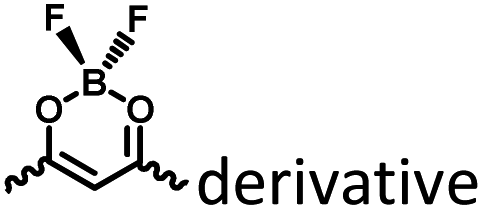DOI:
10.1039/D2SC01770A
(Review Article)
Chem. Sci., 2022,
13, 7976-7989
Supramolecular assembly confined purely organic room temperature phosphorescence and its biological imaging
Received
28th March 2022
, Accepted 6th June 2022
First published on 6th June 2022
Abstract
Purely organic room temperature phosphorescence, especially in aqueous solution, is attracting increasing attention owing to its large Stokes shift, long lifetime, low preparation cost, low toxicity, good processing performance advantages, and broad application value. This review mainly focuses on macrocyclic (cyclodextrin and cucurbituril) hosts, nanoassembly, and macromolecule (polyether) confinement-driven RTP. As an optical probe, the assembly and the two-stage assembly strategy can realize the confined purely organic RTP and achieve energy transfer and light-harvesting from fluorescence to delayed fluorescence or phosphorescence. This supramolecular assembly is widely applied for luminescent materials, cell imaging, and other fields because it effectively avoids oxygen quenching. In addition, the near-infrared excitation, near-infrared emission, and in situ imaging of purely organic room temperature phosphorescence in assembled confinement materials are also prospected.
Introduction
Complex biological processes need to be monitored with optical probes for imaging and sensing that provide information on probe identity, spatial location, and the concentration based on the wavelength and intensity of the signal.1 Therefore, optical imaging plays an indispensable role in the biomedical field.2 However, endogenous background fluorescence or spontaneous fluorescence from cellular organelles has been always a problem, resulting in reduced imaging sensitivity and specificity.3 Given the continuous progress and development of science and technology, the sensitivity, specificity, and achievable spatial and temporal resolution of optical imaging technology have been continuously improved.1a,4 The development of chemiluminescent or Cerenkov probes eliminates the problem of auto-fluorescence because they do not require light excitation, but some problems are still inevitable.2i,5 For example, chemiluminescence usually requires specific enzymes and substrates, which somewhat complicates the imaging process, and Cherenkov luminescence requires radioisotope radiation to be produced, which is also associated with biological safety risks.6 Therefore, it is highly desirable to design new luminescent probes that can overcome the abovementioned drawbacks in biological imaging applications.
Room-temperature phosphorescence (RTP) materials with a longer lifetime, larger Stokes shift, and the involvement of triplet states have garnered attention in the fields of biosensing, bioimaging, and imaging-guided surgery or therapy for providing high signal-to-noise ratio images, because they have persistent luminescence even after removing the light source, allowing in vivo imaging without real-time external excitation.7 However, the RTP effect is common in inorganic compounds containing rare-earth metals or heavy metal complexes and metal–organic compounds, and the high cost and the potential cytotoxicity of metals limit their biological applications.7n,8,9 To avoid the introduction of metals, purely organic RTP materials are good candidates for RTP materials. The high bonding electrons in purely organic materials limit the material ability to decay from the triplet state through the radiation relaxation pathway and are prone to non-radiative relaxation through heat and collision and energy transfer from the triplet state to the triplet state of oxygen.7f,h,n,p,8d,10
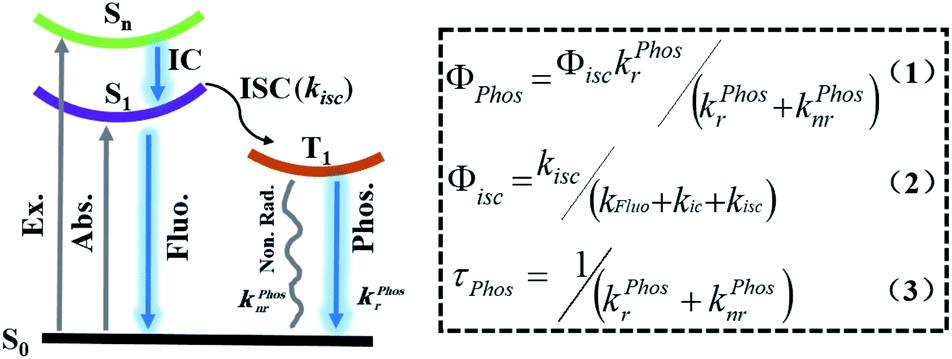
Scheme 1 shows the formulae for the quantum yield of phosphorescence (1), the quantum yield of intersystem crossing (2), and the lifetime of phosphorescence (3).7h,i,n,p,r According to eqn (1) and (2), to obtain high phosphorescence quantum yield, it is necessary to increase the intersystem crossing rate (kisc) and the phosphorescence decay rate (kphosr), and at the same time, reduce the rate of nonradiative transitions (kphosnr) and suppress the influence of quenching groups. On the one hand, kisc and kphosr can be enhanced by adjusting the molecular orbital structure by introducing abundant lone-pair electrons (including O, N, P, S, etc.), introducing heavy atoms (Cl, Br, I, S, Se, etc.), narrowing the energy gap between the singlet and the triplet state (use of donor–acceptor-type molecules for the charge transfer state), and increasing the degree of molecular aggregation. On the other hand, kphosnr and quenching groups can be controlled by improving the rigidity of the phosphorescent structure with the introduction of numerous intermolecular interactions (hydrogen bonding, halogen bonding, ionic bonding, CH–π interaction, π–π interaction, etc.). According to eqn (3), the long-lived RTP needs to minimize kphosnr and kphosr and avoid quencher interference. In addition, the introduction of long-range charge diffusion before the emission of triplet excitons is also a feasible way to improve the lifetime of RTP. Based on eqn (1) and (3), an increase in τphos implies a decrease in Φphos based on the change in kphosr. However, owing to the quenching from the collision of the oxygen dissolved in water and the free molecular motions for nonradiative decay processes, the most efficient purely organic RTP emissions are common in the solid-state, which make them impractical for use in biological applications. Thus, to obtain purely organic RTP in water, three important aspects should be considered: (1) the enhancement of the heavy atom effect to promote intersystem crossing (ISC); (2) a rigid environment to suppress non-radiative transitions in water; (3) avoiding the influence of quenchers such as O2.
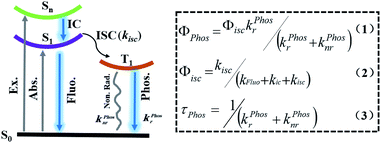 |
| | Scheme 1 Schematic illustration of the Jablonski diagram of the emissive processes of phosphorescence (Φphos: the quantum yield of phosphorescence; Φisc: the quantum yield of intersystem crossing; τphos: the lifetime of phosphorescence; kfluor: the fluorescence decay rate constant; kfluonr: the fluorescence nonradiative rate constant; kisc: the intersystem crossing rate constant from S1 to T1; kphosr: the phosphorescence decay rate constant; kphosnr: the phosphorescence nonradiative rate constant). | |
Unlike traditional covalent interactions, the supramolecular chemistry approach mainly focuses on weak noncovalent interactions, such as hydrogen bonds, π–π stacking, hydrophobic forces, van der Waals forces, and electrostatic interactions; not only do they make the single functional molecule multi-functionalized in a simple way, but they also bring new properties by adjusting the molecular conformation, vibration, energy gap, or electronic distribution.7f,h,i,m,n,10b,11 More importantly, this approach can provide an effective method for the construction of water-phase purely organic RTP. In this mini-review, we briefly introduce the research progress into a supramolecular strategy for the construction of a purely organic aqueous RTP system mediated by macrocyclic hosts (cyclodextrin and cucurbituril), macromolecules (polyether) and nanoassembly confinement, as well as its application in cell imaging. In addition, we provide the key challenges and perspectives for further advances in purely organic aqueous phosphorescence. We hope that this review will bring insight and inspiration to readers and allow the future development of phosphorescence and its biological applications in water. Table 1 presents a list of the chemical structures of the phosphorescent molecules involved in this paper.
Table 1 Chemical structures of phosphorescent molecules
| Structure |
Citation |
Strategy |
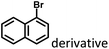
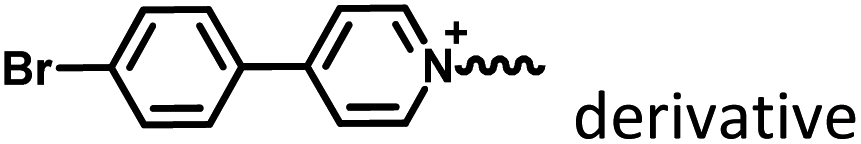
|
Fig. 1–3 and 15 |
Marocyclic confinement |
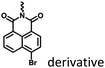
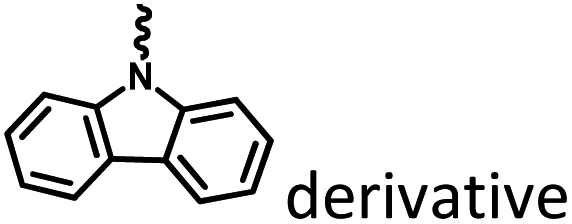
|
Fig. 4, 6 and 19 |
Marocyclic and nanoassembly confinement |
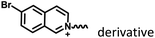
|
Fig. 5 and 7 |
Marocyclic confinement |
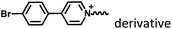
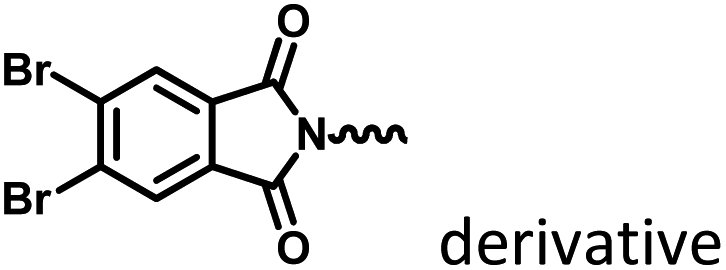
|
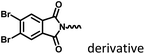
Fig. 8–14
|
Marocyclic confinement |
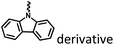
|
Fig. 16 and 21 |
Marocyclic and nanoassembly confinement |
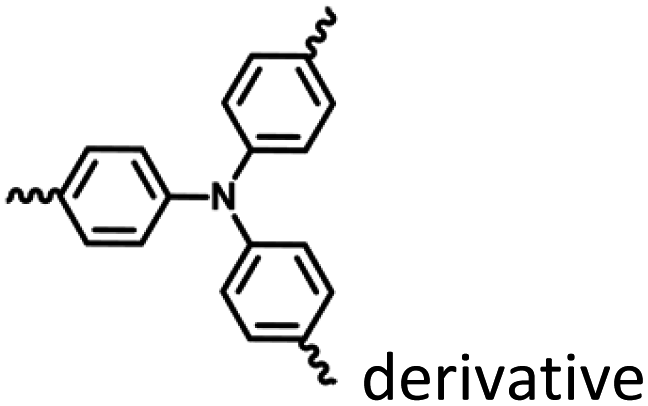
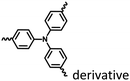
|
Fig. 17 and 18 |
Nanoassembly confinement |
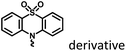
|
Fig. 25 and 28 |
Macromolecular confinement |
|
Fig. 26 and 27 |
Macromolecular confinement |

|
Fig. 18, 23 and 24 |
Nanoassembly and macromolecular confinement |
|
Fig. 20
|
Nanoassembly confinement |
Marocyclic confinement
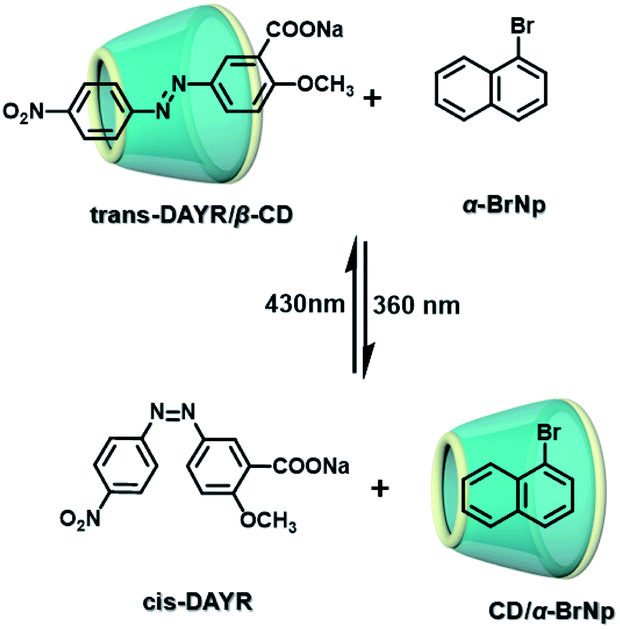
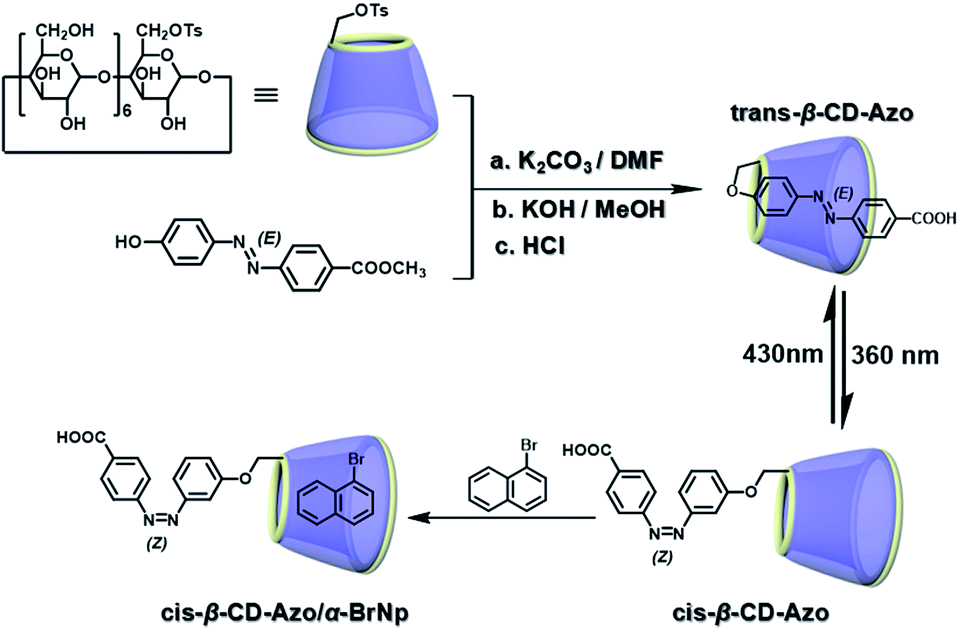
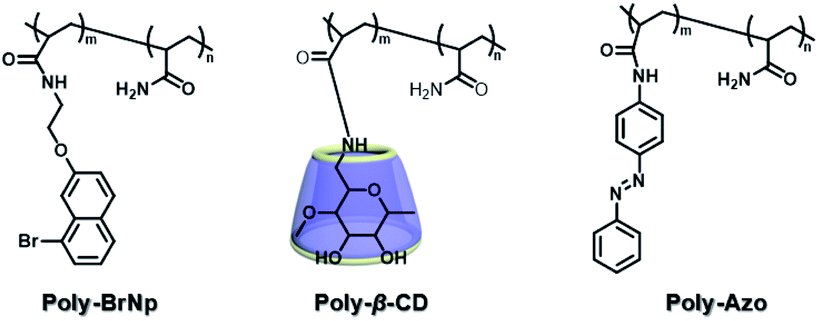
