
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolar-driven H2O2 production via cooperative auto- and photocatalytic oxidation in fine-tuned reaction media†
Byeong Cheul
Moon‡
 a,
Bolormaa
Bayarkhuu‡
bc,
Kai A. I.
Zhang
d,
Dong Ki
Lee
*acef and
Jeehye
Byun
*bc
a,
Bolormaa
Bayarkhuu‡
bc,
Kai A. I.
Zhang
d,
Dong Ki
Lee
*acef and
Jeehye
Byun
*bc
aClean Energy Research Center, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea. E-mail: dnklee@kist.re.kr
bWater Cycle Research Center, Korea Institute of Science and Technology, Seoul 02792, Republic of Korea. E-mail: jbyun@kist.re.kr
cDivision of Energy and Environment Technology, KIST-School, University of Science and Technology, Seoul 02792, Republic of Korea
dDepartment of Materials Science, Fudan University, Shanghai 200433, P. R. China
eGraduate School of Energy and Environment, Korea University, Seoul 02841, Republic of Korea
fDepartment of Chemical and Biomolecular Engineering, Yonsei-KIST Convergence Research Institute, Yonsei University, Seoul 03722, Republic of Korea
First published on 8th November 2022
Abstract
Solar photocatalysis has emerged as a sustainable route to produce H2O2; however, the structural design of photocatalysts makes them expensive, limiting their practical applications. Inspired by the industrial anthraquinone process, we developed a new reaction design for solar H2O2 production using an organic working solution (OWS) comprising aryl alcohols and a metal-free polymeric photocatalyst. The synergistic auto- and photocatalytic oxidation of aryl alcohols in the OWS allowed for the quantitative generation of H2O2 by counter O2 reduction. The fine modulation of the OWS with water enhanced the photocatalytic route, resulting in an unprecedented H2O2 production rate of 46.9 mmol h−1 g−1 with a solar-to-chemical conversion efficiency of 1.1% under simulated sunlight. Pure H2O2 was obtained after extraction and purification by membrane filtration, and was directly used in large-scale water purification as a proof of concept. This study demonstrates the importance of reaction design in photocatalytic applications and will promote future advancements in green, solar-driven H2O2 production.
Broader contextThe industrial synthesis of H2O2 is based on the anthraquinone auto-oxidation process, which requires high-energy consumption and an expensive metal catalyst. Solar photocatalysis has been suggested as a green and sustainable process to produce H2O2. Although there have been significant advances in materials design to improve the H2O2 production efficiency, the photocatalytic H2O2 production performance remains insufficient to meet industrial demands. In this study, we design a new reaction environment using aryl alcohol as an organic photo-oxidation substrate. With a metal-free covalent triazine framework (CTF) as a model photocatalyst, we combine photo-oxidation with O2 reduction under fully organic conditions for the co-production of H2O2 and aryl aldehyde. Furthermore, this organic system allows for the light-activated autocatalytic oxidation of aryl alcohol, facilitating O2 reduction kinetics by photo-initiated radicals. Inspired by the organic working solution of the anthraquinone process, we fine-tune the reaction composition by varying the water–aryl alcohol–solvent ratios to improve the photocatalytic pathway, achieving one of the highest solar-to-chemical conversion efficiencies ever reported. This study demonstrates that tailoring the reaction media is a simple but effective route for promoting overall solar H2O2 productivity, providing a new opportunity in photocatalytic reactions. |
Introduction
Solar photocatalysis provides a green route for the synthesis of hydrogen peroxide (H2O2) from O2 and sunlight.1 Among the different reported classes of photocatalysts, metal-free polymeric photocatalysts have attracted considerable attention because their structures and properties are amenable to modification,2,3 thus allowing significant improvement in the photocatalytic H2O2 production efficiencies. Compared to metallic semiconductors, polymeric photocatalysts enable the stable generation of H2O2, inhibiting undesirable H2O2 decomposition on the catalytic surfaces.4,5 Recent state-of-the-art examples of methods to prepare polymeric photocatalysts include heterojunction formation,6 single-atom anchoring,7,8 heteroatom doping,9,10 donor–acceptor (D–A) modulation,11–13 and defect control.14 Despite the excellent performance, sophisticated synthetic techniques for these photocatalysts are essential to realizing their practical applications.Reaction design is a better alternative for improving the photocatalytic efficiency without synthetic modification.15,16 A typical example of rational design is the integration of photo-oxidation reactions with photocatalytic O2 reduction such that no sacrificial agents are necessary to effectively initiate the O2 reduction.17,18 This cooperative coupling closely resembles the industrial anthraquinone process that generates H2O2 in an oxidation reaction.19 In contrast to the anthraquinone process, the integrated photoredox process does not require a catalytic regeneration cycle that involves expensive noble metals and H2 gas, and will continuously produce two products at each redox reaction.9 Inspired by the concept of an organic working solution (OWS) used in the anthraquinone process, we sought to fine-engineer the reaction media in the integrated photoredox reaction to further accelerate the reactivity.19
Benzyl alcohol (BzOH) is a suitable oxidation substrate for such photoredox reactions because it is a readily available liquid that produces benzaldehyde (BzCHO) as a valuable product.20,21 Although H2O2 is known to form during the photo-oxidation processes, it was generally considered as a byproduct.20,21 Recently, Yamashita et al. reported the photocatalytic production of H2O2 in a BzOH/water two-phase system wherein BzOH was used as an electron donor.17 Despite the frequent use of BzOH in photocatalysis, its autocatalytic conversion under UV irradiation has only recently drawn attention.22,23 The self-catalyzing attributes of photo-excited organic molecules can provide an opportunity to facilitate O2 reduction in the photocatalytic reaction. If sunlight itself activates the oxidation substrates, not only photocatalysis but also autocatalysis may involve O2 activation and proton supply, making O2 reduction much more favorable. Despite this intriguing property, a fully organic reaction environment to provide both, auto- and photocatalytic pathways, has never been considered in solar H2O2 production.
Herein, we report the rational design of an OWS for solar H2O2 production via cooperative auto- and photocatalytic aryl alcohol oxidation. The intrinsic reactivity of a model polymeric photocatalyst was evaluated in organic environments, whereby the reaction composition was modulated using water–alcohol–solvent mixtures for promoting photocatalytic O2 reduction. Simultaneously, the light-driven auto-oxidation of aryl alcohol in the OWS was identified as a pivotal contributor to H2O2 production by generating the photo-initiated free radicals for the O2 activation. In situ electron paramagnetic resonance (EPR) and electrochemical characterization revealed the origin of improved H2O2 production in fine-tuned OWS, proving synergistic auto- and photo-catalytic pathways. The use of the designed OWS led to one of the highest solar-to-chemical conversion efficiencies (1.1%) reported to date, with an H2O2 production rate of 46.9 mmol h−1 g−1. The integrated photoredox reaction with auto-oxidation is demonstrated to be a highly effective method for converting solar energy to valuable chemicals, which emphasizes the significance of reaction design in photocatalytic applications.
