
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanical deformation and multiple thermal restoration of organic crystals: reversible multi-stage shape-changing effect with luminescence-color changes†
Chi
Feng
 a,
Tomohiro
Seki
*b,
Shunichi
Sakamoto
c,
Toshiyuki
Sasaki
c,
Satoshi
Takamizawa
*c and
Hajime
Ito
*ad
a,
Tomohiro
Seki
*b,
Shunichi
Sakamoto
c,
Toshiyuki
Sasaki
c,
Satoshi
Takamizawa
*c and
Hajime
Ito
*ad
aDivision of Applied Chemistry, Graduate School of Engineering Hokkaido University, Sapporo, Hokkaido 060-8628, Japan. E-mail: hajito@eng.hokudai.ac.jp
bDepartment of Chemistry, Faculty of Science, Shizuoka University, Shizuoka 422-8017, Japan. E-mail: seki.tomohiro@shizuoka.ac.jp
cDepartment of Materials System Science, Graduate School of Nanobioscience, Yokohama City University, 22-2 Seto, Kanazawa-ku, Yokohama, Kanagawa 236-0027, Japan. E-mail: staka@yokohama-cu.ac.jp
dInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD) Hokkaido University, Sapporo, Hokkaido 060-8628, Japan
First published on 23rd July 2022
Abstract
Shape-memory materials can be mechanically deformed and subsequently reverse the deformation upon changing the temperature. Shape-memory materials have attracted considerable attention for basic research and industrial applications, and polymer and alloy shape-memory materials have been well studied; however, it is formidably challenging to develop functional shape-memory materials, such as materials with multi-stage and anisotropic shape changes and shape changes accompanied by changes in color and light emission. Here, we found a reversible multi-stage shape-changing effect after mechanical deformation in a molecular crystal induced by multi-step thermal phase transitions with reversible shape changes and luminescence-color changes. Using single-crystal structure and thermal analyses as well as mechanical property measurements, we found that the reversible multi-stage shape-changing effect was achieved by a combination of a twinning deformation and multi-step thermal phase transitions. The changes in the crystal shape and luminescence suggest novel strategies for imparting known shape-memory materials with additional functionalities.
Introduction
Shape memory refers to the phenomenon when a material is mechanically deformed and subsequently reverses the deformation upon a change of temperature. After its first discovery in 1951, the shape-memory effect attracted substantial attention on account of its considerable scientific and technological significance.1,2 To date, Ni–Ti alloys are the most commonly used shape-memory alloys in science and engineering.3 Since the 1980s, the relatively new class of polymeric shape-recovery materials has attracted increasing attention.4–8 Many shape-memory materials are already in practical use in surgical procedures, mechanical engineering, and clothing, and more are expected to find applications in aerospace engineering and robotics.2,3,8–11 In typical shape-memory metal alloys and polymers, the material can be deformed at low temperature and reverts to its pre-deformation shape at high temperature. However, despite significant advances in this area, it remains difficult to impart known shape-memory materials with additional functionalities; for example, multi-stage and anisotropic shape changes,12–24 as well as changes in light absorbance and emission to accompany the shape change remain elusive.25–28 This is due to the fact that the shape-memory properties of metal alloys and polymers strongly depend on their components or chemical structures, and that it is difficult to alter their atomic or molecular structure while retaining the shape-memory-type effect.Dynamic molecular crystals that are responsive to external stimuli have drawn increasing attention in recent years. Upon applying external stimuli, these dynamic molecular crystals show macroscopic dynamic effects such as bending, jumping, crawling, or creeping.29–33 In 2016, the groups of Naumov34 and Takamizawa35 almost simultaneously reported the first examples of organic molecular crystals with shape-memory-type phenomena (Fig. 1) These crystals can be bent mechanically, resulting in a stress-induced martensitic transformation. Upon heating, the deformed crystals regain its straight shape. The mechanism of the shape-memory-type phenomena can be expected to be different from those reported for metal alloys and polymers, and we observed similar “shape-deformation and -restoration (shape-memory)” behavior. These findings suggest that the variety of shape-memory-type materials, which had previously been limited to metal alloys and polymers, could be broadened significantly. In addition, the characteristics of organic crystals could enable functionalities that are unprecedented in conventional shape-memory materials, such as anisotropic or multiple shape changes12–24 and optical-property changes.25–28,36–51
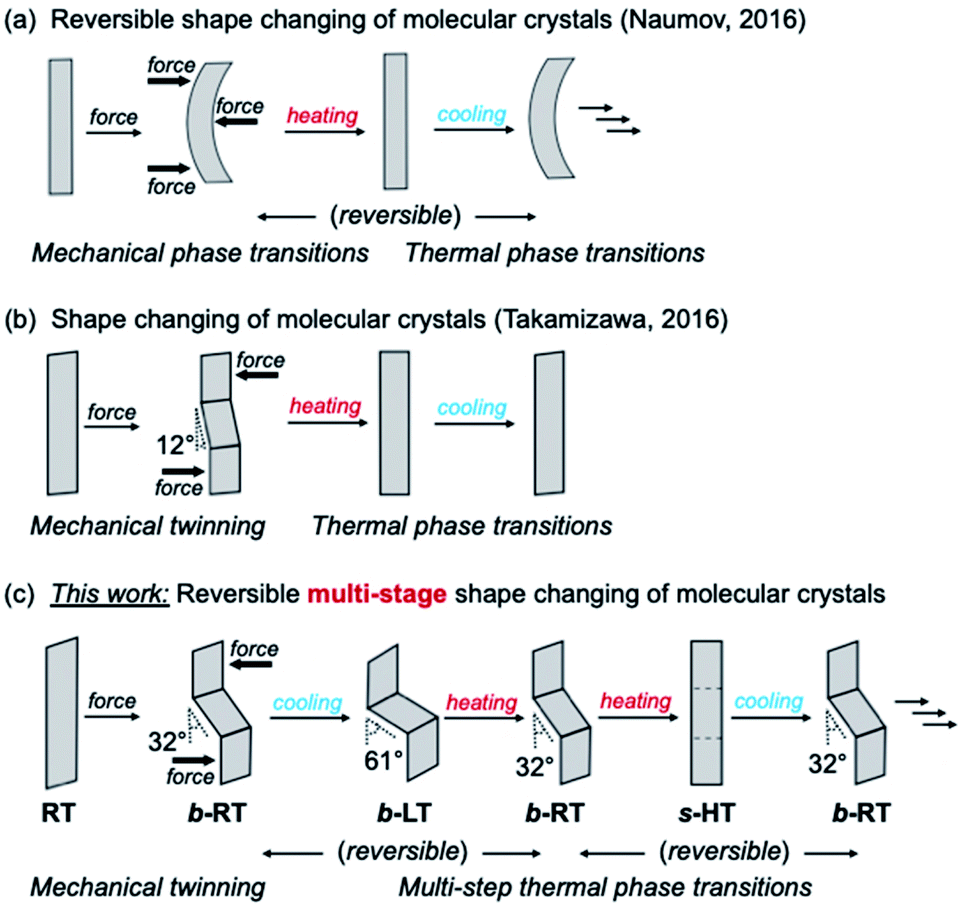 | ||
| Fig. 1 Schematic illustration of the shape-changing effect of molecular crystals after mechanical deformation. (a) Reversible shape changing through a combination of mechanical and thermal phase transitions. (b) Shape changing through a combination of mechanical twinning and thermal phase transitions. (c) Reversible multi-stage shape changing through a combination of mechanical twinning and multi-step thermal phase transitions. | ||