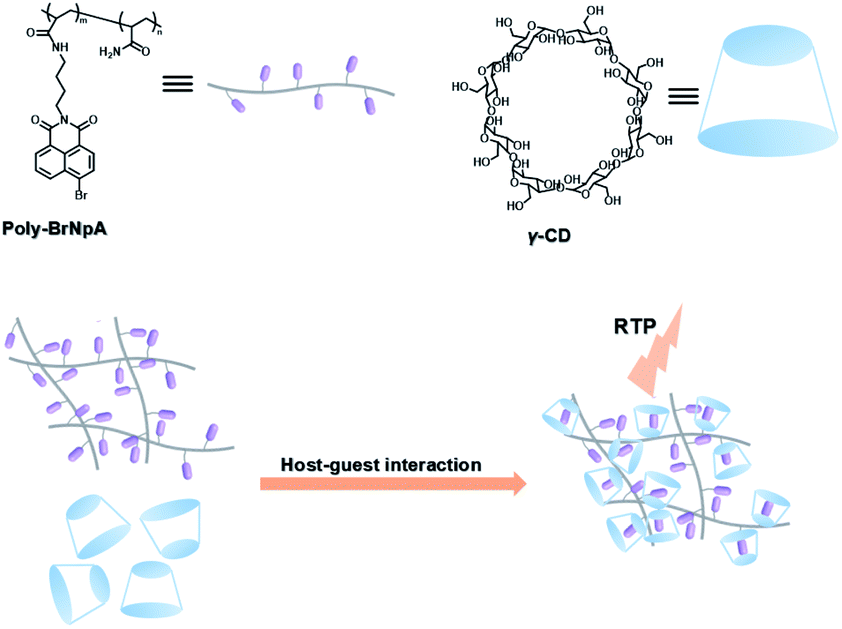
The two main water-soluble biocompatible macrocyclic host compounds, cyclodextrin (CD) and cucurbituril (CB), can provide a hydrophobic cavity for the guest molecule in aqueous solution, which could protect the luminescent guest from the influence of free oxygen and restrict molecular motion for reducing nonradiative decay via host–guest interaction. CDs are a type of cyclic oligosaccharide constructed by D-glucose units through α-1,4-glucose bonds and can form stable host–guest complexes with various neutral or charged phosphorescent molecules.7h,m,11d,12 As early as 1982, Turro et al. achieved phosphorescent emission in nitrogen-purged aqueous solutions based on the inclusion of β-CD with 1-bromonaphthalene and 1-chloronaphthalene.13 Furthermore, the addition of a third component with heavy atoms to the phosphorescent inclusion complex of CD with nitrogen heterocycles or bridged biphenyl compounds could also enhance the phosphorescence effect in water.14 In another case, RTP was enhanced via the formation of the ternary complex 1-bromonaphthalene/glucosyl-modified-β-CD/alcohol, in which the RTP of 1-BrNp is effectively protected from oxygen quenching.15 When 1-BrNp is modified on the secondary OH groups of β-CD, the RTP at 530 nm is achieved in aqueous solutions through the self-inclusion of 1-BrNp into the hydrophobic cavity of β-CD to shield the oxygen.16 Meanwhile, haloalkanes have an external heavy atom enhancement effect on CD-RTP in water in the order Br > Cl ≫ I.17 This is because of the heavy atom effect for enhancing the spin–orbit (S–O) coupling interaction, which can further increase the ISC from the singlet excited state to the triplet state. However, if the heavy atom is too strong, kphosnr may also increase greatly and result in a significant reduction in RTP. Among them, dibromoalkanes are the best of all bromoalkanes, and t-butyl chloride can induce RTP of polycyclic aromatic compounds more effectively. Adding a third part of hexahydropyridine (HHP), cyclohexane (CH), or 1-ethylpiperidine (EP) to 1-BrNp/β-CD aqueous solution could improve the RTP of 1-BrNp/β-CD; in particular, the addition of EP exhibited the strongest RTP due to the large rigidity.18 Furthermore, the photocontrolled phosphorescence of CD-RTP in water can be realized by the assembly and disassembly of cyclodextrin and azobenzene. For example, a photocontrolled reversible RTP pseudorotaxane in aqueous solution was constructed via a β-CD/α-BrNp/sodium 2-methoxy-5-((4-nitrophenyl)diazenyl)benzoate (DAYR) ternary system (Fig. 1).19 In this system, trans-DAYR could form a pseudorotaxane with β-CD. When trans-DAYR converted cis-DAYR via photo-irradiation, the DAYR/β-CD pseudorotaxane was disassembled, and the inclusion complex of α-BrNp/β-CD was formed, achieving RTP, because the order of binding constants was β-CD/trans-DAYR > β-CD/α-BrNp > β-CD/cis-DAYR. In the binary RTP system between the azo modified β-CD and α-BrNp, the RTP of β-CD/α-BrNp could be reversibly controlled via the photoisomerization of the azo group in and out of the cyclodextrin cavity (Fig. 2).20 Moreover, this regulation of phosphorescence can also be achieved in polymers. Three polymers, i.e., poly-BrNp, poly-β-CD, and poly-azo were prepared by the copolymerization of acrylamide and the monomer of α-BrNp, β-CD or azobenzene, respectively (Fig. 3).21 The weak RTP emission was observed upon the addition of β-CD to the polymer poly-BrNp aqueous solution. However, the stronger RTP emission was achieved while the poly-β-CD aqueous solution was added to the poly-BrNp aqueous solution owing to the formation of hydrogel through the host–guest interaction for protecting the BrNp moiety to some extent. When aqueous solutions of three components poly-BrNp, poly-β-CD and poly-Azo were mixed to form a smart hydrogel, photo-stimulated RTP was realized through the trans–cis photo-isomerization of azobenzene units for the threading and de-threading into the cavity of β-CD. Another polycyclic aromatic hydrocarbon 4-bromo-1,8-naphthalic anhydride (BrNpA) was used to investigate RTP in water (Fig. 4),22 and a photo-controlled RTP supramolecular assembly was constructed in aqueous solution through the ternary poly-BrNpA/γ-CD/poly-azo system, where the phosphorescence excitation and emission wavelengths presented large red shifts compared with previously reported systems.
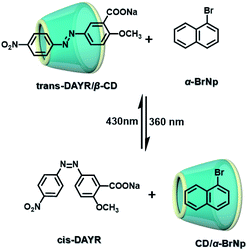 |
| | Fig. 1 Schematic illustration of photo-controlled reversible RTP pseudorotaxane in aqueous solutions. | |
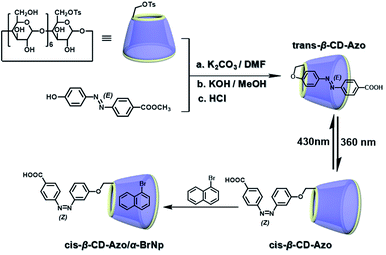 |
| | Fig. 2 Schematic illustration of photo-controlled reversible RTP supramolecular assembly based on azo-β-CD and α-BrNp in aqueous solutions. | |
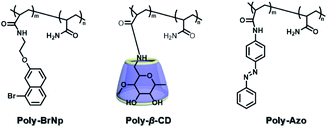 |
| | Fig. 3 Schematic illustration of construction of the compound structure of the photoresponsive supramolecular RTP gel based on β-CD and bromonaphthalene. | |
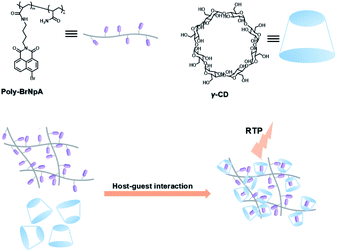 |
| | Fig. 4 Schematic illustration of construction of photocontrolled RTP supramolecular assembly in aqueous solution through the poly-BrNpA/γ-CD/poly-azo system. | |
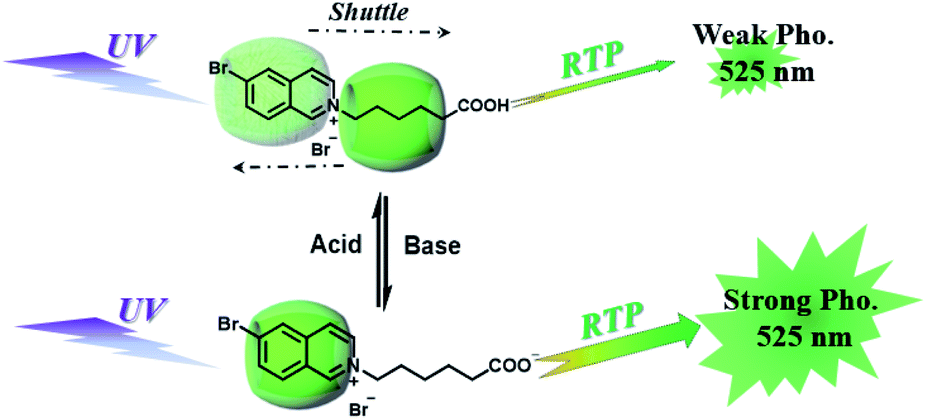
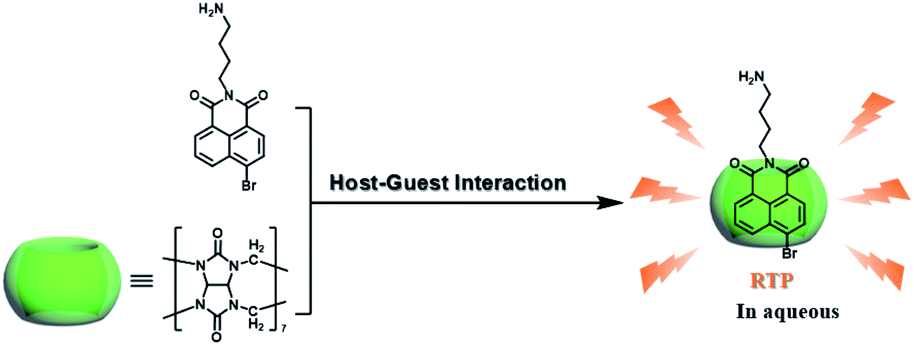
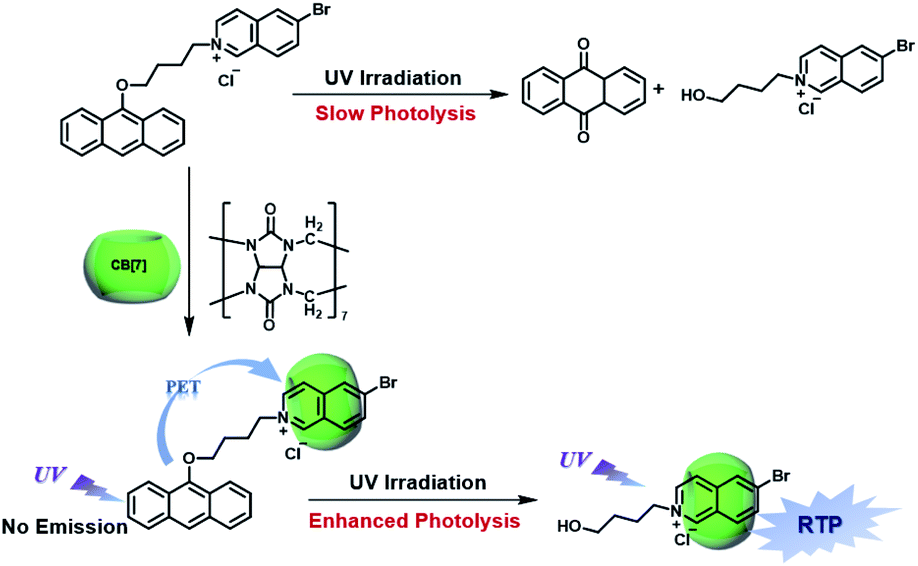
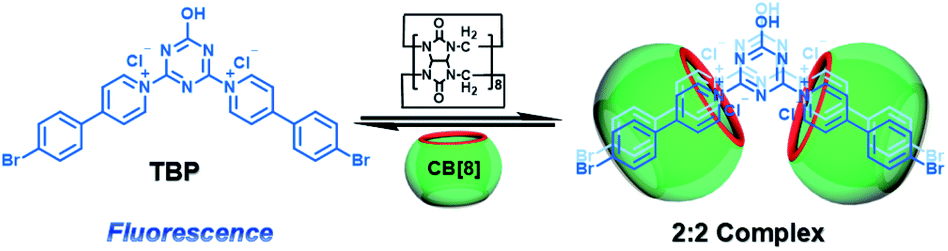
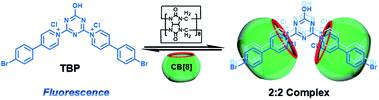
CBs are a class of water-soluble macrocycle compounds with different numbers of glycoluril units linked by methylene bridges that are able to form stable host–guest complexes with positively charged organic or inorganic guests in aqueous solution.11a,d,12a,23 The binding constant between CBs and guests is usually much larger than the corresponding value between CDs and guests due to the strong charge–dipole and hydrogen bonding interactions, as well as the hydrophobic/hydrophilic effect. This strong host–guest interaction can protect and immobilize the phosphor guest efficiently to improve their luminescence efficiency and lifetime in water. The RTP of quinoline and its derivatives could be induced through the formation of inclusion complexes with the different CBs in aqueous solution, and the effect of RTP was affected by the variation in heavy metal ions and pH.24 In this system, the heavy atom effect of the Ti+ cation was stronger than that of the I− anion, and the lower pH could enhance the RTP due to the strong binding constant. The optical properties of inclusion complexes of different CBs with 2,4,6-triphenylpyrylium were studied in water, and the addition of CB[8] could produce strong RTP at 595 nm as a result of the CB carbonyl groups and the conformational immobilization inside the CB.25 Tian and Ma also performed a series of studies on this feature. For example, a pH-controlled molecular shuttle in aqueous solution was prepared based on CB[7] and a 6-bromoisoquinoline derivative (Fig. 5).26 In this shuttle, strong RTP was observed under basic conditions, resulting from the strong electrostatic repulsion between the carboxyl anion of the deprotonated terminal carboxylic group and the high-density electron atmosphere of CB[7], leading to the encapsulation of the quinoline aromatic region in CB[7] for protecting the triplet state. Then, RTP was realized through the host–guest complex of CB[7] and 2-(4-aminobutyl)-6-bromo-1H-benzo[de]isoquinoline-1,3(2H)-dione (ANBrNpA) in water (Fig. 6).27 The RTP was further enhanced by the association of the 4-bromo-1,8-naphthalic anhydride polymer (poly-BrNpA) with CB[7]. Subsequently, a photoresponsive RTP system was constructed in aqueous solution by CB[7] and amphiphilic 6-bromoisoquinoline-functionalized alkoxyanthracene (AnBq).28 In this system, the photoinduced electron transfer (PET) from anthracene to 6-bromoisoquinoline occurred for AnBq@CB[7], and therefore, RTP was not observed. When AnBq@CB[7] underwent photolysis (Fig. 7), 6-bromoisoquinoline@CB[7], which lacked the anthracene group, had no intramolecular PET, resulting in phosphorescence. Recently, visible light-excited water-soluble RTP was achieved via CB[8] and a double-sided modification of 4-(4-bromophenyl)-pyridine with the triazine core for the two-branched molecule TBP.29 In this system, a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 TBP-CB[8] quaternary complex was acquired as CB[8] was added into the TBP aqueous solution (Fig. 8). However, with the gradual increase in CB[8], multicolor emission could be realized by controlling the different proportions of the TBP monomer, and the (TBP)2@CB[8]2 assembly with yellow phosphorescence could also be regulated through the addition of a competitive guest (1-amino-3,5-dimethyladamantane hydrochloride). Meanwhile, a multicolor emission hydrogel was obtained via the mixture of (TBP)2@CB[8]2 aqueous solution with different colors and an agarose gelator; finally, cell imaging was performed in the endosomes by taking advantage of the visible light-excited RTP in water.