Results and discussion
Synthesis and characterization of CTF
By taking BzOH as a representative example of an aryl alcohol substrate, photocatalysts should possess a strong oxidation potential (up to 1.78 V vs. Ag/Ag+)24 and a sufficient band gap for photoinduced electron transfer in O2 reduction (−0.53 V vs. Ag/Ag+).25 To investigate the cooperative coupling of alcohol oxidation and O2 reduction, a covalent triazine framework (CTF) was selected as a model polymeric photocatalyst to achieve fine band alignment by donor–acceptor combination. Typically, CTF-Ph (also known as CTF-126) was synthesized via solid-phase cyclization polymerization, resulting in the quantitative yield of an off-white powder (Fig. 1a). The optical band gap of CTF-Ph was determined to be 2.87 eV with a visible-light absorbance of up to 430 nm (Fig. 1b). Cyclic voltammetry revealed that the reduction potential of CTF-Ph was −0.70 V vs. Ag/Ag+ (Fig. 1c) and the corresponding oxidation potential was estimated to be 2.17 V vs. Ag/Ag+ by subtracting the reduction potential from the band gap. CTF-Ph showed a high oxidation potential due to the electron-donating ability of the phenyl ring, which is adequate for the selective oxidation of BzOH.20 All the characterization data of the CTF photocatalyst are provided in the ESI† (Fig. S1–S5). | ||
| Fig. 1 (a) Solid-phase polymerization of CTF-Ph as an off-white powder. (b) UV-vis diffusion reflectance spectrum of CTF-Ph with an inset of the corresponding Tauc plot. (c) Electronic band structure of CTF-Ph determined by cyclic voltammetry. | ||
Effect of solution composition on photocatalytic H2O2 production
H2O2 production using the CTF-Ph photocatalyst with its suitable redox potentials was demonstrated under aqueous and organic conditions. As shown in Fig. 2a, CTF-Ph affords 4.9 μmol of H2O2 in O2-saturated pure water over 3 h, which is consistent with the results obtained using a previously reported CTF photocatalyst.27 When the reaction was conducted in an Ar-saturated atmosphere, a trace amount of H2O2 was obtained, indicating that H2O2 was primarily generated by the two-electron O2 reduction (Fig. S6, ESI†). The H2O2 production efficiency was further enhanced by the quantitative addition of hole scavengers to water. When the typical hole scavenger isopropanol (IPA) was used in combination with CTF-Ph, a higher amount of H2O2 was produced (34.3 μmol). When BzOH was used as a hole scavenger, the amount of H2O2 produced significantly increased to 377.2 μmol after 3 h of solar irradiation. CTF-Ph is known as a low-performing photocatalyst owing to its large energy band gap;28 hence, the drastic increase in H2O2 production after the addition of BzOH is unexpected. Moreover, the current density–voltage plot of CTF-Ph exhibited a high anodic peak current for BzOH oxidation (Fig. S7, ESI†), indicating that BzOH plays a crucial role as a hole scavenger.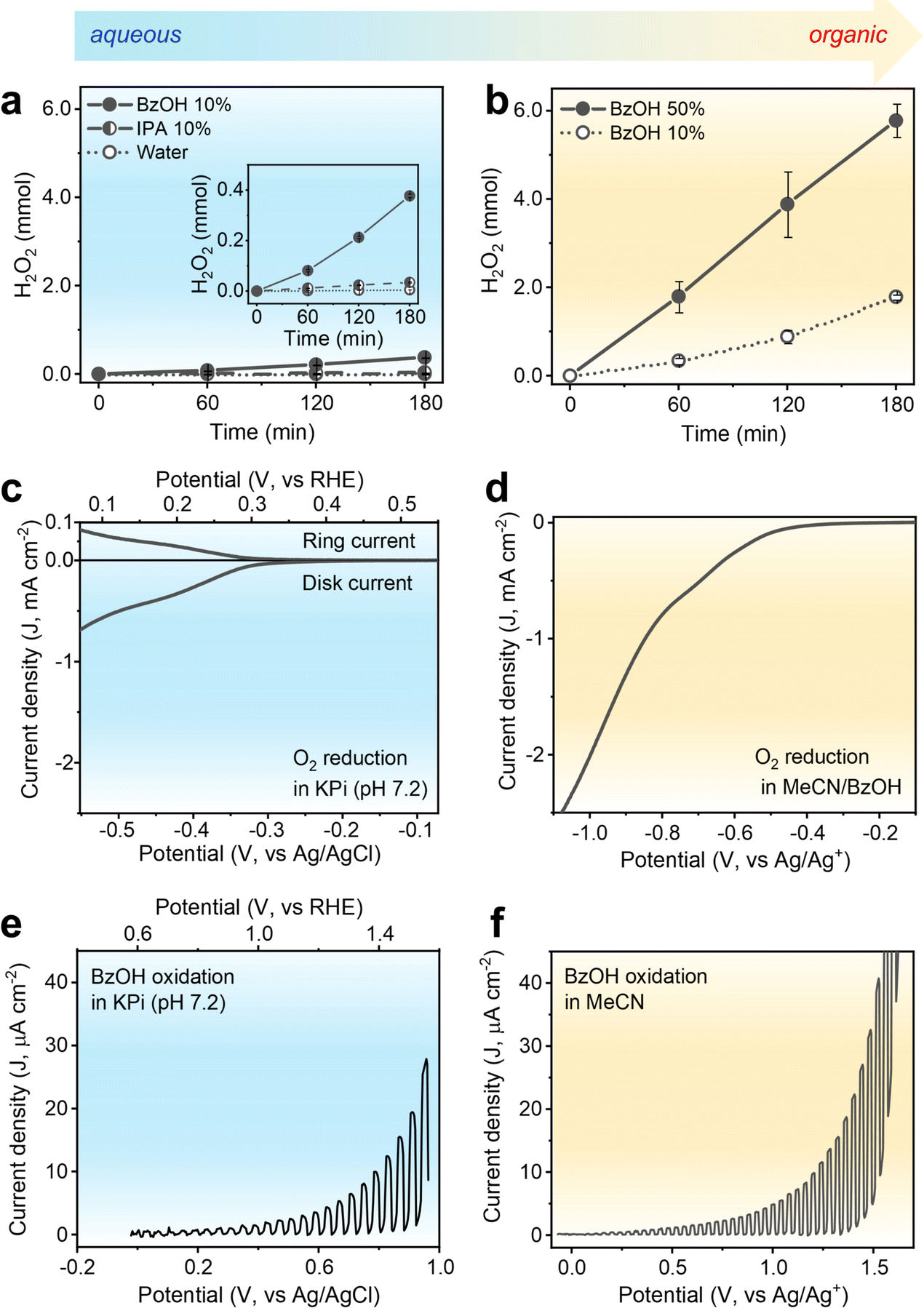 | ||
| Fig. 2 (a) Time-dependent photocatalytic H2O2 production using CTF-Ph under aqueous conditions. Reaction conditions: O2-saturated water (27 mL), 10% hole scavenger (3 mL; benzyl alcohol (BzOH) or isopropanol (IPA)), photocatalyst (50 mg), AM 1.5G simulated sunlight (∼980 W m−2), 298 K. (b) Photocatalytic H2O2 production under organic conditions. Reaction conditions: BzOH (3 mL for 10% or 15 mL for 50%), O2-saturated acetonitrile (MeCN) (27 mL or 15 mL), photocatalyst (50 mg), simulated sunlight, 298 K. (c) Rotating ring-disk electrode (RRDE) voltammogram of O2-saturated 0.1 M BzOH in 0.1 M phosphate buffer (KPi, pH 7.2). The applied potential on the Pt ring electrode was 1.0 V vs. Ag/AgCl. (d) RRDE voltammogram of O2-saturated 0.1 M BzOH in MeCN-0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6). Photoelectrochemical oxidation of 0.1 M BzOH using the CTF-Ph photoelectrode in (e) aqueous 0.1 M KPi and (f) 0.1 M TBAPF6-MeCN under simulated sunlight. All the LSV scans were performed at a scan rate of 10 mV s−1, and the rotating speed of the RDE was 1600 rpm. | ||
The BzOH-rich environment was thus achieved under organic conditions with acetonitrile (MeCN) as the solvent. As shown in Fig. 2b, H2O2 production under organic conditions is one order of magnitude higher than that under aqueous conditions. The reaction in the presence of CTF-Ph and 10 vol% of BzOH produced 1.79 mmol of H2O2 under light irradiation for 3 h. A higher loading of BzOH (up to 50 vol%) increased the H2O2 production up to 5.78 mmol, and thereafter, the BzOH loading was maintained at 50 vol% in further experiments. Other aryl alcohol substrates, such as substituted BzOHs and secondary aryl alcohols, can also be used as hole scavengers in photocatalytic H2O2 production (Fig. S8, ESI†).
