
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, aromaticity, charge transport in OFET devices and nonlinear optical properties of tetrathia/oxa[22]porphyrin(2.1.2.1)s: a decade of progress
Kamaljit
Singh
 * and
Paramjit
Kaur
* and
Paramjit
Kaur
Department of Chemistry, Centre of Advanced Study, Guru Nanak Dev University, Amritsar – 143 005, India. E-mail: kamaljit.chem@gndu.ac.in
First published on 4th October 2022
Abstract
Distinctive physicochemical characteristics, aromaticity–antiaromaticity relations, electrochemistry, charge transport in organic-field effect transistor devices (thin films and single crystals), supramolecular donor–acceptor dyads, photoresponsivity, and linear and nonlinear optical behaviour (2nd and 3rd-order) of rather under-appreciated classes of meso-substituted tetrathia/oxa[22]porphyrin(2.1.2.1)s explored by our group await further exploration to open new possibilities in organic electronics. This personal account discusses the above attributes including the structure dependent p-channel and ambipolar charge transport in tetrathia/oxa[22]porphyrin(2.1.2.1)s and their donor–acceptor dyads with tetracyanoethylene and C60/C70 fullerenes, respectively. Also we discuss the reverse saturation absorption, two-photon absorption and third-order nonlinear polarizability of tetrathia[22]porphyrin(2.1.2.1)s.
 Professor Kamaljit Singh (Right) Professor Paramjit Kaur(Left) | Professor Kamaljit Singh (Right) received PhD degree from Guru Nanak Dev University (GNDU) in 1989. He was postdoctoral fellow at National Institute of Organic Chemistry (CSIC), Madrid, working on carbohydrate chemistry. He has varied research interests spanning from synthetic organic/medicinal chemistry to material chemistry and is author of over 160 research papers, several reviews and book chapters. He has been an INSA-RSC and The British Council visiting fellow at UMIST, UK and Brainpool visiting fellow at Kangwon National University, South Korea. He was awarded Bronze Medal by Chemical Research Society of India (2009). He is also a recipient of Professor K. Venkataraman endowment lecture award (2015). Professor Paramjit Kaur (Left) received her PhD degree from Guru Nanak Dev University in 1990. She did a post-doctorate at the Departmento de Quimica Inorganica at Universidad Complutense de Madrid before joining as a Lecturer in the Department of Chemistry at Guru Nanak Dev University in 1997, where she is now a full Professor. She is author of over 80 research papers, review articles and book chapters. Her research interest focuses on analyte detection using fluorescence probes and nonlinear optical materials. |
1. Introduction
Porphyrinoids have attained undisputed status in supramolecular chemistry, biology and materials science owing to their outstanding structural diversity and properties attributed to their inbuilt physicochemical features, well-defined structures and π-conjugation.1 Such compounds display interesting biological, optical, photophysical, electronic and magnetic properties and find applications in the areas of non-linear optics,2a,b two-photon absorption materials,1a organic light emitting diodes,2c dye sensitized solar cells,2d organic semiconductors,2e and artificial photosynthesis,2f as materials for imaging and photodynamic therapy,2getc. Aromaticity of such porphyrinoids in terms of cyclic delocalization of the mobile electrons is a distinctive feature, which allows switching to antiaromatic structures upon suitable alteration of the macrocyclic conjugation pathway. Synthesis of porphyrinoids through modification of the macrocyclic core, and/or the size and topology of the macrocyclic framework, and substitution pattern has led to the understanding of the structure–property relationship of these important classes of macrocycles.3 Altering the macrocyclic core by replacing one or more pyrrole rings with other heterocyclic or carbocyclic rings constitutes a promising approach to tune the aromaticity, binding ability, and electronic and photochemical properties of porphyrins. Consequently, understanding of the effect of structure modification on aromaticity–antiaromaticity (Huckel and Mobius)4 relationships, and photophysical and functional properties of new classes of porphyrins is of tremendous significance.Among a variety of structurally diverse classes of porphyrins, tetrathia[22]porphyrin(2.1.2.1)s (TTPs hereinafter) and their oxygen analogues, tetraoxa[22]porphyrin(2.1.2.1)s (TOPs hereinafter) have presented themselves as efficient materials owing to flexibility in synthesis and interesting structural features compared to the tetrapyrrolic counterparts. The aromaticity of these systems is not lost even upon ring puckering under the influence of substituents at the meso-positions. Owing to the favourable energies of the frontier molecular orbitals, these porphyrinoids exhibit efficient p-channel charge transport in thin film and/or single crystal based organic field-effect transistor (OFET) devices. Further, being efficient donors, TTPs in combination with strong acceptors produce stable charge transfer type complexes. Overall, these porphyrinoids are purported to be candidates for the next generation organic electronics owing to tailorability of the structure dependent functional properties.
Porphyrins are also endowed with large optical nonlinearities, fast response time, broad-band spectral response and other superior optoelectronic attributes. However, finding new porphyrinoids capable of strong nonlinear optical behaviour for applications in fluorescence microscopy, photodynamic therapy, two-photon excitation, 3D microfabrication, sensor protection, optical data storage and optical limiting has been a very sought after objective. Particularly, there have been intense efforts for improvement of the nonlinear optical properties such as molecular second hyperpolarizability, γ, excited state absorption cross-section, σ, etc. For this purpose, due to extended π-electron conjugation, TTPs/TOPs were expected to show large excited state absorption cross-sections leading to reverse saturation absorption (RSA), high nonlinear refractive index coefficient and nonlinear absorption coefficient values and fast response times.
In this personal account, we intend to discuss various aspects related to the design, synthesis, structure, linear optical properties, aromaticity/antiaromaticity, and redox behaviour of TTPs/TOPs. In addition, we also explore the semiconducting and nonlinear optical (NLO) behaviour of these scantily studied porphyrinoids. For the sake of brevity and objectivity, this account excludes general discussion of the porphyrin chemistry, detailed synthesis and properties of all other classes of porphyrins, for which excellent review articles have already been published.1,5
2. Meso-substituted TTPs/TOPs: charge transport in thin film and single crystal organic field-effect transistor devices
2.1. Synthesis
The first synthesis of the neutral aromatic TTP 5 [also named sulphur bridged[22]annulene(2.1.2.1) in the annulene nomenclature] was reported by Cava6 and is outlined in Scheme 1. The intermediate 5,16-dihydro TTP 4 (Scheme 1) was obtained through a McMurry coupling reaction of 5,5′-methylenebis(thiophene-2-carbaldehyde) 3 using titanium tetrachloride/zinc. Compound 3 in turn was obtained from 2-bromothiophene via intermediate 2. Compound 4 was dehydrogenated6 by 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ)/hydrazine to furnish 5. The aromaticity of 5 was evidenced by its 1H NMR and UV-visible absorption spectra.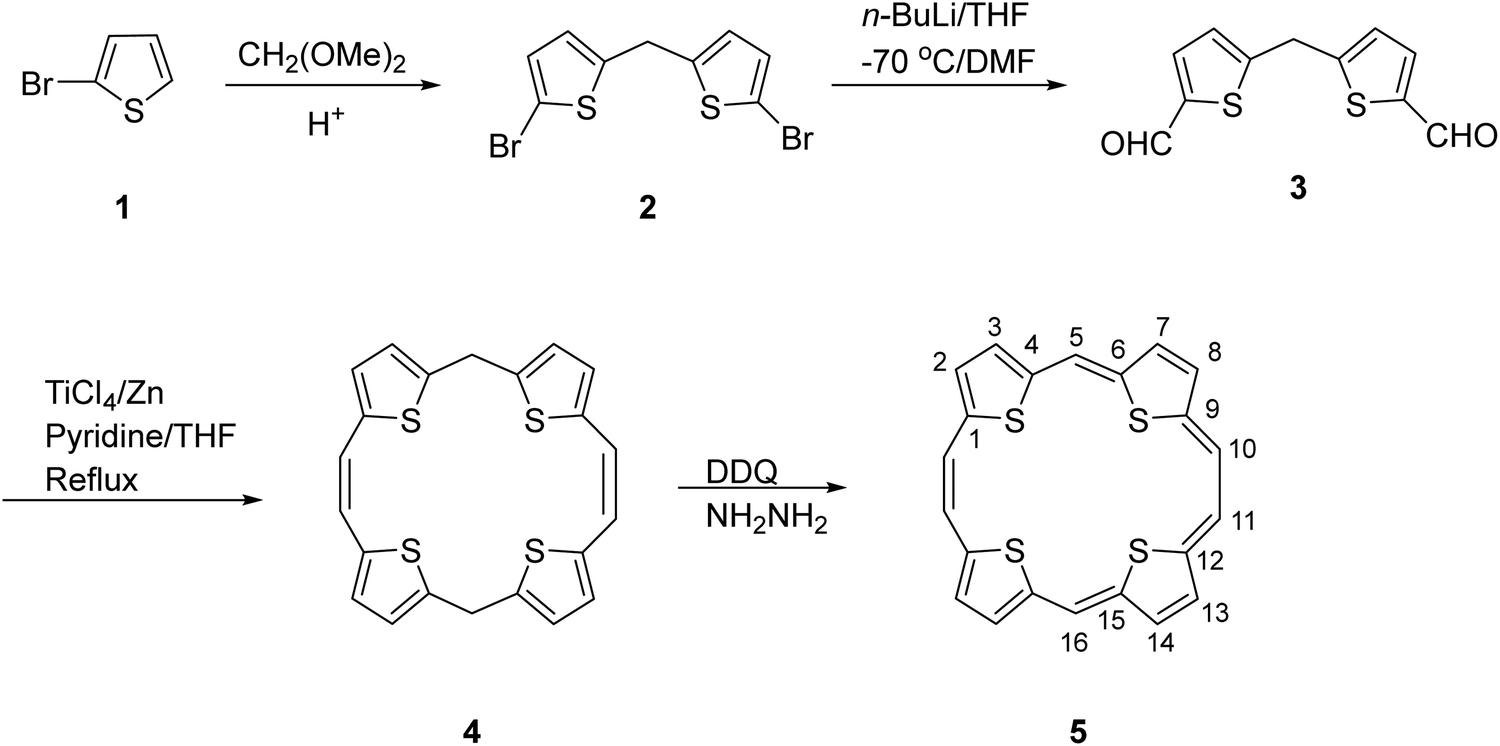 | ||
| Scheme 1 Synthesis of aromatic tetrathia[22]porphyrin(2.1.2.1) 5. | ||
The appearance of two singlets at δ 11.36 and 12.34 ppm corresponding to ethene carbons and an AB splitting pattern of the thiophene protons (δ 10.84 and 10.86 ppm) in the 1H NMR spectrum of 5 suggested the presence of strong aromatic ring currents. The considerably redshifted UV-visible absorption spectrum showed sharp Soret and Q type bands at 417, 503, 540, 579, and 771 nm. This contrasted the broad peaks observed in the absorption spectra of the antiaromatic counterparts.7 Further, 5 is comparatively less crowded, enabling it to have a planar or near-planar structure with the conjugated π-electrons around the periphery.
In the solid state, the molecules of 5 packed into a sandwich-herringbone arrangement. Further, good thermal stability was observed by thermogravimetric analysis.
The revelation of the structural features of 5 stimulated us to design a more practicable, general synthetic route to procure structurally decorated 5 (and its oxygen analogues) and to investigate their structure, optical properties, and electrochemical behaviour in order to develop potential applications as organic semiconductors and NLO materials. We also planned to investigate the charge transport behaviour of these porphyrinoids in organic field-effect transistor (OFET) devices. The reason for this was the fact that OFETs attracted considerable interest due to their potential applications in low-cost, large area and flexible electronics, such as radiofrequency identification (RFID) tags, flexible displays, etc.8 Since the performance of OFETs is influenced by such factors as the molecular structure, solid-state packing, film morphology and material stability, we investigated the molecular structure and its relationship with aromaticity, the influence of geometry of the macrocyclic core on these properties, effect of substituents at the methine carbons (meso-positions) of 5 as well as their corresponding oxygen bridged analogues (vide infra). In this context, the flexibility of appending electron-withdrawing or electron-donating groups at the meso-positions was recognized to be an attractive attribute as it modulated the charge transport properties, especially the field-effect mobility and/or current on/off ratios, of the OFET devices fabricated from these compounds. These properties have been discussed in a later section.
In order to develop a general route to the synthesis of TTPs/TOPs, we envisaged the synthesis of meso-substituted bis(heterocyclyl)methanes so that the key bis-formyl-di(thien/furan-2-yl)methane precursor is prefunctionalized at the meso-position without resorting to the synthesis of a number of meso-elaborated 2 (Scheme 1).
The most general route to the synthesis of di(thien/furan-2-yl)methane would involve acid catalysed condensation of thiophene/furan with a carbonyl compound. However, this route faces limitations in the case of aliphatic aldehydes owing to the limitation of showing side reactions such as polymerization, aldol condensation, and oxidation to carboxylic acids. Additionally the non-availability of many functionalized aldehydes would preclude the synthesis of di(thien/furan-2-yl)methane derivatives substituted with a functionalized chain at the meso-position. To avoid these limitations and realize our objective, we sought to employ a lithiation–substitution protocol9–11 (Scheme 2). This new route furnished access to a number of meso-substituted di(thien-2-yl)methanes 8 and di(furan-2-yl)methanes 10 in a synthetically useful manner.
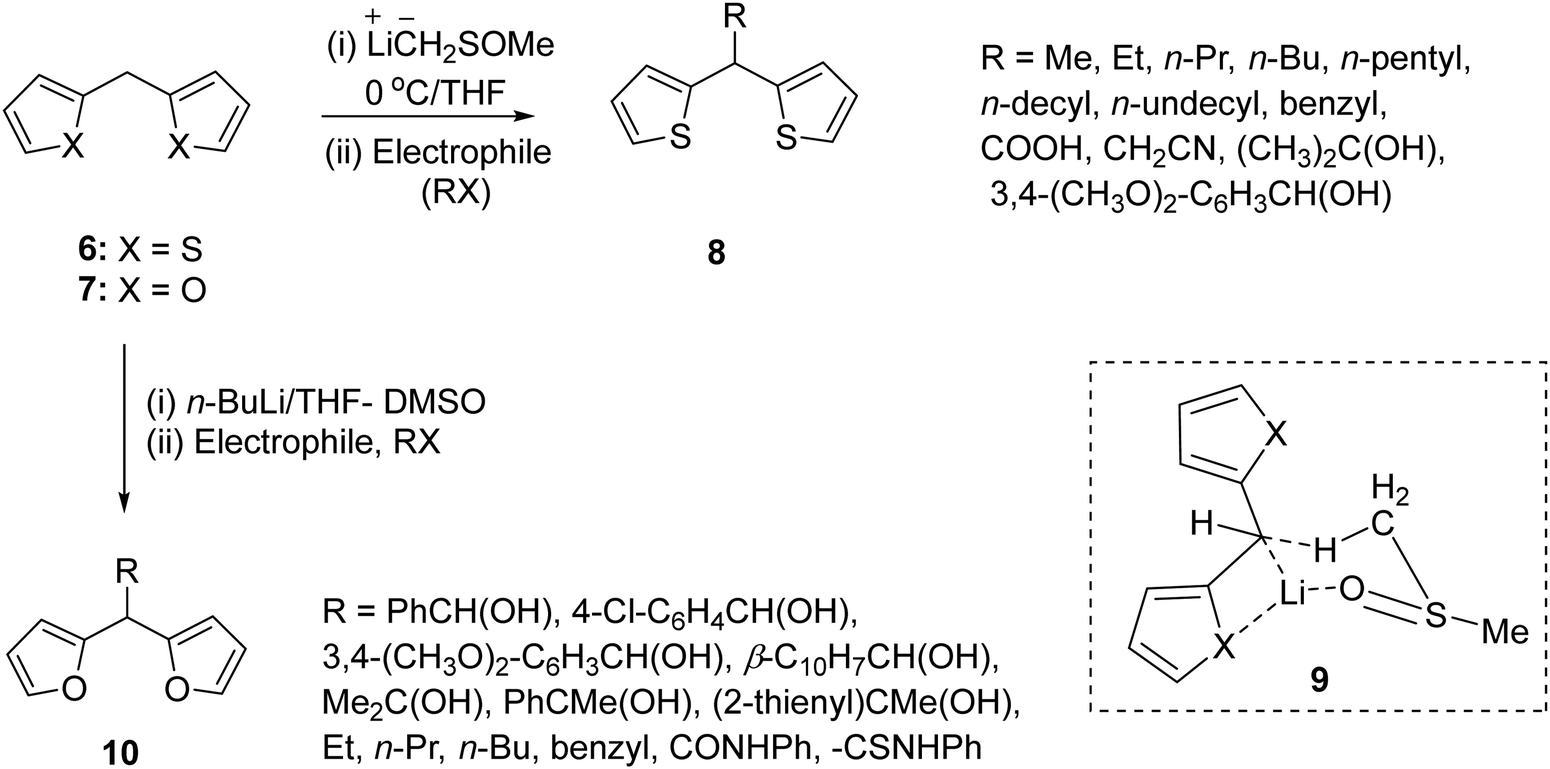 | ||
| Scheme 2 Highly regioselective synthesis of di-(thien-2-yl)methanes and di-(furan-2-yl)methanes employing a lithiation–substitution protocol. | ||
This approach allowed incorporation of those groups at the meso-positions of 8 and 10, which are otherwise difficult to append through an aldehyde condensation approach. The regioselectivity of the deprotonation (Scheme 2) of 6 and 7 was proposed via transition state 9 and the higher acidity of the protons at the meso- (pKa < 30.2) than the C-5 (pKa = 35) position.
Having successfully obtained the putative intermediates 8 and 10, diformylation expeditiously furnished (Scheme 3) the corresponding bis-formyl-(dithien-2-yl)methane 11 or bis-formyl(difuran-2-yl)methane 12 derivatives. Starting from 11, the synthesis of meso-functionalized 5,16-dihydro TTPs 13 (R = Me, Et, n-Pr, n-Bu, n-pentyl, n-decyl, n-undecyl, benzyl, COOH, or CH2CN) was achieved by using standard McMurry coupling reaction conditions. Thus, overall starting from 8, the synthesis of 13 bearing a variety of long aliphatic chains at the 5,16-positions (meso-positions) constituted a useful synthetic sequence. However, we realized that the McMurry coupling reaction was extremely sensitive to moisture and demanded starting materials of very highly purity. Notwithstanding these precautions, the yield of 13/14 was invariably low. Thus, using this synthetic strategy, while the limitations of the aldehydes were obviated, obtaining differently meso-substituted 13/14, following this route was cumbersome. Further, the need to run all the preceding synthetic steps including diformylation of appropriate 8/10 as well as McMurry coupling reactions for each compound rendered the strategy less efficient.