:2 TBP-CB[8] quaternary complex was acquired as CB[8] was added into the TBP aqueous solution (Fig. 8). However, with the gradual increase in CB[8], multicolor emission could be realized by controlling the different proportions of the TBP monomer, and the (TBP)2@CB[8]2 assembly with yellow phosphorescence could also be regulated through the addition of a competitive guest (1-amino-3,5-dimethyladamantane hydrochloride). Meanwhile, a multicolor emission hydrogel was obtained via the mixture of (TBP)2@CB[8]2 aqueous solution with different colors and an agarose gelator; finally, cell imaging was performed in the endosomes by taking advantage of the visible light-excited RTP in water.
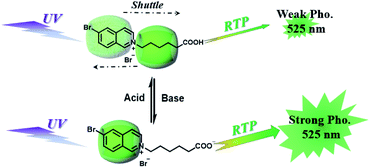 |
| | Fig. 5 Schematic illustration of the construction of a pH-controlled molecular shuttle in aqueous solution based on CB[7] and the 6-bromoisoquinoline derivative. | |
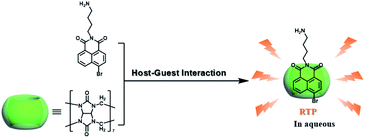 |
| | Fig. 6 Schematic illustration of the construction of supramolecular aqueous RTP assembly based on CB[7] and ANBrNpA. | |
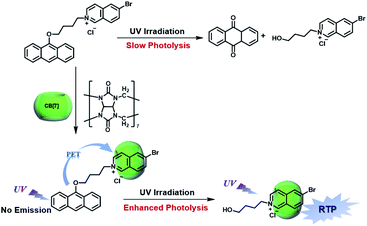 |
| | Fig. 7 Schematic illustration of construction of photoresponsive RTP supramolecular assembly by CB[7] and amphiphilic 6-bromoisoquinoline. | |
 |
| | Fig. 8 Schematic illustration of construction of the TBP-CB[8] 2:2 quaternary RTP supramolecular assembly via CB[8] and TBP. | |
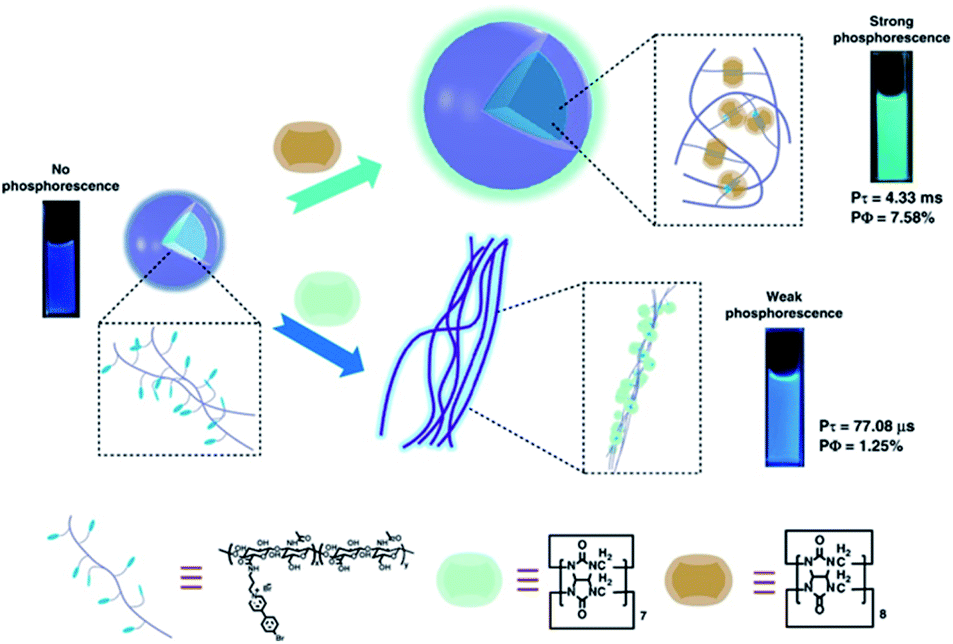
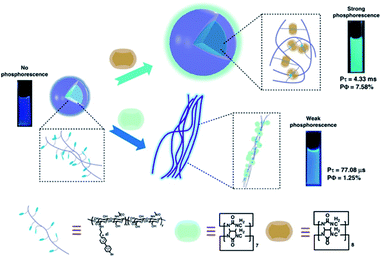
More recently, Liu and coworkers combined the 4-(4-bromophenyl)pyridine-1-ium bromide (BrBP) phosphor with a tumor-targeted hyaluronic acid (HA) polymer chain30 subsequently assembled with cucurbituril (Fig. 9).31 BrBP, a type of organic phosphor, possessed a positive charge that could bind with CB.32 HA is a water-soluble, biocompatible, biodegradable polymer chain that can be used as a targeting reagent owing to specific binding with CD44 and RHAMM receptors that are overexpressed on the surface of cancer cells.30 The encapsulation of CB[8] provided a rigid hydrophobic microenvironment to protect the phosphor guest from the collision of the triplet oxygen and reduce nonradiative decay as well as enhance ISC through the interactions of π–π stacking, halogen bonding, and multiple hydrogen bonding. The BrBP-modified HA formed small spherical aggregates. After complexation with CB[8], larger aggregates were formed and resulted in purely organic RTP in water with a lifetime of up to 4.33 ms (quantum yield 7.58%). In this system, the 1:2 strong inclusion complex of CB[8] with BrBP, as well as the interaction of CB[8]/BrBP complexes with HA, resulted in strong charge–dipole, π–π stacking, and hydrogen bonding interactions, and a hydrophobic effect, which prevented the influence of a quenching agent in aqueous solution, inhibited molecular rotation, and enhanced ISC. Furthermore, cell imaging was performed taking advantage of cancer cell targeting properties of HA via confocal laser scanning microscopy after incubation with the supramolecular polymer HA-BrBP/CB[8] and acquired optical signals of phosphorescence emission. The results in vitro revealed that the supramolecular polymer preferentially targeted the mitochondrion of tumor cells over normal cells and had negligible cytotoxic effects at the concentrations used for imaging. Meanwhile, the problem of UV excitation is expected to be solved by attempts to combine the supramolecular polymer with upconversion nanoparticles (UCNPs).
 |
| | Fig. 9 Schematic illustration of the self-assembly of the ultralong water-soluble purely organic RTP supramolecular polymer by supramolecular and macromolecular effects. | |
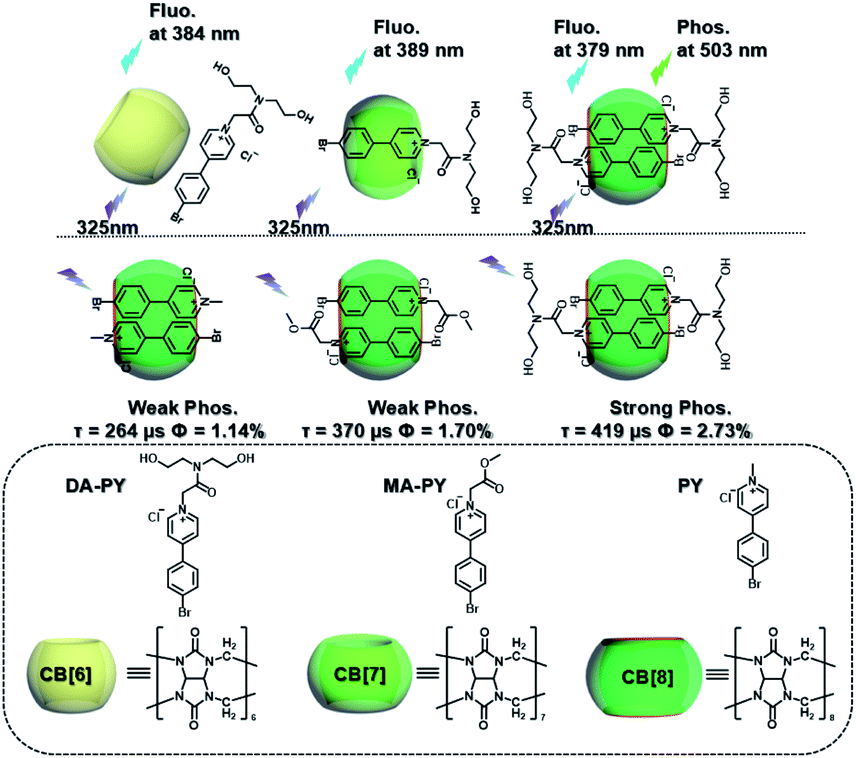
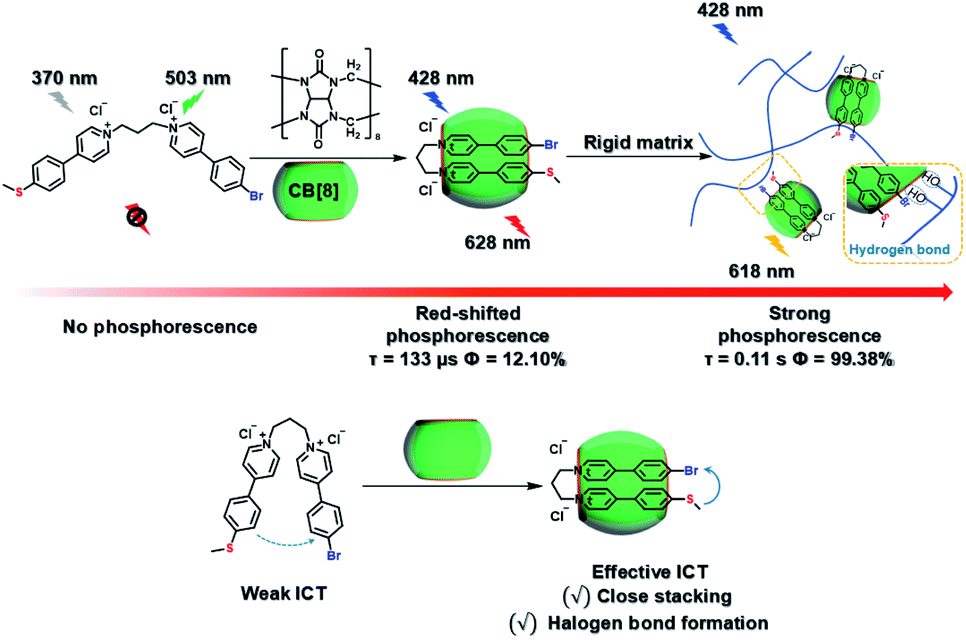
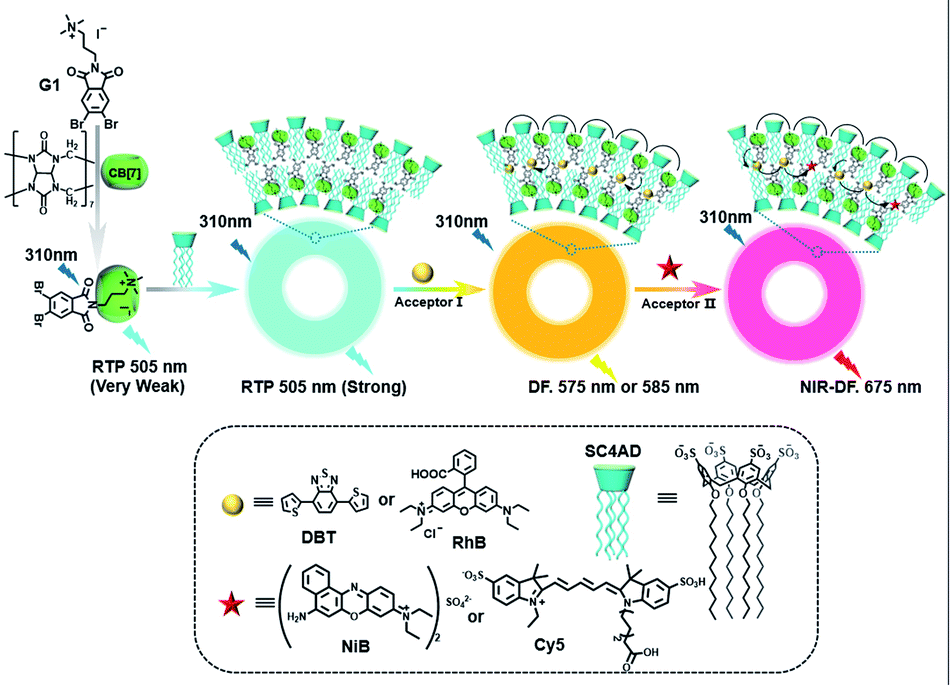
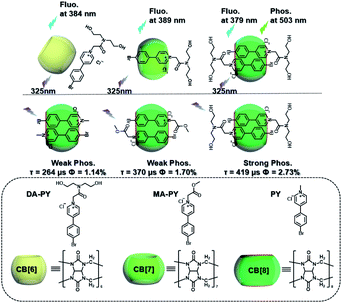
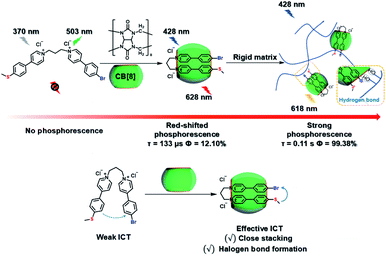
Meanwhile, a “two-end blocked” pseudorotaxane was constructed based on CB[8] and diethanolamine-modified bromopyridine salt (DA-PY), as shown in Fig. 10.33 This special rotaxane structure could not only increase the phosphorescence emission in the water phase via reducing the nonradiative transition and enhancing halogen bonding interaction, but also realized the targeted imaging of cancer cell mitochondria. Additionally, supramolecular phosphorescent pins with afterglow luminescence and ultra-high quantum yield (99.38%) were designed through combining the assembly of alkyl-bridged phenylpyridinium salts and CB[8] into a rigid matrix (Fig. 11).34 This high quantum yield arises from: (1) the suppressed nonradiative decay of the chromophore from the host–guest interaction of the chromophore with CB[8] and flexible chains; (2) the “one host and one guest” folding mode promoting the effective ICT for increasing the rate of ISC; (3) the intramolecular halogen bonding improving the rate of radiative decay. Furthermore, it was revealed that ICT and halogen bonding together promote RTP performance through the spectral comparison of reference compounds with only the ICT structure or only the halogen bond structure. However, there also exists a possibility other than the CT effect, where the main contribution comes from the heavy-atom induced RTP that is enhanced in a CB[8] cavity. Finally, this supramolecular pin was successfully used for phosphorescence imaging located in the mitochondria owing to the red-shift of the absorption.