We have previously reported gold(I) complexes with ferroelastic properties, whose crystal shape can be deformed by an applied force, which induces twinning.52 The gold(I) crystals show luminescence under UV light. Although they are not temperature-responsive, we anticipated that it should be possible to develop a crystalline compound that exhibits both temperature-dependent, reversible crystal-phase changes and ferroelasticity as well as changes to its crystal shape and luminescence. Herein, we report molecular crystals of an arylgold(I) (N-heterocyclic carbene) (NHC) complex with a reversible multi-stage shape-changing effect after mechanical deformation. This material is able to remember at least three different shapes. These shapes can be reproduced in a cyclic fashion among different shapes upon changing the temperature even after the crystal has regained its straight shape.
The reversible multi-stage shape-changing effect was achieved by a combined process involving twinning deformation and multi-step thermal phase transitions among three phases, i.e., a low temperature (LT), room temperature (RT), and high temperature (HT) phase. Ferroelastic behavior,35,52–66i.e., mechanical-stress-induced twinning deformation with spontaneous straining, was found in the RT crystal (Fig. 1c). Under an applied force, the RT crystal undergoes mechanical bending with a zigzag morphology. Upon cooling, the bent RT (b-RT) transforms into a bent LT (b-LT) with an enhanced zigzag morphology (Fig. 1c). Intriguingly, this shape change is reversible upon heating b-LT, the zigzag morphology of b-RT is recovered, which confirms reversible multi-stage shape-changing effect (Fig. 1c). However, when b-RT is heated to give straight HT (s-HT), the zigzag morphology is lost, and the bent crystal regain its straight shape (Fig. 1c). Surprisingly, after ‘memorizing’ the bent shape, the crystals exhibit temperature-induced reversible shape changes even after transformation to the unbent s-HT phase. The zigzag-shaped b-RT was recovered upon cooling straight s-HT (Fig. 1c). This full reversibility is able to vary among distinct shapes without external force by simply changing in temperature. Interestingly, the transformations among the three shapes are accompanied by changes of the luminescence color. To the best of our knowledge, such reversible multi-stage shape-changing materials with luminescence-color changes have not yet been reported. Based on XRD, spectroscopic, and thermal analyses, as well as mechanical-property measurements, we will discuss the origin of this unique stimulus-responsivity.
Results and discussion
Complex 1 was prepared according to our previously reported procedure.52 Typically, pristine crystals of 1, hereafter referred to as RT, are colorless parallelogram plates with corner angles of 74° (Fig. 2c and d and S1†). The RT crystals show weak yellow photoluminescence under UV light. | ||
| Fig. 2 (a) Molecular structure of 1. (b and c) Schematic illustration of the thermal transitions for the low temperature (LT, yellow-green), room temperature (RT, yellow), and high temperature (HT, green) phases of a crystal of 1. (d) Photographs of a crystal of 1 under UV light at cooling/heating rates of 20 K min−1. (e) Photoluminescence spectra of LT, RT, and HT crystals of 1 (λex = 365 nm) (for further details, see ESI, Fig. S5–S8 and Table S1†); scale bars: 100 μm. | ||
Crystals of 1 exhibit simultaneous changes of their luminescence and morphology in response to temperature changes. Upon cooling to ∼170 K, the emission intensity of RT gradually increased (Fig. 2d(ii)). Upon further cooling to ∼160 K, the crystal shape begins to change, and several ordered crystalline domains with pale yellow-green emission appear (Fig. 2d(iii)). Upon further cooling to ∼93 K, the pale yellow-green-emitting domains gradually expand, accompanied by a prominent morphological change (Fig. 2d(iv)). Eventually, a more oblique parallelogram crystal with corner angles of 60° is obtained. The resulting yellow-green emissive crystal will henceforth be referred to as LT. Both the changes in emission color and shape between the LT and RT crystals are reversible upon changing the temperature: upon heating LT, RT is recovered at ∼273 K (Fig. 2d(vi)). Upon further heating of RT, gradual macroscopic morphological changes are observed starting at ∼360 K. At ∼373 K, a rectangular crystal (hereafter: HT) is obtained (Fig. 2d(viii)). The conversion of RT to HT also leads to an emission-color change: HT shows weak green photoluminescence. The photoluminescence intensity enhances upon cooling to ∼303 K (Fig. 2d(ix)). Recovery of the emission color and shape due to the phase transition from HT to RT was confirmed at ∼293 K (Fig. S1 and S2†). The entire sequence can be seen in the ESI (Movie S1†). The reversible shape deformation among LT, RT, and HT was performed for at least 16 heating/cooling cycles without visible cracking of the crystals (Fig. S3†). A differential scanning calorimetry (DSC) analysis confirmed the reversibility of the transitions among the three phases (RT to LT: 191 K; LT to RT: 237 K; RT to HT: 324 K; HT to RT: 296 K; Fig. 2b and S4†). These observations confirm that crystals of 1 show reversible changes of their emission color and macroscopic morphology upon cooling/heating.