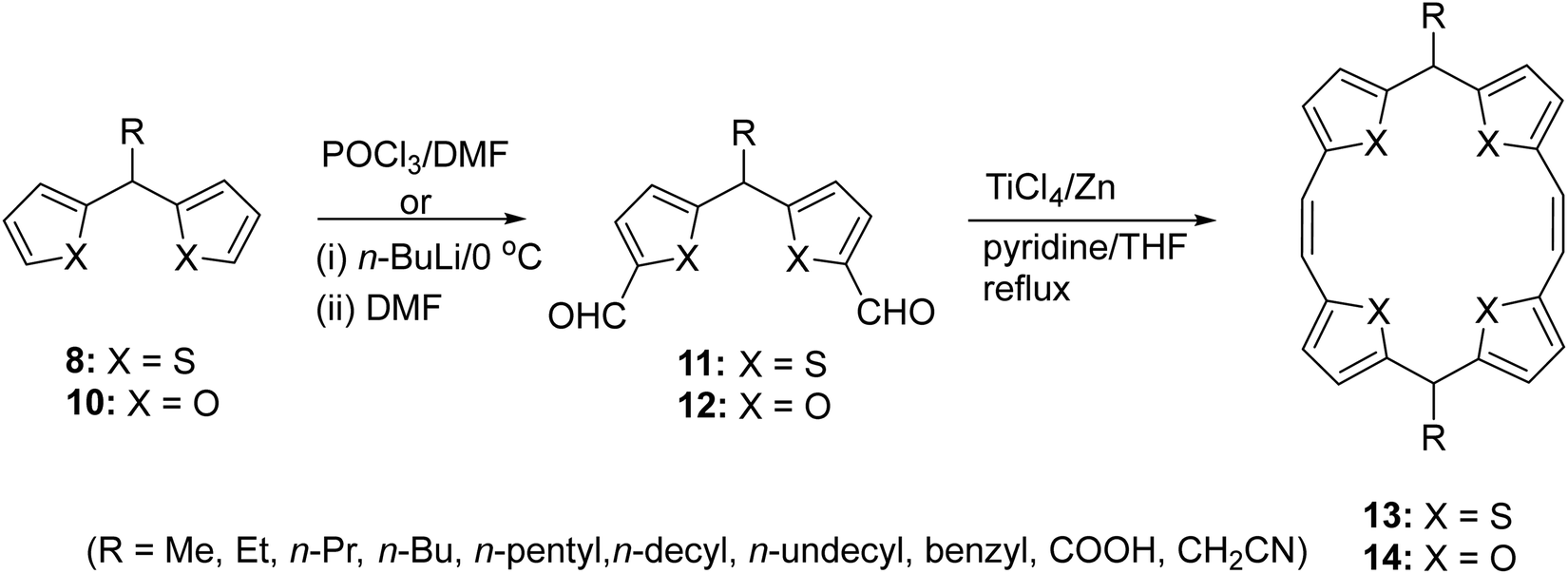 | ||
| Scheme 3 A generalized synthesis of meso-substituted 5,16-dihydro TTPs/TOPs 13/14. | ||
As an alternative approach, we developed a metalation protocol for regioselective generation of carbanion at the meso-position of 5,16-dihydro TTP 4 (Scheme 4)12 and obtained the dianionic analogue 15. Subsequent reaction with an appropriate electrophile paved the way for a number of meso-substituted 5,16-dihydro TTPs 13. This protocol avoided repetitive bis-formylation reactions of 8 as well as the need to perform low-yielding McMurry coupling reaction for each 11.
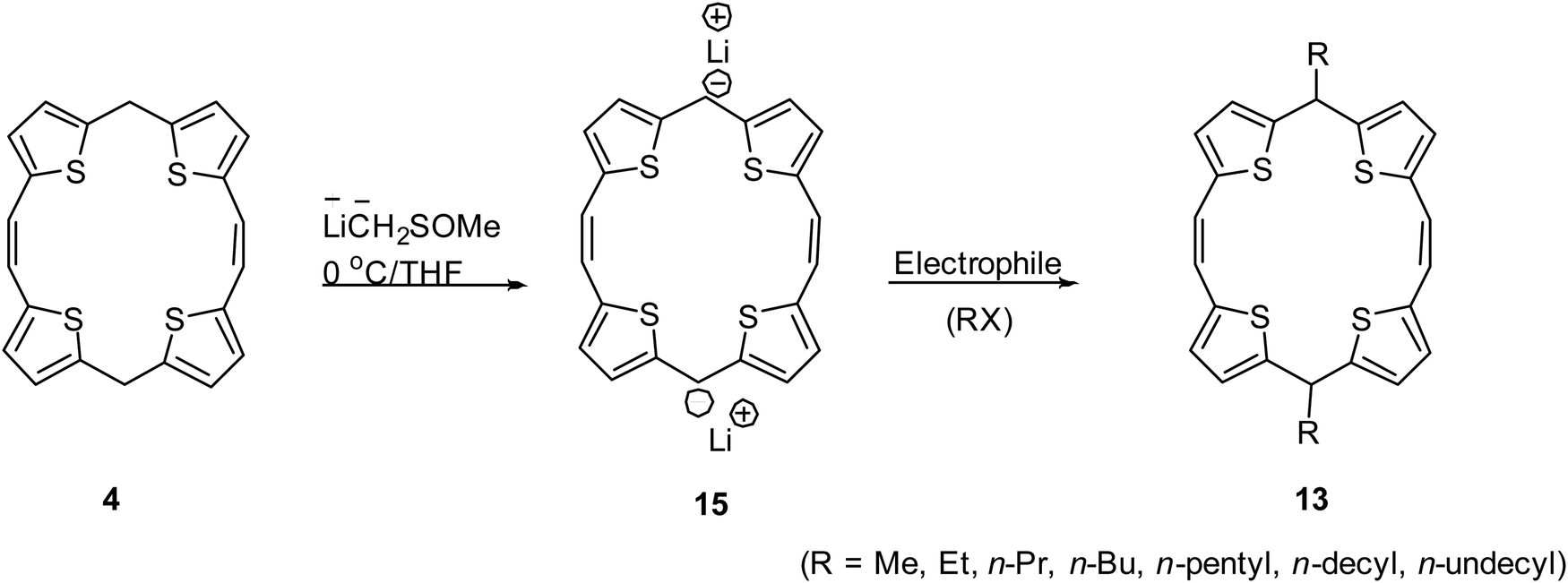 | ||
| Scheme 4 Regioselective lithiation–substitution protocol for the direct synthesis of 5,16-dihydro TTPs. | ||
However, our attempts to oxidize the nonaromatic meso-elaborated 13 (Scheme 4) with DDQ/hydrazine and a number of other reagents met with failure. This is due to the sp3 hybridization at the 5- and 16-positions of 13, and the consequent ring puckering. Presumably, the more flexible alkyl substituents at the meso-positions could not enforce planarity to rein in aromaticity in the macrocycle upon oxidation.
Appending aromatic substituents on the meso-positions of 13 or 14 appeared particularly attractive as it would have furnished derivatives required to understand the influence of meso-substituents on aromaticity. Also it would have allowed access to the hitherto elusive meso-aryl substituted analogues of 22 and 23 upon oxidation of the intermediate 5,16-dihydro analogues. In this context, the synthesis of meso-aryl substituted building blocks 16 and 17 was conveniently achieved through the known aldehyde-thiophene/furan condensation routes as described above. Facile transformation of 16 (and 17) to the corresponding diformyl derivatives 18 (and 19) was achieved by employing a lithiation–formylation sequence of reactions in the former and Vilsmeier Haack reaction conditions in the latter (Scheme 5).13,14 McMurry type coupling of these intermediates using low valent titanium furnished the 5,16-diaryldihydro- TTP 20 or TOP 21 in good yields. Subsequent two-electron oxidation using DDQ/hydrazine resulted in smooth transformation to the corresponding 5,16-diaryl TTPs 22 and TOPs 23 in a synthetically useful manner. As is evident from Scheme 5, a number of meso-aryl substituents with varying steric bulk could be incorporated in 22 and 23.
 | ||
| Scheme 5 Synthesis of neutral, aromatic 5,16-diaryl TTPs/TOPs. | ||
2.2. Structure, aromaticity and redox behaviour
As a representative example,13 in the 1H NMR spectrum of 22a, the methine carbons appeared as a singlet at δ 11.08 ppm. Additionally, the thiophene protons appeared at δ 10.38 and 10.01 ppm as an AB quartet. The coupling constant (Jvic = 4.5–5.0 Hz) of the thiophene protons in 22a was partially equalized, indicating delocalization of electrons. The protons of the meso-aryl group appeared at δ 8.46 and 8.00 ppm. Overall the chemical shifts of the protons of 22a were significantly downfield in analogy to aromatic porphyrinoids. Further, unlike the 5,16-dihydro derivative 20a, none of the protons of 22a appeared upfield to δ 7.39 ppm.On the other hand, the 1H NMR spectrum of oxygen analogue 23a14 was very characteristic as it differed from that of its sulphur analogue 22a. As expected, in the 1H NMR spectrum of 5,16-dihydro TOP 21a, the furan β-protons appeared as a pair of doublets and the singlet for the signal of methine protons was relatively upfield. The magnitude of the vicinal coupling constants was very similar to pristine furan, indicating the thermodynamically more stable double-bond character of the furan units. In contrast, in the 1H NMR spectrum of the (4n+2)π analogue 23a, not only did the AB system of the furan appear significantly downfield compared to 21a, the coupling constant was also partially equalized indicating delocalization and none of the proton signals of the porphyrin framework appeared upfield to δ 9.01 ppm. Interestingly, as a characteristic feature of 23a, one of the methine proton pairs resonated significantly upfield to appear at δ −5.42 ppm, indicating the influence of aromatic ring currents and difference in the topology of the macrocyclic framework.
As a quantitative probe of aromaticity, Nucleus-Independent Chemical Shift, NICS(1), analysis using ab initio quantum mechanical density functional theory (DFT) calculations at the B3LYP/6-31G(d) level, using the Gauge Independent Atomic Orbital (GIAO) method, revealed large negative NICS values. As a representative case, NICS(1)15 values for 23a (δ −13.17) and 22d (δ −13.30; ppm) are shown in Fig. 1. The values at the centre of the macrocyclic ring as well as individual heterocyclic and meso aromatic rings indicated a greater degree of aromaticity owing to the enhanced ring current effects of a planar geometry. The shapes of the plots of the chemical shifts vs. distance of the NICS probe, a ghost atom (termed “bq” after the ghost Banquo in Shakespeare's Macbeth) from the molecular plane furnished a proof of diamagnetic ring currents.
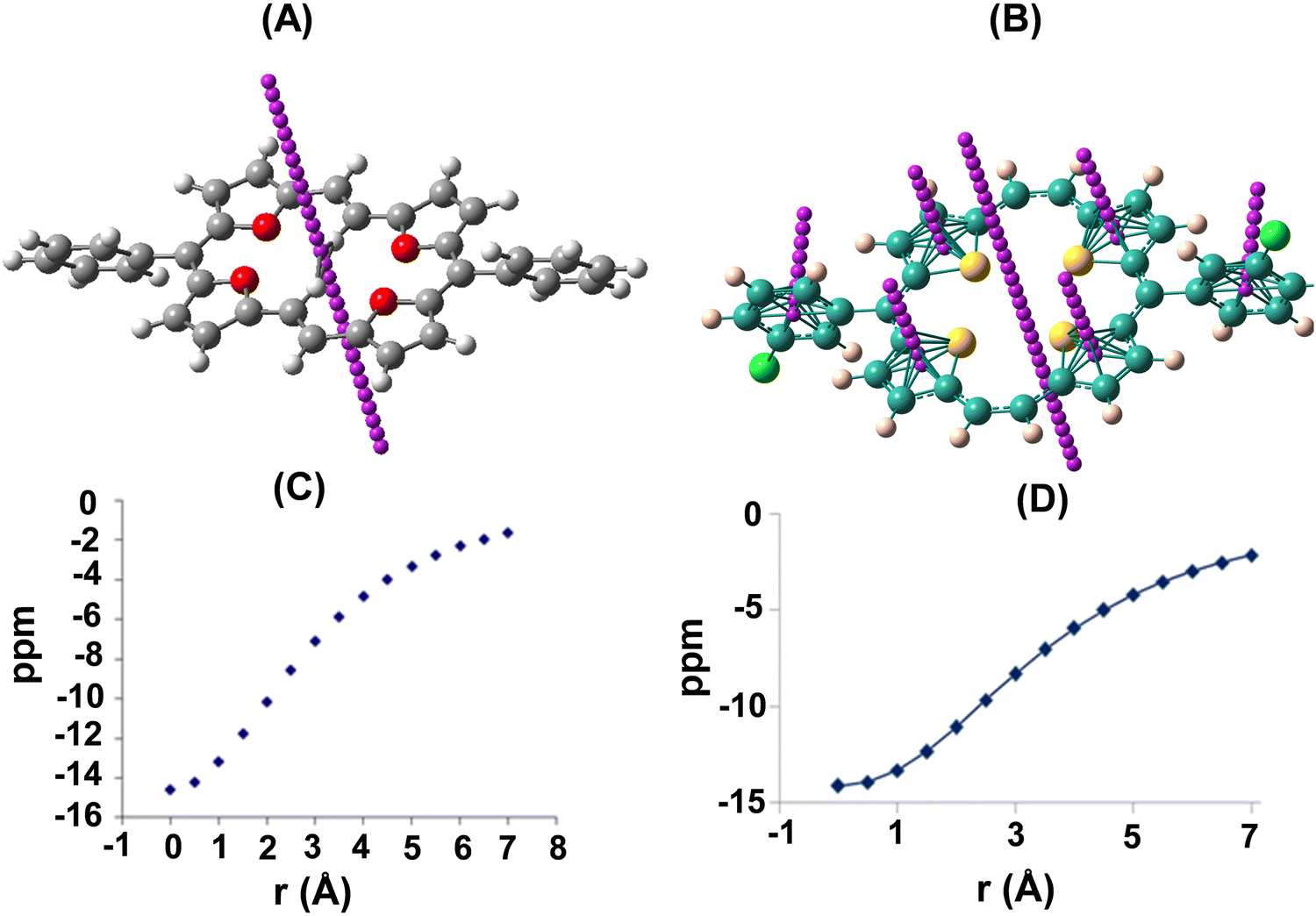 | ||
| Fig. 1 NICS analysis of 23a (A) and 22d (B). Variation of NICS ppm in 23a (C) and 22d (D) with distance (Å) from the centre of the macrocyclic plane. | ||
The FAB mass spectra of 22a and 23a and other members of the series indicated the exceptional stability of these porphyrinoids as these showed only a strong molecular ion peak (100%) without fragmentation. The good thermal stability of 22a, 23a and other analogues was also indicated by the onset thermal decomposition temperature in the thermogravimetric analyses, which was more than 370 °C. Further, these porphyrinoids showed air stability and did not require an inert atmosphere for handling/storage.
The characteristic feature of the UV-visible absorption spectrum of 22a (Fig. 2) was the presence of a sharp and strong absorption band at 429 nm (εmax = 222![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 860 dm3 mol−1 cm−1), which was absent in the corresponding dihydro derivative 20a. In addition, several weaker absorptions at longer wavelengths [519, 557, 600 and 777 nm (εmax = 2900, 9200, 57380 and 1700 dm3 mol−1 cm−1)] were also present. The absorption band at 429 nm in 22a is similar to the Soret band of porphyrins and porphycenes. The absorptions at longer wavelengths are akin to Q-bands, but with a bathochromic shift compared to porphycenes.
860 dm3 mol−1 cm−1), which was absent in the corresponding dihydro derivative 20a. In addition, several weaker absorptions at longer wavelengths [519, 557, 600 and 777 nm (εmax = 2900, 9200, 57380 and 1700 dm3 mol−1 cm−1)] were also present. The absorption band at 429 nm in 22a is similar to the Soret band of porphyrins and porphycenes. The absorptions at longer wavelengths are akin to Q-bands, but with a bathochromic shift compared to porphycenes.
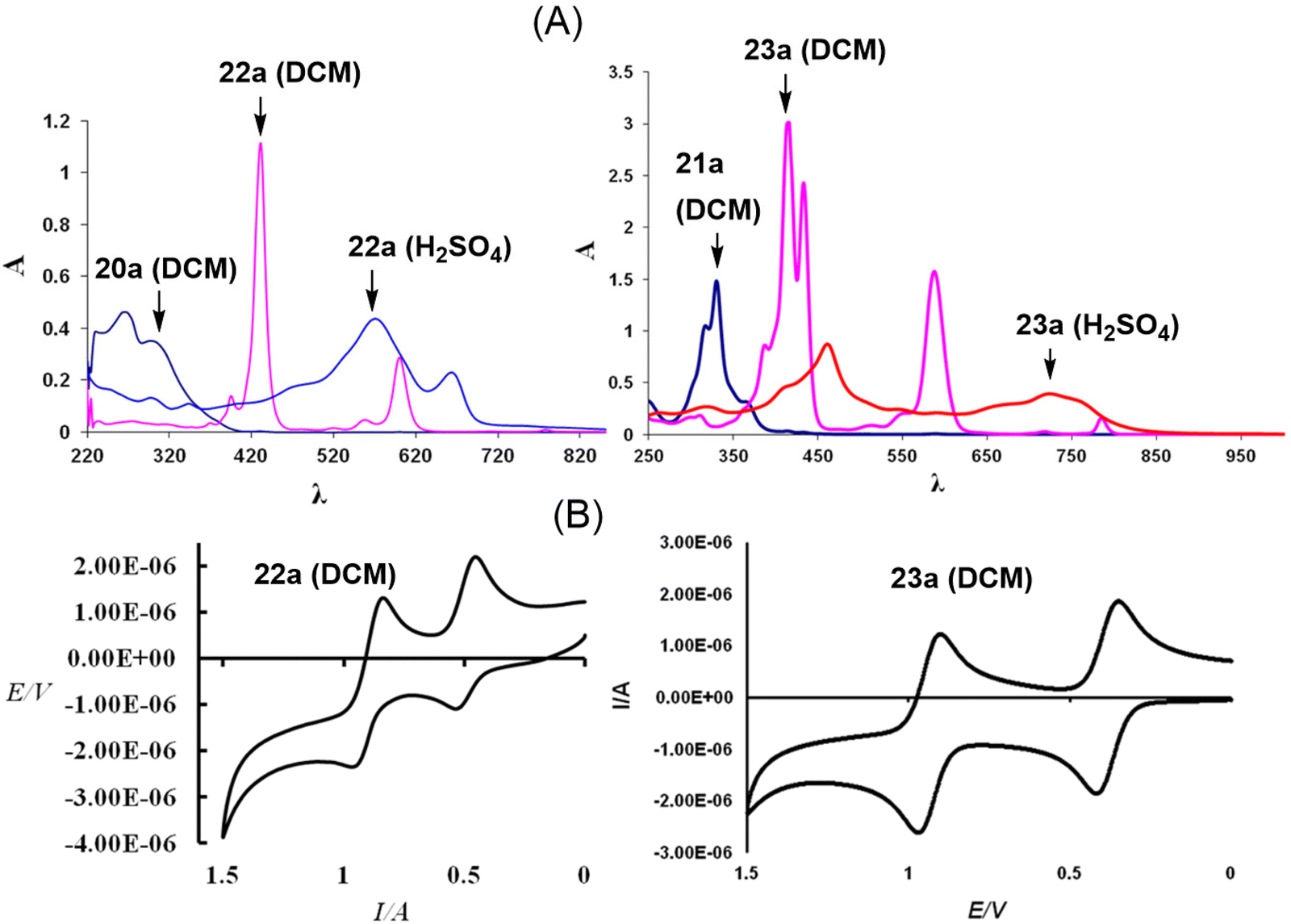 | ||
| Fig. 2 (A) UV-visible absorption spectra (20a/22a in DCM and H2SO4; 21a/23a in DCM) and (B) cyclic voltammograms of 22a and 23a (DCM, electrolyte: TBAPF6; working electrode: Pt; reference electrode: Ag/AgCl; 80 mV s−1). Reproduced with permission from ref. 13 and 14. Copyright 2011 and 2012, respectively, The Royal Society of Chemistry. | ||
On the other hand, compound 23a showed a split Soret band at 415 and 433 nm, suggesting lower symmetry (C2), compared to the analogous unsplit Soret band of the high (D2h) symmetry 22a, while the Q-type bands appeared slightly redshifted than those of porphyrins and porphycenes. Interestingly, both 22a and 23a dissolved in acids (H2SO4 and 70% HClO4) to give a reddish-violet solution, indicative of the formation of a 20π dicationic species 24/25a (Scheme 6).13,14 The UV-visible spectra of 24/25a in acid solutions showed considerable broadening of the peaks (Fig. 1), suggesting the formation of antiaromatic species having significantly positive NICS values [δ 11.29 (24a); δ 19.59 (25a)].