 |
| | Fig. 10 Schematic illustration of the construction of “two-end blocked” pseudorotaxane based on CB[8] and DA-PY. | |
 |
| | Fig. 11 Schematic illustration of construction of supramolecular phosphorescent pins via the assembly of CB[8] and alkyl-bridged phenylpyridinium salts. | |
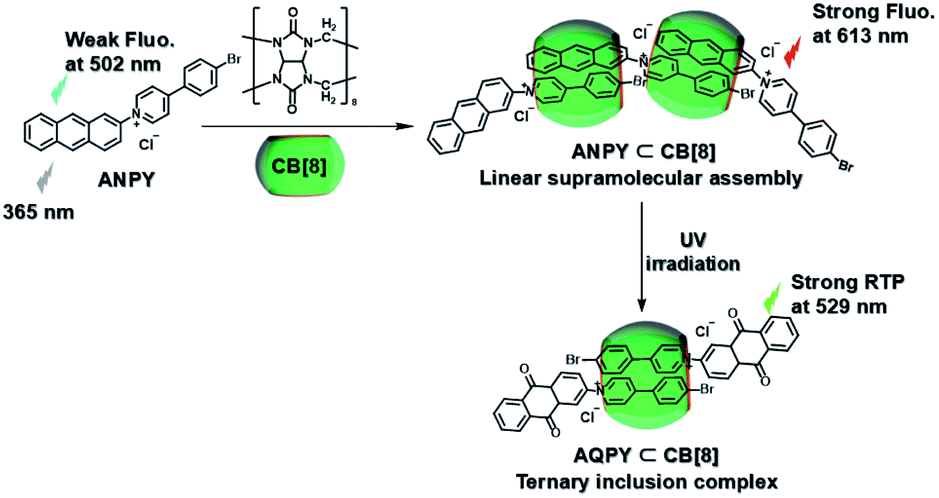
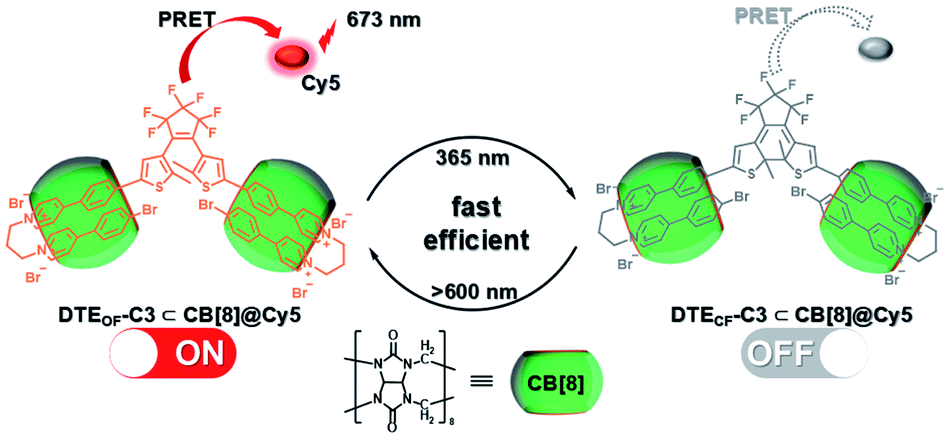
Subsequently, the emission of purely organic RTP could be controlled by introducing photoresponsive groups. Anthracene, a reversible response group that can perform photodimerization and photooxidation, is favored by scientists.35 Moreover, the topological structure and fluorescence emission behavior could be controlled by modifying the anthracene groups on the π–aromatic cores, which prompted us to examine combining it with the phosphorescent groups to achieve the effective regulation of phosphorescent emission. Therefore, a bromophenylpyridinium salt was conjugated at the 2-position of anthracene (Fig. 12),36 and the resultant compound displayed weak fluorescence emission at 500 nm. After adding CB[8], linear supramolecular aggregates were formed, and the fluorescence became stronger and red-shifted to 613 nm. In view of the photoresponse of anthracene, an illumination experiment showed that the morphology of the assembly was transformed from the linear head-to-tail inclusion complex into a head-to-head homoternary complex under 365 nm irradiation, accompanied by the conversion of fluorescence to phosphorescence (529 nm). Unexpectedly, this change in optical properties afforded two-organelle imaging from the nucleus to the lysosome in cells. In addition, diarylethylene derivatives are a class of molecules with photoisomerism and very good fatigue resistance.37 Their configuration changes lead to different absorbances; this is conducive to the realization of optical property regulation. Therefore, the phosphorescent groups 4-(4-bromophenyl)-pyridinium and 4-(4-(5-methylthiophen-2-yl)phenyl)-pyridinium were connected on both sides of diarylethene through three carbon chains (Fig. 13).38 In the presence of CB [7], due to the 1:1 bonding with the pyridine salt, the restriction of molecular motion is weak, and the emission of RTP cannot be achieved. In the presence of CB[8], owing to the strong 1:2 bonding with pyridine salt, π–π stacking could be enhanced to form a single-molecule dual-fold structure, and the RTP emission could be realized. Interestingly, the assembly emission overlapped well with the absorption of the near-infrared dye Cy5 (emission: 673 nm) to produce phosphorescence energy resonance transfer (PRET), and the process was regulated by the open–close ring of the diarylethene. This light-controlled PRET assembly displayed good results in cell imaging.
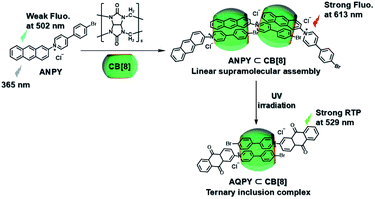 |
| | Fig. 12 Schematic illustration of the construction of photoresponsive linear supramolecular aggregates based on CB[8] and anthracene-modified bromophenylpyridinium salt. | |
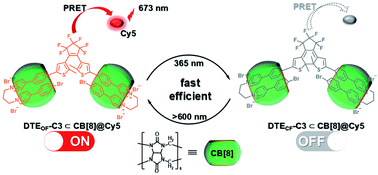 |
| | Fig. 13 Schematic illustration of the construction of photocontrolled supramolecular phosphorescence energy transfer based on the diarylethene-modified bromophenylpyridinium salt. | |
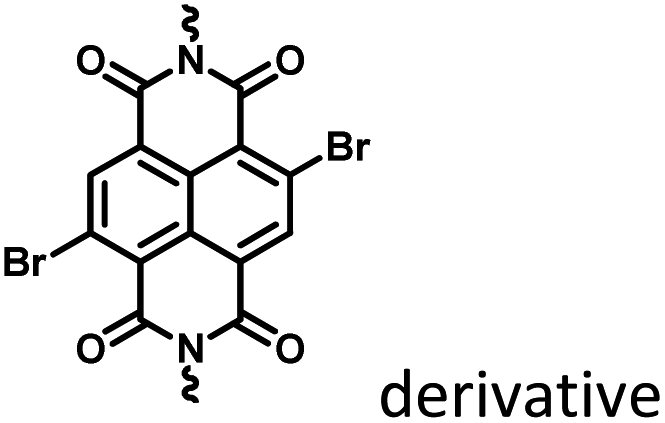
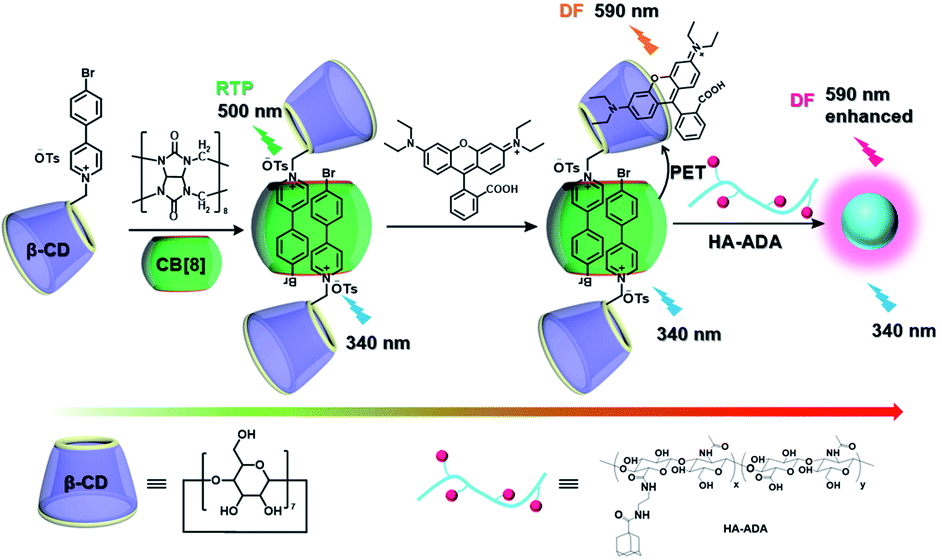
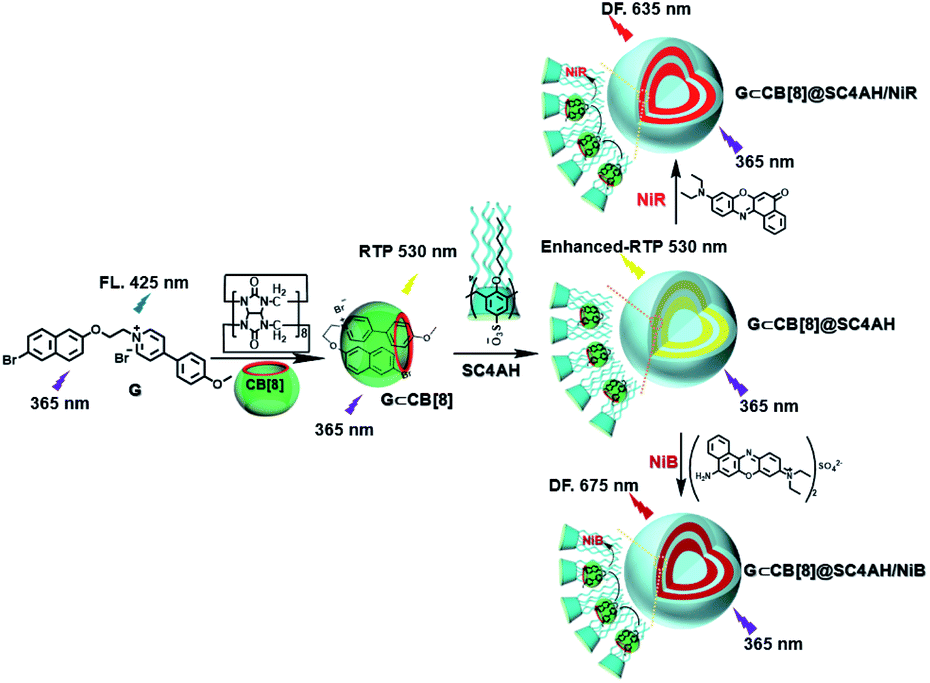
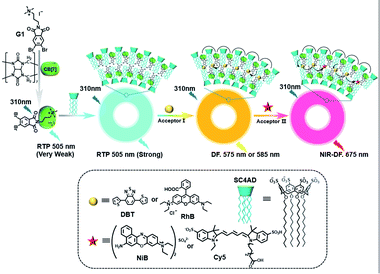
The use of purely organic RTP and near-infrared light emitting dyes can solve the problem of shallow imaging depth caused by most RTP emissions in the UV or visible range through energy transfer.7q,39 In a purely organic light-harvesting phosphorescence energy transfer system (Fig. 14) for mitochondria-targeted imaging constructed from 4-(4-bromophenyl)-pyridine modified β-cyclodextrin (CD-PY), CB[8], rhodamine B (RhB), and adamantine-modified hyaluronic acid (HA-ADA), CD-PY@CB[8] acted as the phosphorescent donor, RhB acted as the phosphorescent acceptor, and HA-ADA acted as the cancer cell targeting agent.40 Free CD-PY has no phosphorescence in water, and the manifested phosphorescence emission was observed at 510 nm as CB[8] was added to form a host–guest inclusion complex. However, the PET process occurred when RhB was gradually added into the above system, and the phosphorescence of CD-PY@CB[8] decreased gradually at 510 nm with a new phosphorescence peak appearing at 590 nm. Interestingly, the addition of HA-ADA/CD into the above system not only further enhanced phosphorescence emission but also successfully presented mitochondria-targeted imaging properties to cancer cells. For the supramolecular pin system, the emission of the folded assembly constructed by CB[8] and bromonaphthalene-modified methoxypyridine changed from fluorescence at 425 nm to strong RTP at 530 nm (lifetime 130.2 μs) as CB[8] enhanced the charge transfer interaction to promote ISC (Fig. 15).41 After the secondary assembly with amphiphilic calixarene (SC4AH), the G1/CB[8] assembly entered the hydrophobic region, and its phosphorescent properties were further improved (luminescence intensity increased by 1.5 times; lifetime increased by 3.3 times). Moreover, a light-harvesting system from phosphorescence to near-infrared (NIR) fluorescence emission was realized using a ternary assembly G1@CB[8]@SC4AH as the donor and Nile red (emission: 635 nm) and Nile blue (emission: 675 nm) as the receptors (antenna effect: 352.9 and 123.5; energy transfer efficiency 64.1% and 49.6%), respectively. This system was successfully applied to the NIR lysosome imaging of A549 cancer cells. Furthermore, by using this secondary assembly method, a two-stage phosphorescence energy transfer system was constructed (Fig. 16).42 First, di-bromophthalimide derivative molecules were pre-assembled with CB[7], which promoted ISC to a certain extent and produced a weak phosphorescence at 505 nm when CB[7] was associated with the alkyl chain. After further assembly with amphiphilic SC4AH, the phosphorescence emission intensity of the assembly increased by 40 times, and the lifetime reached 1.13 ms. Taking the assembly as the donor and RhB or benzothiadiazole (DBT) as the receptor, efficient energy transfer from phosphorescence to fluorescence was achieved, with a transfer efficiency of 84.4% or 76.3% and an antenna effect of 289.4 or 119.5, respectively. Interestingly, after the addition of NIR dye Cy5 or Nile blue, which have better spectral overlap with RhB or DBT, second-level phosphorescence to fluorescence energy transfer was realized, and its emission occurred over a wide range of emission from 425 nm to 800 nm. This secondary assembly was incubated with cells for 12 hours to explore the delay effect, and its emission at 505 nm, 585 nm, and 675 nm showed the corresponding time-resolved fluorescence signals after a delay of 50 μs. Finally, multicolor range imaging was realized in the lysosome of A549 cells.