To understand the origin of the discrepancy between the performances obtained under aqueous and organic conditions, electrochemical evaluation of CTF-Ph was conducted. The electrochemical O2 reduction was analyzed via rotating ring-disk electrode (RRDE) measurements. The linear voltammetry sweep (LSV) curve of CTF (Fig. 2c) in an aqueous potassium phosphate buffer (KPi, pH 7.2) showed that the onset potential for the O2 reduction current was approximately −0.2 V vs. Ag/AgCl and the calculated H2O2 selectivity was ∼60% (Fig. S9, ESI†). In contrast, the LSV curve of CTF-Ph in the BzOH–MeCN solution showed a substantial increase in the O2 reduction current (Fig. 2d) compared to that under aqueous conditions. This demonstrates that the O2 reduction activity of CTF can be significantly enhanced by replacing an aqueous electrolyte with an organic one.
The photoelectrochemical oxidation properties of CTF-Ph, which is the other half of the photoredox system, were examined. The LSV curve under aqueous conditions indicates a clear photo-response for water oxidation with an onset potential shift of ∼0.7 V (from 0.9 to 0.2 V vs. Ag/AgCl) (Fig. S10, ESI†). When 0.1 M BzOH was added to the aqueous electrolyte, CTF-Ph demonstrated a cathodic shift in the onset of the photocurrent (Fig. 2e). This is because the major oxidation reaction changed from water oxidation to BzOH oxidation. The photovoltage of CTF also slightly increased from 0.7 to 1.0 V in the presence of BzOH. The addition of IPA to water, on the other hand, resulted in a minor increase in photocurrent as a less efficient hole scavenger (Fig. S10, ESI†). With the BzOH-rich environment in MeCN, the CTF-Ph photoelectrode exhibited an additional enhancement in both the photovoltage (from 1.0 to 1.5 V) and photocurrent for BzOH oxidation compared to those in the aqueous electrolyte (Fig. 2f). This indicates that BzOH oxidation is thermodynamically and kinetically more facile than water and IPA oxidation, thus enhancing the O2 reduction activity of the CTF by improving the photogenerated charge separation.
Electrochemical assessments indicated that the O2 reduction and BzOH oxidation activities of CTF-Ph were affected by the solution composition. Nonetheless, the intrinsic activity of CTF in different media does not fully account for the enhanced H2O2 production under organic conditions. The potential auto-oxidation of BzOH in the UV region is an important aspect to investigate. The control experiment showed that a quantitative amount of H2O2 was produced under simulated sunlight illumination even without the addition of CTF-Ph (Fig. S11, ESI†). BzCHO was generated in almost the same amount as H2O2, implying that their production is interrelated. Notably, these two products were not detected when the organic solution was irradiated with visible light (>400 nm), demonstrating that the UV-initiated autocatalytic reaction under organic conditions significantly contributed to the generation of H2O2 and BzCHO.
Solar-driven H2O2 production in the designed OWSs
Since organic conditions enhanced the solar H2O2 production, several OWSs were designed by using different molar ratios of water to BzOH (0, 10, 50, 66, and 98.5 mol%) (Table S1, ESI†). The effect of water on the photocatalytic activity of the CTF and the autocatalytic contribution was determined by measuring the amounts of H2O2 and BzCHO produced. Significant amounts of H2O2 and BzCHO were produced without a photocatalyst (denoted as “BLK”), with the maximum amounts of H2O2 and BzCHO (4.90 and 6.17 mmol, respectively) produced under water-free conditions. The performance with BLK rapidly decreased with the addition of water. However, in the presence of CTF-Ph, the overall production of H2O2 and BzCHO was higher than that obtained under BLK conditions. Furthermore, the photocatalytic efficiency of the CTF increased with the addition of small amounts of water to the OWS. For instance, CTF-Ph demonstrated the maximum H2O2 and BzCHO production (7.04 and 7.44 mmol, respectively) in the OWS for 3 h with a 10 mol% addition of water (Fig. 3a and b); however, a small amount of benzyl benzoate (BBA, 0.11 mmol) was generated as a side product29 (Fig. S12, ESI†). The formation of H2 gas by a competing reaction of proton reduction was marginal in the OWS even with the excess water (Fig. S13, ESI†). To the best of our knowledge, our method of solar-driven H2O2 production using an OWS and CTF-Ph resulted in one of the highest recorded solar-to-chemical conversion (SCC) efficiencies of up to 1.1% with a H2O2 production rate of 46.9 mmol h−1 g−1, comparable with the most efficient water splitting photocatalyst (Fig. 3c and Table S2 for a full comparison, ESI†).5,7,30,31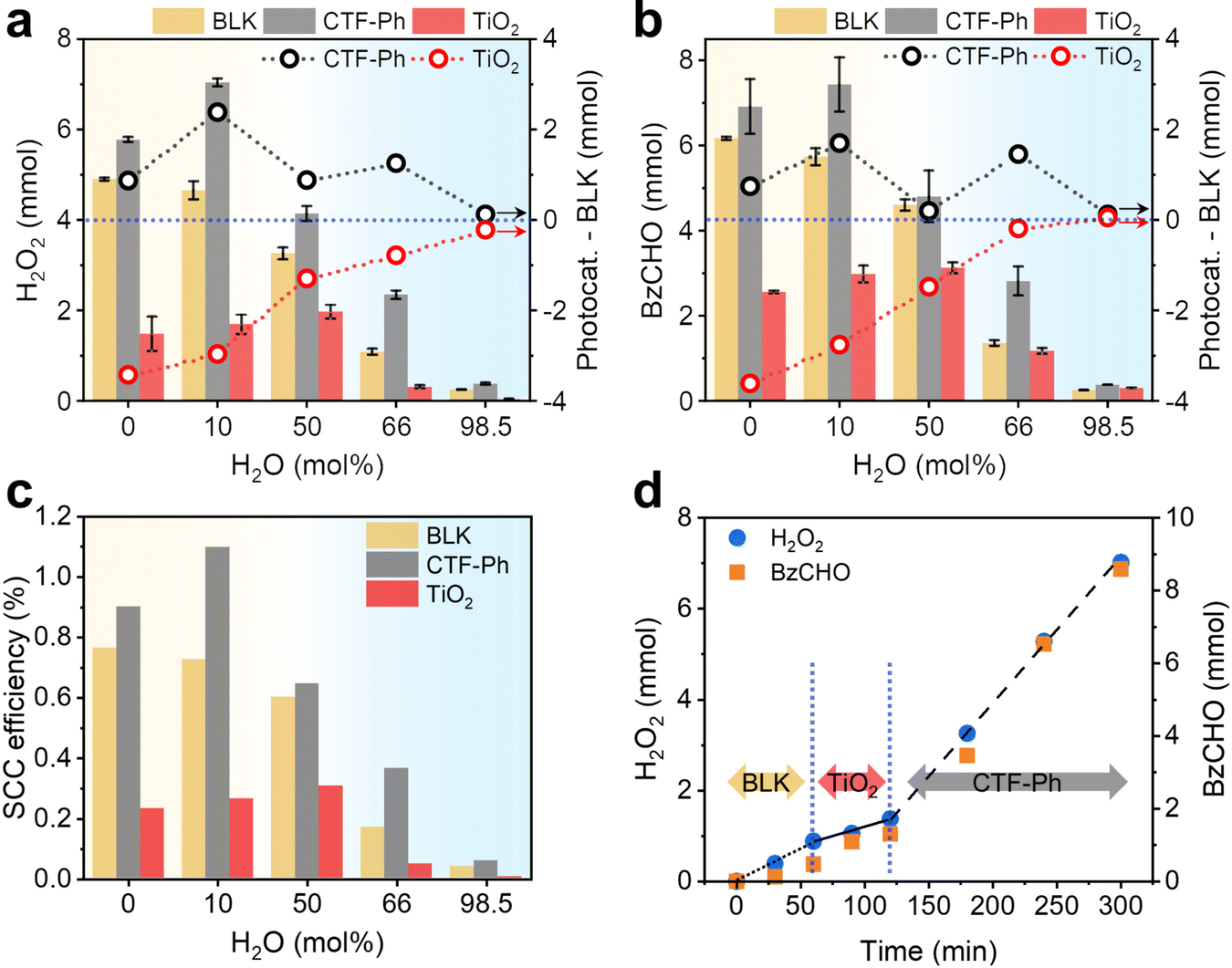 | ||
| Fig. 3 Reaction design with varying molar ratios of water to BzOH in MeCN. Solar-driven production of (a) H2O2 and (b) benzaldehyde (BzCHO) in the organic working solution (OWS) with and without the photocatalyst (BLK). The photocatalytic effect was estimated by subtracting the amount of products obtained under BLK conditions from those obtained using the photocatalysts (Photocat.-BLK). (c) Solar-to-chemical conversion (SCC) efficiencies of H2O2 production under AM 1.5G simulated sunlight irradiation for 3 h. (d) Sequential shift of photocatalysts in the OWS. Reaction conditions: photocatalyst (50 mg), organic solution (30 mL; 50% BzOH, 50% MeCN, 10 mol% H2O), simulated sunlight (∼980 W m−2), O2 (1 bar), 298 K. In the shift, the photocatalyst was filtered through a 0.22 μm hydrophilic PTFE syringe filter and O2 was purged in the reaction solution. | ||
Despite the significant contribution of autocatalytic oxidation toward H2O2 production under solar irradiation, the photocatalytic effect remained evident. For instance, P25 TiO2 as a control photocatalyst with a strong oxidation potential (2.69 V vs. Ag/Ag+, Fig. S1, ESI†) exhibited far lower production efficiencies, generating a maximum of 1.97 and 3.13 mmol of H2O2 and BzCHO, respectively, with a 50 mol% addition of water (Fig. 3a and b). The photocatalytic contribution was determined by subtracting the amounts of H2O2 and BzCHO produced using BLK from those obtained when the photocatalyst was used (Photocat. – BLK in Fig. 3a and b). When TiO2 was used, an adverse effect on H2O2 and BzCHO production was observed, indicating that the reaction was governed by the type of photocatalyst used (Fig. S14, ESI†). Because of this dependence, sequentially changing the photocatalyst in the reaction at each stage changed the reaction performance (Fig. 3d). TiO2 has insufficient potential for O2 reduction (Fig. S1, ESI†) and is likely to decompose H2O2 under both aqueous and organic conditions (Fig. S15 and S16, ESI†). This suggests that metal-free photocatalysts, such as common polymeric photocatalysts, are more suitable for these reactions (Fig. S17, ESI†).
