DOI:
10.1039/D2QO01341J
(Review Article)
Org. Chem. Front., 2022,
9, 6077-6113
Catalysts’ evolution in the asymmetric conjugate addition of nitroalkanes to electron-poor alkenes
Received
23rd August 2022
, Accepted 12th September 2022
First published on 13th September 2022
Abstract
The conjugate addition of nitroalkanes to electron-poor alkenes is a widely used process which only in the late nineties of the last century has efficiently evolved in its asymmetric version. Synthetic protocols based on chiral organocatalytic methods have been largely exploited for the generation of optically pure γ-nitro derivatives through carbon–carbon bond formation. Chiral metal–ligand complexes have also been successfully employed for these conjugate additions, although their use in the synthesis of targeted bioactive compounds still appears rather limited. Most of the practical applications of the obtained adducts are based on the easy conversion of the nitro group into a primary amine directed to the preparation of nitrogen-containing structures. This review aims to provide a journey of the catalyst usage for the enantioselective conjugate addition of nitroalkanes to electron-poor olefins, from the early attempts to the latest achievements. The discussion is categorized according to the nature of different catalytic systems, and a final section reporting selected applications to targeted compounds is provided.
 Roberto Ballini | Roberto Ballini is Full Professor of Organic Chemistry at the University of Camerino (Italy) and Past President of the Organic Chemistry Division of the Italian Chemical Society (SCI). His scientific activity is mainly dedicated to organic synthesis, with a particular interest in the chemistry of nitroalkanes, including the formation of single and double C,C bonds and their conversion into other important functionalities. He is the author of about 300 papers, including a large number of review articles. Furthermore, he is editor of two books in the Green Chemistry Series by the RCS, Eco-Friendly Processes and Procedures of Fine Chemicals and Green Synthetic Processes and Procedures, and the monographic book Nitroalkanes: Synthesis, Reactivity and Applications (Wiley-VCH). |
 Alessandro Palmieri | Alessandro Palmieri obtained his Laurea degree cum laude in Chemistry in 2002 at the University of Camerino (Italy) where, five years later, he received a PhD degree in Chemical Sciences. In 2008 he moved, as a visiting postdoctoral fellow, to the ITC Laboratory at the University of Cambridge (Prof. Steven V. Ley). In 2014 he was appointed associate professor in Organic Chemistry at the University of Camerino. His research interests involve (i) the chemistry of aliphatic nitro compounds, (ii) the realization of new one-pot protocols for generating and derivatizing heterocyclic systems, (iii) the preparation of solid supported reagents, and (iv) the development of new sustainable processes and (v) flow chemical protocols. |
 Marino Petrini | Marino Petrini obtained a Laurea degree in Chemistry in 1980 (University of Camerino, Italy). In 1983 he became a Research Associate at the University of Camerino and during the period 1987–88 he was a visiting scientist at the University of Montreal (Prof. S. Hanessian). In 1992 he was appointed Associate Professor and then Full Professor in Organic Chemistry at the University of Camerino. His research interests mainly deal with the following topics: synthesis and reactivity of aliphatic and aromatic nitro compounds; functionalization of azole derivatives; synthesis and reactivity of imino derivatives. |
1. Introduction
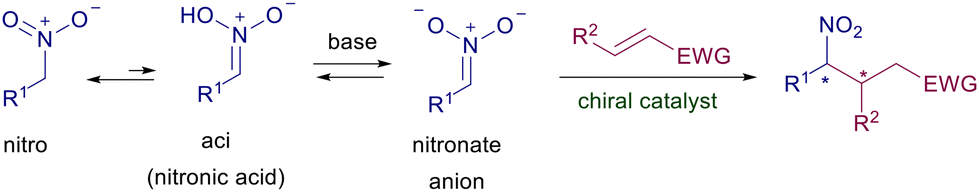
The conjugate addition of stabilized enolate anions to electron-poor alkenes has been firmly established as one of the leading synthetic processes since its discovery by Michael at the end of the nineteenth century. This reaction, essentially promoted by mildly basic conditions, has been largely employed for carbon–carbon bond formation and often represents the cornerstone for the assembling of complex structural frameworks.1 The advent of catalyzed asymmetric synthesis has found in these conjugate additions an ideal benchmark to develop new chiral catalysts endowed with growing levels of efficiency and sustainability.2 Nitroalkanes, by virtue of the tautomeric aci-nitro equilibrium, can be easily converted into nitronate anions which are effective nucleophilic reagents toward several electrophiles including electron-poor alkenes (Scheme 1).3
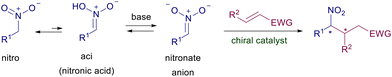 |
| | Scheme 1 Aci-nitro equilibrium and conjugate addition to electron-poor alkenes. | |
In this reaction, one or more stereocenters can be generated, according to the nature of the reactants employed, and thus a diastereo- and/or enantiocontrol can be exerted on the corresponding adduct providing that a suitable chiral environment is applied. The synthetic versatility of the nitro group has been widely demonstrated over many years through its manifold transformations into other functional groups. The reduction of the nitro group into a primary amine is certainly the most practiced transformation and represents a flexible gateway to a plethora of nitrogenated compounds.4 On the other hand, the nitro to carbonyl conversion, also known as the Nef reaction,5 the reductive denitration and the nitrous acid elimination introducing an unsaturation into the molecular framework are complementary processes highlighting the powerful synthetic attributes of the nitro moiety.6 The electrophilic character of the employed Michael acceptors is of crucial importance in leading to fast and efficient additions, and may substantially be tuned through a suitable choice of the nature of the electron-withdrawing group. α,β-Unsaturated carbonyls play a dominant role among Michael acceptors for their remarkable reactivity and for the electronic attributes of the carbonyl group. The electrophilicity of the carbonyl and the oxygen basic center have been conveniently used to generate covalent or electrostatic linkages with chiral catalysts in order to make their action more powerful. Other electron-withdrawing groups such as the cyano,7 the arylsulfonyl and the phosphonyl groups have been used in the conjugate additions with nitroalkanes, but less frequently than their carbonyl counterparts.8 A special mention can be made for nitroalkenes, which are the most reactive Michael acceptors and are used to generate 1,3-dinitro derivatives amenable for a subsequent reductive process leading to optically active 1,3-diamino compounds.9
The asymmetric procedures, which until twenty years ago still were in their infancy for this reaction, have been propelled by the need to ensure increasingly efficient catalytic devices to generate polyfunctionalized chiral compounds as pivotal intermediates in multistep synthetic protocols. This review is not intended to provide a comprehensive coverage of this topic, but aims to critically present the chiral catalyst developments from the early attempts to the latest achievements. The discussion privileges the presentation of the original procedure in which the catalyst was first used. Selected examples of other substrate combinations are reported only on the basis of their envisaged synthetic impact. Finally, the application of the discussed protocols to paradigmatic syntheses of targeted bioactive compounds is reported in the final section.
2. Organocatalyzed reactions
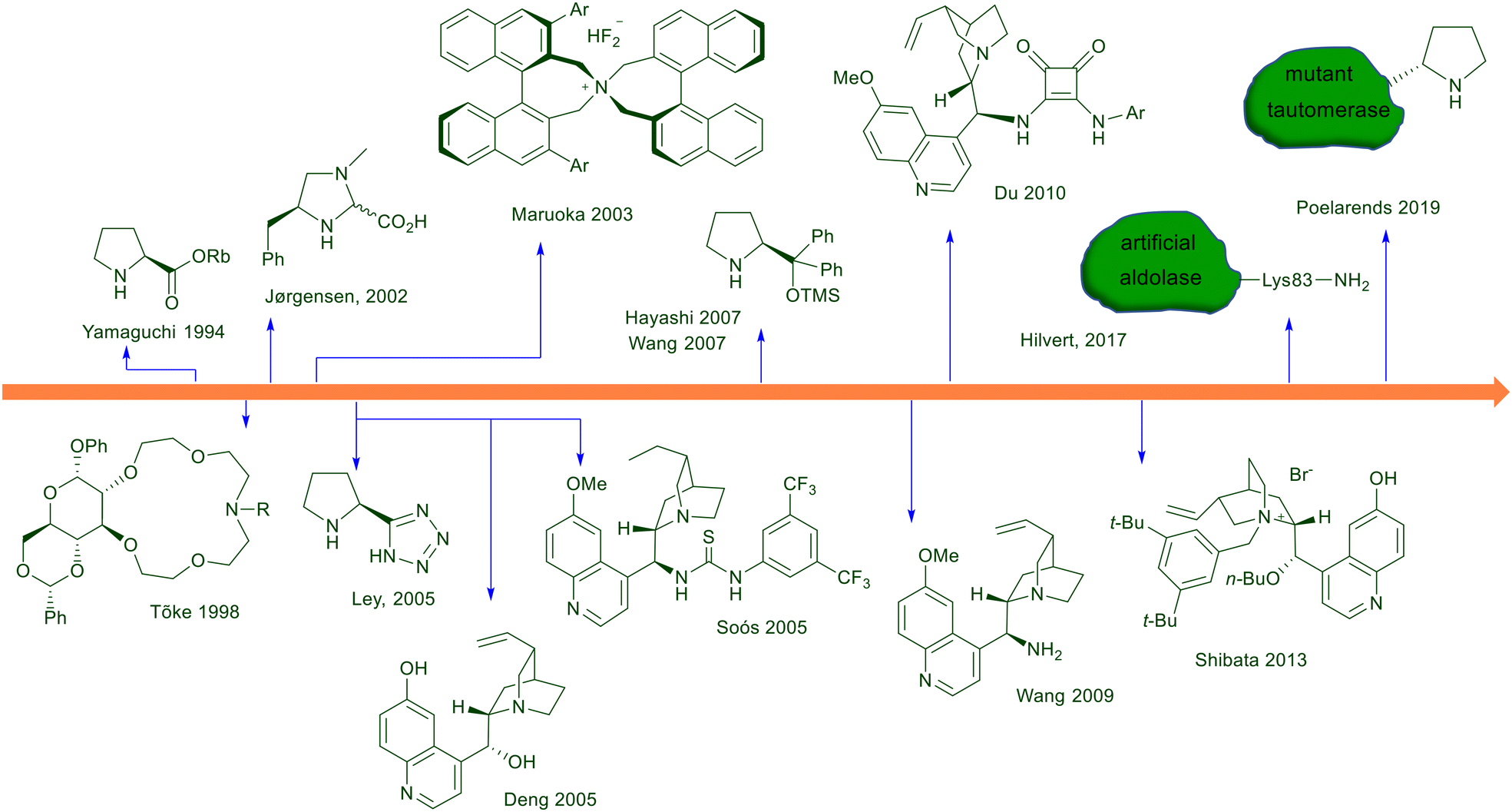
At the dawn of asymmetric conjugate additions involving nitroalkanes, purely organic molecules were the first optically active compounds tested as catalysts or promoters. The first example dates back to 1975, using quinine in the reaction of 1-tosylnitroethane with methyl vinyl ketone.10 Later on, different organic molecules were progressively tested in this reaction, and the most used class of catalysts showing better performances are chiral secondary amines and ureas/amido compounds (Fig. 1). Interestingly, to these classes of nitrogen derivatives pertain complementary ways of action, since primary and secondary amines are known to act through covalent iminium ion catalysis activating the unsaturated carbonyls while ureas/amides enhance the reactivity of nitroalkanes. Compared with other amino derivatives, cinchona-derived chiral primary amines were introduced only at the end of the first decade of this century, including soon after peptide foldamers and aldolase biomimetic catalysts. Phase-transfer catalysts including chiral crown ethers and quaternary ammonium salts have been successfully employed through the years, although their use for practical purposes is totally surpassed by chiral pyrrolidine and thiourea-based catalysts.
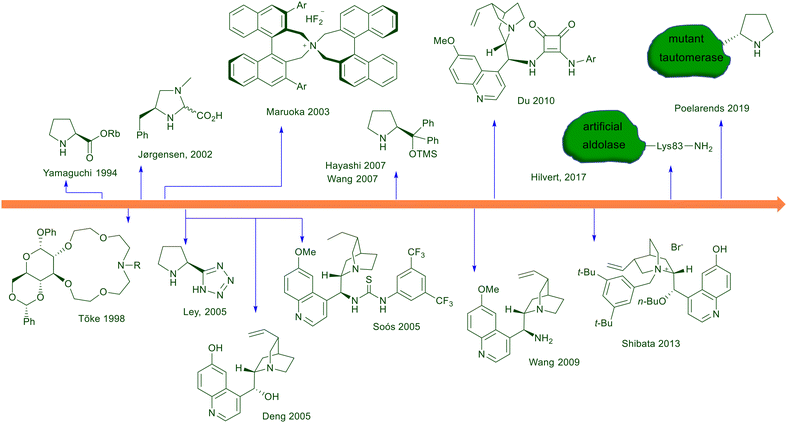 |
| | Fig. 1 Chronological development of organocatalysts employed for the asymmetric conjugate addition of nitroalkanes to electron-poor alkenes. | |
2.1 Pyrrolidines and related derivatives
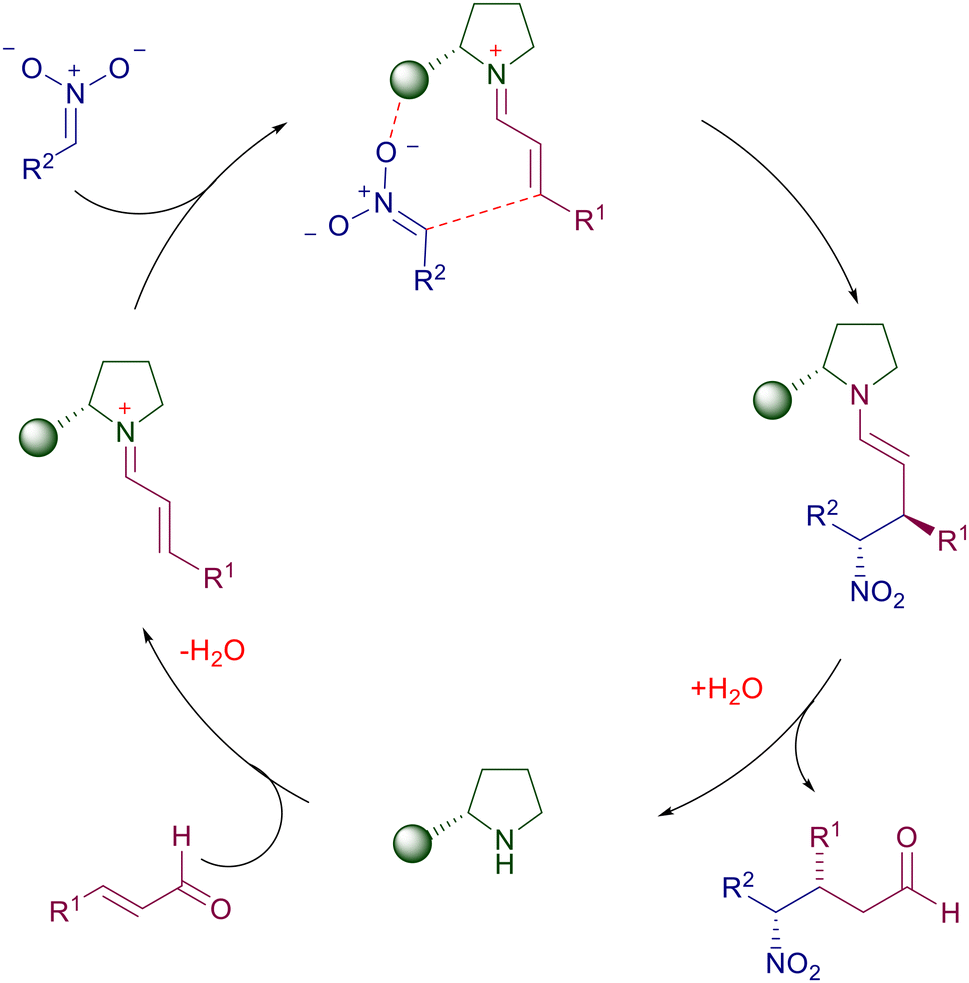
Chiral cyclic secondary amines are effective in the reaction of nitroalkanes with α,β-unsaturated carbonyls via pyrrolidinium ion catalysis which enhances the electrophilic character of the double bond towards the nitronate addition (Scheme 2). The appendage of the pyrrolidine ring may exert a synergistic effect through hydrogen bonding with the oxygen atoms of the nitro moiety, enabling an appropriate stereofacial nucleophilic attack (cooperative intramolecular catalysis).
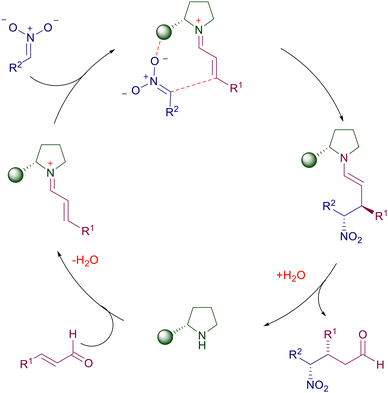 |
| | Scheme 2 Catalytic cycle for conjugate additions using chiral pyrrolidines. | |
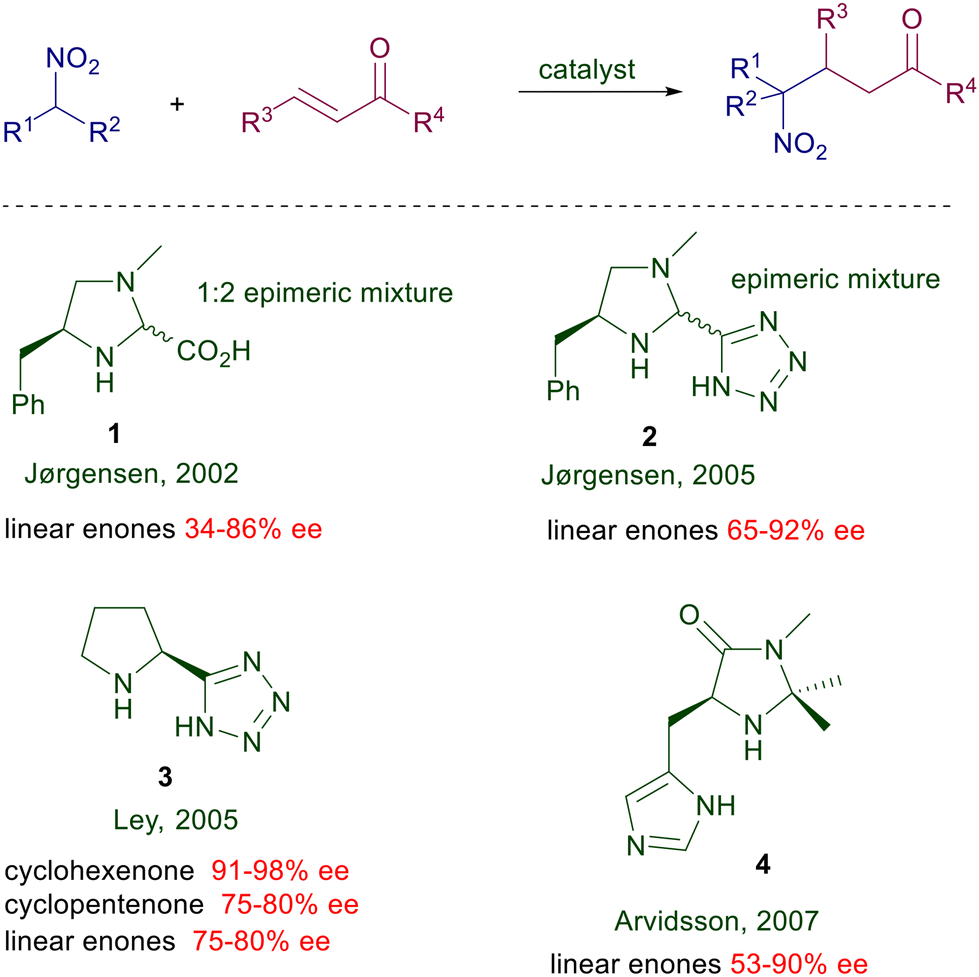
2.1.1 Proline and imidazolidine derivatives.
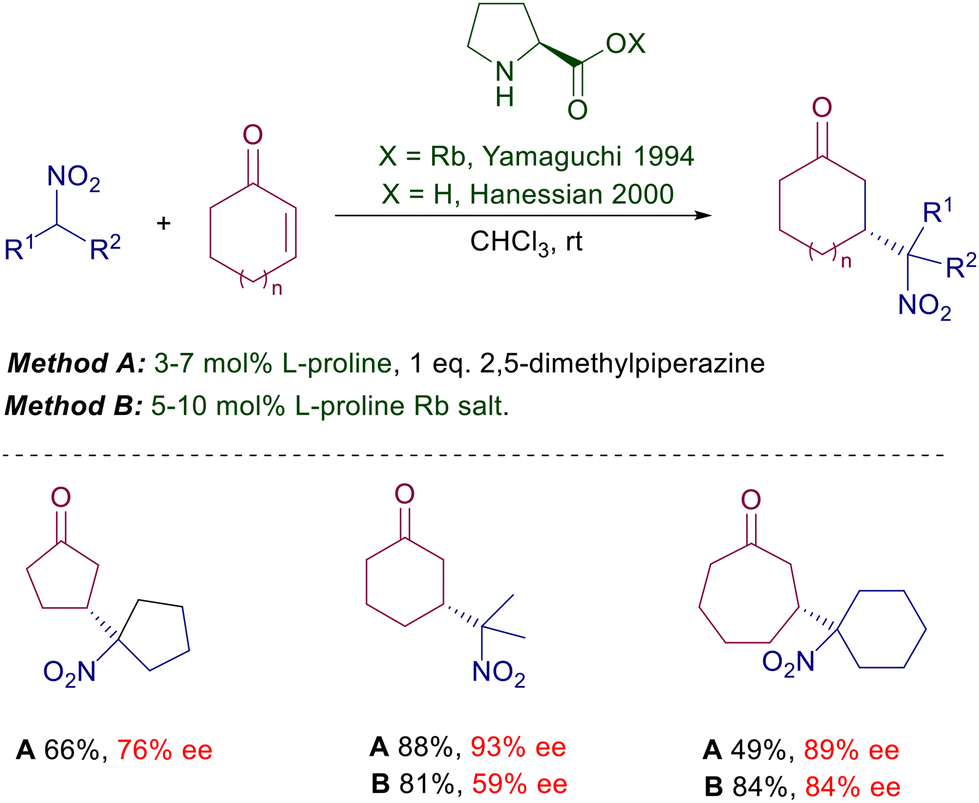
Proline, a natural α-amino acid readily available in both enantiomeric forms, was the first chiral pyrrolidine employed as catalyst for the conjugate addition of nitroalkanes to enones. It was initially used as rubidium salt which was already proved to be effective in the classical Michael addition using malonate esters.11 The preliminary results in the enantioselective reaction of nitroalkanes with linear or cyclic ketones were satisfactory (38–86% ee) considering the state of the art at that time.
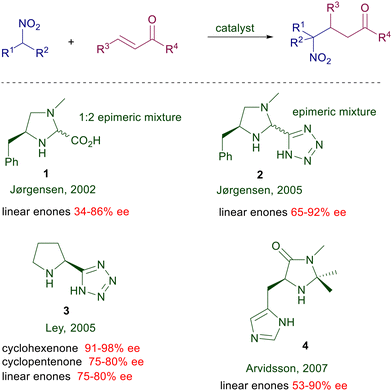
Structural modifications of the pyrrolidine ring have also been attempted using 4-hydroxyproline derivatives and even the rubidium salt of (S)-azetidine-2-carboxylic acid, but the improvements were rather modest. A notable step forward was evidenced in the use of L-proline limitedly to the reaction of nitroalkanes with cycloalkenones introducing trans-2,5-dimethylpiperazine as an equimolar additive (Scheme 3).12 The additive is instrumental in providing superior ee values using proline over its rubidium salt, although its role in the asymmetric process has not been ascertained. Interestingly, nonlinear effects were observed with the use of the proline–dimethylpiperazine couple but not with proline rubidium salt. All these results indicate that proline alone is unable to provide a notable stereochemical bias, probably because of the limited ability of the carboxylate moiety to efficiently coordinate the nitronate anion. This item has been confirmed moving to imidazolidine derivatives bearing a carboxylic group (Scheme 4).13 The Jørgensen's catalyst 1 is used as an epimeric mixture, showing that the effect brought by the carboxylic group is practically irrelevant on the stereochemical outcome since the E iminium ion intermediate is favored over the Z by 3 kcal mol−1 according to PM3 calculations. The tetrazole unit mimics the carboxylic group in several practical applications, and therefore a logical upgrade of the proline-based catalysts has involved the use of these tetrazole-containing catalytic systems. The results obtained using the chiral imidazolidine 2 are superior compared with those recorded with the acid analogue.14 This trend is confirmed by the results obtained by Ley using the proline analogue 3 which is effective both on linear than cycloalkenone substrates.15 The importance of the proximity effect exerted by the tetrazole unit is evidenced by the poor enantioselectivity (28% ee) obtained in the reaction of cyclohexenone with 2-nitropropane using a catalyst homologue of 3 in which the two heterocycles are connected by a methylene bridge.
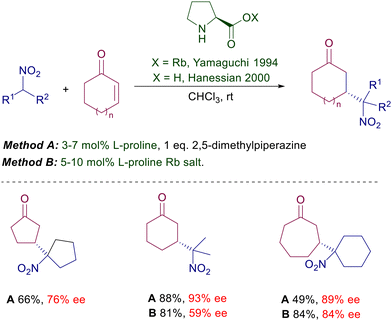 |
| | Scheme 3
L-Proline and its rubidium salt in the reaction with cycloalkenones. | |
 |
| | Scheme 4 Conjugate additions using pyrrolidines and imidazolidine catalysts. | |
These findings have spurred the exploitation of other surrogates of the carboxylic group such as the phosphonate group, but with limited success.16 The behavior of imidazolidinone 4 obtained from L-histidine parallels that of chiral imidazolines also regarding the poor diastereoselectivity evidenced using primary nitroalkanes as reactants.17 In the attempt to increase the stereochemical bias of the catalytic system, a rigid spiropyrrolidine embedding a triazole moiety 5 has been devised for the addition of nitromethane to enals (Scheme 5).18 The nitro alcohol 7 obtained after reduction is formed with high enantioselectivity especially when 3-arylenals are used as substrates.
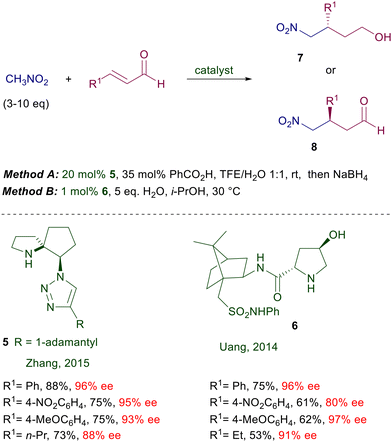 |
| | Scheme 5 Spiropyrrolidines and prolinamide-based organocatalysts. | |
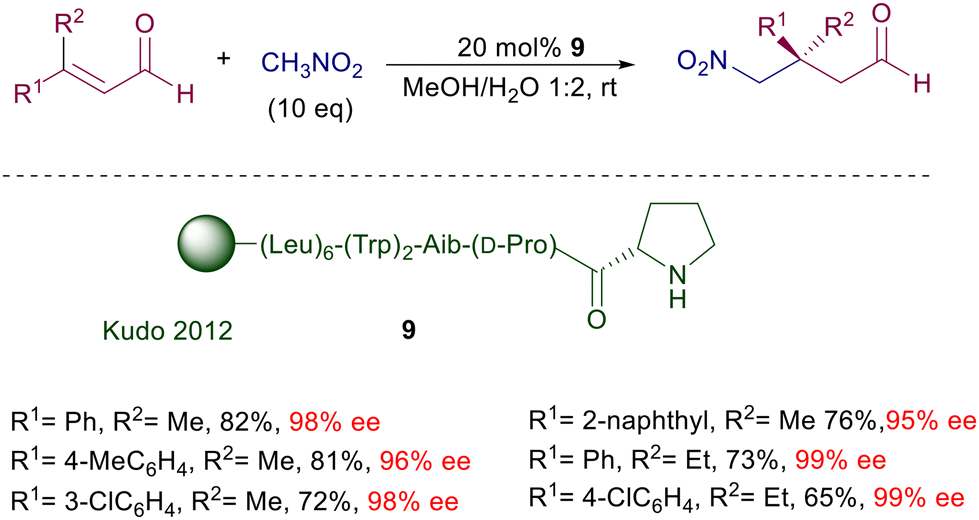
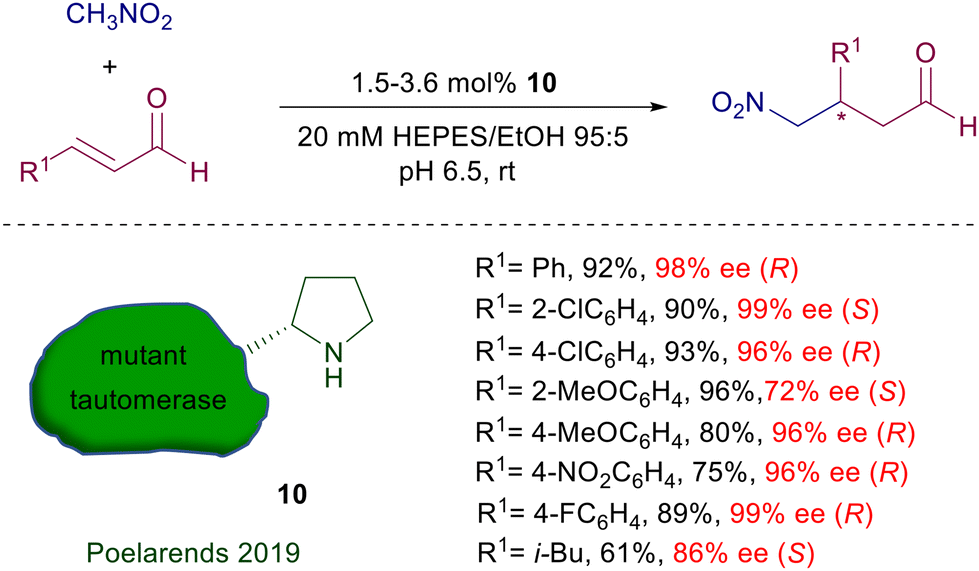
In the same reaction leading to nitro aldehyde 8, the pyrrolidine–camphorsulfonamide catalyst 6 shows enhanced performances considering the very low charge employed.19 Interestingly, addition of a few equivalents of water enables a sensible acceleration of the reaction, as also observed in related processes. According to the general mechanism reported in Fig. 1, a crucial role seems to be played by the acidic sulfonamide NH group in linking by hydrogen bonding the nitronate anion. It is worth noting that also for the latter catalysts the utilization of primary nitroalkanes affords the corresponding adducts with very poor diastereoselectivity, although with catalyst 6 the ee for each diastereomer is satisfactory (88–96% ee). Peptides bearing a terminal proline unit can also be involved in organocatalytic processes. The resin-supported peptide catalyst 9 has been employed for the enantioselective conjugate addition of nitromethane to β,β-disubstituted enals in hydroalcoholic solution (Scheme 6).20 This catalyst features a helical portion made of six leucine units and the non-proteinogenic 2-aminoisobutyric acid (Aib), which enhance the reaction rate and the enantioselectivity. At the end of the reaction the catalyst is just filtered off, but its reuse in further processes led to a notable conversion decrease probably caused by an oxidative deactivation. Enzymes bearing an N-terminal proline are the last developed biocatalytic systems containing this amino acid. The enzyme 4-oxalocrotonate tautomerase, specifically the F50A mutant species 10, is able to catalyze the Michael addition of nitromethane to enals (Scheme 7).21 The reaction is carried out in HEPES buffer (N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid) with a minimum amount of ethanol at room temperature.
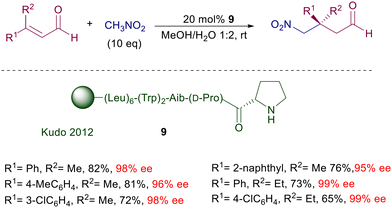 |
| | Scheme 6 Resin-bounded peptide catalyst bearing a terminal proline residue. | |
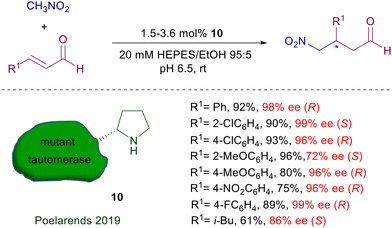 |
| | Scheme 7 Tautomerase catalyst embedding a proline residue. | |
Interestingly, the absolute stereochemistry of the resulting γ-nitroaldehyde is affected by the position of the substituent in the aryl ring, being S with ortho-substituents and R with meta and para substituted aryls. This effect is probably due to a relocation of the aryl group in the enzyme active pocket or a stereofacial shielding effect. The ability of this enzyme to catalyze crossed aldol condensations as well has been exploited in a four-step biocatalytic cascade synthesis of γ-nitrobutyric acids starting from acetaldehyde and arylaldehydes. The initially formed enal reacts with nitromethane, and finally the resulting γ-nitroaldehyde is oxidized to the acid by oxygen catalyzed by the NAD+/NADH redox couple. Engineered tetrameric streptavidin has been recently used for the same purpose, leading to encouraging results for the reaction of nitromethane with cinnamaldehyde.22
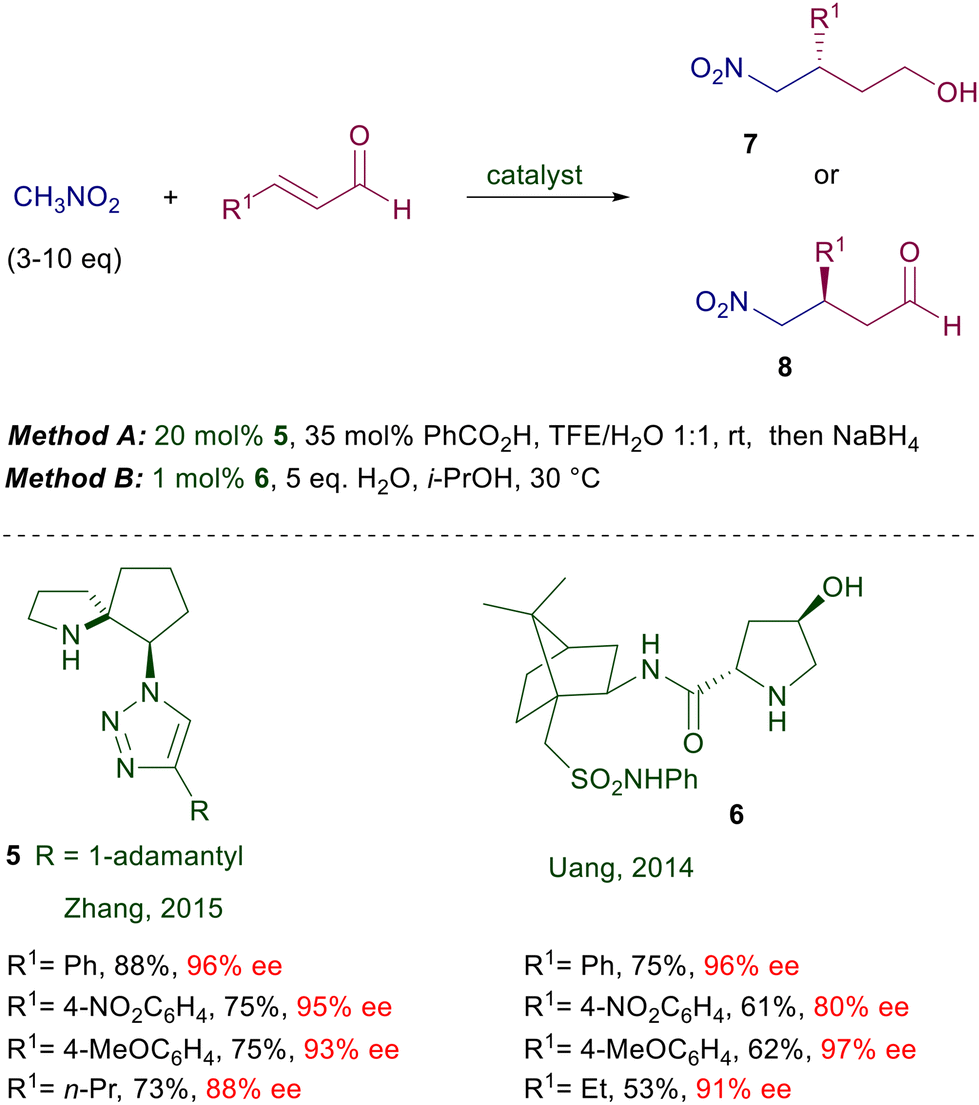
2.1.2 Prolinol derivatives.
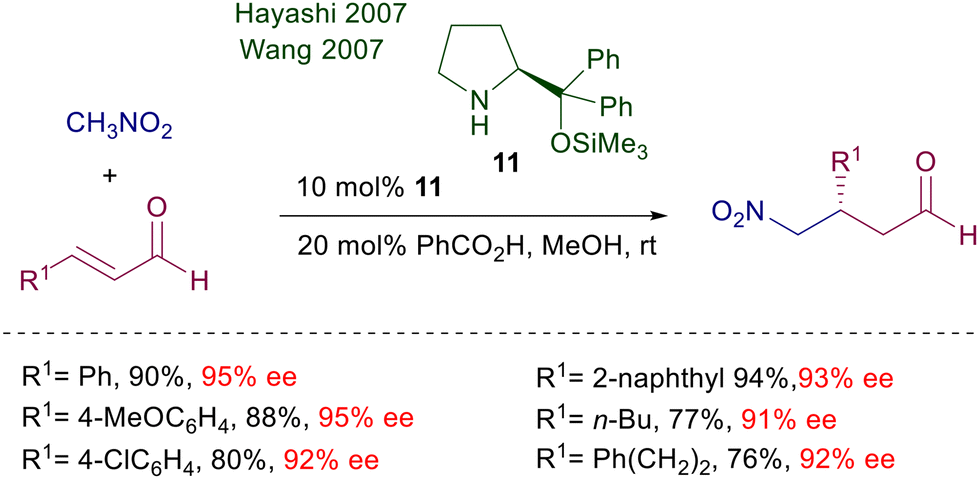
Structural modifications of the proline system also included the synthesis of diarylprolinol derivatives formerly used for the preparation of chiral oxazaborolidines and subsequently employed as catalysts in other processes including conjugate additions.23 Particularly, since its first trials O-trimethylsilyldiphenyl prolinol 11 proved very effective in the conjugate addition of nitromethane to enals (Scheme 8).24,25 Interestingly, neither unprotected diarylprolinol nor trifluoromethylarylprolinol gave results comparable with 11. Furthermore, the reaction efficiency is greatly enhanced using methanol or ethanol as solvents and benzoic acid as additive. The reactivity of primary nitroalkanes also leads to excellent results in terms of enantioselectivity, but no diastereoselectivity can be recorded. Later on, the same catalyst 11 was used under slightly different reaction conditions for the same reaction without any notable improvement.26 The reduced reactivity of α,β-unsaturated esters over enals and the inability to use the iminium ion catalytic approach with them led to the development of synthetic protocols for the one-pot oxidative esterification of the obtained γ-nitroaldehyde to the corresponding γ-nitroesters with NBS in methanol.27
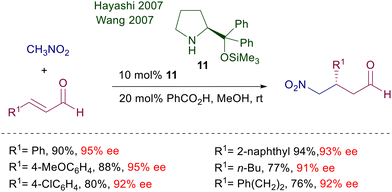 |
| | Scheme 8 The classical and efficient O-trimethylsilyldiphenyl prolinol catalyst. | |
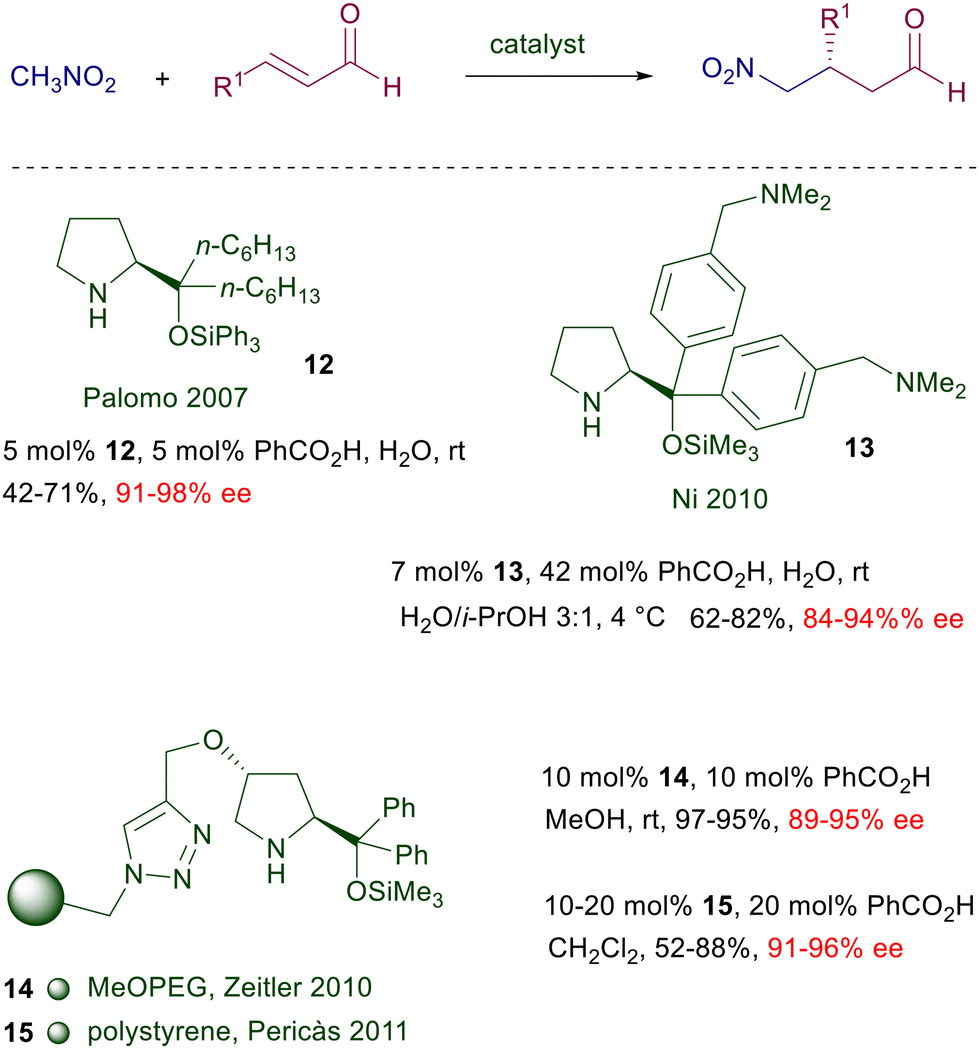
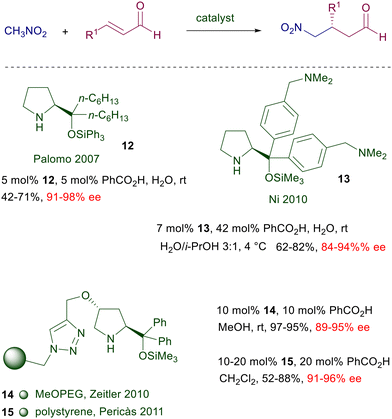
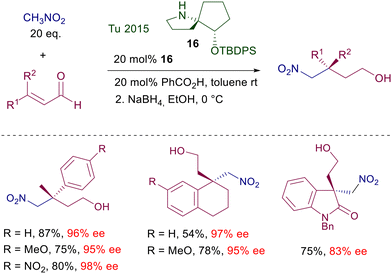
Despite the excellent results obtained with prolinol 11, a number of structural modifications have been proposed in order to carry out the reactions in water, or for recovering and recycling the catalyst (Scheme 9). Catalyst 12 has been designed with the aim of increasing its water compatibility and the steric constraint improving the E geometry of the intermediate iminium ion.28 The hexyl linear chain and the O-triphenylsilyl group provide better results over other shorter or longer alkyl chains in the catalyst. In order to increase the water solubility of the catalyst, a tertiary amino group was applied as substituent of the aryl groups in catalyst 13.29 The superior solubility over other catalysts is produced by the formation of an ammonium salt with benzoic acid, which in this scope is used in a larger amount than usual. The catalyst can also be recycled, keeping the aqueous solution for subsequent reactions, maintaining a good level of enantioselectivity but with prolonged reaction times and a drop in the chemical yield. The immobilized catalyst 14 prepared from 4-hydroxyproline shows good performances in terms of enantioselectivity and recyclability.30 Notably, the crude products obtained after simple work-up do not require further purification. A related polystyrene-bound catalyst 15 gives similar results but its recyclability has not been tested.31 Attempts to improve the catalytic activity by introducing imidazolium salts into the 4-hydroxyprolinol systems, thus generating ionic liquids, have not given significant results.32 The first successful generation of quaternary stereocenters by reaction of nitromethane with β,β-disubstituted enals using proline derivatives was carried out employing a chiral spiropyrrolidine catalyst 16 (Scheme 10).33 Subsequent reduction of the aldehyde ensures stability of the corresponding nitro alcohols and prevents the products from undergoing a retro-Michael reaction.
 |
| | Scheme 9 Structural modifications of chiral prolinol catalysts. | |
 |
| | Scheme 10 Spiropyrrolidine embedding a silyl ether appendage. | |
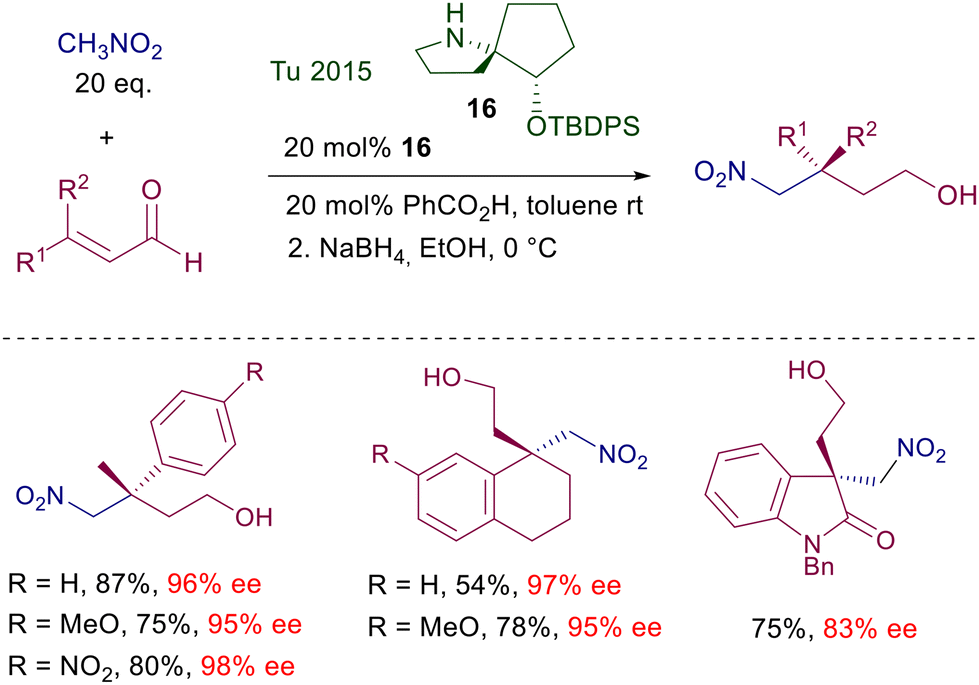
The process shows a notable efficiency level and can be applied to several functionalized substrates. The stereochemical bias offered by the rigid bicyclic framework is probably far superior to simple prolinols, and parallels the behavior further observed with triazole catalyst 5. A notable drawback in this protocol stems from the elaborate multistep synthesis required to prepare catalyst 16 which obviously cannot take advantage of the chiral pool as many other pyrrolidine-based catalysts.
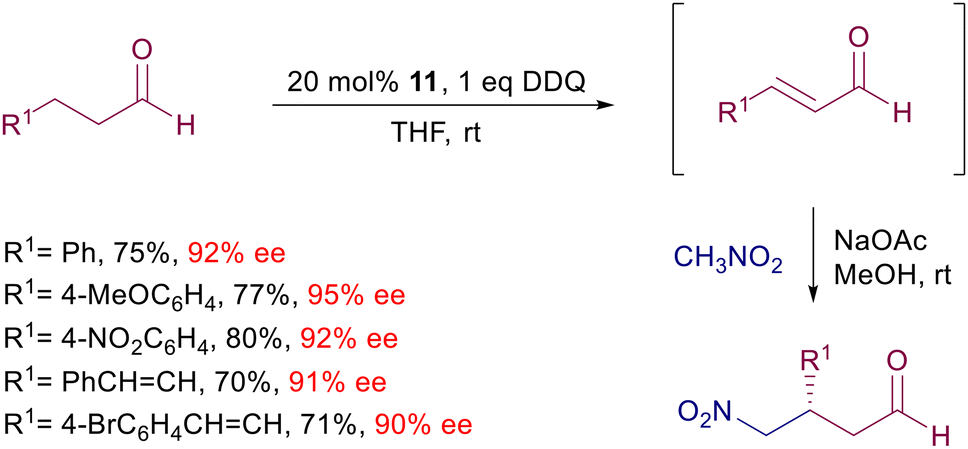
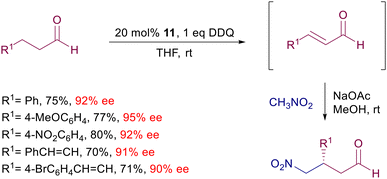
The limited stability of enals led to the development of an interesting protocol involving the one-pot oxidation of alkanals to enals followed by the conjugate addition of nitromethane (Scheme 11).34 Catalyst 11 generates the enamine intermediate of the alkanal, which is promptly oxidized by DDQ to the corresponding enal. Sodium acetate provides the basic milieu for the subsequent addition of nitromethane via the usual iminium ion. Interestingly, application of this procedure to γ,δ-unsaturated aldehydes leads to the β-regioselective addition of nitromethane to the intermediate dienal.
 |
| | Scheme 11 One-pot oxidative conversion of aldehydes into enals followed by conjugate addition of nitromethane catalyzed by prolinol 11. | |
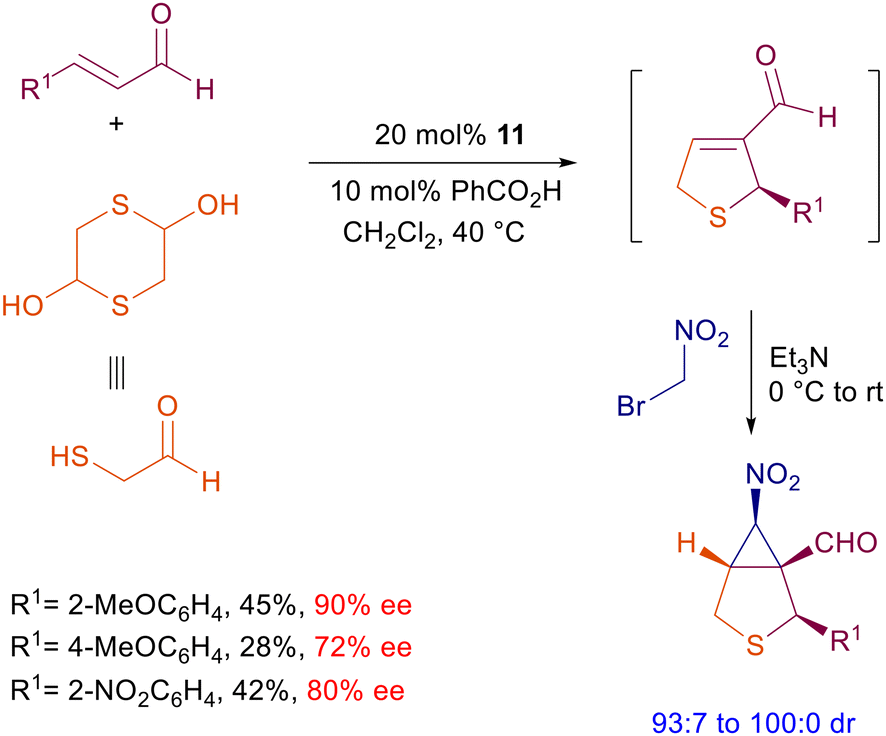
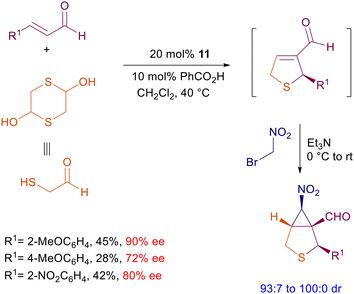
The wide success of prolinol 11 as a pyrrolidine-based catalyst for several applications can be ascribed to its ease of preparation from proline and the rewarding results obtained in terms of chemical yield and enantioselectivity. A still standing problem associated with 11 and related catalysts is practically the lack of diastereoselectivity detected using primary nitroalkanes. However, the involvement of functionalized nitroalkanes in cascade or tandem processes often leads to better stereodefined products, probably because of a thermodynamic equilibration of the α-nitro stereocenter which is more notable in cyclic products. Eventually, this equilibration can be forced toward the more stable stereoisomer by an added base in a further step. Bromonitromethane has been involved in several synthetic processes for the preparation of chiral nitrocyclopropane derivatives.35 Recently, this reagent was used in a one-pot process for the preparation of bicyclic derivatives embedding the nitrocyclopropane moiety (Scheme 12).36 The whole process consists of a preliminary sulfa-Michael reaction of 2-mercaptoacetaldehyde generated by its dimer with enals followed by an intramolecular aldol condensation. The reaction, catalyzed by prolinol 11, affords a chiral dihydrothiophene intermediate which undergoes a tandem conjugate addition-nucleophilic substitution to the final bicyclic derivative. The chiral prolinol is supposed to be active in both steps via iminium ion catalysis. The modest chemical yields recorded seem to be associated with the first step, which also affects the enantioselectivity of the target products which are, however, obtained with high diastereoselectivity. 2-Aryl-3-nitrocyclopentancarboxaldehydes can be prepared by reaction of 3-bromonitropropane with enals using catalyst 11 following the Michael-nucleophilic substitution strategy.37 Remarkably, the reaction conditions enable outstanding control in the formation of three stereocenters including the labile α-nitro one by thermodynamic equilibration.
 |
| | Scheme 12 Synthesis of nitrocyclopropane derivatives. | |
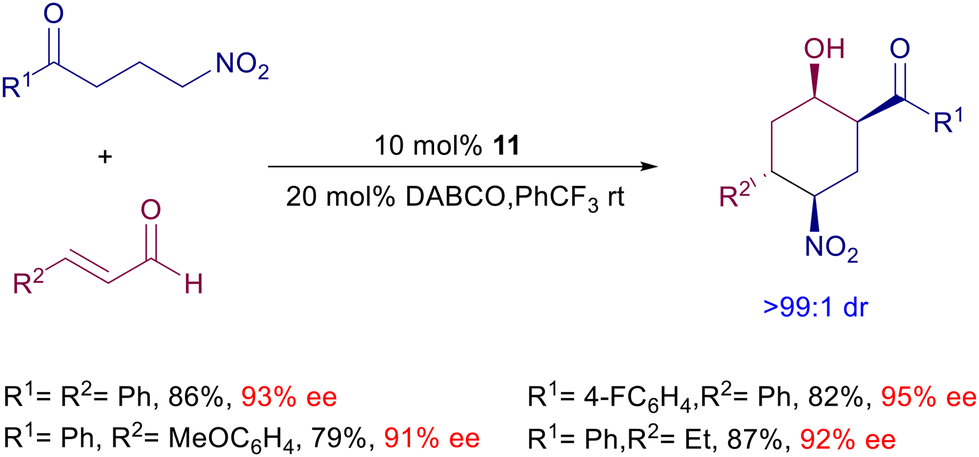
The conjugate addition of γ-nitroketones to enals can be coupled with an intramolecular aldol reaction leading to densely functionalized nitrocyclohexanol derivatives (Scheme 13).38 Four stereocenters are generated in a single synthetic operation with an outstanding level of diastereoselectivity and satisfactory ee values. This process is also possible starting from allylic alcohols as enal precursors exploiting an in situ preliminary oxidation using the TPAP/NMO couple.
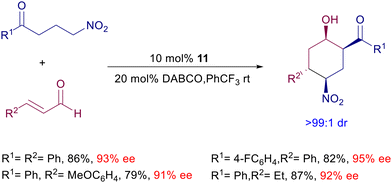 |
| | Scheme 13 Tandem conjugate addition/aldol reactions catalyzed by chiral prolinol 11. | |
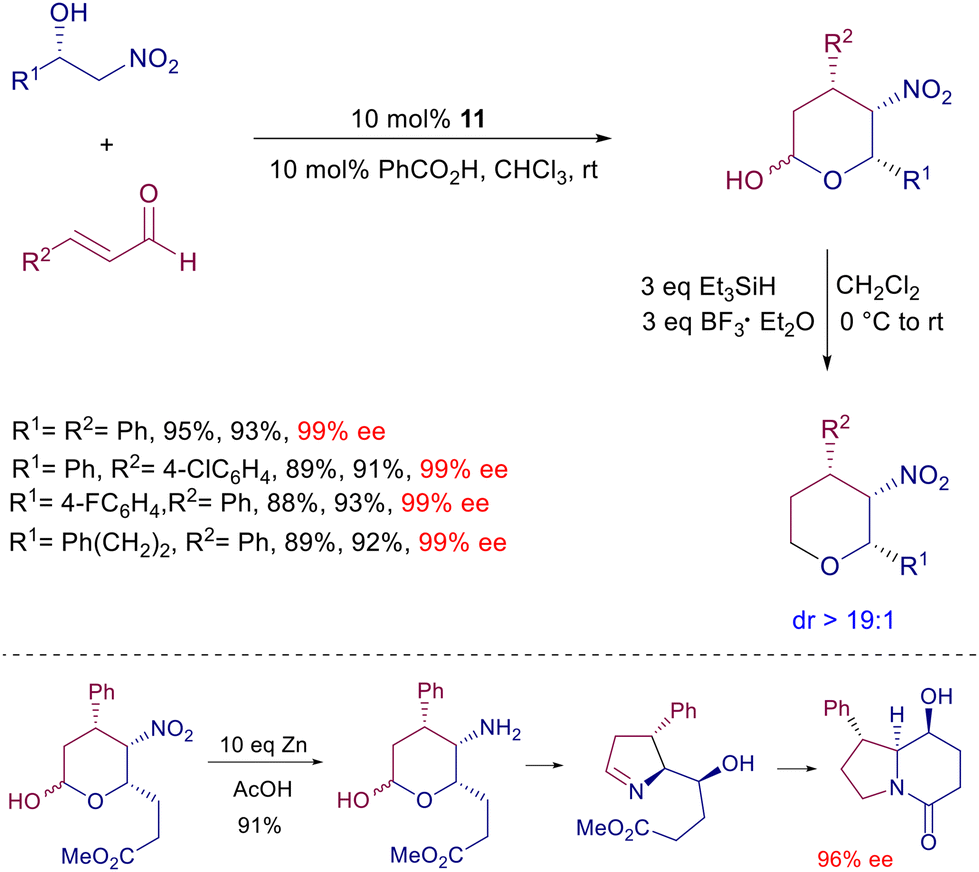
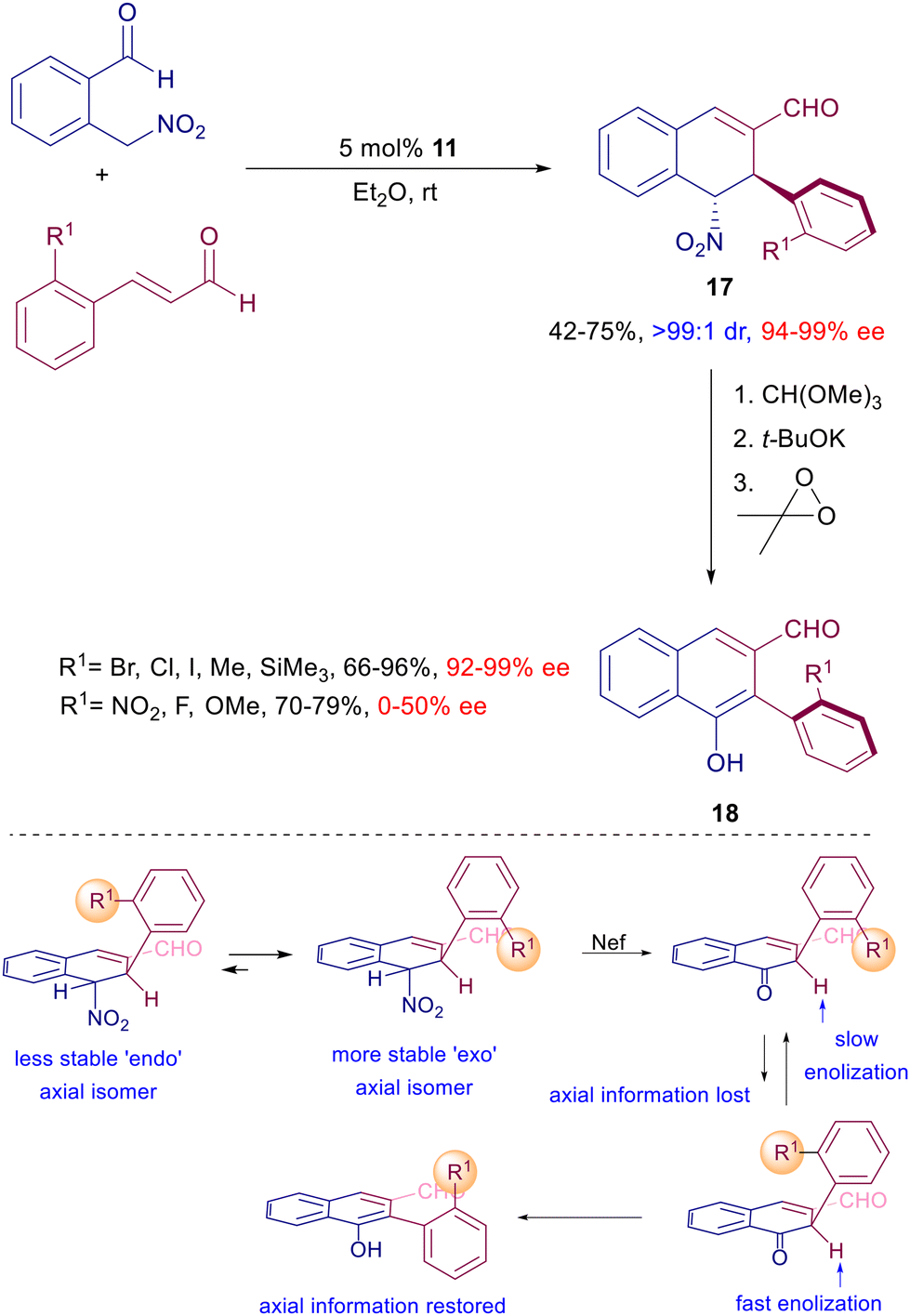
Optically active nitro alcohols obtained by an asymmetric Henry reaction can be used as substrates in the enantioselective reaction with enals catalyzed by prolinol derivative 11 (Scheme 14).39 The initially formed conjugate adducts undergo a spontaneous cyclization leading to an epimeric mixture of the corresponding lactols. The high diastereo- and enantioselectivity of the process is evidenced by the subsequent reductive conversion of the lactols by triethylsilane, via oxonium ion intermediate, giving enantiopure tetrahydropyrans. The synthetic usefulness of these derivatives has been demonstrated converting a specific lactol bearing an ester appendage to a chiral indolizinone compound by zinc reduction under acidic conditions. This very efficient conversion involves a cascade process entailing the nitro group reduction, intramolecular imination, imine reduction and final lactamization. The reaction of 2-nitromethylbenzaldehyde with ortho-substituted 3-arylenals catalyzed by 11 offers an additional intriguing aspect beyond the expected asymmetric induction of the obtained products (Scheme 15).40
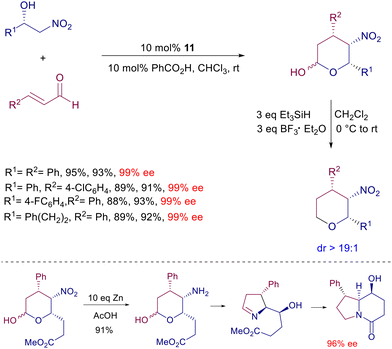 |
| | Scheme 14 Synthesis of nitrolactols and their application to the preparation of optically active indolizinones. | |
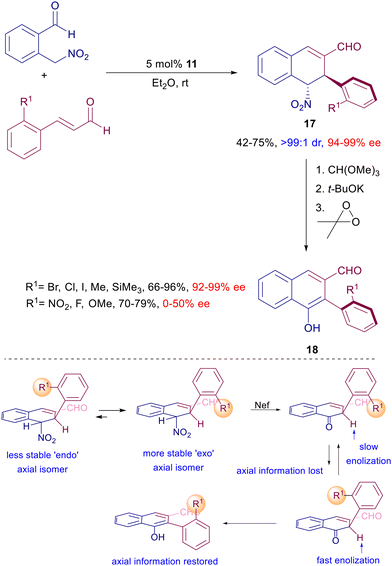 |
| | Scheme 15 Enantioselective synthesis of optically active biaryls catalyzed by 11. | |
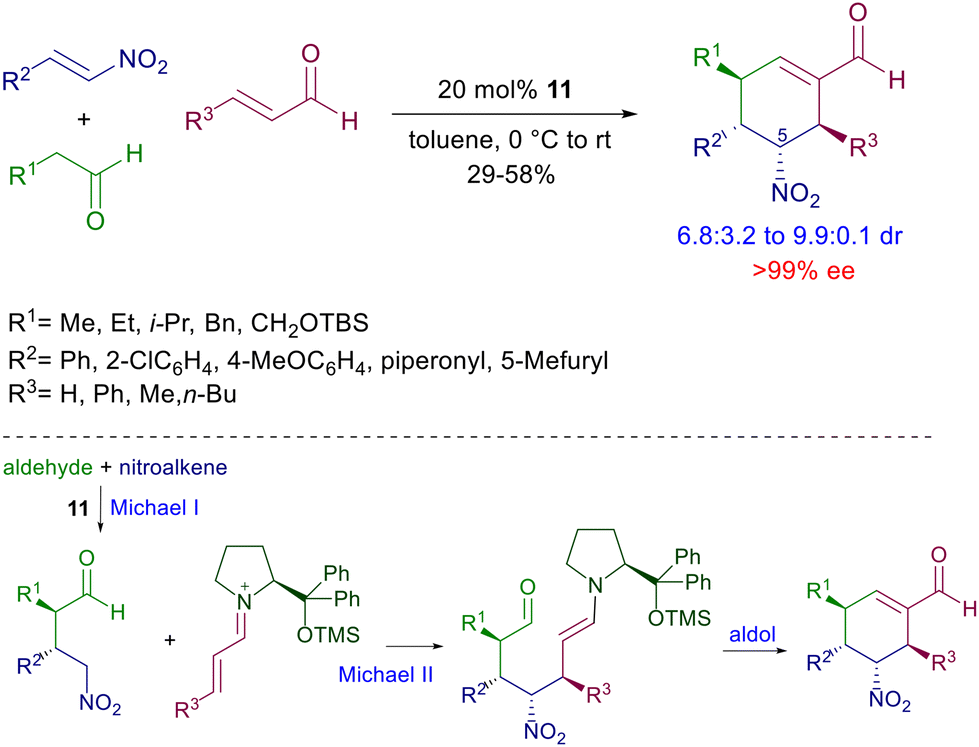
Besides the two formed stereocenters, dihydronaphthalenes 17 feature additional axial chiral information controlled by the thermodynamic stability of the ‘exo’ rotamer over the ‘endo’ one. The nitro to carbonyl conversion (Nef reaction), after carbonyl protection, of compounds 17 affords naphthalenone intermediates in which the axial chiral information is lost, but since the ‘endo’ rotamer is enolized faster than the ‘exo’ one, the axial chiral information is restored in the final biaryls 18. It should be observed that this effect tends to considerably decrease with the reduced dimensions of the ortho substituents. The conjugate addition of nitroalkanes can also be included in multistep processes involving other nitro derivatives as substrates. In a seminal paper, Enders and co-workers elaborated a procedure entailing a three-component coupling of aldehydes, enals and nitroalkenes catalyzed by 11 leading to the formation of cyclohexene carbaldehydes controlling the generation of four stereocenters (Scheme 16).41 The obtained diastereomeric mixture was epimeric at C-5 with an outstanding level of enantioselectivity for each stereoisomer. The cascade process involves a preliminary conjugate addition of the aldehyde to the nitroalkene via enamine catalysis followed by the usual addition of the resulting γ-nitroaldehyde to the enal via iminium ion catalysis. The catalytic cycle ends with the ring closure by aldol condensation. This strategy has been subsequently implemented using other functionalized nitroalkenes for the preparation of polycyclic derivatives.42
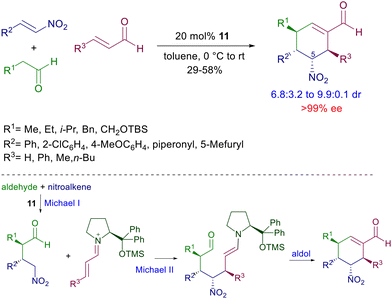 |
| | Scheme 16 Three-component coupling involving a cascade Michael–Michael–aldol condensation catalyzed by 11. | |
2.2 Primary amines and peptides
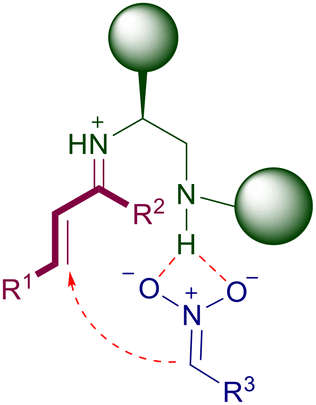
The iminium ion catalysis can also be exploited using primary amines providing that an acid co-catalyst is added.43 These catalysts have been particularly developed for the conjugate additions of nitroalkanes to enones, which require a more reactive amino derivative to generate the active iminium ion. Particularly, the vast majority of the catalysts employed are actually 1,2-diamines in which the second nitrogen atom is a secondary or a tertiary amine. Thus, the catalytic action is jointly provided by the two amino groups according to the general interaction depicted in Fig. 2. The secondary or tertiary amino group interacts with the nitronate anion through hydrogen bonding, enabling the appropriate proximity effect required for an efficient addition.
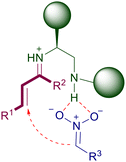 |
| | Fig. 2 Catalytic action of chiral 1,2-diamino compounds in the conjugate addition of nitroalkanes with enones. | |
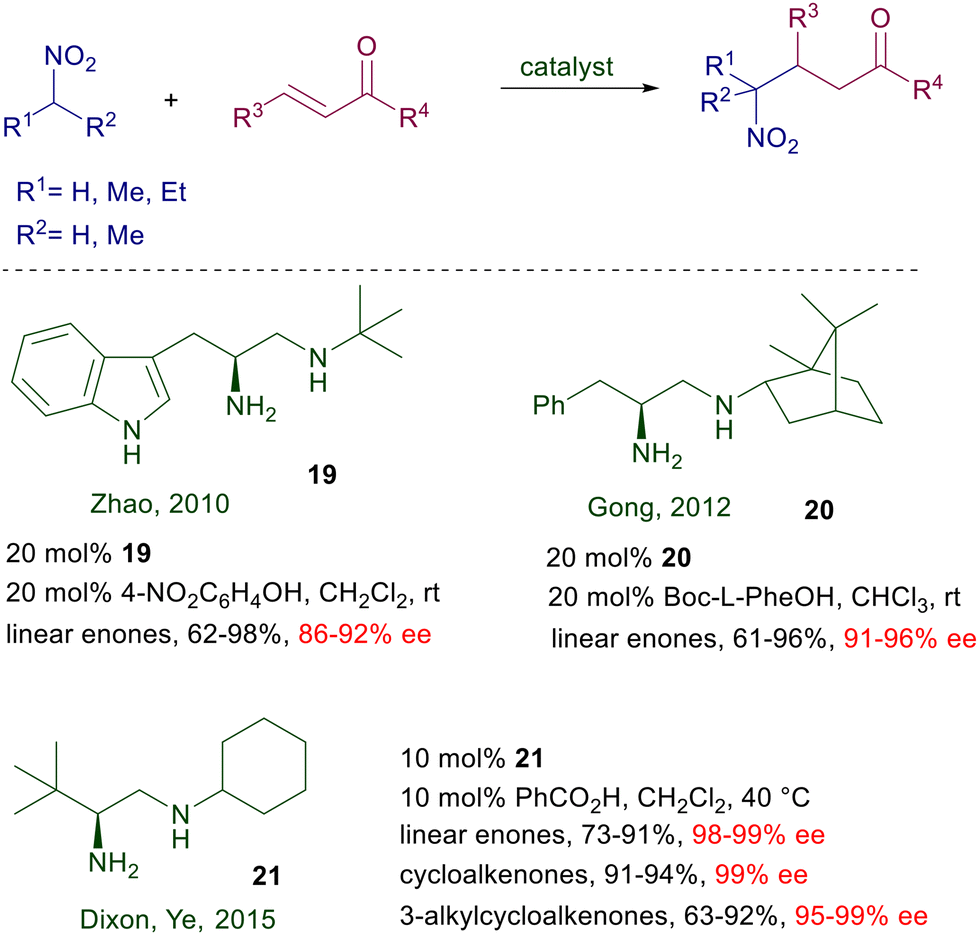
Chiral 1,2-diamines obtained from α-amino acids have been tested for the conjugate addition of nitroalkanes with enones (Scheme 17). The L-tryptophan-derived catalyst 19 was proved to be effective for the reaction of 2-nitropropane with linear enones.44 Enforcement of the chirality by introducing a camphor residue in catalyst 20 and N-Boc-phenylalanine as additive was attempted with only modest improvements.45
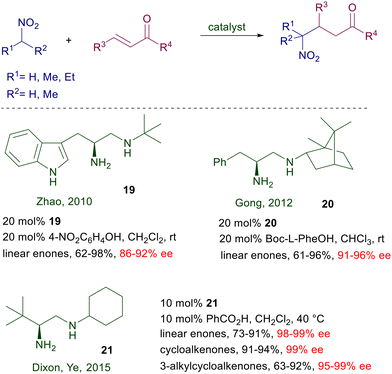 |
| | Scheme 17 Catalytic activity of various chiral 1,2-diamines. | |
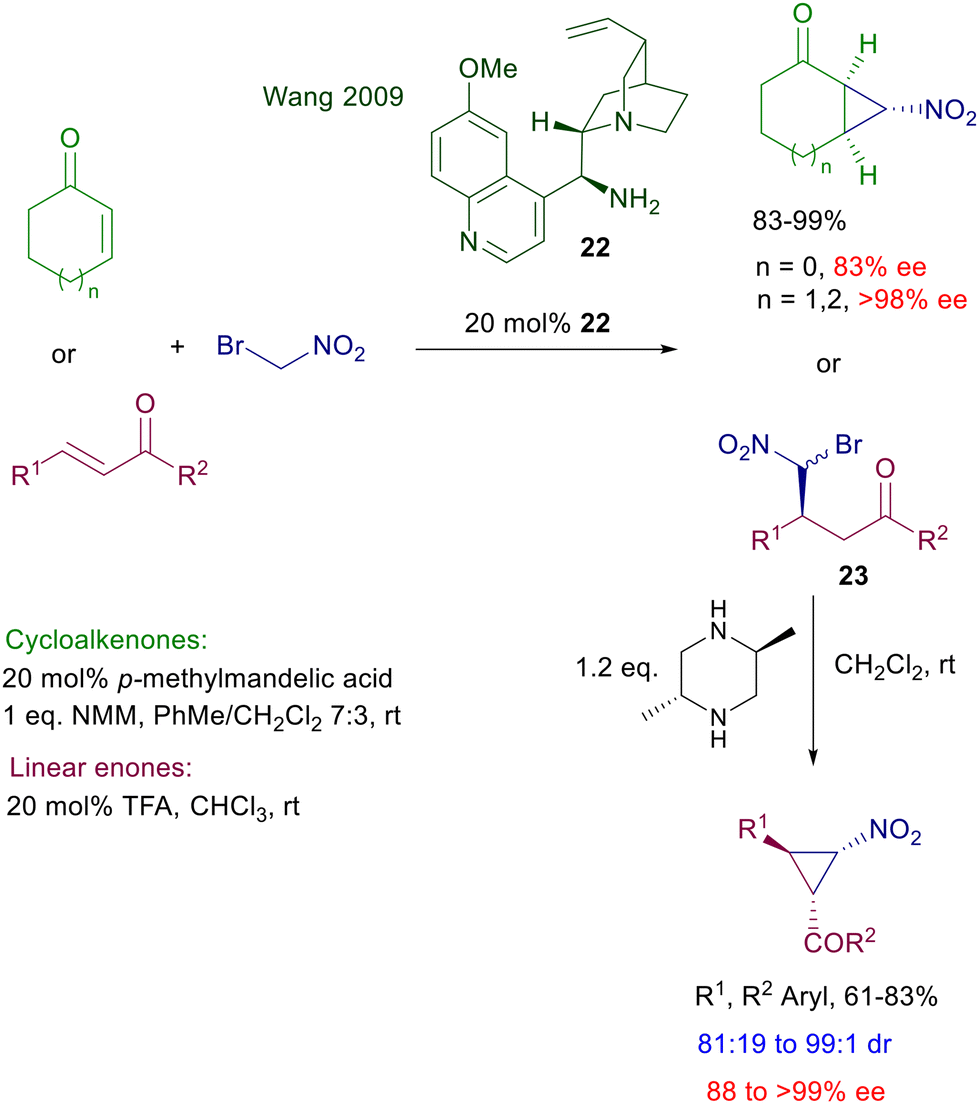
Increasing the steric crowding around the primary amino group in catalyst 21 proved very effective in leading to an excellent enantioselectivity for the reaction of nitromethane with linear enones and cycloalkenones even bearing a C-3 substituent.46 The latter protocol thus allows an efficient entry to γ-nitroketone derivatives featuring quaternary stereocenters to be used as intermediates for the synthesis of sesquiterpenoid skeletons. Primary amines derived from cinchona alkaloids are probably the most employed diamino derivatives for these conjugate additions.47 The pseudoenantiomeric relationship between the quinine and quinidine series enables a versatile access to both enantiomeric forms of the possible adducts. The acidic cocatalyst plays a fundamental role by protonation of the quinuclidine moiety, affecting the rate of formation of the intermediate imine. Furthermore, the cocatalyst can be suitably used to modify the steric shielding through appropriate modulation of its anionic structure. Quinine-derived amine 22 was selected as the most effective catalyst for the nitrocyclopropanation of enones using bromonitromethane (Scheme 18).48 Cycloalkenones are directly converted into bicyclic derivatives in the presence of N-methylmorpholine (NMM), which assists the final ring closure by nucleophilic displacement of the bromide anion. The enantioselectivity is satisfactory for cyclopentenone and excellent for cycloalkenone homologues as substrates.
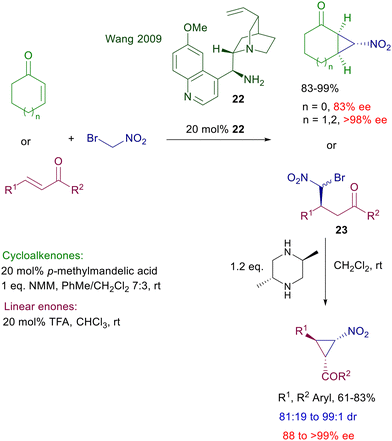 |
| | Scheme 18 Synthesis of chiral nitrocyclopropane derivatives. | |
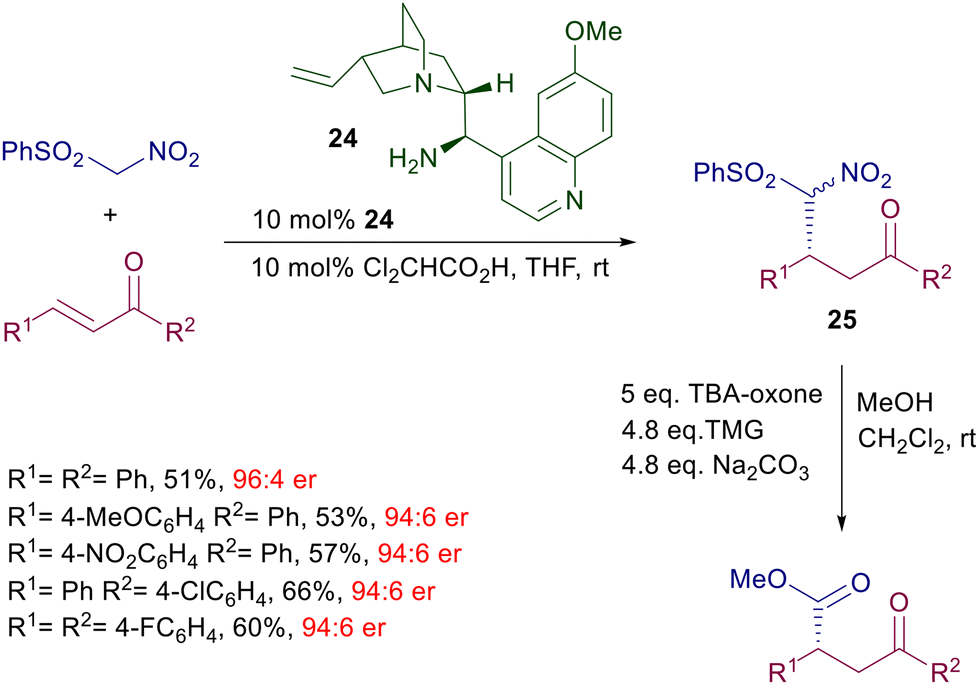
Linear enones require two separate steps to afford the corresponding nitrocyclopropane derivatives with satisfactory enantioselectivity. The initial adduct 23 is obtained with poor diastereoselectivity but excellent enantioselectivity for each formed diastereomer. Among different bases, trans-2,5-dimethylpiperazine has been identified as the best promoter for the ring closure, leading to the cyclopropane derivatives in good diastereo- and enantioselectivity. The reaction of phenylsulfonylnitromethane with enones catalyzed by quinidine derivative 24 has been employed for the two-step synthesis of optically active 2-aryl-γ-ketoesters (Scheme 19).49 Catalyst 24 affords the expected adducts 25 in an enantiocomplementary fashion compared with its pseudoenantiomer 22 for the synthesis of 23, thus confirming that the same mechanism is operative for both functionalized nitromethane derivatives. Conversion of nitro ketone 25 into the target ketoesters is carried out through an oxidative Nef reaction using tetrabutylammonium-oxone, thus realizing a formal conjugate addition of an acyl anion synthon to enones.
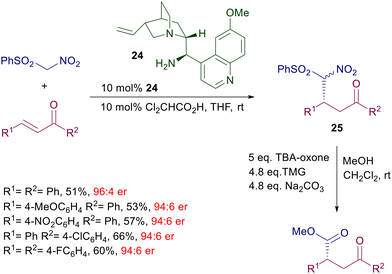 |
| | Scheme 19 Synthesis of chiral 4-oxoesters by conjugate addition/Nef reaction. | |
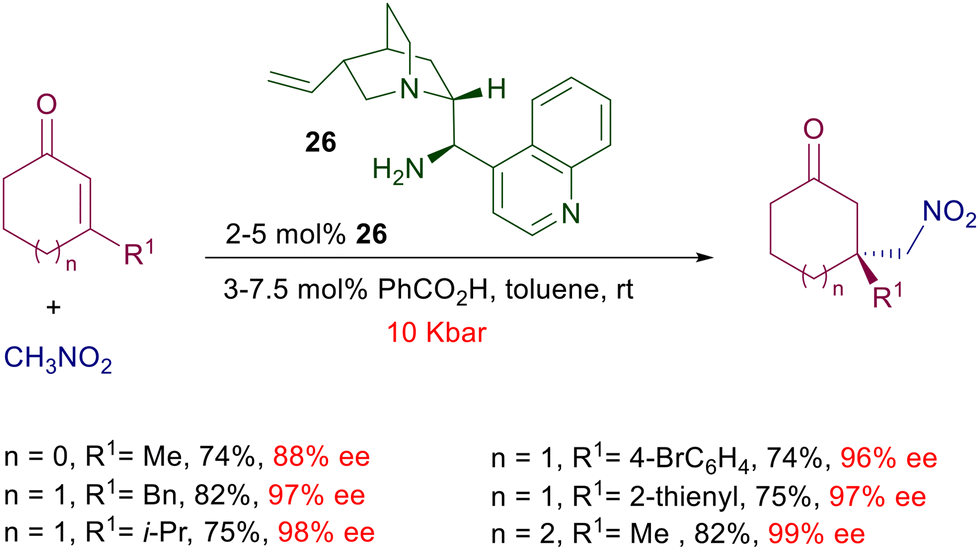
The influence of pressure has been evaluated in the enantioselective addition of nitromethane to 3-substituted cycloalkenones catalyzed by cinchonine-derived amine 26 (Scheme 20).50 At normal pressure (1 bar) the reaction is practically ineffective (1–9% yield), contrary to what was observed in the same process catalyzed by chiral diamine 21 (Scheme 17). However, by increasing the pressure to 10 kbar a notable improvement in the chemical yield was recorded. It should be observed that the pressure applied has no effect on the enantioselectivity of the reaction, which is very high even at 1 bar, but only affects the yield of the reaction. The same effect is evidenced for the nitromethane addition to some β,β-disubstituted linear enones.
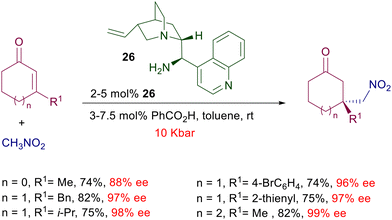 |
| | Scheme 20 Conjugate addition of nitromethane to 3-substituted cycloalkenones catalyzed by cinchonine-derived amine 26. | |
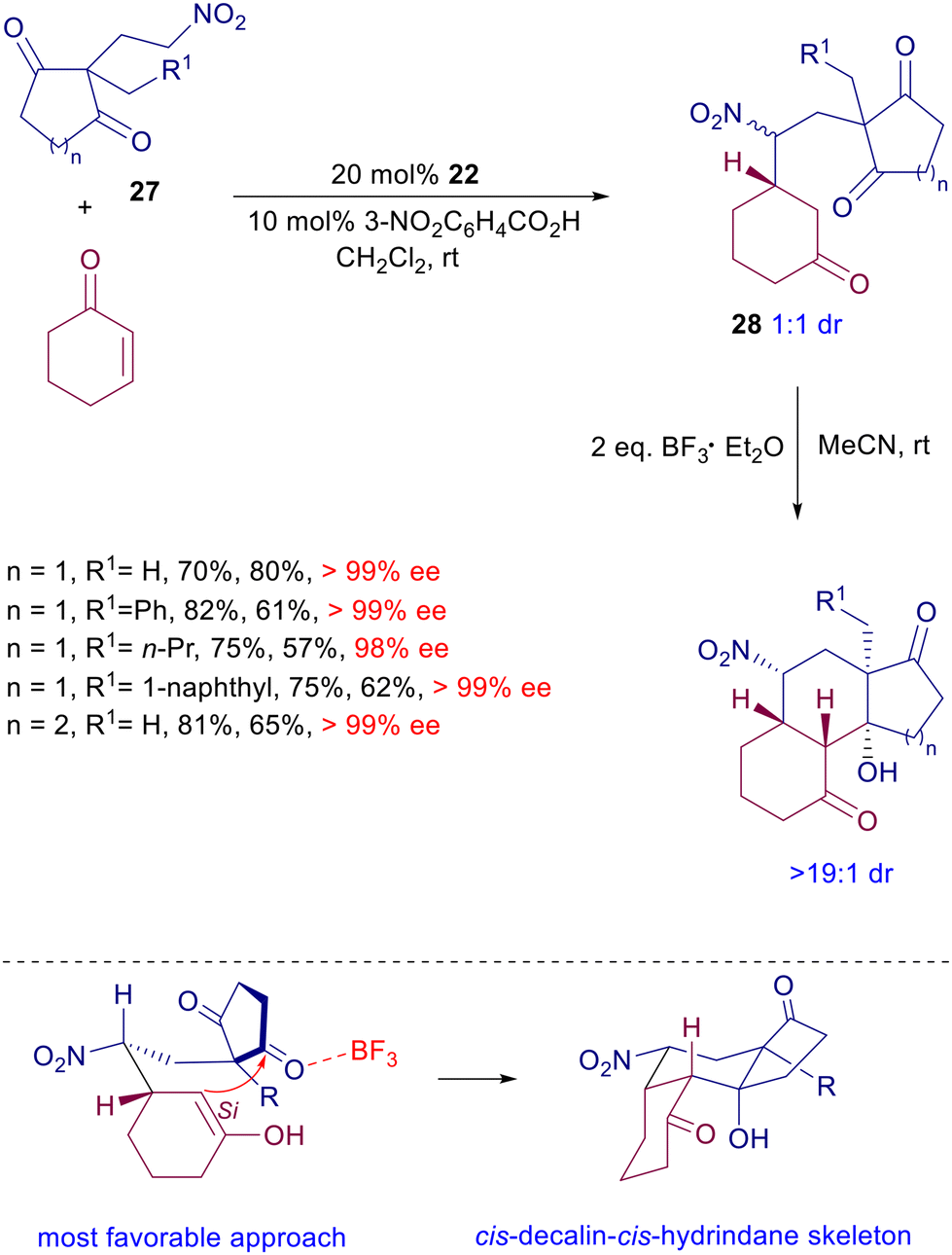
Ready access to the uncommon cis,cis-tricyclic diterpenoid skeleton can be achieved by reaction of functionalized nitroethylcycloalkanediones 27 with cyclohexenone in the presence of chiral diamine catalyst 22 (Scheme 21).51 Adducts 28 are formed as an equimolar epimeric mixture with excellent control of the ring stereocenter. The subsequent intramolecular aldol reaction, catalyzed by boron trifluoride etherate, affords the target tricyclic derivatives with outstanding stereocontrol, considering that in the six-membered ring transition state, the most favorable approach entails a pseudoequatorial position of the nitro group and attack of the enolate Si side to the ketone.
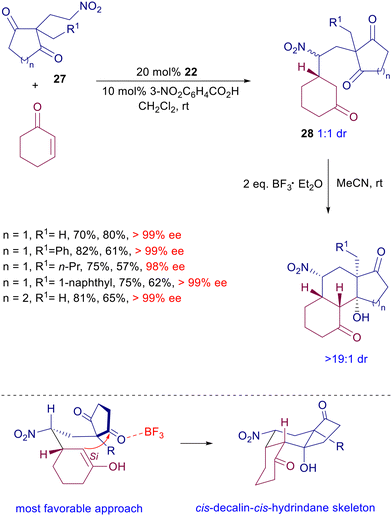 |
| | Scheme 21 Synthesis of cis,cis-tricyclic diterpenoid derivatives by a tandem conjugate addition/aldol condensation catalyzed by quinine-derived amine 22. | |
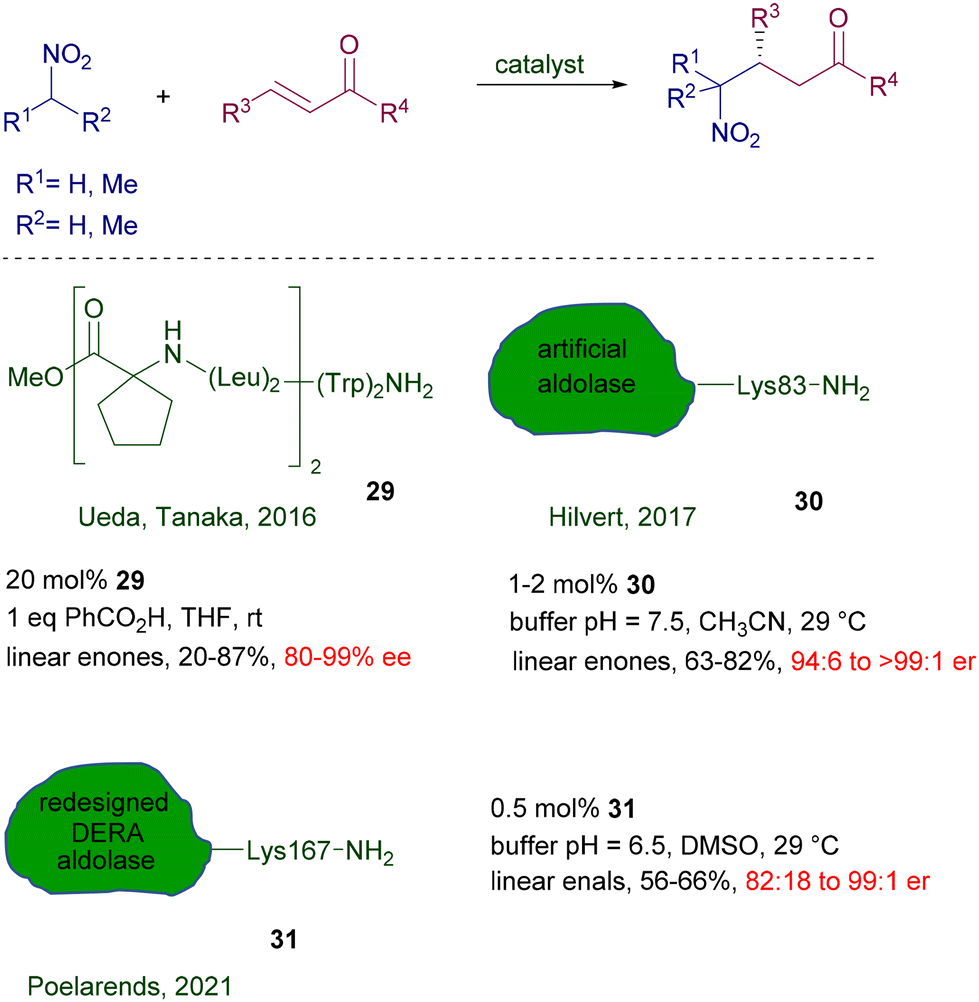
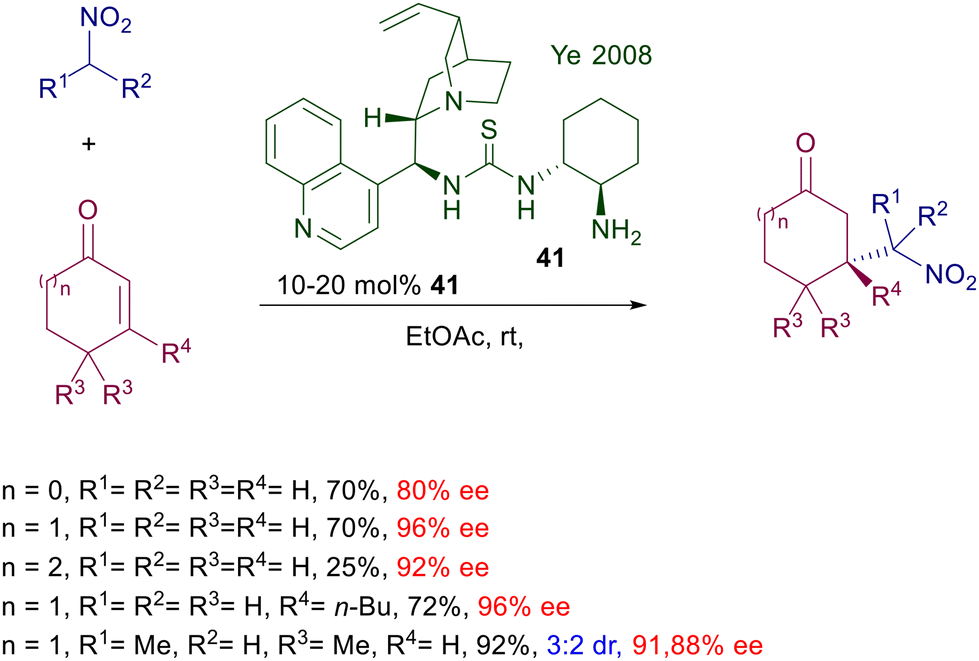
Biocatalytic methods can be applied in these reactions using peptides and enzymes providing that free primary amino residues are present in the molecular framework. Peptide foldamer 29 having a helical structure has been used for the addition of nitroalkanes to linear enones using a terminal tryptophan residue as catalytic site (Scheme 22).52 The catalyst charge parallels that of most organocatalyzed procedures, and the enantioselectivity obtained is high for several of the reported examples. Artificial aldolase RA95.5-8 30 created by computational design is very effective in the reaction of nitromethane with linear enones at low catalyst charge.53 As for many enzymatic procedures, careful pH and temperature controls are mandatory for suitable catalytic activity, which has been evaluated to occur at the level of the lysine-83 residue. A redesigned aldolase 31 from 2-deoxy-D-ribose-5-phosphate aldolase (DERA) has been recently introduced for the addition of nitromethane to enals.54 The new version of the DERA enzyme differs from the original one by twelve amino acids and appears 190-fold more active at a very reduced catalyst charge. Also for this enzyme, the active site is attributable to the primary amino group of the lysine-167 residue.
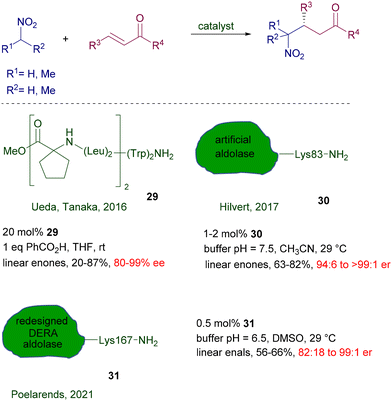 |
| | Scheme 22 Chiral peptide foldamers and aldolases as biocatalysts. | |
2.3 Thioureas as bifunctional catalysts
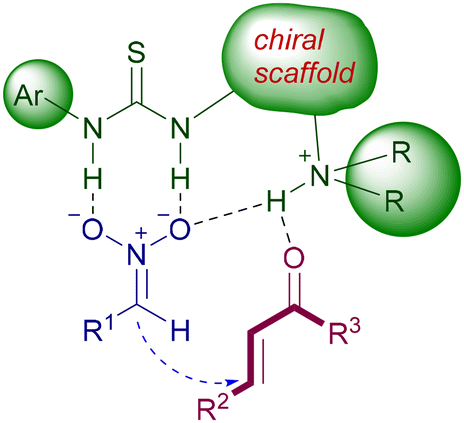
Functionalized chiral ureas exert their catalytic activity in a complementary fashion to amino derivatives, activating the nitroalkane towards the conjugate addition. The activation is due to the strong hydrogen bonding established between the acidic NH of urea and the oxygen atoms of the nitro group. The acidity of these NH bonds is superior in thioureas, which are thus preferred to ureas as catalysts. Furthermore, the reduced electronegativity of sulfur compared with that of oxygen makes the self-association by hydrogen bonding among thiourea molecules less favorable, thus enhancing the catalytic activity. The interactive model is totally based on the hydrogen bonding network among the bifunctional catalyst and the substrate according to Fig. 3. The protonated amino group is instrumental in providing the necessary interaction with the Michael acceptor, generating an electrophilic catalysis and ensuring the required proximity effect.
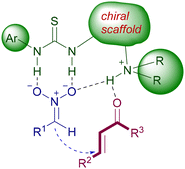 |
| | Fig. 3 Mechanism of activation of the reactants by chiral bifunctional thioureas. | |
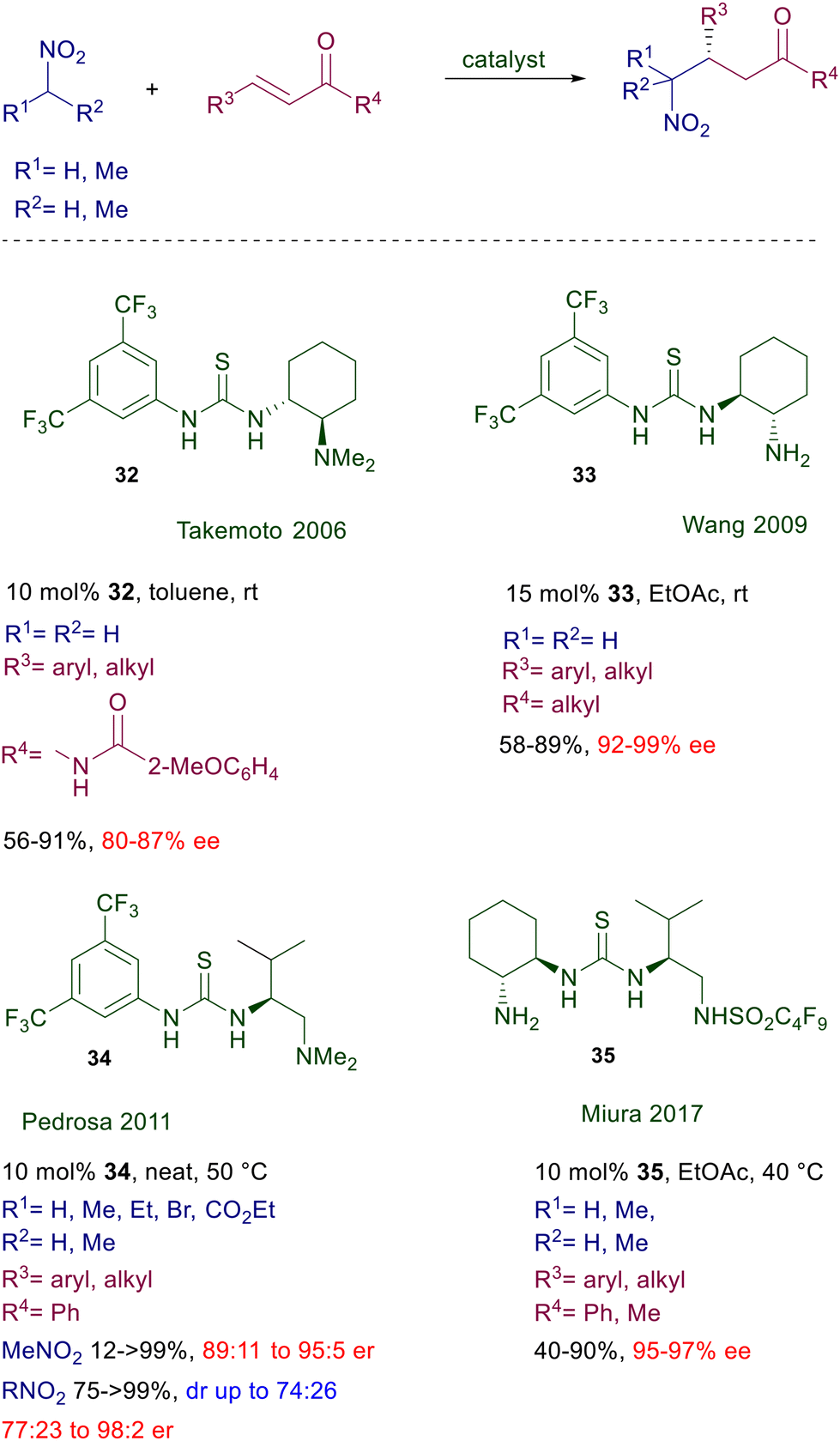
In 2003, a chiral bifunctional thiourea was introduced by Takemoto and co-workers for the conjugate addition of malonates to nitroalkenes,55 but only three years later the same catalyst 32 was used for the addition of nitromethane to α,β-unsaturated imides (Scheme 23).56 Thus, the use of catalyst 32 in these conjugate additions was anticipated by a cinchona-derived thiourea one year before (vide infra).
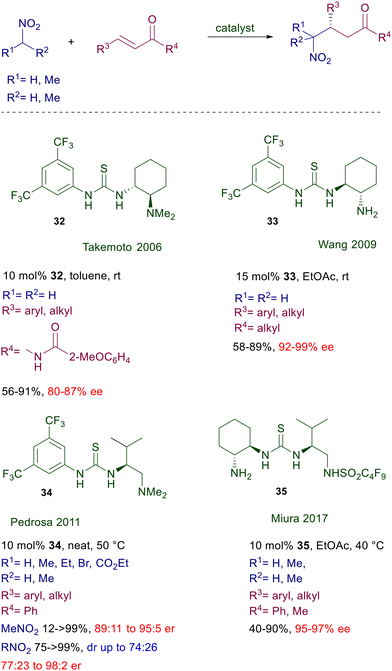 |
| | Scheme 23 Chiral bifunctional thioureas as catalysts. | |
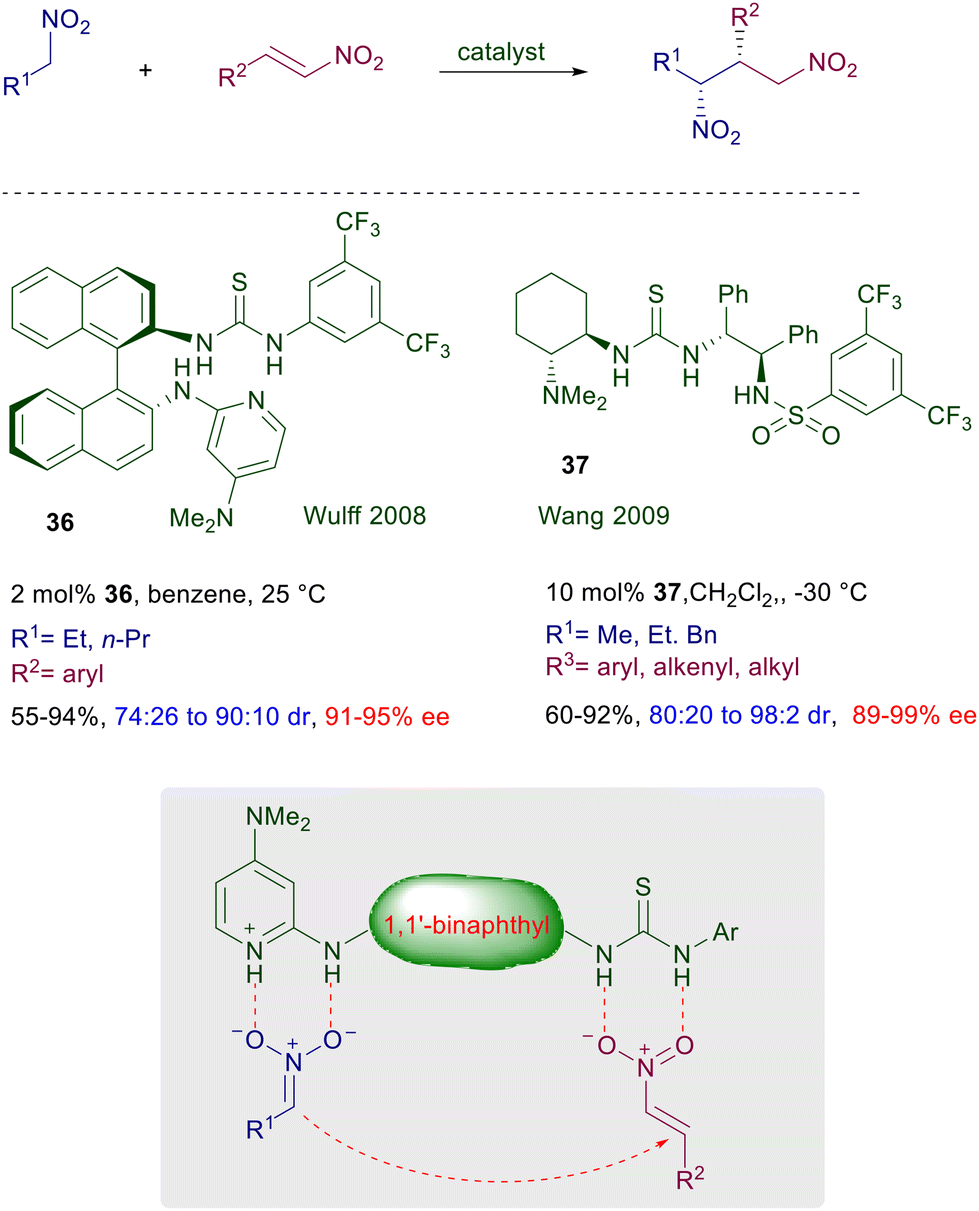
The catalytic activity of 32 was ascribed to the activation of the imide moiety rather than the nitromethane by hydrogen bonding with the NH of thiourea, although no evidence for this assumption has been provided. Furthermore, the presence of a 2-methoxy group in the aryl substituent of the imide was proved to be mandatory for an optimal enantioselectivity. Later on, a structural modification was introduced converting the tertiary amino group into a primary one in catalyst 33.57 The performance of this catalyst is superior compared with its previous analogue, and although activation of the enone carbonyl by hydrogen bonding with the NH groups of the catalyst has been proposed, the concomitant iminium ion formation and nitroalkane activation appears to be the most probable mechanism. In any event, catalyst 34, developed as a further modification of the Takemoto's catalyst 32, is also effective on simple enones and functionalized nitroalkanes.58 Theoretical studies on this catalyst suggest the general assumption that the nitroalkane is activated by the thiourea NH groups and the enone by the protonated tertiary amine as depicted in Fig. 3. The recently developed bifunctional catalyst 35 includes in its structure an acidic sulfonamide group and a primary amine.59 This catalyst affords high ee values in the corresponding adducts, and it is correctly proposed that activation of the enone occurs via iminium ion while activation of the nitroalkane is brought about by a joint action of the three acidic NH groups. The asymmetric conjugate addition of nitroalkanes to nitroalkenes affords 1,3-dinitro derivatives which are direct precursors of optically active 1,3-diamines (Scheme 24). Thiourea catalyst 36, featuring a binaphthyl chiral scaffold, gives good results in the asymmetric reaction of primary nitroalkanes with nitrostyrene derivatives.60 The thiourea and the 2-aminopyridyl catalytic sites jointly provide the activation of the reactants through a closely related hydrogen bonding mechanism. Interestingly, the observed diastereoselectivity is superior compared with that recorded in similar reactions with enones, that barely exceeds the value of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. Satisfactory results are also obtained in the same process using the more traditional catalyst 37, for which generally high values of diastereo- and enantioselectivity are recorded.61
:2. Satisfactory results are also obtained in the same process using the more traditional catalyst 37, for which generally high values of diastereo- and enantioselectivity are recorded.61
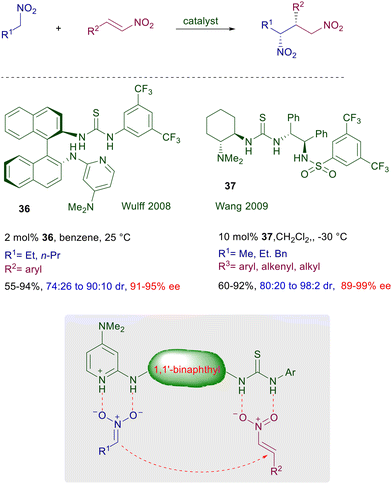 |
| | Scheme 24 Conjugate addition of nitroalkanes to nitroalkenes catalyzed by bifunctional chiral catalysts 36 and 37. | |
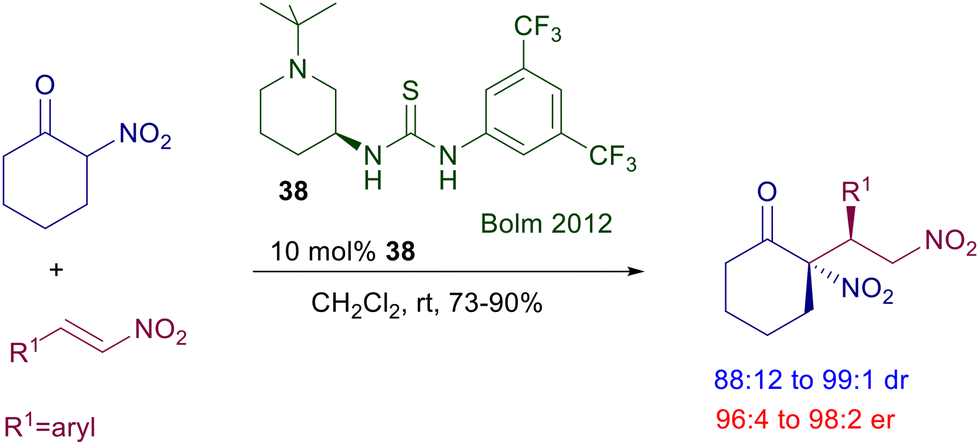
The reaction of 2-nitrocyclohexanone with nitrostyrenes required the elaboration of a different thiourea derivative considering that classical catalysts such as 32 were proved to be poorly effective in this process (Scheme 25).62 Catalyst 38, featuring a piperidine unit, was highly active in this conjugate addition, leading to the corresponding dinitro derivatives with diastereo- and enantioselectivity.
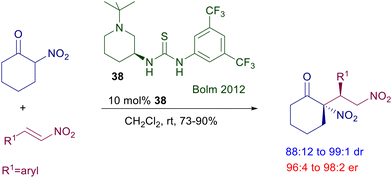 |
| | Scheme 25 Conjugate addition of 2-nitrocyclohexanone to nitroalkenes. | |
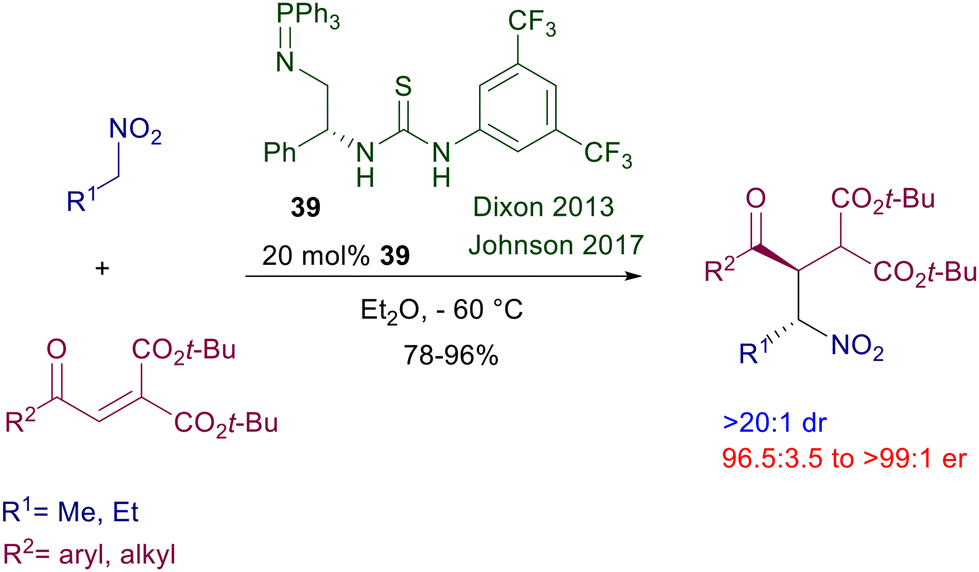
Increasing the basic character of the catalyst has a notable impact on the efficiency of the reactions involving nitroalkanes. Iminophosphorane-based thiourea catalyst 39 was originally designed for enantioselective nitro-Mannich reactions,63 but is also very effective for the conjugate addition of nitroalkanes to enone diesters (Scheme 26).64 The corresponding adducts are practically obtained as single diastereomers and with very good enantioselectivity.
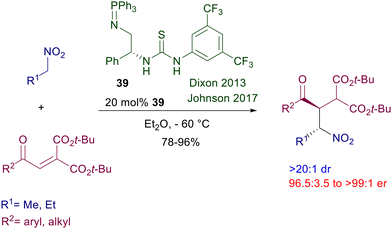 |
| | Scheme 26 Chiral iminophosphorane-thiourea catalyst in the reaction of nitroalkanes with enone diesters. | |
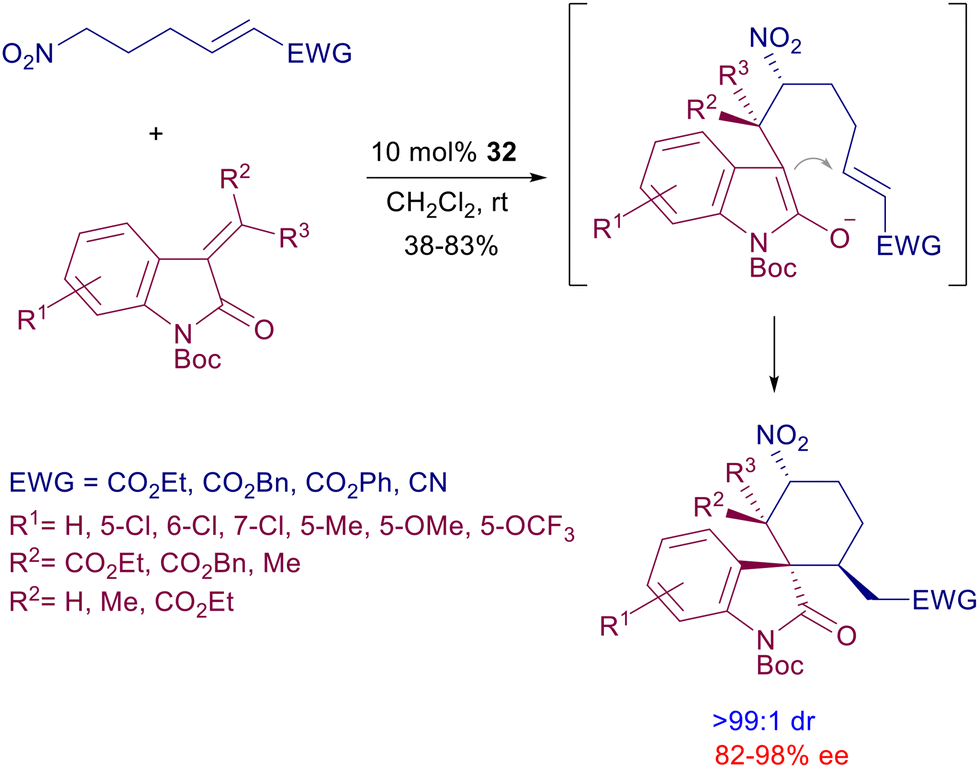
These relatively simple bifunctional catalysts have been involved in the synthesis of complex structural entities, exploiting cascade processes such as that displayed in Scheme 27.65 Nitroalkanes bearing a terminal electron-poor alkene react with 3-alkylideneoxindoles in the presence of catalyst 32 leading to a tricyclic derivative with outstanding diastereoselectivity. The process entails a preliminary nitro-Michael reaction followed by ring closure of the intermediate enolate to the electron-poor alkene. The utilization of the lower homologue (C4) of the nitroalkane in the same reaction occurs with poor diastereoselectivity using catalyst 32, but a notable improvement can be achieved by moving to cinchona-derived thiourea catalysts (vide infra).
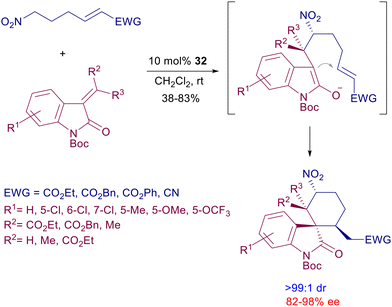 |
| | Scheme 27 Synthesis of chiral spiroxindoles by double conjugate addition. | |
The chiral 1,2-diamino framework required for the preparation of the thiourea catalysts comes from chiral trans-1,2-cyclohexanamine or from synthetic manipulation of amino acid derivatives. As already discussed in Section 2.2, optically active 1,2-diamino derivatives can also be obtained from cinchona alkaloids through a stereospecific conversion of the secondary hydroxy group into a primary amine. The thiourea moiety is then built up on the primary amine, leading to the bifunctional catalyst. In devising the structural elements to be embedded in these cinchona-thiourea catalysts, an essential motif like the 2,4-trifluoromethylphenyl group that proved effective in Takemoto's catalyst was included. Thus dihydroquinine-derived bifunctional catalyst 40 was the first thiourea derivative employed for the reaction of nitromethane to chalcones and α,β-unsaturated-N-acylpyrroles (Scheme 28).66
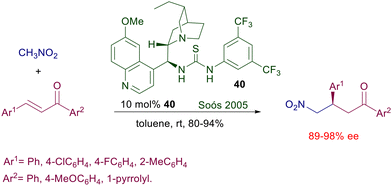 |
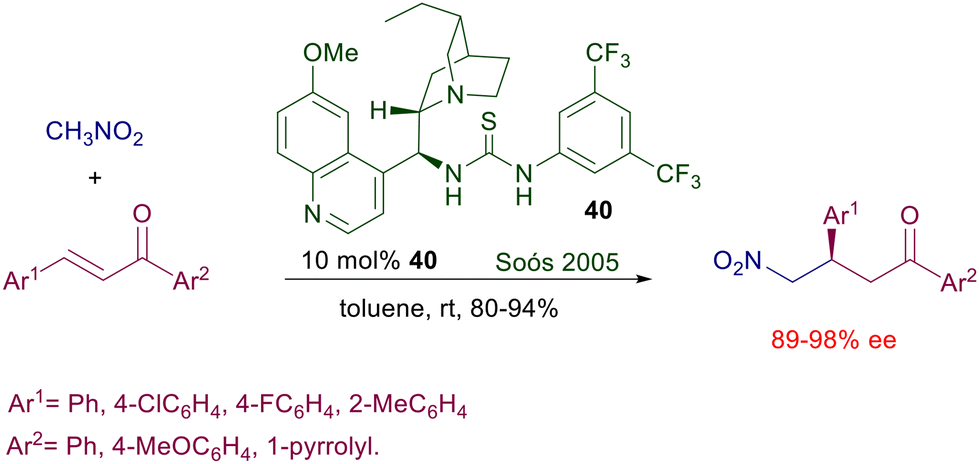
| | Scheme 28 Dihydroquinine-derived bifunctional catalyst in the reaction of nitromethane with chalcones. | |
Interestingly, the features of this catalyst are effective for many synthetic purposes, so that a limited number of structural modifications have been introduced over time. A first important change was made by substituting the bistrifluoromethylphenyl moiety with a (R,R)-cyclohexyldiamino group in catalyst 41 (Scheme 29).67 This catalyst has been employed for the conjugate addition of nitroalkanes to various cycloalkenones.
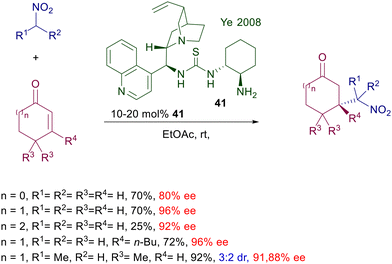 |
| | Scheme 29 Cinchonidine-cyclohexane diamine mixed thiourea catalyst in the conjugate addition of nitroalkanes with cycloalkenones. | |
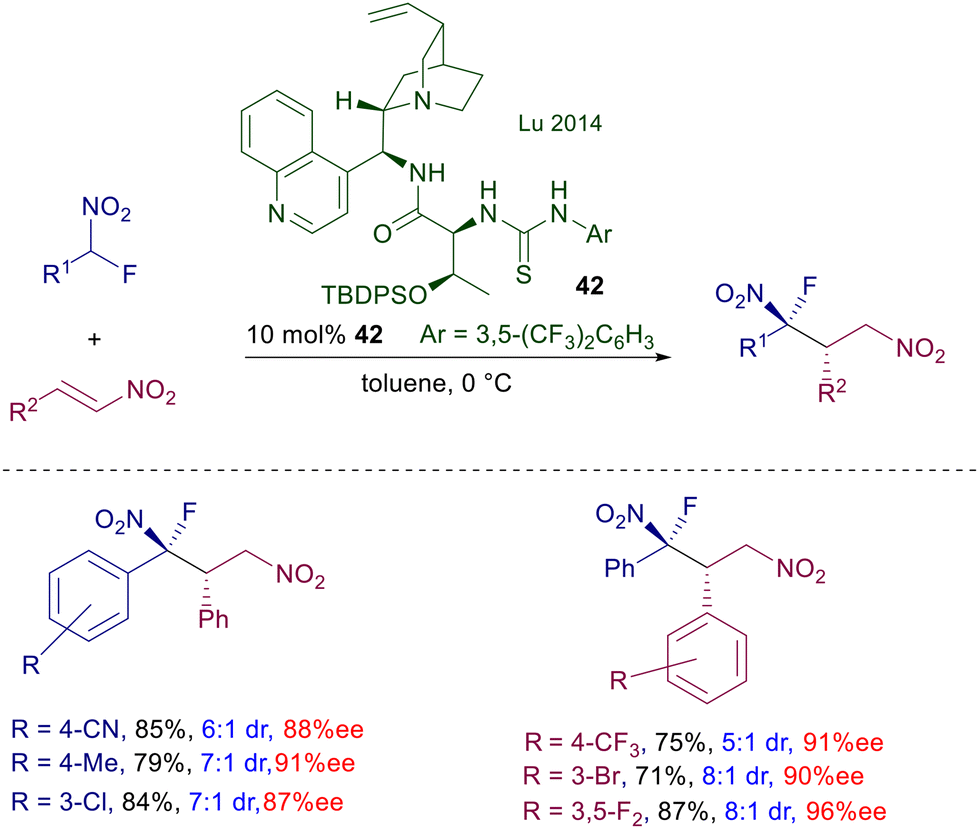
The enantiofacial preference for the attack of nitromethane to cyclohexenone is dictated by the stereochemistry of the (R,R)-cyclohexyldiamino group, considering that the catalyst bearing the S,S unit affords the other enantiomer of the product. This is congruent with the formation of an iminium ion intermediate, as observed with catalysts bearing a primary amino group. Catalyst 41 can also be used on linear enone derivatives, but the enantioselectivity is slightly lower compared with cycloalkenones. Another significant change was introduced in catalyst 42, in which the cinchona system is spaced from the thiourea by a L-threonine moiety (Scheme 30).68 This structural modification allows better performances over similar catalysts in the conjugate addition of 1-fluoronitroalkanes to nitroalkenes. It is not clear what the effect is brought by the threonine unit in which the hydroxy group is protected with a cumbersome t-butyldiphenylsilyl group, except that it can increase the steric constraint around the reactants in the transition state.
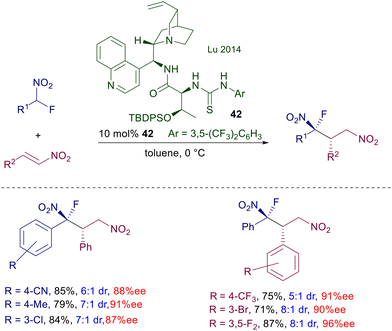 |
| | Scheme 30 Cinchonidine-threonine mixed thiourea catalyst in the conjugate addition of 1-fluoronitroalkanes with nitroalkenes. | |
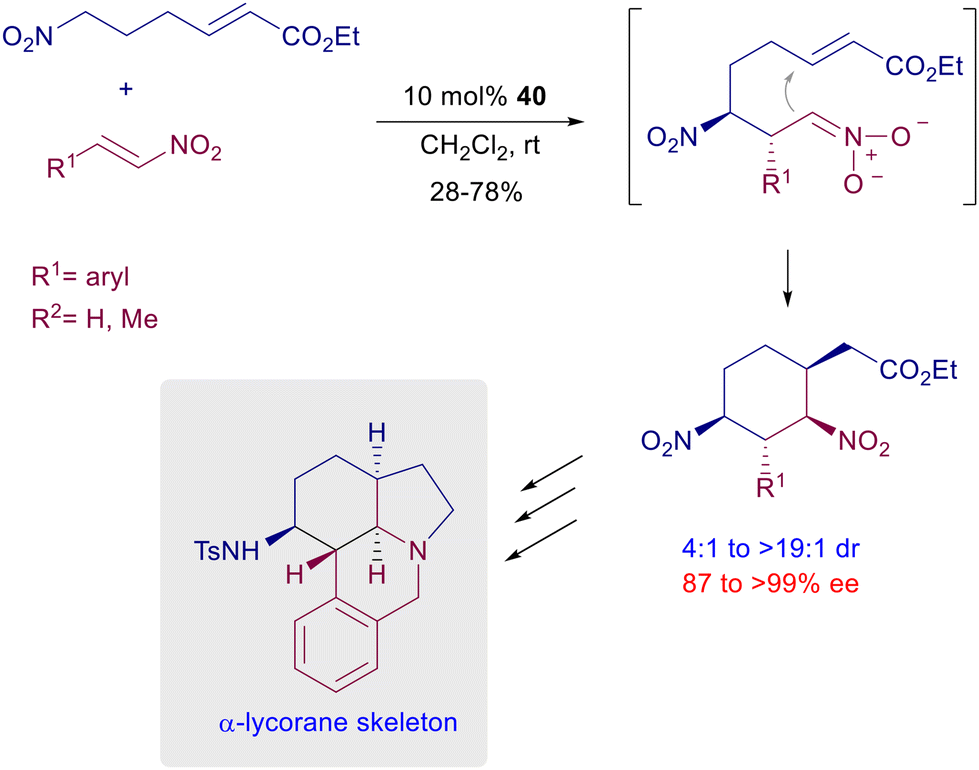
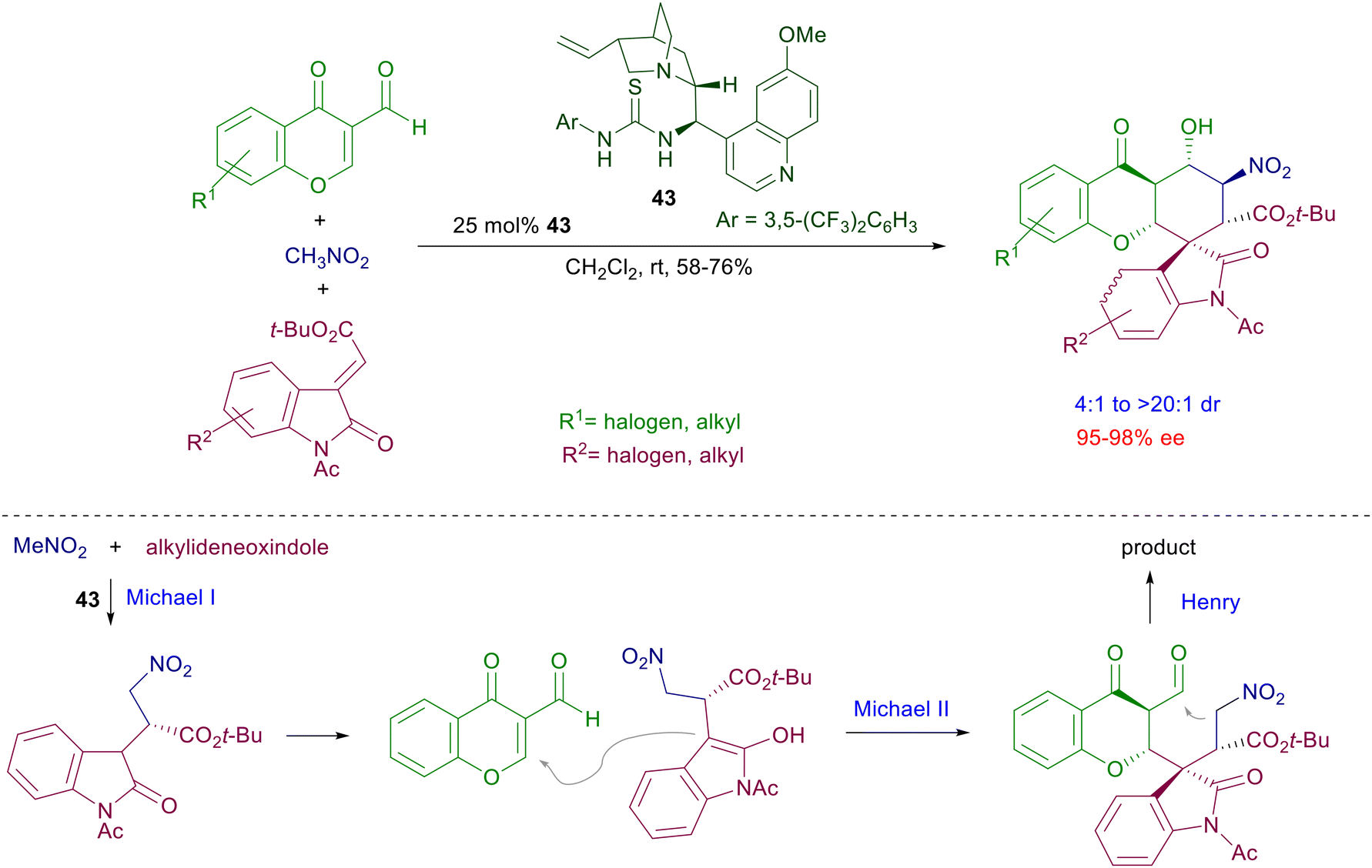
A double Michael reaction of a α,β-unsaturated-ω-nitroester to nitrostyrenes has been envisaged using catalyst 40 under mild reaction conditions (Scheme 31).69 After the first conjugate addition the resulting intermediate nitronate anion undergoes a further Michael addition with the acrylate moiety, leading to a dinitrocyclohexane derivative with satisfactory control of the four generated stereocenters. The formed dinitro derivatives can be converted in a three-step process into tetracyclic compounds that feature the α-lycorane skeleton. The level of architectural complexity can be driven until the formation of polycyclic structures featuring several stereocenters with overall good diastereo- and enantiocontrol. The three-component coupling of nitromethane with chromene and oxindole compounds in the presence of quinidine-thiourea catalyst 43 affords hexahydroxanthone derivatives through a formal [2 + 1 + 3] annulation process (Scheme 32).70 The overall process starts with a conjugate addition of nitromethane to the alkylideneoxindole, thus generating the first stereocenter. A second Michael addition of the nitroxindole enol to the chromone unit provides the installation of three further stereocenters, but the last nitroaldol Henry reaction finally leads to the target hexahydroxanthone featuring six stereocenters.
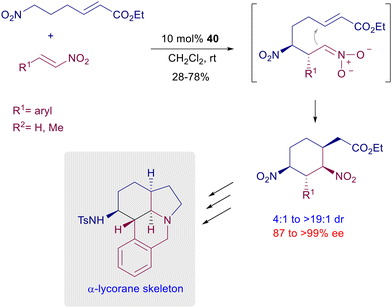 |
| | Scheme 31 Double asymmetric conjugate addition catalyzed by chiral thiourea 40. | |
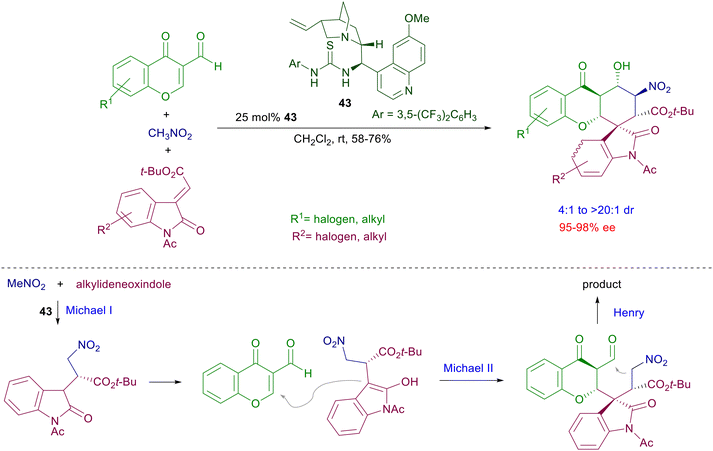 |
| | Scheme 32 Three-component cascade process catalyzed by chiral thiourea 43 leading to spiroxindole derivatives. | |
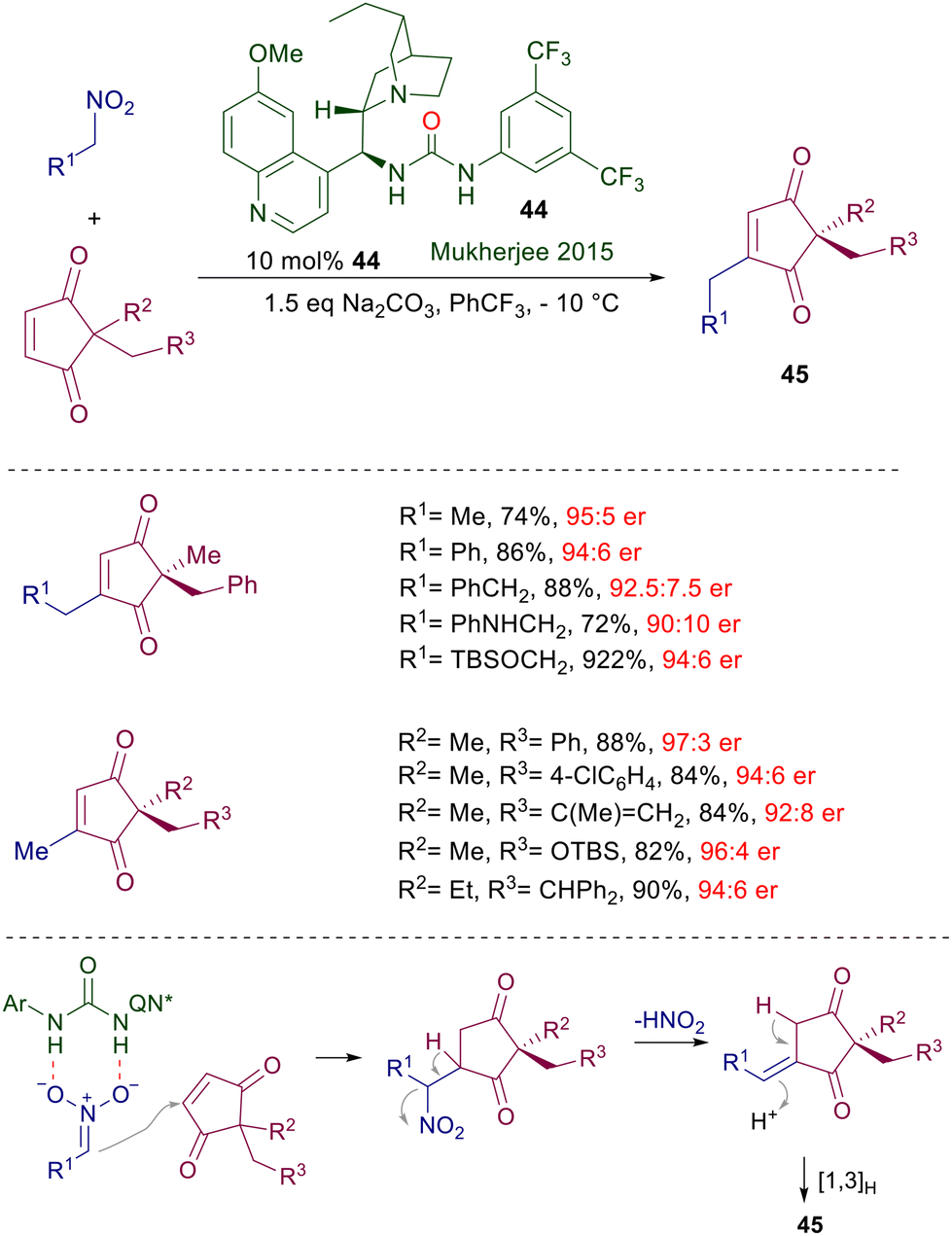
A formal alkylative desymmetrization of prochiral 2,2-disubstituted cyclopentene-1,3-diones has been realized using nitroalkanes as the alkyl source in the presence of quinine-derived urea catalyst 44 (Scheme 33).71 This is one of the rare examples where the urea group shows a superior catalytic activity compared with the thiourea analogue. The large number of examples reported includes the use of nitroalkanes and 1,3-diones embedding useful amino and oxygenated functional groups. The alkylative process is based on the well-known aptitude of β-nitrocarbonyl derivatives in undergoing loss of nitrous acid generating α,β-unsaturated derivatives.6 Thus, the selective enantiofacial attack of the nitronate anion to the enedione substrate affords the expected Michael adduct, which upon loss of nitrous acid initially gives the exocyclic unsaturated cyclopentane-1,3-dione. Formation of the final product 45 is then ensured by a thermodynamically driven hydrogen shift.
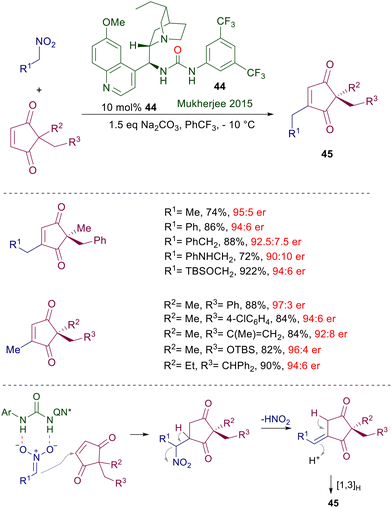 |
| | Scheme 33 Alkylative desymmetrization of cyclopentendione derivatives catalyzed by quinine-derived chiral urea 44. | |
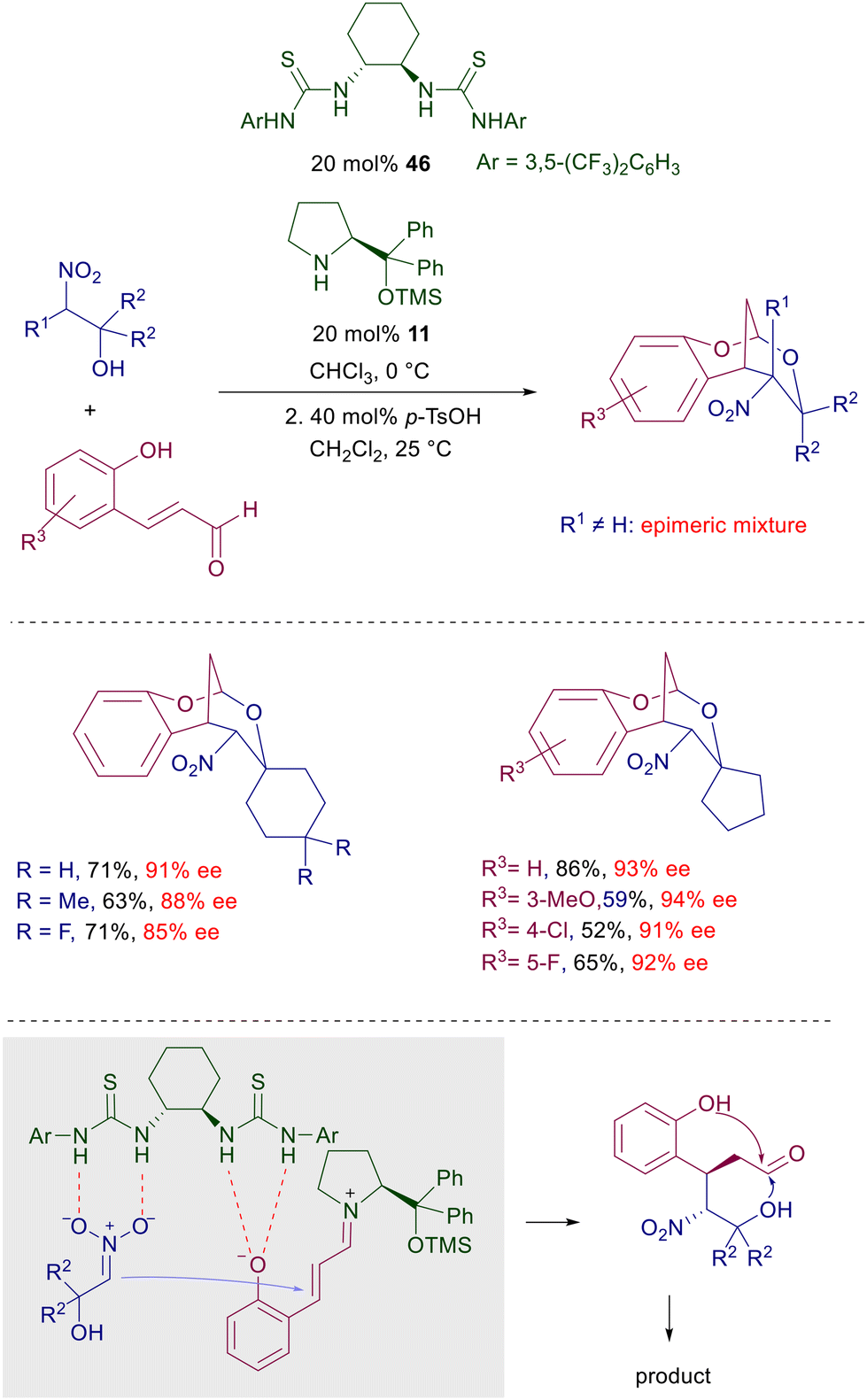
A recent example of cooperative heterocatalysis has been reported in the Michael addition of nitro alcohols with 2-hydroxycinnamaldehydes jointly catalyzed by chiral bisthiourea 46 and prolinol 11 (Scheme 34).72 The resulting products are tricyclic nitroacetal derivatives obtained with good enantioselectivity but variable diastereoselectivity depending on the nature of the substituents on the nitro alcohol. The presence of α-substituents (R1 ≠ H) or small substituents on carbinol [R2 = H or –(CH2)3–] usually affords epimeric mixtures of products. The cooperative action of the catalysts involves the hydrogen bonding interaction of the thiourea NH with the nitronate and the phenolic oxygen atom while the prolinol acts through the usual iminium ion catalysis. The initially formed dihydroxynitroaldehydes can be acetalyzed under acidic conditions to afford the target products.
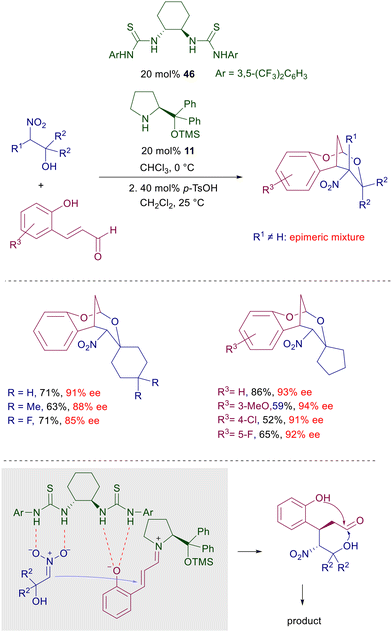 |
| | Scheme 34 Cooperative heterocatalysis in the conjugate addition of nitro alcohols to 2-hydroxycinnamaldehydes. | |
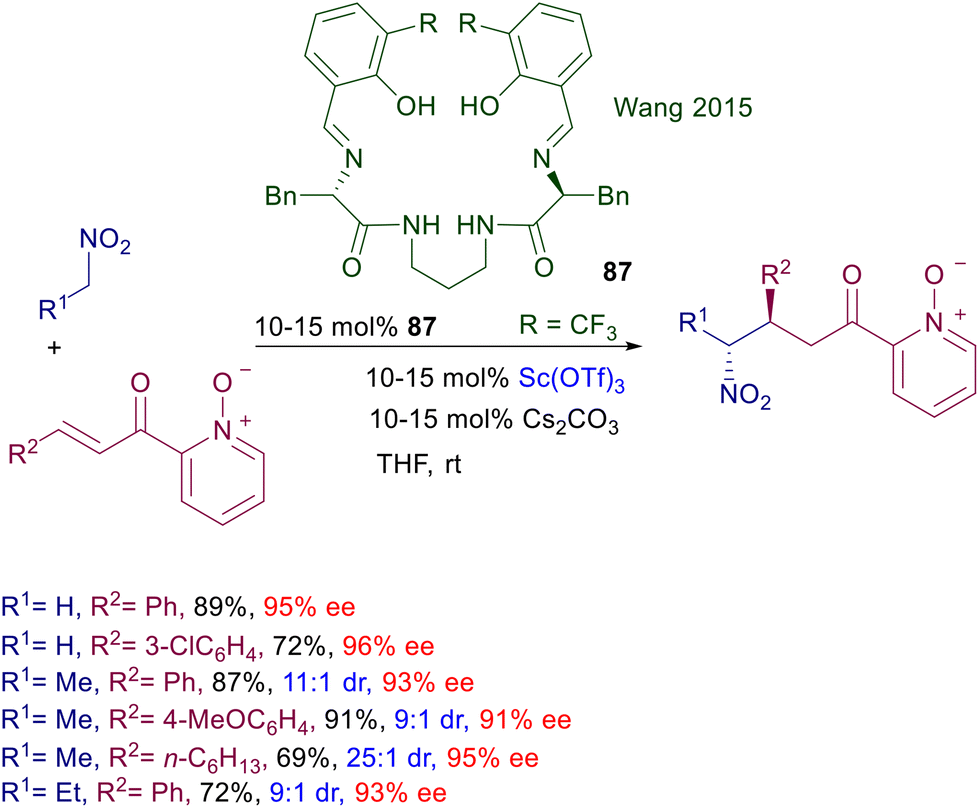
2.4 Squaramide catalysts
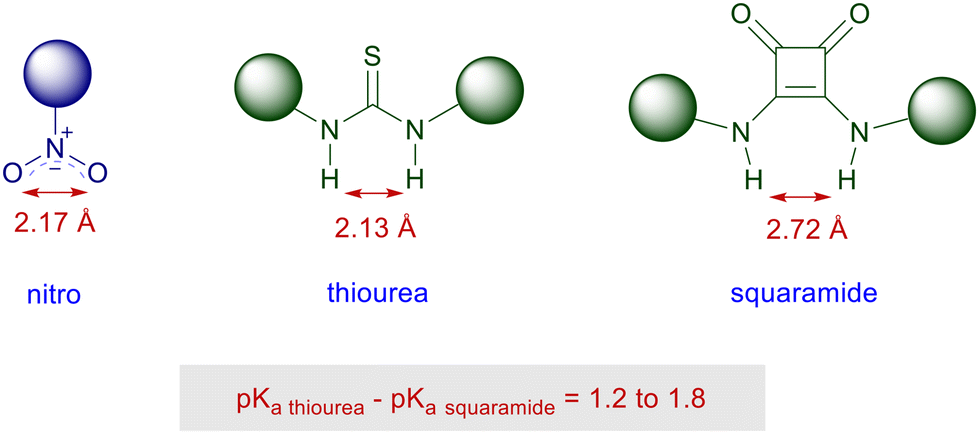
Squaramides are derivatives of squaric acid largely employed in a notable number of applications in material chemistry and drug design as bioisosteres.73 The structural features of squaramides closely resemble those of thioureas, although the H–H interatomic distance is superior in the squaramide system by ∼0.6 Å (Fig. 4).74 A further analogy can be found in the acidity level of the NH bonds which, however, is fairly superior in squaramides than in thioureas.75 This allows squaramide to establish stronger hydrogen bonding with functionalities commonly involved in thiourea-catalyzed reactions such as nitro, carbonyl, carboxylates and imino groups.76
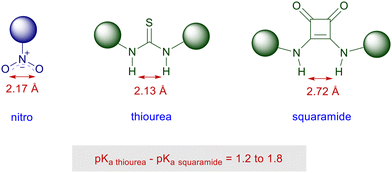 |
| | Fig. 4 Structural features of nitro, thiourea and squaramide derivatives. | |
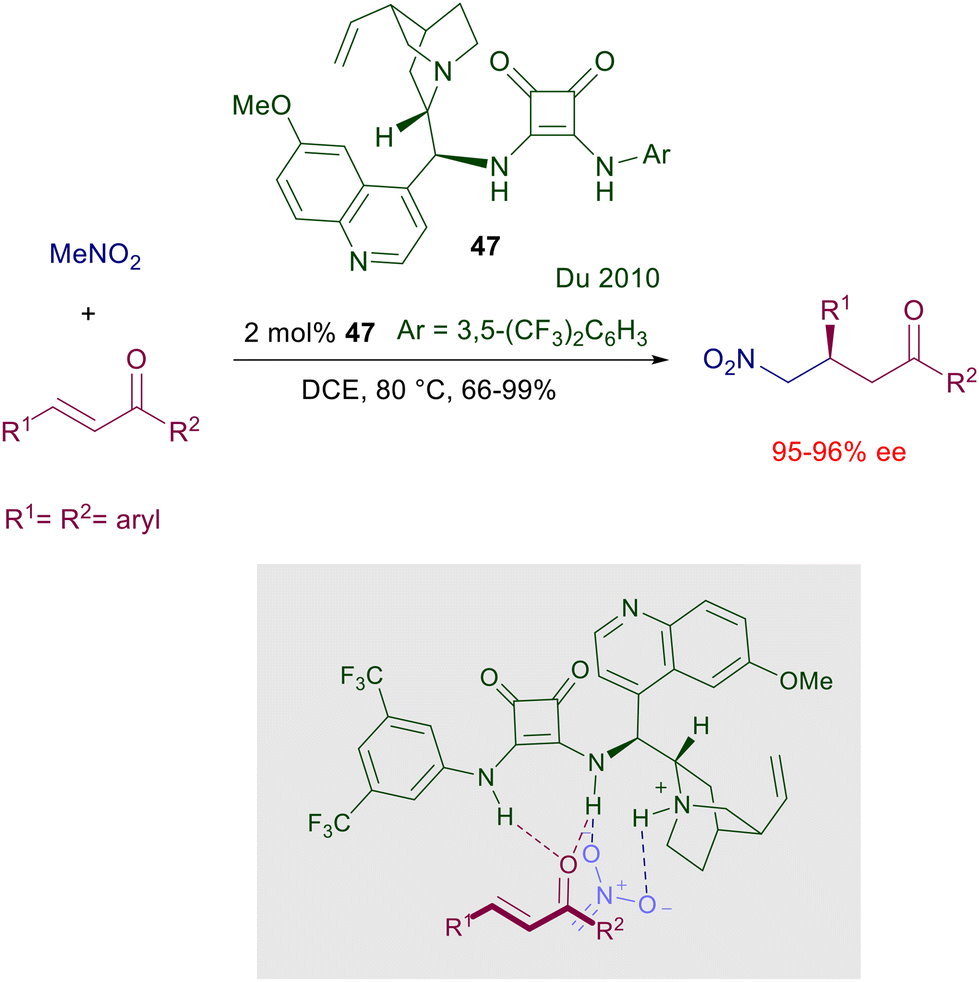
In 2008, the results previously obtained using chiral thioureas inspired the development of a chiral bifunctional squaramide catalyst embedding a cinchonine-derived amine and a substituted benzylamine which was effective in the asymmetric Michael reaction of 1,3-diketones with nitroalkenes.74a A couple of years later, the first enantioselective addition of nitromethane to enones was reported using the related quinine-derived catalyst 47 (Scheme 35).77 The reaction is effective even with a reduced catalyst charge, although a relatively high temperature must be applied. Modest results have been obtained using 2-cyclohexen-1-one as substrate and, as expected, the utilization of a chiral squaramide embedding the pseudoenantiomeric quinidine-derived amine afforded the corresponding (S)-adducts. Interestingly, in the plausible transition state depicted for this process the interaction of the enone carbonyl oxygen with the squaramide is privileged over that of the nitromethane, which appears mostly activated by the ammonium salt.
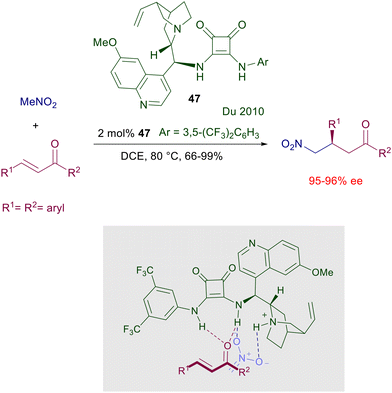 |
| | Scheme 35 The first enantioselective conjugate addition of nitromethane to chalcones catalyzed by a quinine-derived squaramide catalyst 47. | |
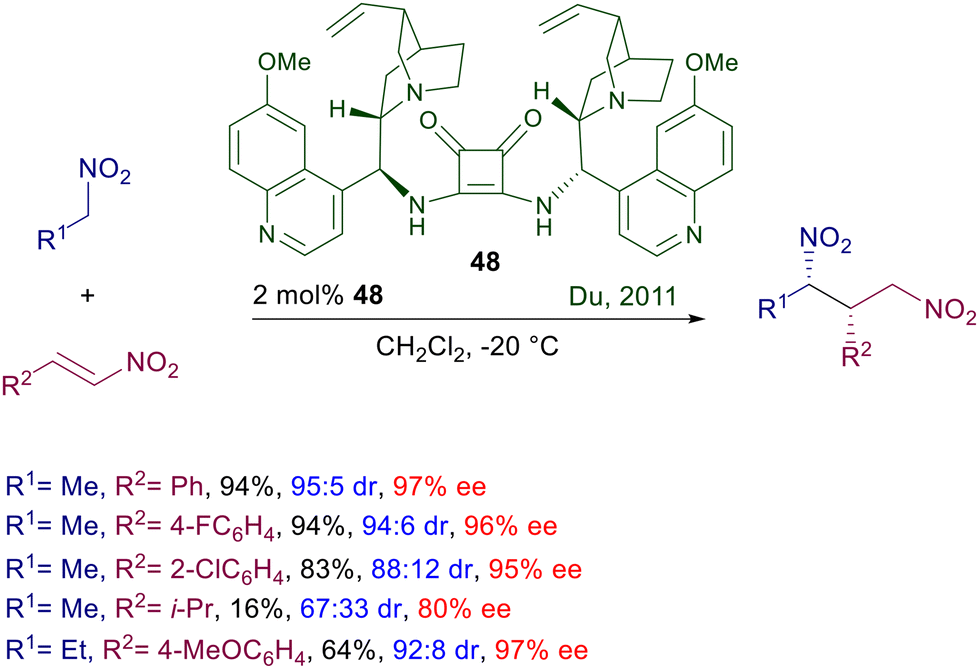
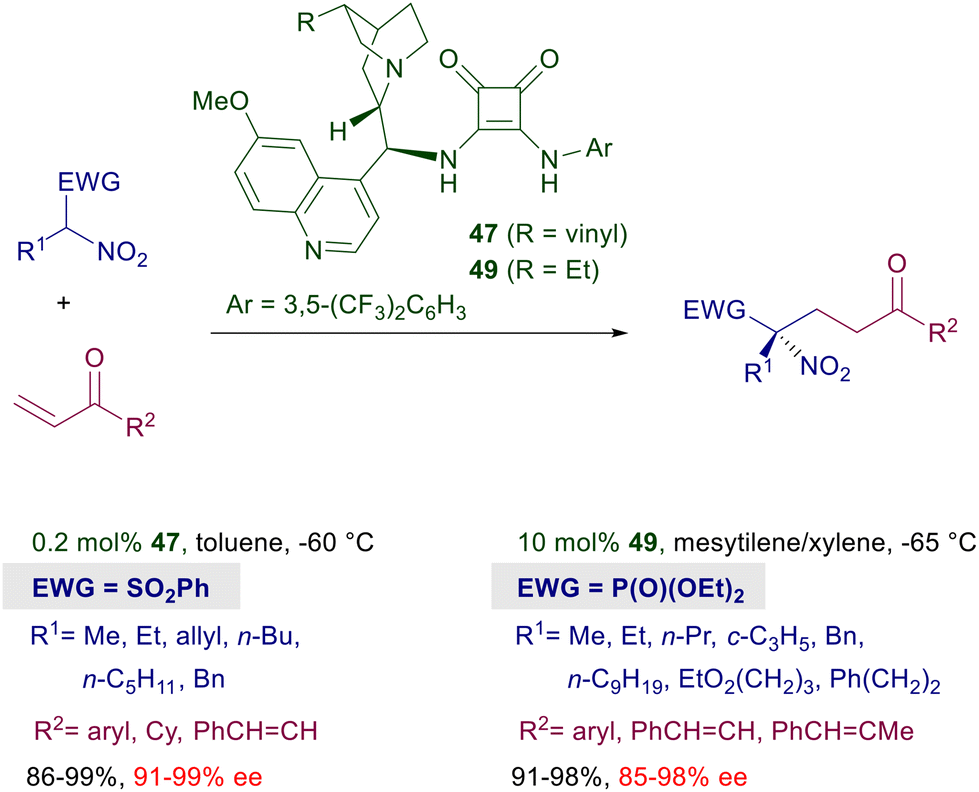
Asymmetric procedures catalyzed by bifunctional squaramides bearing cinchona-derived amines are largely predominant over related methods using different chiral amines. Furthermore, in optimization studies where thiourea and squaramide catalysts are comparatively evaluated for the conjugate addition of nitroalkanes to electron-poor alkenes, the latter catalysts very often show better performances. The catalyst activity can be further increased by moving to biscinchona-squaramide catalyst 48 featuring a C2-symmetry which is effective in the conjugate addition of nitroalkanes to nitroalkenes (Scheme 36).78 The diastereoselectivity observed in this reaction is satisfactory with para-substituted nitrostyrenes, but decreases when the substituent occupies the ortho position. The enantioselectivity is always excellent with all nitrostyrenes employed except with alkyl-substituted nitroalkenes, which provide disappointing results. Nitroalkanes bearing an additional electron-withdrawing group at the α-position can be efficiently added to enones using cinchona-derived squaramides (Scheme 37). A direct comparison of these squaramide catalysts with structurally related thiourea derivatives shows a superior catalytic activity of the former compounds often resulting in a reduced catalyst charge and low reaction temperature. Nitrosulfonyl derivatives react with terminal enones in the presence of just 0.2 mol% of catalyst 47 leading to the corresponding adducts in good yield and excellent enantioselectivity.79 Similarly, nitrophosphonates can be added to terminal enones using catalyst 49 under similar reaction conditions, giving ketophosphonates which upon selective reduction of the nitro group easily afford α,α-disubstituted aminophosphonic acid derivatives.80
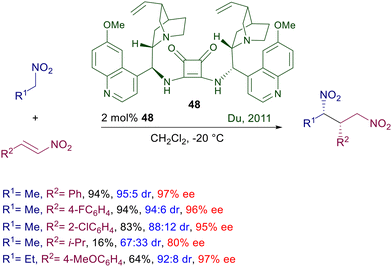 |
| | Scheme 36 Asymmetric conjugate addition of nitroalkanes to nitroakenes catalyzed by biscinchona-squaramide catalyst 48. | |
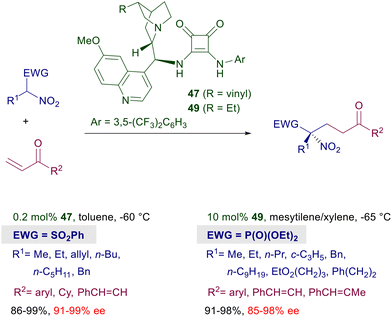 |
| | Scheme 37 Enantioselective conjugate addition of activated nitroalkanes to enones. | |
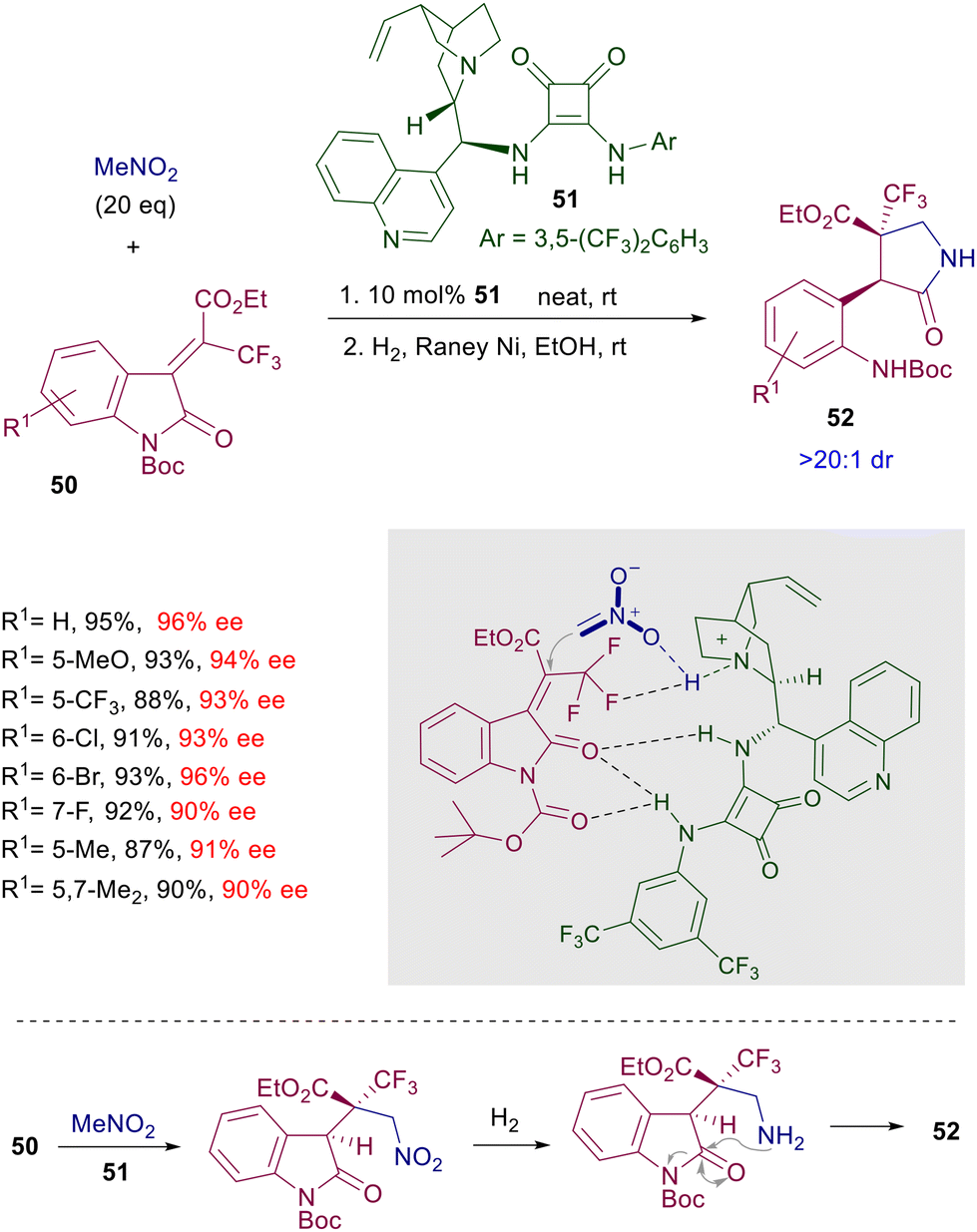
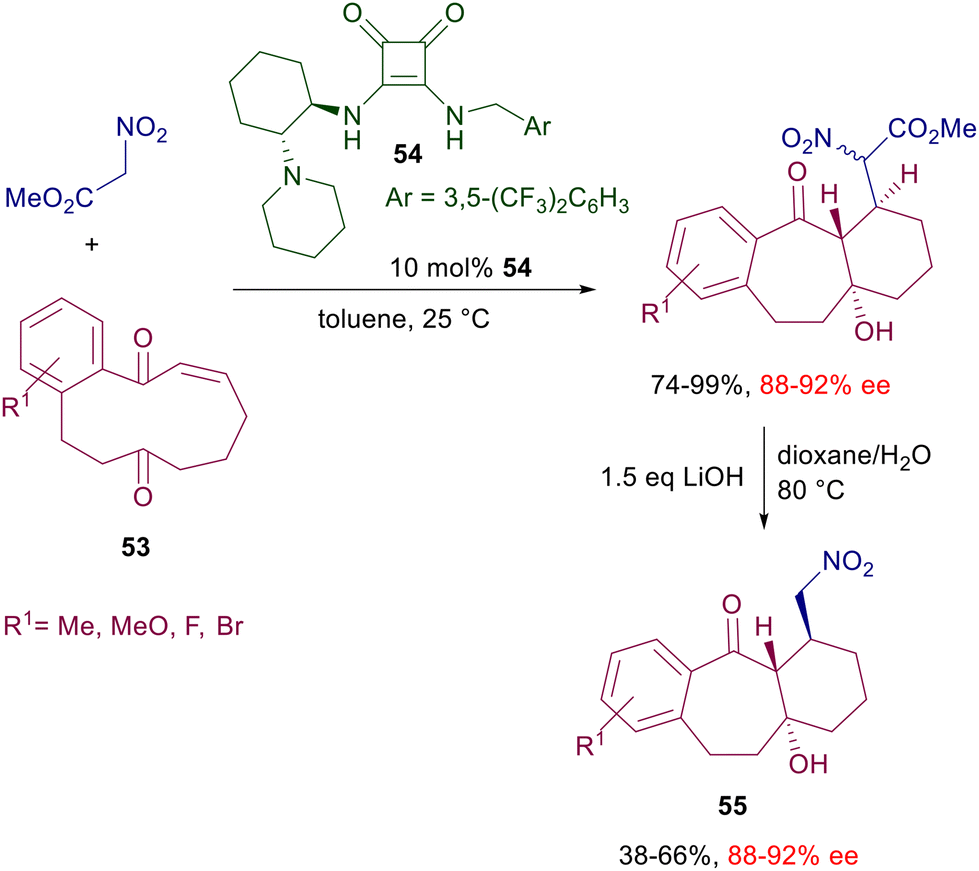
An effective asymmetric synthesis of pyrrolidinones 52 can be realized in a two-step procedure reacting alkylideneoxindoles 50 with nitromethane in the presence of cinchonidine-derived squaramide 51 (Scheme 38).81 The nitro group of the initially formed adduct is reduced by catalytic hydrogenation, and the resulting primary amine is then involved in a thermodynamically controlled transamidation leading to the target pyrrolidinone 52 as a single diastereomer and very good ee value. The trifluoromethyl group in the electrophile seems to play an essential role in enhancing the steric constraint of the transition state through an additional hydrogen bonding with the ammonium ion. As a matter of fact, the oxindole 50 lacking the ethyl ester group still gives a product with high enantioselectivity (95% ee), while the substrates in which the trifluoromethyl is substituted by a methyl or a hydrogen atom evidence a notable drop in the ee value (74% and 51% ee). The reduced reactivity of nitromethane with particular enones can be overcome using nitroacetate derivatives endowed with a higher enolizability. The addition of methyl nitroacetate to cyclic diketoenones 53 occurs smoothly at 25 °C in the presence of diamine-derived squaramide 54, which was found superior to thioureas as well as cinchona-derived squaramides (Scheme 39).82
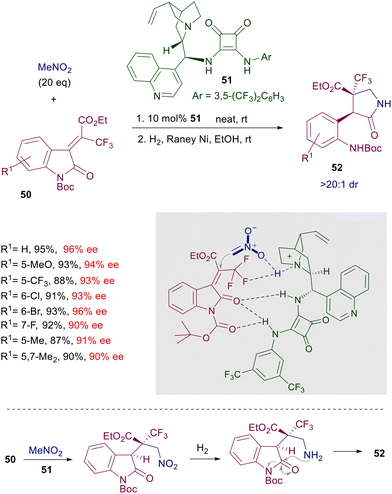 |
| | Scheme 38 Asymmetric synthesis of pyrrolidinones 52. | |
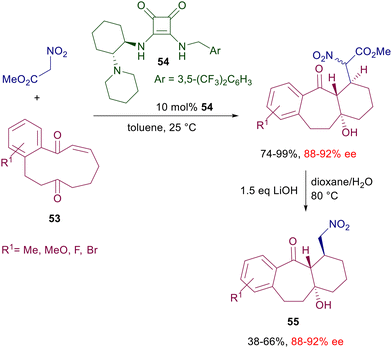 |
| | Scheme 39 Asymmetric tandem conjugate addition/aldol reaction catalyzed by squaramide 54. | |
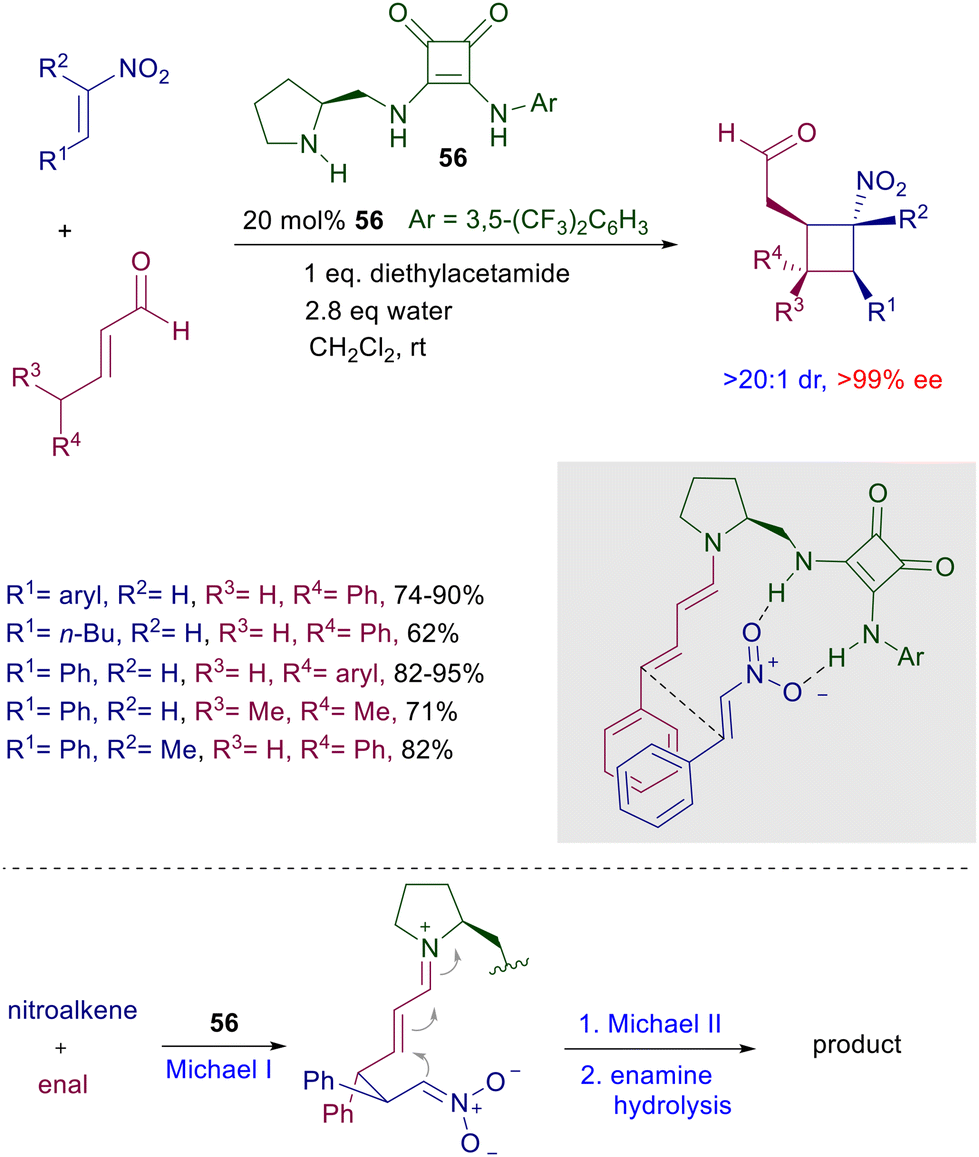
The chiral catalysts ensure a very good enantiocontrol in the conjugate addition, while a substrate control is believed to be operative in the subsequent intramolecular aldol condensation leading to the target tricyclic compounds. As always occurs, no stereocontrol is possible at the rather acidic nitroester stereocenter, so that these products are obtained as a 2:1 epimeric mixture.83 In any event, basic decarboxylative hydrolysis of these compounds finally affords nitromethyl derivatives 55 as a result of a formal addition of nitromethane to diketoenones 53. Reaction of nitroalkenes with enals in the presence of bifunctional pyrrolidine-squaramide catalyst 56 affords nitrocyclobutane derivatives through a formal [2 + 2] cycloaddition (Scheme 40).84 The process occurs with perfect control of the four stereocenters generated in the cyclobutanation reaction involving a double conjugate addition with dienamine-iminium ion intermediates. The reactants’ arrangement leading to the first Michael addition is favored by the hydrogen bonding of the nitro group with the squaramide moiety and by the π-stacking interaction between the aryl groups. The second Michael addition occurs between the nitronate anion and the vinylogous iminium ion leading, after enamine hydrolysis, to the final product.
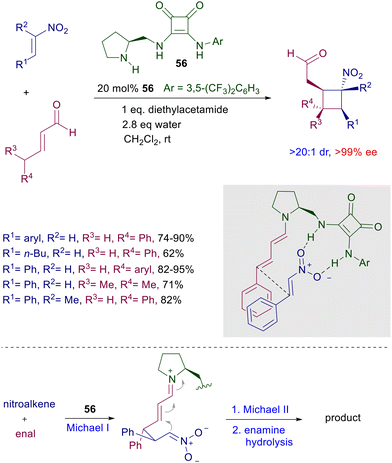 |
| | Scheme 40 Formal [2 + 2] cycloaddition of nitroalkenes to enals catalyzed by bifunctional squaramide 56. | |
2.5 Amidines, guanidines and related catalysts
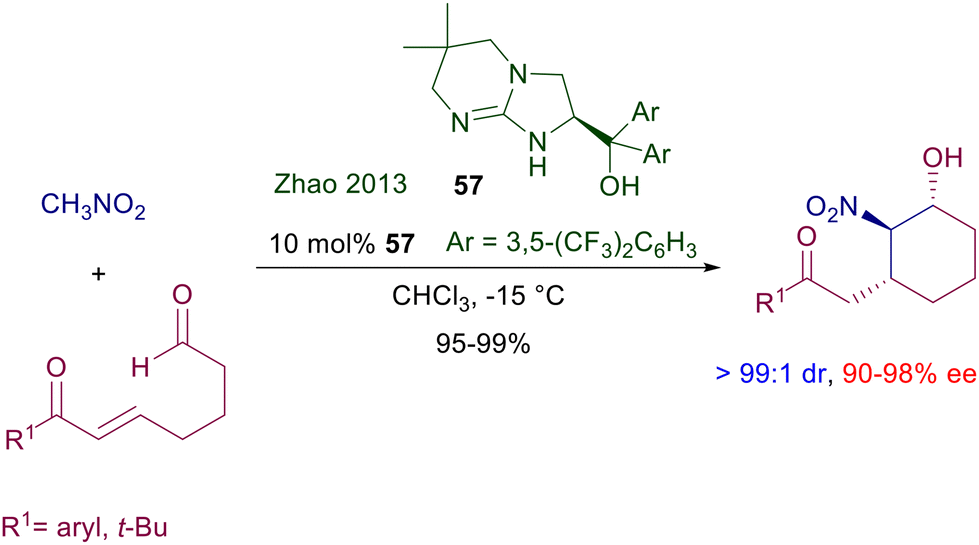
The activation mode of the nitro group by hydrogen bonding with protonated amidines and guanidines closely resembles that with thioureas and squaramides, and has been confirmed by isolation and characterization of the complex between phenylnitromethane and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD).85 The first chiral guanidine catalyst that featured a C2-symmetrical bicyclic structure was devised several years ago and gave low ee values for the reaction of nitroalkanes with methyl vinyl ketone.86 More recently, chiral bicyclic guanidine 57 was revealed to be very effective in a tandem nitroaldol-Michael addition of nitromethane to 7-oxohept-5-enals (Scheme 41).87 Control experiments demonstrated that the nitroaldol process occurs first and is followed by the conjugate addition leading to the ring closure.
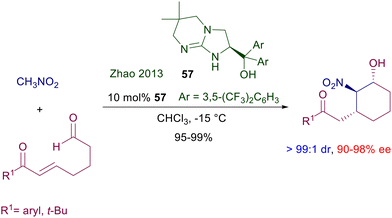 |
| | Scheme 41 Chiral bicyclic guanidine 57 as catalyst for the tandem conjugate addition/aldol condensation of nitromethane to oxoenals. | |
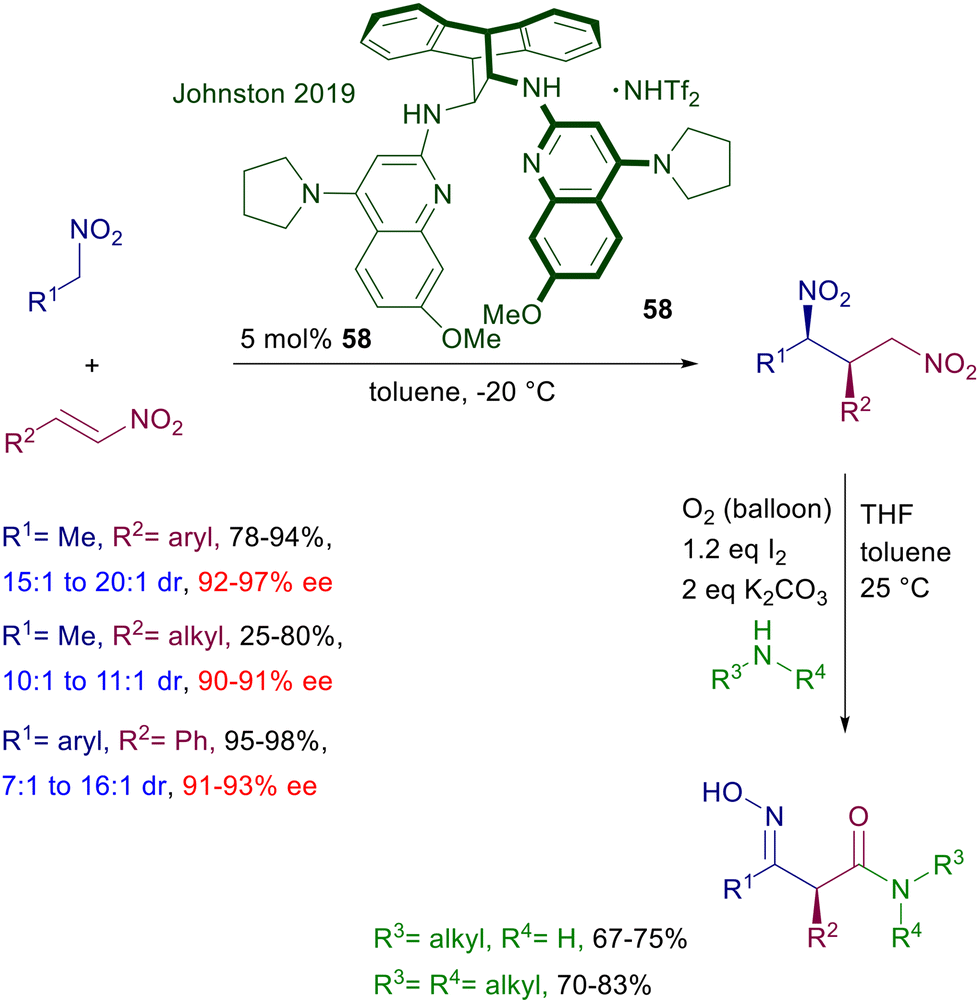
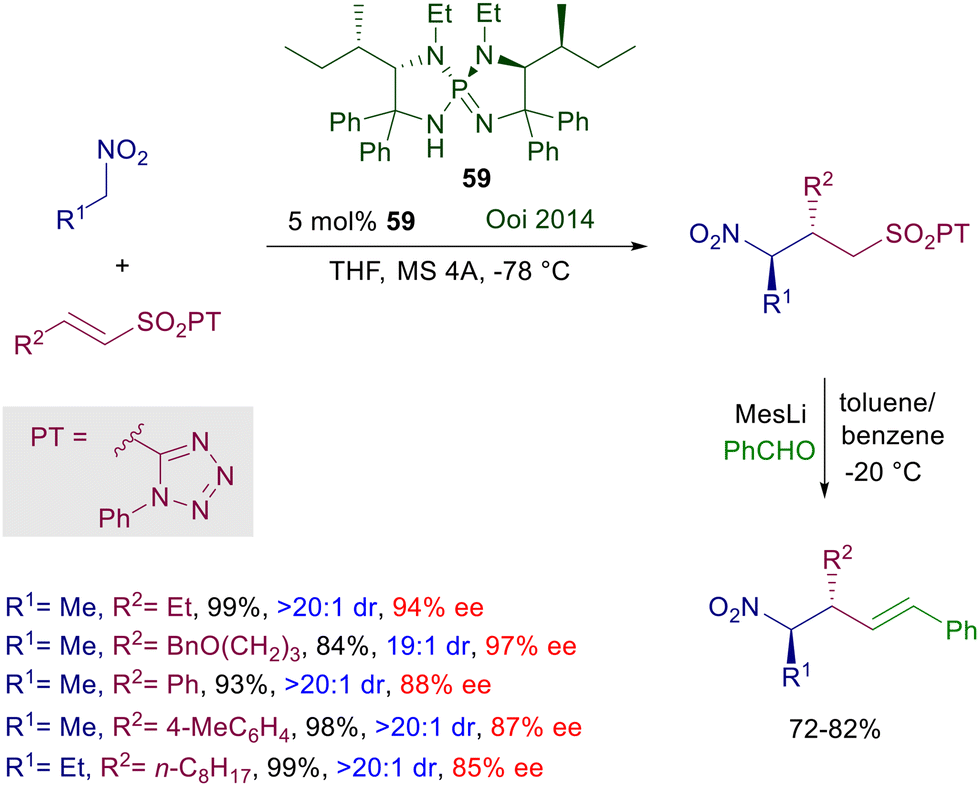
The anthracene-derived chiral bisamidine 58 has been recently introduced as catalyst for the conjugate addition of nitroalkanes to nitroalkenes (Scheme 42).88 The reaction is particularly effective with nitrostyrenes and affords with satisfactory syn diastereoselectivity the corresponding 1,3-dinitro derivatives. Catalyst 58 is used as triflimidic acid salt since the free base does not bring about significant levels of stereoselectivity in the formation of 1,3-dinitro derivatives, and this aspect stresses the bifunctional nature of the catalytic action provided. An original application of the obtained adducts involves a double oxidative Nef reaction,5 using the molecular oxygen–iodine couple in the presence of primary and secondary amines to generate 3-hydroxyiminoalkanamides with full retention of the original C2 configuration. Triaminoiminophosphoranes show an enhanced basic character and similarly to guanidines and amidines are able to deprotonate nitroalkanes, establishing strong hydrogen bonding interactions with the corresponding nitronate anions. Chiral triaminoiminophosphorane 59 catalyzes the conjugate addition of nitroalkanes to vinylsulfones leading to the corresponding nitrosulfonyl derivatives with excellent diastereocontrol (Scheme 43).89 The observed enantioselectivity for the obtained adducts is better for 2-alkyl substituted vinylsulfones compared with their aryl analogues. A structural implementation of the target 3-sulfonylnitro derivatives has been proposed through a Julia–Kocienski olefination generating 3-nitroalkenyl compounds arising from an overall formal allylation of the initially employed nitroalkanes. To this scope, the nitrosulfonyl derivatives were treated with the bulky mesityllithium (MesLi) in order to ensure a regioselective kinetic deprotonation of the less acidic α-sulfonyl hydrogen atoms. Reaction with benzaldehyde resulted in the formation of 3-alkenylnitro compounds with substantial retention of the original configuration of stereocenters.
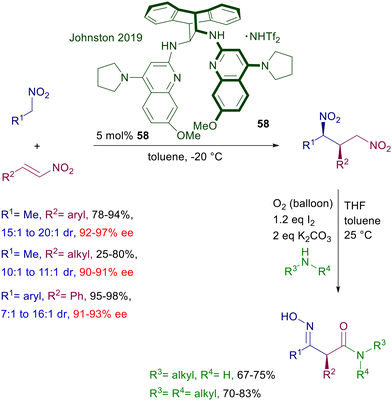 |
| | Scheme 42 Asymmetric synthesis of 3-hydroxyiminoalkanamides. | |
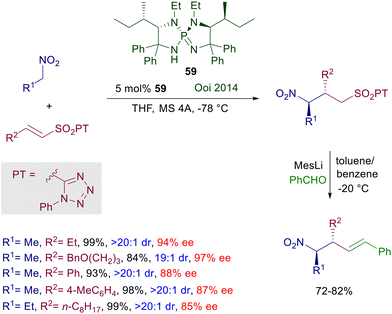 |
| | Scheme 43 Conjugate addition of nitroalkanes to vinyl sulfones catalyzed by chiral triaminoiminophosphorane 59. | |
2.6 Cinchona alkaloids
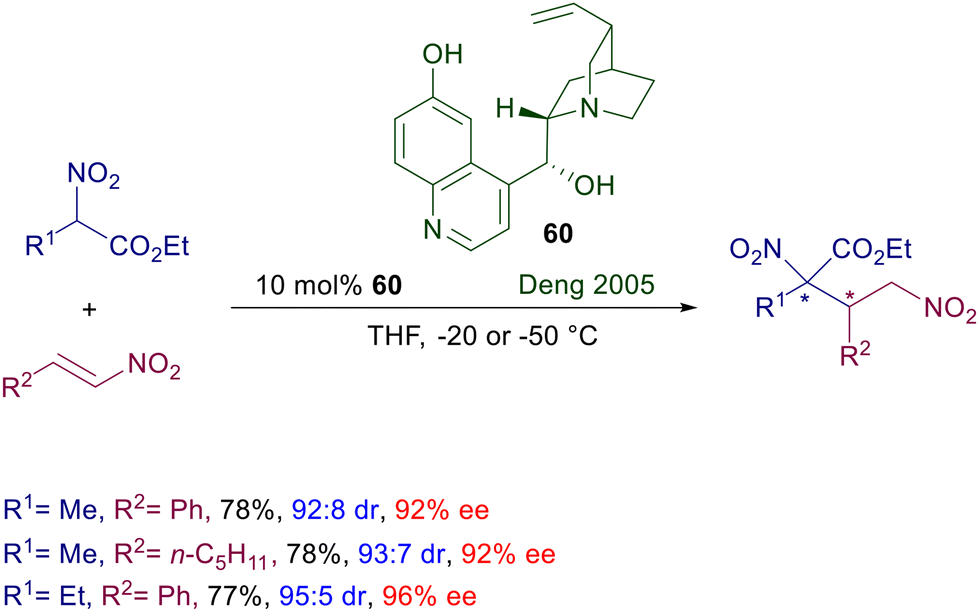
Quinine was the first chiral catalyst employed in the seventies of the last century for the conjugate addition of nitroalkanes to enones,10 but other cinchona alkaloids were soon after used with relatively modest results.90 It is therefore evident that the structural features of cinchona alkaloids hardly allow appropriate interactions with the nitroalkane–enone couple to provide a satisfactory enantiofacial discrimination.91 The first successful application of cinchona alkaloids was found in the utilization of cupreine 60 as catalyst for the conjugate addition of 2-nitroesters to nitroalkenes (Scheme 44).92 Although the absolute configuration of the stereocenters was not evaluated, the obtained adducts were formed with high diastereo- and enantioselectivity. The free hydroxy group in the quinoline ring and the ester group in the nitro derivative seem to play an essential role in establishing an appropriate hydrogen bonding network leading to the target products. As a matter of fact, reaction of unfunctionalized nitroalkanes with nitroalkenes catalyzed by cupreidine, the pseudoenantiomer of 60, leads only to modest results in terms of enantioselectivity (67–86% ee).93
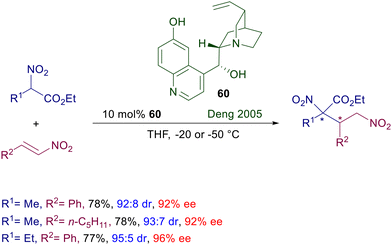 |
| | Scheme 44 Asymmetric conjugate addition of nitroesters to nitroalkenes catalyzed by cupreine 60. | |
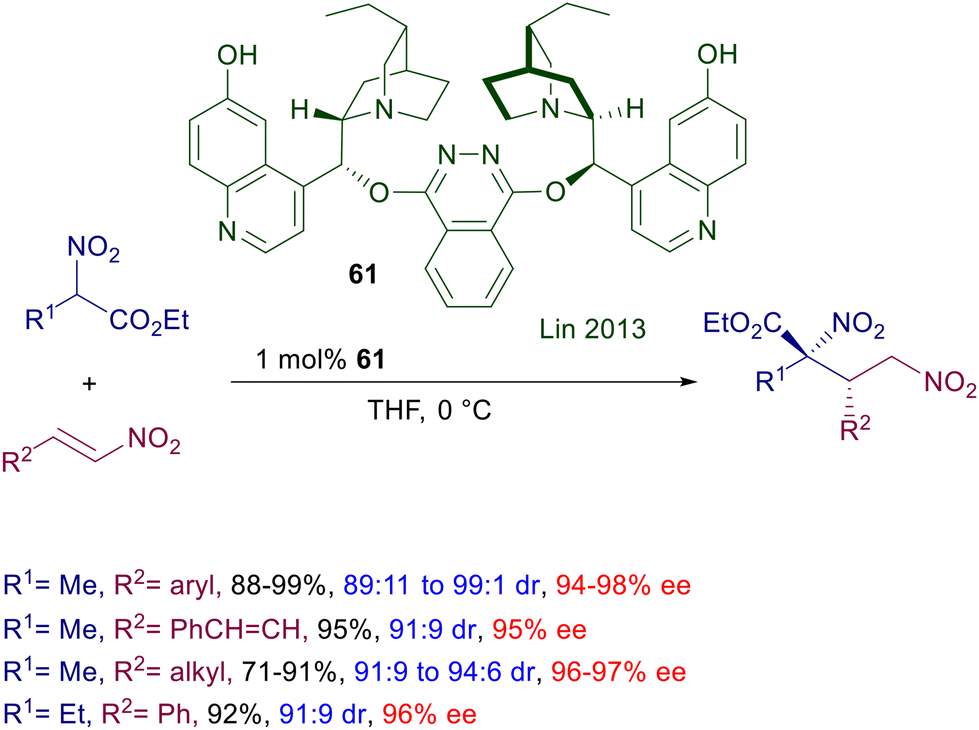
Later on, the same process was carried out using catalyst 61 based on two molecules of demethyldihydroquinine tethered by a phthalazine unit (Scheme 45).94 The catalyst is active even at 1 mol% concentration and gives satisfactory results with nitrostyrenes as well as linear nitroalkenes.
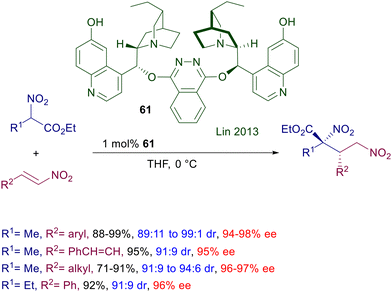 |
| | Scheme 45 Asymmetric conjugate addition of nitroesters to nitroalkenes catalyzed by biscinchona-phthalazine 61. | |
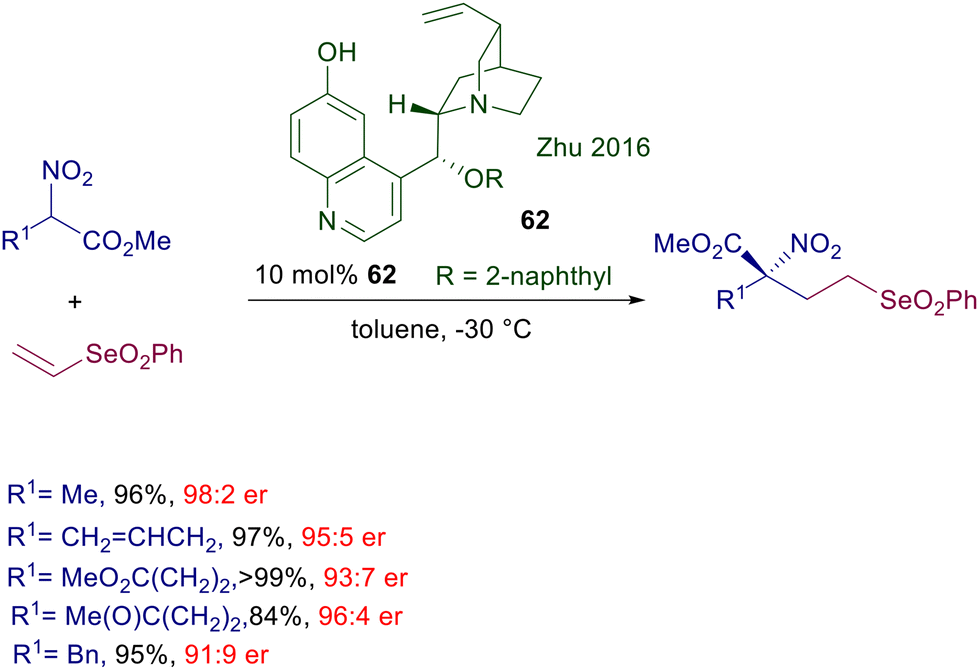
Conversion of the aliphatic hydroxy group of cupreine into the 2-naphthyl ether derivative 62 also provides a notable improvement in the enantioselectivity of the conjugate addition of functionalized nitroesters to vinylselenones (Scheme 46).95 Considering that the phenylselenonyl moiety is an excellent leaving group in eliminations and nucleophilic substitution reactions, the target selenonyl nitro esters are potentially useful building blocks for the preparation of α,α-difunctionalized amino acid derivatives.
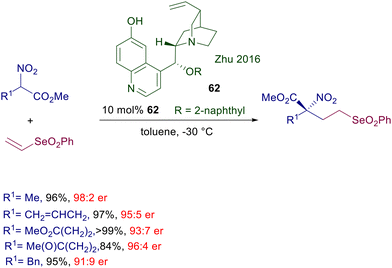 |
| | Scheme 46 Enantioselective conjugate addition of nitroesters to vinyl selenones. | |
2.7 Phase-transfer catalysts
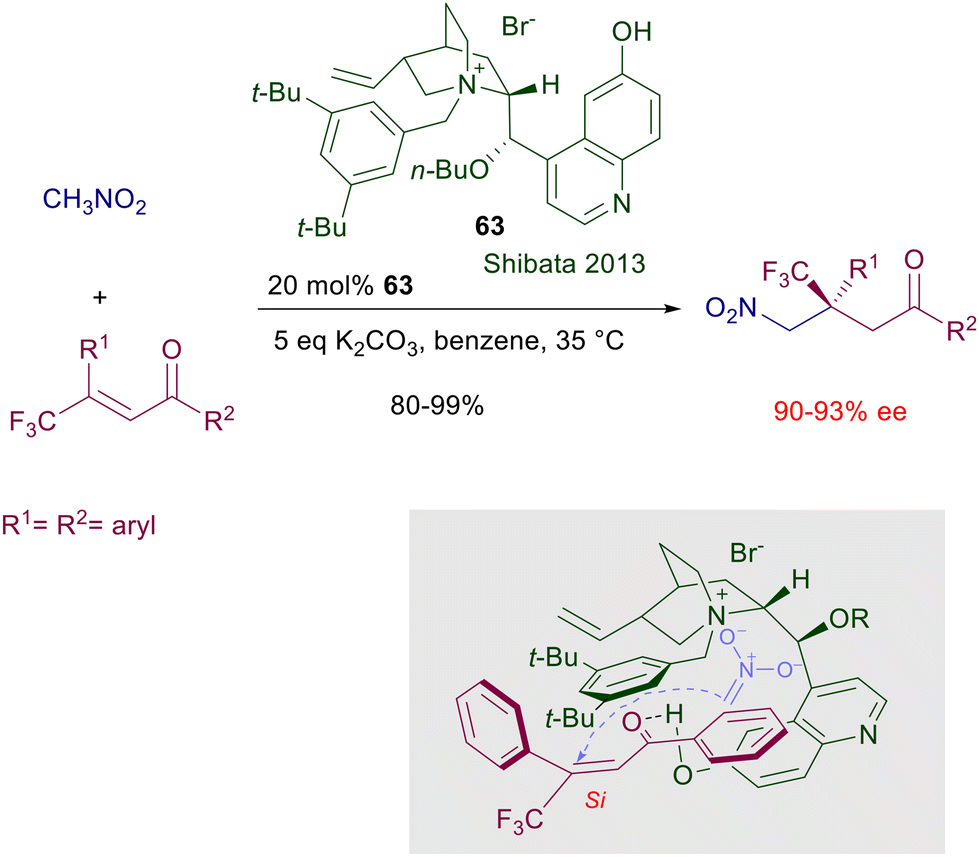
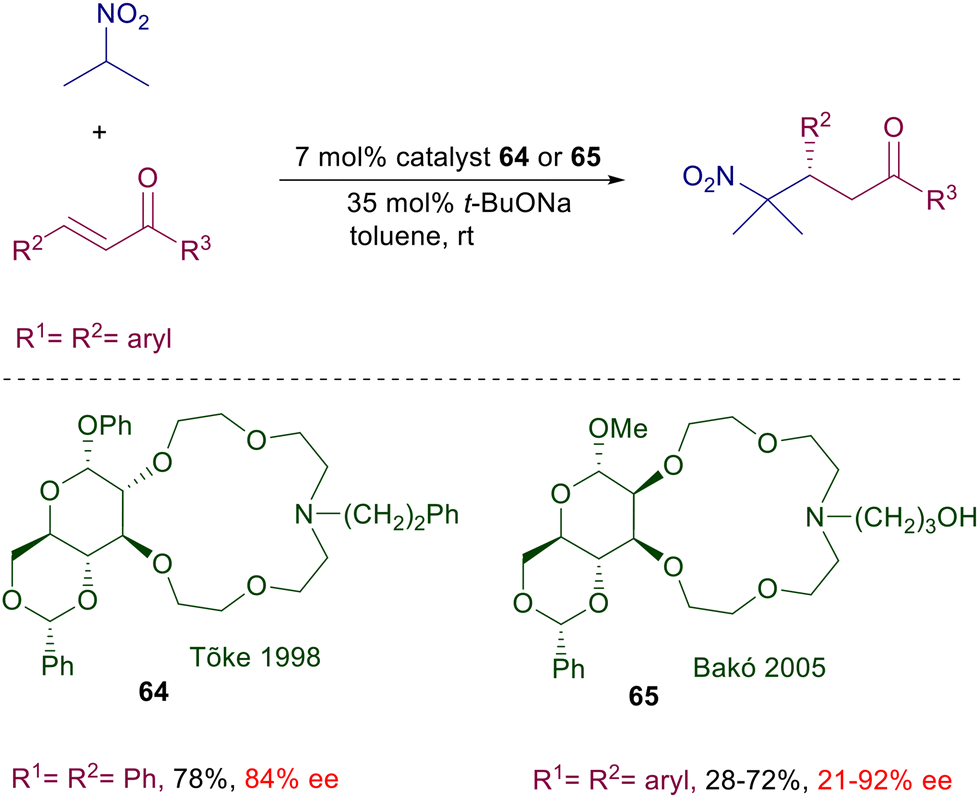
Ammonium and phosphonium salts bearing large substituents were originally designed to act as anion carriers in biphasic systems, enabling a dramatic rate enhancement of the corresponding ionic processes. In asymmetric synthesis, the role played by ammonium salts in phase-transfer catalysis (PTC) is to provide a chiral environment around tightly bonded anionic reactants involved in nucleophilic addition or substitution reactions.96 The main ionic bond provided by the ammonium moiety can be obviously assisted by further hydrogen bonding with additional polar groups present in the catalyst and in the reactants.97 The development of ammonium salts as phase-transfer catalysts for conjugate additions took place in parallel with the use of cinchona alkaloids for the same purpose. Early attempts to use quinine N-benzylic salts for the conjugate addition of nitromethane and chalcones in a biphasic system gave only very modest results.98 Only quite recently cupreidine derivative 63 has been successfully employed in the conjugate addition of nitromethane to 3-trifluoromethyl chalcones (Scheme 47).99 In the proposed transition state the ammonium ion is supposed to interact with the nitronate anion in which enantiofacial attack to the chalcone derivative is biased by π-stacking and hydrogen bonding interactions. Conceptually different phase-transfer catalysts based on monoaza-15-crown-5 ethers derived from D-glucose were introduced in the late nineties for the conjugate addition of 2-nitropropane to chalcones (Scheme 48).100 The activity of catalyst 64 is based on the ability of the azacrown ether in complexating the sodium cation, providing the formation of a ionic pair with the nitronate anion. The reaction gives interesting results with chalcone (R1 = R2 = Ph), but very modest levels of enantioselectivity are recorded with other chalcone derivatives. Attempts to use related azacrown ethers derived from D-mannose such as catalyst 65 did not provide significantly enhanced results except for single chalcones as substrates.101
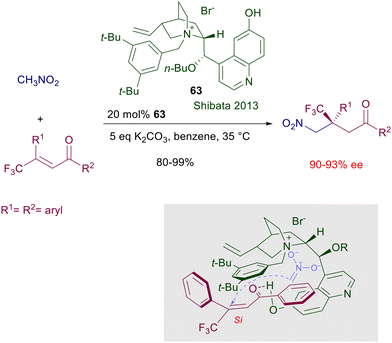 |
| | Scheme 47 Cinchona-derived ammonium salt 63 as phase-transfer catalyst. | |
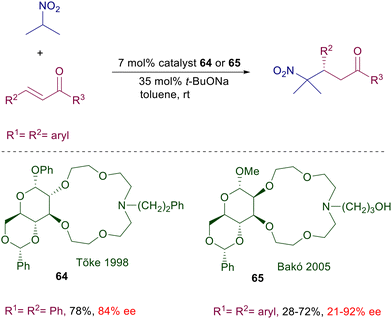 |
| | Scheme 48 Monoaza-15-crown-5 ethers as phase transfer catalysts. | |
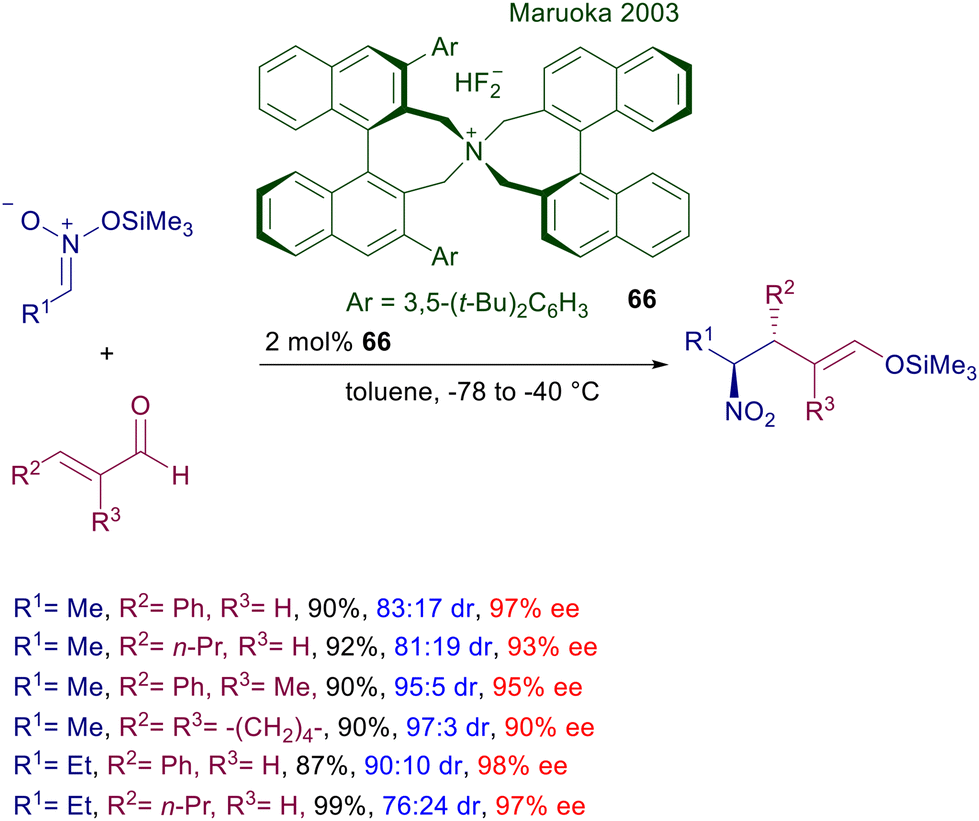
A truly effective quaternary ammonium salt catalyst was introduced by Maruoka in 2003 for the conjugate addition of silyl nitronates to enals (Scheme 49).102 The bisbinaphthylammonium difluoride 66 is particularly active at low charge in toluene and at low temperature, allowing the diastereoselective synthesis of the anti-adducts that can be recovered as the corresponding silyl enol ethers. Mild acidic hydrolysis of these silyl enol ethers allows regeneration of the carbonyl function almost quantitatively without any erosion of the original stereochemical attributes.
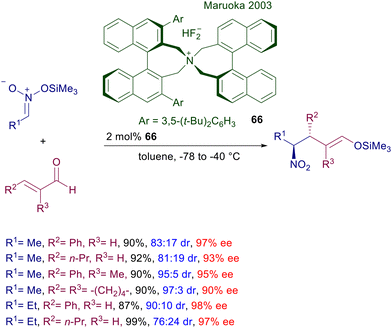 |
| | Scheme 49 Asymmetric conjugate addition of silyl nitronates to enals catalyzed by bisbinaphthylammonium difluoride 66. | |
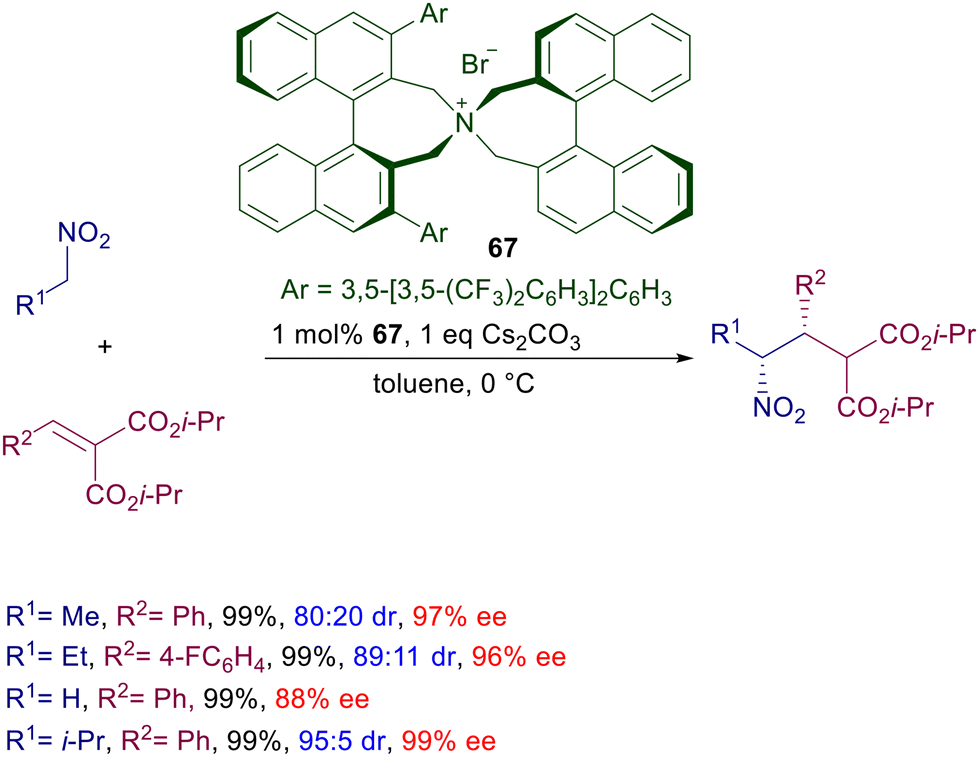
The same catalyst with marginal modifications, mainly regarding the aryl substituents of the binaphthyl system, has been used for the conjugate addition of other functionalized nitroalkanes with various Michael acceptors. In the reaction portrayed in Scheme 50, nitroalkanes have been made to react with diisopropyl arylidenemalonates in the presence of chiral ammonium bromide 67, leading to the corresponding syn-adducts with satisfactory diastereo- and enantioselectivity.103 This process represents an interesting case of matching–mismatching interactions since the ee values recorded for the minor anti stereoisomer are considerably lower compared with those of the syn.
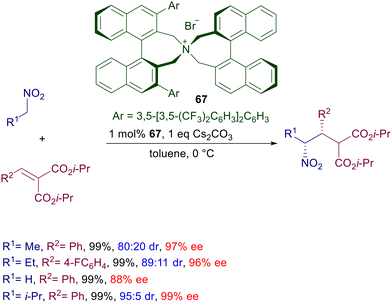 |
| | Scheme 50 Chiral ammonium salt 67 in the conjugate addition of nitroalkanes to arylidene malonates. | |
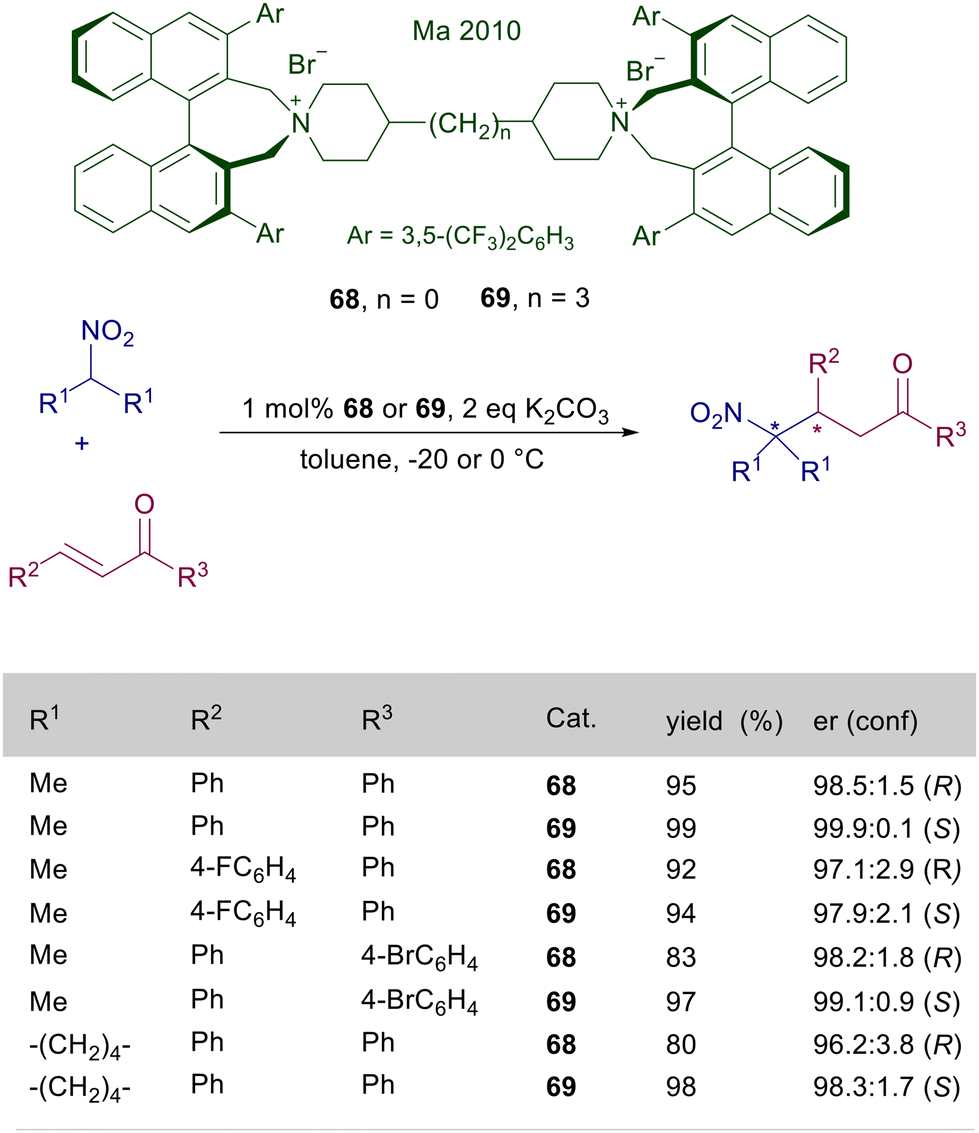
The synthetic versatility and high performances of catalysts of type 66 have been subsequently confirmed in the reaction of nitroalkanes with cycloalkenones,104 and silyl nitronates with nitroalkenes.105 These quaternary ammonium catalysts are monofunctional in nature, only allowing the activation of the nitronate anion thus excluding any interaction with the electron-poor olefin. This aspect has been revealed as problematic in the reaction of secondary nitroalkanes with enones, since the above discussed catalysts fail to provide significant levels of asymmetric induction in these reactions. Thus a bifunctional catalyst embedding two ammonium ions has been devised with the aim to provide the activation of both reactants, overcoming this limitation. The bisammonium bromides 68 and 69, built up by tethering two binaphthylammonium units, are excellent catalysts in the conjugate addition of secondary nitroalkanes with enones (Scheme 51).106 A peculiar aspect in the utilization of these catalysts is that a simple modification of the length of the tether enables a complete reversal of the stereochemical bias, thus allowing the preparation of both enantiomers of the resulting γ-nitroketones in a formal enantiodivergent process. A rationale for this behavior has not been provided, although it could be associated with the rotational rigidity of catalyst 68 featuring a diazaspiroundecane system compared with catalyst 69 tethered by a propyl framework.
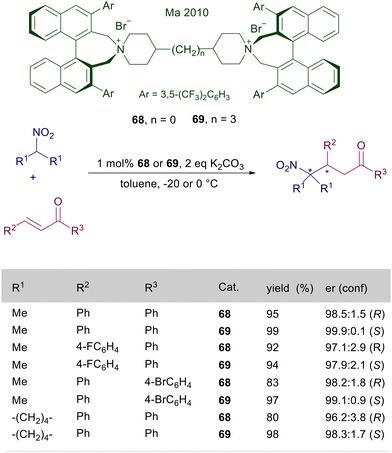 |
| | Scheme 51 Enantiodivergent conjugate addition of tertiary nitroalkanes to chalcones catalyzed by differently tethered ammonium salts 68 and 69. | |
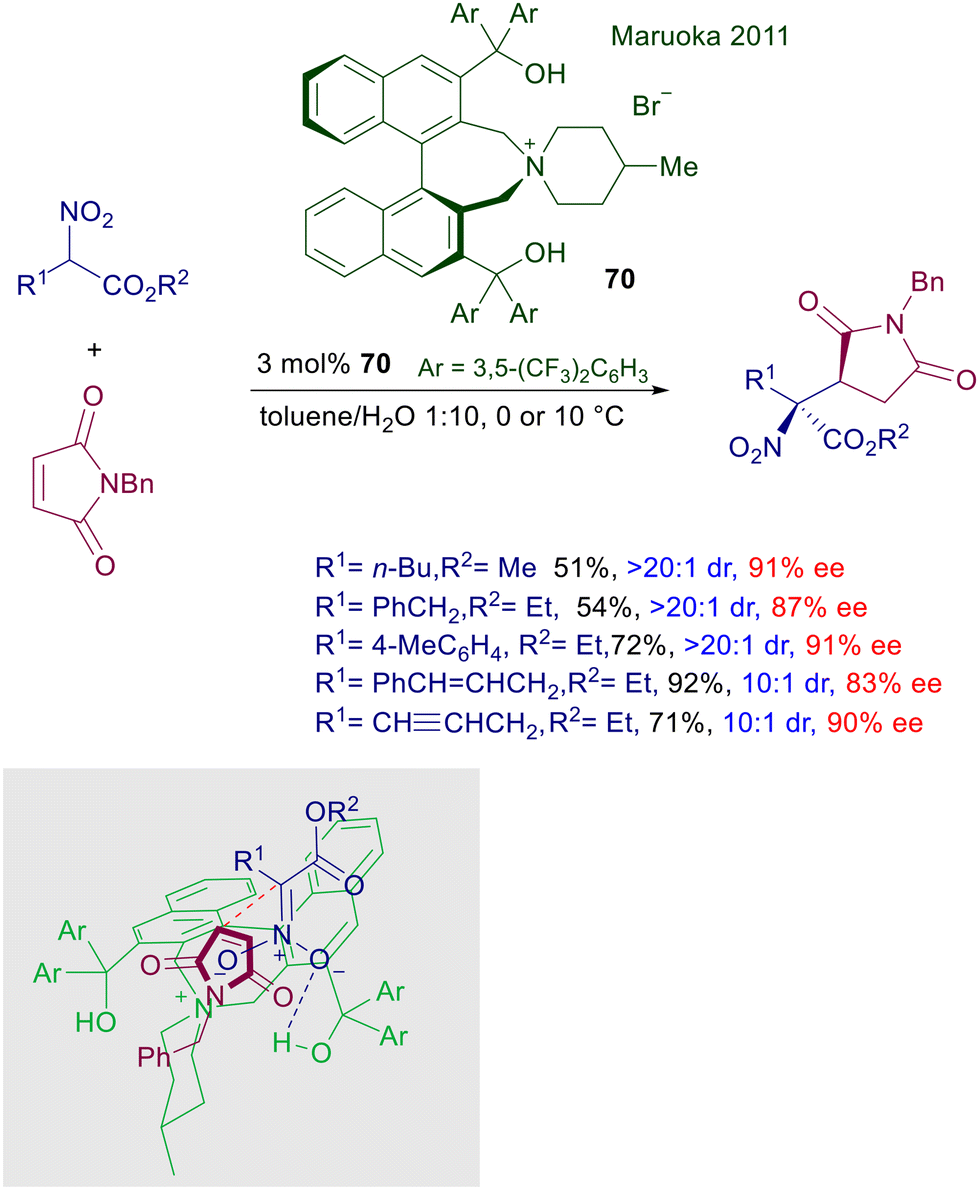
The enantioselective addition of nitroacetate esters to maleimide has been devised using a different version of the old-fashioned catalysts 66 embedding a couple of binaphthyl units (Scheme 52).107 Catalyst 70 includes two diarylmethanol frameworks, which would enhance the interactions with the nitronate anion thus favoring the enolization process. As a matter of fact, this is a base-free protocol carried out under the classical PTC conditions in a toluene/water biphasic system. Interestingly, the use of mild bases is not only unnecessary, but proved to be detrimental for the reaction as well as the use of homogeneous conditions using a single organic phase. In the proposed transition state, the attack of nitronate anion on the prochiral imide occurs selectively on the Re side of the unsaturation in order to minimize the clashing interactions with the left-sided diarylmethanol group.
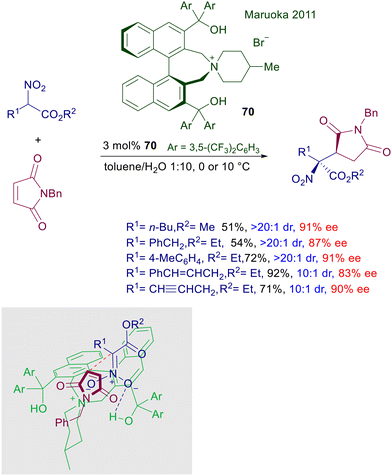 |
| | Scheme 52 Asymmetric conjugate addition of nitroesters to maleimides catalyzed by chiral ammonium salt 70. | |
2.8 Acylammonium ion and NHC catalysis
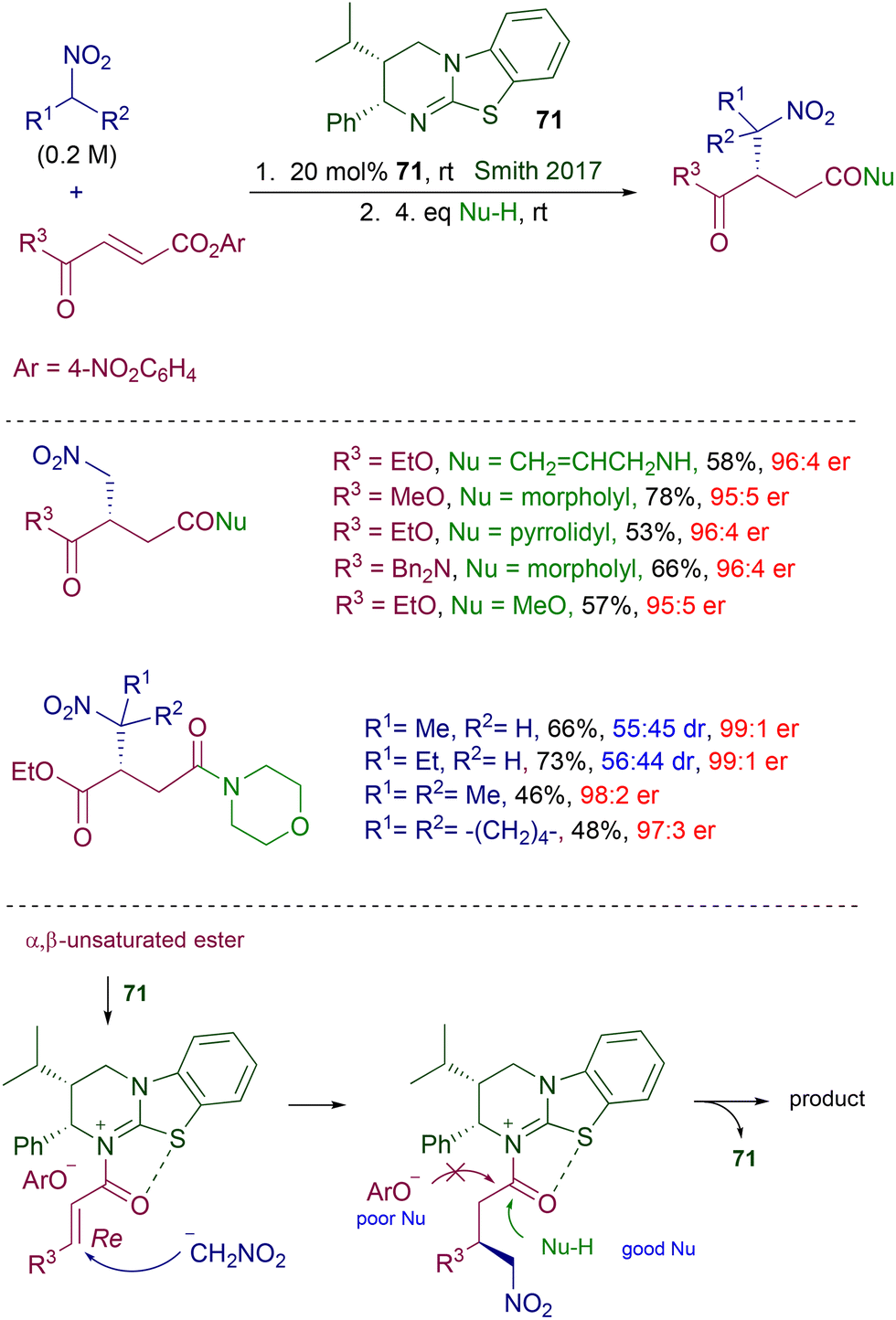
Among Michael acceptors, α,β-unsaturated esters are seldom used in organocatalyzed asymmetric reactions because of their reduced reactivity over the parent carbonyl derivatives and the poor efficiency of the activating modes provided by classical organocatalytic systems. Particularly, the enhancement in the reactivity of nitroalkanes provided by the usual hydrogen bonding with the chiral catalyst is generally inadequate for a suitable reaction with α,β-unsaturated esters. In this context, the activation of the acyl moiety by its temporary conversion into an acylammonium salt using a suitable chiral Lewis base catalyst represents an efficient strategy to allow these esters to be used as electrophiles in conjugate additions.108 The effectiveness of this approach has been demonstrated in the reaction of nitroalkanes with fumaric acid derivatives in the presence of chiral dihydroimidazo-benzothiazole 71 (Scheme 53).109 According to the proposed mechanism, reaction of the fumaric acid diester with catalyst 71 chemoselectively generates an acylammonium salt on the acyl moiety bearing the better leaving group (p-nitrophenoxide). The conformation of the acylammonium ion intermediate is locked by a sulfur–oxygen interaction caused by the electron-withdrawing effect exerted by the positive nitrogen atom on sulfur. Thus, the subsequent attack of the nitronate anion occurs regio- and stereoselectively to the Re face of the alkenyl framework. The phenoxide counteranion is poorly effective as a nucleophile, and thus in order to ensure an appropriate catalyst turnover stronger nucleophiles such as primary or secondary amines or alkanols are subsequently added to obtain the target products. The enantiomeric ratios for the obtained products are satisfactory even when primary nitroalkanes are used as reactants, providing the final products as an almost equimolar mixture of diastereomers. In a related approach, the same catalyst 71 has been used for the conjugate addition of triisopropylsilyl nitronates with anhydrides of fumaric acid monoesters.110 The resulting adducts are obtained as triisopropyl esters, making unnecessary the utilization of external nucleophiles as in the original protocol. However, reaction parameters such as atom economy and process mass intensity (PMI) are not very favorable due to the waste of half of the anhydride reactants.
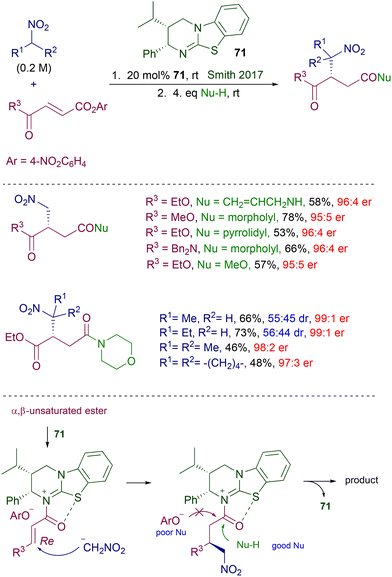 |
| | Scheme 53 Enantioselective conjugate addition of nitroalkanes to fumaric acid derivatives via chiral acylammonium ion intermediate. | |
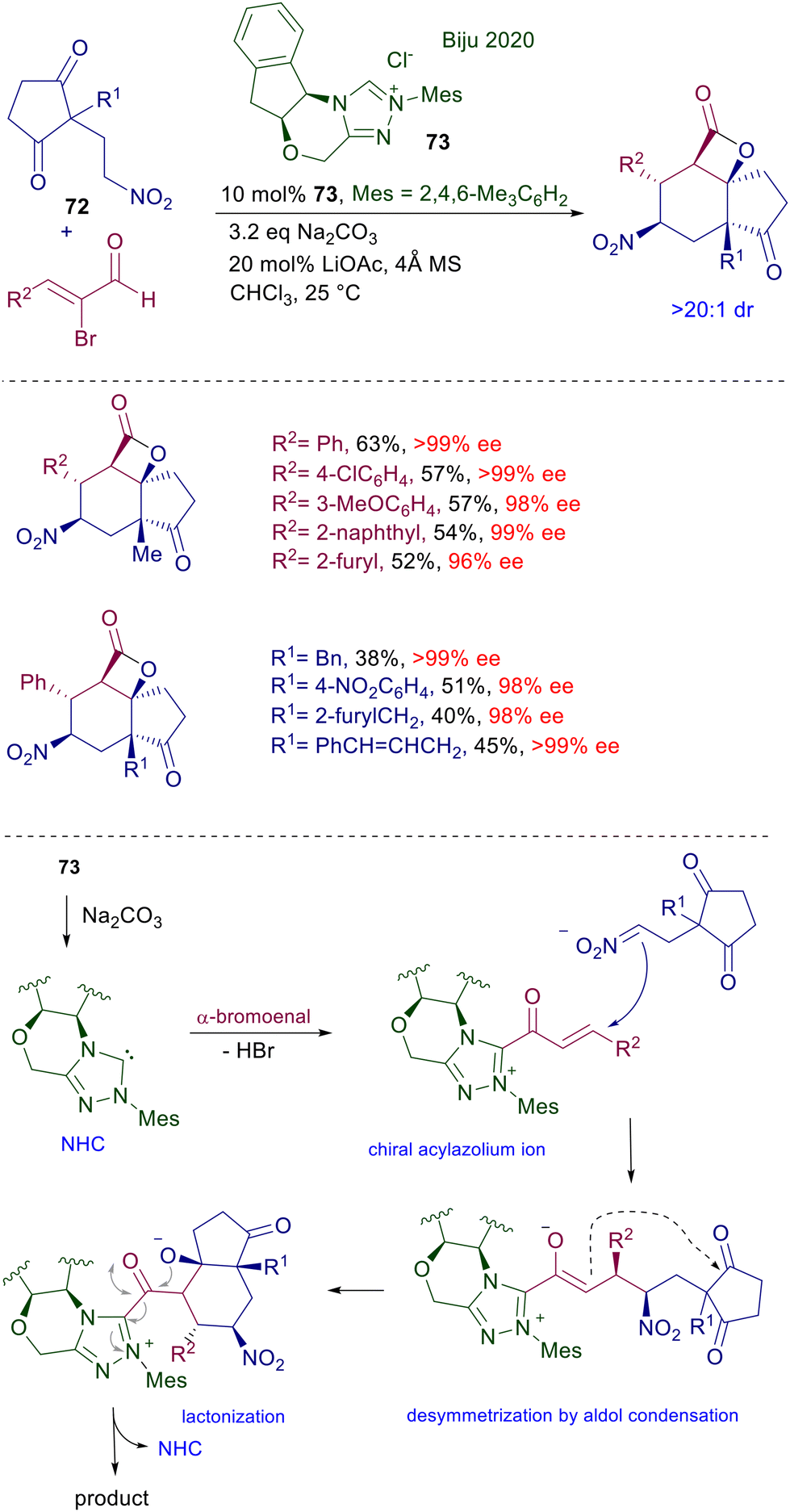
The reaction of N-heterocyclic carbenes (NHC) with α-haloenals represents a powerful method for the preparation of α,β-unsaturated acylazolium salts showing an enhanced reactivity as Michael acceptors towards a large variety of nucleophiles. 2-Nitroethylcyclopentandione 72 reacts with 2-bromoenals in the presence of chiral triazolium salt precatalyst 73, leading to tricyclic lactones with an outstanding control of the five contiguous generated stereocenters (Scheme 54).111 Under basic conditions, triazolium salt 73 is converted into the corresponding NHC, which upon reaction with the α-bromoenal affords the α,β-unsaturated acylazolium salt intermediate. Nitroalkane 72 undergoes a conjugate addition with the acylazolium salt, leading to a chiral enolate anion intermediate which, through an intramolecular aldol condensation, enables a desymmetrization of the 1,3-cyclopentandione moiety. The last step is a lactonization reaction releasing the target tricyclic compound with concomitant regeneration of the NHC, thus closing the catalytic cycle.
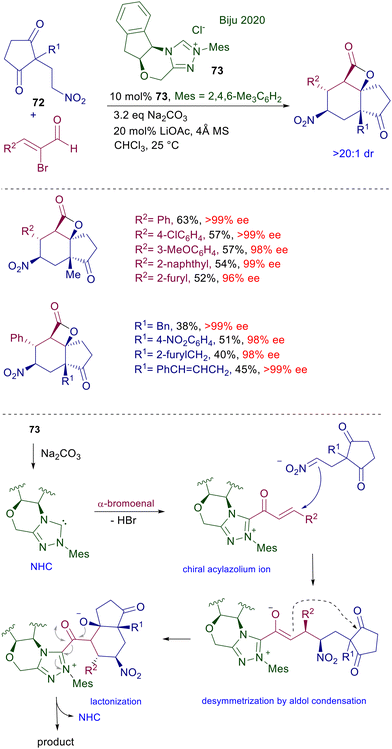 |
| | Scheme 54 Chiral NHC-catalyzed conjugate additions using precatalyst 73. | |
3. Organometallic complexes-catalyzed reactions
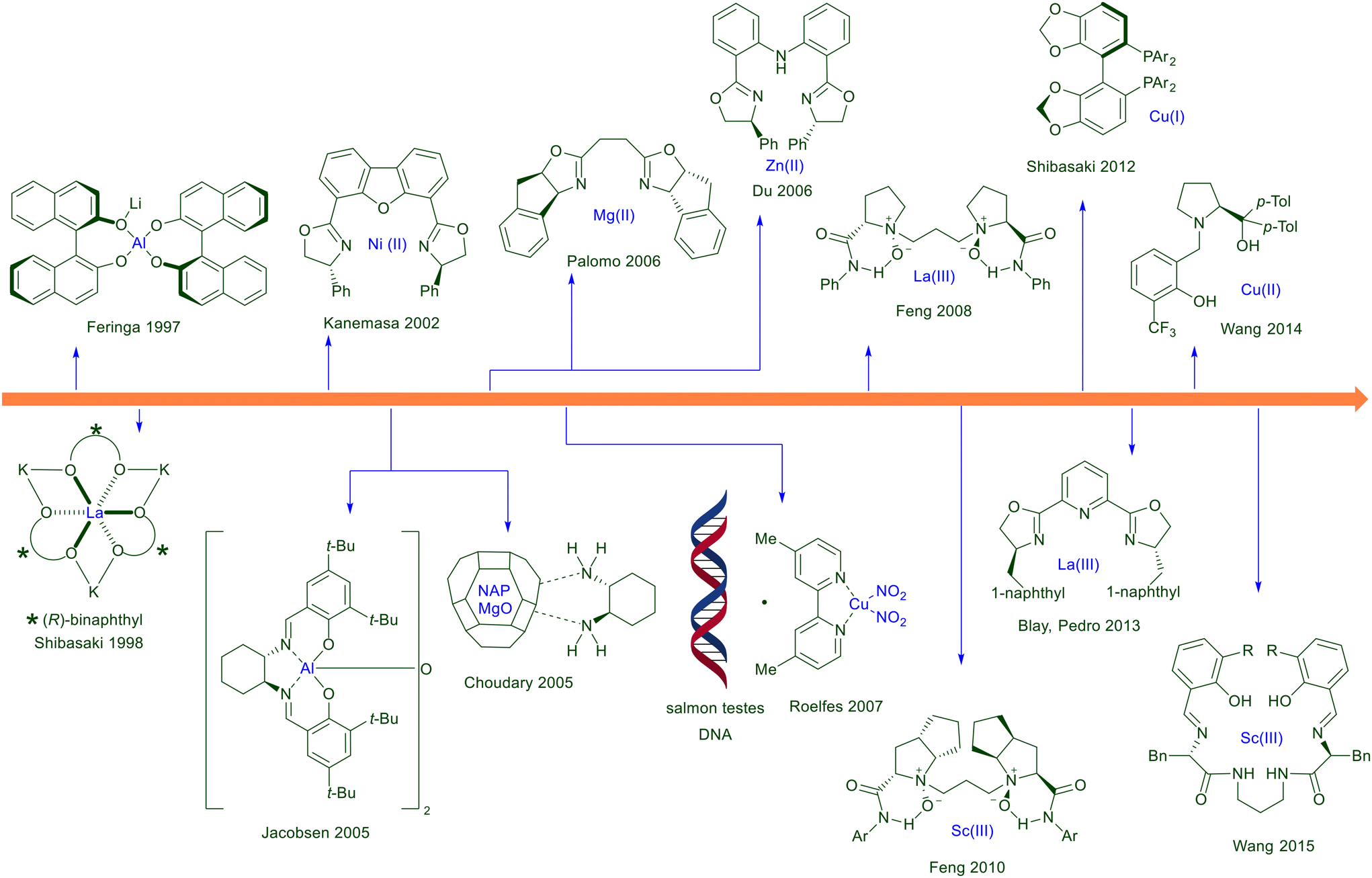
The development of synthetic procedures based on the use of chiral organometallic complexes for the conjugate addition of nitroalkanes to electron-poor alkenes has initially run parallel with that of organocatalytic methods. A selected number of metal–ligand couples have been envisaged for these conjugate additions and since the substrate–catalyst interaction is based on strong coordinating bonding, the results are often rewarding in terms of asymmetric induction (Fig. 5). Notwithstanding, to date the utilization of organometallic catalysts still remains underdeveloped compared with the synthetic protocols based on the use of organocatalysts. Interestingly, there are some metallic complexes based on aluminium which, after an initial success, have been neglected, while those based on lanthanum are rather recurrent throughout the years. The same observation can be made for magnesium, nickel and zinc-based complexes, which more recently have been superseded by copper and scandium complexes. Among chiral ligands bisoxazolines are undoubtedly the most employed derivatives, widely known for their chelating aptitude, although chiral N-oxides, biphosphines and Schiff base ligands have been successfully used in the conjugate addition of nitroalkanes to electron-poor olefins. The discussion of various metal–ligand complexes follows the chronological order in which they first appeared in the literature for these conjugate additions.
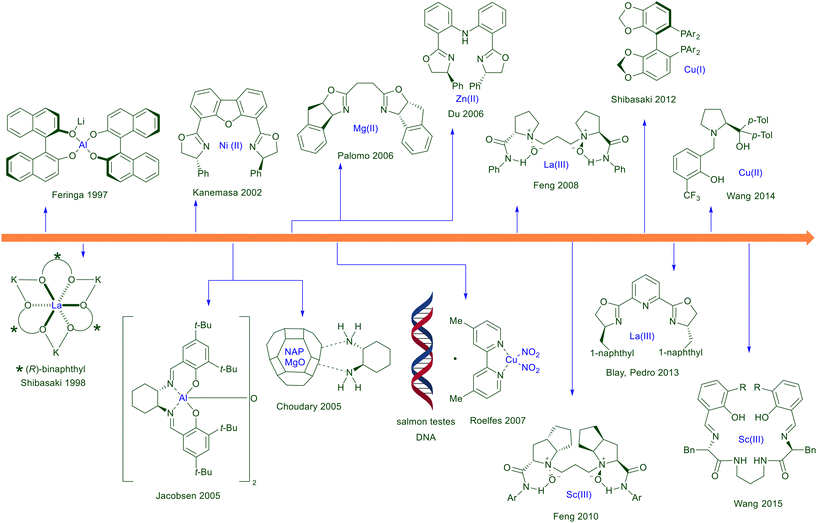 |
| | Fig. 5 Chronological development of metal–chiral ligand complexes employed for the asymmetric conjugate addition of nitroalkanes to electron-poor alkenes. | |
3.1 Nickel and cobalt complexes
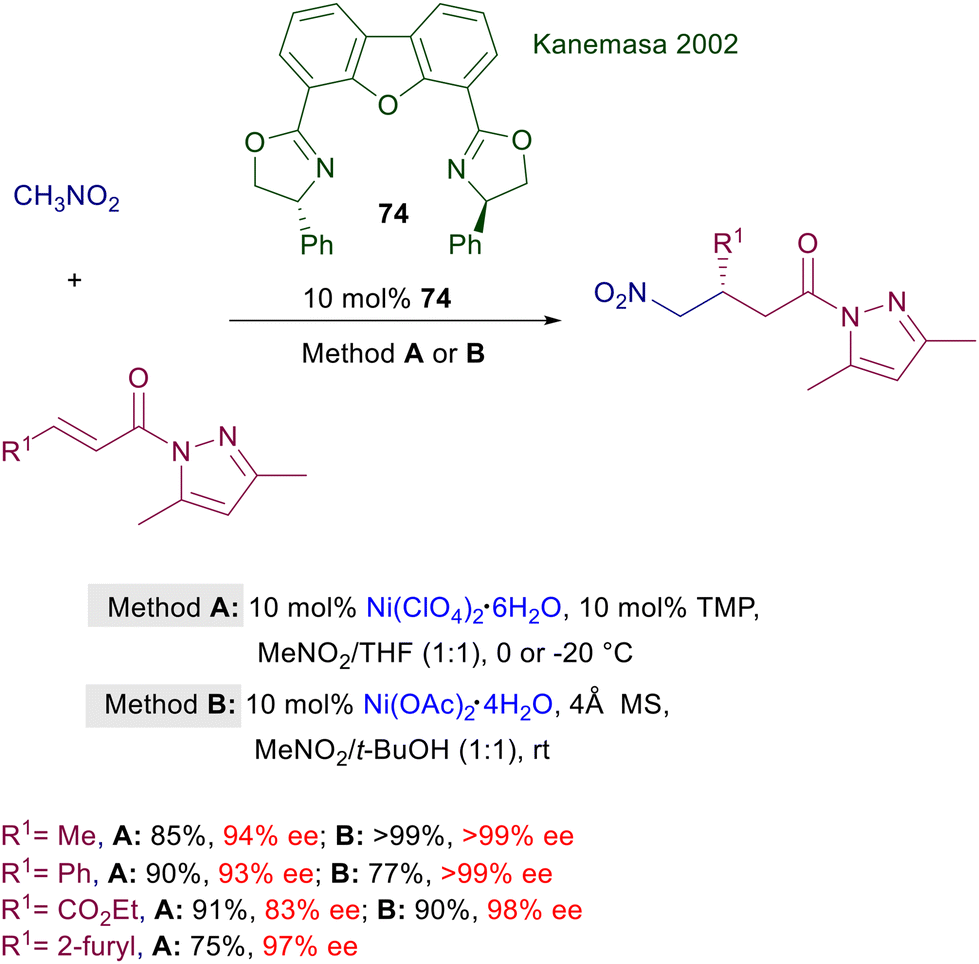
In a couple of papers that appeared more than thirty years ago the first conjugate addition of nitromethane to enones catalyzed by chiral nickel and cobalt complexes was reported.112 Ni(acac)2 and Co(acac)2 were coupled with various ligands including chiral 2,2′-bipyridines, 1,10-phenanthrolines, prolinamdes, prolinamines and prolinol. The nickel complexes were found to be particularly effective in the reaction of nitromethane with chalcones, leading to a maximum value of 60.9% ee evaluated by comparison with the optical rotation of the enantiomerically pure adduct. The first successful method for the addition of nitromethane to α,β-unsaturated acylpyrazoles was accomplished using Ni(II) complexes of bisoxazoline ligand 74 (Scheme 55). In the original procedure,113 the aqua complex obtained using nickel perchlorate hexahydrate (method A) was used in combination with 2,2,6,6-tetramethylpiperidine (TMP) as activator, since the metallic aqua complex alone was catalytically inactive. Later on, a modified procedure (method B) was introduced, changing the nature of the complex by using nickel acetate tetrahydrate and replacing TMP with 4 Å molecular sieves.114 The second procedure was more effective in leading to the target compounds with improved ee values, although both methods employ a very large excess of nitromethane used as cosolvent in the reaction. Recently, a chiral nickel-diamine complex supported on a mesoporous silica composite (MCM-41) has been introduced for the conjugate addition of malonate esters to nitroalkenes.115 This catalyst is also effective in the Michael addition of nitromethane to alkylidene malonates, but the observed enantioselectivity never exceeds the value of 86% ee.
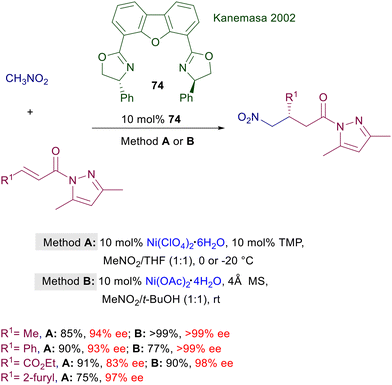 |
| | Scheme 55 Ni(II)-Bisoxazoline complex catalyzes the conjugate addition of nitromethane to α,β-unsaturated acylpyrazoles. | |
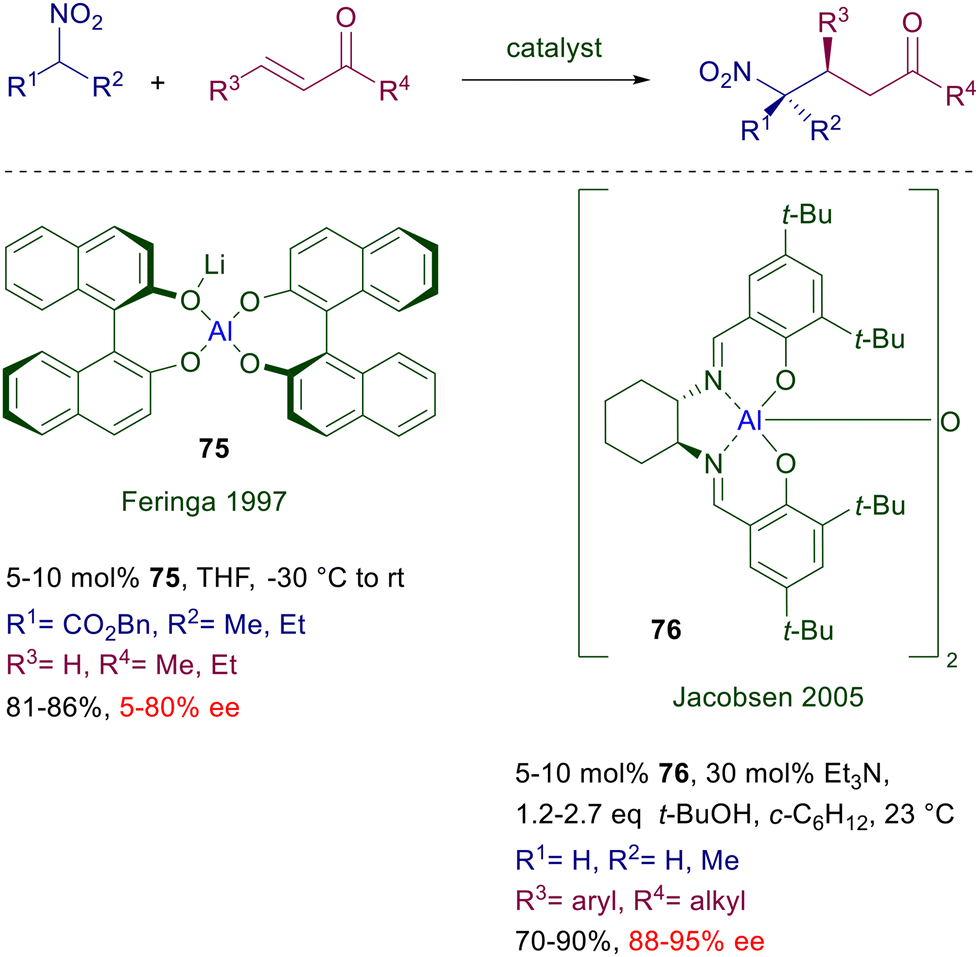
3.2 Aluminium complexes
The first aluminium complex employed as catalyst for the conjugate addition of nitroalkanes to enones was a mixed Li–Al alkoxide 75 obtained by reaction of chiral BINOL with lithium aluminium hydride (Scheme 56).116 This catalyst was used for the reaction of a couple of nitroesters with enones, with generally modest results in term of ee values and, as expected, the enantioselectivity could be improved at low temperature. Spectroscopic studies have revealed that the effective catalyst is actually a mixture of aluminium complexes in solution. A more effective chiral [(salen)Al]2O catalyst 76 has been subsequently developed for the reaction of nitroalkanes with enones at room temperature.117 The use of an added base is necessary for a suitable conversion and, as usually observed, nitroethane affords an equimolar mixture of diastereomers.
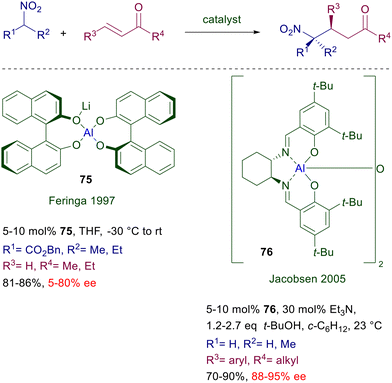 |
| | Scheme 56 Asymmetric conjugate additions catalyzed by Al(III)-complexes. | |
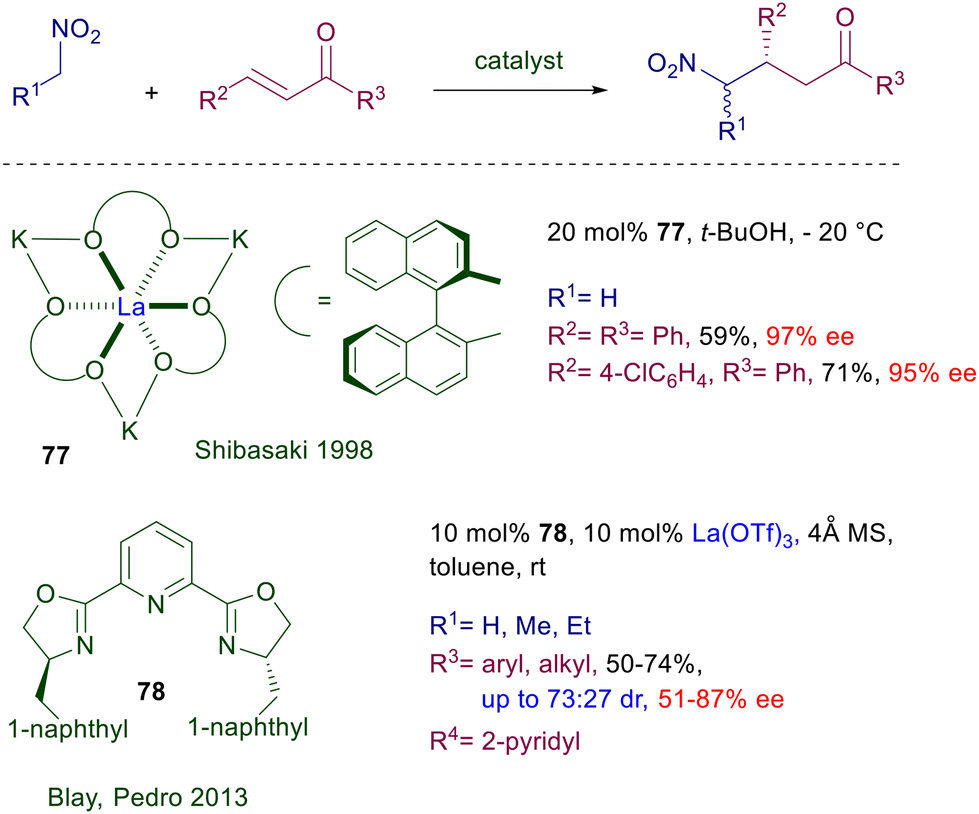
3.3 Lanthanum complexes
Chiral heterobimetallic complexes of La(III)–alkali metals with BINOL are particularly effective in the nitroaldol reaction as well as other nucleophilic addition processes.118 The heterobimetallic complex 77 obtained by reaction of (R)-BINOL with lanthanum(III) triisopropoxide and potassium bis(trimethylsilyl)amide has been used to demonstrate that this catalyst is also potentially useful for the conjugate addition of nitromethane to enones (Scheme 57).119 Curiously, in spite of the excellent results obtained with a couple of enones, the use of this catalyst has not been further expanded to other substrate–reagent combinations. The lanthanum complex obtained using bisoxazoline 78 as chiral ligand has been tested for the same purpose, but the results obtained in the reaction of nitromethane and other primary nitroalkanes with enones are less encouraging.120
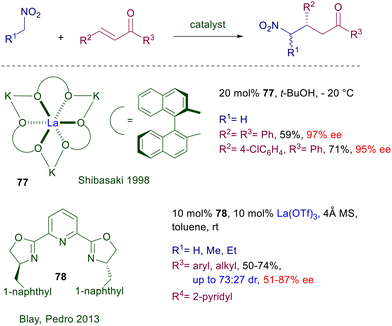 |
| | Scheme 57 La(III)-complexes in the conjugate addition of nitroalkanes to enones. | |
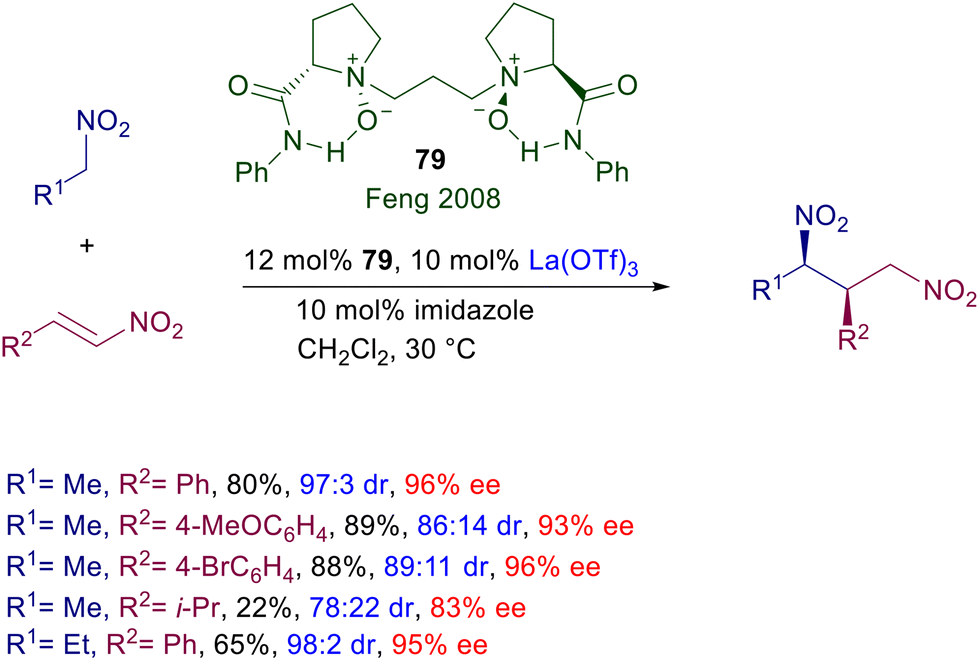
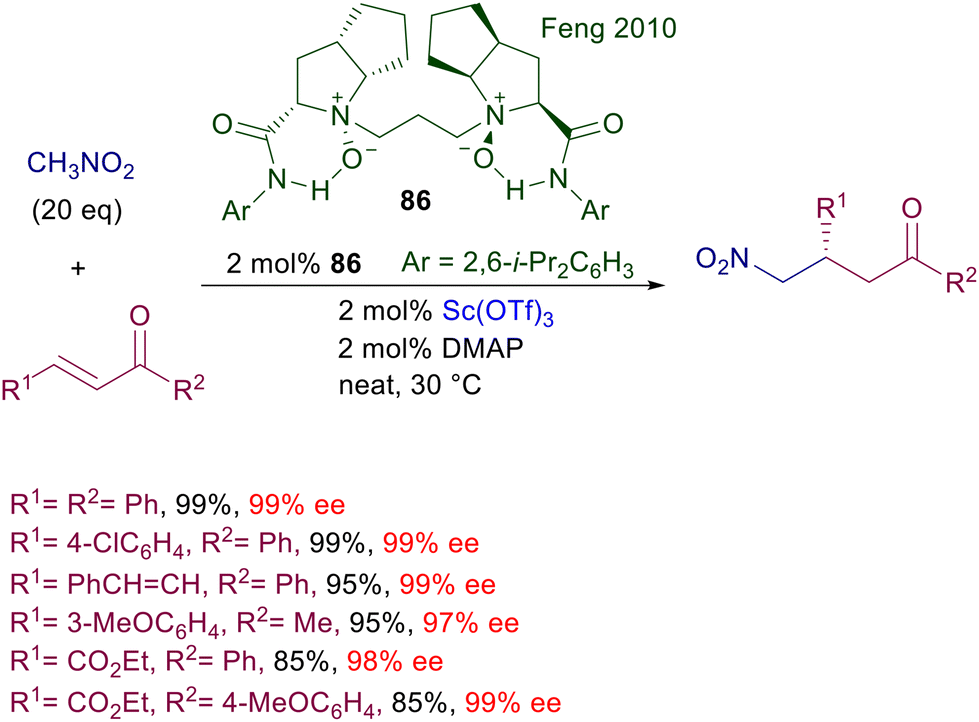
Chiral N,N-dioxides developed by Feng and co-workers are particularly versatile catalysts since they can be used as organocatalysts as well as metal ligands in several asymmetric processes.121 The features of the catalyst can be appropriately tuned by changing the ring size of the two N-oxide subunits and the length of tethering connection. Chiral dioxide 79 acts as an effective ligand for lanthanum(III) triflate in the conjugate addition of nitroalkanes to nitroalkenes (Scheme 58).122 The reaction has been tested with a couple of nitroalkanes and nitrostyrenes, leading to the corresponding adducts with satisfactory diastereoselectivity and generally high ee values. Modest results are evidenced using a linear nitroalkene both in terms of yield and stereoselectivity.
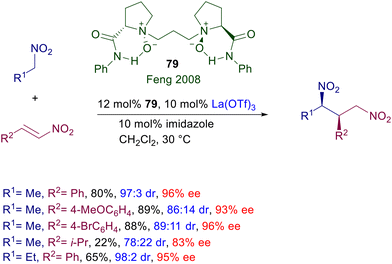 |
| | Scheme 58 Asymmetric conjugate addition of nitroalkanes to nitroalkenes catalyzed by La(III)-dioxide complex. | |
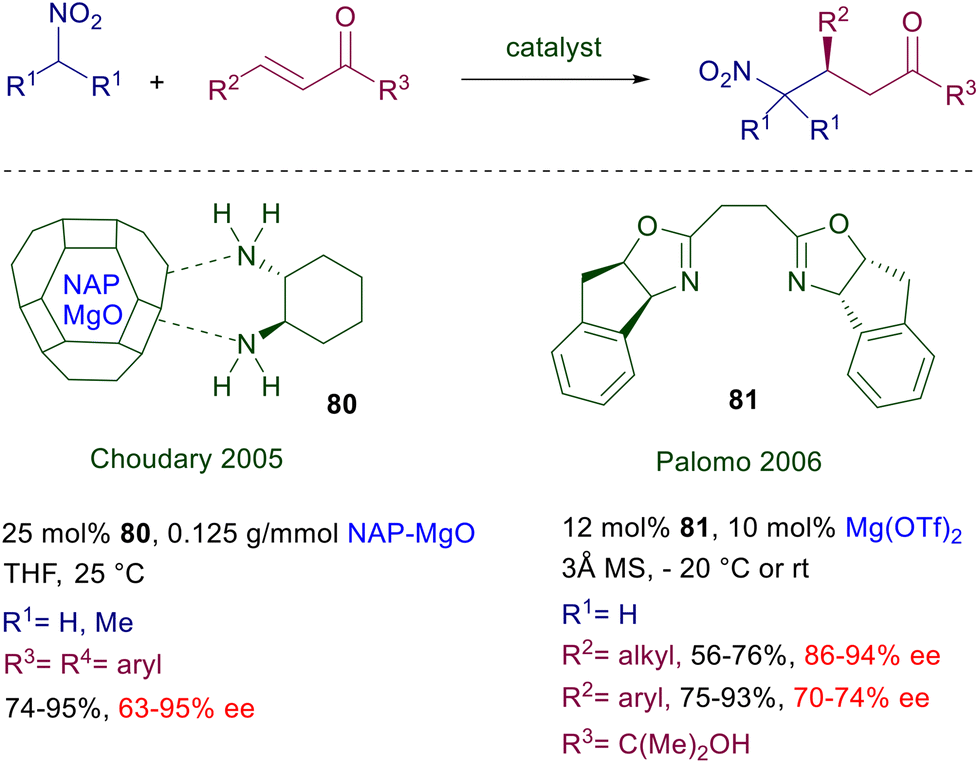
3.4 Magnesium complexes
The chelating ability of the magnesium cation was evaluated in a couple of papers more than fifteen years ago, exploiting two different magnesium-containing compounds interacting with chiral ligands. Nanocrystalline magnesium oxide (NAP-MgO) was used as ligand for (R,R)-1,2-diaminocyclohexane 80 as catalyst for the addition of nitromethane and 2-nitropropane to chalcones (Scheme 59).123 The results obtained using this protocol are not particularly remarkable, with the exception of a single example. The NAP-MgO can be recycled up to five times after heating at high temperature, maintaining the same performance for the best observed process. The complex of chiral bisoxazoline 81 with magnesium triflate is effective in the addition of nitromethane to α′-hydroxy enones (R3 = C(Me)2OH).124 Interestingly, Michael acceptors bearing alkyl substituents give better results compared with aryl-substituted ones. The dimethylcarbinol group in the target adducts acts as a dummy substituent, since by oxidative cleavage with sodium periodate it can provide access to the corresponding γ-nitroacids or γ-nitroaldehydes maintaining the same enantiopurity of the corresponding substrates.
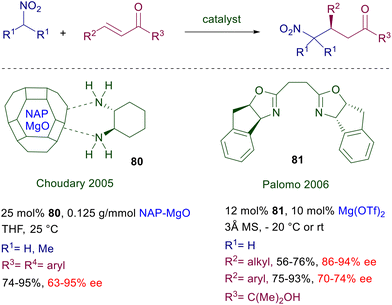 |
| | Scheme 59 Enantioselective conjugate addition of nitroalkanes to enones using Mg(II)-complexes. | |
3.5 Copper and zinc complexes
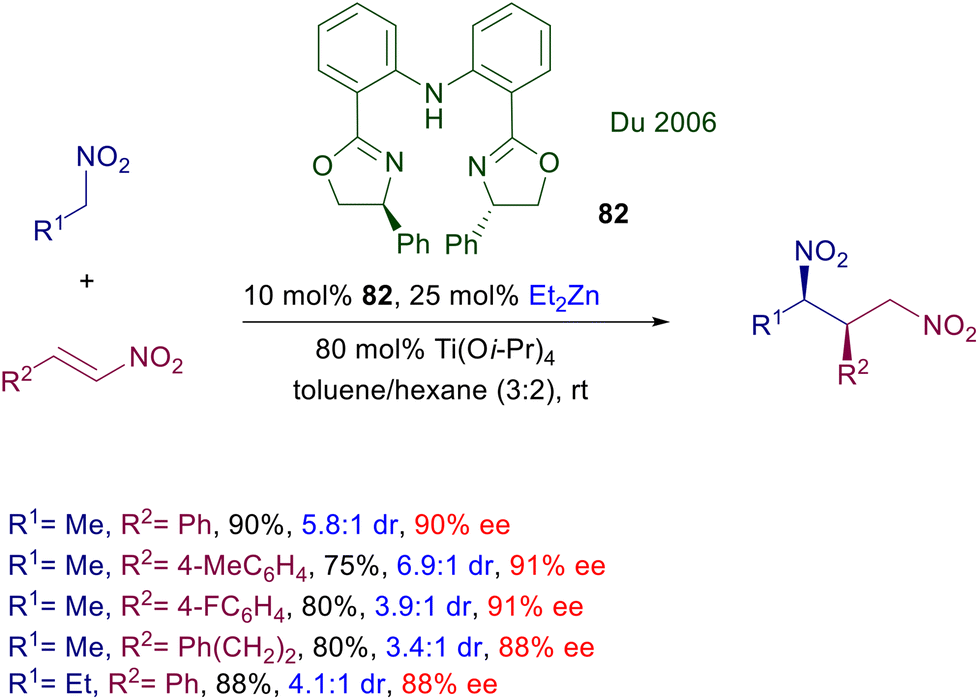
The first enantioselective conjugate addition of nitroalkanes to nitroalkenes was carried out using the zinc(II) complex with bisoxazoline 82 (Scheme 60).125 The required zinc cation was generated by diethylzinc upon activation with titanium tetraisopropoxide. The reaction is syn selective and affords the corresponding 1,3-dinitro derivatives with satisfactory levels of enantioselectivity. To date, this is the only available asymmetric conjugate addition involving nitroalkanes which employs a zinc complex as catalyst.
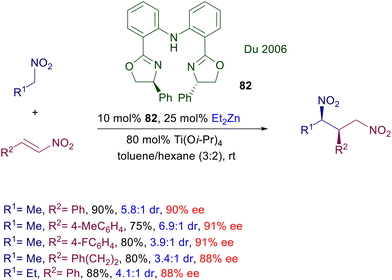 |
| | Scheme 60 Asymmetric synthesis of 1,3-dinitro derivatives by a Zn(II)-bisoxazoline complex. | |
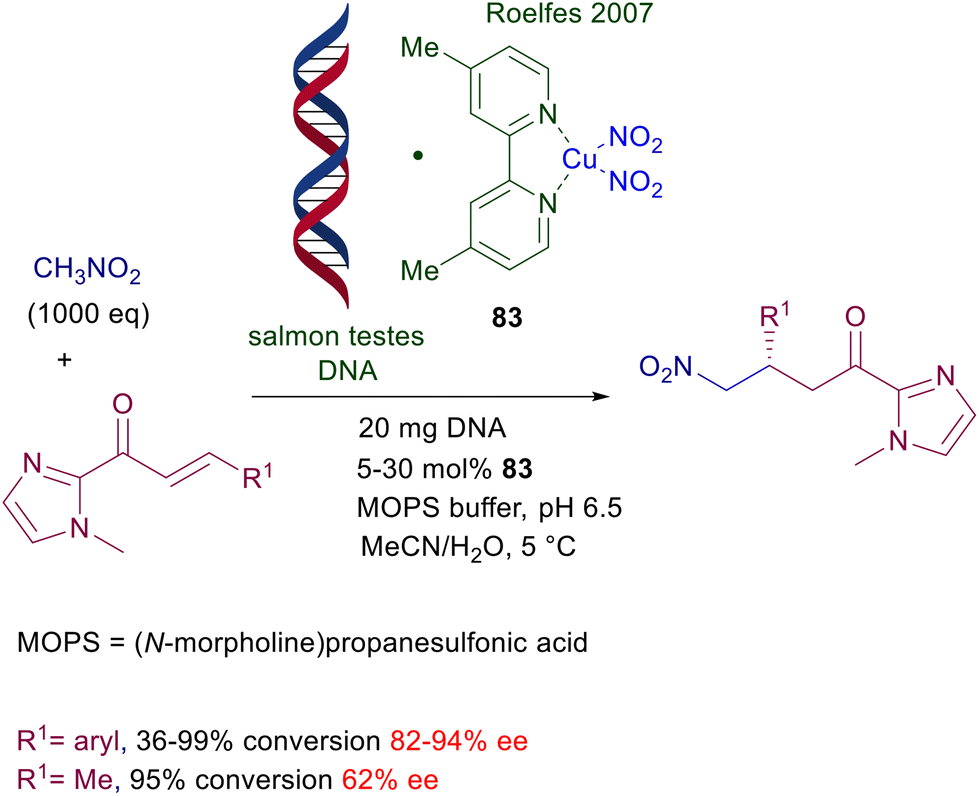
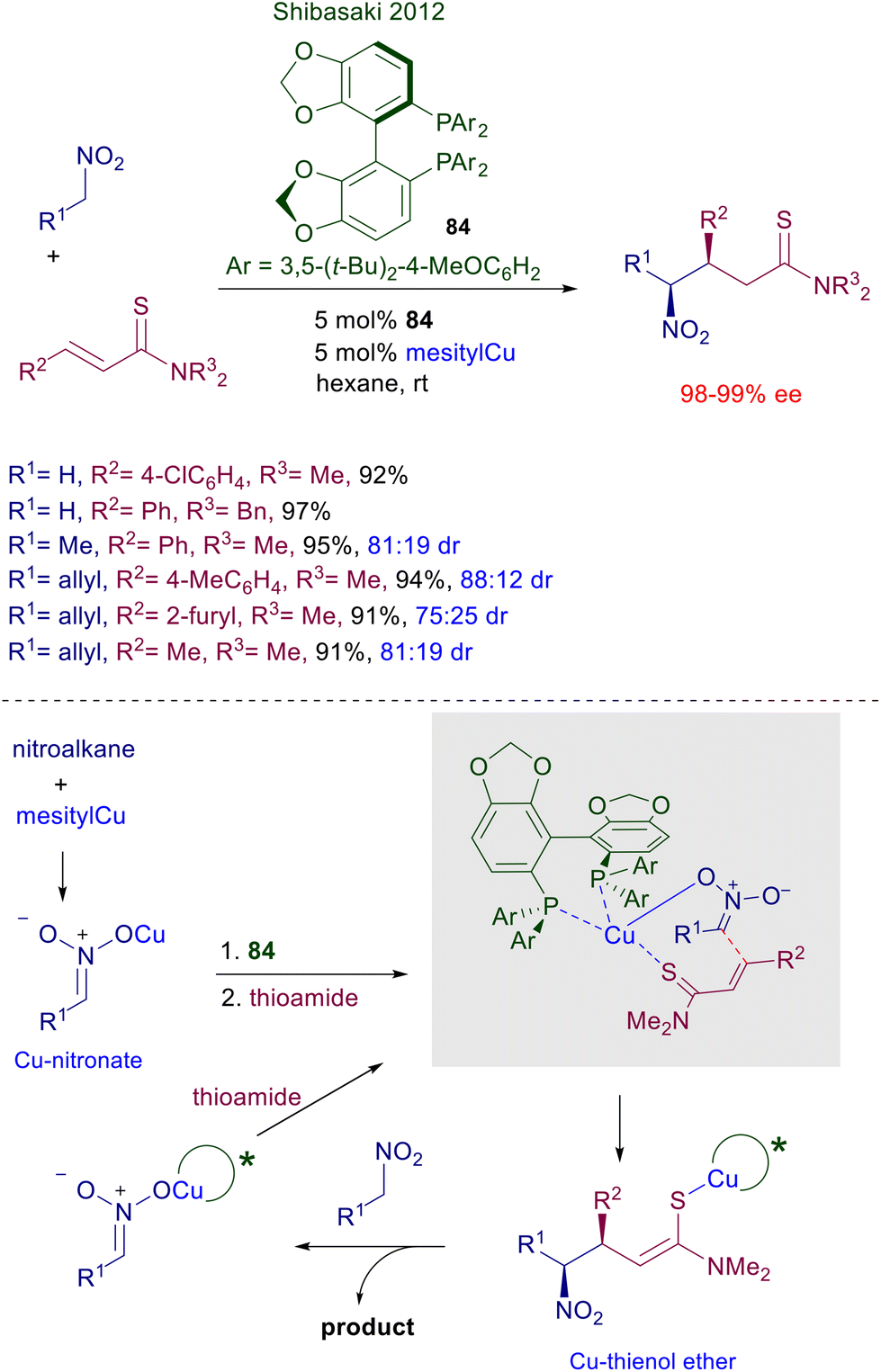
The use of copper complexes in these conjugate additions was introduced with an unusual protocol involving salmon testes DNA as chiral element associated with copper(II)-bipyridine complex 83 (Scheme 61).126 The exact nature of the interaction between DNA and the achiral copper complex is unknown, but the bipyridine ligand is mandatory for a successful reaction since no asymmetric induction was observed using only the DNA/copper(II) nitrate couple. A large excess of nitromethane is required for an effective conversion, and the enantioselectivity is constantly below 90% ee but with just one example (R1 = 2-BrC6H4, 94% ee). A more effective chiral copper complex has been devised using (R)-DTBM-Segphos 84 in combination with mesitylcopper for the conjugate addition of nitroalkanes with α,β-unsaturated thioamides (Scheme 62).127 The mesitylcopper initially reacts with the nitroalkane, generating a copper nitronate which in the presence of the phosphine ligand 84 leads to an organometallic complex. Subsequent interaction of the copper complex with the sulfur atom of the thioamide paves the way for a preferential syn diastereoselective conjugate addition which occurs with outstanding levels of enantioselectivity.
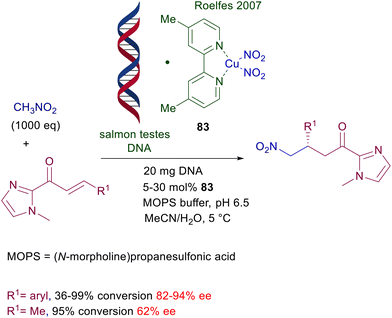 |
| | Scheme 61 Salmon testes DNA/Cu(II)-bipyridine complex as catalyst in conjugate additions of nitromethane to enones. | |
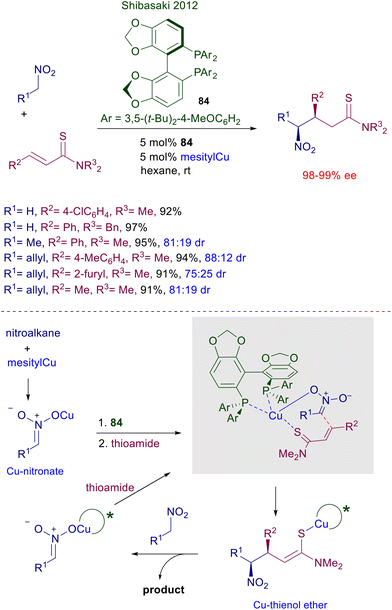 |
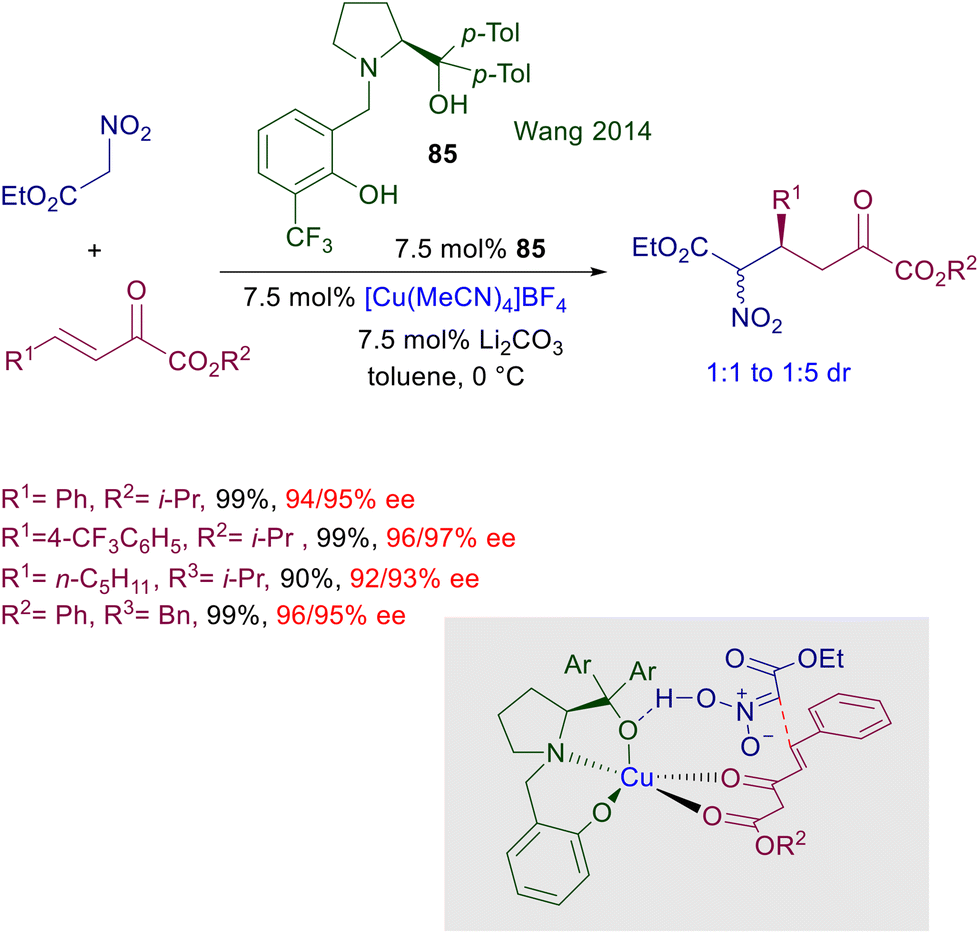
| | Scheme 62 Asymmetric conjugate addition of nitroalkanes to unsaturated thioamides catalyzed by Cu(I)-DBTM-Segphos. | |
The addition reaction results in the formation of an intermediate copper thioenol ether, which upon reaction with the nitroalkane delivers the final product and generates the chiral copper nitronate, thus ensuring an efficient catalyst turnover. It should be observed that this catalytic system is highly chemoselective for α,β-unsaturated thioamide substrates, since the corresponding amides or enones are unaffected in competitive cross experiments. The copper(I) complex with prolinol-derived ligand 85 is a valuable catalyst for the conjugate addition of nitroacetate esters to β,γ-unsaturated α-ketoesters (Scheme 63).128 As is commonly observed, no stereocontrol is possible at the very easily enolizable α-nitro carbon, but the enantioselectivity recorded for both diastereoisomers formed is very good. The hydroxy groups in the ligand are able to coordinate the copper atom with further assistance from the pyrrolidine nitrogen atom. In the plausible transition state, the arrangement of the reactants leading to the observed stereochemical outcome is caused by the β-ketoester coordination with the copper and by the hydrogen bonding of the nitronic acid with the diarylmethanol group. A related copper(II) complex with the same ligand has been subsequently used for the conjugate addition of nitroalkanes to 4-alkylidenpyrrolidin-2,3-diones in water, with excellent stereoselectivity.129
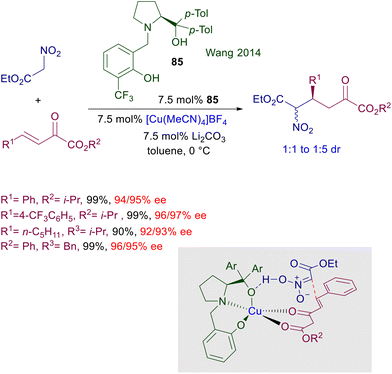 |
| | Scheme 63 Cu(I)–ligand 85 complex for the enantioselective reaction of ethyl nitroacetate with unsaturated ketoesters. | |
3.6 Scandium complexes
The complex between chiral N,N-dioxide 86 and scandium(III) triflate gives superior results over complexes of other metal triflates (La, Sm, Y, Yb) with the same ligand in the reaction of nitromethane with enones and 4-oxo-4-arylbutenoates (Scheme 64).130 The chemical yields and the enantioselectivity obtained for the corresponding adducts are really outstanding for the use of nitromethane, but this protocol has not been evaluated for the same process using primary or secondary nitroalkanes. In any event, chiral N,N-dioxides of type 79 and 86 have been proposed as ligands of gadolinium(III) in the catalyzed reaction of nitroalkanes with enones, giving excellent ee values but an almost complete lack of diastereoselectivity.131
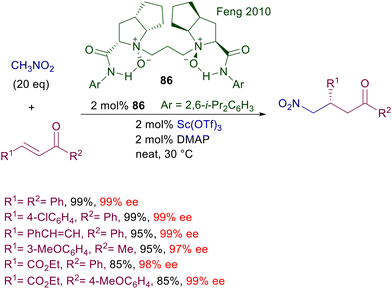 |
| | Scheme 64 Enantioselective conjugate addition of nitromethane to chalcones catyalyzed by Sc(III)-N,N-dioxide complex. | |
With the aim of increasing the level of diastereoselectivity obtainable in the reaction of nitroalkanes with enone derivatives a new chiral ligand 87, particularly suited to accommodate early-transition metals like scandium(III), has been designed (Scheme 65).132 The ligand ability of the C2-symmetric Schiff base 87 is coupled with the insertion of a pyridyl N-oxide moiety in the Michael acceptor, which would ensure a strong coordination with the metal cation in the transition state. These combined effects lead to a satisfactory anti diastereoselectivity in the reaction of primary nitroalkanes with the employed substrate and a generally good level of enantioselectivity including the use of nitromethane as reactant.
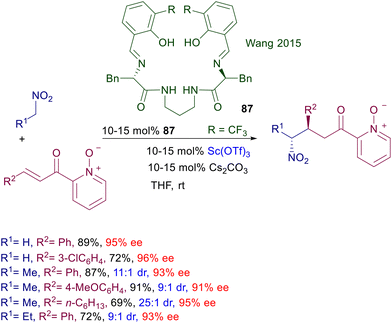 |
| | Scheme 65 Sc(III)-Schiff base 97 complex catalyzes the asymmetric conjugate addition of nitroalkanes to pyridine N-oxide enones. | |
4. Selected synthetic applications
The asymmetric conjugate addition of nitroalkanes to various Michael acceptors has been particularly employed in preliminary stages of multistep processes in order to generate the embryonic element of chirality required to build up the whole optically active molecule. Soon after the introduction of the early protocols for the asymmetric synthesis of γ-nitrocarbonyl derivatives, the preparation of small bioactive compounds such as the GABAB receptor agonist baclofen or the enzyme inhibitor rolipram has been used to highlight the importance of these methods. Recently, the advent of telescoped continuous flow chemistry has provided a technical tool for the efficient synthesis of (S)-rolipram using a polystyrene-bound prolinol-derived catalyst 88 (Scheme 66).133 The enantioselective conjugate addition of nitromethane to the appropriate functionalized enal, catalyzed by the immobilized chiral catalyst 88, is followed by the oxidation–esterification step carried out in a heated coil reactor using the in situ generated persulfuric acid.
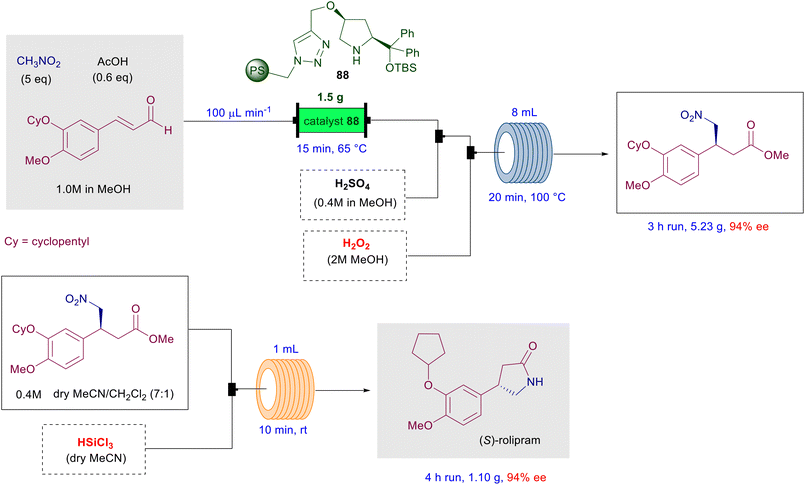 |
| | Scheme 66 Telescoped flow chemical process for the enantioselective synthesis of (S)-rolipram. | |
The intermediate γ-nitroester is obtained in 94% ee and is submitted to a metal-free reductive process in a coil reactor using trichlorosilane, leading to (S)-rolipram after spontaneous lactamization. Efficient and sustainable protocols for the synthesis of bioactive compounds also entail a limitation in the work-up and purification operations, exploiting one-pot transformations. The total synthesis of the spiroxindole (−)-horsfiline has been carried using six synthetic transformations grouped in two one-pot processes and a final cleavage, resulting in an overall yield of 33% of the enantiopure target product starting from the oxindole substrate (Scheme 67).134 The initial aldol condensation between the oxindole and acetaldehyde requires two steps, including the acid-promoted dehydration, which can be realized in a one-pot fashion. The enantioselective conjugate addition of nitromethane to the isolated α,β-unsaturated aldehyde is carried out using chiral diarylprolinol 89 followed by reduction of the nitro group just by adding zinc metal, acetic acid and water to the solution. Formation of the primary amine is coupled with intramolecular imination and further reduction leading to the spirocyclic derivative. The N-methylation of the pyrrolidine ring by aqueous formaldehyde takes advantage of the presence of zinc metal used in the previous step, ultimately leading to the N-benzylated horsfiline in 46% overall yield from the enal substrate. The cleavage of the N-benzyl group is finally carried out by a Birch electron transfer reaction using sodium metal/ammonia, giving (R)-horsfiline in good overall yield. The same approach has been used to prepare the demethoxylated analogue of (−)-horsfiline named (−)-coerulescine in 46% overall yield and 94% ee.
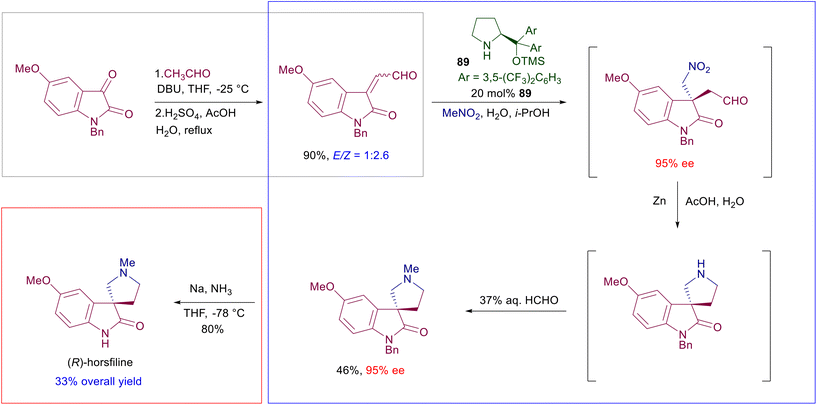 |
| | Scheme 67 Total synthesis of (R)-horsfiline by one-pot processes. | |
The macrocyclic alkaloid (−)-haliclonin A, isolated from a marine sponge, showed promising cytotoxic activity against the K562 leukemia cell line. The core of this natural product is represented by the aza-bicyclic derivative 90, which was obtained starting from 3-(5-hexenyl)-2-cyclohexen-1-one (Scheme 68).135 Reaction of the cyclohexenone with nitromethane employed as a solvent in the presence of the Takemoto's chiral thiourea 32 led to the formation of the corresponding adduct in good yield and enantioselectivity. After the oxidative regeneration of the α,β-unsaturated system, selective reduction of the carbonyl group using the Luche protocol and zinc-promoted reduction of the nitro group gave an amino alcohol intermediate. Protection of the primary amino group as p-methoxybenzyl (PMB) derivative was accomplished by reductive amination followed by amidation with phenylchlorothioformate. After oxidation of the allylic alcohol to the enone moiety, the last step to intermediate 90 was carried out by a Pd(II)-catalyzed cyclization using 1,3-bis(diphenylphosphino)propane (dppp) as ligand. After several steps compound 90 was converted into (−)-haliclonin A, thus confirming the structural assignment of the isolated natural product.
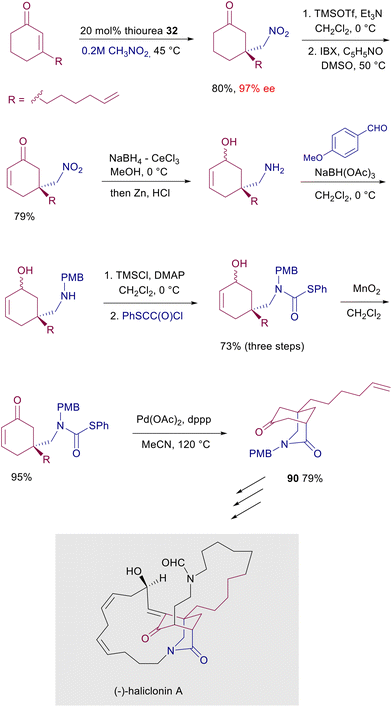 |
| | Scheme 68 Synthesis of the macrocyclic alkaloid (−)-haliclonin. | |
The enhanced pharmacological profile of many alkaloids of the ergot family led to the development of many procedures for their asymmetric synthesis. Considering the common structural motif of these alkaloids, a unifying strategy consists of the synthetic elaboration of 4-substituted 3-(2-nitroethyl) indoles, easily obtainable by Friedel–Crafts reaction of indoles with nitroalkenes (Scheme 69).136 Particularly, (−)-festuclavine and (−)-pyroclavine, which show an epimeric relationship, have been prepared by a preliminary intramolecular conjugate addition of nitroindole 91 in the presence of chiral bifunctional thiourea catalyst 92. This reaction allows the generation of two stereocenters of the three present in the target products, with a satisfactory level of diastereo- and enantioselectivity.
 |
| | Scheme 69 Synthesis of the ergot alkaloids (−)-festuclavine and (−)-pyroclavine. | |
The adopted synthetic strategy entailed the reductive transformation of the nitro group into a protected N-methylamino derivative and the installation of an isopropenyl side chain, which proved rather troublesome by nucleophilic methylation-elimination since a regioisomeric mixture of alkenyl compounds was formed. The required 2-methylallyl regioisomer was submitted to a hydroboration–oxidation followed by O-tosylation with poor stereocontrol which, however, enabled the formation of the epimeric couple of N-tosylated natural products by nucleophilic displacement by the N-deprotected amino group. The target alkaloids were obtained after final deprotection of the indole nitrogen atom. The procedure devised for the alkylative desymmetrization of meso-cyclopenten-1,3-diones portrayed in Scheme 33 has been successfully employed for the total synthesis of (+)-[3]-ladderanol, a component of natural ladderane phospholipids (Scheme 70).137 The cyclohexenedione 93 required for the asymmetric conjugate addition was prepared by [2 + 2] photocycloaddition of cyclohex-2-ene-1,4-dione with bicyclo[2.2.0]hex-2-ene followed by oxidation with selenium dioxide. Reaction of 93 with an appropriate nitroalkane in the presence of bifunctional squaramide catalyst 94 gave the expected optically active cyclohexenedione in 9:1 er. Subsequently, carbonyl reduction of the 1,3-dienone system led to the corresponding cyclohexadiene derivative, which upon a highly diastereoselective hydrogenation using the Crabtree's catalyst afforded the O-protected ladderanol. The natural (+)-[3]-ladderanol was finally obtained by hydrolytic cleavage of the silylated hydroxy group.
 |
| | Scheme 70 Total synthesis of (+)-[3]-ladderanol by alkylative desymmetrization of tetracyclic derivative 93 as a key step. | |
5. Conclusions and outlook
The conjugate addition of nitroalkanes to electron-poor alkenes is an old-fashioned process widely used to generate γ-nitro-functionalized derivatives to be mainly used as a gateway to γ-amino compounds. The asymmetric version of this reaction was introduced almost fifty years ago, but only in the last couple of decades has it achieved notable results in terms of diastereo- and enantioselectivity. Synthetic protocols based on the use of chiral organocatalyst are preeminent over those employing chiral metal–ligand complexes, especially regarding the practical application to targeted compounds. The success of organocatalyst-based procedures is to be found in the stability and easy handling of these purely organic molecules and their good performances even at low catalyst charge. A large majority of the organocatalysts employed for this purpose are obtained by relatively simple structural modifications of natural compounds coming from the chiral pool such as amino acids and alkaloids. Very often, the most effective catalysts are those evidencing a simple structure, as in the case of chiral diarylprolinols easily obtained from proline in a few synthetic steps. A notable advance has been introduced exploiting the use of bifunctional thiourea and squaramide-based catalysts, enabling the efficient activation of both reactants participating in the conjugate addition. The last systems developed are peptide foldamers and engineered aldolase enzymes whose primary amine and pyrrolidine amino acid residues are able to effectively catalyze this conjugate addition. On the side of the metal complexes, new chiral ligands have been developed to ensure better results, with traditional metals used for this purpose like copper, scandium and lanthanum. For some of these metal complexes the presence of additional chelating groups in the Michael acceptor is often required in order to obtain satisfactory results. Among chiral ligands employed, N,N-dioxides, diphosphines and Schiff base derivatives appear to be the most effective systems for these processes. The procedures presented in this report clearly indicate that although a high level of enantiocontrol is very often observed with these methods, the achievable diastereoselectivity is still largely unsatisfactory. The high ee values recorded for each diastereomeric couple indicate that a thermodynamic equilibration involving the acidic α-nitro hydrogen atoms could be responsible for the reduced level of diastereoselectivity observed. As a matter of fact, high diastereomeric ratios are frequently evidenced in cascade processes, leading to cyclic derivatives featuring superior sterical constraints. The selected examples of targeted applications reported in the second-last section are paradigmatic of the synthetic potential of the discussed procedures for these conjugate additions, which are expected to provide new and exciting results in the future.
Author contributions
Roberto Ballini: visualization, writing – original draft and writing – review & editing. Alessandro Palmieri: visualization, writing – original draft and writing – review & editing. Marino Petrini: conceptualization, visualization, writing – original draft and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support from the University of Camerino (FAR program) and MIUR (PRIN 2017, NATURECHEM project, grant no. 2017A5HXFC).The research network CINMPIS-Bari is also gratefully aknowledged.
References
-
W.-Q. Chen and J.-A. Ma, Stabilized nucleophiles with electron deficient alkenes, alkynes, allenes, in Comprehensive Organic Synthesis, ed. G. A. Molander and P. Knochel, Elsevier, Oxford, 2nd edn, 2014, vol. 4, pp. 1–85 Search PubMed.
-
(a)
J. L. Vicario, D. Badia, L. Carrillo and E. Reyes, Organocatalytic enantioselective conjugate addition reactions: a powerful tool for the stereocontrolled synthesis of complex molecules, Royal Society of Chemistry, Cambridge, 2010 Search PubMed;
(b)
J. L. Vicario, E. Reyes, L. Carrillo and U. Uria, Organocatalytic asymmetric nucleophilic addition to electron-deficient alkenes, in Comprehensive Organic Synthesis, ed. G. A. Molander and P. Knochel, Elsevier, Oxford, 2nd edn, 2014, vol. 4, pp. 119–188 Search PubMed.
-
(a)
R. Ballini and A. Palmieri, Nitroalkanes, Synthesis, Reactivity and Applications, Wiley-VCH, Weinheim, 2021 Search PubMed;
(b) R. Ballini, G. Bosica, D. Fiorini, A. Palmieri and M. Petrini, Conjugate additions of nitroalkanes to electron-poor alkenes: recent results, Chem. Rev., 2005, 105, 933–971 CrossRef CAS PubMed.
-
(a) D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Reduction of nitro compounds using 3d-non-noble metal catalysts, Chem. Rev., 2019, 119, 2611–2680 CrossRef CAS PubMed;
(b) M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Recent developments in the reduction of aromatic and aliphatic nitro compounds to amines, Org. Process Res. Dev., 2018, 22, 430–445 CrossRef CAS.
-
(a) R. Ballini and M. Petrini, The nitro to carbonyl conversion (Nef reaction): new perspectives for a classical transformation, Adv. Synth. Catal., 2015, 357, 2371–2402 CrossRef CAS;
(b) R. Ballini and M. Petrini, Recent synthetic developments in the nitro to carbonyl conversion (Nef reaction), Tetrahedron, 2004, 60, 1017–1047 CrossRef CAS;
(c) H. W. Pinnick, The Nef reaction, Org. React., 1990, 38, 655–792 CAS.
-
(a) R. Ballini and A. Palmieri, Formation of carbon-carbon double bonds: recent developments via nitrous acid elimination (NAE) from aliphatic nitro compounds, Adv. Synth. Catal., 2019, 361, 5070–5097 CrossRef CAS;
(b) T. C. Fessard, H. Motoyoshi and E. M. Carreira, Pd-catalyzed cleavage of benzylic nitro bonds: new opportunities for asymmetric synthesis, Angew. Chem., Int. Ed., 2007, 46, 2078–2081 CrossRef CAS.
- F. F. Fleming and Q. Wang, Unsaturated nitriles: conjugate additions of carbon nucleophiles to a recalcitrant class of acceptors, Chem. Rev., 2003, 103, 2035–2078 CrossRef CAS PubMed.
-
(a) Y. Fang, Z. Luo and X. Xu, Recent advances in the synthesis of vinyl sulfones, RSC Adv., 2016, 6, 59661–59676 RSC;
(b) T. Janecki, J. Kędzia and T. Wąsek, Michael additions to activated vinylphosphonates, Synthesis, 2009, 1227–1254 CrossRef CAS.
-
(a) T. Das, S. Mohapatra, N. P. Mishra, S. Nayak and B. P. Raiguru, Recent advances in organocatalytic asymmetric Michael addition reactions to α,β-unsaturated nitroolefins, ChemistrySelect, 2021, 6, 3745–3781 CrossRef CAS;
(b) R. Ballini, S. Gabrielli and A. Palmieri, 1,3-Dinitro alkanes: an emerging class of bidentate compounds, Eur. J. Org. Chem., 2014, 1805–1816 CrossRef CAS.
- H. Wynberg and R. Helder, Asymmetric induction in the alkaloid-catalysed Michael reaction, Tetrahedron Lett., 1975, 46, 4057–4060 CrossRef.
-
(a) M. Yamaguchi, Y. Igarashi, R. S. Reddy, T. Shiraishi and M. Hirama, Asymmetric Michael addition of nitroalkanes to prochiral acceptors catalyzed by proline rubidium salts, Tetrahedron, 1997, 53, 11223–11236 CrossRef CAS;
(b) M. Yamaguchi, T. Shiraishi, Y. Igarashi and M. Hirama, Catalytic asymmetric Michael addition of nitroalkane to enone and enal, Tetrahedron Lett., 1994, 35, 8233–8236 CrossRef CAS.
- S. Hanessian and V. Pham, Catalytic asymmetric conjugate addition of nitroalkanes to cycloalkenones, Org. Lett., 2000, 2, 2975–2978 CrossRef CAS PubMed.
- N. Halland, R. G. Hazell and K. A. Jørgensen, Organocatalytic asymmetric conjugate addition of nitroalkanes to α,β-unsaturated enones using novel imidazoline catalysts, J. Org. Chem., 2002, 67, 8331–8338 CrossRef CAS.
- A. Prieto, N. Halland and K. A. Jørgensen, Novel imidazolidine-tetrazole organocatalyst for asymmetric conjugate addition of nitroalkanes, Org. Lett., 2005, 7, 3897–3900 CrossRef CAS PubMed.
-
(a) C. E. T. Mitchell, S. E. Brenner, J. García-Fortanet and S. V. Ley, An efficient, asymmetric organocatalyst-mediated conjugate addition of nitroalkanes to unsaturated cyclic and acyclic ketones, Org. Biomol. Chem., 2006, 4, 2039–2049 RSC;
(b) C. E. T. Mitchell, S. E. Brenner and S. V. Ley, A versatile organocatalyst for the asymmetric conjugate addition of nitroalkanes to enones, Chem. Commun., 2005, 5346–5348 RSC.
- M. Malmgren, J. Granander and M. Amedjkouh, Asymmetric conjugate addition of nitroalkanes to enones with a chiral α-aminophosphonate catalyst, Tetrahedron: Asymmetry, 2008, 19, 1934–1940 CrossRef CAS.
- L. Hojabri, A. Hartikka, F. M. Moghaddam and P. I. Arvidsson, A new imidazole-containing imidazolidinone catalyst for organocatalyzed asymmetric conjugate addition of nitroalkanes to aldehydes, Adv. Synth. Catal., 2007, 349, 740–748 CrossRef CAS.
- W.-X. Liu, S.-K. Chen, J.-M. Tian, Y.-Q. Tu, S.-H. Wang and F.-M. Zhang, A triazole organocatalyst with spiropyrrolidine framework and its application to the catalytic asymmetric addition of nitromethane to α,β-unsaturated aldehydes, Adv. Synth. Catal., 2015, 357, 3831–3835 CrossRef CAS.
- Y.-C. Huang and B.-J. Uang, Remote sulfonamido group enhances reactivity and selectivity for asymmetric Michael addition of nitroalkanes to α,β-unsaturated aldehydes, Chem. – Asian J., 2014, 9, 2444–2448 CrossRef CAS PubMed.
- K. Akagawa and K. Kudo, Construction of an all-carbon quaternary stereocenter by the peptide-catalyzed asymmetric Michael addition of nitromethane to β-disubstituted α,β-unsaturated aldehydes, Angew. Chem., Int. Ed., 2012, 51, 12786–12789 CrossRef CAS PubMed.
-
(a) C. Guo, M. Saifuddin, T. Saravanan, M. Sharifi and G. J. Poelarends, Biocatalytic asymmetric Michael additions of nitromethane to α,β-unsaturated aldehydes via enzyme-bound iminium ion intermediates, ACS Catal., 2019, 9, 4369–4373 CrossRef CAS PubMed;
(b) G. Xu, A. Kunzendorf, M. Crotti, H. J. Rozeboom, A.-M. W. H. Thunnissen and G. J. Poelarends, Gene fusion and directed evolution to break structural symmetry and boost catalysis by an oligomeric C-C bond-forming enzyme, Angew. Chem., Int. Ed., 2022, 61, e202113970 CAS. For a recent review on the use of enzymes in iminium ion catalysis see:
(c) G. Xu and G. J. Poelarends, Unlocking new reactivities in enzymes by iminium catalysis, Angew. Chem., Int. Ed., 2022, 61, e202203613 CAS.
- A. R. Nödling, N. Santi, R. Castillo, M. Lipka-Lloyd, Y. Jin, L. C. Morrill, K. Świderek, V. Moliner and L. Y. P. Luk, The role of streptavidin and its variants in catalysis by biotinylated secondary amines, Org. Biomol. Chem., 2021, 19, 10424–10431 RSC.
-
(a) M. Marigo, T. Schulte, J. Franzén and K. A. Jørgensen, Asymmetric multicomponent domino reactions and highly enantioselective conjugated addition of thiols to α,β-unsaturated aldehydes, J. Am. Chem. Soc., 2005, 127, 15710–15711 CrossRef CAS;
(b) Y. Hayashi, H. Gotoh, T. Hayashi and M. Shoji, Diphenylprolinol silyl ethers as efficient organocatalysts for the asymmetric Michael reaction of aldehydes and nitroalkenes, Angew. Chem., Int. Ed., 2005, 44, 4212–4215 CrossRef CAS PubMed.
- H. Gotoh, H. Ishikawa and Y. Hayashi, Diphenylprolinol silyl ether as catalyst of an asymmetric, catalytic, and direct Michael reaction of nitroalkanes with α,β-unsaturated aldehydes, Org. Lett., 2007, 9, 5307–5309 CrossRef CAS.
- L. Zu, H. Xie, H. Li, J. Wang and W. Wang, Highly enantioselective organocatalytic conjugate addition of nitromethane to α,β-unsaturated aldehydes: three-step synthesis of optically active baclofen, Adv. Synth. Catal., 2007, 349, 2660–2664 CrossRef CAS.
- Y. Wang, P. Li, X. Liang, T. Y. Zhang and J. Ye, An efficient enantioselective method for asymmetric Michael addition of nitroalkanes to α,β-unsaturated aldehydes, Chem. Commun., 2008, 1232–1234 RSC.
-
(a) K. L. Jensen, P. H. Poulsen, B. S. Donslund, F. Morana and K. A. Jørgensen, Asymmetric synthesis of γ-nitroesters by an organocatalytic one-pot strategy, Org. Lett., 2012, 14, 1516–1519 CrossRef CAS PubMed;
(b) C.-H. Lin, B.-C. Hong and G.-H. Lee, Asymmetric synthesis of functionalized pyrrolizidines by an organocatalytic and pot-economy strategy, RSC Adv., 2016, 6, 8243–8247 RSC.
- C. Palomo, A. Landa, A. Mielgo, M. Oiarbide, Á. Puente and S. Vera, Water-compatible iminium activation: organocatalytic Michael reactions of carbon-centered nucleophiles with enals, Angew. Chem., Int. Ed., 2007, 46, 8431–8435 CrossRef CAS PubMed.
- S. K. Ghosh, Z. Zheng and B. Ni, Highly active water-soluble and recyclable organocatalyst for the asymmetric 1,4-conjugate addition of nitroalkanes to α,β-unsaturated aldehydes, Adv. Synth. Catal., 2010, 352, 2378–2382 CrossRef CAS.
- I. Mager and K. Zeitler, Efficient, Enantioselective iminium catalysis with an immobilized, recyclable diarylprolinol silyl ether catalyst, Org. Lett., 2010, 12, 1480–1483 CrossRef CAS PubMed.
- E. Alza, S. Sayalero, P. Kasaplar, D. Almaşi and M. A. Pericàs, Polystyrene-supported diarylprolinol ethers as highly efficient organocatalysts for Michael-type reactions, Chem. – Eur. J., 2011, 17, 11585–11595 CrossRef CAS PubMed.
-
(a) O. V. Maltsev, A. S. Kucherenko, I. P. Beletskaya, V. A. Tartakovsky and S. G. Zlotin, Chiral ionic liquids bearing O-silylated α,α-diphenyl (S)- or (R)-prolinol units: recoverable organocatalysts for asymmetric Michael addition of nitroalkanes to α,β-enals, Eur. J. Org. Chem., 2010, 2927–2933 CrossRef CAS;
(b) O. V. Maltsev, A. S. Kucherenko and S. G. Zlotin, Ionic polymer-supported O-trimethylsilyl-α,α-diphenyl-(S)-prolinols as recoverable organocatalysts for the asymmetric Michael reactions of carbon acids with α,β-enals, Mendeleev Commun., 2011, 21, 146–148 CrossRef CAS.
- J.-M. Tian, Y.-H. Yuan, Y.-Q. Tu, F.-M. Zhang, X.-B. Zhang, S.-H. Zhang, S.-H. Wang and X.-M. Zhang, The design of a spiro-pyrrolidine organocatalystand its application to catalytic asymmetric Michael addition for the construction of all-carbon quaternary centers, Chem. Commun., 2015, 51, 9979–9982 RSC.
-
(a) Y. Hayashi, T. Itoh and H. Ishikawa, Oxidative and enantioselective cross-coupling of aldehydes and nitromethane catalyzed by diphenylprolinol silyl ether, Angew. Chem., Int. Ed., 2011, 50, 3920–3924 CrossRef CAS;
(b) Y. Hayashi, T. Itoh and H. Ishikawa, Organocatalyst-mediated dehydrogenation of aldehydes to α,β-unsaturated aldehydes, and oxidative and enantioselective reaction of aldehydes and nitromethane catalyzed by diphenylprolinol silyl ether, Adv. Synth. Catal., 2013, 355, 3661–3669 CrossRef CAS.
-
(a) J. Vesely, G.-L. Zhao, A. Bartoszewicz and A. Córdova, Organocatalytic asymmetric nitrocyclopropanation of α,β-unsaturated aldehydes, Tetrahedron Lett., 2008, 49, 4209–4212 CrossRef CAS;
(b) Y. Hayashi, T. Yamazaki, Y. Nakanishi, T. Ono, T. Taniguchi, K. Monde and T. Uchimaru, Asymmetric nitrocyclopropanation of α-substituted α,β-enals catalyzed by diphenylprolinol silyl ether for the construction of all-carbon quaternary stereogenic centers, Eur. J. Org. Chem., 2015, 5747–5754 CrossRef CAS.
- A. Zaghi, T. Bernardi, V. Bertolasi, O. Bortolini, A. Massi and C. De Risi, One-pot, four-step organocatalytic asymmetric synthesis of functionalized nitrocyclopropanes, J. Org. Chem., 2015, 80, 9176–9184 CrossRef CAS PubMed.
- M. Šotolová, M. Kamlar, M. Remeš, P.-Y. Géant, I. Císařová, M. Štícha and J. Veselý, Enantioselective organocatalytic synthesis of 1,2,3-trisubstituted cyclopentanes, Eur. J. Org. Chem., 2021, 5080–5089 CrossRef.
- N. K. Rana, H. Joshi, R. K. Jha and V. K. Singh, Enantioselective tandem oxidation/Michael–aldol approaches to tetrasubstituted cyclohexanes, Chem. – Eur. J., 2017, 23, 2040–2043 CrossRef CAS PubMed.
- Y. Zhou, Q. Yang, J. Shen, X. Chen, Y. Peng and Y. Gong, Merging asymmetric Henry reaction with organocatalytic cascade reaction for the construction of a chiral indolizidine alkaloid skeleton, J. Org. Chem., 2015, 80, 1446–1456 CrossRef CAS PubMed.
- S. Koshino, A. Takikawa, K. Ishida, T. Taniguchi, K. Monde, E. Kwon, S. Umemiya and Y. Hayashi, Inversion of the axial information during oxidative aromatization in the synthesis of axially chiral biaryls with organocatalysis as a key step, Chem. – Eur. J., 2020, 26, 4524–4530 CrossRef CAS PubMed.
-
(a) D. Enders, M. R. M. Hüttl, C. Grondal and G. Raabe, Control of four stereocentres in a triple cascade organocatalytic reaction, Nature, 2006, 441, 861–863 CrossRef CAS PubMed;
(b) X. Zeng, Q. Ni, G. Raabe and D. Enders, A branched domino reaction: asymmetric organocatalytic two-component four-step synthesis of polyfunctionalized cyclohexene derivatives, Angew. Chem., Int. Ed., 2013, 52, 2977–2980 CrossRef CAS PubMed.
-
(a) L.-H. Zou, A. R. Philipps, G. Raabe and D. Enders, Asymmetric synthesis of fully substituted cyclopentane-oxindoles through an organocatalytic triple Michael domino reaction, Chem. – Eur. J., 2015, 21, 1004–1008 CrossRef CAS PubMed;
(b) M. Kumar, P. Chauhan, A. Valkonen, K. Rissanen and D. Enders, Asymmetric synthesis of functionalized tricyclic chromanes via an organocatalytic triple domino reaction, Org. Lett., 2017, 19, 3025–3028 CrossRef CAS PubMed.
- L.-W. Xu, J. Luo and Y. Lu, Asymmetric catalysis with chiral primary amine-based organocatalysts, Chem. Commun., 2009, 1807–1821 RSC.
- Y.-Q. Yang, X.-K. Chen, H. Xiao, W. Liu and G. Zhao, Organocatalyzed enantioselective Michael additions of nitroalkanes to enones by using primary–secondary diamine catalysts, Chem. Commun., 2010, 46, 4130–4132 RSC.
- Y. Zhou, Q. Liu and Y. Gong, Camphor-derived C1-symmetric chiral diamine organocatalysts for asymmetric Michael addition of nitroalkanes to enones, Org. Biomol. Chem., 2012, 10, 7618–7627 RSC.
- X. Gu, Y. Dai, T. Guo, A. Franchino, D. J. Dixon and J. Ye, A general, scalable, organocatalytic nitro-Michael addition to enones: enantioselective access to all-carbon quaternary stereocenters, Org. Lett., 2015, 17, 1505–1508 CrossRef CAS PubMed.
-
(a) J. Duan and P. Li, Asymmetric organocatalysis mediated by primary amines derived from cinchona alkaloids: recent advances, Catal. Sci. Technol., 2014, 4, 311–320 RSC;
(b) P. Melchiorre, Cinchona-based primary amine catalysis in the asymmetric functionalization of carbonyl compounds, Angew. Chem., Int. Ed., 2012, 51, 9748–9770 CrossRef CAS PubMed.
- J. Lv, J. Zhang, Z. Lin and Y. Wang, Enantioselective synthesis of functionalized nitrocyclopropanes by organocatalytic conjugate addition of bromonitroalkanes to α,β-unsaturated enones, Chem. – Eur. J., 2009, 15, 972–979 CrossRef CAS.
- G. K. S. Prakash, F. Wang, Z. Zhang, C. Ni, R. Haiges and G. A. Olah, Enantioselective synthesis of α-stereogenic γ-keto esters via formal umpolung, Org. Lett., 2012, 14, 3260–3263 CrossRef CAS PubMed.
- P. Kwiatkowski, K. Dudziński and D. Łyżwa, Effect of high pressure on the organocatalytic asymmetric Michael reaction: highly enantioselective synthesis of γ-nitroketones with quaternary stereogenic centers, Org. Lett., 2011, 13, 3624–3627 CrossRef CAS PubMed.
- D. Townsend, K. Shankland, A. Weymouth-Wilson, Z. Komsta, T. Evans and A. J. A. Cobb, Highly enantioselective, organocatalytic, and scalable synthesis of a rare cis,cis-tricyclic diterpenoid, Chem. – Eur. J., 2020, 26, 3504–3508 CrossRef CAS PubMed.
- A. Ueda, T. Umeno, M. Doi, K. Akagawa, K. Kudo and M. Tanaka, Helical-peptide-catalyzed enantioselective Michael addition reactions and their mechanistic insights, J. Org. Chem., 2016, 81, 6343–6356 CrossRef CAS.
- X. Garrabou, R. Verez and D. Hilvert, Enantiocomplementary synthesis of γ-nitroketones using designed and evolved carboligases, J. Am. Chem. Soc., 2017, 139, 103–106 CrossRef CAS PubMed.
- A. Kunzendorf, G. Xu, J. J. H. van der Velde, H. J. Rozeboom, A.-M. W. H. Thunnissen and G. J. Poelarends, Unlocking asymmetric Michael additions in an archetypical class I aldolase by directed evolution, ACS Catal., 2021, 11, 13236–13243 CrossRef CAS PubMed.
- T. Okino, Y. Hoashi and Y. Takemoto, Enantioselective Michael reaction of malonates to nitroolefins catalyzed by bifunctional organocatalysts, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS.
- T. Inokuma, Y. Hoashi and Y. Takemoto, Thiourea-catalyzed asymmetric Michael addition of activated methylene compounds to α,β-unsaturated imides: dual activation of imide by intra- and intermolecular hydrogen bonding, J. Am. Chem. Soc., 2006, 128, 9413–9419 CrossRef CAS PubMed.
- K. Mei, M. Jin, S. Zhang, P. Li, W. Liu, X. Chen, F. Xue, W. Duan and W. Wang, Simple cyclohexanediamine-derived primary amine thiourea catalyzed highly enantioselective conjugate addition of nitroalkanes to enones, Org. Lett., 2009, 11, 2864–2867 CrossRef CAS PubMed.
- R. Manzano, J. M. Andrés, R. Álvarez, M. D. Muruzábal, Á. R. de Lera and R. Pedrosa, Enantioselective conjugate addition of nitro compounds to α,β-unsaturated ketones: an experimental and computational study, Chem. – Eur. J., 2011, 17, 5931–5938 CrossRef CAS PubMed.
- M. Kawada, K. Nakashima, S. Hirashima, A. Yoshida, Y. Koseki and T. Miura, Asymmetric conjugate addition of nitroalkanes to enones using a sulfonamide–thiourea organocatalyst, J. Org. Chem., 2017, 82, 6986–6991 CrossRef CAS PubMed.
- C. Rabalakos and W. D. Wulff, Enantioselective organocatalytic direct Michael addition of nitroalkanes to nitroalkenes promoted by a unique bifunctional DMAP-thiourea, J. Am. Chem. Soc., 2008, 130, 13524–13525 CrossRef CAS.
- X.-Q. Dong, H.-L. Teng and C.-J. Wang, Highly enantioselective direct Michael addition of nitroalkanes to nitroalkenes catalyzed by amine-thiourea bearing multiple hydrogen-bonding donors, Org. Lett., 2009, 11, 1265–1268 CrossRef CAS PubMed.
- M. Jörres, I. Schiffers, I. Atodiresei and C. Bolm, Asymmetric Michael additions of α-nitrocyclohexanone to aryl nitroalkenes catalyzed by natural amino acid-derived bifunctional thioureas, Org. Lett., 2012, 14, 4518–4521 CrossRef PubMed.
- M. G. Núñez, A. J. M. Farley and D. J. Dixon, Bifunctional iminophosphorane organocatalysts for enantioselective synthesis: application to the ketimine nitro-Mannich reaction, J. Am. Chem. Soc., 2013, 135, 16348–16351 CrossRef.
- M. A. Horwitz, J. L. Fulton and J. S. Johnson, Enantio- and diastereoselective organocatalytic conjugate additions of nitroalkanes to enone diesters, Org. Lett., 2017, 19, 5783–5785 CrossRef CAS PubMed.
- M. Monari, E. Montroni, A. Nitti, M. Lombardo, C. Trombini and A. Quintavalla, Highly stereoselective [4+2] and [3+2] spiroannulations of 2-(2-oxoindolin-3-ylidene)acetic esters catalyzed by bifunctional thioureas, Chem. – Eur. J., 2015, 21, 11038–11049 CrossRef CAS PubMed.
-
(a) B. Vakulya, S. Varga, A. Csámpai and T. Soós, Highly enantioselective conjugate addition of nitromethane to chalcones using bifunctional cinchona organocatalysts, Org. Lett., 2005, 7, 1967–1969 CrossRef CAS;
(b) B. Vakulya, S. Varga and T. Soós,
Epi-cinchona based thiourea organocatalyst family as an efficient asymmetric Michael addition promoter: enantioselective conjugate addition of nitroalkanes to chalcones and α,β-unsaturated N-acylpyrroles, J. Org. Chem., 2008, 73, 3475–3480 CrossRef CAS PubMed.
- P. Li, Y. Wang, X. Liang and J. Ye, Asymmetric multifunctional organocatalytic Michael addition of nitroalkanes to α,β-unsaturated ketones, Chem. Commun., 2008, 3302–3304 RSC.
- J. Kwiatkowski and Y. Lu, Asymmetric Michael addition of α-fluoro-α-nitroalkanes to nitroolefins: facile preparation of fluorinated amines and tetrahydropyrimidines, Chem. Commun., 2014, 50, 9313–9316 RSC.
- S. Rajkumar, K. Shankland, G. D. Brown and A. J. A. Cobb, Organocatalytic enantioselective construction of nitrocyclohexanes containing multiple chiral centres via a cascade reaction, Chem. Sci., 2013, 3, 584–588 RSC.
- S.-Q. Chang, X. Zou, Y. Gong, X.-W. He, X.-L. Liu and Y. Zhou, Stereocontrolled construction of six vicinal stereogenic centers on a hexahydroxanthone framework through a formal [2+1+3] annulation, Chem. Commun., 2019, 55, 14003–14006 RSC.
- M. S. Manna and S. Mukherjee, Organocatalytic enantioselective formal C(sp2)–H alkylation, J. Am. Chem. Soc., 2015, 137, 130–133 CrossRef CAS PubMed.
- C.-J. Peng, J.-P. Pei, Y.-H. Chen, Z.-Y. Wu, M. Liu and Y.-K. Liu, Enantioselective organocatalytic sequential Michael-cyclization of functionalized nitroalkanes to 2-hydroxycinnamaldehydes: synthesis of benzofused dioxa[3.3.1] and oxa[4.3.1]methylene-bridged compounds, Org. Chem. Front., 2021, 9, 4217–4223 RSC.
- K. A. Agnew-Francis and C. M. Williams, Squaramides as bioisosteres in contemporary drug design, Chem. Rev., 2020, 120, 11616–11650 CrossRef CAS PubMed.
-
(a) J. P. Malerich, K. Hagihara and V. H. Rawal, Chiral squaramide derivatives are excellent hydrogen bond donor catalysts, J. Am. Chem. Soc., 2008, 130, 14416–14417 CrossRef CAS PubMed. The interatomic distance between the oxygen atoms in the nitro group particularly fits with the interatomic H–H distance in thiourea:
(b) E. M. Archer, The crystal structure of meta-dinitrobenzene, Proc. R. Soc. London, Ser. A, 1946, 188, 51–62 CrossRef CAS.
-
(a) X. Ni, X. Li, Z. Wang and J.-P. Cheng, Squaramide equilibrium acidities in DMSO, Org. Lett., 2014, 16, 1786–1789 CrossRef CAS PubMed;
(b) R. I. Storer, C. Aciro and L. H. Jones, Squaramides: physical properties, synthesis and applications, Chem. Soc. Rev., 2011, 40, 2330–2346 RSC.
- For recent reviews on synthetic applications of squaramides see:
(a) A. Biswas, A. Ghosh, R. Shankhdhar and I. Chatterjee, Squaramide catalyzed asymmetric synthesis of five- and six-membered rings, Asian J. Org. Chem., 2021, 10, 1345–1376 CrossRef CAS;
(b) A. M. Faisca Phillips, M. H. G. Prechtl and A. J. L. Pombeiro, Non-covalent interactions in enantioselective organocatalysis: theoretical and mechanistic studies of reactions mediated by dual H-bond donors, bifunctional squaramides, thioureas and related catalysts, Catalysts, 2021, 11, 569 CrossRef;
(c) X.-Q. Hou and D.-M. Du, Recent advances in squaramide-catalyzed asymmetric Mannich reactions, Adv. Synth. Catal., 2020, 362, 4487–4512 CrossRef CAS;
(d) B.-L. Zhao, J.-H. Li and D.-M. Du, Squaramide-catalyzed asymmetric reactions, Chem. Rec., 2017, 17, 994–1018 CrossRef CAS PubMed.
- W. Yang and D.-M. Du, Highly Enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts, Org. Lett., 2010, 12, 5450–5453 CrossRef CAS PubMed.
- W. Yang and D.-M. Du, Chiral squaramide-catalyzed highly diastereo- and enantioselective direct Michael addition of nitroalkanes to nitroalkenes, Chem. Commun., 2011, 47, 12706–12708 RSC.
- K. Bera and I. N. N. Namboothiri, Enantioselective synthesis of α-nitro-δ-ketosulfones via a quinine–squaramide catalyzed conjugate addition of α-nitrosulfones to enones, Chem. Commun., 2013, 49, 10632–10634 RSC.
- K. Bera and I. N. N. Namboothiri, Quinine-derived thiourea and squaramide catalyzed conjugate addition of α-nitrophosphonates to enones: asymmetric synthesis of quaternary
α-aminophosphonates, J. Org. Chem., 2015, 80, 1402–1413 CrossRef CAS PubMed.
- Z. Jia, X. Hu, Y. Zhao, F. G. Qiu, A. S. C. Chan and J. Zhao, One-pot enantioselective synthesis of 2-pyrrolidinone derivatives bearing a trifluoromethylated all-carbon quaternary stereocenter, Org. Lett., 2019, 21, 9584–9588 CrossRef CAS PubMed.
- R. Mato, E. Reyes, L. Carrillo, U. Uria, L. Prieto, R. Manzano and J. L. Vicario, Catalytic enantioselective domino Michael/transannular aldol reaction under bifunctional catalysis, Chem. Commun., 2020, 56, 13149–13152 RSC.
- Differently, the addition of 2-nitro-5-oxo esters to nitroalkenes occurs with high stereocontrol at the generated quaternary center: J. I. Martínez, L. Villar, U. Uria, L. Carrillo, E. Reyes and J. L. Vicario, Bifunctional squaramide catalysts with the same absolute chirality for the diastereodivergent access to densely functionalised cyclohexanes through enantioselective domino reactions. Synthesis and mechanistic studies, Adv. Synth. Catal., 2014, 356, 3627–3648 CrossRef.
- Ł. Albrecht, G. Dickmeiss, F. C. Acosta, C. Rodríguez-Escrich, R. L. Davis and K. A. Jørgensen, Asymmetric organocatalytic formal [2 + 2]-cycloadditions via bifunctional H-bond directing dienamine catalysis, J. Am. Chem. Soc., 2012, 134, 2543–2546 CrossRef PubMed.
-
(a) E. van Aken, H. Wynberg and F. van Bolhuis, Nitroalkanes in C-C bond forming reactions: a crystal structure of a complex of a guanidine catalyst and a nitroalkane substrate, J. Chem. Soc., Chem. Commun., 1992, 629–630 RSC;
(b) P. H. Boyle, M. A. Convery, A. P. Davis, G. D. Hosken and B. A. Murray, Deprotonation of nitroalkanes by bicyclic amidine and guanidine bases; evidence for molecular recognition within a catalytic cycle for C-C bond formation, J. Chem. Soc., Chem. Commun., 1992, 239–242 RSC.
- A. P. Davis and K. J. Dempsey, Synthesis and investigation of a hindered, chiral, bicyclic guanidine, Tetrahedron: Asymmetry, 1995, 6, 2829–2840 CrossRef CAS.
-
(a) Q. Dai, H. Huang and J. C.-G. Zhao, Highly stereoselective synthesis of trisubstituted cyclohexanols using a guanidine-catalyzed tandem Henry–Michael reaction, J. Org. Chem., 2013, 78, 4153–4157 CrossRef CAS PubMed. For the cinchona-thiourea usage in the same reaction see:
(b) Q. Dai, H. Arman and J. C.-G. Zhao, One-pot sequential organocatalysis: highly stereoselective synthesis of trisubstituted cyclohexanols, Chem. – Eur. J., 2013, 19, 1666–1671 CrossRef CAS PubMed.
- M. Vishe and J. N. Johnston, The inverted ketene synthon: a double umpolung approach to enantioselective β2,3-amino amide synthesis, Chem. Sci., 2019, 10, 1138–1143 RSC.
- D. Uraguchi, S. Nakamura, H. Sasaki, Y. Konakade and T. Ooi, Enantioselective formal α-allylation of nitroalkanes through a chiral iminophosphorane-catalyzed Michael reaction-Julia-Kocienski olefination sequence, Chem. Commun., 2014, 50, 3491–3493 RSC.
-
(a) K. Matsumoto and T. Uchida, Asymmetric induction under high-pressure. Michael addition of nitromethane to chalcone catalyzed by chiral alkaloids, Chem. Lett., 1981, 1673–1676 CrossRef CAS;
(b) A. Sera, K. Takagi, H. Katayama and H. Yamada, High-pressure asymmetric Michael additions of thiols, nitromethane, and methyl oxoindancarboxylate to enones, J. Org. Chem., 1988, 53, 1157–1161 CrossRef CAS;
(c) A. Latvala, S. Stanchev, A. Linden and M. Hesse, Unexpected change of absolute configuration in asymmetric Michael addition of methyl vinyl ketone to 2-nitrocycloalkanones, Tetrahedron: Asymmetry, 1993, 4, 173–176 CrossRef CAS.
-
P. J. Boratyński, M. Zielińska-Błajet and J. Skarżewski, Cinchona alkaloids-derivatives and applications, The Alkaloids: Chemistry and Biology, 2019, vol. 92, pp. 29–145 Search PubMed.
- H. Li, Y. Wang, L. Tang, F. Wu, X. Liu, C. Guo, B. M. Foxman and L. Deng, Stereocontrolled creation of adjacent quaternary and tertiary stereocenters by a catalytic conjugate addition, Angew. Chem., Int. Ed., 2005, 44, 105–108 CrossRef CAS PubMed.
- J. Wang, H. Li, L. Zu, W. Jiang and W. Wang, Organocatalytic, enantioselective conjugate addition of nitroalkanes to nitroolefins, Adv. Synth. Catal., 2006, 348, 2047–2050 CrossRef CAS.
- Y.-Z. Li, F. Li, P. Tian and G.-Q. Lin, Catalytic asymmetric conjugate addition of α-substituted nitro acetates to nitro olefins: enantioselective synthesis of highly substituted γ-lactams, Eur. J. Org. Chem., 2013, 1558–1565 CrossRef CAS.
- A. Clemenceau, Q. Wang and J. Zhu, Enantioselective synthesis of quaternary α-amino acids enabled by the versatility of the phenylselenonyl group, Chem. – Eur. J., 2016, 22, 18368–18372 CrossRef CAS PubMed.
-
(a) K. Maruoka, Design of high-performance chiral phase-transfer catalysts with privileged structures, Proc. Jpn. Acad., Ser. B, 2019, 95, 1–16 CrossRef CAS;
(b) S. Shirakawa and K. Maruoka, Recent developments in asymmetric phase-transfer reactions, Angew. Chem., Int. Ed., 2013, 52, 4312–4348 CrossRef CAS PubMed.
- H. Wang, Chiral phase-transfer catalysts with hydrogen bond: a powerful tool in the asymmetric synthesis, Catalysts, 2019, 9, 244 CrossRef.
-
(a) S. Colonna, H. Hiemstra and H. Wynberg, Asymmetric induction in the base-catalysed Michael addition of nitromethane to chalcone, J. Chem. Soc., Chem. Commun., 1978, 238–239 RSC;
(b) S. Colonna, A. Re and H. Wynberg, Asymmetric induction in the Michael reaction by means of chiral phase-transfer catalysts derived from cinchona and ephedra alkaloids, J. Chem. Soc., Perkin Trans. 1, 1981, 547–552 RSC.
- H. Kawai, Z. Yuan, T. Kitayama, E. Tokunaga and N. Shibata, Efficient access to trifluoromethyl diarylpyrrolines and their N-oxides through enantioselective conjugate addition of nitromethane to β,β-disubstituted enones, Angew. Chem., Int. Ed., 2013, 52, 5575–5579 CrossRef CAS PubMed.
-
(a) P. Bakó, K. Vizvárdi, Z. Bajora and L. Tõke, Synthesis and application in asymmetric synthesis of azacrown ethers derivedfrom D-glucose, Chem. Commun., 1998, 1193–1194 RSC;
(b) T. Bakó, P. Bakó, Á. Szöllõsy, M. Czugler, G. Keglevich and L. Tõke, Enantioselective Michael reaction of 2-nitropropane with substituted chalcones catalysed by chiral azacrown ethers derived from α-D-glucose, Tetrahedron: Asymmetry, 2002, 13, 203–209 CrossRef.
- P. Bakó, A. Makó, G. Keglevich, M. Kubinyi and M. Pál, Synthesis of D-mannose-based azacrown ethers and their application in enantioselective reactions, Tetrahedron: Asymmetry, 2005, 16, 1861–1871 CrossRef.
- T. Ooi, K. Doda and K. Maruoka, Highly enantioselective Michael addition of silyl nitronates to α,β-unsaturated aldehydes catalyzed by designer chiral ammonium bifluorides: efficient access to optically active γ-nitro aldehydes and their enol silyl ethers, J. Am. Chem. Soc., 2003, 125, 9022–9023 CrossRef CAS PubMed.
- T. Ooi, S. Fujioka and K. Maruoka, Highly enantioselective conjugate addition of nitroalkanes to alkylidenemalonates using efficient phase-transfer catalysis of N-spiro chiral ammonium bromides, J. Am. Chem. Soc., 2004, 126, 11790–11791 CrossRef CAS PubMed.
- T. Ooi, S. Takada, S. Fujioka and K. Maruoka,
N-Spiro chiral quaternary ammonium bromide catalyzed diastereo- and enantioselective conjugate addition of nitroalkanes to cyclic α,β-unsaturated ketones under phase-transfer conditions, Org. Lett., 2005, 7, 5143–5146 CrossRef CAS PubMed.
- T. Ooi, S. Takada, K. Doda and K. Maruoka, Highly diastereo- and enantioselective formal conjugate addition of nitroalkanes to nitroalkenes by chiral ammonium bifluoride catalysis, Angew. Chem., Int. Ed., 2006, 45, 7606–7608 CrossRef CAS PubMed.
- M.-Q. Hua, H.-F. Cui, L. Wang, J. Nie and J.-A. Ma, Reversal of enantioselectivity by tuning the conformational flexibility of phase-transfer catalysts, Angew. Chem., Int. Ed., 2010, 49, 2772–2776 CrossRef CAS PubMed.
- S. Shirakawa, S. J. Terao, R. He and K. Maruoka, Diastereo- and enantioselective conjugate addition of α-substituted nitroacetates to maleimides under base-free neutral phase-transfer conditions, Chem. Commun., 2011, 47, 10557–10559 RSC.
- S. Vellalath and D. Romo, Asymmetric organocatalysis: the emerging utility of α,β-unsaturated acylammonium salts, Angew. Chem., Int. Ed., 2016, 55, 13934–13943 CrossRef CAS PubMed.
- A. Matviitsuk, M. D. Greenhalgh, D.-J. Barrios Antúnez, A. M. Z. Slawin and A. D. Smith, Aryloxide-facilitated catalyst turnover in enantioselective α,β-unsaturated acyl ammonium catalysis, Angew. Chem., Int. Ed., 2017, 56, 12282–12287 CrossRef CAS PubMed.
- A. Matviitsuk, M. D. Greenhalgh, J. E. Taylor, X. B. Nguyen, D. B. Cordes, A. M. Z. Slawin, D. W. Lupton and A. D. Smith, Unanticipated silyl transfer in enantioselective α,β-unsaturated acyl ammonium catalysis using silyl nitronates, Org. Lett., 2020, 22, 335–339 CrossRef CAS PubMed.
- S. Shee, S. Mukherjee, R. G. Gonnade and A. T. Biju, Enantioselective synthesis of tricyclic β-lactones by NHC-catalyzed desymmetrization of cyclic 1,3-diketones, Org. Lett., 2020, 22, 5407–5411 CrossRef CAS PubMed.
-
(a) A. Schionato, A. Paganelli, C. Botteghi and G. Chelucci, Enantioselective Michael additions of nitromethane to α,β-unsaturated ketones catalyzed by transition metal complexes with chiral nitrogen ligands, J. Mol. Catal., 1989, 50, 11–18 CrossRef CAS;
(b) C. Botteghi, A. Paganelli, A. Schionato, C. Boga and A. Fava, Asymmetric Michael additions catalyzed by Ni(II) and Co(II) complexes with homochiral ligands, J. Mol. Catal., 1991, 66, 7–21 CrossRef CAS.
- K. Itoh and S. Kanemasa, Enantioselective Michael additions of nitromethane by a catalytic double activation method using chiral Lewis acid and achiral amine catalysts, J. Am. Chem. Soc., 2002, 124, 13394–13395 CrossRef CAS PubMed.
- M. Hasegawa, F. Ono and S. Kanemasa, Molecular sieve 4A work to mediate the catalytic metal enolization of nucleophile precursors: application to catalyzed enantioselective Michael addition reactions, Tetrahedron Lett., 2008, 49, 5220–5223 CrossRef CAS.
- H. Ishitani, K. Kanai, W.-J. Yoo, T. Yoshida and S. Kobayashi, A nickel-diamine/mesoporous silica composite as a heterogeneous chiral catalyst for asymmetric 1,4-addition reactions, Angew. Chem., Int. Ed., 2019, 58, 13313–13317 CrossRef CAS PubMed.
- E. Keller, N. Veldman, A. L. Spek and B. L. Feringa, Catalytic enantioselective Michael addition reactions of α-nitroesters to α,β-unsaturated ketones, Tetrahedron: Asymmetry, 1997, 8, 3403–3413 CrossRef CAS.
- M. S. Taylor, D. N. Zalatan, A. M. Lerchner and E. N. Jacobsen, Highly enantioselective conjugate additions to α,β-unsaturated ketones catalyzed by a (salen)Al Complex, J. Am. Chem. Soc., 2005, 127, 1313–1317 CrossRef CAS PubMed.
- M. Shibasaki, H. Sasai and T. Arai, Asymmetric catalysis with heterobimetallic compounds, Angew. Chem., Int. Ed. Engl., 1997, 36, 1236–1256 CrossRef.
- K. Funabashi, Y. Saida, M. Kanai, T. Arai, H. Sasai and M. Shibasaki, Catalytic asymmetric Michael addition of nitromethane to enones controlled by (R)-LPB, Tetrahedron Lett., 1998, 39, 7557–7558 CrossRef CAS.
- G. Blay, C. Incerti, M. C. Muñoz and J. R. Pedro, Enantioselective LaIII-pyBOX-catalyzed nitro-Michael addition to (E)-2-azachalcones, Eur. J. Org. Chem., 2013, 1696–1705 CrossRef CAS.
-
(a) X. Liu, L. Lin and X. Feng, Chiral N,N′-dioxide ligands: synthesis, coordination chemistry and asymmetric catalysis, Org. Chem. Front., 2014, 1, 298–302 RSC;
(b) X. Liu, L. Lin and X. Feng, Chiral N,N′-dioxides: new ligands and organocatalysts for catalytic asymmetric reactions, Acc. Chem. Res., 2011, 44, 574–587 CrossRef CAS.
- X. Yang, X. Zhou, L. Lin, L. Chang, X. Liu and X. Feng, Highly enantioselective direct Michael addition of nitroalkanes to nitroolefins catalyzed by La(OTf)3/N,N′-dioxide complexes, Angew. Chem., Int. Ed., 2008, 47, 7079–7081 CrossRef CAS.
- B. M. Choudary, K. V. S. Ranganath, U. Pal, M. L. Kantam and B. Sreedhar, Nanocrystalline MgO for asymmetric Henry and Michael reactions, J. Am. Chem. Soc., 2005, 127, 13167–13171 CrossRef CAS PubMed.
- C. Palomo, R. Pazos, M. Oiarbide and J. M. García, Catalytic enantioselective conjugate addition of nitromethane to α′-hydroxy enones as surrogates of α,β-unsaturated carboxylic acids and aldehydes, Adv. Synth. Catal., 2006, 348, 1161–1164 CrossRef CAS.
- S.-F. Lu, D.-M. Du, J. Xu and S.-W. Zhang, Asymmetric Michael addition of nitroalkanes to nitroalkenes catalyzed by C2-symmetric tridentate bis(oxazoline) and bis(thiazoline) zinc complexes, J. Am. Chem. Soc., 2006, 128, 7418–7419 CrossRef CAS PubMed.
- D. Coquière, B. L. Feringa and G. Roelfes, DNA-based catalytic enantioselective Michael reactions in water, Angew. Chem., Int. Ed., 2007, 46, 9308–9311 CrossRef PubMed.
- T. Ogawa, S. Mouri, R. Yazaki, N. Kumagai and M. Shibasaki, Intermediate as catalyst: catalytic asymmetric conjugate addition of nitroalkanes to α,β-unsaturated thioamides, Org. Lett., 2012, 14, 110–113 CrossRef CAS PubMed.
- S. Zhang, K. Xu, F. Guo, Y. Hu, Z. Zha and Z. Wang, Enantioselective copper(I/II)-catalyzed conjugate addition of nitro esters to β,γ-unsaturated α-ketoesters, Chem. – Eur. J., 2014, 20, 979–982 CrossRef CAS.
- Y. Huang, Z. Zha and Z. Wang, Copper catalyzed diastereo- and enantioselective 1,4-addition Michael reaction of 2,3-dioxopyrrolidines with nitroalkanes in aqueous media, Org. Lett., 2020, 22, 2512–2516 CrossRef CAS.
-
(a) L. Wang, Q. Zhang, X. Zhou, X. Liu, L. Lin, B. Qin and X. Feng, Asymmetric conjugate addition of nitromethane to enones catalyzed by chiral N,N′-dioxide-scandium(III) complexes, Chem. – Eur. J., 2010, 16, 7696–7699 CrossRef CAS;
(b) Z. Wang, D. Chen, Z. Yang, S. Bai, X. Liu, L. Lin and X. Feng, Highly enantioselective synthesis of α-stereogenic esters through catalytic asymmetric Michael addition of 4-oxo-4-arylbutenoates, Chem. – Eur. J., 2010, 16, 10130–10136 CrossRef CAS PubMed.
- Q. Yao, Z. Wang, Y. Zhang, X. Liu, L. Lin and X. Feng,
N,N′-Dioxide/gadolinium(III)-catalyzed asymmetric conjugate addition of nitroalkanes to α,β-unsaturated pyrazolamides, J. Org. Chem., 2015, 80, 5704–5712 CrossRef CAS PubMed.
- L. Li, S. Zhang, Y. Hu, Y. Li, C. Li, Z. Zha and Z. Wang, Highly diastereo- and enantioselective Michael addition of nitroalkanes to 2-enoyl-pyridine N-oxides catalyzed by scandium(III)/copper(II) complexes, Chem. – Eur. J., 2015, 21, 12885–12888 CrossRef CAS.
-
(a) B. S. Nagy, P. Llanes, M. A. Pericas, C. O. Kappe and S. B. Ötvös, Enantioselective flow synthesis of rolipram enabled by a telescoped asymmetric conjugate addition–oxidative aldehyde esterification sequence using in situ-generated persulfuric acid as oxidant, Org. Lett., 2022, 24, 1066–1071 CrossRef CAS PubMed. For a related preparation of baclofen see:
(b) S. B. Ötvös, P. Llanes, M. A. Pericàs and C. O. Kappe, Telescoped continuous flow synthesis of optically active γ-nitrobutyric acids as key intermediates of baclofen, phenibut, and fluorophenibut, Org. Lett., 2020, 22, 8122–8126 CrossRef PubMed.
- T. Mukaiyama, K. Ogata, I. Sato and Y. Hayashi, Asymmetric organocatalyzed Michael addition of nitromethane to a 2-oxoindoline-3-ylidene acetaldehyde and the three one-pot sequential synthesis of (−)-horsfiline and (−)-coerulescine, Chem. – Eur. J., 2014, 20, 13583–13588 CrossRef CAS PubMed.
- L.-D. Guo, X.-Z. Huang, S.-P. Luo, W.-S. Cao, Y.-P. Ruan, J.-L. Ye and P.-Q. Huang, Organocatalytic, asymmetric total synthesis of (−)-Haliclonin A, Angew. Chem., Int. Ed., 2016, 55, 4064–4068 CrossRef CAS.
- S. Bhunia, S. Chaudhuri and A. Bisai, Total syntheses of pyroclavine, festuclavine, lysergol, and isolysergol via a catalytic asymmetric nitro-Michael reaction, Chem. – Eur. J., 2017, 23, 11234–11238 CrossRef CAS PubMed.
- S. Ray, S. Mondal and S. Mukherjee, Asymmetric catalysis enantioselective total synthesis of [3]-ladderanol through late-stage organocatalytic desymmetrization, Angew. Chem., Int. Ed., 2022, 61, e202201584 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2022 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Alessandro
Palmieri
,
Alessandro
Palmieri