanM. (F) SEC-HPLC and urine metabolite analysis of [89Zr]-DFO-LanM. | ||
To further assess the protein's native distribution in vivo, we conducted the biodistribution analysis of [89Zr]-DFO-LanM. [89Zr]-DFO-LanM resulted in low bone uptake (0.48 ± 0.07% ID/g) compared to the 89Zr4+-oxalate (6.23 ± 4.35% ID/g) and primarily kidney uptake, indicating renal clearance of [89Zr]-DFO-LanM (Fig. 4C). This behavior is comparable to that observed for similarly sized 89Zr-labeled Fab fragments or single-chain variable fragments (scFv's). [89Zr]-DFO-LanM did not result in significant accumulation in liver and bone tissues, indicating that in vivo dissociation of a fraction of 132/135La and, especially, 177Lu from endogenous labeling sites within LanM is likely. Urine metabolite analysis using SEC-HPLC also detected intact, radiometalated protein (Fig. 4F) for the [89Zr]-DFO-LanM species.
Imaging and biodistribution of LanM-PSMA in a mouse xenograft model
Radiolabeling of LanM-PSMA with 132/135La was conducted in accordance with the previously established, natLa3+ carrier-added method to afford [132/135La]-LanM-PSMA. We carried out cell binding experiments, to probe if [132/135La]-LanM-PSMA could retain binding to its target. PC3 PiP (PSMA+ cells) and PC3 Flu (PSMA− cells) were incubated with [132/135La]-LanM-PSMA for two hours.40,41 While significantly higher uptake was observed in PSMA+ cells when compared to PSMA− cells (1.97% and 1.00%, respectively, p < 0.0001, Fig. 5), overall uptake was reduced compared to the binding affinity observed for [68Ga]-PSMA-617 (∼16%).42 The source of this reduced affinity is possibly two-fold: decreased affinity to the target due to interference of the LanM protein with efficient binding to the cell surface antigen and/or the slow dissociation of the 132/135La isotope from the endogenous metal ion binding site over the course of the cell binding experiment.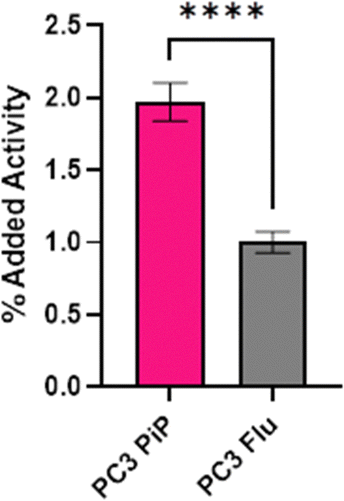 | ||
| Fig. 5 Cell binding [132/135La]-LanM-PSMA to PC3 PiP (PSMA+ cells) and PC3 Flu (PSMA− cells). | ||
As [132/135La]-LanM-PSMA showed enhanced uptake in PSMA-expressing cells, we proceeded to in vivo assessment in a bilateral xenograft model. [132/135La]-LanM-PSMA was administered to NCr athymic nude mice bearing PC3 PiP (PSMA+ cells) and PC3 Flu (PSMA− cells) xenografts on the left and right shoulders, respectively. Biodistribution analysis conducted at the 2 hour post injection time point shows localization of the activity in kidneys and bladder (Fig. S15, ESI†). [132/135La]-LanM-PSMA resulted in 97.74 ± 50.10% ID/g in the kidneys, in comparison with 9.86 ± 3.89% ID/g in the [132/135La]-citrate cohort, mirroring the protein's native renal clearance and indicating that some fraction of 132/135La is likely retained within the protein. However, this study further affirmed that in vivo circulation also resulted in release of 132/135La from the endogenous binding site, as significant high bone uptake for [132/135La]-LanM-PSMA, comparable to 132/135La-citrate (14.06 ± 5.13 and 17.93 ± 2.99% ID g−1, respectively, Fig. S15, ESI†) was observed. The accumulation in tumor tissue remained low and differences between PSMA+ and PSMA− tumors were not statistically significant. Urine metabolite analysis confirmed residual presence of endogenously labeled [132/135La]-LanM-PSMA (Fig. S16, ESI†). The increased deposition in bone for the LanM-PSMA conjugate relative to LanM itself may also indicate possible destabilization of protein folding with respect to the lanthanide-binding sites due to the presence of the PSMA-peptide.