The photophysical properties of the LT, RT, and HT crystals were also investigated. As RT exhibits strong yellow photoluminescence upon moderate cooling (Fig. 2d(ii)), its emission spectrum was measured at 193 K. Under UV light, RT exhibited an emission spectrum with a maximum at 581 nm (Fig. 2e and S5†). The emission spectrum of LT at 93 K showed a maximum at 544 nm, indicating a hypsochromic shift of the emission band upon cooling (Fig. 2e and S5†). Similar to that of RT, the emission spectrum of HT at 373 K is broad. However, its maximum emission wavelength (510 nm) is shorter than those of RT and LT (Fig. 2e and S5†). We also confirmed that the RT crystal recovered from LT or HT showed an emission spectrum similar to that of pristine RT (Fig. S5†), indicating that the photophysical properties are reversible.
Single-crystal X-ray diffraction (XRD) analyses demonstrated that the LT, RT, and HT crystal structures differ from one another. The single-crystal diffraction analysis of LT was performed at 123 K and indicated that LT crystallizes in the monoclinic space group P21/c at this temperature (Table S2†). In LT, the molecules of 1 adopt a flat conformation with a dihedral angle (θ) of 2.5789(2)° between the NHC ring and the halogenated benzene ring on the gold atom (Fig. 3b and S9†). These flat molecules form dimers with a head-to-tail arrangement through π–π stacking interactions between the NHC ring and the benzene ring (heterogeneous stacking) with a longitudinal offset (Fig. S9†). The dimers interact with adjacent dimers through π–π stacking interactions between the NHC rings or benzene rings (homogeneous stacking), allowing the formation of a zigzag stacking motif. To investigate the crystal structure of RT, an XRD analysis of the same piece of the crystal was then performed at 293 K. Importantly, RT exhibits a crystal structure different from that of LT, i.e., RT crystallizes in the monoclinic space group P21/c (Table S2†). In RT, the molecules also adopt flat conformations (θ = 4.1(3)° or 0.2(3)°; Fig. 3b and S10†). These flat molecules form π–π stacking interactions with a head-to-tail arrangement. Stacks of four molecules (tetramer units) interact with adjacent tetramers through homogeneous π–π stacking between NHC rings or benzene rings along the crystallographic a-axis. For the analysis of HT, XRD measurements of the same piece of the crystal were performed at 363 K. HT showed a symmetry change from a monoclinic to an orthorhombic system with the space group Pnma (Table S2†). The molecule has a flat conformation (θ = 0.0(8)°) and engages in heterogeneous π–π stacking in a head-to-tail manner (Fig. 3b and S11†). No homogeneous π–π stacking interactions were observed in HT, indicating that the stacking motif lacks an offset. In their entirety, these results suggest that the LT, RT, and HT crystal structures all involve molecular stacking of molecules with flat conformations. However, the motifs of the molecular stacking differ, which could be the origin of the stimulus-responsivity.
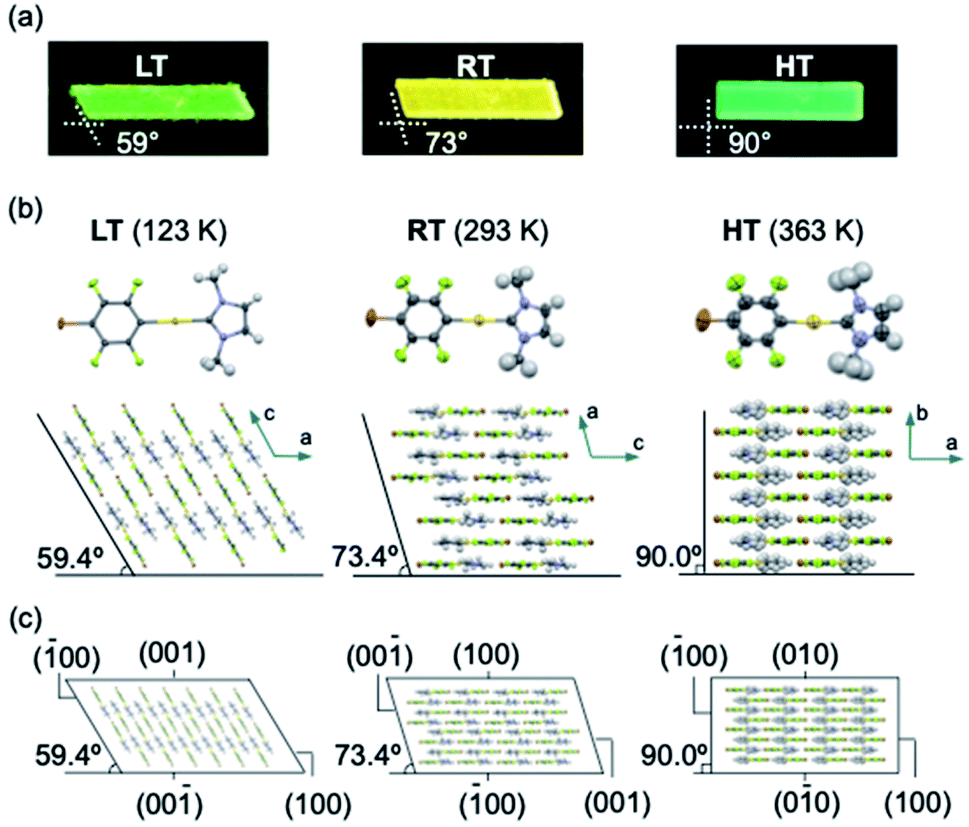 | ||
| Fig. 3 (a) Photographic images of LT, RT, and HT crystals under UV light. (b) Molecular stacking of the LT, RT, and HT crystal structures. (c) Crystal packing with face indices of LT (left), RT (middle), and HT (right). | ||
Remarkably, the macroscopic corner angles of the crystals of LT, RT, and HT are correlated to the microscopic offset angle of their molecular stacking. In LT, molecules stack with a longitudinal displacement along the molecular long axis (Fig. 3b) with an overall offset angle of 59.4°, which coincides with the 59° corner angle of its macroscopic crystal (Fig. 3a and b). In RT, the overall offset angle of molecular stacking is 73.4°, which is in good agreement with the macroscopic corner angle of its crystal (73°; Fig. 3a and b). For HT, the molecules stack without longitudinal displacement along the long molecular axis, producing an offset angle of 90.0°, which also coincides with the corner angle of its rectangular crystal (90°; Fig. 3a and b). Moreover, the results of the face-indexing experiments support the correspondence between the corner and offset angles (Fig. 3c and S12†). These detailed analyses of the crystal structures of 1 thus clarified the origin of their macroscopic morphology changes.
Subsequently, we examined the mechanical properties of the RT crystal at room temperature. For that purpose, the crystal was mechanically bent by applying shear stress to its (100) face (Fig. 4a(i and ii) and Movie S2†). Even after removing the mechanical stress, the bent shape was retained, confirming that it exhibited plastic bending. The bending angle of the bent RT (b-RT) is 32°, and the reproducibility of the bending angle was confirmed (Fig. S13†). Scanning electron microscopy (SEM) observation of b-RT showed a clear bending interface at the boundaries between the bent and unbent regions (Fig. 4b and S14†). Upon applying shear stress in the opposite direction, the bent region of RT was completely restored to its original parallelogram shape (Fig. 4a(iii and iv)). These reversible mechanical deformations are similar to those of the ferroelastic molecular crystals that we have previously reported for a structurally similar gold complex.52
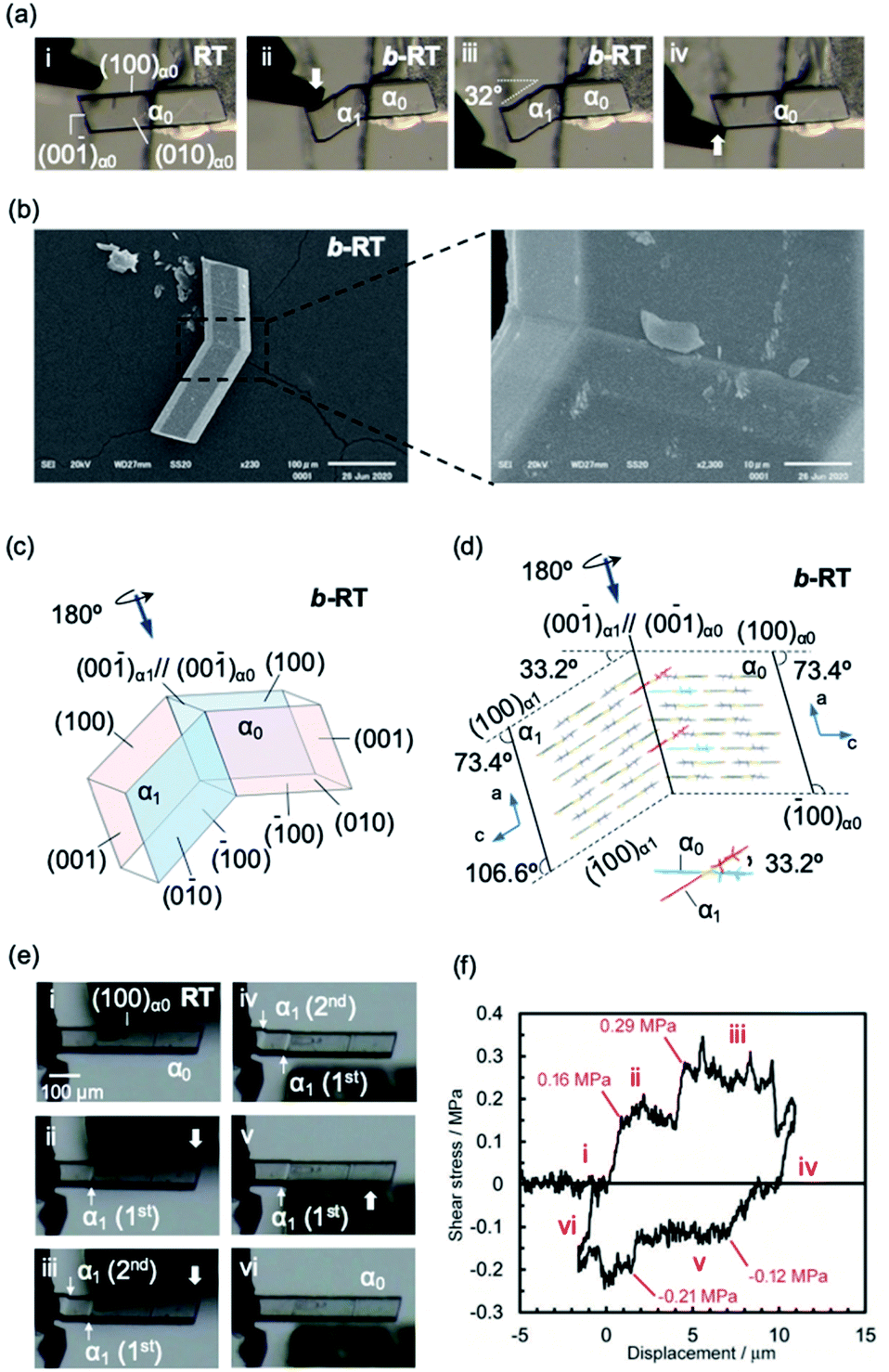 | ||
| Fig. 4 (a and b) Morphology visualized by optical microscopy (a) and SEM (b). An RT crystal was fixed on a base with epoxy glue and bent manually by applying shear stress on the (100)α0 face (a, i and ii) using tweezers. When shear stress was applied on the opposite face, the bent RT (b-RT) was restored to its original shape (a, iii and iv). (b) SEM micrographs showing the bending interface of b-RT generated during the bending process. (c) Crystal-face indices of b-RT determined using single-crystal XRD. (d) Crystal packing of b-RT viewed parallel to the unbent domain (010)α0 and bent domain (0−10)α1. (e) Snapshots of the twinning deformation of the RT crystal (449 × 63 × 26 μm3) upon applying shear stress: (i–iii) forward direction and (iv–vi) reverse direction. (f) Ferroelastic hysteresis loops for the twinning deformation of RT (for further details, see ESI, Fig. S15–S17†). | ||
A single-crystal XRD analysis of b-RT was performed at 293 K and demonstrated a twinning deformation composed of an unbent domain (α0) and a bent domain (α1), which indicates that the crystal structures of the α0 and α1 domains are identical but oriented differently. According to the crystal-face indexing, the α0 and α1 domains are connected via the interface (00−1)α0//(00−1)α1 (Fig. 4c). The lattices of the mother and daughter phase are related by a 180° rotation along the rotational axis (arrow in Fig. 4c and d). The microscopic bending angle between α0 and α1 predicted from these twinning analyses is 33.2° (Fig. 4d), which agrees well with the macroscopic bending angle (32°) confirmed by optical microscopy images (Fig. 4a). The packing diagram shows that a molecular rotation of 33.2° is required to retain continuous boundaries between the α0 and the α1 domain during the deformation (Fig. 4d). Overall, the XRD results demonstrate that macroscopic and microscopic changes occur upon the twinning deformation from the α0 to the α1 domain. The stress–strain relationship during the ferroelastic deformation of the RT crystal was investigated at 303 K. Upon increasing the shear stress on the (100) plane of the RT, the α1 domain was generated at the jig side at 0.16 MPa (Fig. 4e and f(i and ii) and Movie S3†). The α1 domain grew upon applying further, relatively constant shear stress. The α1 domain expanded until another α1 domain was generated at a shear stress of 0.29 MPa(III). The second α1 domain then expanded under almost-constant stress. After removing the stress, these two α1 domains remained and the generation of spontaneous strain was observed (iv). Moreover, upon applying shear stress from the opposite direction, the reversible deformation from the α1 to the α0 domain was observed (v and vi). The second α1 domain began to contract when the stress reached 0.12 MPa. After the second α1 domain disappeared, the α1 domain close to the jig started to contract at 0.21 MPa (v). Subsequently, the last α1 domain disappeared and the bent b-RT was restored to its original straight shape. The forward (0.16 MPa) and reverse (0.12 MPa) critical stress of RT are lower than those in our previous report (0.61 MPa and 0.49, respectively),52 indicating that RT is superior in terms of absorbing weak mechanical shocks. The stress–strain measurement of the RT crystal demonstrates typical ferroelastic behavior, including hysteresis behavior and the build-up of spontaneous strain.