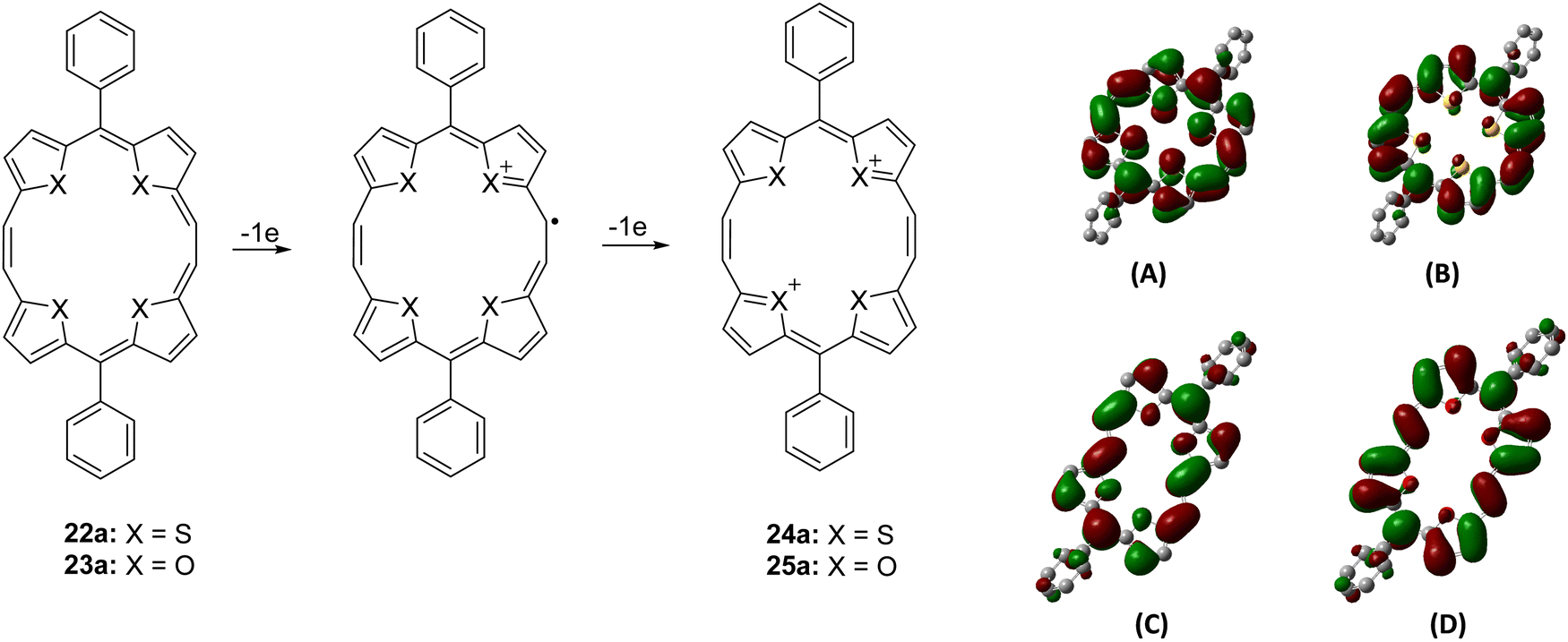 | ||
| Scheme 6 Electrochemical oxidation of 22a/23a to 24a/25a and charge density isosurfaces of the HOMOs and LUMOs of 22a (A: HOMO; B: LUMO) and 23a (C: HOMO; D: LUMO). | ||
Being 22π electron macrocycles, 22a and 22b13 are expected to lose two electrons to form the corresponding 20π dications. The cyclic voltammograms (Fig. 2) of 22a and 23a show two reversible oxidation peaks at 552.6 and 968.2 mV (22a) and 418.1 and 970.0 mV (23a) (vs. SCE), indicating the formation of 20π dicationic species 24a and 25a (Scheme 6). Further, the HOMO–LUMO gaps obtained from density functional theory (DFT) calculations in 22a (2.32 eV) and 23a (2.06 eV) were quite narrow. Visualization of the charge density isosurfaces of HOMOs and LUMOs revealed π-delocalization over the heteroannulene rings supporting the aromatic character with a greater contribution of heteroatoms to HOMOs compared to LUMOs (Scheme 6).
The single crystal X-ray structures of these porphyrinoids revealed interesting structural features. The four sulphur (or oxygen) atoms of the macrocycle 22a (or 23a) lie in one plane (Fig. 3), as were also the four thiophene/furan rings. One of the two sets of sulphur atoms, the ones intercepted by four carbon atoms, are 3.110 Å apart, while the distance between the neighbouring atoms separated by three-carbon atoms is 3.018 Å. Both of these distances were marginally shorter than twice the van der Waals radius of sulphur (3.60 Å). The distance between the two sulphur atoms with a diagonal relationship between each other is 4.333 Å. The planarity of these porphyrinoids is additionally indicated by the torsional angles (0° and 3.97°) between the sulphur (or oxygen) atoms and the mean plane of the respective macrocycles. However, in 23a, the interatomic distance between the two oxygen atoms intercepted by four carbon atoms was 4.590 Å, while the other set intercepted by a two carbon bridge was 2.485 Å apart.
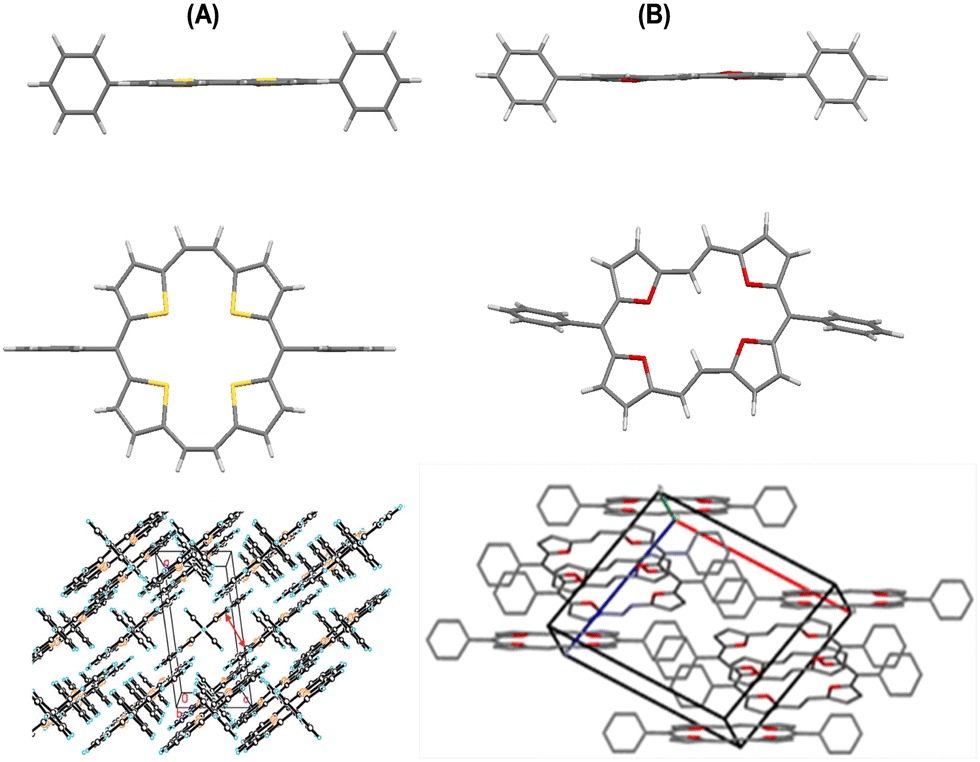 | ||
| Fig. 3 Single crystal X-ray structures: side views, top views and stacking patterns of 22a (A) and 23a (B). Reproduced with permission from ref. 13 and 14. Copyright 2011 and 2012, respectively, The Royal Society of Chemistry. | ||
In contrast to the cis geometrical arrangement of the two carbon bridge linking two thiophene rings and square arrangement of the four sulphur atoms in 22a, the oxygen bridged analogue 23a represented a trans geometry of the two carbon bridge connecting the two neighbouring furan rings as well as a rectangular arrangement of the four oxygen atoms.
Consistent with the electron delocalization in the planar aromatic porphyrinoids, bond alteration was not evident in either of 22a and 23a. The carbon–carbon distances in the thiophene or furan rings of 22a and 23a, respectively, showed the bond length relation Cα–Cβ > Cβ–Cβ, reflecting delocalization in the porphyrin ring. It is worth mentioning that the aromatic tetrathiaporphyrin dication also showed17 the above bond length relation, in spite of the fact that the four thiophene rings were tilted up and down from the mean molecular plane by 22.8° and 3.7°, respectively. The aromaticity was further verified from the NICS(1) values, which, in contrast to the isolated furan (δ −10.35 ppm) in 21a, were significantly negative (δ −20.01, −18.65, −15.15 and −16.61 ppm) for the four furan rings of the fully conjugated 23a.
The molecules of 22a stacked in a face-to-face pattern along the c-axis into columns. The intermolecular π–π interactions could be observed as the interplanar distance between the adjacent molecules in one column is 3.526 Å. Likewise, 23a stacked in a standard layer-by-layer herringbone fashion along the bc-plane with a tilt angle of 52.79° between the macrocyclic core and the bc-plane. Interestingly, the meso-phenyl substituents of 22a and 23a did not induce puckering of the macrocyclic ring, nor do these affected the aromaticity of the porphyrinoids. However, with the increasing bulk at the meso-position, slight macrocyclic ring puckering was observed in other analogues 22c, d, f, and g (Fig. 4) compared to 22a and b (Fig. 3 and 4).18,19 However, the aromaticity of these porphyrinoids was intact.
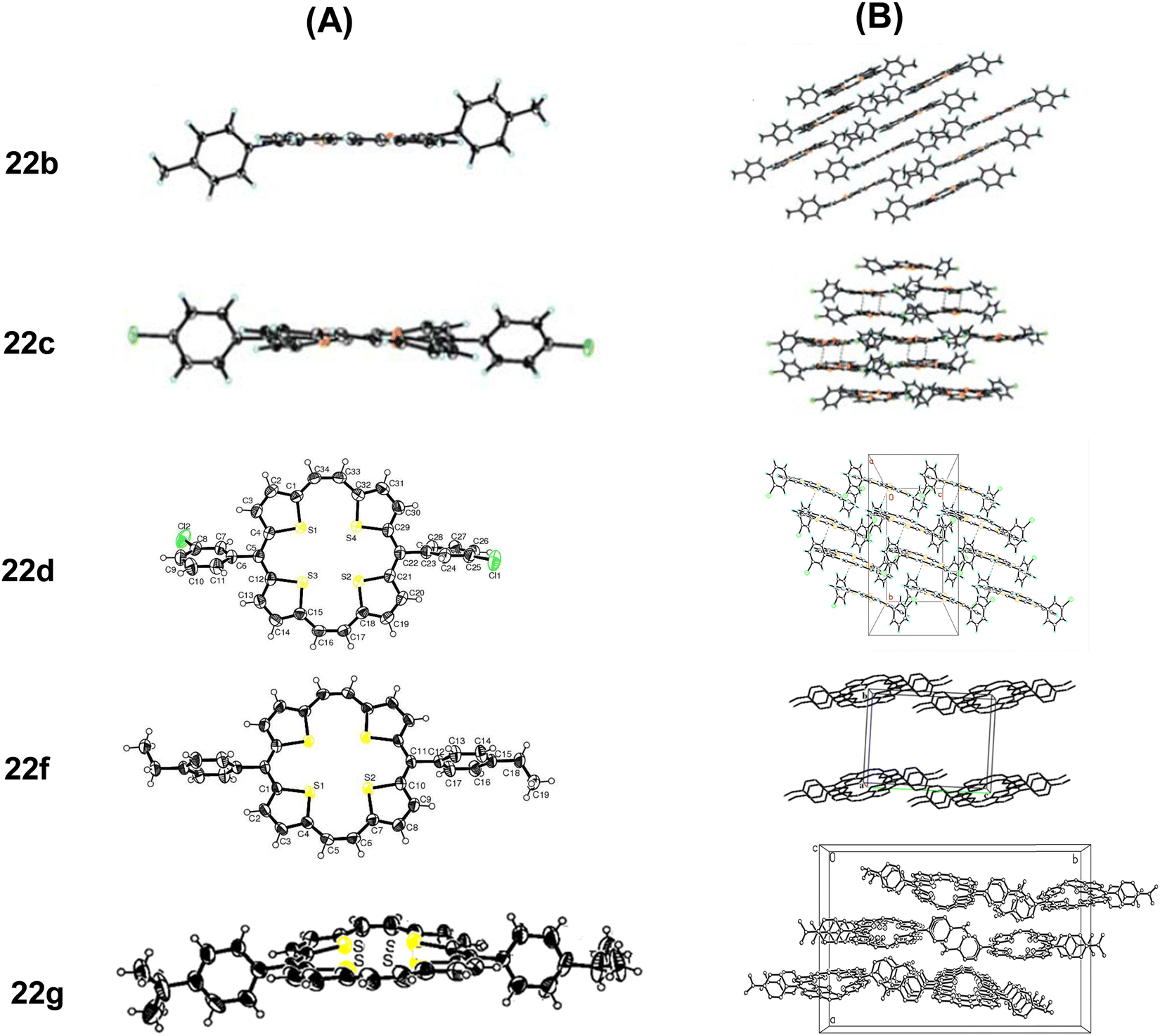 | ||
| Fig. 4 Single crystal X-ray structures (A) and stacking patterns (B) of porphyrins 22b–d, 22f and 22g bearing bulkier substituents on the meso-phenyl group. Reproduced with permission from ref. 16, 18 and 19. Copyright 2013, 2012 and 2014, respectively, The Royal Society of Chemistry. | ||
Likewise, the sulphur atoms of 22g were not coplanar and so are the thiophene rings. In the former, sulphur atoms linked through a meso-bridge lied below the twisted macrocyclic plane, compared to the other set. Although the S–S distances of the thiophene rings intercepted by the meso or the two carbon bridges were unequal, these were marginally shorter than twice the van der Waals radius of S (3.60 Å).19
Comparison of the crystal structures of a number of meso-substituted porphyrinoids revealed that hosting bulkier meso-substituents resulted in ring puckering, but aromaticity was maintained. Face-to-face crystal packing as in 22g provides an efficient π-orbital overlap and interplanar distances that facilitate charge transport. Even molecular twisting/curving did not hamper the transistor behaviour of these porphyrinoids.
2.3. Semiconducting behaviour of TTPs and TOPs
Among organic semiconductors, π-conjugated macrocyclic architectures, such as porphyrins and phthalocyanines, have attracted considerable attention due to their promising electronic, optical, and photophysical properties and self-assembling behavior.19 In recent years, ambipolar (simultaneous p- as well as n-channel charge transport) OFETs have seen unprecedented research activity. This is attributed to their applications in organic circuits and organic light emitting transistors. However, in most cases, ambipolar charge transport was realized by the construction of heterostructures comprising p-type and n-type semiconductors, e.g. bilayer, lateral and bulk heterojunctions. Only in a very few cases, an efficient single component transistor based on an ambipolar charge transport material was reported. An additional advantage of a single component ambipolar charge transport material would be simplification of the fabrication process when two unipolar materials would be replaced with a single ambipolar material.TTPs and TOPs were expected to show charge transport owing to the enhanced π–π interactions of the 22π porphyrins compared to the smaller 18π counterparts. In order to demonstrate their application potential in organic electronics, we investigated the semiconducting behaviour of a number of derivatives of 22 and 23 in thin films and single crystal OFET devices. We also explored the possibility of creating a self-assembled “molecular level heterojunction” obtained through co-crystallization of the electron rich 22 with a strong electron acceptor such as 7,7,8,8-tetracyanoquinodimethane (TCNQ) or tetracyanoethylene (TCNE).
As discussed below, 22 and 23 showed substantial p-channel (hole) transport and functioned as p-type semiconductors in thin film OFET devices. The devices displayed good reproducibility under ambient conditions. A good relationship between the molecular structure and transport properties of these aromatic porphyrinoids has been deduced.20 The charge mobility of different devices was explained in terms of the calculated molecular reorganization energies and the intermolecular transfer integrals. Further, appending substituents at the meso-positions of these porphyrin derivatives provided an understanding of the relationship between the structure and charge transport and the related attributes, such as the device on/off ratio, threshold voltage, etc. Before discussing the semiconducting behaviour of these porphyrinoids, a brief introduction to OFET devices and measurement of charge transport behaviour is presented.