Mechanism of solar-driven H2O2 production via interplay between auto- and photo-catalysis
To reveal the reaction mechanism in the OWS, the radical intermediate was monitored by EPR measurements under simulated sunlight irradiation. The selective O2 reduction on CTF-Ph was evidenced by the 5,5-dimethyl-1-pyrroline N-oxide (DMPO)-hydroperoxyl (˙OOH) adduct (g = 2.00561; AN = 13.5 G; AH,β = 11.0 G; AH,γ = 1.30 G) (Fig. 4a).32,33 In 100% BzOH in air, a clear sextet spectrum was obtained by CTF-Ph, which is attributed to a DMPO-benzyl (OR˙) adduct (g = 2.00561; AN = 15.3 G; AH = 22.2 G) (Fig. 4b),32,33 indicating that BzOH oxidation mainly proceeds via an α-hydroxybenzyl radical intermediate.24,34 Both DMPO-˙OOH and DMPO-OR˙ adducts were also observed in BLK, implying that autocatalysis takes place by free radical reactions. | ||
| Fig. 4 Electron paramagnetic resonance (EPR) spectra of (a) DMPO-hydroperoxyl (˙OOH) and (b) DMPO-benzyl (OR˙) radical with CTF-Ph and without the photocatalyst (BLK) under simulated sunlight irradiation (∼980 W m−2). Reaction conditions: (a) BzOH (1.5 mL), MeCN (1.5 mL), O2 (purged for 30 min), 298 K, DMPO (20 μL); (b) BzOH (3 mL), air (exposed to air for 3 h), 298 K, DMPO (20 μL). EPR spectra were obtained at 0, 30, and 60 min of irradiation. With the photocatalyst, CTF-Ph (10 mg) was added in the reaction vessel (4 mL vial). The asterisk indicates a signal from the Mn marker used for calibration. Time-dependent in situ EPR spectra of (c) CTF-Ph and (d) BLK in an O2-saturated OWS (50% BzOH, 50% MeCN, 5 mol% H2O). (e) Double integral of the in situ EPR signals for CTF-Ph and BLK in the absence and presence of water (5 mol%) in the OWS. (f) Photocatalytic performance of CTF-Ph in the OWS with the addition of radical scavengers. Copper chloride (CuCl2), potassium iodide (KI), and 1,4-benzoquinone (BQ) were tested as photogenerated electron (e−), photoinduced hole (h+), and hydroperoxyl radical (O2˙−) scavengers, respectively. Reaction conditions: CTF-Ph (20 mg), scavenger (166.7 μmol), organic solution (10 mL; 50% BzOH, 50% MeCN), simulated sunlight, O2 (1 bar), 298 K. | ||
In situ EPR profiles further provided evidence of the progress of the photo- and auto-catalytic reactions. In the photocatalytic reaction with water (CTF-Ph + H2O), the strong quartet signals of the DMPO-˙OOH adduct were mainly observed at an early stage (Fig. 4c and Fig. S18, ESI†). The signal intensity reached a maximum within a few minutes and began to decrease with O2 consumption. The bottom projection showed the evolution of the DMPO-OR˙ adduct at 334 and 339 mT after approximately 10 min. In contrast, in the autocatalytic reaction with water (BLK + H2O), the intensity of the DMPO-˙OOH adduct signals was significantly lower and quickly decayed (Fig. 4d). Instead, a strong peak from the DMPO-OR˙ adduct was observed on the projection in BLK + H2O. Time-resolved quantitative analysis showed that the photogenerated radical signal was more intense and long-lived in CTF-Ph + H2O compared to that in BLK + H2O (Fig. 4e). Although a higher level of radical formation was also apparent with CTF-Ph compared to that with BLK in the absence of water (Fig. S19, ESI†), the addition of water further enhanced the radical generation. This observation was consistent with the improved H2O2 production in the water-containing OWS with CTF-Ph.
First, in BLK as autocatalysis, the formation of hydroperoxyl and α-hydroxybenzyl radicals was substantially lower than that in the presence of CTF-Ph (Fig. 4a and b). Direct activation of BzOH under UV irradiation is known to form an alcohol radical cation and subsequently an alkoxyl radical;23 however, this was not mainly observed under solar irradiation. As shown in Fig. S11 (ESI†), the autocatalytic reaction generates H2O2 under irradiation with UV-A light (315–400 nm), which is consistent with the light absorption range of the BzCHO solution (Fig. S20, ESI†). Even in pure BzOH, a minor amount of BzCHO exists at a concentration of 1414.5 ppm (Fig. S21a, ESI†). Under solar irradiation, BzCHO is excited to the 1n,π* state by absorbing light longer than 300 nm, and the singlet state of BzCHO is then transformed to the 3n,π* state by intersystem crossing.35 The excited triplet state of BzCHO can abstract hydrogen from BzOH to generate an α-hydroxybenzyl radical that is responsible for O2 activation.36 The addition of water under the BLK conditions led to a rapid decrease in production efficiency, which could be ascribed to the rate-retarding effect of water in the hydrogen abstraction reaction.37,38
Intentionally spiking the BLK solution with a minimal amount of BzCHO, however, did not always increase the H2O2 production after solar irradiation (Fig. S21b–d, ESI†). By increasing the BzCHO content in BzOH, the formation of the side product BBA could rather increase. Notably, benzoic acid as an over-oxidized product was not detected under both BLK and CTF-Ph conditions, indicating that BBA did not result from the esterification of BzOH with benzoic acid. With a higher initial BzCHO concentration, a larger amount of benzoyl radicals can be produced by the decomposition of the excited triplet state of BzCHO.36 On the other hand, the photocatalytic oxidation of BzOH generated an alkoxyl radical under visible light as a minor intermediate (Fig. S22, ESI†). The coupling between the benzoyl radical and alkoxyl radical led to the production of a larger amount of BBA side product. A catalytic amount of BzCHO in pure BzOH was thereby sufficient to attain the autocatalytic pathway without forming the excessive side product. Nevertheless, the amount of BzCHO can vary depending on the scale of the OWS, which should be a key consideration when scaling up the reaction in future studies.