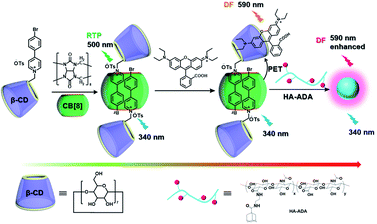 |
| | Fig. 14 Schematic illustration of the construction of light-harvesting phosphorescence energy transfer supramolecular assembly via CD-PY, CB[8], RhB, and HA-ADA. | |
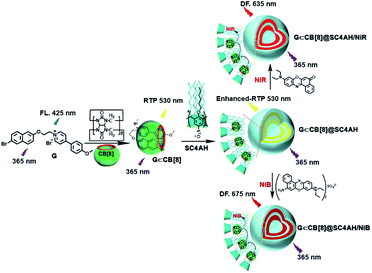 |
| | Fig. 15 Schematic illustration of the construction of light-harvesting phosphorescence energy transfer supramolecular assembly via SC4AH, CB[8], and bromonaphthalene-modified methoxypyridine. | |
 |
| | Fig. 16 Schematic illustration of construction of a two-stage light-harvesting phosphorescence energy transfer supramolecular assembly via SC4AH, CB[8], and di-bromophthalimide derivative. | |
Nanoassembly confinement
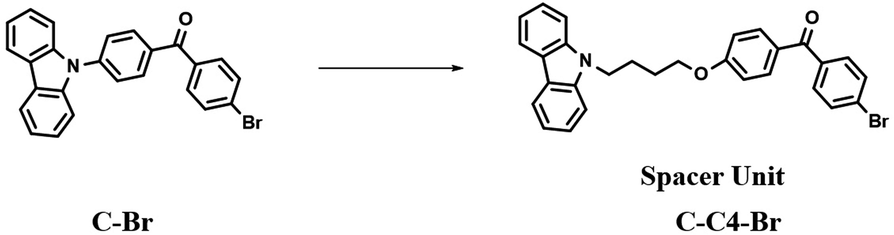
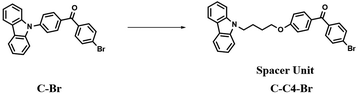
The construction of nanoassemblies, including biocompatible and small size nanocrystals or aggregates, through hydrogen bonding, π–π stacking, and electrostatic interactions in aqueous media is an effective means to achieve the superior phosphorescence imaging of cells.43a,44 The previously reported high-efficiency red phosphorescent compound C–Br was improved by the Liu group (Fig. 17),10a and the carbazole and 4-bromobenzophenone groups were connected through an alkyl chain to give it a flexible conformation. The improved molecule can easily promote the formation of Br–H through a change in the molecular structure stacking mode for increasing the intermolecular electronic coupling (IEC) and the heavy halogen atom effect, thus making the constructed nanocrystals display purely organic red phosphorescent emission in water, which is thus successfully applied in the afterglow imaging of cancer cells.
 |
| | Fig. 17 Schematic illustration of the structures of phosphorescent compounds C–Br and C–C4–Br. | |
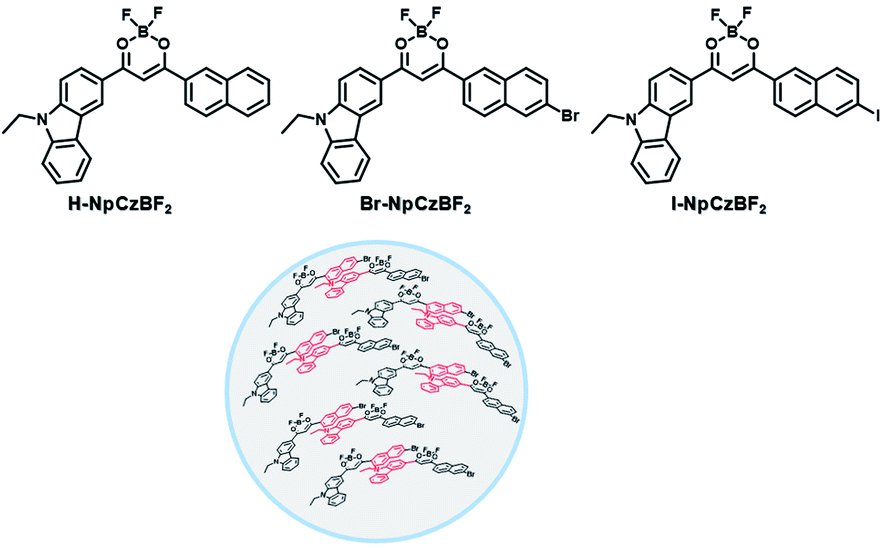
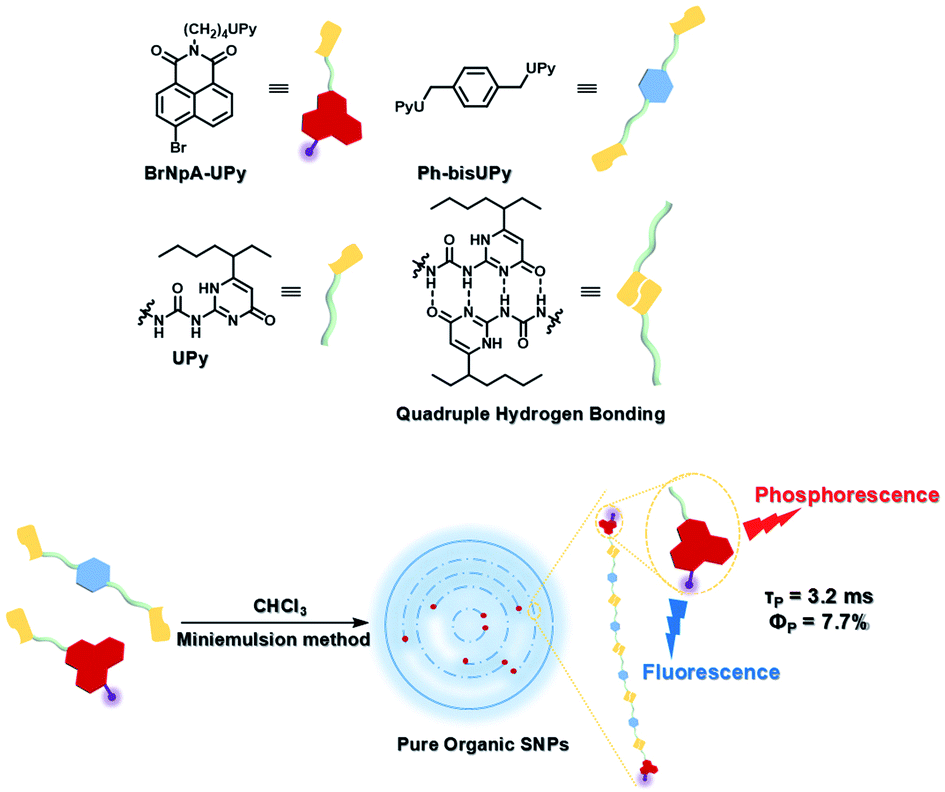
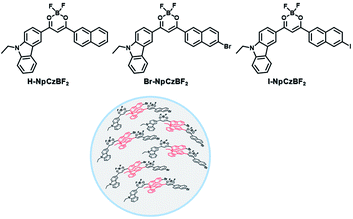
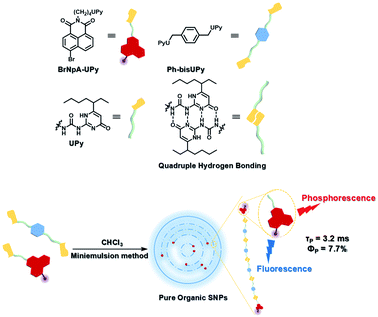
To avoid the problems of UV light-induced phototoxicity and lower light permeability to tissues and cells, three compounds with different substituents (H-NpCzBF2, Br-NpCzBF2, and I-NpCzBF2) were prepared based on difluoroboron β-diketonate (BF2bdk) and carbazole (Fig. 18),7j in which BF2bdk has a two-photon character. In addition, the heavy atoms on the naphthalene molecule and the charge transfer and multiple bond interactions of the dimer could promote ISC and reduce nonradiative transitions for improving RTP with NIR excitation. They then constructed H-NpCzBF2, Br-NpCzBF2, and I-NpCzBF2 self-assembled dimeric nanoparticles in water by a nanoprecipitation method and achieved purely organic room temperature phosphorescence emission at approximately 630 nm by using two types of excitation (visible light at 470 nm or NIR light at 820 nm). Furthermore, the phosphorescence emission of the three molecules was also observed in the cytoplasm of HeLa cells excited by visible or NIR light. However, the phosphorescence phenomenon based on carbazole derivatives was further confirmed by Liu and co-workers in 2021,8i in which the phosphorescence originates from the presence of a small number of carbazole isomers. Among them, two types of structure undergo charge transfer after photoexcitation to form a charge-separated state, and the charge-separated state is trapped and detrapped by defects, resulting in long-lived phosphorescence. However, to realize phosphorescence emission in aqueous solution, it is necessary to prepare nanoassemblies by π–π stacking, hydrogen bonding, and electrostatic interaction, which prevents nonradiative transition and the influence of free oxygen and other quenching groups on the high quantum yield. In addition, based on the sensitivity of phosphorescence to oxygen (O2), the intracellular O2 level could be read out accurately from the optical signals. The degree of hypoxia is an important parameter for the early diagnosis of cancer, cardiovascular disease, and stroke. For the quantification of molecular O2, phosphorescence systems based on transition metal complexes are common; this is not only problematic because of the difficult synthesis, high cost, and cytotoxicity, but is also a challenge regarding stability. Subsequently, a 2-ureido-4-[1H] pyrimidinone (UPy)-functionalized bromine-substituted naphthalimide derivative was designed (BrNpA-UPy) (the UPy part is a quadruple hydrogen bonding unit that can form a supramolecular polymer in chloroform), which could be further dimerized in chloroform with the bis-UPy modified benzyl (Ph-bisUPy) through a microemulsion method, and then assembled in water to form a supramolecular aggregate (Fig. 19).7g Due to the interaction of quadruple hydrogen bonds and the heavy atom effect, the aggregate could emit a dual emission of 396 nm fluorescence and 570 nm phosphorescence at 348 nm under nitrogen conditions, revealing ratio change characteristics, notably, the phosphorescence disappears in air as it is sensitive to oxygen while the fluorescence emission part is unaffected. The aggregate could detect hypoxia in the cytoplasm of HeLa cancer cells by showing different emission colors in different oxygen concentration environments.
 |
| | Fig. 18 Schematic illustration of the structures of the different substituents (H-NpCzBF2, Br-NpCzBF2, and I-NpCzBF2) based on difluoroboron β-diketonate (BF2bdk) and carbazole. | |
 |
| | Fig. 19 Schematic illustration of the phosphorescence self-assembly of UPy-functionalized BrNpA-UPy. | |
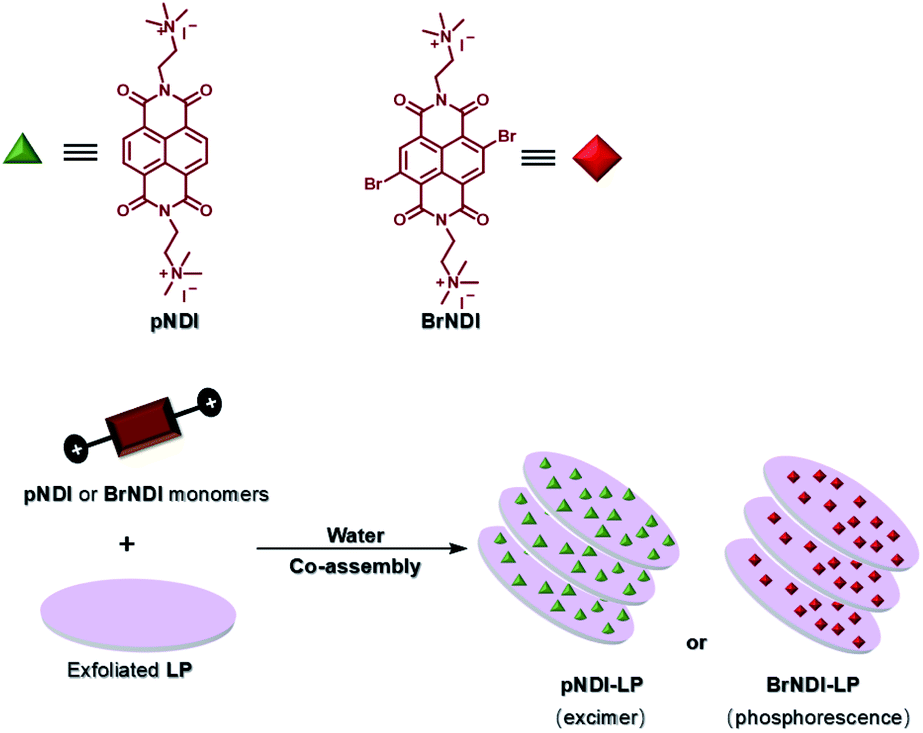
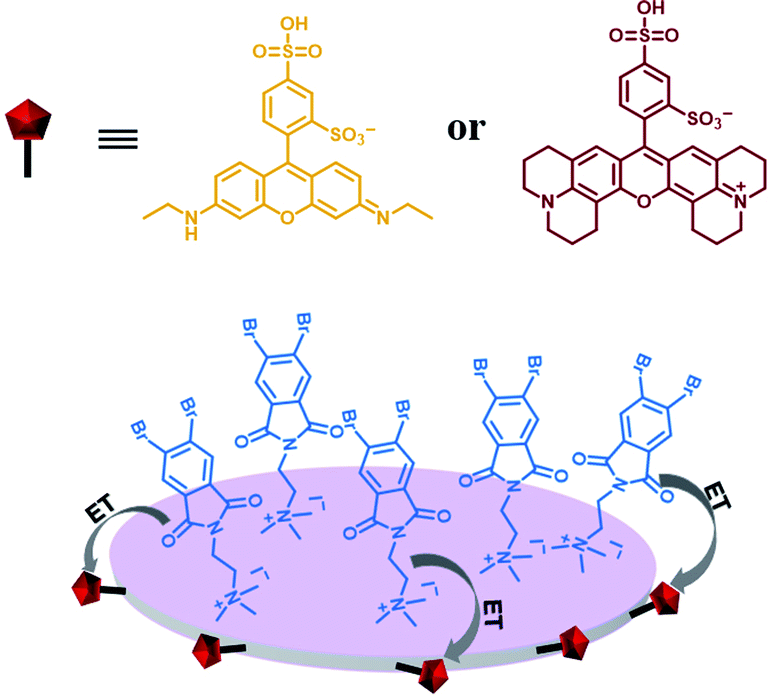
Furthermore, water-soluble LAPONITE® (LP) clay nanoplates are an inorganic scaffold with a negatively charged surface and positively charged edge structure.39 The positively charged edge can be pre-neutralized by the negatively charged polymer chain, which prevents the packing of LP. George et al. utilized the negatively charged surface of LP to assemble with positively charged bromine-substituted naphthalenediimine derivatives on both sides, thereby achieving deep red phosphorescence (emission = 615 nm) in water (Fig. 20).39a Subsequently, they also reported a highly efficient light-harvesting system from phosphorescence to delayed fluorescence, which was assembled by heavy atom-substituted cationic phthalimide derivative/nanoclay hybrids (emission = 530 nm) with the negatively charged fluorescent dyes sulforhodamine G (emission = 560 nm) and sulforhodamine 101 (emission = 610 nm) with energy transfer efficiencies of 32% and 60% (D:A = 100:10), respectively (Fig. 21).39e
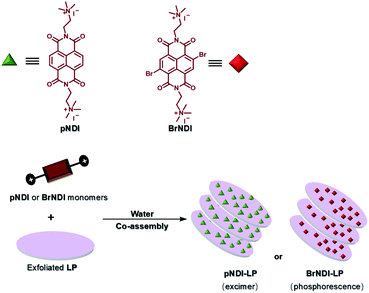 |
| | Fig. 20 Schematic illustration of the proposed LAPONITE® ionic hybrid self-assembly. | |
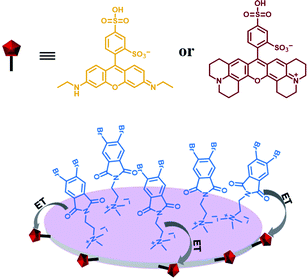 |
| | Fig. 21 Schematic illustration of the LP-based phosphorescence harvesting assembly. | |
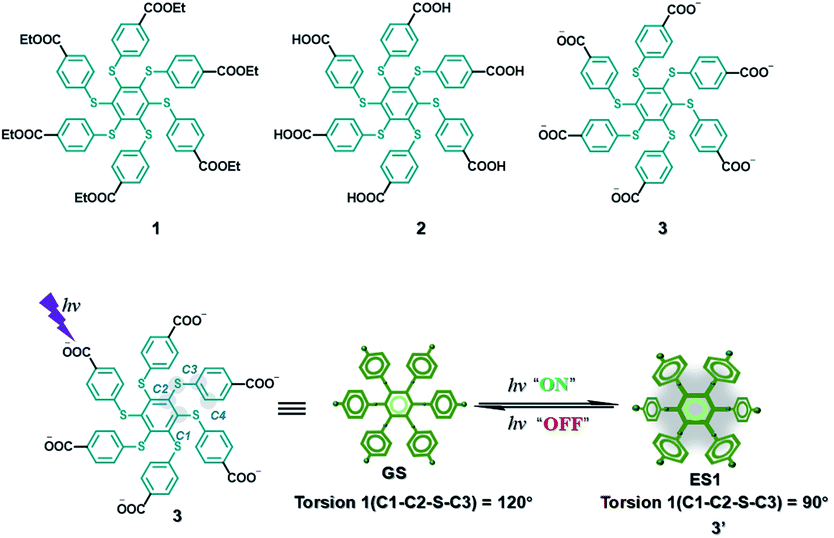
AIE is a phenomenon with high luminescence in an aggregated or confined state.45,46 Molecules with this property could overcome the quenching effect caused by aggregation. This property has a certain promoting effect on the realization of water phase phosphorescence. A hexathiobenzene persulfurated aromatic molecule was designed with six carboxylate groups on the periphery (Fig. 22),47 which facilitated control of the ISC process through environmental changes to achieve RTP. The amphiphilic molecule 3 exhibits photoactivation and self-recovery of aggregation-induced phosphorescence (AIP) in water, and the reciprocation comes from the change in the degree of aggregation caused by the molecular conformational alteration and the dynamics of the dark lowest excited state. Similarly, the design of the two other reference molecules 1 and 2 confirmed this result. When an aqueous solution of molecule 3 was incubated with HeLa cancer cells, the photocontrollable phosphorescence effect was also achieved through rhythmic irradiation.