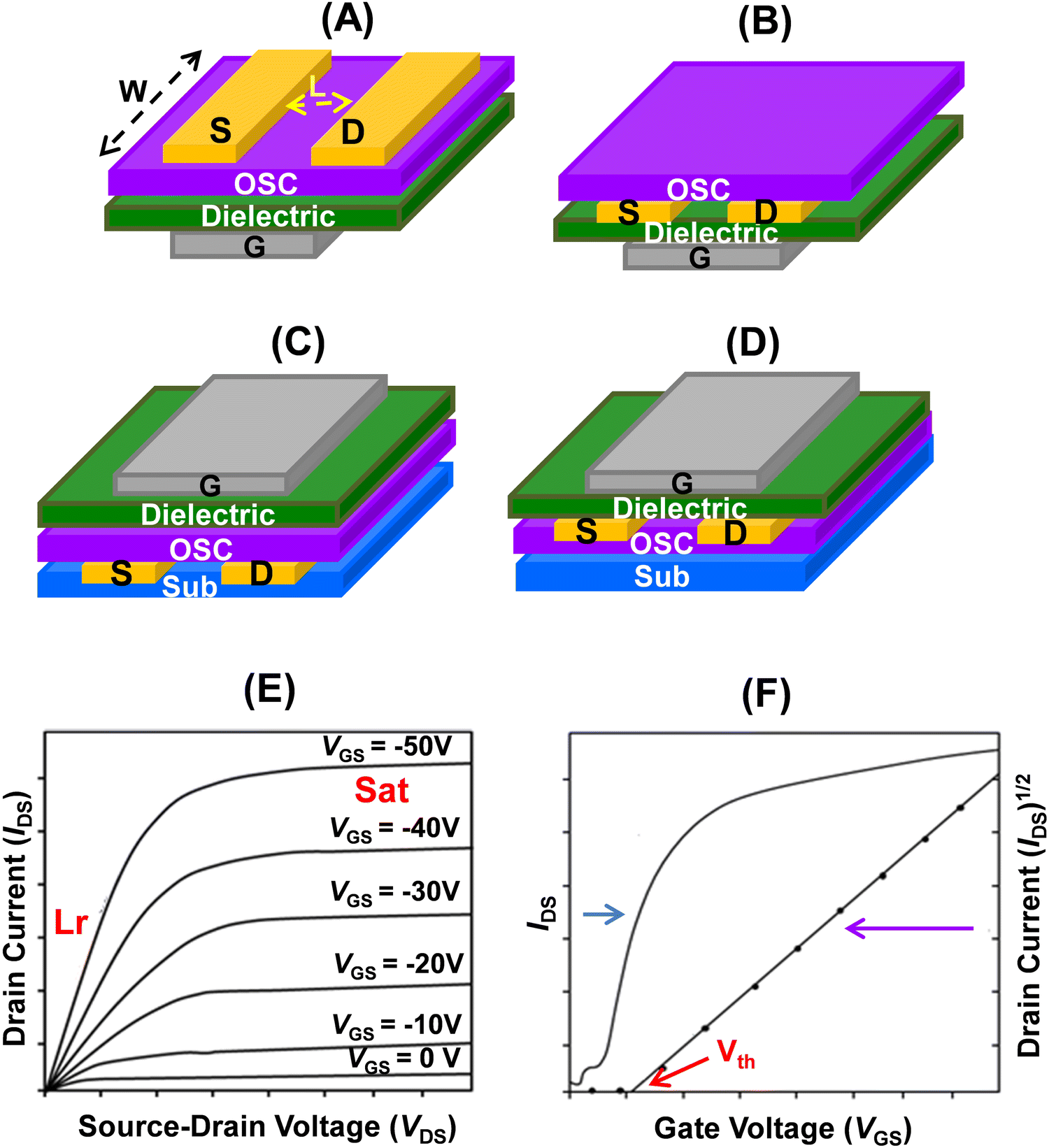 | ||
| Fig. 5 Schematics of OFET configurations. (A): BGTC; (B): BGBC; (C): TGBC; (D): TCTC (S: source; D: drain; OSC: organic semiconductor; G: gate; Sub: substrate; W: width and L: length of the conducting channel). Typical current–voltage (I–V) characteristics using arbitrary VGS values. (E): output curves (Lr: linear and Sat: saturation regimes); (F): transfer curves (Vth: threshold voltage). | ||
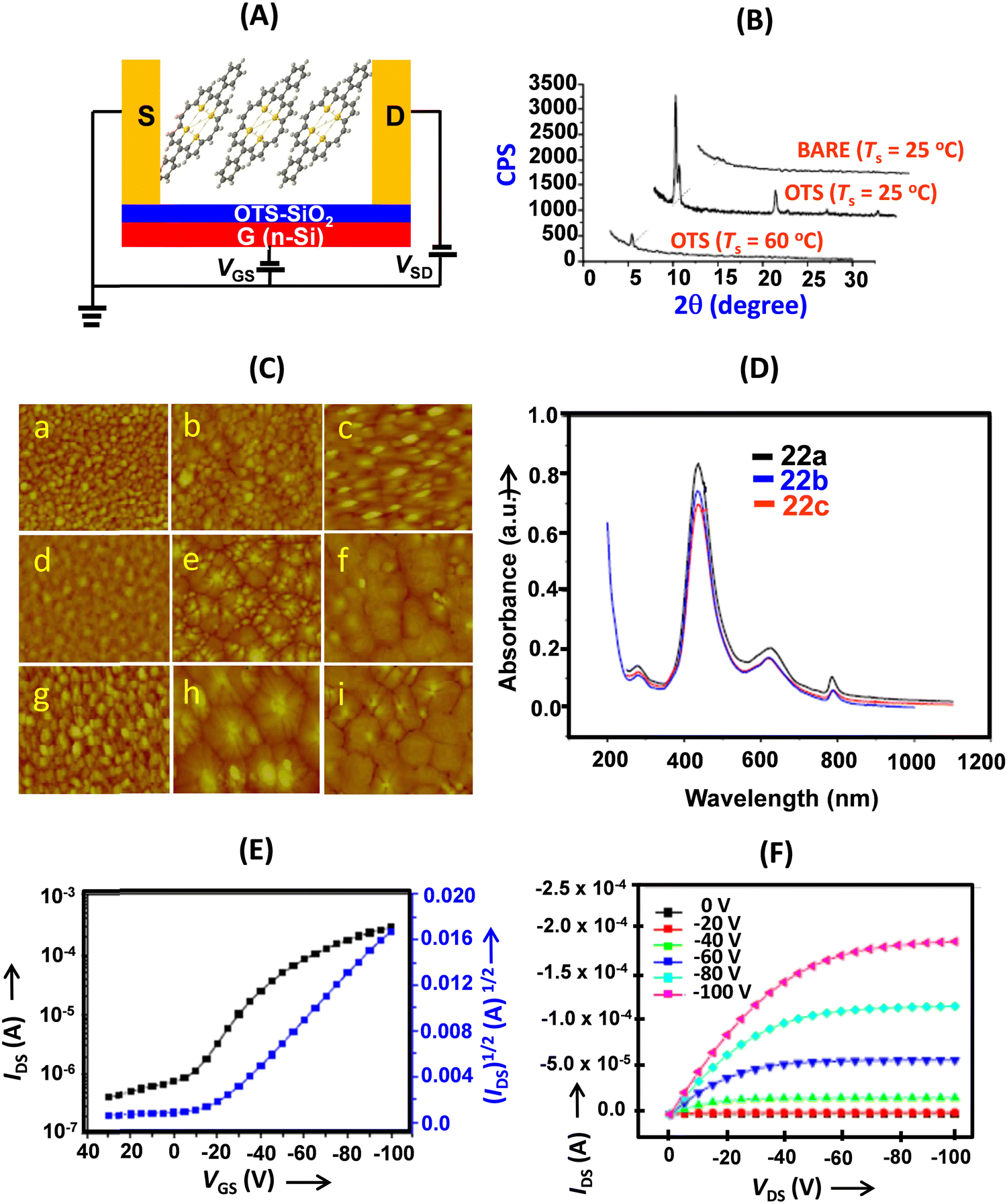 | ||
| Fig. 6 (A) Schematic of the BGTC thin film OFET devices of 22a–c; (B) X-ray diffraction patterns of the thin films of 22a deposited on SiO2/Si (BARE) and OTS/SiO2/Si substrates at different temperatures; (C) AFM images (2 × 2 μm) of 50 nm thick films of 22a (a: BARE Ts = 25 °C; b: OTS Ts = 25 °C; c: OTS Ts = 60 °C); 22b (d: BARE Ts = 25 °C; e: OTS Ts = 25 °C; f: OTS Ts = 60 °C); 22c (g: BARE Ts = 25 °C; h: OTS Ts = 25 °C; i: OTS Ts = 60 °C); (D) normalized UV-vis absorption spectra of thin films of 22a–c at room temperature; (E) transfer and (F) output characteristics of the thin film OFET devices (deposited on an OTS treated SiO2/Si substrate (Ts = 25 °C) based on 22a. Reproduced with permission from ref. 13. Copyright 2011, The Royal Society of Chemistry. | ||
Thin films of 22a and 22b were highly crystalline and the first intense reflection was observed at 2θ = 5.43, 5.09 and 5.111 (Fig. 6) for 22a, 22b and 22c, respectively, corresponding to d-spacings of 1.62, 1.73 and 1.73, which corresponded to the molecular lengths along the longer axis (1.86 Å, 2.02 Å and 2.00 Å nm for 22a–c, respectively). We deduced that the porphyrins were aligned on the substrate with their long axes inclined at angles of 60.6, 59.0 and 59.0 degrees, respectively. This was in stark contrast to the meso-unsubstituted 5, which aligned nearly perpendicular to the substrate.22,23 Furthermore, the UV-visible absorption spectra of the thin films were identical to the solution spectrum (Fig. 6).
The thin film OFET devices of 22a–c revealed them to be p-type semiconductors (Fig. 6). The highest performance (field-effect mobility) was shown by 22b in thin film devices deposited on OTS-modified SiO2 at a substrate temperature of 25 °C. It recorded field-effect mobility as high as 0.65 cm2 V−1 s−1 and the value was among the highest of thin film OFETs.
Compared to the meso-unsubstituted derivative 5, the meso-phenyl substituents (electron-withdrawing as well as electron-donating, vide infra) significantly improved the transport properties of these materials. This could presumably be attributed to the change of the molecular stacking pattern of 22a–c in the thin films. The substituents at the meso-aryl groups modulated the charge transport properties, especially the current on/off ratios, of the thin film devices. The superior charge transport was attributed to the extended π-conjugation of these cyclic conjugated molecules, which lowers the reorganization energy and leads to enhanced intermolecular π–π overlap. Compared with 5, which showed herringbone (edge-to-face) stacking in the solid state, the meso-substituents in 22 (and 23, vide infra) altered the molecular stacking pattern to face-to-face or shifted face-to-face and significantly improved the charge transport as well as switching properties of the materials based on thin films. Interestingly, the charge (hole current) transport values of these porphyrin derivatives were considerably superior to those of the corresponding α-oligofurans (mobility: 0.05–0.066 cm2 V−1 s−1) and α-oligothiophenes (0.09 cm2 V−1 s−1).24
Thin-film OFETs of 23a and 23b were fabricated on OTS or polymethylmethacrylate (PMMA)/SiO2/Si substrates in a BGTC configuration using Au source and drain electrodes. The UV-visible absorption spectrum of the thin films (Fig. 7A) corresponded to the solution spectra. The morphology of the thin films changed with temperature. Upon increasing the temperature from 20 °C to 60 °C, the grain size increased and the films turned more ordered; however, at 100 °C, cracks appeared in the films (Fig. 7B). The best charge transport (Fig. 7C and D) with a mobility as high as 0.40 cm2 V−1 s−1 was observed on thin films of 23a deposited on OTS modified SiO2 at a substrate temperature of 60 °C. The more positive value of the threshold voltage (Vth), observed for the OTS modified devices, was attributed to the assembly of charges on the OTS surface during the operation. The device of 23a based on a PMMA surface modified SiO2/Si substrate yielded a rather higher on/off ratio and a favorable Vth.
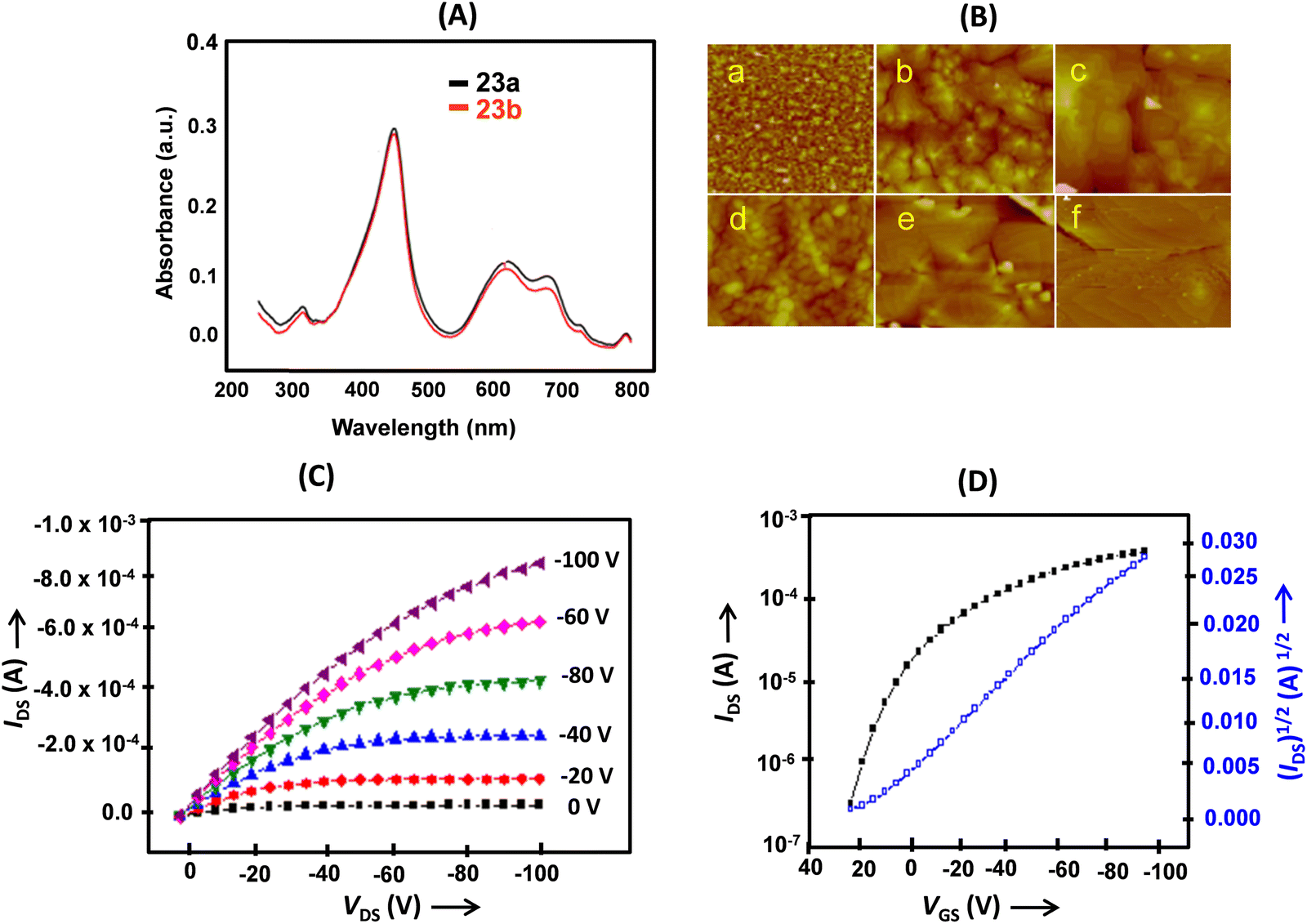 | ||
| Fig. 7 (A) Normalized UV-visible absorption spectra of thin films of 23a and 23b at room temperature; (B) AFM images of 50 nm thick films deposited on an OTS modified SiO2/Si substrate of 23a (5× 5 μm) at 25 °C (a), 60 °C (b) and 100 °C (c) and 23b (2 × 2 μm) at 25 °C (d), 60 °C (e) and 100 °C (f); (C) transfer and (D) output characteristics of the thin film OFET devices deposited on an OTS treated SiO2/Si substrate (Ts = 60 °C). Reproduced with permission from ref. 14. Copyright 2012, The Royal Society of Chemistry. | ||
Compound 22d crystalized in an orthorhombic unit cell and, unlike 22a, the sulphur atoms of the porphyrin macrocycle are not in the same plane. The two sulphur atoms linked through the meso-bridge are positioned below the plane of the otherwise twisted macrocycle. The set of sulphur atoms intercepted by a four carbon bridge is well above the mean macrocyclic plane. The shifted face-to-face stacking pattern in the layered supramolecular structure and the interplanar distance (3.4–3.6 Å) between the adjacent molecules suggested intermolecular π–π interactions and the consequent electron transport.
The thin films (50 nm) of 22d and 22e were vacuum-deposited on OTS treated SiO2/Si wafers by thermal evaporation under a pressure of 8 × 10−4 Pa. The deposition rate was gradually increased from 0.1 Å s−1 to 0.4 Å s−1 up to the first 20 nm and then maintained 0.5 Å s−1 until the thickness of the film was 50 nm. The films deposited at a substrate temperature of 20 °C essentially consisted of small grains, which grew in size at 60 °C, and resulted in a more ordered film. However, increasing the substrate temperature to 100 °C, the films cracked and lamellar crystalline grains were observed. In contrast to the thin films of 22a–b, no obvious diffraction peaks could be observed in the XRD patterns of 22d and 22e, attesting to the amorphous character of the thin films.
The band gap of 22d and 22e was estimated to be 1.57 eV from the absorption onsets. The HOMO levels of 22d and 22e were estimated to be −5.04 and −4.98 eV, respectively, by CV measurements which were comparable to the HOMO level of gold (−4.9 eV), suggesting an effective hole mobility between the electrode and the semiconductor leading to improved device performance. Visualization of the HOMOs and LUMOs (Fig. 8) of 22d and 22e revealed π-delocalization over the porphyrin macrocycle supporting the observed aromatic character and greater contribution of sulphur atoms to HOMOs compared to LUMOs. The AFM morphologies of the thin (50 nm) films of 23d and 23e vacuum-deposited on OTS treated SiO2/Si wafers are shown in Fig. 8. The films essentially consisted of small grains when the substrate temperature was 20 °C. However, at elevated temperature (60 °C), these grains turned bigger in size and resulted in more ordered thin films on the OTS/SiO2/Si substrate. Finally, when the substrate temperature was further increased to 100 °C, lamellar crystalline grains were observed. However, no obvious diffraction peaks could be observed in the XRD patterns, which indicated the amorphous character of these thin films.
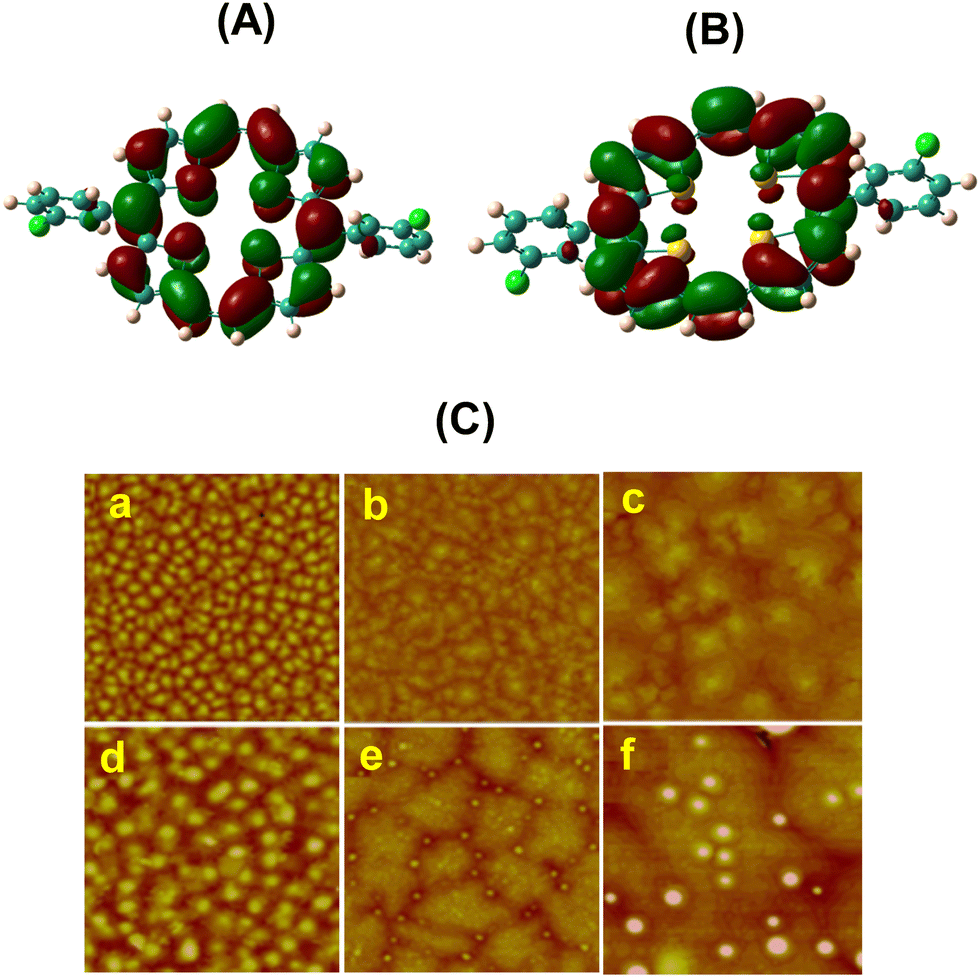 | ||
| Fig. 8 The electron densities of the HOMO (A) and LUMO (B) of 23d (isosurface value = 0.04); (C) AFM images: 50 nm thick films (2 × 2 μm) of 23d deposited on an (a) OTS/SiO2/Si substrate, Ts = 20 °C; (b) OTS/SiO2/Si substrate, Ts = 60 °C; and (c) OTS/SiO2/Si substrate, Ts = 100 °C; and 50 nm thick films (5 μm × 5 μm) of 23e deposited on an (d) OTS/SiO2/Si substrate, Ts = 20 °C; (e) OTS/SiO2/Si substrate, Ts = 60 °C; and (f) OTS/SiO2/Si substrate, Ts = 100 °C. Reproduced with permission from ref. 18. Copyright 2012, The Royal Society of Chemistry. | ||
Devices of 23d fabricated at 100 °C exhibited the best OFET performance (μ = 0.23 cm2 V−1 s−1) with a much improved on/off ratio of 5 × 105. Interestingly, the mobility of these devices did not change significantly upon exposure to air even up to ten days, although the on/off ratio decreased by one order of magnitude. Small molecule based amorphous thin films with a relatively high mobility are still rare. We believe that the extended π-conjugation in these porphyrinoids contributes to efficient intermolecular coupling in the thin films lacking molecular structural ordering.