Along with autocatalysis, the photocatalytic role of CTF-Ph in the OWS was further investigated. Control experiments with radical scavengers proved the importance of photogenerated electrons and holes in the O2 reduction and alcohol oxidation processes (Fig. 4f). The H2O2 and BzCHO production efficiencies of CTF-Ph were both affected by the addition of electron and hole scavengers compared to those in the standard reaction (STD). In particular, the BzCHO production significantly decreased by the addition of a h+ scavenger. The quenching of the O2˙− radical also decreased the BzCHO production of CTF-Ph, indicating that the oxidation of the BzOH radical intermediate by the O2˙− radical is a critical pathway for BzCHO formation. Although the formation of the BBA side product was insignificant in STD, it was somewhat facilitated when the O2 reduction was suppressed by scavenging photoinduced electrons and O2˙−. Therefore, the selective formation of H2O2 and BzCHO only occurs through cooperative O2 reduction and BzOH oxidation reactions.
The positive effect of water in the respective O2 reduction and BzOH oxidation reactions with CTF-Ph was electrochemically evaluated using H2O–MeCN mixtures (Table S3, ESI†). Notably, the amount of BzOH in the electrochemical studies was adjusted to be relatively smaller than that under the photocatalytic reaction conditions to circumvent inaccurate readings of the reaction currents on the electrodes as a result of the limited water miscibility of BzOH and possible surface-impeding effects. When the water content was in the range of 0–50 vol%, the current density of BzOH oxidation was in the range of 0.9–1.4 mA cm−2, which then decreased to 0.7 mA cm−2 when the water content was 60–100 vol% (Fig. 5a). Moreover, the O2 reduction current was significantly affected by the addition of a small amount of water to the electrolyte. An approximately 15-fold increase in the O2 reduction current was observed when the water content in the electrolyte increased from 0 to 10 vol%, followed by a rapid decrease in the current density when the water content was further increased from 50 to 100 vol%, indicating a volcano-like trend. The onset potentials for the O2 reduction and alcohol oxidation reactions shifted rapidly with the addition of water and became saturated at a water content of 10 vol% (Fig. 5b). The shifting direction was beneficial for both reactions: a cathodic shift for BzOH oxidation and an anodic shift for O2 reduction. The onset potential differences between the two opposite reactions (ΔVonset) show that the addition of water up to 10 vol% decreases the overpotential from 1.9 to 1.1 V, which is significantly less than the band gap of CTF-Ph.
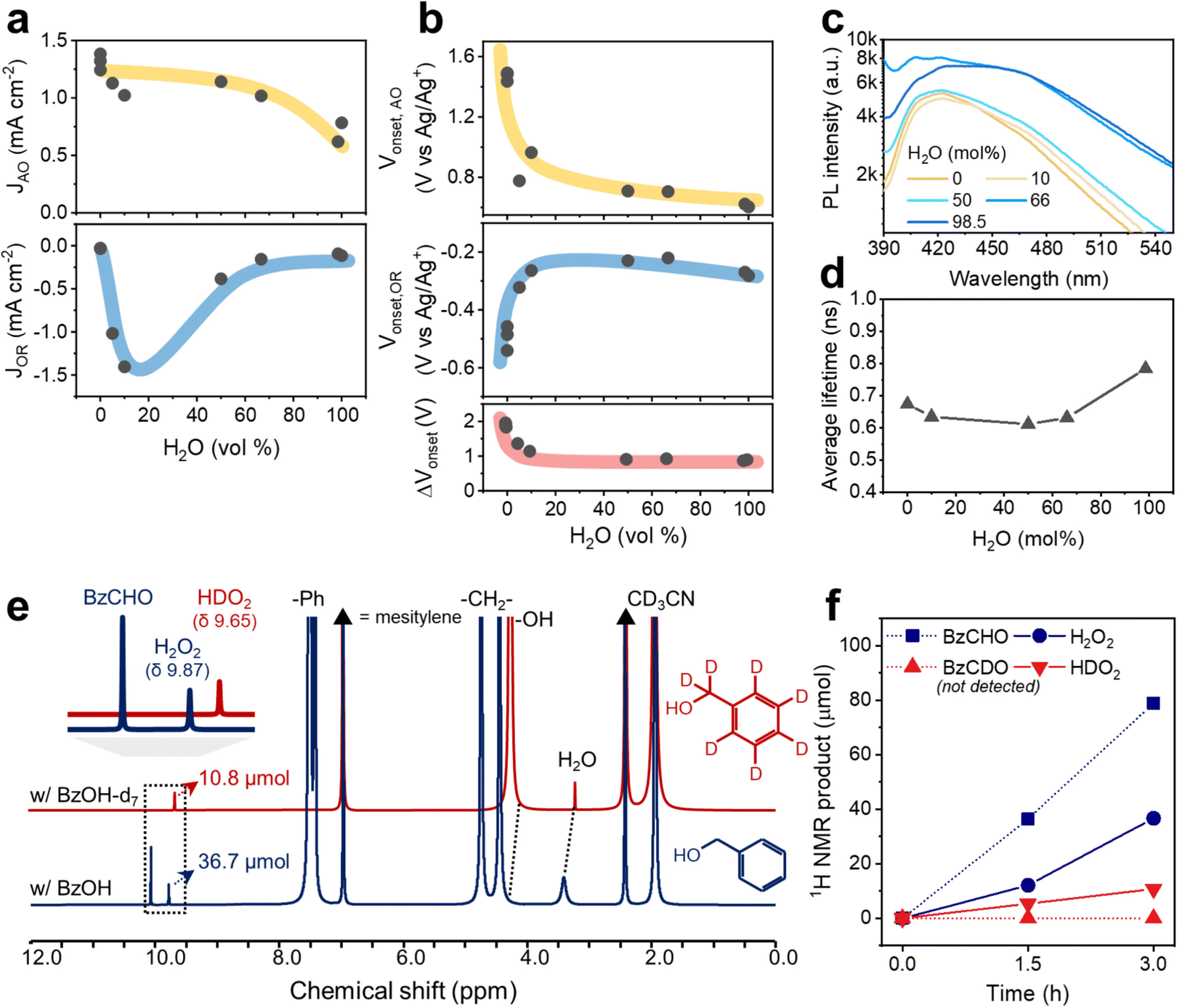 | ||
| Fig. 5 (a) Reaction currents and (b) onset voltage changes of CTF-Ph for O2 reduction (OR) and alcohol oxidation (AO) with different water contents in the H2O–MeCN mixture with 0.1 M BzOH. The reaction current for O2 reduction and BzOH oxidation was determined to be −1.5 V and +2.0 V vs. Ag/Ag+, respectively, and the onset potentials for both reactions were determined at the voltages when the absolute reaction current reaches 10 μA cm−2. 0.1 M LiClO4 was used as the supporting electrolyte. (c) Photoluminescence spectra and (d) average lifetimes of CTF-Ph dispersed in the OWS under O2-saturated conditions. (e) 1H NMR isotope experiment using BzOH-d7 and BzOH under O2-saturated conditions for 3 h (CD3CN, 298 K), and (f) the corresponding products quantified from the spectra using mesitylene as the internal standard. Reaction conditions: BzOH(-d7) (1.5 mL), CTF-Ph photocatalyst (15 mg), H2O (26 μL), CD3CN (1.5 mL), O2 (1 bar), simulated sunlight (∼990 W m−2), 298 K. | ||
In proportion to the decrease in overpotential, the charge separation of CTF-Ph increased in the H2O–BzOH–MeCN mixtures with higher O2 concentrations (Fig. S23, ESI†). From Fig. 5c, the steady-state photoluminescence (PL) peaks of CTF are largely quenched under the organic conditions, with a lower PL intensity under conditions from 0 to 50 mol% water. The photo-oxidation of BzOH was facilitated in the presence of a small amount of water, corresponding to the PL quenching of CTFs in the water–organic mixtures. The lifetime of the excited state in the OWS was estimated using time-correlated single-photon counting (Fig. S24, ESI†). The average lifetimes of the CTF were slightly shortened using 0 to 66 mol% water, in accordance with the PL measurements (Fig. 5d). The CTF photocatalysts are well dispersed in the OWSs, as evidenced by the low transmittance (<20%) (Fig. S25, ESI†), leading to a low likelihood of error from the polymer concentration in the PL measurement. The electrochemical and PL results indicate that the addition of water to the OWS provides a thermodynamically and kinetically favorable environment for the O2 reduction and alcohol oxidation reactions, enabling CTF-Ph to enhance the photocatalytic production of H2O2 and BzCHO.