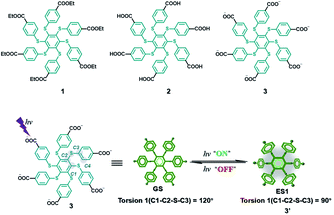 |
| | Fig. 22 Schematic illustration of the phosphorescent molecular structure. | |
Macromolecular confinement
Influence factors of purely organic room temperature phosphorescence mainly have two aspects: one is the non-radiative transition, and the other is the quenching caused by the collision of oxygen.7f,h,i,10b In the solid state, the non-radiative transition can be minimized, and the collision with oxygen can also be reduced, so the design of solid-state purely organic room temperature phosphorescent molecules is easier to implement. In addition, amphiphiles are compounds or polymers with hydrophilic and hydrophobic fragments that can self-assemble to form well-defined structures in aqueous solutions, such as micelles and vesicles.48 The hydrophobic drugs or molecules are encapsulated in hydrophobic fragments of amphiphiles through non-covalent interactions (π–π stacking, hydrophobic forces and hydrogen-bonding), and the hydrophilic segment could not only increase the water-solubility and stability of the hydrophobic carrier but also improve the EPR effect in the tumor part for enhancing the targeting.49 Therefore, it is possible to combine the hydrophobic phosphorescent molecules with a supramolecular amphiphilic system, so that their π–π stacking can be deposited in the hydrophobic region to prevent oxygen attack, which may retain excellent phosphorescent properties in the solid state.
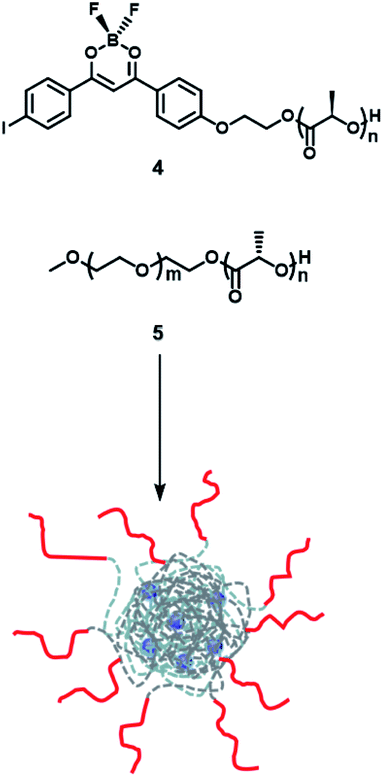
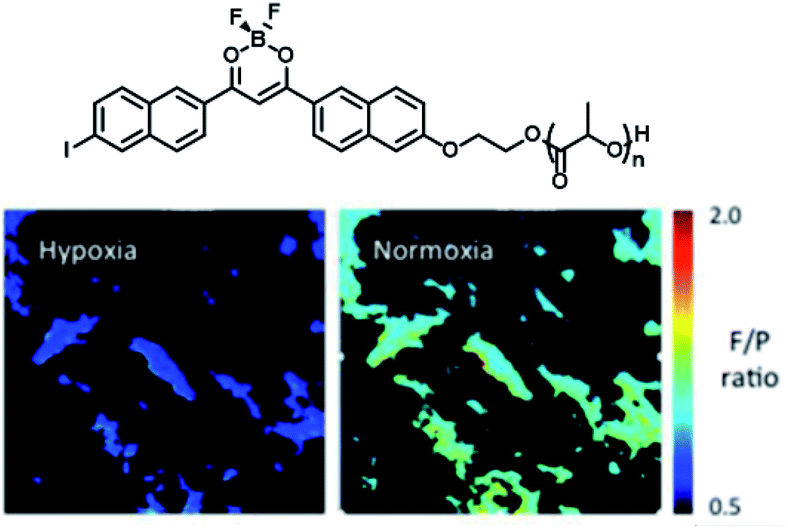
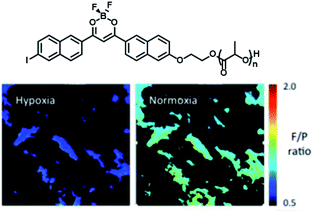
Frase et al.50 reported the phosphorescence phenomena in water through a nanoprecipitation method using iodide-substituted difluoroborondibenzoylmethane-poly(L-lactide) (BF2dbm(I)PLLA, 4) and methoxy-terminated poly(ethylene glycol)-b-poly(D-lactide) (mPEG-PDLA, 5) (Fig. 23). Compounds 1 and 2 could be assembled by stereo-complexation of hydrophobic PLA segments in DMF or mixed solvents (DMF/THF), with hydrophilic PEG chains outside. The constructed amphiphilic assembly dispersed in water, which could not only promote uptake of cancer cells by the EPR effect but also realize phosphorescence/fluorescence colorimetric detection in tumor cells by using phosphorescence sensitivity to oxygen. The emission in the UV region is easily absorbed or scattered by absorbers in tissues, which limits biological imaging. Therefore, the conjugated structure of the dye was extended by substituting naphthalene to the benzene ring unit to red-shift the emission and reduce the scattering and absorption of the luminescence by the tissue structure; this consequently increased tissue penetration and enhanced the phosphorescence/fluorescence sensing and imaging efficiency of oxygen in living cells (Fig. 24).51
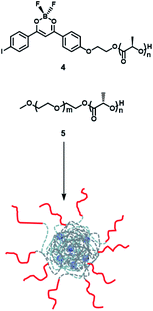 |
| | Fig. 23 Schematic illustration of the structure of pegylated BNPs based on BF2dbm(I)PLLA (4) and mPEG-b-PDLA (5). | |
 |
| | Fig. 24 Schematic illustration of the phosphorescent molecular structure and cell imaging. | |
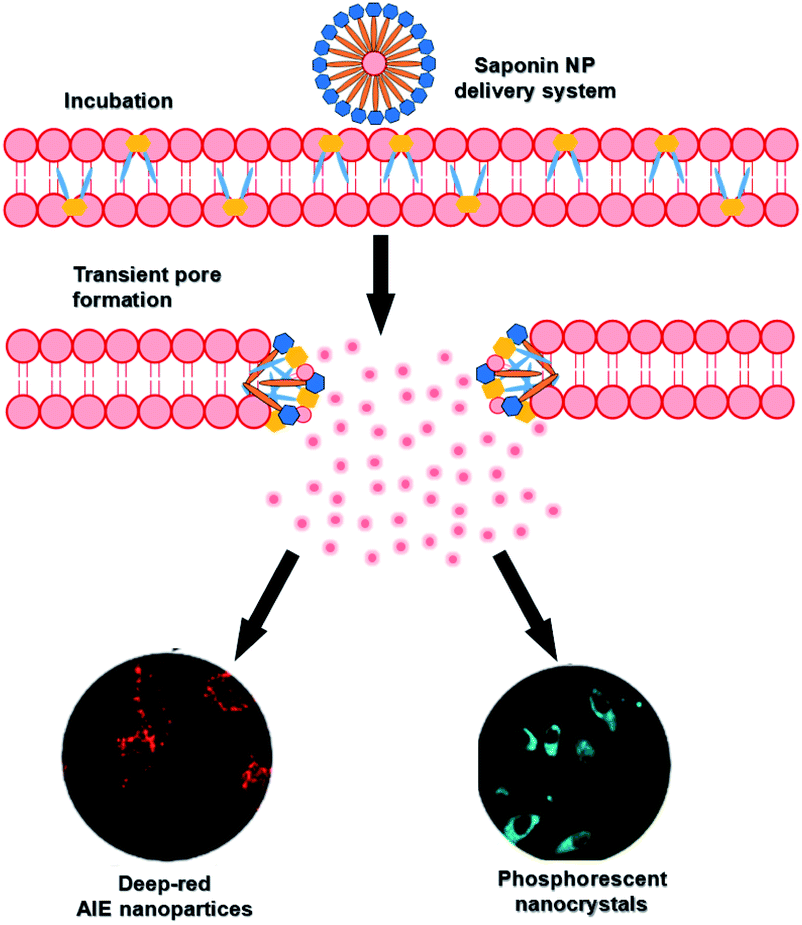
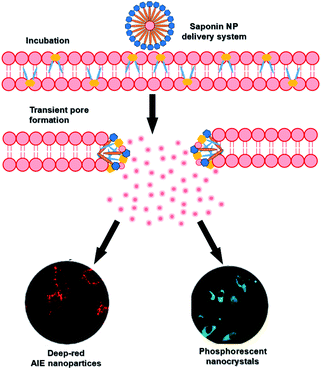
Saponins, a class of amphiphilic sterols and triterpene glycosides with biological activity and biocompatibility that occur naturally in plants, have good membrane permeabilization properties owing to the formation of transient pores by interacting with the cholesterol of the membrane above the critical micelle concentration (CMC).52 After combining RTP with aggregation-induced luminescence (AIE) molecules, the assembly had very good cell permeability and could enter cells very quickly (96 times faster than unencapsulated AIE molecules) (Fig. 25).45 Furthermore, the long-lived purely organic RTP nanocrystals (NCs) 1-(dibenzo[b,d]furan-2-yl)phenylmethanone (BDBF) were encapsulated into nanorods to obtain an amphiphilic assembly that could quickly enter the cancer cells for long-lived luminescence imaging. The permeability of the unencapsulated monomer BDBF NCs in cells is very poor, but the amphiphilic assembly had better bleaching resistance compared with commercial dyes. Therefore, this amphiphilic assembly of purely organic room-temperature phosphorescence has very good prospects in biological applications.
 |
| | Fig. 25 Schematic illustration of the cell permeabilization mechanism and AIE molecular structure. | |
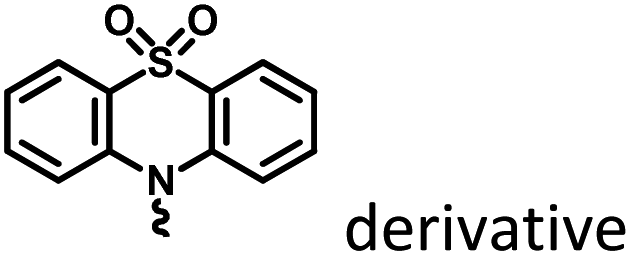
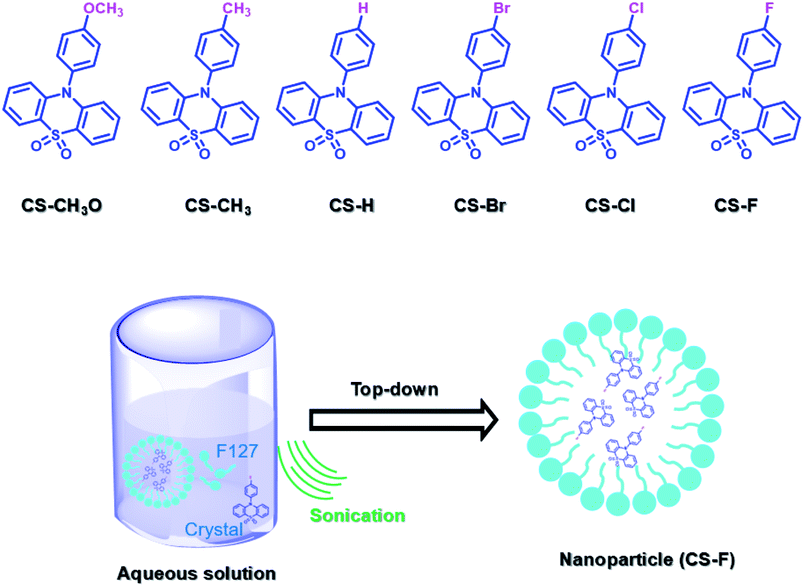
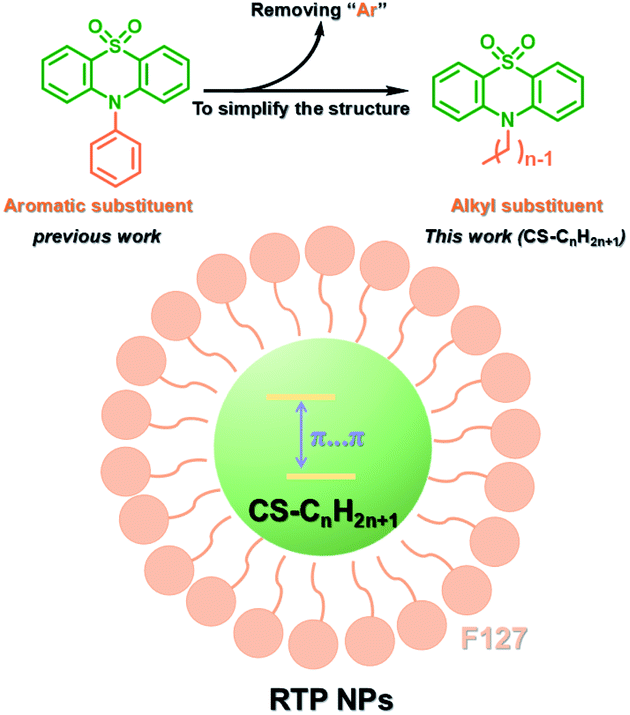
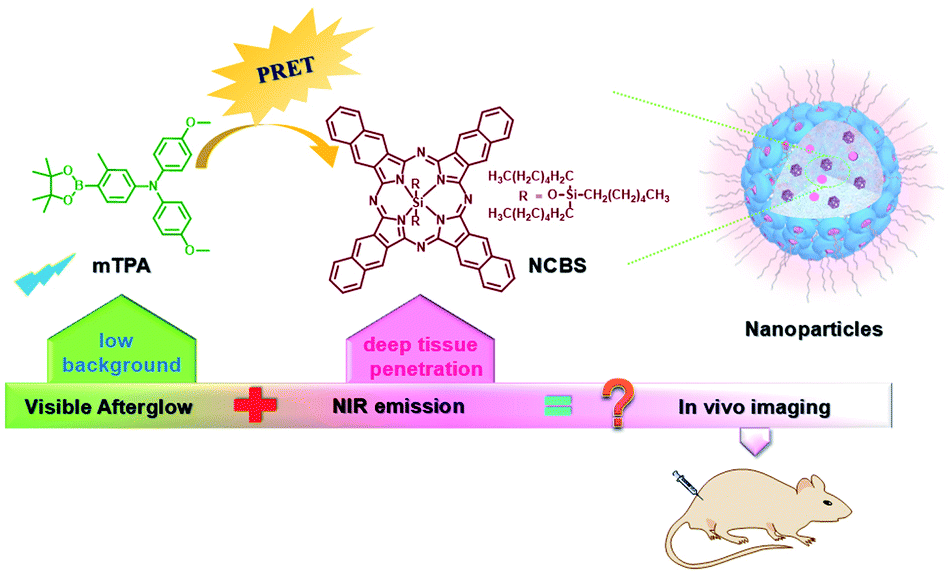
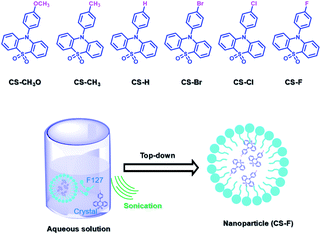
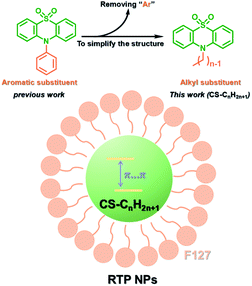
PEG-b-PPG-b-PEG (F127), an amphiphilic block polymer composed of hydrophobic PPG and hydrophilic PEG, can form micelles through the aggregation of hydrophobic groups in aqueous solution.53 Therefore, F127 can encapsulate the long-lived hydrophobic phosphorescent group in its hydrophobic portion in water. Six 10-phenyl-10H-phenothiazine-5,5-dioxide-based derivatives (i.e. CS–CH3O, CS–CH3, CSH, CS–Br, CS–Cl, and CS–F) were designed, in which different substituent groups were introduced onto the 10-phenyl ring, including the electron-donating groups –CH3O and –CH3 and the electron-withdrawing groups –Br, –Cl, and –F (Fig. 26).54 The integration of theoretical calculations of electron cloud distribution, electric potential, etc., demonstrated the influence of substituent group changes on their phosphorescent properties. CS–F had the greater electron-withdrawing effect, which resulted in a shorter distance among the phenothiazine units and stronger π–π interaction that could stabilize the excited triplet state efficiently to achieve ultralong RTP. Phosphorescent nanoparticles were then constructed with F127 and CS–F by the top-down approach in water to perform afterglow phosphorescence imaging of lymph nodes in living mice. Subsequently, six new compounds were constructed by replacing the benzene ring with a non-aromatic alkyl chain on the phenothiazine (Fig. 27).55 Through experiments and theoretical calculations, the influence of alkyl chain length on phosphorescence was studied. It was found that the designed molecule had an odd/even effect; that is, odd-numbered chains had stronger π–π interactions compared with even-numbered chains, leading to a longer phosphorescence effect. Among them, CS–C3H7, which has the longest lifetime RTP, was further combined with F127 and encapsulated in its hydrophobic region in water in a top-down manner. The phosphorescence imaging of human cancer cells in living mice has been achieved. In order to avoid problems with imaging depth encountered by UV or visible light emission, the method of PRET was adopted to achieve afterglow purely organic RTP with NIR emission (Fig. 28).56N,N-Bis(4-methoxyphenyl)-3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) aniline (mTPA) emitted at 530 nm and silicon 2,3-naphthalocyanine bis(trihexylsilyloxide) (NCBS) emitted at 780 nm were also co-assembled with F127 in the hydrophobic region in a top-down manner. Since the emission of mTPA and the absorption of NCBS partially overlapped, the NIR phosphorescence afterglow was realized in water by virtue of PRET. Furthermore, it was revealed that the effect of NIR afterglow imaging was shown by the aggregation in the lymph nodes of live mice.