As a logical extension of this work, we planned to investigate the influence of steric bulk at the meso-positions of TTPs. Thus, we synthesized porphyrins 22f–h bearing p-Et-C6H4, p-iPr-C6H4, and p-tBu-C6H4 substituents at 5- and 16-positions.19 We observed that the solubility of these porphyrinoids was highly dependent on the meso-substitution. The solubility significantly decreased from 22f to 22h bearing p-Et-C6H4, p-iPr-C6H4, and p-tBu-C6H4 substituents at the two meso-positions. This provided a guideline for designing meso-substituted porphyrins with the possibility of solution processible OFETs.
Among 22f–h, the thin film OFETs (Fig. 9) of 22f exhibited the highest OFET performance at 100 °C. While the hole mobility averaged at 0.32 cm2 V−1 s−1 (100 °C), the on/off ratio was 1 × 106 (25 °C). It also showed a very favourable threshold voltage (VT) of −14 V.
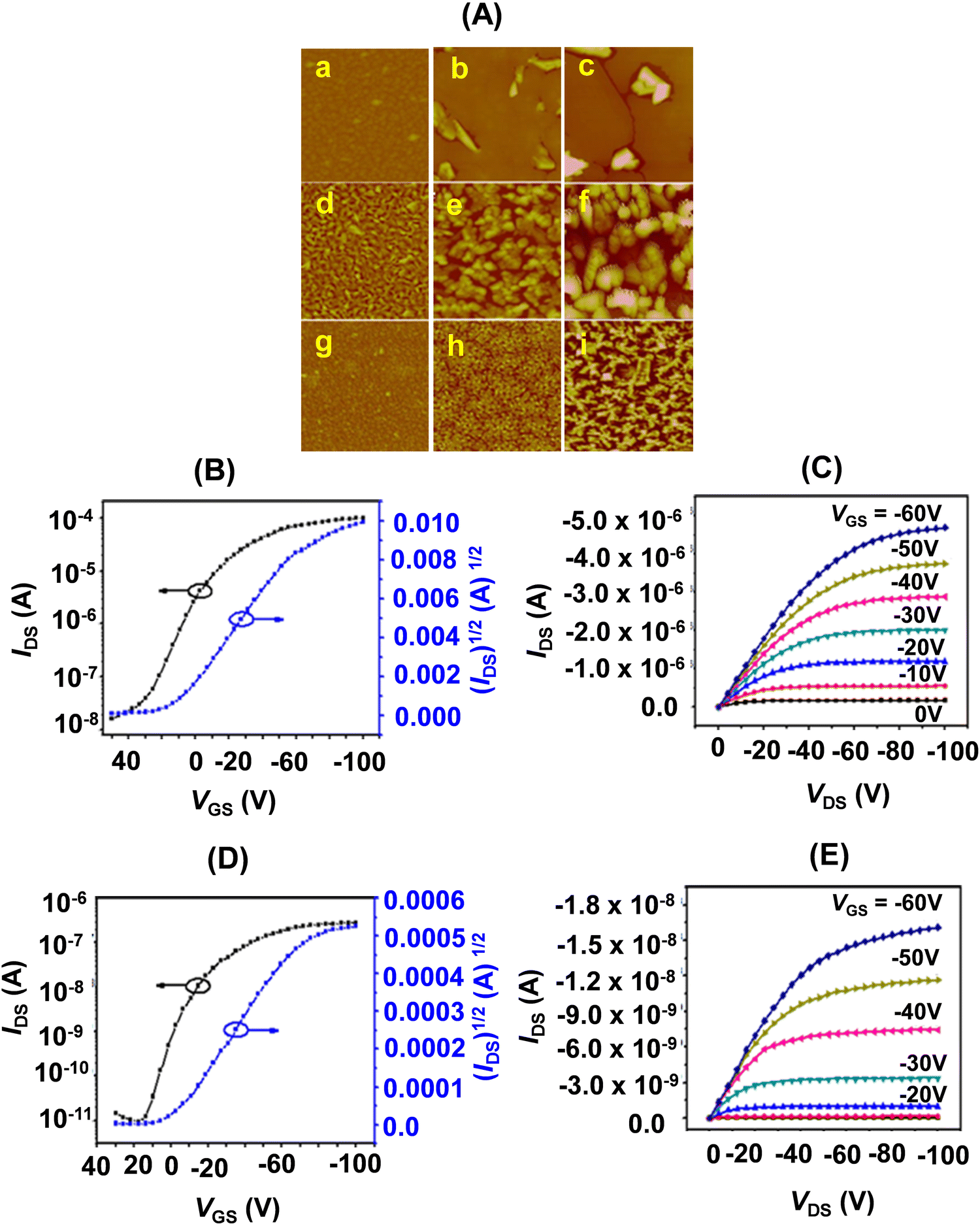 | ||
| Fig. 9 (A) AFM images (5 × 5 μm) of 50 nm thick films of 22f–h, (a, d and g) Ts = 25 °C; (b, e and h) Ts = 60 °C; (c, f and i) Ts = 100 °C. (B/D) Transfer and (C/E) output characteristics of the thin film OFET devices of 22f/22g (deposited on OTS treated SiO2/Si substrate (Ts = 25 °C). Reproduced with permission from ref. 19. Copyright 2014, The Royal Society of Chemistry. | ||
Interestingly, a comparison of the field-effect mobility (Table 1) of these porphyrinoids revealed that substituting a phenyl group at the meso position of 5 to produce 22a (Ar = Ph) and 22b (Ar = p-Me-Ph) led to nearly 12.5- and 31.5-fold, respectively, increases in the field effect mobility. This is attributed to the difference in the crystal packing pattern, leading to effective π–π interactions. Further, substituting the meso-phenyl group with p-Et, p-iPr, and p-tBu to produce 22f, 22g, and 22h, respectively, led to significant deterioration of the field-effect mobility. The molecules of these compounds were arranged in a shifted face-to-face fashion in the crystal packing structures. Likewise, a p-F, p-Cl or m-Cl substituent on the meso-phenyl group led to a decrease in the field-effect mobility compared to the unsubstituted Ph 22a or p-Me-Ph 22b. Except for 5 (meso-phenyl) and 22b/22f (meso-p-Me-Ph/p-Et-Ph), the thin films of all TTPs were amorphous (Table 1), which seems to be a distinctive feature affecting the field-effect mobility of this class of porphyrinoids.
| Ar | Crystal packing mode | Thin film type | T sub (°C) | μ | I on/Ioff | V T (V) | |
|---|---|---|---|---|---|---|---|
| a cm2 V−1 s−1. b Dimers arranged in a herringbone fashion. c Herringbone. | |||||||
| 5 | — | Edge-to-faceb | Crystalline | 18 | 0.0222 | 103 | — |
| 22a | Ph | Face-to-face | Crystalline | 25 | 0.2913 | 1.34 × 103 | −12.9 |
| 22b | p-Me-Ph | Slipped face-to-face | Crystalline | 25 | 0.6313 | 3 × 102 | −7.47 |
| 22f | p-Et-Ph | Face-to-face | Crystalline | 100 | 0.3219 | 6 × 104 | −14.0 |
| 22g | p-iPr-Ph | Shifted face-to-face | Amorphous | 25 | 0.001419 | 3 × 104 | 3.0 |
| 22h | p-tBu-Ph | Shifted face-to-face | Amorphous | 25 | 0.008719 | 9 × 102 | −5.8 |
| 22e | p-F-Ph | Shifted face-to-face | Amorphous | 100 | 0.01218 | 2.4 × 105 | −56.1 |
| 22c | p-Cl-Ph | Shifted face-to-face | Amorphous | 60 | 0.024518 | 3.03 × 104 | −50.1 |
| 22d | m-Cl-Ph | Shifted face-to-face | Amorphous | 100 | 0.2318 | 5.27 × 105 | −17.1 |
| 23a | Ph | Layer by layerc | Amorphous | 60 | 0.4016 | 3.5 × 103 | −20 |
| 23b | p-Me-Ph | Layer by layerc | Amorphous | 100 | 0.11 | 1.0 × 102 | −24.4 |
The oxygen analogue 23a (meso-Ph), due to a layered herringbone crystal packing, seems to maintain rather amorphous thin films and yielded high field-effect mobility (Table 1), while 23b (meso-p-Me-Ph) showed less, yet good field-effect mobility.
To examine the relationship between the structure and charge transport behaviour of these porphyrinoids, with an eye on the rational design of organic semiconductors from this category of compounds, single crystal organic field effect transistors (SCOFETs) based on representative members (22a–c) of these porphyrinoids were fabricated and their charge transport behaviour was analysed and corroborated by theoretical calculations of their molecular reorganization energies and the maximum intermolecular transfer integrals. Fabricating SCOFET devices ensured elimination of possible defects and morphological issues of the thin film devices as discussed in the preceding sections.
While the porphyrin ring of 22a, having meso-Ph groups maintaining coplanar conformation with S atoms and four thiophene rings, deviates from the mean plane only by less than 0.1 Å (Fig. 3), the deviation in 22b having p-Me-Ph groups at the meso positions was greater (dihedral angle of 3–4°) (Fig. 3). In 22c, however, the bulkier p-Cl-Ph groups at the meso positions made it to adopt significantly twisted conformation (dihedral angle of 13.7–22.2°) (Fig. 4). These compounds showed face-to-face packing in the crystals, which was distinctively different from the dimerized herringbone packing of the meso-unsubstituted analogue 5.23
The theoretical calculations on the two structural isomers of these compounds, namely, the parallel and staggered forms (meso substituents w.r.t. the macrocyclic ring), revealed only a small stabilization (10 meV) of the latter relative to the former, which meant that 22a and b may adopt a parallel conformation, whereas 22c would prefer a staggered conformation. However, the HOMOs and LUMOs of 22a–c (Fig. 10) are mainly localized on the cyclic conjugated pathway, negating the effect of the meso-Ph substituents on the HOMO and LUMO charge densities. The stability of these porphyrins was demonstrated from the calculated ionization energies (both adiabatic and vertical). These were in the range (from 5.68 eV to 6.78 eV) recorded for air stable p-channel materials.25 The calculated reorganization energy for both parallel and staggered conformations of the compounds revealed a greater influence of the molecular conformation than the modification of the meso-substituent. The latter however seems to be the major factor affecting the charge transport properties through alteration of the molecular packing and transfer integrals.
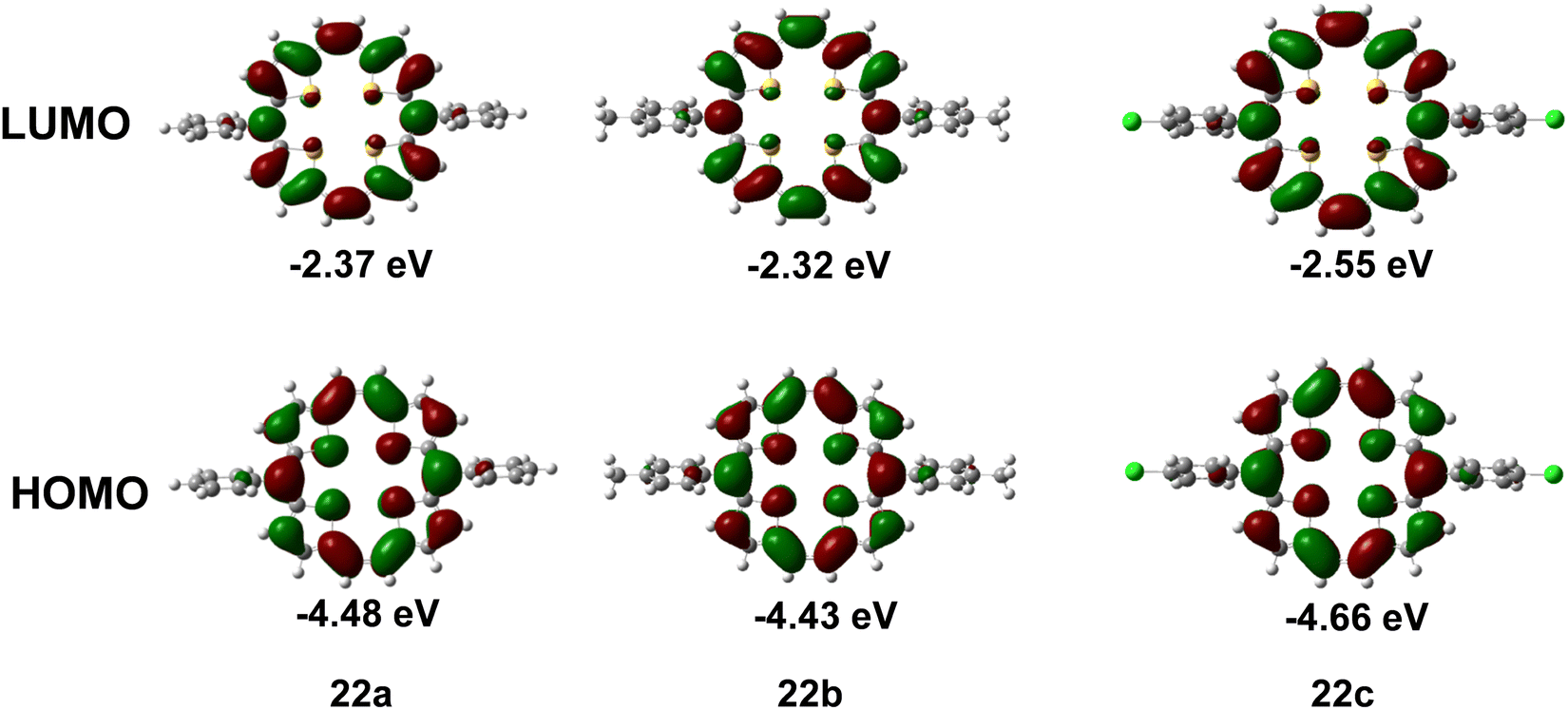 | ||
| Fig. 10 HOMO and LUMO energies of 22a–c. | ||
The SCOFETs (Fig. 11) of 22a–c exhibited typical p-type characteristics under ambient conditions. Single crystal (top contact) devices of 22a fabricated using the drop casting method showed the highest mobility of 0.7 cm2 V−1 s−1 with an average mobility of 0.4 cm2 V−1 s−1, while the field-effect mobility was in the range of 0.03 to 0.4 cm2 V−1 s−1 for 22b, and for 22c the mobility was in the range of 0.005 to 0.2 cm2 V−1 s−1.16
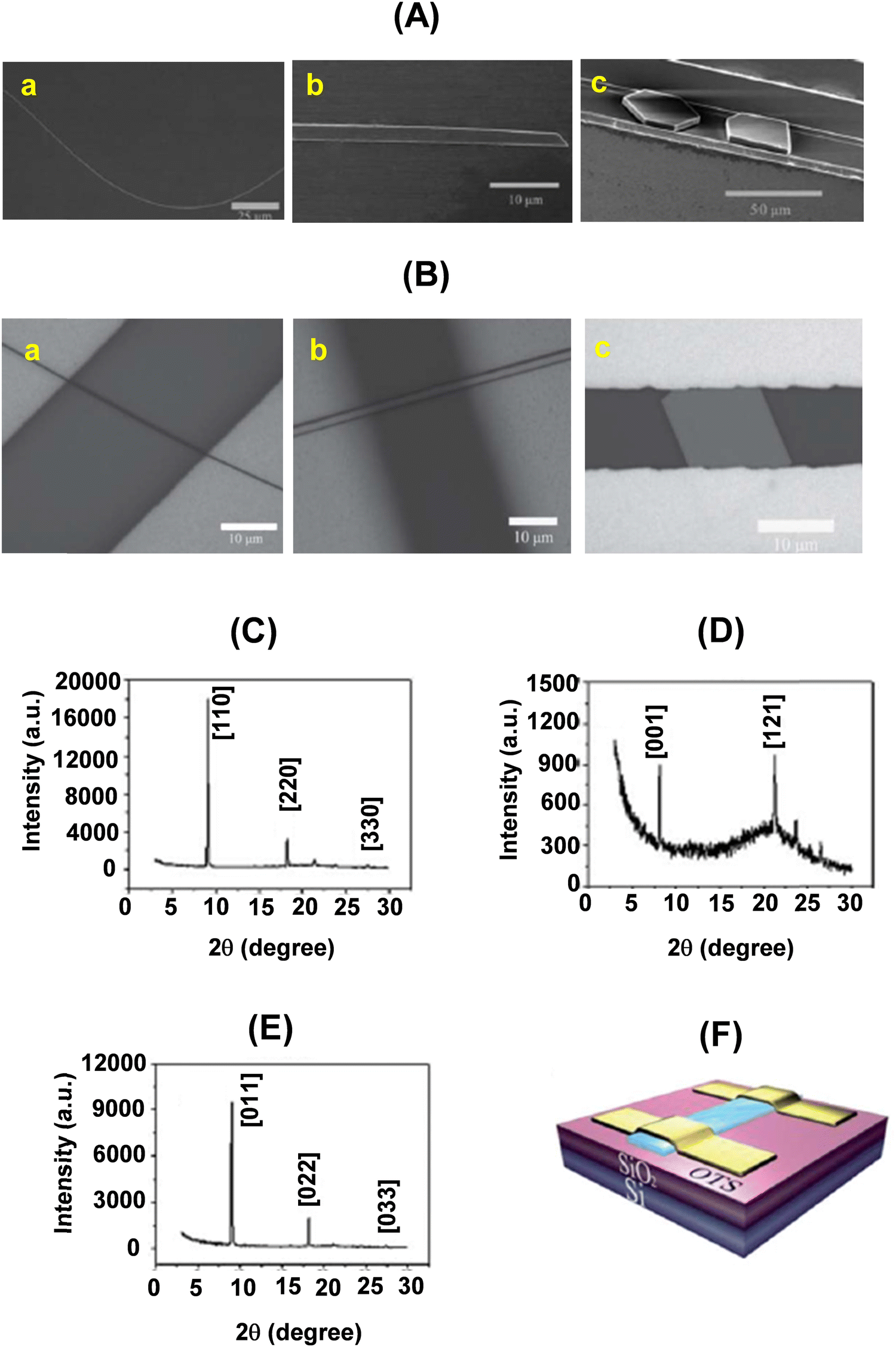 | ||
| Fig. 11 (A) SEM images of an individual wire (a), ribbon (b) and plate (c) of 22a–c, respectively; (B) optical images (a, b and c) of the SCOFET devices of 22a–c, respectively; (C–E) powder diffraction patterns of the wires, ribbons and plates of 22a–c, respectively. The peaks are indexed with lattice constants of the bulk crystals; (F) schematic structure of a BGTC SCOFET device. Reproduced with permission from ref. 16. Copyright 2013, The Royal Society of Chemistry. | ||
Based on the crystal structures of 22a–c and the theoretically calculated (site energy correction method: PW91PW91/6-31G* level)26 transfer integrals, the major intermolecular charge transfer pathways are shown in Fig. 12. It is obvious that 22a displayed one-dimensional transport character along the c-axis (i.e. along P3 and P4). The larger intermolecular distance along the other pathways (P1 and P2) yielded very small transfer integrals. However, 22b and 22c displayed two-dimensional transport behaviour, although charge transfer integrals along all directions were small. Further, since 22c adopts twisted conformation due to the presence of bulky p-Cl-Ph groups at 5- and 15-positions, π–π stacking is inhibited. Consequently, the charge transport integrals along P3 and P4 were unusually small (−1.803 and −9.362 meV). The smaller transfer integrals have been rationalized in terms of molecular displacement along the molecular axis in the dimeric arrangement and the shape of HOMOs that do not favour molecular overlap. This analysis further suggested that the reorganization energy is a superior contributor than the intermolecular electronic coupling (transfer integrals) for these classes of porphyrinoids.
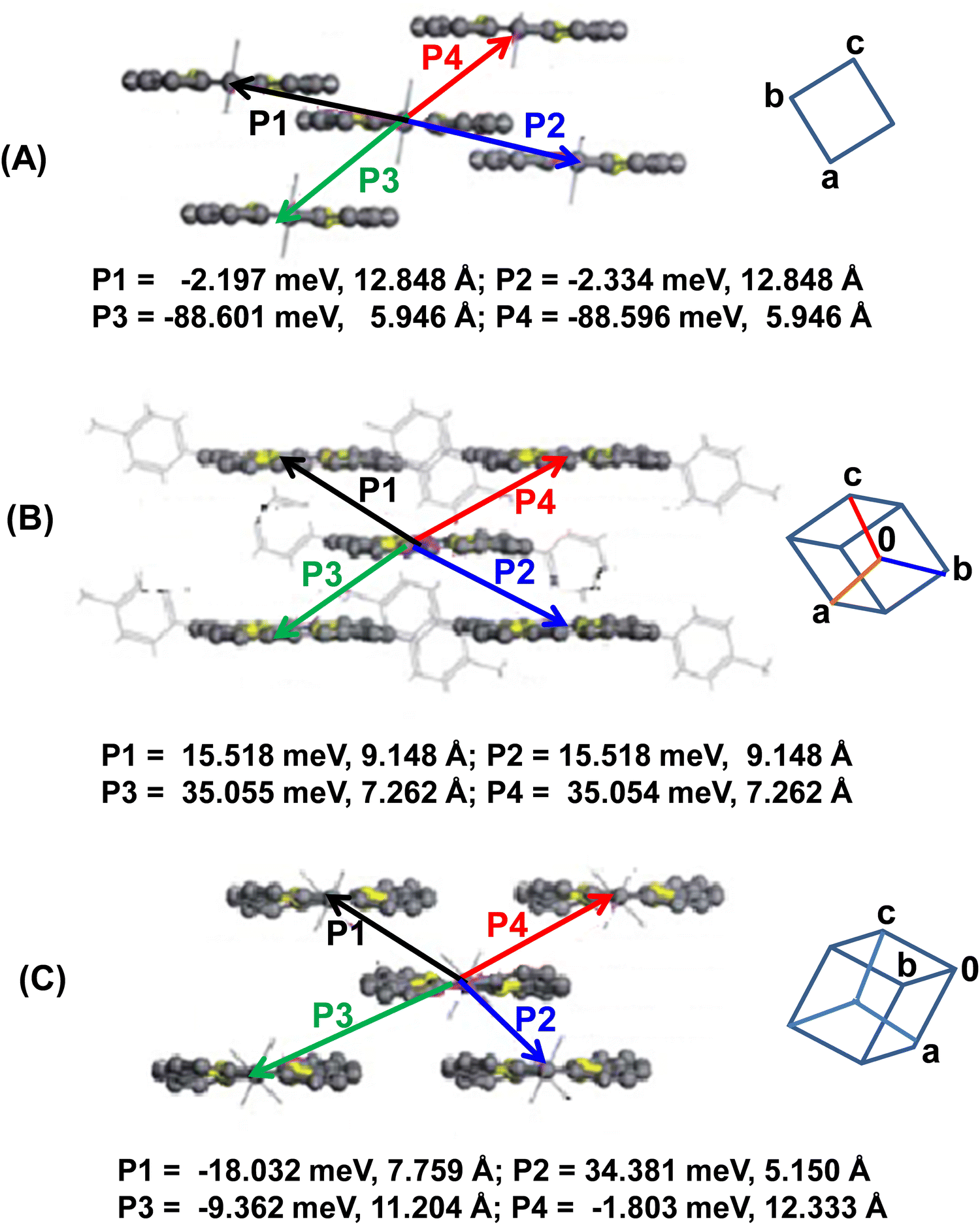 | ||
| Fig. 12 Schematics depicting the distances of the centroids and major transfer integrals between the two molecules of 22a–c (A–C, respectively). Reproduced with permission from ref. 16. Copyright 2013, The Royal Society of Chemistry. | ||
Thus, the nature of the substituents at the 5- and 15-positions of the TTPs play a significant role in molecular packing (from herringbone to face-to-face) and π–π stacking in addition to the solubility, crystallinity and thus the performance of SCOFET devices. Suffice to say, we have demonstrated that chemical modification at the meso-position of TTPs/TOPs constitutes a powerful tool in designing efficacious organic semiconductors. In the succeeding section, we have presented our work on ambipolar charge transport materials.
2.4. Ambipolar charge transport materials based on TTPs
Materials wherein both p-type and n-type channels transport holes and electrons, simultaneously, are called ambipolar charge transport materials. These are of tremendous significance because of their potential applications in complementary-like circuits, organic light-emitting transistors, etc.27 Despite the significant progress in unipolar (p- or n-type) semiconductors, designing unimolecular compounds that show efficient ambipolar transport behaviour (both stable p-type and n-type) under ambient conditions is still a challenge. Unimolecular ambipolar charge transport materials avoid the complexity in the fabrication processes of ambipolar devices where two unipolar (p- and n-type) materials are used. Alternatively, ambipolar transistors have been fabricated using heterostructures with p-type and n-type semiconductors as exemplified by bilayer heterojunctions,28 bulk heterojunctions29 and lateral heterostructures.30a,b This approach allowed construction of heterojunctions with molecular level self-assembly through cocrystallization of p-type and n-type organic semiconductors. Recently, theoretical evaluation of the superexchange couplings for donor–acceptor cocrystals and copolymers has been introduced and rationalized to elucidate the design principles to obtain high-mobility organic semiconductors.30cIn view of the superior p-type semiconducting behaviour of 22 as discussed in the preceding sections, we considered creating molecular level heterojunctions through self-assembling of the donor-acceptor (D–A) dyads using 22a as a p-type component in combination with an established n-type component such as tetracyanoquinodimethane (TCNQ)31 or fullerenes (C60 and C70).32 Using cocrystals of these dyads would have also cultivated the advantage of SCOFETs, which have been proven to be one of the most powerful tools for probing the relationship between intrinsic electrical transport properties, molecular structures and their stacking patterns. The superiority of such devices lies in the fact that there are fewer defects and morphological issues compared to thin film devices.33
The donor component 22a with a HOMO energy level of −4.88 eV showed a hole mobility of 0.29 cm2 V−1 s−1 (Table 1) in thin film OFET devices. On the other hand TCNQ is an efficient n-type semiconductor with a high electron mobility of up to 1.6 cm2 V−1 s−1 in single crystals.
The single crystal of 22a-TCNQ was obtained by slow evaporation of a chlorobenzene solution of a 1:1 mixture of 22a (1 mg mL−1) and TCNQ. In the molecular structure of 22a-TCNQ (Fig. 13A), 22a adopted a nearly coplanar structure with the four S atoms deviating from the mean plane by 0.05, 0.07, −0.05 and −0.07, respectively. The dihedral angle between the molecular planes of 22a and TCNQ was of the order of 3.46(4)°. Further, the intermolecular distance between the two components of the crystal was 3.4 Å, which was slightly shorter than the typical van der Waals interaction, while a strong non-bonding interaction between the two components was 3.158 Å. Further, in the solid state of the complex, 22a and TCNQ stacked alternatively (Fig. 13B) into a one-dimensional column structure along the a-axis. The face-to-face stacking allowed overlap of TCNQ with half of the plane of 22a, leading to lateral interactions between 22a and TCNQ in adjacent columns, comprising conducting channels for both holes and electrons, respectively. The powder X-ray diffraction pattern of the micro-ribbons (Fig. 12C) revealed intense peaks (001 and 002). A very interesting feature of the 22a–TCNQ complex was the lack of charge transfer between the two constituting components. This was inferred from the stretching frequency of CN of TCNQ in the complex (2224 cm−1), which matched favourably with the pristine, neutral TCNQ (2223 cm−1), and from the geometry of TCNQ in the complex. Thus, the ratio of the three conjugating bonds c/b + d (Fig. 13A) for the 22a–TCNQ complex as well as neutral TCNQ was identical (0.476).
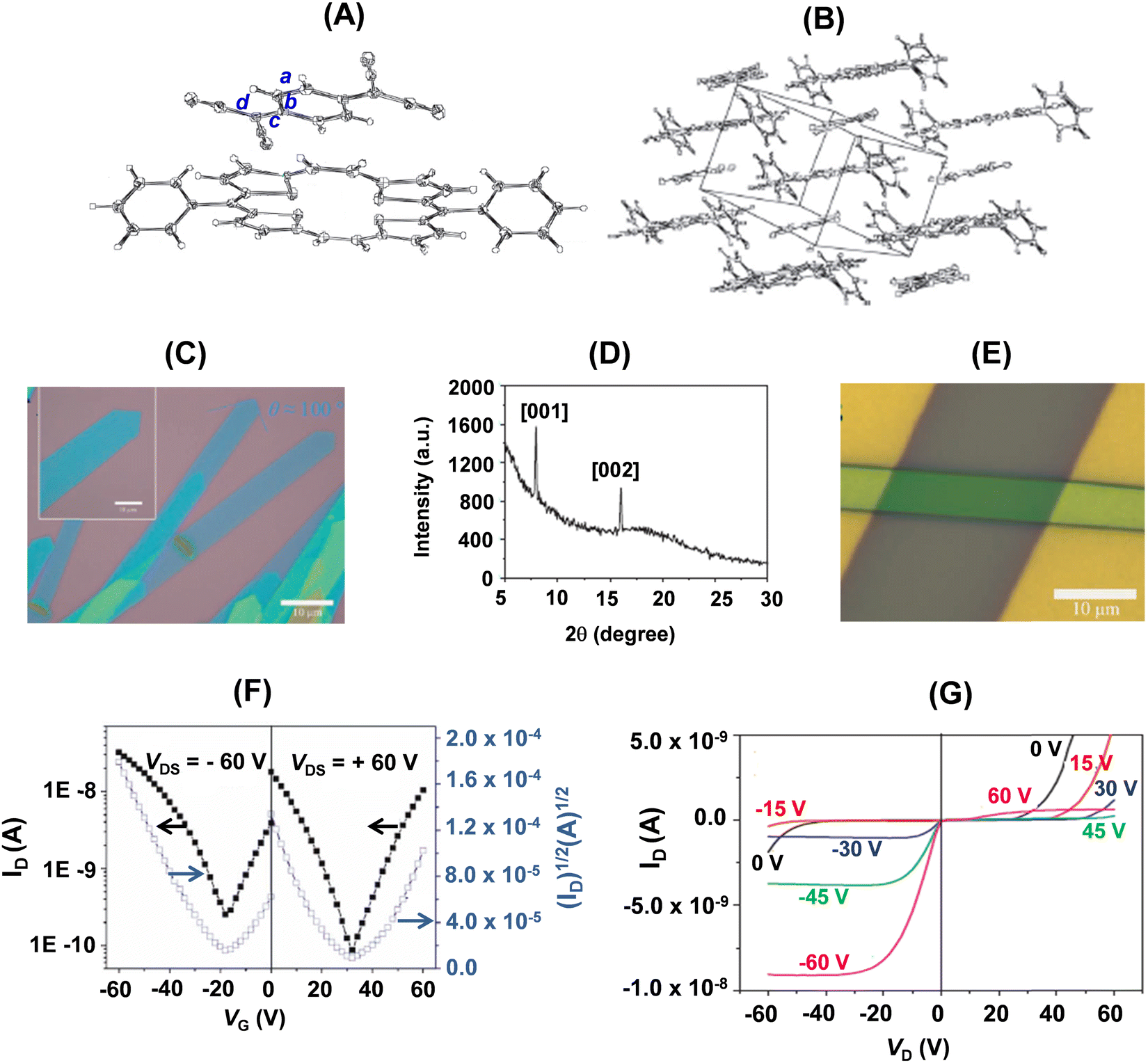 | ||
| Fig. 13 (A) Molecular structure of the 22a–TCNQ cocrystal with 50% probability ellipsoids; (B) alternative stacking pattern of 22a and TCNQ; (C) optical images of a single and several cocrystal micro-ribbons of 22a–TCNQ obtained by the drop-casting method through evaporation of a drop of a mixture of 22a and TCNQ in chlorobenzene on an OTS/SiO2/Si substrate; (D) powder diffraction pattern of the micro-ribbons; (E) optical image of the SCOFET BGTC device of 22a–TCNQ with an individual single crystal; (F) transfer and (G) output characteristics of the device of 22a–TCNQ (channel length, L = 43.7 μm, channel width, W = 6.0 μm). Reproduced with permission from ref. 31. Copyright 2012, Wiley-VCH. | ||
The devices of 22a–TCNQ exhibited ambipolar behaviour with stable balanced hole mobility of 0.04 cm2 V−1 s−1 and electron mobility of 0.03 cm2 V−1 s−1 in ambient atmosphere and represented a novel and easy-to-process approach for realizing ambipolar transport. Further, complementary metal-oxide-semiconductor (CMOS)-like inverters have been constructed by using two identical transistors built on a single crystal using a common gate (Si) for both the transistors. The inverter worked with a maximum gain of 10. The 22a–TCNQ cocrystal constituted the first example of a neutral, D–A type self-assembled complex that displayed a balanced ambipolar charge transport and application in CMOS-like circuits based on micro-scale crystals. Since this mixed D–A stacking complex represented a heterostructure with molecular level order, it was termed as a “molecular level heterojunction”.
The superiority of the electrical and optical properties of the self-assembled D–A dyad of 22a–TCNQ, and the distinctive electronic and photophysical properties of TTPs led us to investigate relationship between the electrical performance and molecular structures. We reported32 the first example of formation of self-assembled D–A cocrystals of the donor 22a with the fullerene (C60 and C70) acceptor as a two-dimensional (2D) segregated alternating layer structure, which displayed ambipolar charge transport as well as photoresponsivity. Additionally, the crystal structures of the D–A dyads provided an unambiguous understanding of the relationship between the solid state molecular packing and charge-transport properties.
The molecular structures (Fig. 14) of the crystals of 22a–C60 and 22a–C70 revealed a slight deviation of the planarity of 22a in a way that half of the porphyrin was bent up, while the other half was bent down which allowed fitting of the curvature of the fullerenes. Both 22a and fullerene partners were stacked in linear column structures supported by nonbonding interactions between the D and A components. For 22a–C60, both the components are packed, individually, into layers along the bc plane. A tetragonal array with a centroid distance of 10.02 Å was formed by the spheroidal C60, which was in fact larger than the distance (9.94 Å)34 in the crystal of pristine C60. Further, the shortest C–C contacts between the two molecules of C60 were 3.325–3.388 Å. On the other hand, in the layer of the donor 22a, the shortest C–C distance was 3.315 Å, which is less than twice the van der Waals radius of carbon (3.40 Å). Such intermolecular distances in the layers of both 22a and C60 suggested strong π–π interactions conducive to the transport of holes and electrons in the individual layers of 22a and C60. In the case of crystals of 22a-C70 a similar layer structure was observed. The single crystal structure analysis provided a good opportunity to understand the relationship between the molecular structure and charge transport and separation properties.