Furthermore, we observed these enhanced O2 reduction and alcohol oxidation reactions facilitated by the addition of water by 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. S26, ESI†). Notably, the peaks derived from water and the hydroxyl group of BzOH were shifted downfield owing to the formation of a hydrogen bond between the two molecules because water behaves as the proton acceptor.39 This activated BzOH effectively, which is consistent with the aforementioned decrease in overpotentials (Fig. 5b).40,41 The photocatalytic contribution of BzOH oxidation to H2O2 formation was studied using deuterated BzOH-d7 (Fig. 5e). BzOH and BzOH-d7 produced 36.7 and 10.8 μmol of H2O2 (9.87 ppm) and HDO2 (9.65 ppm), respectively (Fig. 5f). The abstraction of the α-hydrogen atom from BzOH was faster than that of the α-deuterium from BzOH-d7,21 resulting in the formation of a lower amount of HDO2. This indicates that BzOH provides more protons for the formation of H2O2 than water18 because if water provided all the required protons, similar amounts of H2O2 and HDO2 would have been formed in both cases. In addition, isotope experiments indicated that proton abstraction from the –CH2OH group on BzOH is the rate-determining step (RDS). Therefore, the autocatalytic generation of α-hydroxybenzyl radicals strongly contributes to the overall alcohol oxidation reaction, helping to overcome the RDS of the photocatalytic routes.
The plausible mechanism of solar H2O2 production in OWSs is proposed in Fig. 6. Under solar irradiation, photo-excitation of BzCHO in BzOH forms a higher excited triplet state of BzCHO, which can initiate the conversion of BzOH to the α-hydroxybenzyl radical by hydrogen atom transfer (HAT)42 (autocatalysis, step 1). The reaction between the α-hydroxybenzyl radical and O2 (step 2) produces BzCHO and ˙OOH, possibly via the formation of the unstable benzoylperoxyl radical43 (step 3). Simultaneously, in the presence of CTF-Ph, the photocatalytic route further improves the reaction kinetics. For instance, under light illumination, the photogenerated electrons in the CTF react with O2 to form O2˙−, which is a proton acceptor (photocatalysis, step a). BzOH is oxidized by the photoinduced hole to afford the BzOH radical cation (step b), which is followed by α-C–H abstraction by O2˙− to form ˙OOH and α-hydroxybenzyl radicals (step c). The successive photo-oxidation and proton abstraction of the benzyl radicals formed via auto- and photocatalysis by ˙OOH yield H2O2 and BzCHO as the final products (step d).
 | ||
| Fig. 6 Schematic of the mechanism of solar-driven H2O2 production showing the interplay between auto- and photo-catalysis in an OWS. | ||
The autocatalytic route efficiently produces α-hydroxybenzyl radicals that can activate O2 molecules to ˙OOH radicals. The subsequent conversion of ˙OOH to H2O2, though, is technically challenging due to the lack of proton and electron supply. On the other hand, the photocatalytic pathway shows the enhanced O2 reduction under organic conditions, however, the oxidation of BzOH to α-hydroxybenzyl radical is the RDS. Therefore, the α-hydroxybenzyl and ˙OOH radicals generated from the autocatalysis can be successfully converted into BzCHO and H2O2, respectively, by the photocatalytic redox reactions on CTF-Ph. Furthermore, autocatalysis is sensitive to water, showing a drastic degradation of the performance in the presence of water. In the photocatalytic pathway, water lowers the activation barriers of redox reactions and promotes proton transfer, balancing out the autocatalytic reaction. Therefore, in the designed OWS systems, the interplay between auto- and photo-catalysis effectively converts O2 into H2O2 under solar light with BzCHO as a value-added co-product.
Conceptual feasibility test
The solar-driven production of H2O2 and BzCHO was tested under long-term conditions to demonstrate the feasibility of the OWS. As shown in Fig. 7a, the amounts of H2O2 and BzCHO increase almost linearly during the reaction (50 h). No additional reactant but O2 was injected every 9 h. After the reaction, the OWS was extracted with water to separate H2O2 and BzCHO. The final amount of H2O2 obtained was 18.5 mmol, equivalent to 3.7 mmol g−1 h−1 and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 488 ppm, which demonstrates the excellent capability of our new OWS to continuously produce H2O2.44 The SCC efficiency was maintained at 0.32% even after 50 h of reaction. The extracted H2O2 was diluted and purified by reverse osmosis membrane filtration, which is a highly energy-effective technique compared to the thermal distillation used in the industrial anthraquinone process.19 With a permeate flux of 1.79 L m−2 h−1 bar−1, the membrane filtration afforded a few liters of aqueous H2O2 at a concentration of 103.4 ppm (Fig. 7b). The purity of the H2O2 solution was determined by 1H NMR spectroscopy, which exhibited a clean signal without impurities (Fig. S27a, ESI†). A high reduction in the total organic carbon of up to 98.4% was attained after membrane filtration (Fig. S27b, ESI†). The corresponding BzCHO concentration in the organic phase reached 29.6 mmol during light irradiation for 50 h, indicating a production efficiency of 5.9 mmol g−1 h−1. No apparent structural changes were observed in the 13C NMR and Fourier-transform infrared spectra of the recovered CTF-Ph after 50 h of reaction time (Fig. S28, ESI†). A slight extension of light absorbance in the UV-vis diffuse reflectance spectrum indicated the formation of coke due to the addition of α-hydroxybenzyl radicals on CTF-Ph during the prolonged light irradiation (Fig. S29, ESI†). Highly durable CTF-Ph could keep producing highly concentrated H2O2 solution in repeated cycles (Fig. S30, ESI†).
488 ppm, which demonstrates the excellent capability of our new OWS to continuously produce H2O2.44 The SCC efficiency was maintained at 0.32% even after 50 h of reaction. The extracted H2O2 was diluted and purified by reverse osmosis membrane filtration, which is a highly energy-effective technique compared to the thermal distillation used in the industrial anthraquinone process.19 With a permeate flux of 1.79 L m−2 h−1 bar−1, the membrane filtration afforded a few liters of aqueous H2O2 at a concentration of 103.4 ppm (Fig. 7b). The purity of the H2O2 solution was determined by 1H NMR spectroscopy, which exhibited a clean signal without impurities (Fig. S27a, ESI†). A high reduction in the total organic carbon of up to 98.4% was attained after membrane filtration (Fig. S27b, ESI†). The corresponding BzCHO concentration in the organic phase reached 29.6 mmol during light irradiation for 50 h, indicating a production efficiency of 5.9 mmol g−1 h−1. No apparent structural changes were observed in the 13C NMR and Fourier-transform infrared spectra of the recovered CTF-Ph after 50 h of reaction time (Fig. S28, ESI†). A slight extension of light absorbance in the UV-vis diffuse reflectance spectrum indicated the formation of coke due to the addition of α-hydroxybenzyl radicals on CTF-Ph during the prolonged light irradiation (Fig. S29, ESI†). Highly durable CTF-Ph could keep producing highly concentrated H2O2 solution in repeated cycles (Fig. S30, ESI†).