 |
| | Fig. 26 Schematic illustration of 10-phenyl-10H-phenothiazine-5,5-dioxide-based derivative structures, and the photograph of the corresponding molecular crystalline phosphorescence. | |
 |
| | Fig. 27 Schematic illustration of the molecular design strategy, and the preparation method of phosphorescent systems. | |
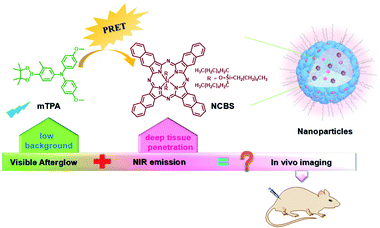 |
| | Fig. 28 Schematic illustration of PRET for in vivo imaging. | |
Outlook and perspectives
In conclusion, compared with fluorescence, aqueous RTP is becoming increasingly popular in the biochemical field owing to the advantages of a larger Stokes shift, longer luminescence lifetime, and the involvement of triplet states, which can avoid the interference of background fluorescence or auto-fluorescence in complex biological systems. In particular, purely organic RTP, which has low cost, low toxicity, and easy modification, is favored by scientists. Additionally, a variety of weak interactions (π–π stacking, hydrogen bonding, and hydrophobicity) can provide a simple supramolecular strategy in water that can not only adjust the molecular configuration or electronic structure to change the properties of luminescent materials but also endow them with dynamic intelligence. Supramolecular strategies may exhibit synergistic effects by combining two or more functional groups to avoid the disadvantages of a single component; this is widely used in the biological and medical fields to meet current needs in the precision medicine field. Therefore, in this review, we systematically introduced the construction of purely organic RTP in the water phase by supramolecular means based on the application of cell imaging, including “macrocyclic confinement,” “nanoassembly confinement,” and “macromolecular confinement.” It is worth noting that although purely organic RTP in water has been developed to a certain extent, the quantum yield and long lifetime in water still need to be improved, as the afterglow effect in water can better avoid the interference of background fluorescence in organisms. To achieve this, triplet excitons could be influenced by modulating the molecular structure, molecular aggregation morphology, or doping with other molecules, thereby improving the phosphorescence yield and lifetime of the molecule. However, UV light is highly phototoxic and has low penetration ability for cells and tissues, so the UV excitation as well as UV or visible light emission of phosphorescence in water is another shortcoming in the application of phosphorescence that is presently unresolved. However, the realization of purely organic RTP in water with visible light and even NIR light excitation as well as NIR light emission is the direction pursued by researchers. In this regard, we believe that the excitation or emission redshift, or even a non-excitation light source, may be achieved by extending the degree of molecular conjugation, triplet–triplet energy transfer, or combining upconversion nanoparticles or chemiluminescence. Additionally, when combined with a variety of supramolecular noncovalent interactions through macrocyclic confinement, nanoconfinement and macromolecular confinement in water is expected to be an effective way to develop purely organic RTP with visible light to NIR excitation and NIR light emission, or even in the absence of an excitation light source, and thus to overcome this current barrier. Moreover, the stability of luminescent materials constructed by noncovalent interaction in living organisms still needs to be optimized and improved. Therefore, the development of purely organic RTP in water requires further effort. It is hoped that this review can provide some reference and inspiration for readers, as well as be a resource for the construction new aqueous purely organic RTP in biology.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Author contributions
Y. L. conceived and designed the idea for the manuscript. W. L. Z. wrote the manuscript. W. L. searched the literature and created the images. Y. L. and Y. C. edited and modified the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22101143, 22131008 and 21971127), the China Postdoctoral Science Foundation (2021M691661), the National Natural Science Foundation of Inner Mongolia (2021BS02014), and the Young Scientific and Technological Talents of Inner Mongolia (NJYT22050).
Notes and references
-
(a) W. Li, S. Wu, X. Xu, J. Zhuang, H. Zhang, X. Zhang, C. Hu, B. Lei, C. F. Kaminski and Y. Liu, Chem. Mater., 2019, 31, 9887–9894 CrossRef CAS;
(b) G. D. Luker and K. E. Luker, J. Nucl. Med., 2008, 49, 1–4 CrossRef PubMed;
(c) T. C. Voss, I. A. Demarco and R. N. Day, BioTechniques, 2005, 38, 413–424 CrossRef CAS PubMed.
-
(a) J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed;
(b) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed;
(c) Y. Fan, P. Wang, Y. Lu, R. Wang, L. Zhou, X. Zheng, X. Li, J. A. Piper and F. Zhang, Nat. Nanotechnol., 2018, 13, 941–946 CrossRef CAS PubMed;
(d) Kenry, Y. Duan and B. Liu, Adv. Mater., 2018, 30, 1802394 CrossRef PubMed;
(e) Z. Lei and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 16294–16308 CrossRef CAS PubMed;
(f) J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC;
(g) J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS PubMed;
(h) K. J. McHugh, L. Jing, A. M. Behrens, S. Jayawardena, W. Tang, M. Gao, R. Langer and A. Jaklenec, Adv. Mater., 2018, 30, 1706356 CrossRef PubMed;
(i) Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS PubMed;
(j) G. M. van Dam, G. Themelis, L. M. A. Crane, N. J. Harlaar, R. G. Pleijhuis, W. Kelder, A. Sarantopoulos, J. S. de Jong, H. J. G. Arts, A. G. J. van der Zee, J. Bart, P. S. Low and V. Ntziachristos, Nat. Med., 2011, 17, 1315–1319 CrossRef CAS PubMed;
(k) H. Wan, J. Yue, S. Zhu, T. Uno, X. Zhang, Q. Yang, K. Yu, G. Hong, J. Wang, L. Li, Z. Ma, H. Gao, Y. Zhong, J. Su, A. L. Antaris, Y. Xia, J. Luo, Y. Liang and H. Dai, Nat. Commun., 2018, 9, 1171 CrossRef PubMed;
(l) J. Zhang, X. Zhen, P. K. Upputuri, M. Pramanik, P. Chen and K. Pu, Adv. Mater., 2017, 29, 1604764 CrossRef PubMed.
-
(a) J. Huang, J. Li, Y. Lyu, Q. Miao and K. Pu, Nat. Mater., 2019, 18, 1133–1143 CrossRef CAS PubMed;
(b) B. Wang, Y. Wang, Y. Wang, Y. Zhao, C. Yang, Z. Zeng, S. Huan, G. Song and X. Zhang, Anal. Chem., 2020, 92, 4154–4163 CrossRef CAS PubMed;
(c) K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- X. Zhen, Y. Tao, Z. An, P. Chen, C. Xu, R. Chen, W. Huang and K. Pu, Adv. Mater., 2017, 29, 1606665 CrossRef PubMed.
-
(a) H. Liu, X. Zhang, B. Xing, P. Han, S. S. Gambhir and Z. Cheng, Small, 2010, 6, 1087–1091 CrossRef CAS PubMed;
(b) M.-K. So, C. Xu, A. M. Loening, S. S. Gambhir and J. Rao, Nat. Biotechnol., 2006, 24, 339–343 CrossRef CAS PubMed.
-
(a) Y. Chen, H. Zhang, Z. Lei and F. Zhang, Small Struct., 2020, 1, 2000036 CrossRef;
(b) O. Green, S. Gnaim, R. Blau, A. Eldar-Boock, R. Satchi-Fainaro and D. Shabat, J. Am. Chem. Soc., 2017, 139, 13243–13248 CrossRef CAS PubMed;
(c) S. Gross, S. T. Gammon, B. L. Moss, D. Rauch, J. Harding, J. W. Heinecke, L. Ratner and D. Piwnica-Worms, Nat. Med., 2009, 15, 455–461 CrossRef CAS PubMed;
(d) C. Zhang and K. Pu, Small Struct., 2020, 1, 2000026 CrossRef.
-
(a) H. Gao and X. Ma, Aggregate, 2021, 2, e38 Search PubMed;
(b) F. Gu and X. Ma, Chem.–Eur. J., 2022, e202104131 CAS;
(c) L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef CAS PubMed;
(d) L. Gu, W. Ye, X. Liang, A. Lv, H. Ma, M. Singh, W. Jia, Z. Shen, Y. Guo, Y. Gao, H. Chen, D. Wang, Y. Wu, J. Liu, H. Wang, Y.-X. Zheng, Z. An, W. Huang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 18527–18535 CrossRef CAS PubMed;
(e) Z. He, H. Gao, S. Zhang, S. Zheng, Y. Wang, Z. Zhao, D. Ding, B. Yang, Y. Zhang and W. Z. Yuan, Adv. Mater., 2019, 31, 1807222 CrossRef PubMed;
(f) Z. Huang and X. Ma, Cell Rep. Phys. Sci., 2020, 1, 100167 CrossRef CAS;
(g) X.-Q. Liu, K. Zhang, J.-F. Gao, Y.-Z. Chen, C.-H. Tung and L.-Z. Wu, Angew. Chem., Int. Ed., 2020, 59, 23456–23460 CrossRef CAS PubMed;
(h) X. Ma, J. Wang and H. Tian, Acc. Chem. Res., 2019, 52, 738–748 CrossRef CAS PubMed;
(i) X.-K. Ma and Y. Liu, Acc. Chem. Res., 2021, 54, 3403–3414 CrossRef CAS PubMed;
(j) X.-F. Wang, H. Xiao, P.-Z. Chen, Q.-Z. Yang, B. Chen, C.-H. Tung, Y.-Z. Chen and L.-Z. Wu, J. Am. Chem. Soc., 2019, 141, 5045–5050 CrossRef CAS PubMed;
(k) J. Yang, M. Fang and Z. Li, Acc. Mater. Res., 2021, 2, 644–654 CrossRef CAS;
(l) Y. You, K. Huang, X. Liu, X. Pan, J. Zhi, Q. He, H. Shi, Z. An, X. Ma and W. Huang, Small, 2020, 16, 1906733 CrossRef CAS PubMed;
(m) X. Yu, W. Liang, Q. Huang, W. Wu, J. J. Chruma and C. Yang, Chem. Commun., 2019, 55, 3156–3159 RSC;
(n) T. Zhang, X. Ma, H. Wu, L. Zhu, Y. Zhao and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 11206–11216 CrossRef CAS PubMed;
(o) Y. Zhang, Y. Su, H. Wu, Z. Wang, C. Wang, Y. Zheng, X. Zheng, L. Gao, Q. Zhou, Y. Yang, X. Chen, C. Yang and Y. Zhao, J. Am. Chem. Soc., 2021, 143, 13675–13685 CrossRef CAS PubMed;
(p) W. Zhao, Z. He and B. Z. Tang, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS;
(q) W.-L. Zhou, W. Lin, Y. Chen, X.-Y. Dai, Z. Liu and Y. Liu, Chem. Sci., 2022, 13, 573–579 RSC;
(r) A. D. Nidhankar, Goudappagouda, V. C. Wakchaure and S. S. Babu, Chem. Sci., 2021, 12, 4216–4236 RSC.
