
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupport stabilized PtCu single-atom alloys for propane dehydrogenation†
Xiaohe
Liu
a,
Xianhui
Wang
a,
Shiyu
Zhen
a,
Guodong
Sun
a,
Chunlei
Pei
ab,
Zhi-Jian
Zhao
 ab and
Jinlong
Gong
*abcd
ab and
Jinlong
Gong
*abcd
aKey Laboratory for Green Chemical Technology of Ministry of Education, School of Chemical Engineering and Technology, Tianjin University, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, China. E-mail: jlgong@tju.edu.cn
bZhejiang Institute of Tianjin University Ningbo, Zhejiang 315201, P. R. China
cHaihe Laboratory of Sustainable Chemical Transformations, Tianjin 300192, China
dJoint School of National University of Singapore and Tianjin University, International Campus of Tianjin University, Binhai New City, Fuzhou 350207, China
First published on 22nd July 2022
Abstract
PtCu single-atom alloys (SAAs) open an extensive prospect for heterogeneous catalysis. However, as the host of SAAs, Cu suffers from severe sintering at elevated temperature, resulting in poor stability of catalysts. This paper describes the suppression of the agglomeration of Cu nanoparticles under high temperature conditions using copper phyllosilicate (CuSiO3) as the support of PtCu SAAs. Based on quasi in situ XPS, in situ CO-DRIFTS, in situ Raman spectroscopy and in situ XRD, we demonstrated that the interfacial Cu+–O–Si formed upon reduction at 680 °C serves as the adhesive between Cu nanoparticles and the silicon dioxide matrix, strengthening the metal–support interaction. Consequently, the resistance to sintering of PtCu SAAs was improved, leading to high catalytic stability during propane dehydrogenation without sacrificing conversion and selectivity. The optimized PtCu SAA catalyst achieved more than 42% propane conversion and 93% propylene selectivity at 580 °C for at least 30 hours. It paves a way for the design and development of highly active supported single-atom alloy catalysts with excellent thermal stability.
Introduction
Single-atom alloys (SAAs) with a unique geometry play a privileged role in heterogeneous catalysis due to the facile dissociation of target products and weak binding of reaction intermediates.1,2 However, the widespread application of SAAs in high-temperature reactions is limited by the well-known severe sintering of a metal host, such as copper.3 Propane dehydrogenation (PDH) is currently an instance of potential yet challenging chemistry, meeting the increasing cosmopolitan demand for propylene.4–6 On reviewing the development of Pt-based catalysts,7–9 introducing a second metal (M) to form Pt–M alloys, such as Pt–Sn,10–13 Pt–Zn,14–16 Pt–Fe,17 Pt–Cu,18,19 Pt–Ga20 and Pt–In,21 is a practical approach to improving the activity and selectivity, accompanied by reducing the Pt usage. Among them, the Pt–Cu alloy catalyst as has been extensively investigated as an effective candidate for PDH.19,22 Miller and co-workers pointed out that the propylene selectivity of Pt–Cu solid solution is improved with increasing Cu content due to the modification of energy levels of Pt 5d orbitals via surrounding Pt by inactive metallic Cu.23 By further increasing the Cu/Pt atom ratio to fully isolate Pt atoms on the surface layer of Cu, Sykes and co-workers first demonstrated how PtCu SAA catalysts can efficiently activate C–H bonds in alkanes.24 Due to the existence of the scaling relationship for Pt3M (M = 3d and 4d transition metals) in PDH, a low dehydrogenation barrier (i.e., high activity) normally accompanies with a high propylene desorption barrier and a low propylene dehydrogenation barrier (i.e., low selectivity). Indeed, PtCu SAAs were demonstrated to break the scaling relationship, with a great improvement in propylene selectivity and only a slight loss in intrinsic activity.25 Although the supported PtCu SAA exhibits impressive PDH activity and selectivity, it is susceptible to sintering due to the low Tammann temperature nature of copper, leading to severe deactivation.To tackle these challenges, enhancing the interaction between the metal and support can be a pivotal strategy to immobilize nanoparticles (NPs) on the support and control their spatial distribution.26 It has been proved that an enlarged interface between inert oxides (Al2O3 and SiO2) and copper NPs, resulting in the encapsulation of NPs by a thin layer of oxides to coat under-coordinated Cu atoms, profoundly promotes the stability of NPs and, consequently, the catalytic performance.27,28 Note that the synthesis process of catalyst precursors has a crucial influence on activated catalysts. Therefore, copper phyllosilicate (CuSiO3) with a lamella structure derived from the synthesis protocol of the ammonia evaporation hydrothermal method (AEM) can be an ideal support for the stabilization of Cu NPs.29 Moreover, the copper loading of copper phyllosilicate is adjustable to adapt a variety of catalytic reactions.30,31 Additionally, the post-synthetic heat treatment procedure is a simple but critical means to validly modify the interaction between copper and the support. On the one hand, suitable temperature treatment can weaken the strong metal–support interaction between Cu atoms and ceria to form Cu1O3 species leading to a greatly promoted catalytic activity.32 On the other hand, high temperature can effectively activate the metal–support interaction between Cu and inert oxide (SiO2).33 However, until now, the resistance-sintering role of CuSiO3 as a SAA support is still elusive.
Herein, thermally stable PtCu SAA catalysts were synthesized through the impregnation method with copper phyllosilicate as a support (denoted as 0.1Pt7CuSiO3 with 0.1 wt% Pt and 7 wt% Cu in the as-prepared catalyst which ensures that the Cu/Pt atomic ratio is more than 200. For the details of the synthesis protocol, please see the ESI†) followed by reduction at elevated temperatures beyond the reaction temperature. In situ X-ray diffraction (XRD) and transmission electron microscopy (TEM) presented conclusive experimental evidence for the sintering-resistance of PtCu SAAs supported on CuSiO3. As confirmed by quasi in situ X-ray photoelectron spectroscopy (XPS), in situ diffuse reflectance infrared spectroscopy with CO probe molecules (CO-DRIFTS) and in situ Raman spectroscopy, we defined Cu+–O–Si created at 680 °C as an adhesive of the Cu–SiO2 interface which significantly promotes the interaction between the alloy and support. The achieved PtCu SAAs with a highly dispersed and uniform size exhibited more than 42% propane conversion and 93% propylene selectivity at 580 °C for at least 30 hours, as a result of inhibiting the sintering of NPs.