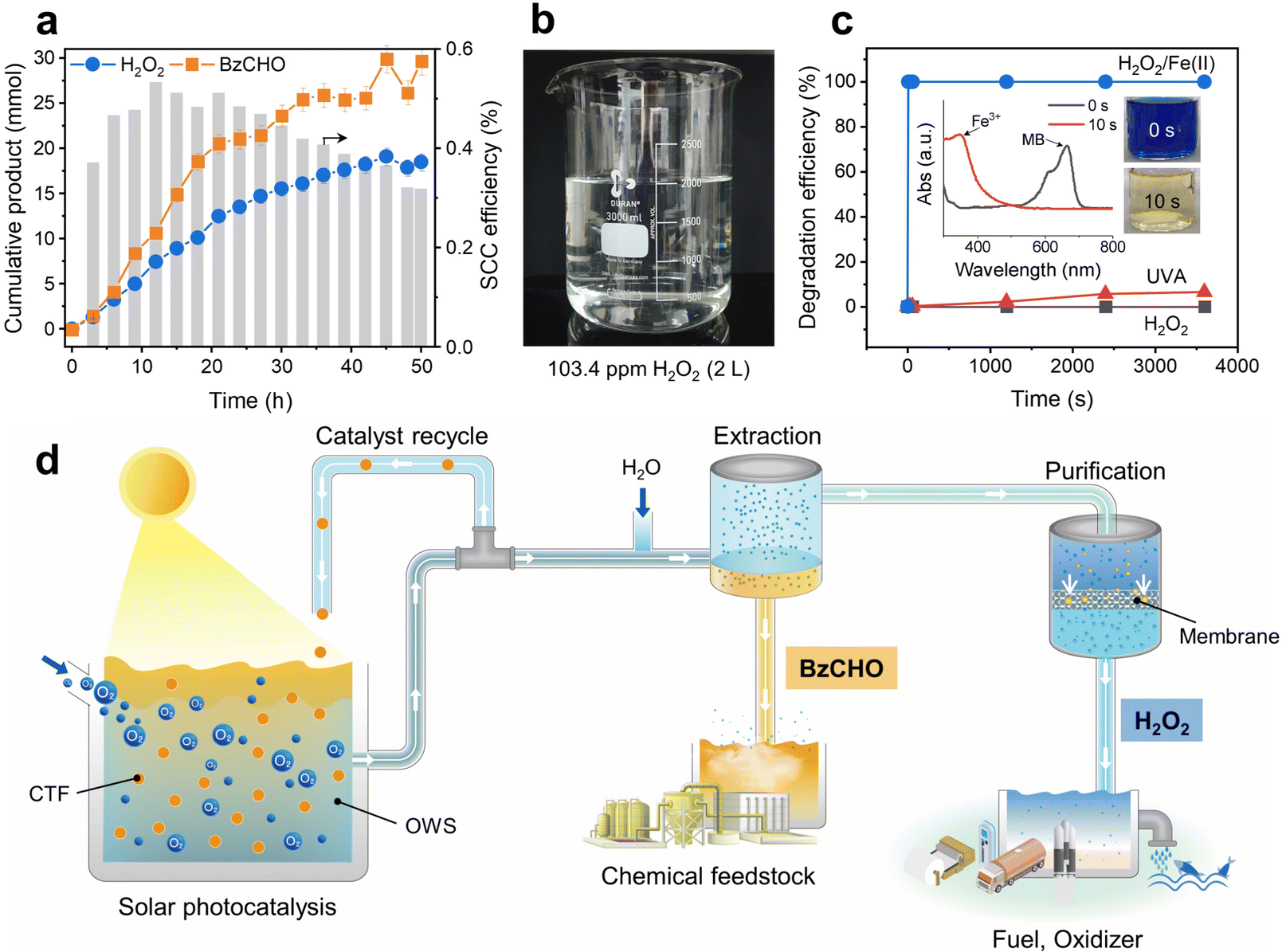 | ||
| Fig. 7 (a) Long-term solar-driven production of H2O2 and BzCHO during 50 h of light irradiation. The corresponding SCC efficiency for H2O2 production was calculated every 3 h. Reaction conditions: BzOH (30 mL), trifluorotoluene (TFT, 29.42 mL), water (0.58 mL), CTF-Ph (100 mg), O2 purging every 9 h, simulated sunlight (∼980 W m−2), 298 K. TFT was chosen for the long-term experiment not only to promote O2 dissolution in the media but also to extract H2O2 using water. (b) Purified H2O2 aqueous solution with an average concentration of 103.4 ppm after reverse osmosis filtration in the 2-L scale. (c) Fenton process for the degradation of organic pollutants in water using the purified H2O2 from the long-term solar H2O2 reaction. Inset: UV-visible spectra of the reaction of methylene blue (MB) solution with H2O2/Fe(II) in the first 10 s and the corresponding photographs of decolorization. Reaction conditions: MB (50 ppm), Fe(II) (3 mM), H2O2 (3 mM), pH 3. (d) Schematic of solar photocatalysis for the co-production of H2O2 and BzCHO and on-site applications. | ||
The high concentration of purified H2O2 from the long-term reaction can be directly utilized for large-scale water treatment applications. Organic pollutants can be degraded by adopting the Fenton process, in which the reaction between H2O2 and Fe(II) forms hydroxyl radicals as a strong oxidant.45Fig. 7c shows the degradation profile of methylene blue (MB) in a mixture of purified H2O2 and Fe(II) solution. The MB molecules rapidly degraded in only 10 s, demonstrating enhanced removal kinetics compared to the photolysis of MB under UV-A or normal H2O2 treatment. Despite the industrial production of H2O2 at high concentrations (27.5–70%), dilute H2O2 solutions are required for on-site applications such as advanced oxidation processes in wastewater treatment (10–100 ppm).46 Solar H2O2 production in the OWS can meet this requirement by producing dilute H2O2 solutions with high purity, thereby providing a practical alternative for decentralized H2O2 production (Fig. 7d).47
Conclusions
This study demonstrates a new reaction design of an organic working solution that can promote the solar production process of H2O2 using a model polymeric photocatalyst. The organic working solution afforded the successful integration of alcohol oxidation with O2 reduction for electron donor-free photocatalysis. Furthermore, the use of the organic solution resulted in the discovery of autocatalytic alcohol oxidation by photo-excitation of benzaldehyde impurity existing in fresh benzyl alcohol, significantly enhancing both H2O2 and benzaldehyde production. The photocatalytic pathway was improved by fine-engineering the reaction composition using water, demonstrating that tailoring the reaction media is a simple but effective route for promoting overall H2O2 productivity. Future work is suggested to improve the performance of the organic working solution by exploring autocatalytic organic molecule candidates. Photo-excitation of aromatic ketones would hold a key to enhancing solar-to-chemical conversion efficiency further. In addition to autocatalysis, structural controls on polymeric photocatalysts are required for selective O2 reduction and extended light absorbance, but in a cost-effective manner. Finally, a system design that combines a scaled solar reactor with a membrane purification setup is needed. We believe that this study provides a breakthrough in photocatalytic reactions, bringing solar photocatalysis a step closer to industrial standards.Author contributions
D. K. L. and J. B. conceived and designed the project; B. C. M. and B. B. performed the experiments; B. C. M., D. K. L., and J. B. analyzed the data; K. A. I. Z., D. K. L., and J. B. discussed the results; D. K. L. and J. B. wrote the manuscript with contributions from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2021R1C1C1005774 to J. B.; No. 2020M3H4A1A02084590 and No. 2022M3A9F3082336 to D. K. L.). The authors acknowledge support from the National Research Council of Science and Technology (NST) grant (CPS21051-100) and institutional programs of KIST (2E31932 to J. B. and 2E31831 to D. K. L.). The authors sincerely thank Dr Chanhee Boo (KIST) for the reverse osmosis membrane filtration experiments, Dr Yeongran Hong (KIST) for the 1H NMR spectroscopy studies, and Sukyoun Son (KIST) for the GC-MS analysis.References
- Y. Sun, L. Han and P. Strasser, Chem. Soc. Rev., 2020, 49, 6605–6631 RSC
.