We then investigated the reversible multi-stage shape-changing effect of crystal 1. First, the bent shape was ‘memorized’ in the pristine RT crystal by mechanical bending to give b-RT (Fig. 5a). Upon cooling b-RT for the transition to LT, the bending angle increases remarkably from 32° to 61° (photographs B and C in Fig. 5a and Movie S4†), whereby b-LT shows a more pronounced zigzag shape than b-RT. The corner angle of the b-LT crystal is 60°, which is similar to that of LT. Upon reheating b-LT to induce the phase transition to b-RT, the bending angle again changed from 61° to 32° at ∼235 K (photographs C and D in Fig. 5a), i.e., the bent shape of b-RT was restored by heating. Upon further heating b-RT to ∼330 K , the bend completely disappeared via phase transition to the HT phase to give straight-shaped s-HT (Fig. 5a(E)). Although HT (Fig. 2d) and s-HT (Fig. 5a) have almost the same morphology and s-HT presented no obvious damage, remarkably, s-HT can reproduce the zig-zag shape; upon cooling to room temperature, the crystal again bent with an angle of 32° (Fig. 5a(F)). This indicates that the ‘memorized’ b-RT shape (Fig. 5a(F)) was recovered from s-HT (Fig. 5a(E)) without applying additional mechanical stress. As mentioned above, HT can be obtained by heating pristine unbent RT; the resulting HT crystal did not undergo bending upon cooling (Fig. S1 and S2†). The b-LT and s-HT shapes could also be reproduced via phase transition upon cooling or heating, respectively. As expected, the morphology changes of the bent crystals of 1 upon cooling/heating were accompanied by emission-color changes (photographs O–R in Fig. 5a). And the reproducibility of the reversible multi-stage shape changing effect was confirmed (Fig. S18 and S19†). These results indicate that the pristine crystal of 1 can take on three different shapes, b-LT, b-RT, and s-HT, which exhibit a reversible multi-stage shape-changing effect upon cyclic cooling and heating.
 | ||
| Fig. 5 Reversible multi-stage shape-changing effect and mechanism of crystal 1. (a) Reversible multi-stage shape-changing. A straight RT crystal is mechanically bent (A and B). The bent crystal will move during the shape changing process. For better observation, one end of the bent RT (b-RT) crystal was fixed on a base with epoxy glue. The b-RT can be transformed into bent LT (b-LT) and straight HT (s-HT) upon cooling or heating at rates of 20 K min−1, respectively (B–E). The shapes of b-LT, b-RT, and s-HT were reproduced in cooling/heating cycles, and the reversible multi-stage shape-changing effect was observed over four consecutive cooling/heating cycles (B–N: under visible light; O–R: under UV light). (b) Mechanism of the reversible multi-stage shape-changing effect. The microscopic bending angles estimated based on the XRD analysis of the crystals of b-LT, b-RT, and s-HT correspond to their macroscopic bending angles; scale bar: 100 μm. | ||
Detailed XRD analyses were carried out to explain the mechanism of the shape memorization and reproduction. Similar to that of b-RT, the observed bending angles of b-LT and s-HT can be reasonably interpreted in terms of their XRD results. An XRD analysis of b-LT was performed at 123 K, and its crystal structure was found to be twinned (Table S3†). Both the α0 and α1 domains have crystal structures that are identical to that of the LT crystal and the interface between the α0 and α1 domains is (−100)α0//(−100)α1 (Fig. S20†). The lattices of the α1 domain are obtained from a 180° rotation around the rotational axis, which is indicated by the arrow parallel to the twinning interface (−100). Its microscopic bending angle was estimated to be 61.2°, which coincides with the macroscopic bending angle of 61° confirmed by optical microscopy (Fig. 5 and S18†). For s-HT, twinning was not observed, and it could thus be modeled as a single component (Table S3 and Fig. S21†). As the offset angle of the molecular stack of s-HT is 90.0°, its microscopic bending angle is estimated to be 0°. This causes the straight shape of s-HT, whereby the zigzag shape caused by the twinning in b-RT and b-LT disappears (Fig. 5a and b). As mentioned above, SEM images of b-RT show bending interfaces at the boundaries between the bent and unbent regions (Fig. 4b). The traces of these bending interfaces remained even after b-RT transformed into s-HT (Fig. S22†). Although the interface seems to disappear via optical observation, the retained post-bending interface in s-HT can be expected to be important for the recovery of the b-RT shape. In contrast, HT, which was obtained starting from unbent RT, showed a clear surface, and no traces were found on s-HT surface via SEM (Fig. S23†). Upon cooling HT to room temperature, it does not bend (Fig. S2†). The macroscopic bending angles of b-LT, b-RT, and s-HT can thus be reasonably rationalized based on their crystal structures. The key to the present reversible multi-stage shape-changing effect in the crystal of complex 1 is that 1 shows both multiple temperature-dependent phase changes (HT, RT, and LT) and mechanical deformation with ferroelasticity (Fig. 6). Moreover, crystals of 1 show simultaneously changes in morphology and luminescence color in response to temperature changes. The crystal of 1 can be mechanically bent by application of stress at room temperature (RT), and subsequently undergo a reversible multi-stage shape-changing effect upon cooling/heating (s-HT, b-RT, and b-LT).
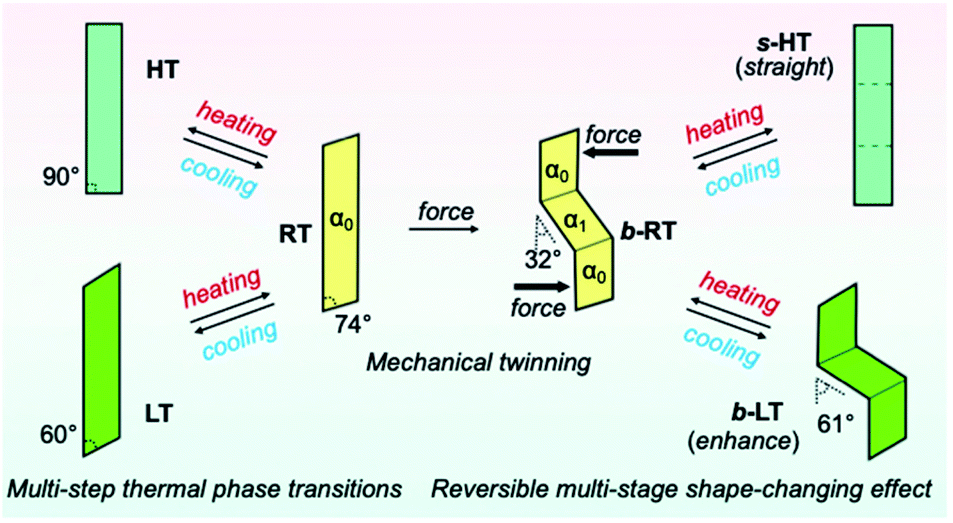 | ||
| Fig. 6 Schematic illustration of the reversible multi-stage shape-changing effect. | ||
Conclusions
We have reported a reversible multi-stage shape-changing material based on an arylgold(I) (N-heterocyclic carbene) (NHC) complex (1) that is characterized by the reproduction of three different shapes and luminescence-color changes (Fig. 6). The reversible multi-stage shape-changing effect is achieved by a combination of twinning deformation and multi-step thermal phase transitions. The twinning deformation causes lattice rotation in a crystal of 1 at room temperature (RT), resulting in a zigzag shape (b-RT) with different orientations of the crystal structures. When RT is mechanically bent, the bending direction of the crystal structure is memorized. Intriguingly, the interconversion among the three different shapes is accompanied by changes of the luminescence color. The discovery of this reversible multi-stage shape-changing material can be expected to result in novel strategies for imparting known shape-memory materials with additional functionalities.Experimental
Preparation of the materials
NHC gold complex 1 was prepared according to a previously reported procedure.52 From 1,4-dibromo-2,3,5,6-tetrafluorobenzene, 1 was synthesized via a two-step reaction (ESI Scheme 1†). Crystals of 1 were obtained by recrystallization from CH2Cl2 and hexane at room temperature.Thermal analysis
Differential scanning calorimetry (DSC) profiles were measured on a DSC Q2000 V24.11 calorimeter using heating and cooling rates of 5 K min−1.Force measurements
Stress–strain tests were carried out at 303 K on a universal testing machine equipped with a hot stage (Tensilon RTG-1210, A&D Co. Ltd). For the stress–strain experiment, one end of the crystal surface was fixed to a glass stage with epoxy glue, and a glass jig was loaded on the plane of the crystal at a speed of 2 μm s−1. When the jig reached the crystal surface, the shear stress was detected.X-ray diffraction analyses
Single-crystal X-ray diffraction analyses were carried out on a Rigaku XtaLAB PRO MM007 diffractometer using graphite-monochromated Mo-Kα radiation. Structures were solved by direct methods and expanded using Fourier techniques. Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined using the riding model. All calculations were performed using the Olex2 crystallographic software package except for the refinement, which was performed using SHELXT-2018. The structures were solved by direct methods with (SHELXT) and refined by full-matrix least-squares techniques against F2 (SHELXL-2018/3) using the Olex2 software package.Data availability
For full characterization data including NMR spectra of all new compounds, single-crystal XRD structure analyses, and experimental details, see the ESI.† All relevant data underlying the results of this study are available from the corresponding authors upon reasonable request.Author contributions
T. Seki, S. T., and H. I. conceived and designed the study. C. F., T. Seki, S. T., and H. I. analyzed the data and co-wrote the paper. S. S., T. Sasaki, and S. T. performed the force measurement and analyzed the data. C. F. and T. Seki recorded the DSC data. C. F. performed the chemical experiments and other characterization procedures. All authors discussed the results and the manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was financially supported by JSPS KAKENHI grants JP17H06370, JP19H02784, JP19H04555, JP22K19058, and JP22H02155 and JST/PRESTO Grant Number JPMJPR21AB as well as by the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) via the Program for Leading Graduate Schools (Hokkaido University ‘Ambitious Leaders’ Program’). This study was also supported by the Institute for Chemical Reaction Design and Discovery (ICReDD), established by the World Premier International Research Initiative (WPI) of MEXT, Japan.Notes and references
- L. C. Chang and T. A. Read, Trans. AIME, 1951, 189, 47–52 Search PubMed.
- L. Sun, W. M. Huang, Z. Ding, Y. Zhao, C. C. Wang, H. Purnawali and C. Tang, Mater. Des., 2012, 33, 577–640 CrossRef CAS.
- J. M. Jani, M. Leary, A. Subic and M. A. Gibson, Mater. Des., 2014, 56, 1078–1113 CrossRef.
- H. Koerner, G. Price, N. A. Pearce, M. Alexander and R. A. Vaia, Nat. Mater., 2004, 3, 115–120 CrossRef CAS PubMed.
- W. M. Huang, B. Yang, L. An, C. Li and Y. S. Chan, Appl. Phys. Lett., 2005, 86, 114105 CrossRef.
- A. Lendlein, H. Jiang, O. Jünger and R. Langer, Nature, 2005, 434, 879–882 CrossRef CAS PubMed.
- R. Mohr, K. Kratz, T. Weigel, M. Lucka-Gabor, M. Moneke and A. Lendlein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3540–3545 CrossRef CAS PubMed.
- C. Liu, Q. Qin and P. T. Mather, J. Mater. Chem., 2007, 17, 1543–1558 RSC.
- C. T. Dotter, R. W. Buschmann, M. K. McKinney and J. Rösch, Radiology, 1983, 147, 259–260 CrossRef CAS PubMed.
- A. Lendlein and R. Langer, Science, 2002, 296, 1673–1676 CrossRef PubMed.
- C. M. Yakacki, R. Shandas, D. Safranski, A. M. Ortega, K. Sassaman and K. Gall, Adv. Funct. Mater., 2008, 18, 2428–2435 CrossRef CAS PubMed.
- D. Das, T. Jacobs and L. J. Barbour, Nat. Mater., 2010, 9, 36–39 CrossRef CAS PubMed.
- S. C. Sahoo, S. B. Sinha, M. S. R. N. Kiran, U. Ramamurty, A. F. Dericioglu, C. M. Reddy and P. Naumov, J. Am. Chem. Soc., 2013, 135, 13843–13850 CrossRef CAS PubMed.
- Z.-S. Yao, M. Mito, T. Kamachi, Y. Shiota, K. Yoshizawa, N. Azuma, Y. Miyazaki, K. Takahashi, K. Zhang, T. Nakanishi, S. Kang, S. Kanegawa and O. Sato, Nat. Chem., 2014, 6, 1079–1083 CrossRef CAS PubMed.
- S.-Q. Su, T. Kamachi, Z.-S. Yao, Y.-G. Huang, Y. Shiota, K. Yoshizawa, N. Azuma, Y. Miyazaki, M. Nakano, G. Maruta, S. Takeda, S. Kanegawa and O. Sato, Nat. Commun., 2015, 6, 8810 CrossRef PubMed.
- P. Commins, I. T. Desta, D. P. Karothu, M. K. Panda and P. Naumov, Chem. Commun., 2016, 52, 13941–13954 RSC.
- H. Chung, D. Dudenko, F. Zhang, G. D'Avino, C. Ruzié, A. Richard, G. Schweicher, J. Cornil, D. Beljonne, Y. Geerts and Y. Diao, Nat. Commun., 2018, 9, 278 CrossRef PubMed.
- T. Seki, T. Mashimo and H. Ito, Chem. Sci., 2019, 10, 4185–4191 RSC.
- P. Gupta, D. P. Karothu, E. Ahmed, P. Naumov and N. K. Nath, Angew. Chem., Int. Ed., 2018, 57, 8498–8502 CrossRef CAS PubMed.
- L. Li, P. Commins, M. B. Al-Handawi, D. P. Karothu, J. M. Halabi, S. Schramm, J. Weston, R. Rezgui and P. Naumov, Chem. Sci., 2019, 10, 7327–7332 RSC.
- E. Ahmed, D. P. Karothu, M. Warren and P. Naumov, Nat. Commun., 2019, 10, 3723 CrossRef PubMed.
- Y. Duan, S. Semin, P. Tinnemans, H. Cuppen, J. Xu and T. Rasing, Nat. Commun., 2019, 10, 4573 CrossRef PubMed.
- V. K. Srirambhatla, R. Guo, D. M. Dawson, S. L. Price and A. J. Florence, Cryst. Growth Des., 2020, 20, 1800–1810 CrossRef CAS.
- S. K. Park and Y. Diao, Chem. Soc. Rev., 2020, 49, 8287–8314 RSC.
- S. Kobatake, S. Takami, H. Muto, T. Ishikawa and M. Irie, Nature, 2007, 446, 778–781 CrossRef CAS PubMed.
- M. K. Panda, T. Runčevski, S. C. Sahoo, A. A. Belik, N. K. Nath, R. E. Dinnebier and P. Naumov, Nat. Commun., 2014, 5, 4811 CrossRef CAS PubMed.
- M. Jin, S. Yamamoto, T. Seki, H. Ito and M. A. Garcia-Garibay, Angew. Chem., Int. Ed., 2019, 58, 18003–18010 CrossRef CAS PubMed.
- T. Mutai, T. Sasaki, S. Sakamoto, I. Yoshikawa, H. Houjou and S. Takamizawa, Nat. Commun., 2020, 11, 1824 CrossRef CAS PubMed.
- D. Kitagawa, H. Tsujioka, F. Tong, X. Dong, C. J. Bardeen and S. Kobatake, J. Am. Chem. Soc., 2018, 140, 4208–4212 CrossRef CAS PubMed.
- S. Saha, M. K. Mishra, C. M. Reddy and G. R. Desiraju, Acc. Chem. Res., 2018, 51, 2957–2967 CrossRef CAS PubMed.
- S. Das, A. Mondal and C. M. Reddy, Chem. Soc. Rev., 2020, 49, 8878–8896 RSC.
- P. Naumov, D. P. Karothu, E. Ahmed, L. Catalano, P. Commins, J. M. Halabi, M. B. Al-Handawi and L. Li, J. Am. Chem. Soc., 2020, 142, 13256–13272 CrossRef CAS PubMed.
- D. P. Karothu, J. M. Halabi, E. Ahmed, R. Ferreira, P. R. Spackman, M. A. Spackman and P. Naumov, Angew. Chem., Int. Ed., 2022, e202113988 CAS.
- D. P. Karothu, J. Weston, I. T. Desta and P. Naumov, J. Am. Chem. Soc., 2016, 138, 13298–13306 CrossRef CAS PubMed.
- S. Takamizawa and Y. Takasaki, Chem. Sci., 2016, 7, 1527–1534 RSC.
- Y. Abe, S. Karasawa and N. Koga, Chem. - Eur. J., 2012, 18, 15038–15048 CrossRef CAS PubMed.
- X. Gu, J. Yao, G. Zhang, Y. Yan, C. Zhang, Q. Peng, Q. Liao, Y. Wu, Z. Xu, Y. Zhao, H. Fu and D. Zhang, Adv. Funct. Mater., 2012, 22, 4862–4872 Search PubMed.
- T. Seki, T. Ozaki, T. Ohkura, K. Asakura, A. Sakon, H. Uekusa and H. Ito, Chem. Sci., 2015, 6, 2187–2195 RSC.
- T. Seki, K. Sakurada, M. Muromoto, S. Seki and H. Ito, Chem. - Eur. J., 2016, 22, 1968–1978 Search PubMed.
- T. Seki, Y. Takamatsu and H. Ito, J. Am. Chem. Soc., 2016, 138, 6252–6260 CrossRef CAS PubMed.
- X. Du, F. Xu, M.-S. Yuan, P. Xue, L. Zhao, D.-E. Wang, W. Wang, Q. Tu, S.-W. Chen and J. Wang, J. Mater. Chem. C, 2016, 4, 8724–8730 CAS.
- Y. Abe, V. Savikhin, J. Yin, A. C. Grimsdale, C. Soci, M. F. Toney and Y. M. Lam, Chem. Mater., 2017, 29, 7686–7696 CAS.
- M. Jin, T. S. Chung, T. Seki, H. Ito and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2017, 139, 18115–18121 Search PubMed.
- M. Jin, T. Sumitani, H. Sato, T. Seki and H. Ito, J. Am. Chem. Soc., 2018, 140, 2875–2879 CAS.
- T. Seki, K. Ida and H. Ito, Mater. Chem. Front., 2018, 2, 1195–1200 CAS.
- T. Yamakado, K. Otsubo, A. Osuka and S. Saito, J. Am. Chem. Soc., 2018, 140, 6245–6248 CAS.
- C. Ge, J. Liu, X. Ye, Q. Han, L. Zhang, S. Cui, Q. Guo, G. Liu, Y. Liu and X. Tao, J. Phys. Chem. C, 2018, 122, 15744–15752 CAS.
- M.-S. Jiang, Y.-H. Tao, Y.-W. Wang, C. Lu, D. J. Young, J.-P. Lang and Z.-G. Ren, Inorg. Chem., 2020, 59, 3072–3078 CAS.
- H. Sun, J. H. Wei, L.-H. Xu, Y. Jiang, B.-X. Miao and Z.-H. Ni, Dyes Pigm., 2020, 177, 108299 CAS.
- Y. Liu, A. Li, S. Xu, W. Xu, Y. Liu, W. Tian and B. Xu, Angew. Chem., Int. Ed., 2020, 59, 15098–15103 CAS.
- M. Jin, R. Ando, M. J. Jellen, M. A. Garcia-Garibay and H. Ito, J. Am. Chem. Soc., 2021, 143, 1144–1153 CAS.
- T. Seki, C. Feng, K. Kashiyama, S. Sakamoto, Y. Takasaki, T. Sasaki, S. Takamizawa and H. Ito, Angew. Chem., Int. Ed., 2020, 59, 8839–8843 CAS.
- S. H. Mir, Y. Takasaki, E. R. Engel and S. Takamizawa, Angew. Chem., Int. Ed., 2017, 56, 15882–15885 CAS.
- S. H. Mir, Y. Takasaki and S. Takamizawa, Phys. Chem. Chem. Phys., 2018, 20, 4641–4645 Search PubMed.
- E. R. Engel, Y. Takasaki, S. H. Mir and S. Takamizawa, R. Soc. Open Sci., 2018, 5, 171146 Search PubMed.
- S. H. Mir, Y. Takasaki, E. R. Engel and S. Takamizawa, CrystEngComm, 2018, 20, 3807–3811 CAS.
- S. H. Mir, Y. Takasaki, E. R. Engel and S. Takamizawa, RSC Adv., 2018, 8, 21933–21936 CAS.
- E. R. Engel and S. Takamizawa, Angew. Chem., Int. Ed., 2018, 57, 11888–11892 CAS.
- S. Takamizawa and Y. Takasaki, Cryst. Growth Des., 2019, 19, 1912–1920 CAS.
- S. Sakamoto, T. Sasaki, A. Sato-Tomita and S. Takamizawa, Angew. Chem., Int. Ed., 2019, 58, 13722–13726 CAS.
- T. Sasaki, S. Sakamoto, Y. Takasaki and S. A. Takamizawa, Angew. Chem., Int. Ed., 2020, 59, 4340–4343 CAS.
- T. Sasaki, S. Sakamoto and S. Takamizawa, Cryst. Growth Des., 2020, 20, 1935–1939 CAS.
- T. Sasaki, S. Sakamoto, K. Nishizawa and S. Takamizawa, Cryst. Growth Des., 2020, 20, 3913–3917 CAS.
- S. K. Park, H. Sun, H. Chung, B. B. Patel, F. Zhang, D. W. Davies, T. J. Woods, K. Zhao and Y. Diao, Angew. Chem., Int. Ed., 2020, 59, 13004–13012 CAS.
- T. Sasaki, S. Sakamoto and S. Takamizawa, Cryst. Growth Des., 2020, 20, 4621–4626 CAS.
- Y. Takasaki, T. Sasaki and S. Takamizawa, Cryst. Growth Des., 2020, 20, 6211–6216 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data, optical microscopy images, thermal data, emission spectra and other additional information. CCDC 2081347–2081352. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03414j |
| This journal is © The Royal Society of Chemistry 2022 |