Results and discussion
Sintering test of PtCu SAAs derived from two types of catalysts
Ostwald ripening plays an important role in the sintering of supported metal catalysts, which generally manifests as interparticle transport of Ostwald ripening species by diffusion.34,35 In order to prove the stabilization of PtCu SAAs supported on copper phyllosilicate, in situ XRD is employed to monitor the particle growth for 0.1Pt7CuSiO3 and 0.1Pt7Cu/SiO2-IM (prepared by co-impregnation using SiO2 as a support) at different reduction temperatures (Fig. S1†). The maximum test temperature is 680 °C, which excludes the effects of the propane dehydrogenation reaction on particle growth.As shown in the in situ XRD patterns (Fig. 1a), the developed 0.1Pt7CuSiO3 exhibited a completely different particle growth model compared with that of the 0.1Pt7Cu/SiO2-IM. For 0.1Pt7CuSiO3 sample, and a weak diffraction of Cu (111) can be detected at 380 °C due to the reduction and growth of Cu species. As 380 °C is close to the Tammann temperature, copper phyllosilicate cannot inhibit the growth of nanoclusters and/or ultra-small nanoparticles, but it can effectively inhibit the further growth of NPs, since Cu NPs remain stable as the temperature increases. The unique lamella structure which is derived from the synthesis protocol of AEM may be a factor for particle stabilization (Fig. S2†). Moreover, the crystallite size of copper was calculated by the X-ray broadening technique using Scherrer's equation (Fig. 1b). The particle size of Cu in 0.1Pt7CuSiO3 slowly increases from 7.9 to 8.9 nm by increasing the temperature from 380 to 680 °C. And the size of particles remained constant for 2 h at 680 °C without obvious growth. Note that the Pt loading is much lower than the detection limit of XRD, and no diffraction peak of Pt species can be detected.
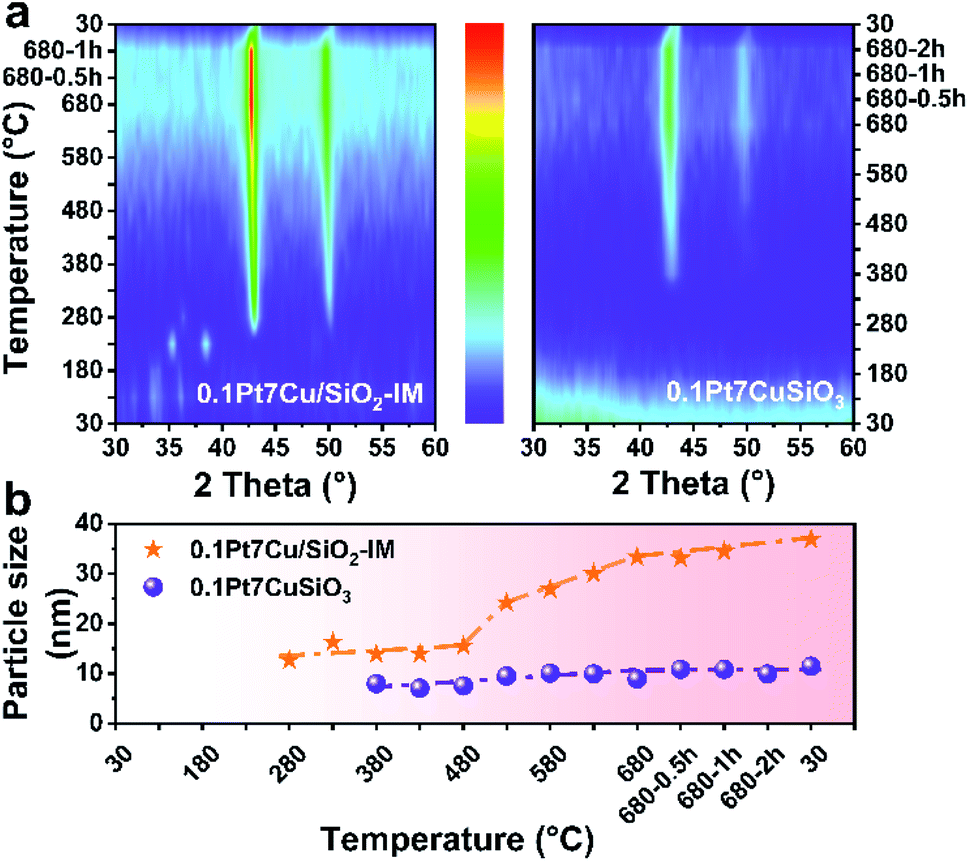 | ||
| Fig. 1 (a) In situ XRD patterns of 0.1Pt7Cu/SiO2-IM and 0.1Pt7CuSiO3 under a reductive atmosphere. (b) The evolution of the size of PtCu NPs versus temperature for reduction of 0.1Pt7Cu/SiO2-IM and 0.1Pt7CuSiO3 in 10 vol% H2/Ar. | ||
However, particle growth of the 0.1Pt7Cu/SiO2-IM sample was different. The CuO NPs, derived from the decomposition of copper nitrate hydrate (Cu2(OH)3NO3), was first observed at 230 °C. When the temperature was increased to 280 °C, accompanied by the intermediate phase of Cu2O, Cu NPs appeared with approximately 12.6 nm. Furthermore, the size of Cu NPs obviously increased from 15.5 nm to 33.3 nm when the reduction temperature exceeded 480 °C. The weak interaction between Cu and SiO2 cannot restrain the growth of Cu NPs.
The reducibility and copper species of 0.1Pt7CuSiO3 and 0.1Pt7Cu/SiO2-IM were identified by H2 temperature-programmed reduction (TPR) (Fig. S3†). The TPR profiles of 0.1Pt7CuSiO3 shows two hydrogen consumption peaks, a sharp main peak centered at ca. 191 °C and a broad weak peak centered at ca. 277 °C, which reveals the presence of highly uniform Cu species without agglomeration in accordance with the result of in situ XRD.36 Whereas, when 0.1Pt7Cu/SiO2-IM was subjected to H2-TPR, three peaks appeared at 175, 209, and 255 °C, respectively. We ascribe the first consumption peak at 175 °C to the decomposition of Cu2(OH)3NO3,37 which is confirmed by the result of in situ XRD. And the other peaks at 209 and 255 °C belong to CuO clusters and larger CuO nanoparticles, respectively, which indicates that Cu species of 0.1Pt7Cu/SiO2-IM is not homogeneously dispersed in the SiO2 matrix.
The difference in the particle size of Cu derived from two kinds of catalysts after reduction at 680 °C (donated as -680R) can be characterized by TEM. As shown in Fig. 2a and b, based on particle size distribution (PSD), the average particle size (APS) of 0.1Pt7Cu/SiO2-IM-680R is 28.0 nm, whereas the APS of 0.1Pt7CuSiO3-680R is 10.5 nm. Moreover, the 0.1Pt7CuSiO3 catalyst exhibits a lamella structure while the 0.1Pt7Cu/SiO2-IM catalyst does not (Fig. S2†). Consequently, the evidence extracted from in situ XRD and TEM provides information that the stronger interaction between Cu and SiO2 and unique lamella structure may play significant roles in restraining the growth of PtCu SAA in 0.1Pt7CuSiO3 with the increase in reduction temperature, compared with 0.1Pt7Cu/SiO2-IM. Furthermore, the TEM images of 0.1Pt7CuSiO3 after reduction at different temperatures and the corresponding PSDs are shown in Fig. S5.† The uniform size of PtCu NPs can be regulated from 8.7 nm at 580 °C to 10.5 nm at 680 °C and further to 14.2 nm at 780 °C. Additionally, detailed structural analysis of 0.1Pt7CuSiO3-680R shows the existence of a half-encapsulation interface between Cu and the SiO2 matrix (Fig. 2c and S6†). And the mapping of Cu species indicates that almost all Cu species from the framework of copper phyllosilicate had been transformed into metallic Cu NPs after high-temperature reduction (Fig. 2e). Whereas, the interface is missing in samples with low reduction temperature, such as 0.1Pt7CuSiO3-580R (Fig. S7†). By examining the EDS maps and line profiles as complemental evidence, the PtCu SAA was partially embedded in a support or/and the surface of the SAA was coated by SiO2 (Fig. 2c–g and S6†). This interaction between Cu and silica may be responsible for the stabilization of SAAs.
 | ||
| Fig. 2 Typical TEM images and particle size distributions of (a) 0.1Pt7Cu/SiO2-IM-680R and (b) 0.1Pt7CuSiO3-680R. (c) Representative TEM images of 0.1Pt7CuSiO3-680R. (d) Corresponding HAADF-STEM images including elemental maps of (e) Cu, (f) Si, and (g) O of 0.1Pt7CuSiO3-680R acquired by EDS. PtCu SAA is highlighted by the yellow dotted circle. | ||
Identification of the Pt single atom
The existence of isolated Pt atoms can be proved by AC-HAADF-STEM and CO-DRIFTS. As a result of the difference in Z-contrast between Pt and Cu, a Pt single-atom existing on the surface of Cu nanoparticles can be well distinguished.38Fig. 3a and b show that individual brighter Pt atoms are highly dispersed on the surface of Cu(111) and Cu(200) with different lattice spacings (0.21 nm for Cu(111) and 0.18 nm for Cu(200)). Moreover, the insets, exhibiting the atom arrangement on the surface of PtCu SAAs, show that the signal intensity of an individual Pt atom is stronger compared with the signal of a Cu atom, indicating the well diluted dispersion of Pt atoms in Cu NPs.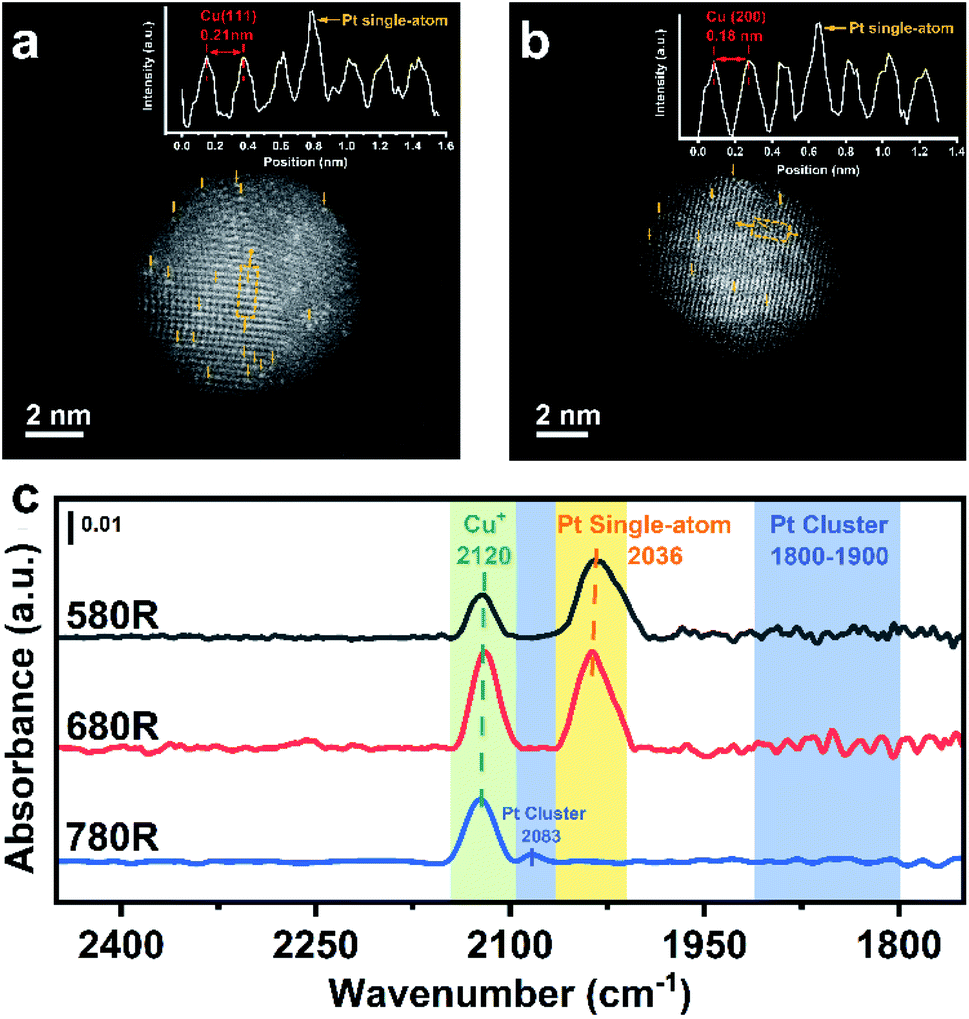 | ||
| Fig. 3 AC-HAADF-STEM images with typical regions of (a) Cu(111) and (b) Cu(200) in the 0.1Pt7CuSiO3-680R catalyst; the inset shows the atom arrangement on the surface of the PtCu SAA where the Pt atom exhibits stronger intensity compared with the Cu atom. Pt atoms were highlighted by yellow arrows. (c) In situ CO-DRIFTS for 0.1Pt7CuSiO3 catalysts with different reduction temperatures (reduction at 580 °C denoted as 580R, reduction at 680 °C denoted as 680R, and reduction at 780 °C denoted as 780R). | ||
According to previous literature,25,39–42in situ DRIFTS with CO as a molecule probe has been widely utilized to analyse the local environment of various metallic Pt species. In order to clearly distinguish the overlapping peaks between CO–Cu species and CO–Pt species, the spectra of the catalysts were recorded at 200 °C to make CO–Cu0 completely desorb at such temperature after purging in inert gas. Although the peaks of CO–Cu+ centered at 2120 cm−1 did not totally vanish due to CO σ-donation interaction being enhanced by Cu+ with lower electron density, the observation of Pt species is not affected. As shown in Fig. 3c, an apparent band centered at 2036 cm−1 appeared for 0.1Pt7CuSiO3-580R and 0.1Pt7CuSiO3-680R, which is identical to linearly bonded carbon monoxide on isolated Pt atoms in the metallic state.43 Whereas, for the 0.1Pt7CuSiO3-780R sample, this peak was missing and another peak centered at 2083 cm−1 appeared due to the aggregation of Pt atoms reduced at a temperature higher than the Tammann temperature of Pt (approximately 703 °C). Moreover, we failed to find the obvious absorption peak appearing at around 1800–1900 cm−1, which belongs to the bridge-adsorbed CO on two adjacent Pt atoms,44 for 0.1Pt7CuSiO3-580R and 0.1Pt7CuSiO3-680R, indicating the absence of dimers or clusters of Pt.
In situ derived Cu+–O–Si species
The interfacial structure between Cu and SiO2 can be further proved by in situ CO-DRIFTS. According to the previous report, both Cu+ and Cu0 appear at the interface between Cu NPs and SiO2 derived from copper phyllosilicate.45 Meanwhile, the CO adsorption characteristic peaks of different valence states of copper are distinguishable.46 As shown in the time-dependent desorption infrared spectra (Fig. S9†), the PtCu SAA catalysts with different reduction temperatures exhibited different adsorption intensities and curve configurations in the CO–Cu adsorption region (Fig. 4a and S10†). This indicates that the composition of surface Cu species varied with reduction temperature. The relative fractions of the Cu0 and Cu+ species on the 0.1Pt7CuSiO3 catalysts with different reduction temperatures were analysed through deconvolution of the recorded spectra using the Gaussian peak fitting method. Three bands that appeared at 2125–2113, 2105–2099, and 2093–2089 cm−1 are ascribed to CO absorption on Cu+ and Cu0 on step sites and Cu0 on terrace sites, respectively.39,47,48 Note that the top-absorbed CO stretching frequencies predicted by our DFT calculations matched with experimentally observed locations (Fig. S18 and Table S3†). Based on the analysis of the relative intensity ratio of Cu+ to Cu0, the percentage of the Cu+ site increased from 19.9 to 28.8% with the pre-treatment temperature increasing from 580 to 680 °C, indicating that the absolute number of Cu+ also improved (Fig. 4b). And then the percentage of Cu+ decreased to 17.7% by further increasing the temperature to 780 °C. At the same time, the relative intensity ratio of Cu0 on step sites significantly reduced with increasing temperature, which demonstrates that SiO2 maybe decorate the step or edge sites of Cu NPs.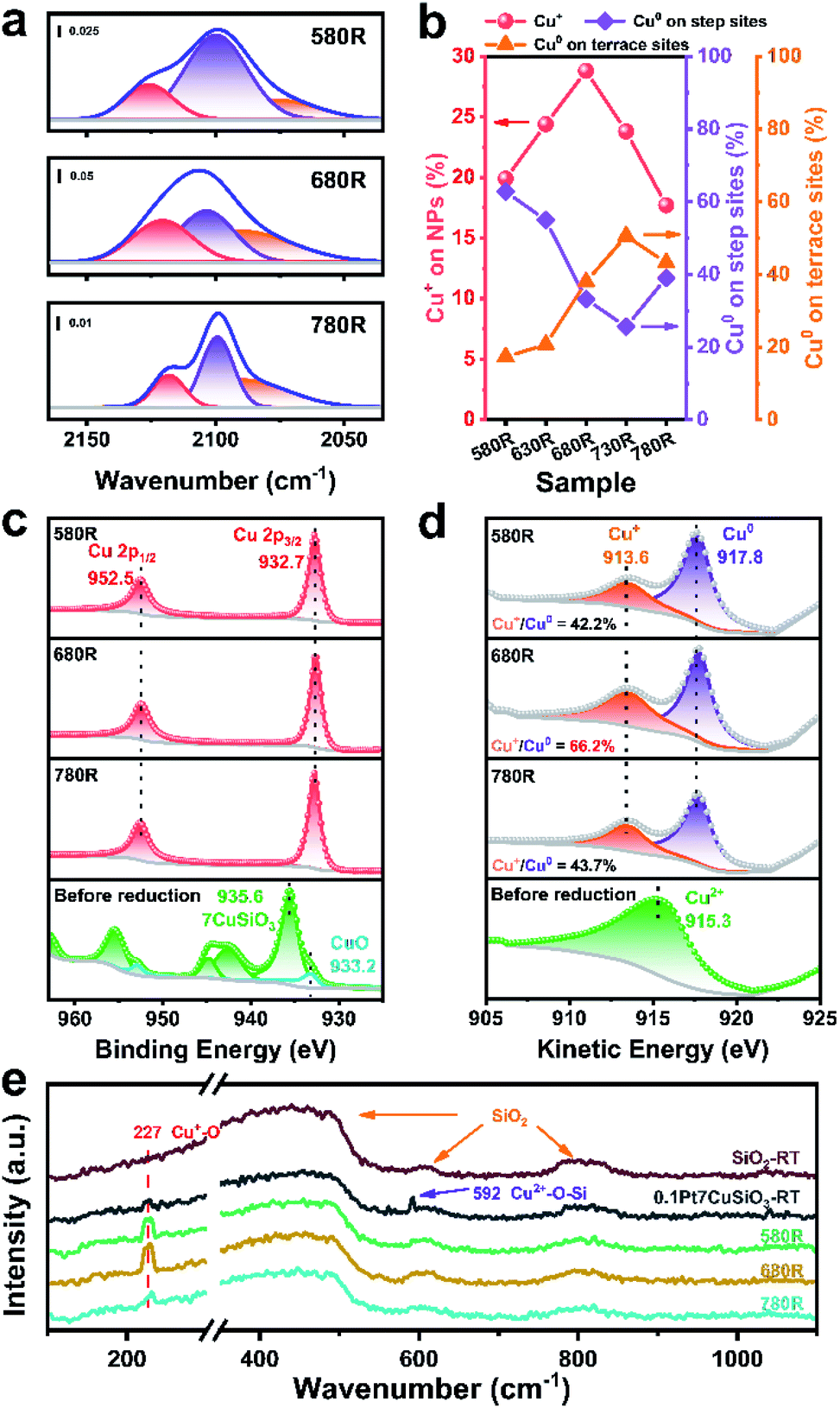 | ||
| Fig. 4 (a) CO-DRIFTS spectra of the 0.1Pt7CuSiO3 samples with different reduction temperatures. (b) Intensity evolution of the infrared bands that result from CO bound to surface Cu+ and Cu0 on step sites and Cu0 on terrace sites. Quasi in situ XPS of (c) Cu 2p and (d) Cu LMM spectra of reduced samples at different temperatures and those of the non-reduced sample. (e) Raman spectra of the pure SiO2 and 0.1Pt7CuSiO3 samples with different reduction temperatures. (RT stands for room temperature). | ||
The chemical states of Cu, Si and O in the 0.1Pt7CuSiO3 catalysts reduced at different temperatures were further investigated by quasi in situ X-ray photoelectron spectroscopy (Fig. 4c, d, S11 and S12†). The Cu 2p3/2 peak for the non-reduced sample centered at 935.6 eV and the Cu 2p1/2 peak located at 955.4 eV, accompanied by the 2p to 3d satellite peak between 940 and 945 eV, demonstrate that the Cu oxidation state is positively bivalent with a d9 electron configuration.30 Meanwhile, the shoulder peak appears in the Cu 2p3/2 envelope, indicating the existence of two different Cu species. Further, this peak can be deconvoluted into a main peak at 935.6 eV and a weak shoulder peak at 933.2 eV, corresponding to copper phyllosilicate and well dispersed CuO, respectively.45,49 It is also confirmed by the result of H2-TPR of 0.1Pt7CuSiO3.
After reducing at different temperatures, all of the Cu2+ was reduced to Cu+ or Cu0, as evidenced by the disappearance of the Cu 2p satellite peak and the emergence of the Cu 2p3/2 peak at 932.7 eV (Fig. 4c). However, Cu+ and Cu0 cannot be distinguished from Cu 2p spectra due to the overlap of the Cu+ and Cu0 peaks in this region. The X-ray induced Auger electron spectra (XAES) of the Cu LMM region was employed to differentiate Cu+, Cu0 as well as Cu2+.50,51 As shown in Fig. 4d, the kinetic energy (KE) peaks at 917.8 eV represented metallic Cu while the KE peaks at 913.6 eV belonged to Cu+,52 which means that Cu+ can survive under a reducing atmosphere at extremely elevated temperatures. Moreover, based on a semi-qualitative analysis of the XPS patterns, the Cu+/Cu0 ratio of the 0.1Pt7CuSiO3-680R sample is 66.2%, which is higher than that of 0.1Pt7CuSiO3-580R (42.2%) and 0.1Pt7CuSiO3-780R (43.7%), which is consistent with the CO-DRIFTS results. Meanwhile, the interfacial Si and O also exhibited the same tendency (Fig. S12†). Accordingly, combining the quasi in situ XPS result and in situ CO-DRIFTS, the Cu+ species existing in 0.1Pt7CuSiO3-680R created during the reduction at an elevated temperature plays a vital role in stabilizing PtCu SAA nanoparticles, presenting a half-encapsulation phenomenon of Cu NPs into the support. Moreover, the decreasing Cu/Si atomic ratios from the results of XPS has also proved the migration of SiO2 onto the surface of PtCu NPs with increasing temperature (Table S1†). But the half-encapsulation of Cu NPs by SiOx did not make a significant impact on the exposed surface of metallic Cu, which was confirmed by N2O surface oxidation experiments, likely due to the porousness of SiO2 coated on the upper surface of Cu NPs.
In situ Raman scattering experiments, which are a powerful tool for the characterization of Cu oxides at the nanoscale, were performed to further study the chemical structures of the surface Cu species in the support and catalysts (Fig. 4e and S20†). The Raman spectrum of pristine SiO2 presents three type of peaks, a strong and broad peak centered at 439 cm−1, the defect peaks at 486 (D1 peak) and 605 cm−1 (D2 peak), and an intrinsic peak centered at around 810 cm−1, which could be assigned to the symmetric stretching mode ω1 of SiO2, symmetric stretching modes of vibrationally isolated four-fold and three-fold rings of SiO2 tetrahedra, and intrinsic SiO2 bands (ω3 modes), respectively.53,54 After the introduction of Cu species through synthesis protocol of AEM, Cu species of the 0.1Pt7CuSiO3 catalyst precursor exhibited a distinctive Raman peak at 591 cm−1 rather than the peak of crystalline bulk CuO (630 cm−1), indicating the high dispersion of Cu species existing in a bivalent state which is in good consistence with the result of XPS.55 After reduction at different temperatures, the Raman peak of bivalent Cu species completely disappeared, accompanied by the appearance of the characteristic peak of Cu+–O species (227 cm−1) which belongs to the Raman allowed mode of the second-order overtone 2Γ12−.33,56 As shown in Fig. 4e, the intensity of the Raman peak of Cu+–O species was significantly enhanced with the pre-treatment temperature increasing from 580 to 680 °C, andby then decreased further increasing the temperature to 780 °C, which corresponds well with the much higher amounts of Cu+ in 0.1Pt7CuSiO3-680R than in 0.1Pt7CuSiO3-580R and 0.1Pt7CuSiO3-780R, detected by in situ CO-DRIFTS and quasi in situ XPS. As mentioned above, increasing amounts of Cu+–O–Si existing on the surface of Cu NPs maybe a key reason for particle stabilization.
Catalytic performance correlated with Cu+–O–Si
The promoting effect of Cu+–O–Si for NP stabilization was further proved in the propane dehydrogenation reaction (Fig. 5a, b and S16†). And the developed 0.1Pt7CuSiO3-680R catalyst exhibited a superior performance. Although 0.1Pt7CuSiO3-580R with the least average NP size (8.8 nm) exhibited high propane conversion at the beginning of the reaction (52% conv., at 5 min), the sintering tendency of Cu NPs seriously restricted the stability. As proved by the TME images, countable Cu NPs of the 0.1Pt7CuSiO3-580R catalyst used dramatically grew from 8.8 to 14.3 nm (Fig. S13 and S14†), suggesting that Cu+–O–Si formed during pre-treatment at 580 °C was insufficient to stabilize Cu NPs (Fig. S7†). Thus, one of deactivation origins of the 0.1Pt7CuSiO3-580R catalyst is the severe sintering of Cu nanoparticles. In addition, 0.1Pt7CuSiO3-780 exhibited a much inferior performance because of the significant sintering of Pt atoms and the inner diffusion of Pt into Cu NPs at such a high temperature beyond the Tammann temperature of Pt (749 °C). Conversely, 0.1Pt7CuSiO3-680R retained high propane conversion and excellent propylene selectivity (>42% conv., >93.6% sel.) for at least 30 h under the harsh reaction condition (Fig. 5c). Thus, the Cu+–O–Si species derived by unusual temperature significantly improved the stability of the PtCu SAA catalyst. After a 30 h long-term test, the 0.1Pt7CuSiO3-680R catalyst still featured well-dispersed NPs and the particle size slightly increased to 13.1 nm, as indicated by the TEM images of the used catalyst (Fig. S15†). At the same time, in situ thermogravimetric (TG) analysis was employed to monitor the formation of coke for 0.1Pt7CuSiO3 reduction at different temperatures during PDH (Fig. S19†). The amount of deposited coke of catalysts was obviously different, among which, coking is the main reason for the deactivation of the 0.1Pt7CuSiO3-780R catalyst due to the facile formation of a large amount of coke, which is a structure sensitive reaction, on Pt–Pt sites caused by aggregation of Pt atoms. Meanwhile, we realize that another deactivation origin of the 0.1Pt7CuSiO3-580R catalyst may be the formation of coke. Under the work condition with elevated temperature, the PtCu SAA nanoparticles of the 0.1Pt7CuSiO3-580R catalyst underwent severe sintering which perhaps caused the reconstitution of Pt species followed by the decrease of the selectivity as shown in Fig. 5a.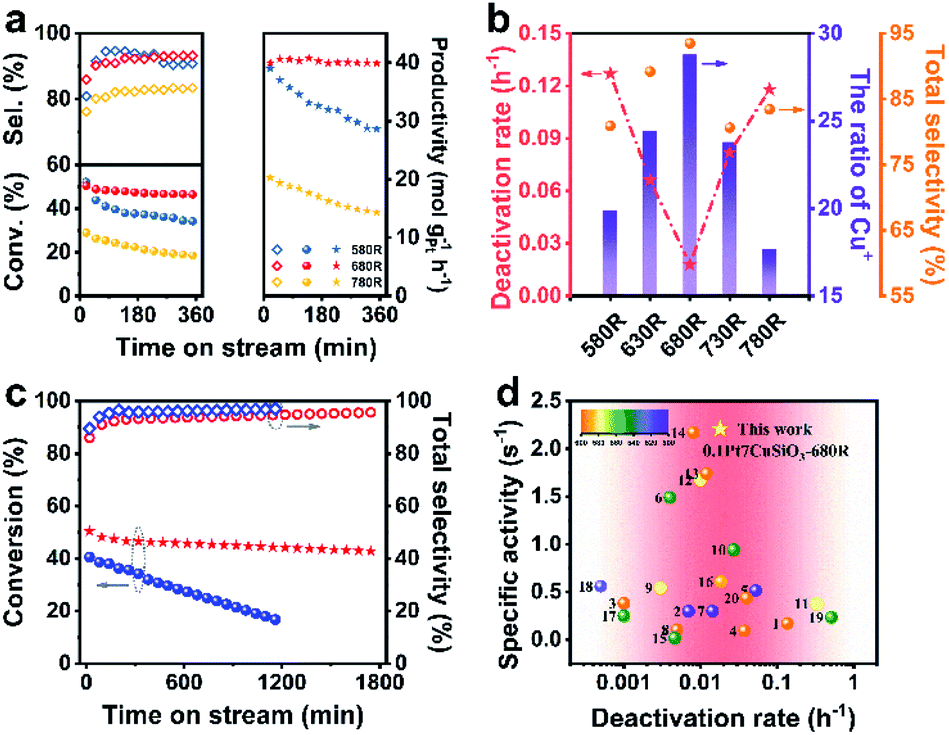 | ||
| Fig. 5 (a) The propane conversion (solid circle), propylene selectivity (hollow rhombus) and the formation rate of C3H6 (solid pentagram) as a function of reaction time over the prepared 0.1Pt7CuSiO3 reduced at 580, 680, and 780 °C for 1 h. (b) Reduction temperature effect of 0.1Pt7CuSiO3 on the ratio of Cu+, deactivation rates and total selectivity. (c) Stability test as a function of reaction time over 0.1Pt7CuSiO3-680R (red symbol) and 0.1Pt7Cu/SiO2-680R-IM (blue symbol). (d) Normalized activities of the formation rate of C3H6 by the specific content of Pt for the catalysts described in this work and in the literature (Table S4†). Catalytic conditions: atmospheric pressure, C3H8/H2 = 1/1, with N2 dilution, total flow rate 50 mL min−1, WHSV of propane = 4.7 h−1 and 200 mg of the sample. | ||
Moreover, to demonstrate the universality of the unusual temperature pre-treatment method, the 0.1Pt7Cu/SiO2-IM catalysts with different reduction temperatures were also tested (Fig. S17†). Interestingly, 0.1Pt7Cu/SiO2-IM-680R exhibits relative stability which is completely different from other reduction temperatures, suggesting that the Cu+–O–Si bond is significant for the stabilization of NPs. However, the long-term stability tests for 0.1Pt7CuSiO3-680R and 0.1Pt7Cu/SiO2-IM-680R catalysts demonstrate that the PtCu SAA derived from the copper phyllosilicate support can effectively stabilize Cu NPs, as the host of SAAs, under a high-temperature heterogeneous catalytic environment (Fig. 5c).
Conclusions
This paper describes an instance of rational design and fabrication of an anti-sintering PtCu SAA catalyst. In situ XRD and TEM show that copper phyllosilicate is an efficient support for limiting the mobilization of Cu NPs. After elevated temperature reduction (680 °C), the growth of Cu nanoparticles is inhibited by the derived Cu+–O–Si which is confirmed by in situ CO-DRIFTS, quasi in situ XPS and in situ Raman spectroscopy. A notable enhancement in the stabilization of PtCu SAAs is observed on propane dehydrogenation at an elevated temperature for at least 30 h without the loss of initial catalytic performance. Our discovery breaks the stereotyped impression that Cu, as the host of SAA catalysts, is severely deactivated upon sintering in a high temperature reaction. Especially, propane dehydrogenation, an extremely energy–intensive reaction under industry-related conditions, usually operates at 550–620 °C to break through the limitation of thermodynamics due to the high positive reaction enthalpy (ΔrH° = 124.3 kJ mol−1). Such a high temperature presents grand challenges for the selectivity of alkene and the stability of nanoscale catalysts. So, the exploitation of copper-host SAA catalysts to enhance sinter-resistance at elevated temperatures is valuable and expected. The concept of high-temperature endurable Cu-host single-atom alloy catalysts can be extended to the application of other energy intensive catalytic reactions which suffer from significant sintering.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Author contributions
J. G. conceived and coordinated the research. X. L. contributed to catalyst synthesis, catalytic experiments, characterization and wrote the draft. S. Z. conducted the density functional theory calculations. X. W., G. S., C. P. and Z. Z. analysed the data. X. W., G. S., Z. Z. and J. G. wrote the manuscript. All authors discussed, commented on and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Key R&D Program of China (2021YFA1501302), the National Natural Science Foundation of China (No. 22121004, U1862207, and 22122808), the Haihe Laboratory of Sustainable Chemical Transformations, and the Program of Introducing Talents of Discipline to Universities (BP0618007) for financial support. This work is supported by the XPLORER PRIZE.References
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- R. T. Hannagan, G. Giannakakis, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Chem. Rev., 2020, 120, 12044–12088 CrossRef CAS PubMed.
- R. T. Hannagan, G. Giannakakis, R. Reocreux, J. Schumann, J. Finzel, Y. C. Wang, A. Michaelides, P. Deshlahra, P. Christopher, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Science, 2021, 372, 1444–1447 CrossRef CAS.
- D. Zhao, X. X. Tian, D. E. Doronkin, S. L. Han, V. A. Kondratenko, J. D. Grunwaldt, A. Perechodjuk, T. H. Vuong, J. Rabeah, R. Eckelt, U. Rodemerck, D. Linke, G. Y. Jiang, H. J. Jiao and E. V. Kondratenko, Nature, 2021, 599, 234–238 CrossRef CAS PubMed.
- H. Yan, K. He, A. Samek Izabela, D. Jing, G. Nanda Macy, C. Stair Peter and M. Notestein Justin, Science, 2021, 371, 1257–1260 CrossRef CAS PubMed.
- R. Ryoo, J. Kim, C. Jo, S. W. Han, J. C. Kim, H. Park, J. Han, H. S. Shin and J. W. Shin, Nature, 2020, 585, 221–224 CrossRef CAS PubMed.
- T. Otroshchenko, G. Y. Jiang, V. A. Kondratenko, U. Rodemerck and E. V. Kondratenko, Chem. Soc. Rev., 2021, 50, 473–527 RSC.
- M. Monai, M. Gambino, S. Wannakao and B. M. Weckhuysen, Chem. Soc. Rev., 2021, 50, 11503–11529 RSC.
- S. Chen, X. Chang, G. D. Sun, T. T. Zhang, Y. Y. Xu, Y. Wang, C. L. Pei and J. L. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- A. H. Motagamwala, R. Almallahi, J. Wortman, V. O. Igenegbai and S. Linic, Science, 2021, 373, 217–222 CrossRef CAS PubMed.
- J. L. Wang, X. Chang, S. Chen, G. D. Sun, X. H. Zhou, E. Vovk, Y. Yang, W. Y. Deng, Z. J. Zhao, R. T. Mu, C. L. Pei and J. L. Gong, ACS Catal., 2021, 11, 4401–4410 CrossRef CAS.
- H. F. Xiong, S. Lin, J. Goetze, P. Pletcher, H. Guo, L. Kovarik, K. Artyushkova, B. M. Weckhuysen and A. K. Datye, Angew. Chem., Int. Ed., 2017, 56, 8986–8991 CrossRef CAS PubMed.
- H. Liu, X. Dong, J. Xia, X. Li, Y. Sun and Q. Cai, Trans. Tianjin Univ., 2019, 26, 362–372 CrossRef.
- Q. M. Sun, N. Wang, Q. Y. Fan, L. Zeng, A. Mayoral, S. Miao, R. O. Yang, Z. Jiang, W. Zhou, J. C. Zhang, T. J. Zhang, J. Xu, P. Zhang, J. Cheng, D. C. Yang, R. Jia, L. Li, Q. H. Zhang, Y. Wang, O. Terasaki and J. H. Yu, Angew. Chem., Int. Ed., 2020, 59, 19450–19459 CrossRef CAS PubMed.
- S. Chen, Z. J. Zhao, R. T. Mu, X. Chang, J. Luo, S. C. Purdy, A. J. Kropf, G. D. Sun, C. L. Pei, J. T. Miller, X. H. Zhou, E. Vovk, Y. Yang and J. L. Gong, Chem, 2021, 7, 387–405 CAS.
- L. Rochlitz, K. Searles, J. Alfke, D. Zemlyanov, O. V. Safonova and C. Coperet, Chem. Sci., 2020, 11, 1549–1555 RSC.
- W. T. Cai, R. T. Mu, S. J. Zha, G. D. Sun, S. Chen, Z. J. Zhao, H. Li, H. Tian, Y. Tang, F. Tao, L. Zeng and J. L. Gong, Sci. Adv., 2018, 4, eaar5418 CrossRef CAS PubMed.
- Y. Li, J. Li, X. Yang, X. Wang, Y. Xu and L. Zhang, Trans. Tianjin Univ., 2018, 25, 169–184 CrossRef.
- G. Q. Ren, G. X. Pei, Y. J. Ren, K. P. Liu, Z. Q. Chen, J. Y. Yang, Y. Su, X. Y. Liu, W. Z. Li and T. Zhang, J. Catal., 2018, 366, 115–126 CrossRef CAS.
- G. Siddiqi, P. P. Sun, V. Galvita and A. T. Bell, J. Catal., 2010, 274, 200–206 CrossRef CAS.
- S. J. Zha, G. D. Sun, T. F. Wu, J. B. Zhao, Z. J. Zhao and J. L. Gong, Chem. Sci., 2018, 9, 3925–3931 RSC.
- Z. P. Han, S. R. Li, F. Jiang, T. Wang, X. B. Ma and J. L. Gong, Nanoscale, 2014, 6, 10000–10008 RSC.
- Z. Ma, Z. Wu and J. T. Miller, Catal., Struct. React., 2017, 3, 43–53 CAS.
- M. D. Marcinkowski, M. T. Darby, J. L. Liu, J. M. Wimble, F. R. Lucci, S. Lee, A. Michaelides, M. Flytzani-Stephanopoulos, M. Stamatakis and E. C. H. Sykes, Nat. Chem., 2018, 10, 325–332 CrossRef CAS PubMed.
- G. Sun, Z. J. Zhao, R. Mu, S. Zha, L. Li, S. Chen, K. Zang, J. Luo, Z. Li, S. C. Purdy, A. J. Kropf, J. T. Miller, L. Zeng and J. Gong, Nat. Commun., 2018, 9, 4454 CrossRef PubMed.
- T. W. van Deelen, C. H. Mejia and K. P. de Jong, Nat. Catal., 2019, 2, 955–970 CrossRef CAS.
- B. J. O'Neill, D. H. K. Jackson, A. J. Crisci, C. A. Farberow, F. Y. Shi, A. C. Alba-Rubio, J. L. Lu, P. J. Dietrich, X. K. Gu, C. L. Marshall, P. C. Stair, J. W. Elam, J. T. Miller, F. H. Ribeiro, P. M. Voyles, J. Greeley, M. Mavrikakis, S. L. Scott, T. F. Kuech and J. A. Dumesic, Angew. Chem., Int. Ed., 2013, 52, 13808–13812 CrossRef PubMed.
- C. F. Xu, G. X. Chen, Y. Zhao, P. X. Liu, X. P. Duan, L. Gu, G. Fu, Y. Z. Yuan and N. F. Zheng, Nat. Commun., 2018, 9, 3367 CrossRef PubMed.
- Z. Q. Wang, Z. N. Xu, S. Y. Peng, M. J. Zhang, G. Lu, Q. S. Chen, Y. M. Chen and G. C. Guo, ACS Catal., 2015, 5, 4255–4259 CrossRef CAS.
- J. L. Gong, H. R. Yue, Y. J. Zhao, S. Zhao, L. Zhao, J. Lv, S. P. Wang and X. B. Ma, J. Am. Chem. Soc., 2012, 134, 13922–13925 CrossRef CAS PubMed.
- X. T. Yuan, S. Chen, D. F. Cheng, L. L. Li, W. J. Zhu, D. Z. Zhong, Z. J. Zhao, J. K. Li, T. Wang and J. L. Gong, Angew. Chem., Int. Ed., 2021, 60, 15344–15347 CrossRef CAS PubMed.
- W. Z. Yu, W. W. Wang, S. Q. Li, X. P. Fu, X. Wang, K. Wu, R. Si, C. Ma, C. J. Jia and C. H. Yan, J. Am. Chem. Soc., 2019, 141, 17548–17557 CrossRef CAS PubMed.
- S. H. Wang, K. Feng, D. K. Zhang, D. R. Yang, M. Q. Xiao, C. C. Zhang, L. He, B. H. Yan, G. A. Ozin and W. Sun, Adv. Sci., 2022, 9, 2104972 CrossRef CAS PubMed.
- R. van den Berg, T. E. Parmentier, C. F. Elkjaer, C. J. Gommes, J. Sehested, S. Helveg, P. E. de Jongh and K. P. de Jong, ACS Catal., 2015, 5, 4439–4448 CrossRef CAS.
- Y. Q. Dai, P. Lu, Z. M. Cao, C. T. Campbell and Y. N. Xia, Chem. Soc. Rev., 2018, 47, 4314–4331 RSC.
- X. Dong, X. Ma, H. Xu and Q. Ge, Catal. Sci. Technol., 2016, 6, 4151–4158 RSC.
- C. J. G. Van Der Grift, A. Mulder and J. W. Geus, Appl. Catal., 1990, 60, 181–192 CrossRef CAS.
- F. R. Lucci, J. L. Liu, M. D. Marcinkowski, M. Yang, L. F. Allard, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Nat. Commun., 2015, 6, 8550 CrossRef PubMed.
- X. Zhang, G. Q. Cui, H. S. Feng, L. F. Chen, H. Wang, B. Wang, X. Zhang, L. R. Zheng, S. Hong and M. Wei, Nat. Commun., 2019, 10, 5812 CrossRef CAS PubMed.
- J. Liu, F. R. Lucci, M. Yang, S. Lee, M. D. Marcinkowski, A. J. Therrien, C. T. Williams, E. C. H. Sykes and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2016, 138, 6396–6399 CrossRef CAS.
- L. DeRita, S. Dai, K. Lopez-Zepeda, N. Pham, G. W. Graham, X. Pan and P. Christopher, J. Am. Chem. Soc., 2017, 139, 14150–14165 CrossRef CAS PubMed.
- L. Deng, H. Miura, T. Shishido, Z. Wang, S. Hosokawa, K. Teramura and T. Tanaka, J. Catal., 2018, 365, 277–291 CrossRef CAS.
- M. J. Kappers and J. H. Vandermaas, Catal. Lett., 1991, 10, 365–373 CrossRef CAS.
- B. T. Qiao, A. Q. Wang, X. F. Yang, L. F. Allard, Z. Jiang, Y. T. Cui, J. Y. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- K. Ma, Y. Tian, Z. J. Zhao, Q. P. Cheng, T. Ding, J. Zhang, L. R. Zheng, Z. Jiang, T. Abe, N. Tsubaki, J. L. Gong and X. G. Li, Chem. Sci., 2019, 10, 2578–2584 RSC.
- A. L. Chen, X. J. Yu, Y. Zhou, S. Miao, Y. Li, S. Kuld, J. Sehested, J. Y. Liu, T. Aoki, S. Hong, M. F. Camellone, S. Fabris, J. Ning, C. C. Jin, C. W. Yang, A. Nefedov, C. Woll, Y. M. Wang and W. J. Shen, Nat. Catal., 2019, 2, 334–341 CrossRef CAS.
- A. Dandekar and M. A. Vannice, J. Catal., 1998, 178, 621–639 CrossRef CAS.
- G. J. Millar, C. H. Rochester and K. C. Waugh, J. Chem. Soc., Faraday Trans., 1991, 87, 1467–1472 RSC.
- T. Toupance, M. Kermarec, J.-F. Lambert and C. Louis, J. Phys. Chem. B, 2002, 106, 2277–2286 CrossRef CAS.
- S. Poulston, P. M. Parlett, P. Stone and M. Bowker, Surf. Interface Anal., 1996, 24, 811–820 CrossRef CAS.
- M. C. Biesinger, Surf. Interface Anal., 2017, 49, 1325–1334 CrossRef CAS.
- J. P. Espinos, J. Morales, A. Barranco, A. Caballero, J. P. Holgado and A. R. Gonzalez-Elipe, J. Phys. Chem. B, 2002, 106, 6921–6929 CrossRef CAS.
- S. K. Sharma, J. F. Mammone and M. F. Nicol, Nature, 1981, 292, 140–141 CrossRef CAS.
- N. Chiodini, F. Meinardi, F. Morazzoni, A. Paleari, R. Scotti and G. Spinolo, Solid State Commun., 1999, 109, 145–150 CrossRef CAS.
- Y. L. Deng, A. D. Handoko, Y. H. Du, S. B. Xi and B. S. Yeo, ACS Catal., 2016, 6, 2473–2481 CrossRef CAS.
- S. Kumar, Mater. Chem. Phys., 2021, 258, 123929 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc03723h |
| This journal is © The Royal Society of Chemistry 2022 |