Conclusions
In this study, we established protocols for the carrier-added, tracer-level radiolabeling of the lanthanide-binding protein, LanM, with medicinally relevant radioisotopes 177Lu, 132/135La, and 89Zr at room tempera ture using endogenous and exogenous, chemically introduced metal-binding sites. The resulting radiolabeled constructs were synthesized with 20-83% radiochemical yields and exhibited good in vitro stability. In vivo biodistribution and metabolite analysis studies revealed partial release of 132/135La and 177Lu from endogenous metal binding sites, resulting in accumulation in bone and liver tissues. 89Zr was efficiently retained by chelation to the exogenous DFO chelator tag, resulting in protein-mediated renal clearance.Taken together, our data indicate that LanM constructs can be efficiently radiolabeled under desirable, mild conditions and the LanM protein exhibits pharmacokinetics comparable to other small proteins such as antibody fragments, clearing predominantly renally. Future work will focus on improving the in vivo stability of endogenously radiolabeled LanM to prevent dissociation of the endogenously incorporated radioisotope in vivo. We anticipate that overall affinity, kinetic inertness, and selectivity for Lu3+ relative to La3+ could be tuned by a combination of site-directed mutagenesis and characterization of other naturally occurring LanMs that may have distinct properties from the M. extorquens protein that was the subject of the present study.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Brett Vaughn is acknowledged for help with animal experiments. K.E.M. acknowledges the SBU Chemistry-Biology interface training program (TM32GM136572). Tatiana Laremore is thanked for assistance with mass spectrometry experiments and analysis, which was performed at the Penn State Proteomics and Mass Spectrometry Core Facility, University Park, PA.References
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- B. M. Zeglis and J. S. Lewis, Dalton Trans., 2011, 40, 6168–6195 RSC.
- C. J. Anderson and M. J. Welch, Chem. Rev., 1999, 99, 2219–2234 CrossRef CAS PubMed.
- C. J. Anderson, F. Dehdashti, P. D. Cutler, S. W. Schwarz, R. Laforest, L. A. Bass, J. S. Lewis and D. W. McCarthy, J. Nucl. Med., 2001, 42, 213–221 CAS.
- N. M. Di Bartolo, A. M. Sargeson, T. M. Donlevy and S. V. Smith, J. Chem. Soc., Dalton Trans., 2001, 2303–2309, 10.1039/b103242a.
- J. L. J. Dearling, S. D. Voss, P. Dunning, E. Snay, F. Fahey, S. V. Smith, J. S. Huston, C. F. Meares, S. T. Treves and A. B. Packard, Nucl. Med. Biol., 2011, 38, 29–38 CrossRef CAS PubMed.
- R. Rossin, P. Renart Verkerk, S. M. van den Bosch, R. C. M. Vulders, I. Verel, J. Lub and M. S. Robillard, Angew. Chem., Int. Ed., 2010, 49, 3375–3378 CrossRef CAS PubMed.
- P. Adumeau, K. E. Carnazza, C. Brand, S. D. Carlin, T. Reiner, B. J. Agnew, J. S. Lewis and B. M. Zeglis, Theranostics, 2016, 6, 2267–2277 CrossRef CAS PubMed.
- J.-P. Meyer, J. L. Houghton, P. Kozlowski, D. Abdel-Atti, T. Reiner, N. V. K. Pillarsetty, W. W. Scholz, B. M. Zeglis and J. S. Lewis, Bioconjugate Chem., 2016, 27, 298–301 CrossRef CAS PubMed.
- S. M. J. van Duijnhoven, R. Rossin, S. M. van den Bosch, M. P. Wheatcroft, P. J. Hudson and M. S. Robillard, J. Nucl. Med., 2015, 56, 1422 CrossRef CAS PubMed.
- D. J. Vugts and G. A. M. S. Van Dongen, in Radiopharmaceutical Chemistry, ed. J. S. Lewis, A. D. Windhorst and B. M. Zeglis, Springer International Publishing, Cham, 2019, pp. 163–179 DOI:10.1007/978-3-319-98947-1_9.
- R. Chakravarty, S. Chakraborty, H. D. Sarma, K. V. V. Nair, A. Rajeswari and A. Dash, J. Labelled Compd. Radiopharm., 2016, 59, 354–363 CrossRef CAS PubMed.
- A. Majkowska-Pilip and A. Bilewicz, J. Inorg. Biochem., 2011, 105, 313–320 CrossRef CAS PubMed.
- A. Hu, E. Aluicio-Sarduy, V. Brown, S. N. MacMillan, K. V. Becker, T. E. Barnhart, V. Radchenko, C. F. Ramogida, J. W. Engle and J. J. Wilson, J. Am. Chem. Soc., 2021, 143, 10429–10440 CrossRef CAS PubMed.
- E. Aluicio-Sarduy, N. A. Thiele, K. E. Martin, B. A. Vaughn, J. Devaraj, A. P. Olson, T. E. Barnhart, J. J. Wilson, E. Boros and J. W. Engle, Chem. – Eur. J., 2020, 26, 1238–1242 CrossRef CAS PubMed.
- E. P. Abel, H. K. Clause, J. Fonslet, R. J. Nickles and G. W. Severin, Phys. Rev. C, 2018, 97, 034312 CrossRef CAS.
- D. Emfietzoglou and H. Nikjoo, Radiat. Res., 2007, 167, 110–120 CrossRef CAS PubMed.
- J. J. Wilson, E. R. Birnbaum, E. R. Batista, R. L. Martin and K. D. John, Inorg. Chem., 2014, 54, 97–109 CrossRef PubMed.