-
(a) A. Abdukayum, J.-T. Chen, Q. Zhao and X.-P. Yan, J. Am. Chem. Soc., 2013, 135, 14125–14133 CrossRef CAS PubMed;
(b) Z. An, C. Zheng, Y. Tao, R. Chen, H. Shi, T. Chen, Z. Wang, H. Li, R. Deng, X. Liu and W. Huang, Nat. Mater., 2015, 14, 685–690 CrossRef CAS PubMed;
(c) R. E. Andrews, K. M. Shah, J. M. Wilkinson and A. Gartland, Bone, 2011, 49, 717–723 CrossRef CAS PubMed;
(d) O. Bolton, K. Lee, H.-J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed;
(e) P. Collery, B. Keppler, C. Madoulet and B. Desoize, Crit. Rev. Oncol. Hemat., 2002, 42, 283–296 CrossRef;
(f) Q. le Masne de Chermont, C. Chanéac, J. Seguin, F. Pellé, S. Maîtrejean, J.-P. Jolivet, D. Gourier, M. Bessodes and D. Scherman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9266 CrossRef CAS PubMed;
(g) Y. Li, M. Gecevicius and J. Qiu, Chem. Soc. Rev., 2016, 45, 2090–2136 RSC;
(h) T. Maldiney, A. Lecointre, B. Viana, A. Bessière, M. Bessodes, D. Gourier, C. Richard and D. Scherman, J. Am. Chem. Soc., 2011, 133, 11810–11815 CrossRef CAS PubMed;
(i) C. Chen, Z. Chi, K. C. Chong, A. S. Batsanov, Z. Yang, Z. Mao, Z. Yang and B. Liu, Nat. Mater., 2021, 20, 175–180 CrossRef CAS PubMed.
- X. Yang and D. Yan, Chem. Sci., 2016, 7, 4519–4526 RSC.
-
(a) S. M. A. Fateminia, Z. Mao, S. Xu, Z. Yang, Z. Chi and B. Liu, Angew. Chem., Int. Ed., 2017, 56, 12160–12164 CrossRef CAS PubMed;
(b) D. Li, J. Wang and X. Ma, Adv. Opt. Mater., 2018, 6, 1800273 CrossRef.
-
(a) S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed;
(b) D. Dai, Z. Li, J. Yang, C. Wang, J.-R. Wu, Y. Wang, D. Zhang and Y.-W. Yang, J. Am. Chem. Soc., 2019, 141, 4756–4763 CrossRef CAS PubMed;
(c) Z. Li and Y.-W. Yang, Acc. Mater. Res., 2021, 2, 292–305 CrossRef CAS;
(d) G. Yu, K. Jie and F. Huang, Chem. Rev., 2015, 115, 7240–7303 CrossRef CAS PubMed;
(e) W.-L. Zhou, Y. Chen and Y. Liu, Acta Chim. Sin., 2020, 78, 1164–1176 CrossRef CAS;
(f) Y. Zhang, L. Wang, J. Wang, S. Xin and X. Sheng, Chin. Chem. Lett., 2021, 32, 1902–1906 CrossRef CAS.
-
(a) Y. Chen, F. Huang, Z.-T. Li and Y. Liu, Sci. China: Chem., 2018, 61, 979–992 CrossRef CAS;
(b) G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed;
(c) W.-F. Lai, A. L. Rogach and W.-T. Wong, Chem. Soc. Rev., 2017, 46, 6379–6419 RSC;
(d) J. Szejtli, Chem. Rev., 1998, 98, 1743–1754 CrossRef CAS PubMed;
(e) W.-L. Zhou, Y. Chen, W. Lin and Y. Liu, Chem. Commun., 2021, 57, 11443–11456 RSC.
- N. J. Turro, J. D. Bolt, Y. Kuroda and I. Tabushi, Photochem. Photobiol., 1982, 35, 69–72 CrossRef CAS.
- S. Scypinski and L. J. C. Love, Anal. Chem., 1984, 56, 322–327 CrossRef CAS.
- A. Ponce, P. A. Wong, J. J. Way and D. G. Nocera, J. Phys. Chem., 1993, 97, 11137–11142 CrossRef CAS.
- M. A. Mortellaro, W. K. Hartmann and D. G. Nocera, Angew. Chem., Int. Ed., 1996, 35, 1945–1946 CrossRef.
- Y.-S. Wei, W.-J. Jin, R.-H. Zhu, G.-W. Xing, C.-S. Liua, S.-S. Zhang and B.-L. Zhou, Spectrochim. Acta, Part A, 1996, 52, 683–690 CrossRef.
- Y.-X. Zhu, J.-H. Peng and Y. Zhang, Anal. Chim. Acta, 2007, 583, 364–369 CrossRef CAS PubMed.
- X. Ma, J. Cao, Q. Wang and H. Tian, Chem. Commun., 2011, 47, 3559–3561 RSC.
- J. Cao, X. Ma, M. Min, T. Cao, S. Wu and H. Tian, Chem. Commun., 2014, 50, 3224–3226 RSC.
- H. Chen, X. Ma, S. Wu and H. Tian, Angew. Chem., Int. Ed., 2014, 53, 14149–14152 CrossRef CAS PubMed.
- H. Chen, L. Xu, X. Ma and H. Tian, Polym. Chem., 2016, 7, 3989–3992 RSC.
-
(a) K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267–279 RSC;
(b) X.-L. Ni, X. Xiao, H. Cong, Q.-J. Zhu, S.-F. Xue and Z. Tao, Acc. Chem. Res., 2014, 47, 1386–1395 CrossRef CAS PubMed;
(c) J. Tian, L. Zhang, H. Wang, D.-W. Zhang and Z.-T. Li, Supramol. Chem., 2016, 28, 769–783 CrossRef CAS;
(d) Z.-J. Yin, Z.-Q. Wu, F. Lin, Q.-Y. Qi, X.-N. Xu and X. Zhao, Chin. Chem. Lett., 2017, 28, 1167–1171 CrossRef CAS.
- L. Mu, X.-B. Yang, S.-F. Xue, Q.-J. Zhu, Z. Tao and X. Zeng, Anal. Chim. Acta, 2007, 597, 90–96 CrossRef CAS PubMed.
- P. Montes-Navajas and H. Garcia, J. Phys. Chem. C, 2010, 114, 2034–2038 CrossRef CAS.
- Y. Gong, H. Chen, X. Ma and H. Tian, ChemPhysChem, 2016, 17, 1934–1938 CrossRef CAS PubMed.
- L. Xu, L. Zou, H. Chen and X. Ma, Dyes Pigm., 2017, 142, 300–305 CrossRef CAS.
- T. Li and X. Ma, Dyes Pigm., 2018, 148, 306–312 CrossRef CAS.
- J. Wang, Z. Huang, X. Ma and H. Tian, Angew. Chem., Int. Ed., 2020, 59, 9928–9933 CrossRef CAS PubMed.
-
(a) S. Jaracz, J. Chen, L. V. Kuznetsova and I. Ojima, Bioorg. Med. Chem., 2005, 13, 5043–5054 CrossRef CAS PubMed;
(b) Q. Yu, Y.-M. Zhang, Y.-H. Liu, X. Xu and Y. Liu, Sci. Adv., 2018, 4, eaat2297 CrossRef CAS PubMed.
- W.-L. Zhou, Y. Chen, Q. Yu, H. Zhang, Z.-X. Liu, X.-Y. Dai, J.-J. Li and Y. Liu, Nat. Commun., 2020, 11, 4655 CrossRef CAS PubMed.
-
(a) Z.-Y. Zhang, Y. Chen and Y. Liu, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed;
(b) Z.-Y. Zhang and Y. Liu, Chem. Sci., 2019, 10, 7773–7778 RSC.
- X.-K. Ma, Y.-M. Zhang, Q. Yu, H. Zhang, Z. Zhang and Y. Liu, Chem. Commun., 2021, 57, 1214–1217 RSC.
- X.-K. Ma, W. Zhang, Z. Liu, H. Zhang, B. Zhang and Y. Liu, Adv. Mater., 2021, 33, 2007476 CrossRef CAS PubMed.
-
(a) G. Collet, T. Lathion, C. Besnard, C. Piguet and S. Petoud, J. Am. Chem. Soc., 2018, 140, 10820–10828 CrossRef CAS PubMed;
(b) M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed;
(c) W. Zhou, Y. Chen, Q. Yu, P. Li, X. Chen and Y. Liu, Chem. Sci., 2019, 10, 3346–3352 RSC;
(d) Y. Zhou, H.-Y. Zhang, Z.-Y. Zhang and Y. Liu, J. Am. Chem. Soc., 2017, 139, 7168–7171 CrossRef CAS PubMed.
- H.-J. Yu, Q. Zhou, X. Dai, F.-F. Shen, Y.-M. Zhang, X. Xu and Y. Liu, J. Am. Chem. Soc., 2021, 143, 13887–13894 CrossRef CAS PubMed.
-
(a) X. Dai, X. Dong, Z. Liu, G. Liu and Y. Liu, Biomacromolecules, 2020, 21, 5369–5379 CrossRef CAS PubMed;
(b) M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed;
(c) Z. Li, G. Davidson-Rozenfeld, M. Vázquez-González, M. Fadeev, J. Zhang, H. Tian and I. Willner, J. Am. Chem. Soc., 2018, 140, 17691–17701 CrossRef CAS PubMed;
(d) G. Liu, X. Xu, X. Dai, C. Jiang, Y. Zhou, L. Lu and Y. Liu, Mater. Horiz., 2021, 8, 2494–2502 RSC;
(e) D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543–7588 CrossRef CAS PubMed.
- C. Wang, X.-K. Ma, P. Guo, C. Jiang, Y.-H. Liu, G. Liu, X. Xu and Y. Liu, Adv. Sci., 2021, 2103041 Search PubMed.
-
(a) S. Kuila, K. V. Rao, S. Garain, P. K. Samanta, S. Das, S. K. Pati, M. Eswaramoorthy and S. J. George, Angew. Chem., Int. Ed., 2018, 57, 17115–17119 CrossRef CAS PubMed;
(b) S. Kuila and S. J. George, Angew. Chem., Int. Ed., 2020, 59, 9393–9397 CrossRef CAS PubMed;
(c) L. Zhang, R. Zhang, P. Cui, W. Cao and F. Gao, Chem. Commun., 2013, 49, 8102–8104 RSC;
(d) B. Zhou and D. Yan, Adv. Funct. Mater., 2019, 29, 1807599 CrossRef;
(e) S. Garain, B. C. Garain, M. Eswaramoorthy, S. K. Pati and S. J. George, Angew. Chem., Int. Ed., 2021, 60, 19720–19724 CrossRef CAS PubMed;
(f) X.-Y. Dai, Y.-Y. Hu, Y. Sun, M. Huo, X. Dong and Y. Liu, Adv. Sci., 2022, 9, 2200524 CrossRef CAS PubMed.
- F.-F. Shen, Y. Chen, X. Dai, H.-Y. Zhang, B. Zhang, Y. Liu and Y. Liu, Chem. Sci., 2021, 12, 1851–1857 RSC.
- M. Huo, X.-Y. Dai and Y. Liu, Small, 2022, 2104514 CrossRef CAS PubMed.
- M. Huo, X.-Y. Dai and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 27171–27177 CrossRef CAS PubMed.
-
(a) S. M. A. Fateminia, Z. Wang, C. C. Goh, P. N. Manghnani, W. Wu, D. Mao, L. G. Ng, Z. Zhao, B. Z. Tang and B. Liu, Adv. Mater., 2017, 29, 1604100 CrossRef PubMed;
(b) A. Pfister, G. Zhang, J. Zareno, A. F. Horwitz and C. L. Fraser, ACS Nano, 2008, 2, 1252–1258 CrossRef CAS PubMed;
(c) Y. S. Zhao, J. Xu, A. Peng, H. Fu, Y. Ma, L. Jiang and J. Yao, Angew. Chem., Int. Ed., 2008, 47, 7301–7305 CrossRef CAS PubMed.
- X. Chen, C. Xu, T. Wang, C. Zhou, J. Du, Z. Wang, H. Xu, T. Xie, G. Bi, J. Jiang, X. Zhang, J. N. Demas, C. O. Trindle, Y. Luo and G. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9872–9876 CrossRef CAS.
- A. Nicol, R. T. K. Kwok, C. Chen, W. Zhao, M. Chen, J. Qu and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 14792–14799 CrossRef CAS.
-
(a)
B. He, J. Huang, J. Zhang, H. H. Y. Sung, J. W. Y. Lam, Z. Zhang, S. Yan, D. Wang, J. Zhang, B. Z. Tang, Angew. Chem., Int. Ed., 2022, 61, e2021177 Search PubMed;
(b) X. Liu, C. Zhu and B. Z. Tang, Acc. Chem. Res., 2022, 55, 197–208 CrossRef CAS PubMed;
(c) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS;
(d) J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed;
(e) Z. Zhang, W. Yan, D. Dang, H. Zhang, J. Z. Sun and B. Z. Tang, Cell Rep. Phys. Sci., 2022, 100716 CrossRef CAS.
- X. Jia, C. Shao, X. Bai, Q. Zhou, B. Wu, L. Wang, B. Yue, H. Zhu and L. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 4816 CrossRef CAS PubMed.
-
(a) H.-W. Tian, Y.-C. Liu and D.-S. Guo, Mater. Chem. Front., 2020, 4, 46–98 RSC;
(b) C. Wang, Z. Wang and X. Zhang, Acc. Chem. Res., 2012, 45, 608–618 CrossRef CAS PubMed;
(c) X. Zhang and C. Wang, Chem. Soc. Rev., 2011, 40, 94–101 RSC.
-
(a) L. Chen, Z. Xie, J. Hu, X. Chen and X. Jing, J. Nanopart. Res., 2007, 9, 777–785 CrossRef CAS;
(b) M. R. Dreher, W. Liu, C. R. Michelich, M. W. Dewhirst, F. Yuan and A. Chilkoti, J. Natl. Cancer Inst., 2006, 98, 335–344 CrossRef CAS.
- F. R. Kersey, G. Zhang, G. M. Palmer, M. W. Dewhirst and C. L. Fraser, ACS Nano, 2010, 4, 4989–4996 CrossRef CAS PubMed.
- C. A. DeRosa, J. Samonina-Kosicka, Z. Fan, H. C. Hendargo, D. H. Weitzel, G. M. Palmer and C. L. Fraser, Macromolecules, 2015, 48, 2967–2977 CrossRef CAS PubMed.
-
(a) I. G. Barr, A. Sjölander and J. C. Cox, Adv. Drug Delivery Rev., 1998, 32, 247–271 CrossRef CAS;
(b) V. R. Dayeh, S. L. Chow, K. Schirmer, D. H. Lynn and N. C. Bols, Ecotoxicol. Environ. Saf., 2004, 57, 375–382 CrossRef CAS PubMed.
-
(a) P. Alexandridis and T. Alan Hatton, Colloids Surf., A, 1995, 96, 1–46 CrossRef CAS;
(b) S. Kerkhofs, T. Willhammar, H. Van Den Noortgate, C. E. A. Kirschhock, E. Breynaert, G. Van Tendeloo, S. Bals and J. A. Martens, Chem. Mater., 2015, 27, 5161–5169 CrossRef CAS;
(c) J. E. Matthew, Y. L. Nazario, S. C. Roberts and S. R. Bhatia, Biomaterials, 2002, 23, 4615–4619 CrossRef CAS PubMed;
(d) M. D. Shin, J. D. Hochberg, J. K. Pokorski and N. F. Steinmetz, ACS Appl. Mater. Interfaces, 2021, 13, 59618–59632 CrossRef CAS PubMed.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- J. Yang, H. Gao, Y. Wang, Y. Yu, Y. Gong, M. Fang, D. Ding, W. Hu, B. Z. Tang and Z. Li, Mater. Chem. Front., 2019, 3, 1391–1397 RSC.
- Q. Dang, Y. Jiang, J. Wang, J. Wang, Q. Zhang, M. Zhang, S. Luo, Y. Xie, K. Pu, Q. Li and Z. Li, Adv. Mater., 2020, 32, 2006752 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a