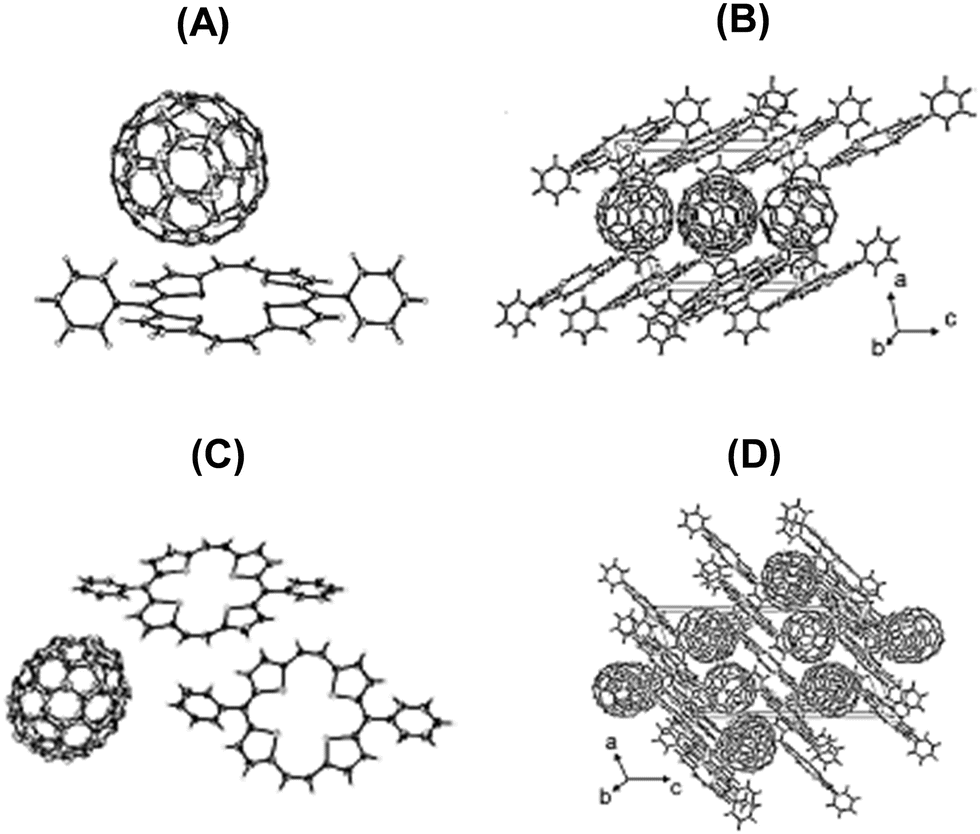 | ||
| Fig. 14 (A and C) ORTEP drawings of the asymmetric units with thermal ellipsoids set at 50% probability level and (B and D) stacking patterns viewed along the b-axis for 22a–C60 (A and B) and 22a–C70 (C and D). Reproduced with permission from ref. 32. Copyright 2013, The American Chemical Society. | ||
Parallelogram shaped D–A cocrystals of 22a with C60/C70 (Fig. 15A and B) representing alternating layer-by-layer packing were obtained by the drop-casting method. The crystal structures of these cocrystals were identical to their bulk counterparts as revealed by their X-ray diffraction patterns (Fig. 15C and D). The strong peak at 6.71° in the XRD pattern of 22a–C60 corresponded to a d spacing of 1.30 nm, which was consistent with the a-axis, meaning thereby that the crystals grew with the bc plane perpendicular to the substrate, while in the case of 22a–C70, the growth of the crystal was along the ab plane parallel to the substrate. The BGTC devices (Fig. 15E) of microcrystals of these D–A dyads showed ambipolar charge transport behaviour. The saturated electron and hole mobilities were 0.01 cm2 V−1 s−1 and 0.3 cm2 V−1 s−1, respectively, for the nanosheets of 22a–C60, while for 22a–C70, a more balanced ambipolar transport behaviour with electron and hole mobilities of 0.05 cm2 V−1 s−1 and 0.07 cm2 V−1 s−1, respectively, was noticed.32 Further, the latter device showed a sensitive photoresponse35 (Fig. 15F), when a large VG was applied. Upon the application of large VG, more charges accumulate in the conducting channel. The light responsivity of the device under an optical power of 5.51 mW cm−2 was 300 A W−1.32
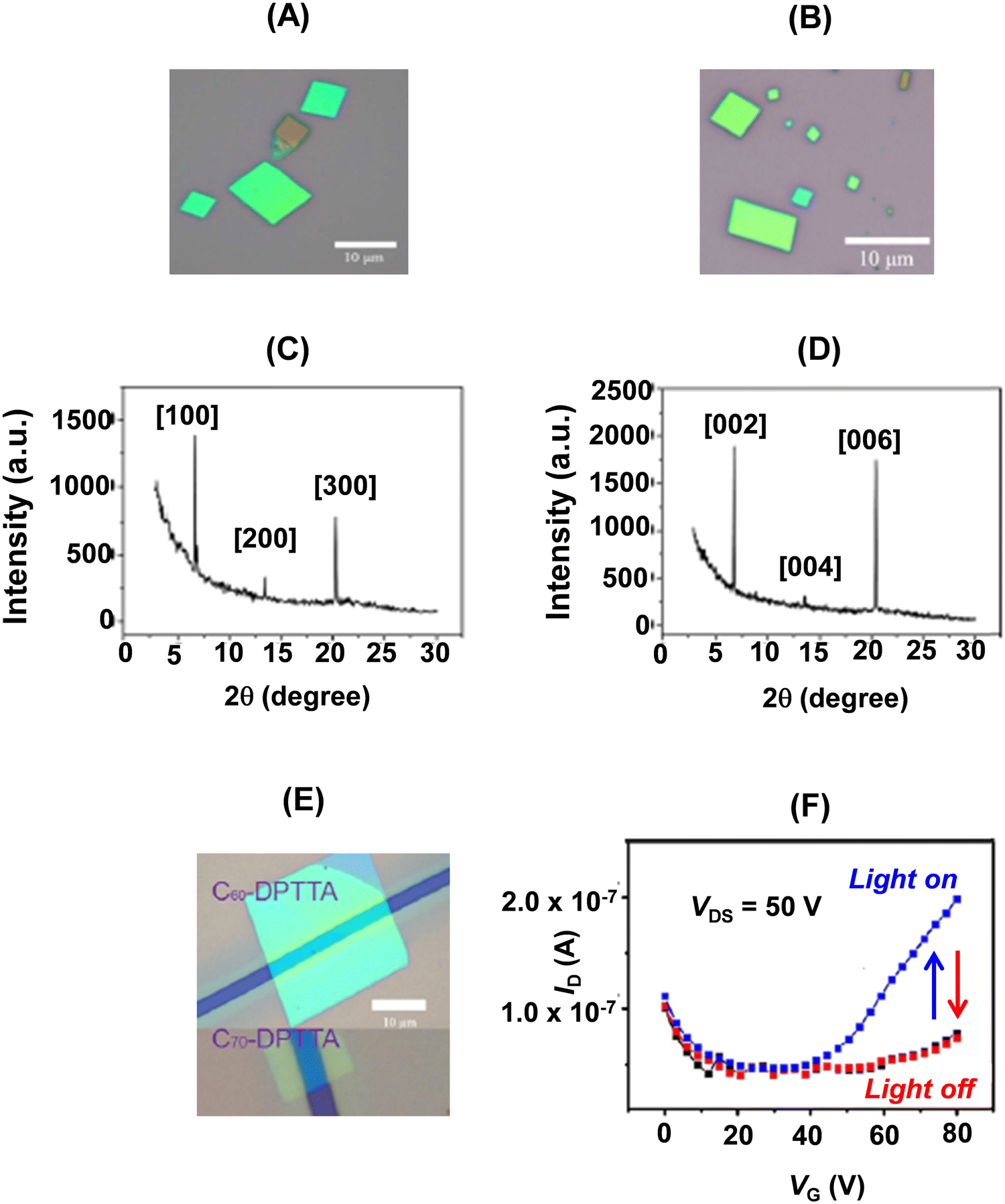 | ||
| Fig. 15 Optical micrographs of self-assembled 22a–C60 (A) and 22a–C70 (B); XRD patterns of 22a–C60 (C) and 22a–C70 (D); (E) optical image of the single crystal BGTC devices; (F) photocurrent responses of the 22a–C70 microcrystal based transistor under light irradiation. Reproduced with permission from ref. 32. Copyright 2013, The American Chemical Society. | ||
As discussed earlier, the reorganization energy and electronic coupling (λ) are the two important factors that influence the charge-transport properties of organic semiconductors. The charge transfer decreases with reorganization energy and increases with electronic coupling. The calculated reorganization energies of C60 and C70 were 135 and 142 meV, respectively, while for 22a it was 201 meV.
The electron and hole transport pathways of the fullerene and 22a components were similar, and for 22a–C60, these are depicted in Fig. 16. The calculated effective transfer integrals were 34 and 12.3 meV, respectively, for electrons and holes, along all pathways indicating isotropy of both the charges in 22a–C60. However, in 22a–C70, the effective integrals for electrons were 34 meV for paths P1 and P2, while for P3 and P4 these were 27.9 meV. Likewise, for holes, only P1 and P3 showed significant coupling. This indicates a higher electron transport compared to hole transport, which was in contrast to the experimental values (0.01 cm2 V−1 s−1 and 0.3 cm2 V−1 s−1, respectively, for electron and hole mobilities), especially in 22a–C60. This has been attributed to the greater sensitivity of electron transport to traps at the semiconductor–dielectric interface and internal defects in the devices.36
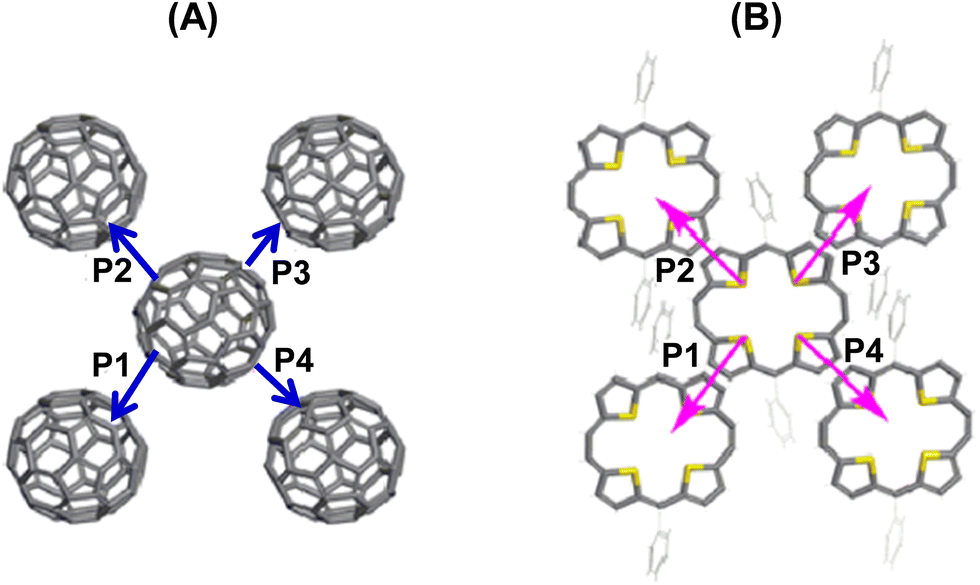 | ||
| Fig. 16 Charge hopping pathways of the (A) acceptor (C60) and (B) donor (22a) components in the individual molecular layers. Reproduced with permission from ref. 32. Copyright 2013, The American Chemical Society. | ||
3. Nonlinear optical behaviour of meso-substituted TTPs and TOPs
Nonlinear optical (NLO) materials have attracted considerable attention owing to their extensive applications in data storage,37a light modulation, optical switching,37b,c optical limiting,37d sensors,37eetc. Therefore, finding new and stable NLO materials with strong NLO response constitutes an important area of investigation. TTPs/TOPs exhibit high stability owing to extensive conjugation and rich redox properties; therefore, these could be recognized as promising NLO materials. A number of reports are available on the third order NLO properties of porphyrins which could be tuned by metal complexation,13,14 functionalization and π-conjugation.37f,g Owing to centrosymmetric nature,37h however, the second order NLO properties of porphyrins and their derivatives have rarely been investigated.In continuation of our interest in developing NLO active materials,38 we initially recorded39 the excited state cross sections of photostable 22a, 22c and 22g in DCM using the open aperture Z-scan technique using 527 nm and 100 ns laser pulses at a low repetition rate of 250 Hz. Since the compounds were almost transparent at 527 nm, correction for resonance enhancement was not needed. While the excited state absorption cross section σex values were calculated using a simple five-level model (Fig. 17), the ground state absorption cross section σgr values were calculated from the linear absorption coefficient.39 Very high cross-section ratios (σex/σgr) of 250, 498 and 197 (Table 2) were obtained for 22a, 22c and 22g, respectively.
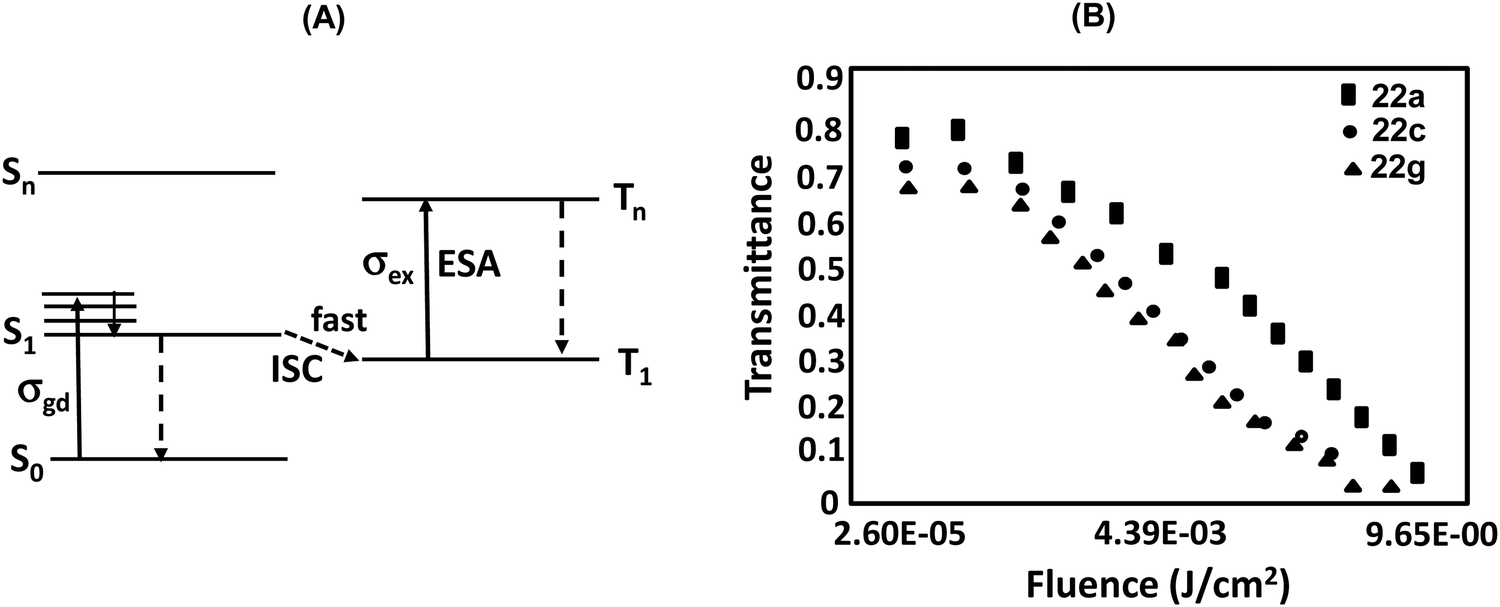 | ||
| Fig. 17 (A) Proposed energy-level model depicting photophysical processes (ISC: intersystem crossing, ESA: excited state absorption leading to RSA); (B) normalized transmittance of 22a, 22c and 22g as a function of the fluence of laser pulse (527 nm). Reproduced with permission from ref. 39. Copyright 2016, The Royal Society of Chemistry. | ||
| σ ex (cm2) | σ g (cm2) | σ ex/σgc | ε H (eV) | ε L (eV) | ε H − εLd (eV) | η | τ s (ps) | K (s−1) | Fluence threshold (J cm−2) | |
|---|---|---|---|---|---|---|---|---|---|---|
| a Excited-state cross-section. b Ground-state cross-section. c Ratio of excited to ground-state absorption cross-section. d Energy of HOMO and LUMO levels. e Derived from Koopman's theorem: η = (εH − εL)/2. f Singlet excited-state lifetime. g Decay constant. | ||||||||||
| 22a | 8.32 × 10−17 | 3.32 × 10−19 | 250 | −4.86 | −2.54 | −2.32 | 1.16 | 170 | 5.9 × 109 | 0.310 |
| 22c | 1.50 × 10−16 | 3.01 × 10−19 | 498 | −4.89 | −2.78 | −2.11 | 1.05 | 1500 | 6.6 × 108 | 0.531 |
| 22g | 6.27 × 10−17 | 3.18 × 10−19 | 197 | −4.86 | −2.55 | −2.31 | 1.15 | 140 | 7.1 × 109 | 0.217 |
The HOMO–LUMO gap of these 22π porphyrins is marginally smaller in 22c compared to 22a and 22c due to stabilization of LUMOs due to the substitution of the meso-position with the electron-withdrawing p-Cl–C6H4 group. Consequently, the larger molecular polarizability and hence global softness (as inferred from the inverse of molecular hardness, η) results in the observed higher excited state cross-section value. Further, the excited state decay profiles (Table 2) of 22a, 22c and 22g yielded the lowest singlet excited state lifetimes of 170, 1500 and 140 ps, respectively (Table 2), which correlated well with the excited state cross-section values. A large (1500 ps) singlet excited state lifetime of 22c indicated slow excited state relaxation, i.e. a decelerated internal conversion between S1 and S0 states in spite of a marginally lower optical band gap. Porphyrins 22a, 22c and 22g demonstrated very good optical limiting potential (Fig. 17) at 527 nm using dilute (1 mM) solution excluding the effect of any aggregation. The fluence threshold was found to be 0.310, 0.53 and 0.217 J cm−2, respectively, for 22a, 22c and 22g, which were quite low. Thus these porphyrins showed reverse saturable absorption (RSA), which was attributed to excited state absorption.
The observed nonlinearities in these porphyrins are attributed to the population redistribution among the accessible energy states under the wavelengths used, which gives rise to absorptive nonlinearities (changes in absorption) as well as refractive nonlinearities (changes in the refractive index). In this context, we studied40 the ultrafast dynamics and third-order nonlinear optical properties of TOPs 23a, i and b using a single beam Z-scan technique with femtosecond (fs) and nanosecond (ns) pulses at respective near-IR and visible wavelengths. Porphyrins 23a, b and i possess unique third-order nonlinear optical behaviour with a wide spectral range and show both ESA and TPA in the same solvent, but at different wavelengths and pulse widths. While ESA mediated RSA was observed at 527 nm, two-photon absorption (TPA) was observed at 800 nm. The high nonlinear refractive index coefficient and nonlinear absorption coefficient values and the fast response times were attributed to the polarizable π-electrons of the cyclic conjugated aromatic porphyrin framework. Our proposed mechanism of third-order nonlinearity was verified using ultrafast transient absorption spectroscopy. The TPA cross-section values (at 800 nm, Z-scan) with 50 fs laser pulses were in the range of 10 to 30 GM and are given in Table 3. Furthermore, since the π-conjugation pathway of 23a, i and b is identical, the H–L energies (Table 3) as well as the H–L gaps were small and of the same order in parallel with the TOPs 23a, i and b. The values of the TPA cross-sections (Table 3) are in agreement with the calculated molecular polarizability (converse of molecular hardness, Z). Among the three compounds studied the presence of p-BrC6H4 substituents at meso-positions in 23i seems to render more polarizability leading to the largest TPA cross-section value (Table 3). The reduction in transmittance (Fig. 18) for these porphyrins as measured by the open aperture (OA) Z-scan technique was independent of nonlinear refraction and used to determine β (Table 3). The OA traces further inferred that absorption was intensity dependent and the maximum absorption occurred at the focal point of the lens, where the on-axis intensity of the beam is maximum. In order to account for the mechanism of nonlinear absorption, occurring in 23a, b and i, the cross sections of the ground and excited states were calculated.
| ε H/εLa (eV) | σ ex × 10−21b (m2) | σ g × 10−23 (m2)b | σ ex/σgb | σ (2) (GM) | α × 10−41d (cm2 V−1) | β × 10−8e (m W−1) | γ ×10−15f (m2 W−1) | τ s (ps) | K (s−1) | |
|---|---|---|---|---|---|---|---|---|---|---|
| a H–L energies (TD-DFT). b Excited-state and ground-state absorption cross-section values at 527 nm and the ratio of excited to ground-state absorption cross-section. c TPA cross-section values at 800 nm in SI units, where 1 GM = 10−50 cm4 s photon−1. d Average polarizability (Gaussian 09). e Calculated in DCM at 527 nm; the values in parenthesis were determined at 800 nm. f Calculated in DCM at 527 nm; the values in parenthesis were determined at 800 nm. g Singlet excited-state lifetime. h Decay constant. | ||||||||||
| 23a | −4.49/−2.43 | 24.9 | 3.43 | 725 | 19.4 (±0.3) | 1121.49 | 2.77 (4.66 × 10−10) | −5.91 (−4.78) | 417 | 1.63 × 109 |
| 23i | −4.53/−2.52 | 7.49 | 9.96 | 75 | 29.0 (±0.4) | 1141.10 | 2.90 (7.01 × 10−10) | −1.29 (−7.92) | 513 | 1.50 × 108 |
| 23b | −4.43/−2.38 | 7.60 | 6.64 | 114 | 13.0 (±0.3) | 1096.48 | 2.18 (3.97 × 10−10) | −5.27 (−5.57) | 336 | 1.52 × 109 |
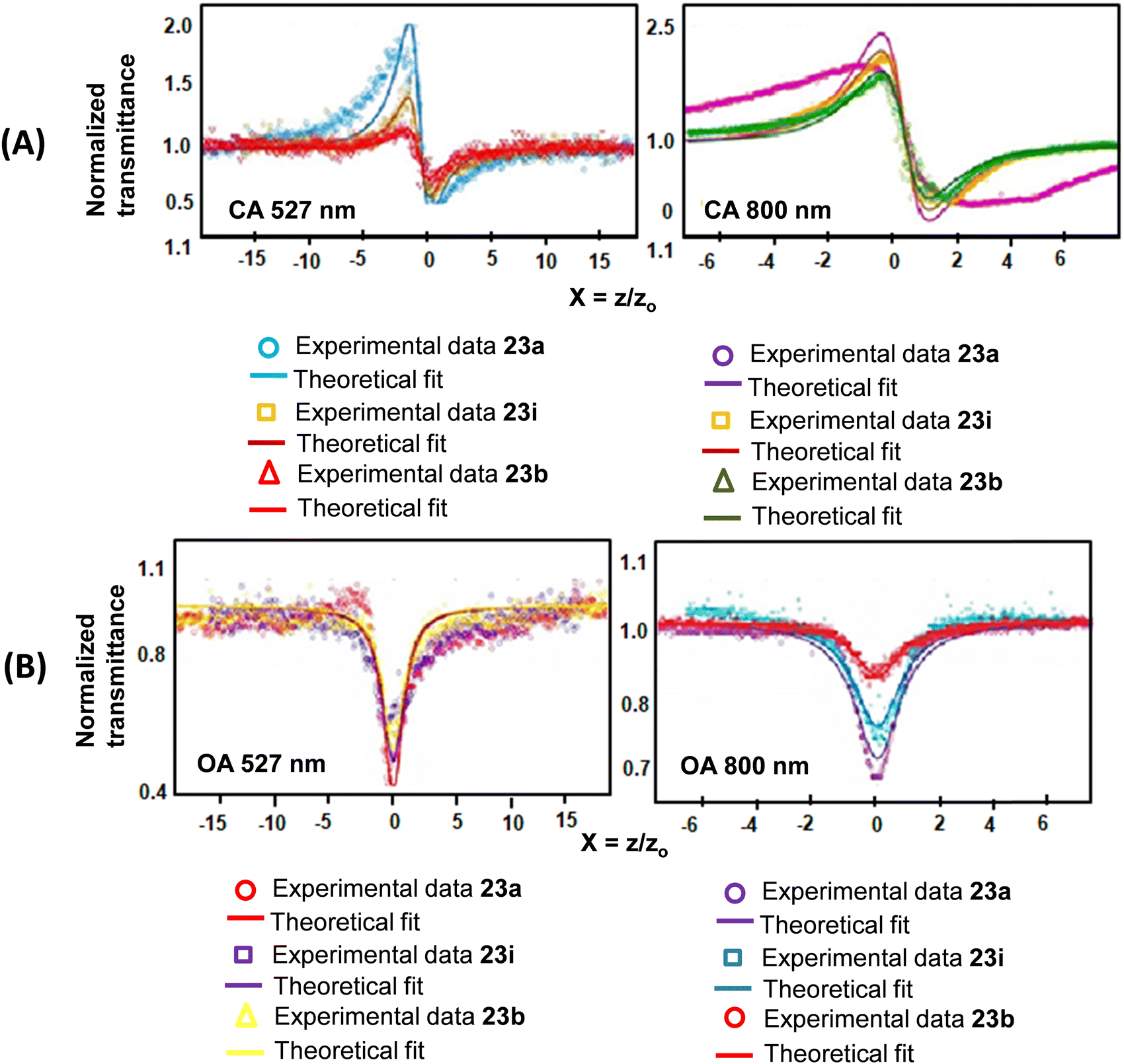 | ||
| Fig. 18 Z-scan traces of 23a, 23i and 23b in DCM under (A) closed aperture (CA) and (B) open aperture (OA) conditions at (i) 527 nm and at (ii) 800 nm. Reproduced with permission from ref. 40. Copyright 2016, The Royal Society of Chemistry. | ||
The excited to ground state absorption cross-section ratio (σex/σgd) (Table 3) was the maximum for 23a having an unsubstituted phenyl ring at the meso position of the porphyrin, while 23b bearing a p-BrC6H4 group at the meso position recorded the minimum value. The population of the excited states after ns (100 ns) pulse excitations in the case of 23a was nearly 750 times greater (due to faster ISC) in the excited state compared to the ground state. This indicated the initial build-up of population at the singlet excited state, which is transferred to the triplet states and is responsible for ESA leading to RSA. Furthermore, RSA is not due to TPA because if the nonlinear absorption was due to TPA then β should have remained constant by varying (increasing) the axis fluence. However, we observed a decrease in β with increasing axis fluence.40 The nonlinear absorption coefficients β determined through both OA and close aperture (CA) techniques at 527 nm and 800 nm for 23a, b and i were identical. While the positive values of β indicated RSA, the negative value of nonlinear refractivity (γ) was attributed to the self-defocusing effect.
4. Concluding remarks
The exciting photophysical properties, charge transport and nonlinear optical behaviour of organic semiconductors such as tetrathia/tetraoxa[22]porphyrin(2.1.2.1)s await further exploration. This is clearly indicated by the development of chemistry and applications of these meso-substituted porphyrinoids by us in the past decade. Refinement in the synthetic approaches is much desired to improve the yield of these porphyrinoids as well as to incorporate aliphatic substituents at the 5- and 16-positions of these porphyrinoids. This has constituted a severe limitation of the work reported by us as we could not oxidize the 5-, 15-dihydro counterparts to obtain the corresponding fully aromatized porphyrinoids. Electrochemical switching of these systems between 22π aromatic and 20π antiaromatic states is another interesting attribute and isolation of stabilized salts of the dicationic antiaromatic species would be interesting. Simultaneously, placing suitable substituents at the macrocyclic rim would provide immense opportunities for obtaining donor–acceptor type porphyrins for applications related to semiconducting charge transport and other optoelectronic applications such as redox switchable nonlinear optical materials. Additionally, the rich collection of porphyrinoids discussed in this personal account will provide an opportunity to understand the interplay between aromaticity and structure. On a more fundamental level, various porphyrin analogues presented in this article serve to highlight what is still relatively “unexplored territory” in porphyrin chemistry. We thus predict that the area of meso-substituted porphyrins will constitute an area of considerable growth in the years to come offering immense opportunities in the area of synthesis, physicochemical analysis, metal complexation chemistry, anion recognition, catalysis, redox switching and organic electronics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. S. thanks SERB (DST, New Delhi) for the grant EMR/2017/000520 and Guru Nanak Dev University, Amritsar for facilities. We express our sincere gratitude to the colleagues with whom we have been collaborating over the years on the device fabrication etc. as well as to our skilled students.References
- (a) The Porphyrin Handbook , ed. K. Kadish, K. M. Smith and R. Guilard, Academic Press, London, 2000, vol. 1–20 Search PubMed; (b) S. Hiroto, Y. Miyaki and H. Shinokubo, Chem. Rev., 2017, 117, 2910 CrossRef CAS PubMed; (c) S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 44342 Search PubMed; (d) Handbook of Porphyrin Science , ed. K. Kadish, K. M. Smith and R. Guilard, World Scientific, London, 2010 Search PubMed; (e) T. K. Chandrashekar and S. Venkataraman, Acc. Chem. Res., 2003, 36, 676 CrossRef CAS PubMed; (f) R. Misra and T. K. Chandrashekar, Acc. Chem. Res., 2008, 41, 265 CrossRef CAS PubMed; (g) M. O. Senge, N. N. Sergeeva and K. J. Hale, Chem. Soc. Rev., 2021, 50, 4730 RSC; (h) B. Sekaran and R. Misra, Coord. Chem. Rev., 2022, 453, 214312 CrossRef CAS.
-
(a) M. O. Senge, M. Fazekas, E. G. A. Notaras, W. J. Blau, M. Zawadzka, O. B. Locos and E. M. NiMhuircheartaigh, Adv. Mater., 2007, 19, 2737 CrossRef CAS;
(b)
A. Chaudhary, A. Srinivasan and T. K. Chandrashekar, in Handbook of Porphyrin Science, ed. K. Kadish, K. M. Smith and R. Guilard, World Scientific, London, 2014, p. 271 Search PubMed;
(c) M. Janghouri and M. Adineh, J. Photochem. Photobiol., A, 2017, 341, 31 CrossRef CAS;
(d)
J. V. Krishna, P. Mrinalini, S. Prasanthkumar and L. Giribabu, Dye-Sensitized Solar Cells, Academic Press, New York, NY, USA, 2019, ch. 7, pp. 231–284 Search PubMed;
(e) J. Kesters, P. Verstappen, M. Kelchtermans, L. Lutsen, D. Vanderzande and W. Maes, Adv. Energy Mater., 2015, 5, 1500218 CrossRef;
(f)
A. Tebo, C. Herrero and A. Aukauloo, in Handbook of Porphyrin Science, ed. K. Kadish, K. M. Smith and R. Guilard, World Scientific, London, 2014, p. 196 Search PubMed;
(g)
T. W. Liu, E. Huynh, T. D. MacDonald and G. Zheng, Cancer Theranostics, 2014, ch. 14, p. 229 Search PubMed.
- (a) T. Samra and P. K. Panda, Chem. Rev., 2017, 117, 2785 CrossRef PubMed; (b) T. Chatterjee, V. S. Shetti, R. Sharma and M. Ravikanth, Chem. Rev., 2017, 117, 3254 CrossRef CAS PubMed; (c) J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134 CrossRef CAS PubMed; (d) J. L. Sessler, A. Gebauer and E. Vogel, in The Porphyrin Handbook, ed. K. Kadish, K. M. Smith and R. Guilard, Academic Press, London, 2000. vol. 2, p. 1 Search PubMed; (e) S. Xue, N. Liu, P. Mei, D. Kuzuhara, M. Zhou, J. Pan, H. Yamada and F. Qiu, Chem. Commun., 2021, 57, 12808 RSC.
- (a) N. Jux, Angew. Chem., Int. Ed., 2008, 47, 2543 CrossRef CAS PubMed; (b) D. Yu, C. Rong, T. Lu, P. Geerlings, F. De Proft, M. Alonso and S. Liu, Phys. Chem. Chem. Phys., 2020, 22, 4715 RSC.
- (a) C.-H. Lee, V. Roznyatovskiy, S.-J. Hong and J. L. Sessler, in Handbook of Porphyrin Science, ed. K. Kadish, K. M. Smith and R. Guilard, World Scientific, London, 2010. vol 13, ch. 60 Search PubMed; (b) K. Singh, A. Sharma and S. Sharma, Heteroporphyrins: Synthesis and Structure Modifications, in Advances in Heterocyclic Chemistry, ed. A. R. Katritzky, Elsevier, Amsterdam, 2012, pp. 111–184. and references cited therein Search PubMed; (c) K. Singh, A. Abebayehu, E. Mulugeta, D. Sareen and C. H.-Lee, J. Porphyrins Phthalocyanines, 2016, 20, 1 CrossRef.
- Z. Hu, J. L. Atwood and M. P. Cava, J. Org. Chem., 1994, 59, 8071 CrossRef CAS.
- G. Markl and U. Striebl, Angew. Chem., Int. Ed. Engl., 1993, 32, 1333 CrossRef.
- In Organic Field-effect Transistors, ed. Z. Boa and J. Locklin, CRC Press, Taylor and Francis Group, Florida, 2007 Search PubMed.
- K. Singh and A. Sharma, Tetrahedron Lett., 2008, 49, 6234 CrossRef CAS.
- K. Singh and A. Sharma, Tetrahedron Lett., 2007, 48, 227 CrossRef CAS.
- K. Singh and A. Sharma, Tetrahedron, 2010, 66, 3682 CrossRef CAS.
- K. Singh, S. Sharma, A. Sharma and P. Kaur, Eur. J. Org. Chem., 2014, 381 CrossRef CAS.
- K. Singh, A. Sharma, J. Zhang, W. Xu and D. Zhu, Chem. Commun., 2011, 47, 905 RSC.
- K. Singh, T. S. Virk, J. Zhang, W. Xu and D. Zhu, Chem. Commun., 2012, 48, 121 RSC.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. V. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- J. Zhang, J. Ma, Q. Zhang, T. S. Virk, H. Geng, D. Wang, W. Xu, Z. Shuai, K. Singh, W. Hu and D. Zhu, J. Mater. Chem. C, 2013, 1, 5765 RSC.
- E. Vogel, P. Rohrig, M. Sicken, B. Knipp, A. Herrmann, M. Pohl, H. Schmickler and J. Lex, Angew. Chem., Int. Ed. Engl., 1989, 28, 1651 CrossRef.
- K. Singh, T. S. Virk, J. Zhang, W. Xu and D. Zhu, Chem. Commun., 2012, 48, 12174 RSC.
- T. S. Virk, K. Singh, Y. Qin, W. Xu and D. Zhu, RSC Adv., 2014, 4, 37503 RSC.
- Q. Tang, L. Jiang, Y. Tong, H. Li, Y. Liu, Z. Wang, W. Hu, Y. Liu and D. Zhu, Adv. Mater., 2008, 20, 2947 CrossRef CAS.
- (a) H. H. Choi, K. Cho, C. D. Frisbie, H. Sirringhaus and V. Podzorov, Nat. Mater., 2018, 17, 2 CrossRef CAS PubMed; (b) Z. A. Lamport, H. F. Haneef, S. Anand, M. Waldrip and O. D. Jurchescu, J. Appl. Phys., 2018, 124, 071101 CrossRef; (c) W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC; (d) G. Horowitz, Adv. Mater., 1998, 10, 365 CrossRef CAS; (e) M. S. Kang and C. D. Frisbie, ChemPhysChem, 2013, 14, 1547 CrossRef CAS PubMed.
- P. Ma, Y. Chen, X. Cai, H. Wang, Y. Zhang, Y. Gao and J. Jiang, Synth. Met., 2010, 160, 510 CrossRef CAS.
- T. Zhao, Z. Wei, Y. Song, W. Xu, W. Hu and D. Zhu, J. Mater. Chem., 2007, 17, 4377 RSC.
- O. Gidron, A. Dadvand, E. W.-H. Sun, I. Chung, L. J. W. Shimon, M. Bendikov and D. F. Perepichka, J. Mater. Chem. C, 2013, 1, 4358 RSC.
- C. Liu, S. W. Mao and M. Kuo, J. Phys. Chem. C, 2010, 114, 22316 CrossRef CAS.
- E. Valeev, V. Coropceanu, D. da Silva Filho, S. Salman and J. Brédas, J. Am. Chem. Soc., 2006, 128, 9882 CrossRef CAS PubMed.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296 CrossRef CAS PubMed.
- (a) R. Ye, M. Baba and K. J. Mori, J. Appl. Phys., 2005, 44, 581 CrossRef; (b) J. Wang, H. B. Wang, X. J. Yan, H. C. Huang, D. Jin, J. W. Shi, Y. H. Tang and D. H. Yan, Adv. Funct. Mater., 2006, 16, 824 CrossRef CAS.
- (a) C. Rost, S. Karg, W. Riess, M. A. Loi, M. Murgia and M. Muccini, Appl. Phys. Lett., 2004, 85, 1613 CrossRef CAS; (b) M. Shkunov, R. Simms, M. Heeney, S. Tierney and I. McCulloch, Adv. Mater., 2005, 17, 2608 CrossRef CAS.
- (a) J.-F. Chang, M. C. Gwinner, M. Caironi, T. Sakanoue and H. Sirringhaus, Adv. Funct. Mater., 2010, 20, 2825 CrossRef CAS; (b) Z. Wei, W. Xu, W. Hu and D. Zhu, J. Mater. Chem., 2008, 18, 2420 RSC; (c) H. Geng, L. Zhu, Y. Yi, D. Zhu and Z. Shuai, Chem. Mater., 2019, 31(17), 6424 CrossRef CAS.
- J. Zhang, H. Geng, T. S. Virk, Y. Zhao, J. Tan, C.-A. Di, W. Xu, K. Singh, W. Hu, Z. Shuai, Y. Liu and D. Zhu, Adv. Mater., 2012, 24, 2603 CrossRef CAS PubMed.
- J. Zhang, J. Tan, Z. Ma, W. Xu, G. Zhao, H. Geng, C. Di, W. Hu, Z. Shuai, K. Singh and D. Zhu, J. Am. Chem. Soc., 2013, 135, 558 CrossRef CAS PubMed.
- Z. Wei, W. Hong, H. Geng, C. Wang, Y. Liu, R. Li, W. Xu, Z. Shuai, W. Hu, Q. Wang and D. Zhu, Adv. Mater., 2010, 22, 2458 CrossRef CAS PubMed.
- M. Makha, A. Purich, C. L. Raston and A. N. Sobolev, Eur. J. Inorg. Chem., 2006, 507 CrossRef CAS.
- N. Marjanović, T. B. Singh, G. Dennler, S. Günes, H. Neugebauer, N. S. Sariciftci, R. Schwödiauer and S. Bauer, Org. Electron., 2006, 7, 188 CrossRef.
- H. T. Nicolai, M. Kuik, G. A. H. Wetzelaer, B. de Boer, C. Campbell, C. Risko, J. L. Brédas and P. W. M. Blom, Nat. Mater., 2012, 11, 882 CrossRef CAS PubMed.
- (a) Y. Zhai, J.-Q. Yang, Y. Zhou, J.-Y. Mao, Y. Ren, V. A.-L. Roy and S.-T. Han, Mater. Horiz., 2018, 5, 641 RSC; (b) B. Champagne, A. Plaquet, J.-L. Pozzo, V. Rodriguez and F. Castet, J. Am. Chem. Soc., 2012, 134, 8101 CrossRef CAS PubMed; (c) B. P. Biswal, S. Valligatla, M. Wang, T. Banerjee, N. A. Saad, B. M.-K. Mariserla, N. Chandrasekhar, D. Becker, M. Addicoat, I. Senkovska, R. Berger, D. N. Rao, S. Kaskel and X. Feng, Angew. Chem., Int. Ed., 2019, 131, 6970 CrossRef; (d) M. S.-S. Bharati, S. Bhattacharya, J. V.-S. Krishna, L. Giribabu and S. V. Rao, Opt. Laser Technol., 2018, 108, 418 CrossRef CAS; (e) S. A. Taya, M. M. Shabat, H. M. Khalil and D. S. Jäger, Sens. Actuators, A, 2008, 147, 137 CrossRef CAS; (f) H. Rath, J. Sankar, V. PrabhuRaja, T. K. Chandrashekar, A. Nag and D. Goswami, J. Am. Chem. Soc., 2005, 127, 11608 CrossRef CAS PubMed; (g) C. Wang, G. Shi, Z. Zhu, S. Luo, W. Sun, W. Jiao, R. Gao, G. Fan and Y. Song, Opt. Mater., 2020, 100, 109621 CrossRef CAS; (h) S. M. LeCours, H.-W. Guan, S. G. DiMagno, C. H. Wang and M. J. Therien, J. Am. Chem. Soc., 1996, 118, 1497 CrossRef.
- P. Kaur and K. Singh, Chem. Rec., 2022, e202200024 CAS.
- K. Singh, S. Arora, K. Makhal, P. Kaur and D. Goswami, RSC Adv., 2016, 6, 22659 RSC.
- K. Makhal, P. Kaur, S. Arora, D. Goswami and K. Singh, J. Mater. Chem. C, 2016, 4, 9445 RSC.
| This journal is © The Royal Society of Chemistry 2022 |