- J. A. Cotruvo Jr, E. R. Featherston, J. A. Mattocks, J. V. Ho and T. N. Laremore, J. Am. Chem. Soc., 2018, 140, 15056–15061 CrossRef PubMed.
- J. A. Mattocks, J. V. Ho and J. A. Cotruvo Jr, J. Am. Chem. Soc., 2019, 141, 2857–2861 CrossRef CAS PubMed.
- G. J.-P. Deblonde, J. A. Mattocks, Z. Dong, P. T. Wooddy, J. A. Cotruvo Jr and M. Zavarin, Sci. Adv., 2021, 7, eabk0273 CrossRef CAS PubMed.
- E. R. Featherston, E. J. Issertell and J. A. Cotruvo Jr, J. Am. Chem. Soc., 2021, 143, 14287–14299 CrossRef CAS PubMed.
- Z. Dong, J. A. Mattocks, G. J.-P. Deblonde, D. Hu, Y. Jiao, J. A. Cotruvo Jr and D. M. Park, ACS Cent. Sci., 2021, 7, 1798–1808 CrossRef CAS PubMed.
- J. A. Mattocks, J. A. Cotruvo and G. J.-P. Deblonde, Chem. Sci., 2022, 13, 6054–6066 RSC.
- G. J.-P. Deblonde, J. A. Mattocks, D. M. Park, D. W. Reed, J. A. Cotruvo Jr and Y. Jiao, Inorg. Chem., 2020, 59, 11855–11867 CrossRef CAS PubMed.
- E. C. Cook, E. R. Featherston, S. A. Showalter and J. A. Cotruvo Jr, Biochemistry, 2018, 58, 120–125 CrossRef PubMed.
- G. J.-P. Deblonde, J. A. Mattocks, H. Wang, E. M. Gale, A. B. Kersting, M. Zavarin and J. A. Cotruvo Jr, J. Am. Chem. Soc., 2021, 143, 15769–15783 CrossRef CAS PubMed.
- H. Singer, B. Drobot, C. Zeymer, R. Steudtner and L. J. Daumann, Chem. Sci., 2021, 12, 15581–15587 RSC.
- E. R. Featherston, J. A. Mattocks, J. L. Tirsch and J. A. Cotruvo, in Methods in Enzymology, ed. J. A. Cotruvo, Academic Press, 2021, vol. 650, pp. 119–157 Search PubMed.
- Z. Dong, J. A. Mattocks, G. J.-P. Deblonde, D. Hu, Y. Jiao, J. A. Cotruvo and D. M. Park, ACS Cent. Sci., 2021, 7, 1798–1808 CrossRef CAS PubMed.
- S. R. Banerjee, M. Pullambhatla, C. A. Foss, S. Nimmagadda, R. Ferdani, C. J. Anderson, R. C. Mease and M. G. Pomper, J. Med. Chem., 2014, 57, 2657–2669 CrossRef CAS PubMed.
- H. M. Shallal, I. Minn, S. R. Banerjee, A. Lisok, R. C. Mease and M. G. Pomper, Bioconjugate Chem., 2014, 25, 393–405 CrossRef CAS PubMed.
- J. N. Whetter, B. A. Vaughn, A. J. Koller and E. Boros, Angew. Chem., Int. Ed., 2022, 61, e202114203 CrossRef CAS PubMed.
- B. A. Vaughn, C. S. Loveless, S. J. Cingoranelli, D. Schlyer, S. E. Lapi and E. Boros, Mol. Pharmaceutics, 2021, 18, 4511–4519 CrossRef CAS PubMed.
- C. A. Umbricht, M. Benešová, R. M. Schmid, A. Türler, R. Schibli, N. P. van der Meulen and C. Müller, EJNMMI res., 2017, 7, 9 CrossRef PubMed.
- D. S. Abou, T. Ku and P. M. Smith-Jones, Nucl. Med. Biol., 2011, 38, 675–681 CrossRef CAS PubMed.
- J. P. Holland, V. Divilov, N. H. Bander, P. M. Smith-Jones, S. M. Larson and J. S. Lewis, J. Nucl. Med., 2010, 51, 1293–1300 CrossRef CAS PubMed.
- S. H. Ahn, B. A. Vaughn, W. A. Solis, M. L. Lupher, T. J. Hallam and E. Boros, Bioconjugate Chem., 2020, 1177–1187, DOI:10.1021/acs.bioconjchem.0c00100.
- R. S. Israeli, C. T. Powell, J. G. Corr, W. R. Fair and W. D. W. Heston, Cancer Res., 1994, 54, 1807–1811 CAS.
- D. A. Silver, I. Pellicer, W. R. Fair, W. D. W. Heston and C. Cordon-Cardo, Clin. Cancer Res., 1997, 3, 81–85 CAS.
- C. A. Umbricht, M. Benešová, R. M. Schmid, A. Türler, R. Schibli, N. P. van der Meulen and C. Müller, EJNMMI res., 2017, 7, 9 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00020f |
| This journal is © The Royal Society of Chemistry 2023 |