- J. Lee, O. Buyukcakir, T.-W. Kwon and A. Coskun, J. Am. Chem. Soc., 2018, 140, 10937–10940 CrossRef PubMed
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef
- J. H. Lee, H. Cho, S. O. Park, J. M. Hwang, Y. Hong, P. Sharma, W. C. Jeon, Y. Cho, C. Yang, S. K. Kwak, H. R. Moon and J.-W. Jang, Appl. Catal., B, 2021, 284, 119690 CrossRef CAS
- Y. Shiraishi, T. Takii, T. Hagi, S. Mori, Y. Kofuji, Y. Kitagawa, S. Tanaka, S. Ichikawa and T. Hirai, Nat. Mater., 2019, 18, 985–993 CrossRef CAS
- S. Wang, B. Cai and H. Tian, Angew. Chem., Int. Ed., 2022, 61, e202202733 CAS
- Z. Teng, Q. Zhang, H. Yang, K. Kato, W. Yang, Y.-R. Lu, S. Liu, C. Wang, A. Yamakata, C. Su, B. Liu and T. Ohno, Nat. Catal., 2021, 4, 374–384 CrossRef CAS
- T. Sun, S. Zhao, W. Chen, D. Zhai, J. Dong, Y. Wang, S. Zhang, A. Han, L. Gu, R. Yu, X. Wen, H. Ren, L. Xu, C. Chen, Q. Peng, D. Wang and Y. Li, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12692–12697 CrossRef CAS
- P. Zhang, Y. Tong, Y. Liu, J. J. M. Vequizo, H. Sun, C. Yang, A. Yamakata, F. Fan, W. Lin, X. Wang and W. Choi, Angew. Chem., Int. Ed., 2020, 59, 16209–16217 CrossRef CAS PubMed
- Y. Zhao, P. Zhang, Z. Yang, L. Li, J. Gao, S. Chen, T. Xie, C. Diao, S. Xi, B. Xiao, C. Hu and W. Choi, Nat. Commun., 2021, 12, 3701 CrossRef CAS PubMed
- Z.-A. Lan, M. Wu, Z. Fang, X. Chi, X. Chen, Y. Zhang and X. Wang, Angew. Chem., Int. Ed., 2021, 60, 16355–16359 CrossRef CAS
- P. B. Pati, G. Damas, L. Tian, D. L. A. Fernandes, L. Zhang, I. B. Pehlivan, T. Edvinsson, C. M. Araujo and H. Tian, Energy Environ. Sci., 2017, 10, 1372–1376 RSC
- C. Krishnaraj, H. Sekhar Jena, L. Bourda, A. Laemont, P. Pachfule, J. Roeser, C. V. Chandran, S. Borgmans, S. M. J. Rogge, K. Leus, C. V. Stevens, J. A. Martens, V. Van Speybroeck, E. Breynaert, A. Thomas and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 20107–20116 CrossRef CAS PubMed
- X. Zhang, P. Ma, C. Wang, L.-Y. Gan, X. Chen, P. Zhang, Y. Wang, H. Li, L. Wang, X. Zhou and K. Zheng, Energy Environ. Sci., 2022, 15, 830–842 RSC
- Q. Guo, F. Liang, X.-B. Li, Y.-J. Gao, M.-Y. Huang, Y. Wang, S.-G. Xia, X.-Y. Gao, Q.-C. Gan, Z.-S. Lin, C.-H. Tung and L.-Z. Wu, Chem, 2019, 5, 2605–2616 CAS
- M. G. Allan, M. J. McKee, F. Marken and M. F. Kuehnel, Energy Environ. Sci., 2021, 14, 5523–5529 RSC
- Y. Isaka, Y. Kawase, Y. Kuwahara, K. Mori and H. Yamashita, Angew. Chem., Int. Ed., 2019, 58, 5402–5406 CrossRef
- B. Wu, L. Zhang, B. Jiang, Q. Li, C. Tian, Y. Xie, W. Li and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 4815–4822 CrossRef
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef PubMed
- W. Huang, B. C. Ma, H. Lu, R. Li, L. Wang, K. Landfester and K. A. I. Zhang, ACS Catal., 2017, 7, 5438–5442 CrossRef CAS
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS
- M. J. Pavan, H. Fridman, G. Segalovich, A. I. Shames, N. G. Lemcoff and T. Mokari, ChemCatChem, 2018, 10, 2541–2545 CrossRef CAS
- P. K. Walia, M. Sharma, M. Kumar and V. Bhalla, RSC Adv., 2019, 9, 36198–36203 RSC
- X. Xiao, J. Jiang and L. Zhang, Appl. Catal., B, 2013, 142–143, 487–493 CrossRef CAS
- J. P. Torella, C. J. Gagliardi, J. S. Chen, D. K. Bediako, B. Colón, J. C. Way, P. A. Silver and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2337–2342 CrossRef
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef PubMed
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, 1904433 CrossRef PubMed
- C. B. Meier, R. S. Sprick, A. Monti, P. Guiglion, J.-S. M. Lee, M. A. Zwijnenburg and A. I. Cooper, Polymer, 2017, 126, 283–290 CrossRef
- X. Liu, B. Xu, J. Haubrich, R. J. Madix and C. M. Friend, J. Am. Chem. Soc., 2009, 131, 5757–5759 CrossRef CAS PubMed
- H. Hou, X. Zeng and X. Zhang, Angew. Chem., Int. Ed., 2020, 59, 17356–17376 CrossRef CAS PubMed
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed
- G. R. Buettner, Free Radical Biol. Med., 1987, 3, 259–303 CrossRef CAS PubMed
- A. Bosnjakovic and S. Schlick, J. Phys. Chem. B, 2006, 110, 10720–10728 CrossRef CAS PubMed
- S. Higashimoto, N. Suetsugu, M. Azuma, H. Ohue and Y. Sakata, J. Catal., 2010, 274, 76–83 CrossRef CAS
- H. Murai and K. Obi, J. Phys. Chem., 1975, 79, 2446–2450 CrossRef CAS
- M. A. Theodoropoulou, N. F. Nikitas and C. G. Kokotos, Beilstein J. Org. Chem., 2020, 16, 833–857 CrossRef
- R. E. Galian, G. Litwinienko, J. Pérez-Prieto and K. U. Ingold, J. Am. Chem. Soc., 2007, 129, 9280–9281 CrossRef PubMed
- M. J. Kamlet and R. W. Taft, J. Am. Chem. Soc., 1976, 98, 377–383 CrossRef
- M. Liu, Z. Zhang, H. Liu, Z. Xie, Q. Mei and B. Han, Sci. Adv., 2018, 4, eaas9319 CrossRef PubMed
- R. F. Duan, L. Cheng, Q. C. Zhang, L. S. Ma, H. Y. Ma and J. C. Yang, RSC Adv., 2015, 5, 83976–83984 RSC
- Y. Li, Y. Wei and W. Zhang, J. Electroanal. Chem., 2020, 870, 114251 CrossRef
- M. Cocivera and A. M. Trozzolo, J. Am. Chem. Soc., 1970, 92, 1772–1774 CrossRef
- G. Palmisano, G. Scandura, V. Augugliaro, V. Loddo, A. Pace, B. S. Tek, S. Yurdakal and L. Palmisano, J. Mol. Catal. A: Chem., 2015, 403, 37–42 CrossRef
- R. J. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308 CrossRef
- O. Primo, M. J. Rivero and I. Ortiz, J. Hazard. Mater., 2008, 153, 834–842 CrossRef PubMed
- S. L. Ulliman, D. B. Miklos, U. Hübner, J. E. Drewes and K. G. Linden, Environ. Sci.: Water Res. Technol., 2018, 4, 1321–1330 RSC
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ee02504c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |