
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Silver(I) complexes bearing heterocyclic thioamide ligands with NH2 and CF3 substituents: effect of ligand group substitution on antibacterial and anticancer properties†
Despoina
Varna
a,
Elena
Geromichalou
b,
Antonios G.
Hatzidimitriou
a,
Rigini
Papi
c,
George
Psomas
 a,
Panagiotis
Dalezis
b,
Paraskevas
Aslanidis
a,
Theodora
Choli-Papadopoulou
c,
Dimitrios T.
Trafalis
*b and
Panagiotis A.
Angaridis
*a
a,
Panagiotis
Dalezis
b,
Paraskevas
Aslanidis
a,
Theodora
Choli-Papadopoulou
c,
Dimitrios T.
Trafalis
*b and
Panagiotis A.
Angaridis
*a
aLaboratory of Inorganic Chemistry, Department of Chemistry, Aristotle University of Thessaloniki, 54124 Thessaloniki, Greece. E-mail: panosangaridis@chem.auth.gr
bLaboratory of Pharmacology, Medical School, National and Kapodistrian University of Athens, 75 Mikras Asias Street, 11527 Athens, Greece. E-mail: dtrafal@med.uoa.gr
cLaboratory of Biochemistry, Department of Chemistry, Aristotle University of Thessaloniki, Thessaloniki 54124, Greece
First published on 30th May 2022
Abstract
In recent years, there has been an increasing interest in the study of Ag(I) coordination compounds as potent antibacterial and anticancer agents. Herein, a series of Ag(I) complexes bearing phosphines and heterocyclic thioamide ligands with highly electronegative NH2- and CF3-group substituents, i.e. [AgCl(atdztH)(xantphos)] (1), [Ag(μ-atdztH)(DPEphos)]2(NO3)2 (2), [Ag(atdzt)(PPh3)3] (3), [Ag(μ-atdzt)(DPEphos)]2 (4), and [Ag(μ-mtft)(DPEphos)]2 (5), where atdztH = 5-amino-1,3,4-thiadiazole-2-thiol, mtftH = 4-methyl-5-(trifluoromethyl)-1,2,4-triazol-3-thiol, xantphos = 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene, and DPEphos = bis(2-diphenylphosphino-phenyl)ether, were synthesized, and their in vitro antibacterial and anticancer properties were evaluated. Complexes 1–4 bearing the NH2-substituted thioamide exhibited moderate-to-high activity against S. aureus, B. subtilis, B. cereus and E. coli bacterial strains. A high antiproliferative activity was also observed for 1–3 against SKOV-3, Hup-T3, DMS114 and PC3 cancer cell lines (IC50 = 4.0–11.7 μM), as well as some degree of selectivity against MRC-5 normal cells. Interestingly, 5 bearing the CF3-substituted thioamide is completely inactive in all bioactivity studies. Binding of 1–3 to drug-carrier proteins BSA and HSA is reasonably strong for their uptake and subsequent release to possible target sites. The three complexes show a significant in vitro antioxidant ability for scavenging free radicals, suggesting likely implication of this property in the mechanism of their bioactivity, but a low potential to destroy the double-strand structure of CT-DNA by intercalation. Complementary insights into possible bioactivity mechanisms were provided by molecular docking calculations, exploring the ability of complexes to bind to bacterial DNA gyrase, and to the overexpressed in the aforementioned cancer cells Fibroblast Growth Factor Receptor 1, affecting their functionalities.
1. Introduction
The introduction of platinum-based drugs in the treatment of cancer in the late 1980s marked the beginning of a new era in cancer therapies, establishing transition metal complexes as feasible therapeutic agents.1 However, despite their high efficacy, these compounds frequently cause a number of side effects, imposing limits in their clinical use.2–5 Nevertheless, the continuing and strong interest for the discovery of novel and low toxicity metal-based therapeutic agents led to the development of a large number of coordination compounds with chemotherapeutic potential, some of which have already been under clinical trials or approved for clinical use.6,7Ag(I) coordination compounds are currently attracting significant attention, owing to their recently recognized effective anticancer activity.8–15 Moreover, antiseptic and antibacterial properties of Ag(I) compounds have been known since ancient times.16–19 In general, the bioactivity potential of Ag(I) compounds is suggested to arise from their unique mechanisms of action. In particular, the mechanism of their antiproliferative activity differs from that of the DNA-targeting Pt-based drugs, as it is suggested to involve their interaction with the mitochondrial membrane and inhibition of thioredoxin reductase,20 leading to mitochondria initiated programmed cell death.21–23 Regarding their antimicrobial activity, the proposed mechanism of action involves their initial binding onto the membrane of the cells, followed by membrane damage and entrance into the cell (either in the form of Ag(I) complexes or solvated Ag(I) ions), and finally disruption of cell metabolism via interaction with cell enzymes or binding to subcellular components.24–28 Other attractive characteristics of Ag(I) complexes that stimulate research in this field include their low toxicity (compared to coordination compounds of other medicinally relevant metals), selectivity effects they occasionally exhibit,29 as well as their ability to overcome antibiotic drug resistance.30 Furthermore, considering that Ag(I) coordination compounds can be photoluminescent (in the presence of appropriate ligands), bioactive Ag(I) complexes can also be used for both therapeutic and imaging applications, as theranostic agents.23
The biological efficacy of Ag(I) complexes is strongly associated with their solubility, thermodynamic and kinetic stability, lipophilicity, redox properties, and ability to release Ag(I) ions. These properties are determined, and can appropriately be tuned, by the set of ligands embracing the metal center. Specifically, the employment of ligands with steric and electronic properties that allow strong coordination to the Ag(I) ions – therefore providing sufficient stability and protection (e.g., avoiding their precipitation in the presence of free Cl− ions) and ensuring their slow release into the cells – is of great importance as drug designing strategy towards the discovery of efficient Ag(I)-based pharmaceuticals.
Among the different types of ligands used for the synthesis of bioactive Ag(I) complexes, phosphines appear to be an interesting case, as they have resulted in a number of cases that exhibit significant in vitro activity against several types of microbes and cancers.31,32 Their favorable coordination to the soft Lewis acid Ag(I) ions, in combination with their intermediate lipophilicity, properly adjusts the kinetic stability and the hydrophilicity/lipophilicity balance of the complexes, resulting in enhanced antibacterial and cytotoxic efficiency, as well as, sometimes, selectivity towards specific cancer cells and pathogens.33 For example, [Ag(d3pype)2]NO3 where d3pype = 1,2-bis(di-3-pyridylphosphino)ethane was found to exert a strong cytotoxic effect against carcinoma (41M and 41McisR) cells due to its enhanced lipophilic character, whereas the more hydrophilic complex [Ag(PTA)4]PF6 showed an improved activity towards adenocarcinoma cells (e.g. MCF-7, SKOV-3) compared to carcinoma and lymphoma cells.34,35 Apart from phosphines, heterocyclic thioamides, which can coordinate to the Ag(I) ions through their exocyclic soft S donor atom, constitute another family of ligands that have also resulted in a number of biologically active Ag(I) complexes. Recent studies on the in vitro biological potential of some thioamide-containing Ag(I) complexes revealed a high efficiency against a variety of bacterial strains as well as high anticancer activity – even higher than that of cisplatin – against certain tumors.36–39 For example, the water soluble cluster compound {[Ag6(μ3-Hmna)4(μ3-mna)2][(Et3NH)]2·(DMSO)2·(H2O)} (H2mna = 2-mercapto-nicotinic acid) was found to be effective disinfectants against P. aeruginosa and S. aureus bacterial strains in contact lenses and showed in vitro toxicity against HEC T human corneal epithelial cells.40,41
Considering the above, mixed ligand phosphine/heterocyclic thioamide Ag(I) complexes appear to be an interesting class of compounds for investigation of their bioactivity, as they provide the opportunity to combine the beneficial properties of both types of ligands. Indeed, recent literature reports confirm the high bioactivity potential of complexes of this type. For example, Ag(I) complexes comprising PPh3 and N-substituted imidazolidine-2-thiones were found to exhibit moderate to high anti-microbial activity against a series of Gram-(+) and Gram-(−) bacterial strains, while showing IC50 values in the range of 6–33 μM against MG63 human osteosarcoma cells.42 In another case, cationic complex [Ag2(totp)(3-benzyl-1,3-thiazolidine-2-thione)2](NO3)2, where totp = tris(o-tolyl)phosphine, was found to inhibit the in vitro growth of MDA-MD-231 breast cancer cells but not HT-29 human colorectal adenocarcinoma cells.38 We also recently reported a number of heteroleptic Ag(I) complexes bearing the thioamides 5-methyl-1,3,4-thiadiazole-2(3H)-thione (mtdztH), 2-pyrimidinethiol (pymtH), 4-phenyl-imidazole-2-thione (phimtH) and 2,2,5,5-tetramethyl-imidazolidine-4-thione (tmimdtH) in combination with different phosphines, such as PPh3, bis[(2-diphenylphosphino)phenyl]ether (DPEphos), 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene (xantphos), and 1,2-bis(diphenylphosphino)ethane (dppe), which showed moderate-to-high antibacterial properties and cytotoxicity, and revealed the impact of the thioamide nature on the observed bioactivity.43,44
Aiming to extend our studies in this field, herein we present a series of Ag(I) complexes bearing a phosphine ligand in combination with a small size heterocyclic ring thioamide with highly electronegative substituents, i.e. 5-amino-1,3,4-thiadiazole-2(3H)-thione (atdztH) and 4-methyl-5-(trifluoromethyl)-1,2,4-triazol-3-thiol (mtftH), and their in vitro antibacterial and anticancer activity assessment. The choice of the particular thioamides was based on previously made observations that ligands with high-electronegativity substituents, such as NH2 and CF3 groups, have a significant impact on the biological properties of their complexes. In particular, it has been suggested that this type of functional groups promote the development of strong intermolecular interactions with several biological entities, such as cell membrane and enzyme active sites, augmenting the bioactivity of their complexes.45–49 Moreover, analogues of atdztH have been found to demonstrate significant in vitro and in vivo antitumor activity against a variety of tumor cells.50,51 The selection of mtftH was based on reports on the high potential of F-substituted organic compounds as efficient antimicrobial, anticancer, analgesic and antipyretic agents.52 In fact, the introduction of CF3-group substituents into the molecular structures of ligands of coordination compounds has been a popular strategy in medicinal inorganic chemistry, as it often enhances the biological efficacy of the complexes,53–55 by promoting the development of favorable electrostatic interactions, improving permeability of cell membrane, and affecting the metabolic profile of the complexes.56 Nevertheless, it should also be noted that there are reports that describe the effect of the CF3 group substitution as controversial.57 Finally, the use of phosphine co-ligands in these thioamide-containing complexes is anticipated to improve their kinetic stability in biological environments, while providing a balanced lipophilic/hydrophilic character, which is essential for efficient biological activity. The in vitro antibacterial activity of the complexes was evaluated against a series of Gram-(+) and Gram-(−) bacterial strains, while their in vitro cytotoxicity was explored against the well-established human cancer ovarian, cell lung cancer, prostate adenocarcinoma, and pancreatic adenocarcinoma cells. Finally, in silico molecular docking calculations on the crystal structures of E. coli and S. aureus DNA gyrases, as well as on the overexpressed in the aforementioned cancer cells Fibroblast Growth Factor Receptor 1 (FGFR1) were employed in order to shed light into the possible mechanism of antibacterial and anticancer activity of the complexes.
2. Results and discussion
2.1 Synthesis and characterization
For heteroleptic phosphine/heterocyclic thioamide Ag(I) complexes, the electronic and steric properties of the ligands, the protonation state of the thioamide as well as the coordinating ability of the X− anion of the AgX salts (used as starting materials for their syntheses) play important roles in the determination of their structural characteristics (e.g. metal coordination number/geometry and nuclearity), which, in turn, have a significant impact on their biological activity. Aiming to investigate potential structure/bioactivity relations within this family of complexes, in this work we focus in Ag(I) complexes with small-size heterocyclic ring thioamide ligands bearing high electronegativity substituents. In particular, by employing reactions of AgX salts (X = Cl−, NO3−) with the heterocyclic thioamides atdztH and mtftH (having NH2- and CF3-group substituents, respectively), either in their neutral or deprotonated form, in the presence of different phosphines, we synthesized the series of neutral or cationic, mono- and binuclear heteroleptic Ag(I) complexes 1–5 shown in Scheme 1. | ||
| Scheme 1 Syntheses of 1–5. (a) CH2Cl2 50 °C for 2 h, (b) CH3CN/CH3OH 70 °C for 2 h (c) CH3CN/MeOH, 65 °C for 2 h, (d) CH2Cl2, 22 °C for 2 h, (e) CH2Cl2/MeOH, 22 °C, 2 h. | ||
All complexes were synthesized following a one-pot/two-step experimental procedure that included treatment of an appropriate AgX salt with a phosphine in a suitable solvent, followed by addition of the corresponding thioamide or thioamidate (see S1.1 in ESI† for detailed synthetic procedures). In brief, addition of the neutral heterocyclic thioamide atdztH to a mixture of equimolar amounts of AgCl and xantphos in MeOH/CH3CN, followed by reflux for 2 h, afforded the neutral mononuclear complex [AgCl(atdztH)(xantphos)] (1). Using AgNO3 instead of AgCl in an analogous reaction with atdztH and DPEphos at room temperature, the dicationic binuclear complex [Ag(μ-atdztH)(DPEphos)]2(NO3)2 (2) was isolated. Addition of 1 equiv. of K+atdzt− to a mixture of equimolar amounts of AgNO3 and DPEphos in CH2Cl2 gave [Ag(μ-atdzt)(DPEphos)]2 (4), a neutral binuclear complex with the same structural motif as the cation of 2. When K+mtft− was used in a similar reaction, the structurally homologous complex [Ag(μ-mtft)(DPEphos)]2 (5) was obtained. Finally, addition of 1 equiv. of K+atdzt− to a mixture of AgNO3 in the presence of 3 equiv. of PPh3 resulted in the mononuclear complex [Ag(atdzt)(PPh3)3] (3). All complexes were isolated in the form of white or off-white microcrystalline solids in good yields (50–65%) after filtration of the corresponding reaction mixtures and subsequent crystallization from the respective filtrates. Compounds 1–5 are stable in air, both in the solid state and in solutions in organic solvents, for long periods of time. In addition, all compounds are thermally stable up to ca. 155–220 °C, while above this temperature range they decompose.
The structures of the complexes in the solid state were determined by single crystal X-ray diffraction (see Table S1† for details of the relevant crystal data). Views of their molecular structures are depicted in Fig. 1 and Fig. S1–S5,† while selected geometrical parameters are summarized in Tables S2 and S3† (the crystal structure of 3 was reported in one of our earlier works58 and it is not discussed herein).
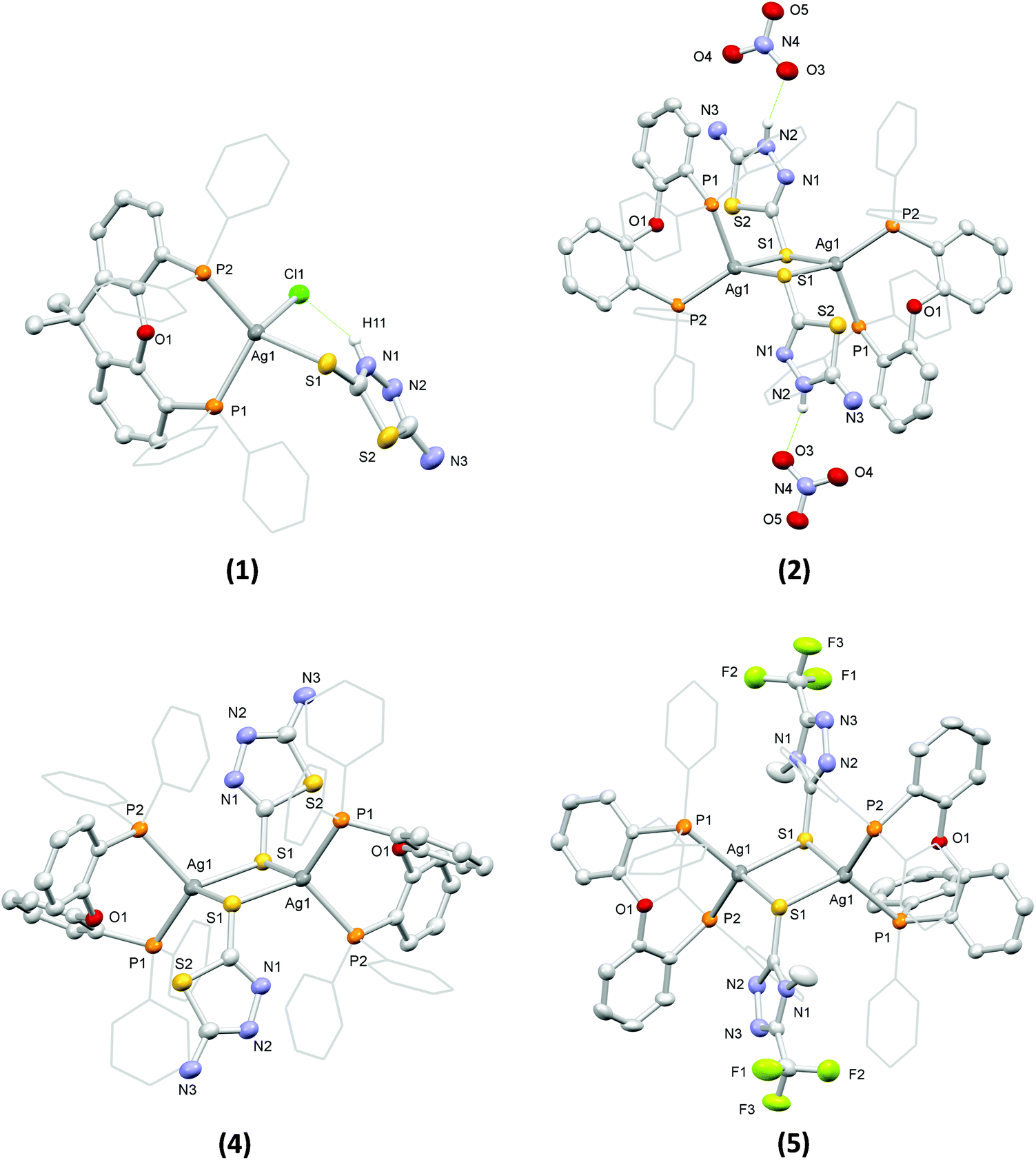 | ||
| Fig. 1 Views of the molecular structures of 1, 2, 4 and 5 with displacement ellipsoids shown in the 30% probability level. In all cases, selected C atoms are drawn in wireframe for clarity reasons, while H atoms are omitted, except from the H atoms of the NH groups of the thioamide ligands in 1 and 2. | ||
Complex 1 comprises a tetrahedrally coordinated Ag(I) ion surrounded by the P atoms of a chelating xantphos, the exocyclic S atom of a terminally bound atdztH, and a Cl− ion. Bond angles around the metal center, falling between 99.97(5)° and 123.44(5)°, indicate distortions from the ideal tetrahedral geometry. The Ag–P bond lengths are slightly different from each other, at 2.471(1) Å and 2.517(2) Å. The Ag–S bond length was found to be 2.647(1) Å, while the Ag–Cl bond length is at 2.612(2) Å. Analogous bond lengths and angles are observed in similar Ag(I) complexes that have been reported in the literature.34,43,59 The molecular structure of 1 is stabilized by an intramolecular hydrogen-bonding interaction between the halogen atom and the NH group of the heterocyclic ring of atdztH, which is evidenced by the relative orientation of thioamide ligand with respect to the halogen atom and the short NH⋯Cl distance (ca. 2.240 Å).
Complex 2 is a dicationic centrosymmetric dimer with two Ag(I) ions doubly bridged by the exocyclic S atoms of two neutrally charged thioamide ligands atdztH, forming a central Ag2(μ-S)2 core. The coordination sphere of each metal center is completed by the P atoms of a chelating DPEphos ligand. Significant distortions from the ideal tetrahedral coordination geometry are observed for both metal centers, as revealed by the relevant bond angles, e.g. P1–Ag1–S1′ = 87.97(4)° and P2–Ag1–S1′ = 132.83(5)°. The Ag–P bond lengths are equal to 2.465(1) Å and 2.536(1) Å and they are similar to those observed in 1. The Ag–S bond lengths are equal to 2.612(2) Å and 2.683(1) Å and, therefore, lead to a distorted rhombic Ag2(μ-S)2 core, with pairs of equal Ag–S bond lengths at opposite sides and angles of 83° and 97°. Noticeably, the Ag2(μ-S)2 unit is perfectly planar with the thioamide rings in a trans orientation to each other. The Ag⋯Ag separation of 3.513 Å is larger than the sum of the van der Waals radii for two neighboring Ag(I) ions (∼3.40 Å) and therefore the existence of argentophilic interactions between the two closed-shell metal ions is excluded. The two NO3− counter ions are held in close proximity to the bridging thioamide ligands through hydrogen-bonding interactions, developed between one of their O atoms and the NH groups of the thioamides (with NH⋯O distances of ca. 1.936 Å and 2.143 Å).
Complexes 4 and 5 are neutral centrosymmetric dimers, isostructural to the cationic part of 2, both having two Ag(I) ions bridged by the exocyclic S atoms of two thioamidate ligands, atdzt− and mtft−, respectively. Each Ag(I) ion is surrounded by a set of P2S2 donors in distorted tetrahedral coordination environments with the relevant bond angles falling in the ranges of 95.00(4)°–121.10(4)° for 4 and 96.57(4)°–122.21(4)° for 5. The Ag–P bond angles of the two complexes do not differ significantly from each other and they are similar to those of the dicationic complex 2. However, their Ag–S bond distances are shorter than the corresponding distances in 2 (at 2.580(1) Å and 2.620(1) Å for 4, 2.587(1) Å and 2.679(2) Å for 5), apparently due to the anionic nature of their S-bound thioamidate ligands compared to the neutral thioamide ligand in 2. As a result, the central Ag2(μ-S)2 cores of the two complexes display distorted rhombic geometries and planar conformations with Ag⋯Ag interatomic distances at 3.514(1) Å and 3.341(1) Å, respectively, indicating the presence of weak intermetallic interactions in the latter.
As a general comment on the molecular structures of the complexes presented herein, one should mention a tendency for the formation of binuclear structural motifs. This should be mainly attributed to the steric properties of the ligands and, in particular, the flexibility of the DPEphos backbone (due to the presence of a single O atom at a pivotal position) and the small size of the five-membered heterocyclic ring of the thioamides. Analogous dimeric structures have also been reported for Ag(I) complexes with DPEphos and other thioamides of similar size.60 Noticeably, when a diphosphine with a more rigid backbone, such as xantphos, was used in similar reactions, mononuclear complexes with three- or four-coordinate Ag(I) ions were obtained.44,61
Examination of the solid-state structures of the complexes reveals the presence of intermolecular hydrogen-bonding interactions and short atom contacts, which result in the formation of extended molecular architectures. For example, intermolecular hydrogen-bonding is developed between pairs of neighboring molecules of 3, and in particular between the NH2 group of the atdzt− ligand of one molecule with the closest N atom of the heterocyclic ring of the atdzt− ligand of the other molecule, with the interatomic N–H⋯N distances being equal to 2.930 Å (Fig. S5a†). Analogous intermolecular interactions are also observed in 4. However, in the latter, each dimer is connected with two neighboring molecules, overall resulting in an infinite linear chain of hydrogen-bonded Ag(I) dimers that extends along the crystallographic a axis (Fig. S5b†). In 5, short intermolecular C–H⋯F contacts are observed. These are developed between the F atoms of the CF3 groups of the two thioamidate ligands of one molecule with the H atoms of the phenyl groups of two different neighboring molecules (Fig. S5c†). The relevant H⋯F interatomic distances are measured at 2.567 Å and 2.659 Å. These contacts extend over a large number of neighboring molecules resulting in a 3-dimensional architecture. Overall, these observations suggest that the complexes have the capacity to develop favorable intermolecular interactions with hydrophilic or hydrophobic functional groups of biological entities, such as cell membrane and enzyme active sites, which would possibly affect their bioactivity.
Infrared spectroscopy confirmed the identity of 1–5 obtained as microcrystalline solids in bulk form. Their FTIR spectra in the 4000–400 cm−1 range are dominated by characteristic bands due to the presence of their respective phosphine and thioamide/thioamidate ligands. While absorption bands attributed to vibrations of their phosphine ligands remain unchanged with respect to phosphines in free form and, therefore, provide no evidence for coordination, thioamide-group vibrations of their thioamide ligands are more informative as they appear to be shifted to higher or lower frequencies, with respect to those of the corresponding ligands in their free form, giving indication for coordination. For example, an intense band at 1499 cm−1 in the spectrum of the uncoordinated atdztH, assigned to the C–N bond stretching vibration, appears to be shifted to 1472 cm−1 in the spectra of [AgCl(atdztH)(xantphos)] (1) and [Ag(μ-atdztH)(DPEphos)]2(NO3)2 (2). Another characteristic band of free atdztH observed at 1328 cm−1 (attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S bond stretching vibration) is shifted to 1318 cm−1 in the spectrum of 1, in agreement with the S-coordination of atdztH. Similarly, the strong band of atdztH at 755 cm−1 undergoes a shift to 743 cm−1. Furthermore, the broad absorption bands at 3270 and 3255 cm−1 in the spectra of 1 and 2, respectively, which bear the same thioamide in its neutral form, might be attributed to the stretching vibration of the N–H bond of their thioamide group. Interestingly, 3 and 4, containing the same thioamide ligand but in its deprotonated form, do not show such an absorption band in this frequency region.
S bond stretching vibration) is shifted to 1318 cm−1 in the spectrum of 1, in agreement with the S-coordination of atdztH. Similarly, the strong band of atdztH at 755 cm−1 undergoes a shift to 743 cm−1. Furthermore, the broad absorption bands at 3270 and 3255 cm−1 in the spectra of 1 and 2, respectively, which bear the same thioamide in its neutral form, might be attributed to the stretching vibration of the N–H bond of their thioamide group. Interestingly, 3 and 4, containing the same thioamide ligand but in its deprotonated form, do not show such an absorption band in this frequency region.
1H NMR spectra of 1–5 in CDCl3 solutions exhibit signals that correspond to the H's of their respective ligands. For example, in the 1H NMR spectrum of [AgCl(atdztH)(xantphos)] (1), signals of H's of the phenyl groups of xantphos appear in the 7.55–7.19 ppm region as a set of multiplets, whereas H's of its CH3 groups (6H) give a singlet in the high field region at 1.67 ppm (probably due to the fast inversion motion of diphosphine's backbone). Similarly, signals of the corresponding phosphine/diphosphine ligands were observed in the spectra of 2–5 (Experimental section S1.2 and Results section S2.2.1). Interestingly, signals of H's of the NH2 group of the thioamide or thioamidate ligands of 1–4 were not observed, apparently due to fast H/D exchange process between the NH2 group and the deuterated solvent. In general, all assignments are in accordance with the crystal structure data of the corresponding complexes, suggesting the retention of their molecular structures in solution.
We also examined the long-term stability of 1–5 in solution. Measurements of the 1H NMR spectra of the five complexes over a period of 3 days revealed that the initial resonances and multiplicities remained unchanged, suggesting the retention of their structures in solution. An analogous study was also conducted by UV-vis spectroscopy, using solutions of the compounds in DMSO/PBS (PBS = phosphate-buffered saline at pH = 7.4 and >0.7% DMSO) as well as DMSO/acidic phosphate buffer at pH = 6.0, at concentrations of ∼10−6 M, over a period of 3 days. Stability assay's results (Fig. S6†) showed no changes in the pattern of the initial UV-vis absorption spectra of all complexes and those recorded after 48 h, suggesting the preservation of the structural integrity of the complexes in aqueous media.
2.2 Photophysical properties
UV-Vis electronic absorption spectra of 1–5 in DMSO solutions in the high energy region display intense absorption bands with wavelength absorption maxima λmax(abs) between 260 nm and 290 nm and molar absorption coefficients ε between 1.0 × 104 and 2.0 × 104 M−1 cm−1 (Fig. 2, Table 1). Since both phosphines and heterocyclic thioamides in their free form also absorb in the particular wavelength region showing similar λmax(abs), these bands can be attributed to intraligand π* ← π transitions of the ligands of the complexes. Additional absorption bands of lower intensity appear in the lower energy region of the spectra of the compounds, in the form of broad shoulders or long tails. For the binuclear complex 2, which contains two bridging thioamides μ-κS-atdztH, a distinct absorption band of medium intensity is observed at λmax(abs) = 318 nm. Complex 4, which is isostructural to 2 but it contains two bridging thioamidates μ-κS-atdzt−, absorbs in the same wavelength region but with higher intensity (absorption band appears as shoulder). In the case of the mtft-containing complex 5, a low intensity band (as shoulder) also appears in this wavelength region but shifted to higher energy. These lower energy bands probably arise from charge transfer electronic transitions involving the thioamide ligands.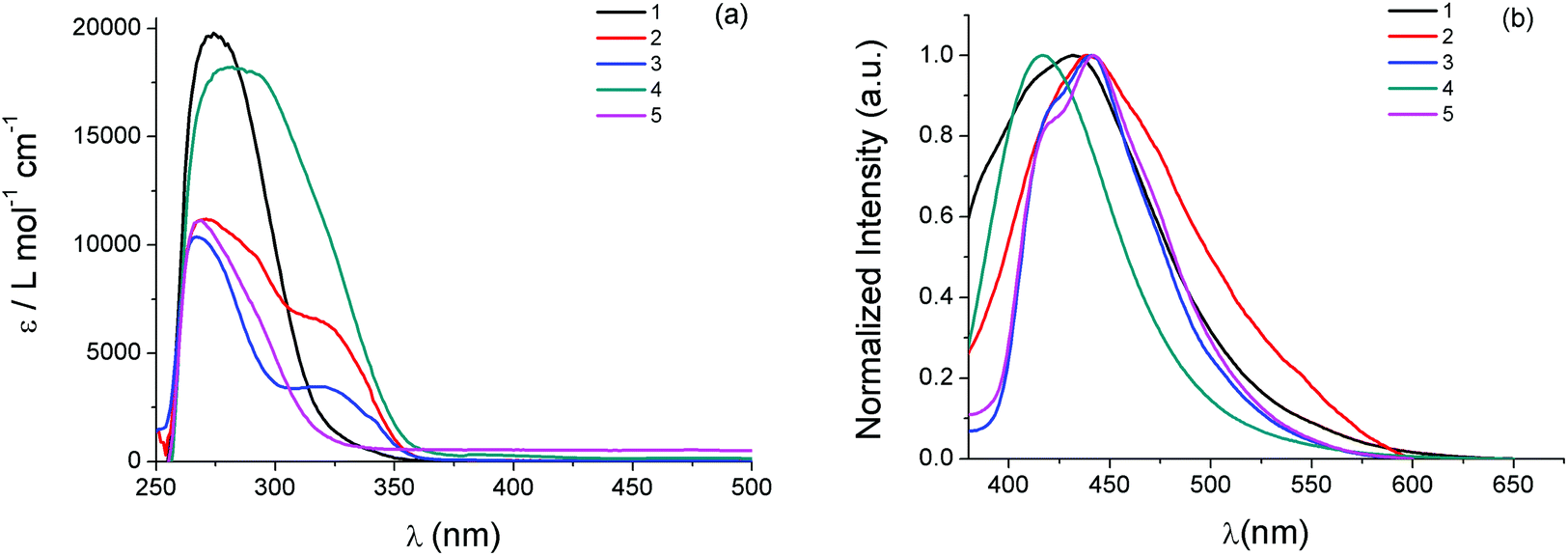 | ||
| Fig. 2 (a) UV-Vis electronic absorption spectra and (b) emission spectra (excitation at 320 nm) of 1–5 in DMSO solutions (∼5 × 10−5 M). | ||
| Complex | λ max(abs)/nm (ε/M−1 cm−1) | λ max(em)/nm |
|---|---|---|
| 1 | 278 (19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800), 320 sh (1900) 800), 320 sh (1900) |
431 |
| 2 | 270 (11200), 292 (9500), 318 (6400) |
438 |
| 3 | 260 (10400), 340 br (3500) |
440 |
| 4 | 280 (18300) |
417 |
| 5 | 260 (11100), 290 (7000) |
442 |
The emission properties of 1–5 were also investigated in DMSO solutions. All complexes, when excited at 320 nm, display broad emission bands with their emission band maxima λmax(em) falling in the violet-blue region of the visible spectrum (Fig. 2, Table 1). In general, complexes comprising atdztH in its neutral form display emission bands which are red shifted compared to those that contain the same thioamide ligand in its deprotonated form. Specifically, [AgCl(atdztH)(xanthphos)] (1) and [Ag(μ-atdztH)(DPEphos)]2(NO3)2 (2) exhibit λmax(em) at 431 nm and 438, respectively (a difference that might be attributed to their different diphosphine ligands and/or their different structural motifs). In contrast, the emission maximum of the binuclear complex [Ag(μ-atdzt)(DPEphos)]2 (4) appears to be blue shifted (with respect to the analogous binuclear dicationic complex 2) at 417 nm, revealing that the luminescent character of these complexes is strongly affected by the protonation state of the thioamide. Binuclear complex [Ag(μ-mtft)(DPEphos)]2 (5) exhibits a red-shifted λmax(em) with respect to that of its homologous complex [Ag(μ-atdzt)(DPEphos)]2 (4) (442 nm vs. 417 nm, respectively). This difference can be attributed to the strong electron withdrawing effect of the CF3-group substituent of the thioamidate ligand of the former, compared to the NH2-group substituent of the thioamidate ligand of the latter, suggesting that the emitting excited states of these complexes have a distinct thioamidate-based character. Finally, in the case of the mononuclear neutral complex [Ag(atdzt)(PPh3)3] (3), the emission maximum is located at λmax(em) = 440 nm, which is shifted to lower energies compared to the emission maxima of the neutral complex 4 bearing the same thiomidate ligand. This might be a result of the presence of three PPh3 ligands in the molecule. However, a direct comparison cannot be made due to the different structural motifs of the two complexes. Overall, it can be suggested that the luminescent properties of 1–5 generally arise from intraligand charge-transfer (ILCT) electronic transitions involving their thioamide/thioamidates and phosphine ligands. In addition, all complexes exhibit appropriate emission characteristics that allow their utilization, on the condition that they demonstrate effective antiproliferative activity, as bifunctional cell imaging agents.
2.3 In vitro antibacterial activity
The in vitro antibacterial efficacy of 1–5 was investigated against the Gram-(+) S. aureus, B. subtilis, and B. cereus as well as the Gram-(−) E. coli bacterial strains. Studies were conducted using a range of different concentrations of the five complexes (100, 50, 25, 12.5, 6.25 μg mL−1) and calculating the half-maximal inhibitory concentrations (IC50) as well as the minimal inhibitory concentrations (MIC). The antibacterial activity of the ligands of the five complexes in their free form was also evaluated for comparison, while ampicillin, owing to its known ability to prevent or treat various bacterial infections, was used as reference. The results of this study are presented in Fig. 3 and Table S8.†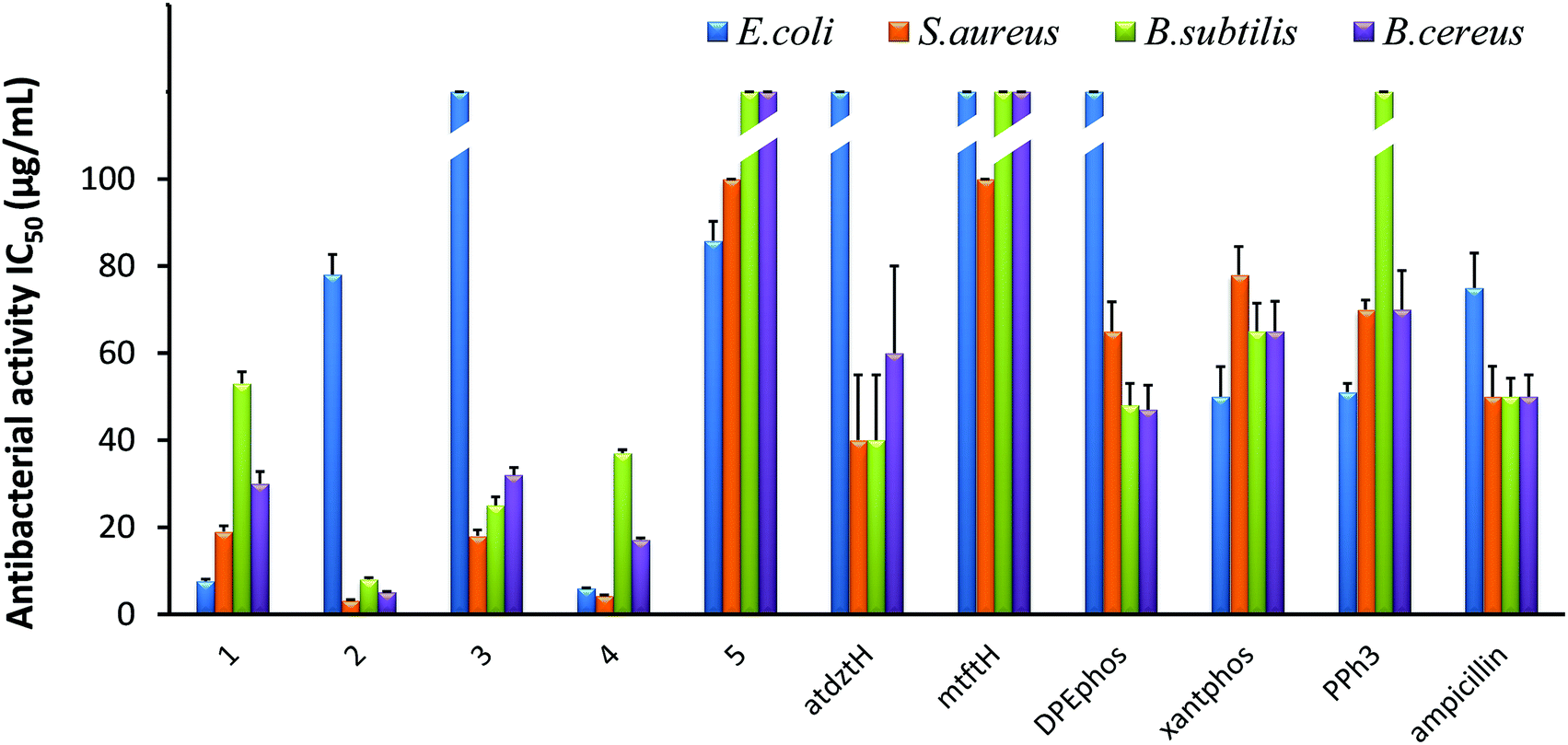 | ||
| Fig. 3 In vitro antibacterial activity of 1–5, and their ligands atdztH, mtftH, DPEPhos, xantphos, and PPh3 in free form, expressed as half-minimum inhibitory concentration (IC50) values (μg mL−1) provided by a nonlinear curve fit-growth/sigmoidal-dose response on the experimental optical density data. Values are expressed as mean ± standard deviation (SD) of three replicate measurements (with the exception of values higher than 100 μg mL−1). | ||
Generally, all complexes display moderate-to-high antibacterial activity against the studied bacterial strains, which is higher than that of their respective ligands in free form. Differences in the bioactivity of the complexes were observed, which are ascribed to alterations of their ligands and their structural characteristics. In particular, [Ag(μ-atdztH)(DPEphos)]2(NO3)2 (2) and [Ag(μ-atdzt)(DPEphos)]2 (4) were found to be the most effective ones, inducing significant growth inhibition almost on all bacterial strains. In particular, dicationic complex 2 appeared to be the most effective against the Gram-(+) S. aureus, B. subtilis, and B. cereus bacterial strains giving very low IC50 values of 3.2 μg mL−1 (2 ± 0.2 μM), 8 μg mL−1 (5 ± 0.4 μM), and 5.1 μg mL−1 (3.3 ± 0.15 μM), respectively. Neutral complex 4 showed an increased inhibition activity against the Gram-(+) S. aureus bacterial strain with an IC50 value of 7.6 μg mL−1 (8.9 ± 0.5 μM). Interestingly, 4, as well as 1, appear to be particularly effective against the Gram-(−) E. coli bacterial strain showing IC50 values of 6 μg mL−1 (3.9 ± 0.1 μM) and 7.6 μg mL−1 (8.9 ± 0.5 μM), respectively. On the contrary, [Ag(atdzt)(PPh3)3] (3) showed negligible activity against the particular bacterial strain. Noticeably, very low antibacterial activity was observed for the neutral binuclear complex [Ag(μ-mtft)(DPEphos)]2 (5) against all bacterial strains.
In general, Ag(I) complexes with the NH2-substituted thioamide presented herein exhibit high antibacterial activity against the studied bacterial strains, which is higher (or comparable) than that of analogous heteroleptic Ag(I) complexes bearing other thioamides or thioamidates that we recently reported.43,44,58 In particular, cationic complex 2 was found to induce a significantly strong antibacterial effect against the Gram-(+) bacterial strains. Similarly, in one of our recent studies, we found that cationic Ag(I) complexes bearing the neutral thioamide 2,2,5,5-tetramethyl-imidazolidine-4-thione and phosphines also exhibit significant antibacterial activity against Gram-(+) bacterial strains.44 Similarly, in another recent study, cationic Ag(I) complexes bearing the neutral thioamide dmp2SH (= 4,6-dimethylpyrimidine-2-thiol) [Ag(dmp2SH)(PPh3)2]NO3 and [Ag(dmp2SH)(xantphos)]NO3 were also found to exhibit a higher potency to inhibit the growth of Gram-(+) bacteria compared to analogous neutral complexes.62 These results suggest a potential correlation between structure and bioactivity in this family of Ag(I) complexes which is related to the total charge of the complexes. Considering that, in general, the efficiency of an antibacterial agent depends on its cellular uptake, which is determined by its interaction with the bacterial membrane,63 it seems reasonable to suggest that the cationic Ag(I) complexes have a higher ability, compared to neutral complexes, to interact strongly with the bacterial membrane of Gram-(+) bacteria (e.g. through electrostatic interactions with highly electronegative O atoms of the building blocks of their thick peptidoglycan layer), penetrating it and, ultimately, leading to cell death. Analogous findings for the high potential of positively charged metal complexes to selectively restrain or inhibit the growth of Gram-(+) strains have also been reported by others, and it has been correlated with their potentially strong interaction with the negatively charged bacterial membranes.64,65 However, the exact mechanism of the antibacterial activity of the studied complexes is still unknown and under investigation.
The extremely low efficacy of 5 against both Gram-(+) and Gram-(−) bacterial strains can be clearly ascribed to its CF3-substituted thioamidate, since its isostructural complex 4 with atdzt− ligands exhibits a much higher antibacterial activity. These data provide an indication for a second potential structure/bioactivity correlation in the particular family of Ag(I) complexes, which is related to the nature of the thioamide ligands. Specifically, it appears that the substituents of the thioamidates of 4 and 5 play an important role not only in their potential to develop strong intermolecular interactions with various functionalities on the cell membrane of bacteria, but also in the lipophilicity of their complexes. It has been suggested that for a high antibacterial activity of a compound, an appropriate lipophilicity/hydrophilicity balance is also crucial, as it defines the binding and subsequent internalization of the compound to the bacterial cell.42 Therefore, the increased lipophilic character of 5, together with its low potential for the development of favorable intermolecular interactions, is detrimental for its antibacterial activity. Moreover, 5 provides an example of a compound for which the presence of CF3-substitution is not beneficial for its bioactivity, in contrast to what has been claimed for many CF3-substituted compounds in the literature,66,67 rendering its influence controversial indeed.
The effect of the lipophilicity of the phosphine/thioamide (or thioamidato) complexes on their antibacterial activity is also observed in the case of the neutral complex [Ag(atdzt)(PPh3)3] (3). Compared to the analogous [Ag(atdzt)(PPh3)2] that we reported earlier to be an effective antibacterial agent against Gram-(+) bacteria,583, exhibiting a greater lipophilic character due to the presence of three PPh3 ligands, demonstrates, as expected, a lower activity against the same bacteria.
2.4 In vitro anticancer activity
The growth inhibition/cytostatic and cytocidal/cytotoxic effects induced by 1–5 against SKOV-3, Hup-T3, DMS114, and PC3 human cancer lines, as well as MRC-5 human normal cell line, are presented in Table S9† and reveal that all complexes caused a dose-dependent inhibition of cell proliferation. The dose-effect curves for all treated cell lines are illustrated in Fig. 4, demonstrating the different chemosensitivity of the cell lines to the studied complexes. The order of potency of the complexes was revealed to be 3 ≥ 2 > 1 > 5 > 4 for all tested cell lines, while the order of cell line chemosensitivity for all complexes was found to be SKOV-3 > DMS114 > PC3 > Hup-T3 > MRC-5. In particular, 1–3 exhibited a very potent cytostatic and cytotoxic effect against all tested cell lines, displaying low micromolar IC50 values. In contrast, 4 and 5 exhibited lower anticancer potency. Complex 5 was found to be slightly more active than 4 in most cell lines exhibiting IC50 values under the threshold of 100 μM, with the exception of Hup-T3 and MRC-5 cell lines. It is important to notice that for 1–3 human normal lung MRC-5 cells were found to be more resistant compared to human cancer cells that were found to be more sensitive (an exception is applied only in 3 for which similar cytotoxicity was documented for all cell lines). Examining the potency of 1–3 against all tested cell lines, it can be concluded that 2 and 3 revealed to be similar in potency and two or three times more potent than 1. It is interesting to notice that among 1–3, exhibiting the lowest cytotoxic activity, the two (1 and 3) are mononuclear complexes and one (2) is a dicationic binuclear complex. A significant conclusion can be drawn from the comparison of the cytotoxic activity between the homologous binuclear complexes 2 and 4, which differ only in their total charge, revealing that the dicationic charge of 2 augments its cytotoxic potency. An analogous conclusion was also reached from the study of the antibacterial activity of these complexes, which revealed 2 to be more active than 4 for the Gram-(+) bacterial strains tested (Fig. 3 and Table S8†). On the other hand, comparing the structures of the complexes relatively to their cytotoxic potency, it can be concluded that the less bulky complexes 1 and 3, as well as the dicationic complex 2, seem to be more active against all tested cells.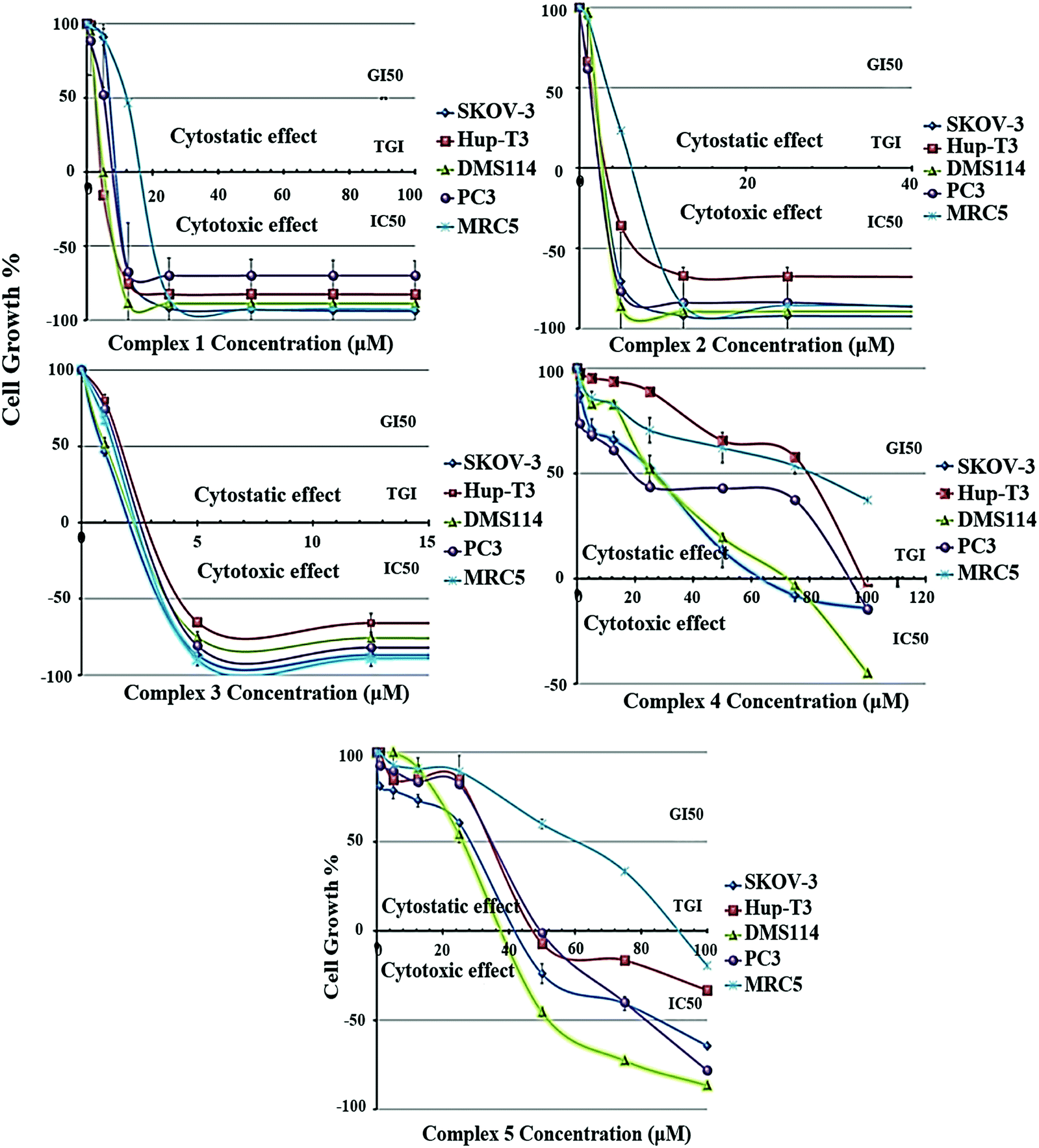 | ||
| Fig. 4 Growth inhibition/cytostatic (GI50 and TGI, in μM) and cytocidal/cytotoxic (IC50, in μM) effects induced by 1–5 against SKOV-3, Hup-T3, DMS114, and PC3 human cancer, and MRC5 human normal cell lines (each point represents mean ± SEM of five measurements. Where error bar is not visible, its size is smaller than that of the corresponding symbol). | ||
It is interesting to compare the results of the cytostatic/cytotoxic activity of 1–5 with those that we recently reported for heteroleptic Ag(I) complexes bearing analogous ligands.44 In particular, [Ag(μ-phimtH)(phimtH)(PPh3)]2(NO3)2 was reported to exhibit an IC50 value of 4.50 ± 0.20 μM against Hup-T3 cells, a value that is very similar to the IC50 value against the same cell line for [Ag(atdzt)(PPh3)3] (3), also bearing PPh3 ligands. Similar IC50 values were found for the two complexes against the two other cell lines, i.e. 4.50 ± 0.20 μM and 4.00 ± 0.20 μM for SKOV-3 cells, and 4.60 ± 0.20 μM and 4.20 ± 0.20 μM for PC3 cells, respectively. Furthermore, [Ag(phimtH)(xantphos)]BF4 was reported to exhibit similar IC50 values with [AgCl(atdztH)(xantphos)] (1), also bearing a xantphos ligand, with IC50 values of 4.60 ± 0.05 μM and 9.20 ± 0.20 μM against Hup-T3 cells, and 13.00 ± 0.20 μM and 11.30 ± 0.20 μM against PC3 cells, for the two complexes respectively. Only SKOV-3 cells were found to be more sensitive to [Ag(phimtH)(xantphos)]BF4 compared to 1 (with IC50 values of 1.80 ± 0.20 μM and 11.70 ± 0.80 μM, respectively). The similarity in the antiproliferative activity between these complexes could be attributed, at least to a first approximation, to their structural similarity regarding their ligands. Further studies to understand the mechanism underlying the antiproliferative activity induced by these complexes, including target proteins involved in cancer growth, may be warranted.
2.5 In vitro antioxidant activity
Antioxidants are substances that prevent oxidative damage of specific biomolecule-targets. Generally, although in the literature there are many reports on the antioxidant activity of a large number of coordination compounds of different metals, Ag(I) complexes have only scarcely been investigated. Therefore, having established the high antibacterial and anticancer potential of the complexes presented in this work, we investigated their in vitro antioxidant capacity by evaluating their H2O2 reducing power and DPPH, ABTS free radical scavenging ability. Studies were conducted in comparison to the ligands of the five complexes in free form and selected reference compounds (NDGA, BHT, Trolox and L-ascorbic acid). The results of this study are depicted in Fig. 5 and Table S10.†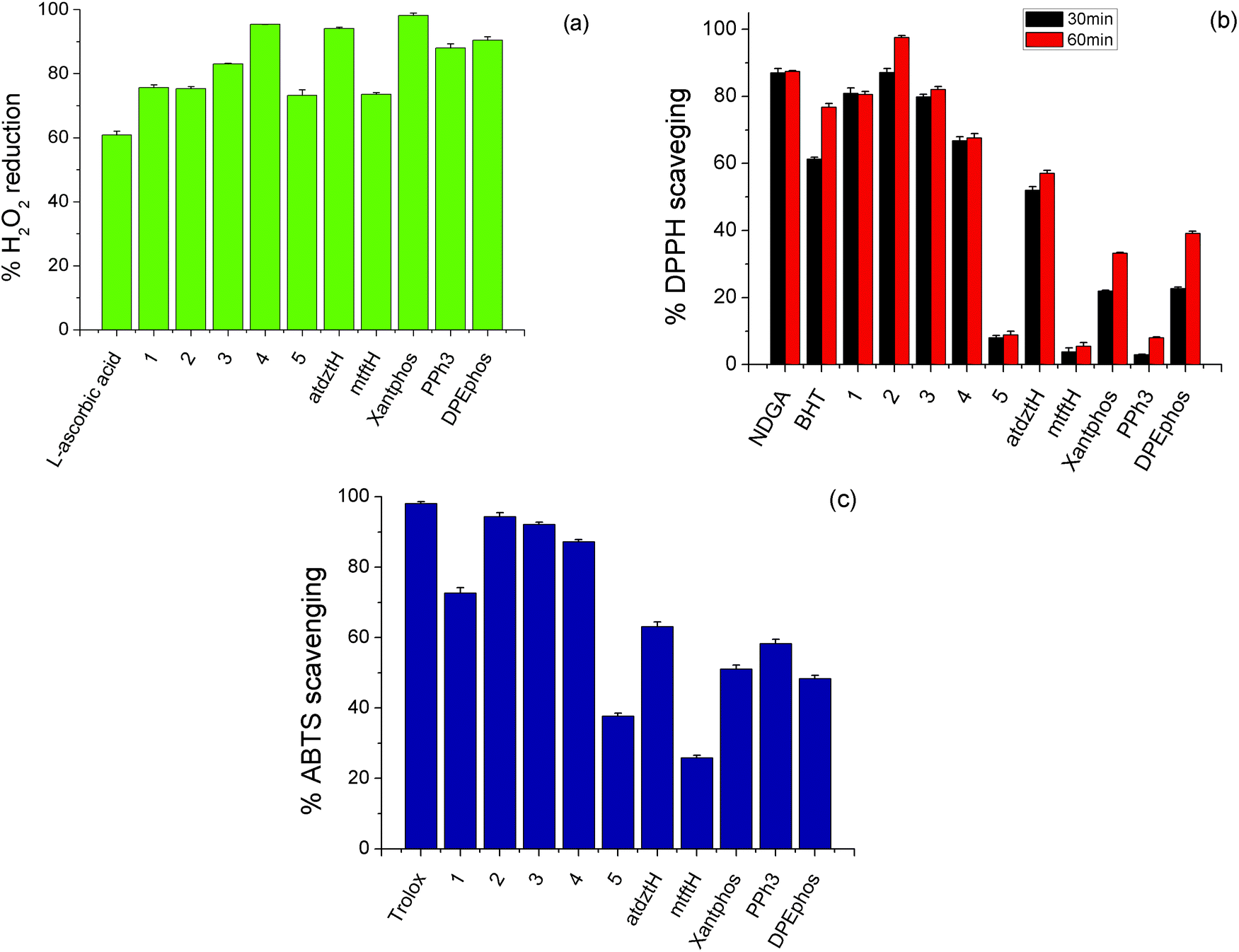 | ||
| Fig. 5 (a) H2O2 reducing power, (b) DPPH free radical and (c) ABTS free radical scavenging activity of 1–5 and their ligands in free form, as well as selected reference compounds (L-ascorbic acid, NDGA, BHT, and Trolox). | ||
H2O2 can act as source of oxygen-based free radicals, such as hydroxyl radicals (˙OH), which can induce DNA damage leading to cancer initiation and progression.68 Therefore compounds with possible ability of hydroxyl radical scavenging can be considered as potential anti-proliferative agents. As shown in Fig. 5a, the studied complexes exhibit high ability to cause reduction of H2O2 (higher than the reference compound L-ascorbic acid), while among them, the highest percentage of H2O2 reduction activity was induced by 4. DPPH scavenging activity of a compound is attributed to its capacity to act as H+ or electron acceptor, and therefore it is related to its potential antiaging, anticancer and anti-inflammatory activity. Herein, DPPH-scavenging activity of 2 was found to be higher than that of the other compounds, which displayed moderate activity, as well as the reference compounds BHT and NDGA, and it appears to be time-dependent, as it increases over time (Fig. 5b). The total antioxidant activity of the studied complexes was evaluated by the ABTS method. As shown in Fig. 5c, 2, 3 and 4 exhibited the stronger antioxidant effect, although their scavenging values are lower to that of the reference compound, Trolox.
Based on the abovementioned results, it can be suggested that the high anticancer activity of the atdztH-containing complexes 1–3 against the tested cell lines might be related, at least in part, to their significant antioxidant ability.
2.6 CT-DNA interaction
DNA is a very common target of many metal-based compounds exhibiting antibacterial and anticancer properties. Considering the high biological efficacy of 1–3, we investigated their potential to interact with this biomolecule69,70 and their mode of interaction.43,44,71The interaction of 1–3 with CT-DNA was initially studied by UV-Vis absorption spectroscopy. In particular, we followed the changes in specific UV absorption bands of the three complexes in DMSO solutions, upon addition of increasing amounts of CT-DNA.72 As shown in Fig. S7,† UV spectra of 1 and 2 display a strong absorption band with λmax(abs) ∼ 270 nm which appeared to undergo a slight hypochromism upon addition of CT-DNA. In contrast, the absorption band of 3 with λmax(abs) = 267 nm was found to show a moderate hyperchromism.53 Another absorption band with maxima in the 318–322 nm region for all complexes exhibited a more significant hypochromism and finally it disappeared. In the case of 2, a third absorption band with λmax(abs) = 294 nm showed a decrease in its intensity (up to 12%). The observed spectral changes are indicative of the formation of conjugates between 1–3 and CT-DNA, but they are not sufficient enough to derive concrete conclusions regarding their exact mode of interaction with this bio-macromolecule (i.e., intercalation, electrostatic interactions or groove-binding73,74). Nevertheless, the data allowed us to get an estimate of the strength of their interaction. In particular, the binding constants Kb of 1–3 on CT-DNA, evaluated using the Wolfe–Shimer equation (eqn (S1)†) and the corresponding plots (Fig. S8†),75 were calculated and they are listed in Table S11.†Kb constants for the interaction of the ligands of 1–3 with CT-DNA were also calculated, for comparison reasons. In general, the three complexes display Kb constants of 104–107 M−1, which are of similar orders of magnitude with previously reported analogous Ag(I) complexes bearing the same types of ligands.43 In particular, these values are indicative of rather strong interactions with CT-DNA, definitely stronger than the interaction of their ligands with CT-DNA, with the mononuclear complex 3 displaying the strongest interaction.
To get a better idea on the possible binding mode of 1–3 with CT-DNA, we monitored the changes of the viscosity of a CT-DNA solution upon addition of increasing amounts of the three complexes. In general, viscosity of a CT-DNA solution is sensitive to changes of its length. Therefore, changes in the CT-DNA viscosity occurring upon its interaction with a compound may reveal the possible mode of interaction (i.e., DNA viscosity shows a significant increase upon intercalation, while in case of groove-binding or electrostatic interaction, DNA viscosity decreases slightly or remains unchanged76). As shown in Fig. S9,† upon addition of increasing amounts of 1 and 2 to a buffered CT-DNA solution, the relative CT-DNA viscosity remained practically unchanged up to r = 0.30 (despite of the observed fluctuations), whereas an increase of the relative viscosity was observed when higher concentrations of the two complexes were added (r > 0.30). These findings suggest non-classical intercalation for 1 and 2 in the low concentration range, i.e., a combination of groove binding and electrostatic interaction (for the cationic complex 2), which apparently cause only a slight kink at CT-DNA double helix. The conspicuous increase of CT-DNA viscosity observed at higher concentrations for the two complexes (r > 0.3) is an indication of their classical intercalation between the CT-DNA base pairs, which leads to an increase of CT-DNA length. In contrast, addition of increasing amounts of 3 had a negligible effect on the relative CT-DNA viscosity throughout the whole range of studied concentrations, suggesting clearly a non-intercalating binding mode, i.e., groove binding.
To further investigate the intercalating ability of 1–3 between CT-DNA strands, competitive binding studies with ethidium bromide (EB), a common DNA intercalator, were conducted. To a buffered solution of EB-DNA conjugate, which shows an intense emission band at λmax(em) = 592 nm (upon excitation at 540 nm), increasing amounts of 1–3 were added (up to r < 0.15). A low-to-moderate quenching of the EB-DNA emission band (∼30%) was observed for 1 and 2, while a moderate-to-high quenching (∼62%) was found for 3 (Fig. S10†). Since the compounds do not exhibit emission maxima in the particular wavelength region, the observed decreasing fluorescence can be ascribed to the competitive behavior of the complexes with EB for the intercalation sites of DNA. A quantification of the ability of the three complexes to replace EB was carried out using the Stern–Volmer equation (eqn (S2)†) and the corresponding plots (Fig. S11†), which resulted in KSV constants of 104–105 M−1 (Table S12†).77 From these data the EB-DNA quenching constants kq were calculated (eqn (S3)†) and found to be >1010 M−1 s−1 (Table S12†), providing an indication that quenching of EB-DNA fluorescence induced by the three complexes occurs via a static mechanism that entails the formation of a new conjugate, i.e., compound/CT-DNA.78 The relatively low KSV constants of the complexes (compared to the KSV constants reported of recently reported analogous Ag(I) complexes43) together with the observed moderate emission quenching of the EB-DNA solution suggest the low efficacy of the three complexes for intercalation into CT-DNA58 (at least in the low concentration range with r < 0.15).
According to the abovementioned results, it can be concluded that 1–3 have the ability to interact fairly strongly with CT-DNA, using different interaction modes which could be related to their particular structural characteristics (e.g. total charge). Specifically, groove binding and/or electrostatic interactions appear to be the most probable modes of interaction (especially at low concentrations), while they show a rather low efficacy to intercalate between the CT-DNA strands and break its double-strand structure.
2.7 Serum albumins binding
Serum albumins are highly abundant proteins in the circulatory system of mammals which are responsible for drug transportation and distribution.79,80 Aiming to get an idea of the pharmacokinetic properties of the compounds presented herein related to their absorption and distribution, we investigated the ability of the most potent compounds 1–3 to interact with bovine serum albumin (BSA) and human serum albumin (HSA) proteins.BSA and HSA solutions, when photoexcited at 295 nm, display high intensity emission bands with λmax(em) at 350 nm and 338 nm, respectively. Upon addition of increasing amounts of 1–3 to BSA and HSA solutions, quenching of the respective emission bands was observed (Fig. S12†), which can be attributed to the binding of the complexes on the albumins that changes their tertiary structures. In particular, a moderate decrease of BSA fluorescence to 66%, 24% and 55% of the initial fluorescence intensity was observed for 1, 2 and 3, respectively, whereas the corresponding HSA emission quenching reached the values of 55%, 81% and 50%, respectively (Table S13†). The experimental data reveal a different pattern of fluorescence quenching for 2, compared to 1 and 3. This could possibly be due to differences in the binding of the three complexes on BSA and HSA (e.g. binding to different sites), which can be related to their different total charge, as 2 is cationic but 1 and 3 are neutral complexes.
The fluorescence quenching constants kq for BSA and HSA, calculated using Stern–Volmer equation (eqn (S4)†) and the corresponding plots (Fig. S13 and S14†), were found to be in the range of 1012–1013 M−1 s−1 (Table S13†), indicating the existence of a static emission quenching mechanism and the formation of conjugates between each compound and the albumins. The corresponding binding constants K of 1–3 to BSA and HSA, obtained using Scatchard equation (eqn (S5)†) and the corresponding plots (Fig. S15 and S16†), were found to be of the order 105 M−1 (Table S13†). These values suggest a tight but reversible non-covalent binding of the three complexes to albumins (for comparison, binding constants of ∼1015 M−1 have been calculated for the strongest non-covalent interaction of diverse ligands to avidin protein), which would allow their successful binding and transfer to their target sites where they could be released.
2.8 Molecular docking calculations
In an effort to get complementary insights on the understanding of the mechanisms of the observed bioactivity of 1–5, a series of molecular docking calculations was performed. In particular, we evaluated the ability of the complexes to bind to bio-macromolecules E. coli and S. aureus DNA gyrase and the overexpressed in the studied cancer cells Fibroblast Growth Factor Receptor 1 (FGFR1). The results of our calculations, in terms of the global binding energies of 1–5 to the three bio-macromolecules, are shown in Table 2.| Complex | Global binding energy (kcal mol−1) | ||
|---|---|---|---|
| Bio-macromolecule | |||
| E. coli DNA gyrase (PDB ID 1KZN) | S. aureus DNA gyrase (PDB ID 5CDM) | FGFR1 (PDB ID 4 V04) | |
| 1 | −24.32 | −40.57 | −41.57 |
| 2 | −22.11 | −31.03 | −37.57 |
| 3 | −23.64 | −45.81 | −47.50 |
| 4 | −27.40 | −36.91 | −50.88 |
| 5 | −14.36 | −12.35 | −42.11 |
| CBN | −18.26 | — | — |
| MFX | — | −42.18 | — |
| QPT-1 | — | −45.66 | — |
 | ||
| Fig. 6 (a) (lower panel) Docking pose orientation of best (lower energy) bound complex 4 and second-best bound complex 1, in both in silico and in vitro studies (rendered in stick representation and colored according to atom type in light pink and olive-green C atoms, respectively), superimposed with co-crystallized drug chlorobiocin (CBN) (hot pink C atoms rendered in stick model) in the crystal structure of E. coli DNA gyrase (PDB ID: 1kzn). (upper panel) A close-up view of the ATP-binding site architecture of the best binding pose of both compounds. Target protein in both structure models is illustrated as cartoon colored in brown with depth cue in the ray-tracing rendering of the whole structure, with additional depiction of opaque surface colored according to cartoon. Selected contacting amino acid residues of the binding pocket are rendered in stick model and colored in split pea green and violet purple for 4 and 1, respectively, also highlighting the mapped surface of the corresponding part of the interacting residues in the same colors. The ligand-binding site of both 4 and 1, as determined by the computation process, is appeared to be the same with that occupied by CBN. All binding contacts of both complexes 4 and 1, illustrated in light pink and violet purple labeling, respectively, are found to be common with those of CBN. An exception is observed only for two binding contacts labeled in split pea green (histidine H95 and alanine A96). Interacting residues common between 1 and 4 are denoted in light pink labeling. Molecular docking simulations were performed individually. (b) (lower panel) Docking pose orientation of 3 and 2, best bound in in silico studies and with best in vitro activity, respectively, in the crystal structure of S. aureus DNA gyrase (PDB ID: 5cdm). Superimposed are also illustrated the docked, active against S. aureus DNA-gyrase, drugs moxifloxacin (MFX) and QPT-1 (co-crystallized and docked, alike). MFX is docked in two binding poses (one lower and one higher binding energy) at symmetrical binding pockets of the protein at the edge of DNA. Target protein is illustrated as cartoon with sub-domains color-coded in split pea green, deep teal, deep purple, and yellow orange colors for chains A, B, C, and D, respectively. An artificially nicked double-stranded DNA interacting with DNA gyrase is also depicted in cartoon colored in salmon and orange for complimentary strands E and N, and white, slate blue for complimentary strands F and I. Docked molecules rendered in sphere model are colored according to atom type in marine blue and warm pink (3 and 2, respectively), light pink and salmon (lowest and higher binding energy poses of MFX, respectively), and orange and hot pink (co-crystallized and docked QPT-1, respectively) C atoms. (upper panel) A close-up view of the binding of complexes 3 and 2 in both DNA and DNA gyrase complex structure depicting the extent of the binding pockets as determined by the computation process and the crystal structure as well. Double stranded DNA and DNA gyrase are depicted in cartoon colored in the same scheme as in lower panel. Complex 3 seems to be anchored in close proximity to docked QPT-1 in a binding cleft located at the edge of the artificially nicked DNA strand, while 2 is shown to be stabilized in a corresponding symmetrical pocket, opposite to co-crystallized QPT-1 position. Complex 3 is shown to penetrate deep in the double helix of DNA, binding to both strands of the DNA (chains I and N), making also contacts with nearby binding residues of chain C and A of the enzyme, while 2 binds only to one strand (chain N) and contacting residues of chain A of the enzyme. Molecular docking simulations of all molecules were performed individually. Binding contacts are shown as dotted lines in yellow and light pink color (a) and dotted yellow lines (b). Heteroatom color-code: O: red, N: blue, S: yellow, P: orange, Cl: green, and Ag: grey. Hydrogen atoms are omitted from all molecules for clarity. The final structure was ray-traced and illustrated with the aid of PyMol Molecular Graphics Systems. Binding interactions are shown in Table S14.† | ||
The best-fitted docking poses of the best bound complex 4, exhibiting the lowest global binding energy, and second-best bound complex 1, in both in silico and in vitro studies, in the crystal structure of E. coli DNA gyrase superimposed with CBN, are depicted in the lower panel of Fig. 6a. Both complexes are shown to be stabilized inside the ATP-binding site of DNA gyrase anchored in the same place with CBN. We chose to use for docking experiments the DNA gyrase in complex with bound co-crystallized drug CBN, which include only the B subunit exhibiting the crucial ATPase activity (A subunit is mainly involved in DNA breakage and reunion).84,85 All molecules are shown to be stabilized inside the same binding pocket of the protein, occupied by the co-crystallized drug CBN.86 Binding interactions of 1 and 4, in their binding pockets are reported in Table S14.†
Terminal methyl-pyrrol ring of CBN is deep buried in the binding pocket while the other terminal moiety [4-hydroxy-3-(3-methylbut-2-enyl)benzoyl]amino is anchored in the entrance of the pocket. Similarly, 1 is shown to be inserted in the binding cleft with one of the phenyl rings of xantphos, and atdztH ligand, anchored in the entrance of the crevice. Chlorine atom is shown to protrude out of the crevice not making any contact with the residues of the entrance. On the contrary, 4, due to its bulkier size, is shown to be stabilized in the pocket with the four phenyl rings of one of the DPEphos ligands, as well as the atdzt− ligand, marginally anchored at the entrance, slightly protruding from it. These moieties are shown to lie at the opening of the catalytic pocket, partially covering the ATP-binding site. Comparing the three molecules, CBN is revealed to be inserted in the crevice along its whole deep, with one terminal moiety buried inside the end of the pocket. In contrast, both 1 and 4 cannot reach the end of the pocket, obviously due to their bulkier size. Nevertheless, the type of their binding mode ensure better binding capacity compared to CBN. The ligand-binding site of all molecules depicting the extent of the pocket as determined by the computation process, labeling the critical residues interacting with the molecules is shown in the upper panel of Fig. 6a. The docking procedure predicts the formation of a variety of interactions between both complexes and the amino acid residues Asn (N46), Asp (D49), Glu (E50), Ala (A53), Arg (R76), Gly (G77), Ile (I78), Pro (P79), Ala (A86), Ile (I90), and Arg (R136) for 1, and Asn (N46), Asp (D49), Glu (E50), Arg (R76), His (H95), Ala (A96), Ile (I78), and Pro (P79) for 4 (upper panel of Fig. 6 and Table S14†). Among these contacts of both 1 and 4, not common with those of CBN are found to be Ala (A53), His (H95), and Ala (A96). Stabilization of both 1 and 4 may be attributed to hydrogen bonding, hydrophobic, mixed π-alkyl type hydrophobic, polar, π-polar, and π-anion and π-cation charged electrostatic interactions, inside the ATP-binding site of DNA gyrase protein (upper panel of Fig. 6a and Table S14†). Complex 1 is further stabilized in the protein as hydrophobic protein atoms of Ile (I78) and Pro (P79) residues enclose the hydrophobic region of two phenyl rings of one of the xantphos ligands. Similarly, complex's 4 phenyl rings of DPEphos ligand are also enclosed by I78, P79, and I90 residues, reinforcing thus its stabilization. Special interactions that attract both complexes to the protein include π-charge electrostatic interactions, including π-cation between the negative charge of phenyl aromatic rings of xantphos and DPEphos ligands with the positive charge of arginines R76 and R136, as well as π-anion between the same aromatic rings with the negative charge of glutamate E50 and aspartate D49.
The best-fitted docking poses of 2 and 3, exhibiting the best in vitro activity and the highest in silico binding capacity, respectively, in the crystal structure of S. aureus DNA gyrase superimposed with MFX and QPT-1 are depicted in the lower panel of Fig. 6b. Binding interactions of the two complexes in their binding pockets are reported in Table S14.† The ligand-binding site architecture for both complexes is shown in the upper panel of Fig. 6b. The docking procedure predicts the formation of a variety of interactions between both complexes and the amino acid residues Arg (R272), Gly (G115), Asp (D116), and Asn (N269) (all in chain A) for 2, and Asp (D83), Ser (S84), Tyr (Y87), Glu (E88), Ala (A120), Met (M121), and Arg (R122) (belonging to chains A and C) for 3. It is obvious that 3 extends its binding to both opposite strands of DNA (chains I in slate blue color and chain N in orange color) at an adjacent position to co-crystallized drug QPT-1 which is bound at the artificial nick of one of the DNA strands. The DNA used for this structure was doubly nicked, with artificial breaks in the DNA backbone at both cleavage sites. Complex 3 is shown to be docked at the same place occupied by QPT-1, possibly exhibiting a similar with this inhibitor activity, inducing double-strand cleavage of DNA mediated by DNA gyrase. On the other hand, QPT-1 is bound between the +1 and −1 bases at the DNA cleavage site, inhibiting DNA re-ligation. Complex 3 seems to interrupt the interstrand classical hydrogen bonding DNA base pairing stabilizing the double helix structure, between N4 atom of pyrimidine's ring amino group of cytosine nucleotide dC′2012 of chain I and the pyrimidine's ring carbonyl group O6 of guanine dG′2009 of chain N, dC′2011/N4 (of chain I) and dG′2010/O6 (of chain N), dG′2010/O6 (of chain I) and dC′2011/N4 (of chain N) (it bids with both atoms of the opposite strand nucleotides), and between dG′2009/O6 (of chain I) and dC′2012/N4 (of chain N) (binds only with O6). On the contrary, 2 binds only to one strand of DNA (chain N) interrupting the hydrogen bonding between dC′2012/N4 (chain N) with dG′2009/O6 (chain I) (it binds only with N4). Additionally, it also binds to one more DNA strand (chain E in salmon color) and especially to dG′3/C8, but not being able to interrupt its interstrand hydrogen bonding. Complex 3 interferes also into three intrastrand base steps of chain I: dG′2009pdG′2010, dG′2010pdC′2011, and dC′2011pdC′2012.
The in vitro antibacterial activity of 1–5 against E. coli and S. aureus bacterial strains was found to be 4 > 1 > 2 > 5 > 3 for E. coli and 2 ≈ 4 > 3 > 1 > 5 for S. aureus (Fig. 3 and Table S8†). It is obvious that there is a consistency between in vitro and in silico studies, indicating both 1 and 4 to possess the highest antibacterial activity and binding capacity for E. coli DNA gyrase, and to a lesser extend for S. aureus DNA gyrase. In the particular case of S. aureus DNA gyrase, 2 possessed the second worst binding capacity though exhibited the best in vitro antibacterial activity. Additionally, 5 which was the least active compound against S. aureus strain, presented the lowest binding capacity among all complexes. However, overall, the predicted binding energy values are correlated well with the observed biological data.
DMS114 has mainly FGFR1 with a small amount of FGFR3 expression.91 It has been shown that BGJ398 compound inhibits FGFR signaling with the levels of total FGFR1/FGFR3 and FGFR substrate 2 (FRS2) being significantly decreased in a dose-dependent manner. Previous studies have shown that the FGFR inhibitor PD173074 potentiated the effects of cisplatin in small cell lung cancer thus showing by a different mechanism that inhibition of FGFRs can augment the effects of cisplatin.92
Similarly, SKOV3 cells also found to express FGFR193 along with FGFR2, 5, and 7. This study showed a reduction in the expression of FGFR2, FGFR3, FGFR4, FGF3 and FGF7 using shRNAi, significantly inhibiting SKOV3 ovarian cancer cell line proliferation by 40–80%. inhibition of FGFR2 and/or FGF3 and 7 impacted significantly on SKOV3 proliferation and reduced significantly the IC50 of cisplatin in vitro.
Furthermore, FGFRs found to represent an important therapeutic target in the fight against the pancreatic ductal adenocarcinoma (PDAC). In pancreatic cancer, aberrations in the FGFR pathway, particularly FGFR1 overexpression, have been reported. The clinical significance of FGFR1 expression in pancreatic cancer has been recently investigated94 showing that the FGFR1-based subclassification of pancreatic cancer may lead to new therapeutic approaches for the FGFR1-positive subtype. Recently, a study on the inhibition of FGF/FGFR signaling on this very aggressive form of cancer in vitro, showed that knocking down FGFR1 and FGFR2 decreased their tumorigenesis abilities in vivo.95 Deregulation of the FGF/FGFR axis is involved in oncogenesis, tumor progression and resistance to anti-cancer treatment across multiple types of tumors.96 Over-expression of FGFs and their receptors is a feature of pancreatic cancer and correlates with poor prognosis.97 In a previous study, the inhibition of FGFR2 attenuated proliferation and invasion of pancreatic cancer.98 In this study, FGF1, FGF2, FGF5, and FGF7 found to be overexpressed in PDAC. FGFRs may thus constitute novel therapeutic targets for PDAC.
Additionally, the elucidation of the role of FGFR1 induction in acquired resistance to MET and VEGFR2 inhibition by cabozantinib in prostate cancer (PCa) was studied recently.99 The researchers concluded that the FGFR1 overexpression mediated acquired resistance to MET/VEGFR2 inhibition, leveraging this understanding to improve therapy outcomes. They revealed that the molecular basis of resistance to MET inhibition in prostate cancer is FGFR1 activation through a YAP/TBX5-dependent mechanism. In other studies, it was found that PCa cells with high FGFR1 expression increased the bone metastatic progression of the PCa and that FGFR1 accelerates PCa metastatic dissemination100 and FGFR blockade with dovitinib (TKI258) has clinical activity in a subset of men with castration-resistant PCa (CRPC) and bone metastases.101 These findings suggest that targeting FGFRs has therapeutic activity in advanced PCa and provide direction for the development of therapies with FGFR inhibitors.
In order to elucidate the anticancer activity of 1–5 on different human cancer cells, we adopted molecular docking studies of the complexes on FGFR1 target protein. As shown in Table 2, the order of decreasing binding capacity (from lower to higher global binding energy) of 1–5 to the crystal structure of FGFR1 was calculated to be 4 > 3 > 5 > 1 > 2. The binding of 3 and 4 in the catalytic site of FGFR1 kinase domain is depicted in the lower panel of Fig. 7. Complex 3 (exhibited the best anticancer activity against all tested cancer cells and also the second-best binding capacity in the computational studies) seems to be bound in the active catalytic site of FGFR1 kinase domain, at the same place occupied by the FGFR1 inhibitor drug ponatinib, and especially at the region of ATP-binding pocket of the enzyme. Ponatinib occupies the cleft between the N- and C-terminal lobes where ATP would otherwise bind.102 It is interesting to notice that although 4 exhibits a better binding affinity (lower binding energy) compared to 3, it seems to be bound in a pocket that is out of the ATP-binding pocket of the enzyme, in contrast to 3 which is localized in close proximity to it. This may also explain the better antitumor activity of 3 compared to 4, in terms of their IC50 values. Ligand binding contacts of 3 in the active site of the kinase domain include N568, R570, and E571 of αD helix, G490 at the starting point of β2 sheet, L484 and G485 of the terminal region of β1 sheet, E486 of the loop following β1 sheet, R570 of the loop connecting β7 sheet with αE helix, and residues F642, T658, and N659 of the activation loop (a-Loop) (upper panel of Fig. 7 and Table S15†). Among the binding contacts of 3, three residues seem to be common with those of the binding of ponatinib, namely, Phe (F642), Leu (L484), and Val (V492). The phenyl ring of one of the PPh3 ligands of complex 3 being in contact to F642 residue, appears also to be in close proximity to the imidazopyridazine ring of ponatinib which also makes contact with F642. Notably, F642 residue belongs to the activation loop (a-Loop) of the protein. The other two residues, L484 and V492, are part of beta strands β1 and β2, respectively.
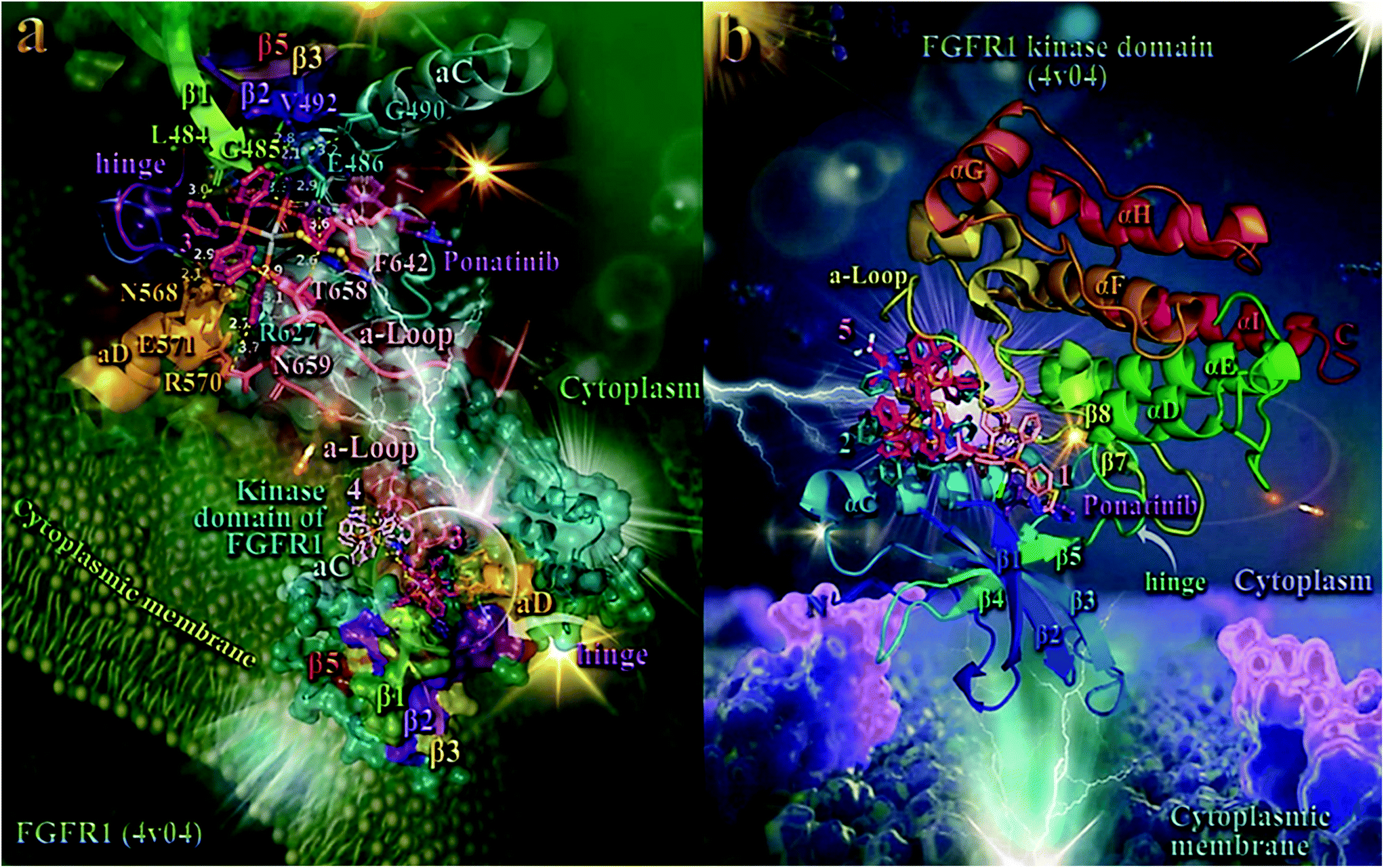 | ||
| Fig. 7 (a) (lower panel) Docking pose orientation of 4 (best bound complex in in silico studies) and 3 (exhibiting the highest in vitro anticancer activity), in the crystal structure of the catalytic domain of fibroblast growth factor receptor 1 (FGFR1) kinase in complex with its inhibitor drug ponatinib (PDB accession number: 4V04). FGFR1 is localized in the cytoplasmic membrane towards the intracellular cytoplasm (transmembrane and extracellular domains are not shown). (upper panel) A close-up view of the ATP-binding site architecture of the second-best binding complex 3. Target protein in both structure models is illustrated as cartoon colored in deep teal with depth cue in the ray-tracing rendering of the whole structure, with additional depiction of semi-transparent surface colored according to cartoon. Docked complexes 3 and 4, as well as the superimposed co-crystallized inhibitor ponatinib, all rendered in ball-and-stick mode and colored according to atom type in warm pink, light pink, and violet purple, respectively. Selected contacting amino acid residues of the binding pocket are rendered in stick model and colored according to protein's segments: activation loop (a-Loop) (deep salmon), aC and aD helices (pale cyan and bright orange, respectively), β1, β2, β3, and β5 antiparallel beta sheets (split pea green, purple blue, yellow-orange, and firebrick red, respectively), and hinge (deep purple), highlighting also the mapped surface of the corresponding part of the interacting residues in the same colors. A-loop is depicted in its active (open) conformation. Complex 3 is found to be anchored in the vicinity of the ATP-binding cleft, at the same place with ponatinib. (b) Docking pose orientation of 1, 2, and 5 in the crystal structure of the catalytic domain of fibroblast growth factor receptor 1 (FGFR1) kinase in complex with its inhibitor drug ponatinib (PDB accession number: 4V04). FGFR1 is localized in the cytoplasmic membrane towards the intracellular cytoplasm (transmembrane and extracellular domains are not shown). Target protein is illustrated as cartoon colored in rainbow. Docked complexes 1, 2, and 5, as well as the superimposed co-crystallized inhibitor ponatinib, all rendered in stick mode and colored according to atom type in salmon, teal, hot pink, and violet purple, respectively. FGFR1 protein's segments: activation loop (a-Loop), aC and aD helices, β1, β2, β3, and β5 antiparallel beta sheets, and hinge, are depicted in the structure colored according to cartoon. A-loop is depicted in its active (open) conformation. Complex 1 is found adjacent to ponatinib at the ATP-binding cleft of the protein. Molecular docking simulations of all molecules were performed individually. Binding contacts are shown as dotted yellow lines. Heteroatom color-code: O: red, N: blue, S: yellow, P: orange, F of ponatinib: cyan, F: cyan, and Ag: grey. Hydrogen atoms are omitted from all molecules for clarity. The final structure was ray-traced and illustrated with the aid of PyMol Molecular Graphics Systems. Binding interactions are shown in Table S15.† | ||
As shown in the upper panel of Fig. 7, atdzt− ligand of 3 extends away from the hinge region making limited interactions with the enzyme, involving only two binding contacts with threonine T658 and glutamate E486. All binding contacts are reported in Table S15.† Critical binding contact of ponatinib, F642, seems to be also one of the most important contacts of 3, stabilizing its molecule in the active site by π–π displaced (offset) and also T-shape with two phenyl rings of a PPh3 ligand. One of the phenyl rings is found in close proximity to imidazopyridazine ring of ponatinib that mediate its binding to F642 via also π–π displaced type interaction. The rest binding contacts of ponatinib (A512, K514, E531, M534, M535, I538, I544, I545, V559, Y563, A564, G567, C619, I620, H621, R622, L630, D641, and L644, do not seem to be involved in direct interactions with 3, indicating thus a more stable binding of ponatinib.
As shown in Fig. 7a, 4 is accommodated in a binding pocket of the kinase domain of FGFR1 formed by αEF, αF, αC helices, β1, β2 beta sheets, and α-Loop. It seems that 3 and 4, along with ponatinib, form a circle enclose in its periphery by the activation loop (a-Loop) and the hinge region, at diametrically opposed positions. Ponatinib (flanked by β2, β3, and β5 sheets and also positioned between the loop formed by β4 sheet and αC helix) and 4 are flanked by αC helix and a-Loop, while 3 by a-Loop, hinge region, β1 and β2 beta sheets, and αD helix. The ascending loop starting from sheet β8 towards the αG helix, is centered in the imaginable formed circle by the docked molecules and the co-crystallized inhibitor, making contact with all three complexes. This ascending loop is also making contacts with 5 (Fig. 7b) and especially with the CF3 group of the mtft− ligand of 5, but not in close proximity to the corresponding trifluoromethylphenyl ring of ponatinib (about 10 Å away from it), with access only to the periphery of the ATP-binding pocket of the kinase. This may explain, in part, the lower binding capacity of 5 compared to 3 and 4. The same is also observed for the antibacterial activity of the similarly structured 4 and 5, revealing 4 to be more active than 5, which suggests that the presence of a CF3-group substituent in 5 does not contribute to its antibacterial activity.
Complex 3 is also flanked by αD helix and the part of loop starting from sheet β7 and in proximity to it, and also by sheets β1 and β2, making contacts with some of their residues. The loop formed between αI helix and β7 sheet is found in close proximity to the binding sites of 4 (the portion in the vicinity of αI) and 3 (the portion in the vicinity of β7). Further stabilization of 3 in the catalytic active site of the kinase is achieved by the interaction of one of its PPh3 ligands with the proximal end of αD helix. It is of interest that one of the PPh3 ligands of 4 is also in close proximity to the imidazopyridazine ring of ponatinib, sharing common binding contacts with it.
The best scored poses of docked complexes 1, 2, and 5 are depicted in Fig. 7b. Binding of 1 in the catalytic active site of FGFR1 clearly indicates that it is located in a pocket of the ATP-binding site, similarly to 3 and at the same place occupied by ponatinb.
3. Conclusions
In this work, we report a series of Ag(I) complexes bearing as ligands a phosphine and a small size heterocyclic ring thioamide with highly electronegative NH2- or CF3-group substituents (atdztH and mtftH, respectively), and the assessment of their in vitro antibacterial and anticancer efficacy. Complexes with the NH2-substituted heterocyclic thioamide were found to display a moderate-to-high potential to inhibit the growth of a series of Gram-(+) and Gram-(−) bacterial strains. A high antiproliferative effect was also observed for these complexes against ovarian, pancreatic, lung and prostate cancer cell lines, while maintaining their cytotoxicity against normal fibroblast cells at lower levels. In contrast, CF3-substitution on the heterocyclic thioamide was found to have a detrimental effect on the bioactivity in this type of Ag(I) complexes. Complexes with the NH2-substituted ligand are able to bind reversibly to BSA and HSA proteins and transport through serum. The complexes display a significant in vitro antioxidant ability for scavenging free radicals and a very low efficacy to destroy the double-strand structure of CT-DNA by intercalation, which provide some hints for possible mechanisms of their in vitro bioactivity. A molecular docking study was also conducted to get further insight into possible mechanisms of the bioactivity of the complexes, both as antibacterial and anticancer agents, suggesting a possible role of DNA gyrase and FGFR1 target proteins.Abbreviations
| a-Loop | Activation loop |
| CBN | Chlorobiocin |
| FGFs | Fibroblast growth factors |
| FGFR1 | Fibroblast growth factor receptor 1 |
| FGFRs | FGF receptors |
| Glide | Grid based Ligand docking with energetics |
| GUI | Graphical user interface |
| IFD | Induced-fit docking |
| MFX | Moxifloxacin |
| OPLS3 | Optimized potential for liquid simulation |
| Pca | Prostate cancer |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDB | Protein data bank |
| RCSB | Research collaboratory for structural bioinformatics |
| RTKs | Receptor tyrosine kinases |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research is co-financed by Greece and the European Union (European Social Fund-ESF) through the Operational Programme “Human Resources Development, Education and Lifelong Learning” in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research” (MIS-5000432), implemented by the State Scholarships Foundation (IKY). The authors would like to thank the Department of Chemistry of Aristotle University of Thessaloniki for financial support of this work. The authors would like to acknowledge the Large Research Instrumentation NMR and single crystal-XRD Facilities of the Chemistry Department of the Aristotle University of Thessaloniki for providing access to the facilities.Notes and references
- S. Dasari and P. B. Tchounwou, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- W. H. Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 20, 3993–3993 CrossRef.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F. Wang, W. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- F. Arnesano, M. Losacco and G. Natile, in Binding, Transport and Storage of Metal Ions in Biological Cells, ed. W. Maret and A. Wedd, Royal Society of Chemistry, Cambridge, United Kingdom, 2014, ch. 15, pp. 429–460 Search PubMed.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481–489 CrossRef CAS PubMed.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- S. Medici, M. Peana, G. Crisponi, V. M. Nurchi, J. I. Lachowicz, M. Remelli and M. A. Zoroddu, Coord. Chem. Rev., 2016, 327–328, 349–359 CrossRef CAS.
- X. Liang, S. Luan, Z. Yin, M. He, C. He, L. Yin, Y. Zou, Z. Yuan, L. Li, X. Song, C. Lv and W. Zhang, Eur. J. Med. Chem., 2018, 157, 62–80 CrossRef CAS PubMed.
- R. A. Haque, N. Hasanudin, M. A. Hussein, S. A. Ahamed and M. A. Iqbal, Inorg. Nano-Met. Chem., 2017, 47, 131–137 CrossRef CAS.
- J. K. Aulakh, T. S. Lobana, H. Sood, D. S. Arora, V. A. Smolinski, C. E. Duff and J. P. Jasinski, J. Inorg. Biochem., 2018, 178, 18–31 CrossRef CAS PubMed.
- R. Algotiml, A. Gab-Alla, R. Seoudi, R. Seoudi, H. H. Abulreesh, M. Z. El-Readi and K. Elbann, Sci. Rep., 2022, 12, 2421 CrossRef CAS PubMed.
- M. Quintana, A. Rodriguez-Rius, A. Vellé, S. Vives, P. J. Sanz Miguel and G. Triola, Bioorg. Med. Chem., 2022, 67, 116814 CrossRef CAS PubMed.
- G. Bresciani, N. Busto, V. Ceccherini, M. Bortoluzzi, G. Pampaloni, B. Garcia and F. Marchetti, J. Inorg. Biochem., 2022, 227, 111667 CrossRef CAS PubMed.
- S. P. Gajare, P. A. Bansode, P. V. Patil, A. D. Patil, D. M. Pore, K. D. Sonawane, M. J. Dhanavade, V. M. Khot and G. S. Rashinkar, ChemistrySelect, 2021, 6, 1958–1968 CrossRef CAS.
- W. R. Hill and D. M. Pillsbury, Argyria: The Pharmacology of Silver, Williams & Wilkins, Baltimore, 1939 Search PubMed.
- H. J. Klasen, Burns, 2000, 26, 131–138 CrossRef CAS PubMed.
- C. N. Banti, M. Kapetana, C. Papachristodoulou, C. P. Raptopoulou, V. Psycharis, P. Zoumpoulakis, T. Mavromoustakos and S. K. Hadjikakou, Dalton Trans., 2021, 50, 13712–13727 RSC.
- A. Sánchez, C. J. Carrasco, F. Montilla, E. Álvarez, A. Galindo, M. Pérez-Aranda, E. Pajuelo and A. Alcudia, Pharmaceutics, 2022, 14, 748 CrossRef PubMed.
- M. Pellei, V. Gandin, M. Marinelli, C. Marzano, M. Yousufuddin, H. V. Dias and C. Santini, Inorg. Chem., 2012, 51, 9873–9882 CrossRef CAS PubMed.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS PubMed.
- Z. Engelbrecht, R. Meijboom and M. J. Cronjé, BioMetals, 2018, 31, 189–202 CrossRef CAS PubMed.
- R. Visbal, V. Fernández-Moreira, I. Marzo, A. Laguna and M. C. Gimeno, Dalton Trans., 2016, 45, 15026–15033 RSC.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- S. Silver, FEMS Microbiol. Rev., 2003, 27, 341–353 CrossRef CAS PubMed.
- W. J. Youngs, A. R. Knapp, P. O. Wagers and C. A. Tessier, Dalton Trans., 2012, 41, 327–336 RSC.
- R. B. Thurman, C. P. Gerba and G. Bitton, Crit. Rev. Environ. Control, 1989, 18, 295–315 CrossRef.
- G. McDonnell and A. D. Russell, Clin. Microbiol. Rev., 1999, 12, 147–179 CrossRef CAS PubMed.
- R. A. Khan, K. Al-Farhan, A. de Almeida, A. Alsalme, A. Casini, M. Ghazzali and J. Reedijk, J. Inorg. Biochem., 2014, 140, 1–5 CrossRef PubMed.
- H. Wang, M. Wang, X. Xu, P. Gao, Z. Xu, Q. Zhang, H. Li, A. Yan, R. Y.-T. Kao and H. Sun, Nat. Commun., 2021, 12, 3331 CrossRef CAS PubMed.
- M. J. Mckeage, L. Maharaj and S. Berners-Price, Coord. Chem. Rev., 2002, 232, 127–135 CrossRef CAS.
- M. J. Mckeage, L. Maharaj, S. Berners-Price, P. Galettis, R. J. Bowen, W. Brouwer, L. Ding, L. Zhuang and B. C. Baguley, Cancer Chemother. Pharmacol., 2000, 46, 343–350 CrossRef CAS PubMed.
- C. N. Banti and S. K. Hadjikakou, Metallomics, 2013, 5, 569–596 CrossRef CAS PubMed.
- S. J. Berners-Price, R. J. Bowen, P. J. Harvey, P. C. Healy and G. A. Koutsantonis, J. Chem. Soc., Dalton Trans., 1998, 1743–1750 RSC.
- C. Santini, M. Pellei, G. Papini, B. Morresi, R. Galassi, S. Ricci, F. Tisato, M. Porchia, M. P. Rigobello, V. Gandin and C. Marzano, J. Inorg. Biochem., 2011, 105, 232–240 CrossRef CAS PubMed.
- L. Kyros, N. Kourkoumelis, M. Kubicki, L. Male, M. B. Hursthouse, I. I. Verginadis, E. Gouma, S. Karkabounas, K. Charalabopoulos and S. K. Hadjikakou, Bioinorg. Chem. Appl., 2010, 2010, 386860 Search PubMed.
- T. Fatima, R. A. Haque, A. Ahmad, L. Elsir, A. Hassan, M. B. Khadeer Ahamed, A. M. S. A. Majid and M. R. Razali, J. Mol. Struct., 2020, 1222, 128890 CrossRef CAS.
- D. Żyro, A. Śliwińska, I. Szymczak-Pajor, M. Stręk and J. Ochocki, Cancers, 2020, 12, 3848 CrossRef PubMed.
- N. R. F. Mohd Sofyan, F. J. Nordin, M. R. Mohd Abd Razak, S. N. A. Abdul Halim, N. A. F. Mohd Khir, A. Muhammad, N. F. Rajab and R. Sarip, J. Chem., 2018, 2018, 8395374 Search PubMed.
- M. P. Chrysouli, C. N. Banti, I. Milionis, D. Koumasi, C. P. Raptopoulou, V. Psycharis, I. Sainis and S. K. Hadjikakou, Mater. Sci. Eng., C, 2018, 93, 902–910 CrossRef CAS PubMed.
- A. K. Rossos, C. N. Banti, A. G. Kalampounias, C. Papachristodoulou, K. Kordatos, P. Zoumpoulakis, T. Mavromoustakos, N. Kourkoumelis and S. K. Hadjikakou, Mater. Sci. Eng., C, 2020, 111, 110770 CrossRef CAS PubMed.
- J. K. Aulakh, T. S. Lobana, H. Sood, D. S. Arora, R. Kaur, J. Singh, I. Garcia-Santos, M. Kaure and J. P. Jasinski, RSC Adv., 2019, 9, 15470–15487 RSC.
- D. Varna, D. I. Zainuddin, A. G. Hatzidimitriou, G. Psomas, A. A. Pantazaki, R. Papi, P. Angaridis and P. Aslanidis, Mater. Sci. Eng., C, 2019, 99, 450–459 CrossRef CAS PubMed.
- D. Anastasiadou, E. Geromichalou, E. Tsavea, G. Psomas, A. G. Hatzidimitriou, S. Kalogiannis, G. Geromichalos, D. Trafalis, P. Dalezis and P. Aslanidis, J. Inorg. Biochem., 2020, 210, 111167 CrossRef CAS PubMed.
- L. H. Takahashi, R. Radhakrishnan, R. E. Rosenfield Jr., E. F. Meyer Jr. and D. A. J. Trainor, J. Am. Chem. Soc., 1989, 111, 3368–3374 CrossRef CAS.
- R. H. Abeles and T. A. J. Alston, Biol. Chem., 1990, 265, 16705–16708 CrossRef CAS.
- T. Kovács, A. Pabuccuoglu, K. Lesiak and P. F. Torrence, Bioorg. Chem., 1993, 21, 192–208 CrossRef.
- C. Mattos, B. Rasmussen, X. Ding, G. A. Petsko and D. Ringe, Struct. Biol., 1994, 1, 55–58 CrossRef CAS PubMed.
- D. O'Hagan and H. S. Rzepa, Chem. Commun., 1997, 7, 645–652 RSC.
- I. H. Krakoff and M. D. Balis, J. Clin. Invest., 1959, 38, 907–915 CrossRef CAS PubMed.
- R. Asbury, J. A. Blessing and D. Moore, Am. J. Clin. Oncol., 1996, 19, 400–402 CrossRef CAS PubMed.
- K. Haranahalli, T. Honda and I. Ojima, J. Fluorine Chem., 2019, 217, 29–40 CrossRef CAS PubMed.
- B. M. Johnson, Y. Z. Shu, X. Zhuo and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed.
- R. Buratto, D. Mammoli, E. Canet and G. Bodenhausen, J. Med. Chem., 2016, 59, 1960–1966 CrossRef CAS PubMed.
- J. B. I. Sap, N. J. W. Straathof, T. Knauber, C. F. Meyer, M. Medebielle, L. Buglioni, C. Genicot, A. A. Trabanco, T. Noel, C. W. am Ende and V. Gouverneur, J. Am. Chem. Soc., 2020, 142, 9181–9187 CrossRef CAS PubMed.
- X. Deng, S. Kokkonda, F. El Mazouni, J. White, J. N. Burrows, W. Kaminsky, S. A. Charman, D. Matthews, P. K. Rathod and M. A. Phillips, J. Med. Chem., 2014, 57, 5381–5394 CrossRef CAS PubMed.
- A. Abula, Z. Xu, Z. Zhu, C. Peng, Z. Chen, W. Zhu and H. A. Aisa, J. Chem. Inf. Model., 2020, 60(12), 6242–6250 CrossRef CAS PubMed.
- D. Varna, E. Kapetanaki, A. Koutsari, A. G. Hatzidimitriou, G. Psomas, P. Angaridis, R. Papi, A. A. Pantazaki and P. Aslanidis, Polyhedron, 2018, 151, 131–140 CrossRef CAS.
- P. A. Papanikolaou, A. G. Papadopoulos, E. Andreadou, A. Hatzidimitriou, P. J. Cox, A. Pantazaki and P. Aslanidis, New J. Chem., 2015, 39, 4830–4844 RSC.
- G. Christofidis, P. J. Cox and P. Aslanidis, Polyhedron, 2012, 31, 502–505 CrossRef CAS.
- M. Calvo, O. Crespo, M. C. Gimeno, A. Laguna, M. T. Oliván, V. Polo, D. Rodríguez and J.-M. Sáez-Rocher, Inorg. Chem., 2020, 59, 14447–14456 CrossRef CAS PubMed.
- D. Varna, E. Geromichalou, E. Papachristou, R. Papi, A. G. Hatzidimitriou, E. Panteris, G. Psomas, G. D. Geromichalos, P. Aslanidis, T. Choli-Papadopoulou and P. A. Angaridis, J. Inorg. Biochem., 2022, 228, 111695 CrossRef CAS PubMed.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 23–24, 1089–1097 CrossRef PubMed.
- L. Conti, A. Mengoni, G. E. Giacomazzo, L. Mari, M. Perfetti, C. Fagorzi, L. Sorace, B. Valtancoli and C. Giorgi, J. Inorg. Biochem., 2021, 220, 11467 CrossRef PubMed.
- Y. Feng, W. Z. Sun, X. S. Wang and Q. X. Zhou, Chemistry, 2019, 25, 13879–13884 CrossRef CAS PubMed.
- H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Chem. – Eur. J., 2019, 25, 11797–11819 CrossRef CAS PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- M. Wettasinghe and F. Shahidi, Food Chem., 2000, 70, 17–26 CrossRef CAS.
- K. Gurova, Future Oncol., 2009, 5, 1685–1704 CrossRef CAS PubMed.
- E. Boros, P. J. Dyson and G. Gasser, Chem, 2020, 6, 41–60 CAS.
- D. P. Ašanin, S. Skaro Bogojevic, F. Perdih, T. P. Andrejević, D. Milivojevic, I. Aleksic, J. Nikodinovic-Runic, B. D. Glišić, I. Turel and M. I. Djuran, Molecules, 2021, 26, 1871 CrossRef PubMed.
- M. Sirajuddin, S. Ali and A. Badshah, J. Photochem. Photobiol., B, 2013, 124, 1–19 CrossRef CAS PubMed.
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617–3630 RSC.
- L. Strekowski and B. Wilson, Mutat. Res., 2007, 623, 3–13 CrossRef CAS PubMed.
- A. Wolfe, G. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS PubMed.
- J. L. Garcia-Gimenez, M. Gonzalez-Alvarez, M. Liu-Gonzalez, B. Macias, J. Borras and G. Alzuet, J. Inorg. Biochem., 2009, 103, 923–934 CrossRef PubMed.
- D. P. Heller and C. L. Greenstock, Biophys. Chem., 1994, 50, 305–312 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Boston, MA, 2006 Search PubMed.
- H. Qi, Y. Wang, X. Wanga, L. Su, Y. Wanga and S. Wang, Spectrochim. Acta, Part A, 2021, 259, 119809 CrossRef CAS PubMed.
- F. Moreno, M. Cortijo and J. González-Jiménez, Photochem. Photobiol., 1999, 69, 8–15 CrossRef CAS PubMed.
- M. Gellert, K. Mizuuchi, M. H. O'Dea and H. A. Nash, Proc. Natl. Acad. Sci. U. S. A., 1976, 73, 3872–3876 CrossRef CAS PubMed.
- L. L. Shen and D. T. W. Chu, Curr. Pharm. Des., 1996, 2, 195–208 CAS.
- A. C. Gentry and N. Osheroff, in Encyclopedia of Biological Chemistry, ed. W. J. Lenarz and M. Daniel Lane, Academic Press, 2nd edn, 2013, ch. DNA Topoisomerase: Type II, pp. 163–168 Search PubMed.
- J. A. Ali, A. P. Jackson, A. J. Howells and A. Maxwell, Biochemistry, 1993, 32, 2717–2724 CrossRef CAS PubMed.
- J. A. Ali, G. Orphanides and A. Maxwell, Biochemistry, 1995, 34, 9801–9808 CrossRef CAS PubMed.
- D. Lafitte, V. Lamour, P. O. Tsvetkov, A. A. Makarov, M. Klich, P. Deprez, D. Moras, C. Briand and R. Gilli, Biochemistry, 2002, 41, 7217–7223 CrossRef CAS PubMed.
- N. Turner and R. Grose, Nat. Rev. Cancer, 2010, 10, 116–129 CrossRef CAS PubMed.
- D. M. Ornitz and N. Itoh, Wiley Interdiscip. Rev.: Dev. Biol, 2015, 4, 215–266 Search PubMed.
- I. S. Babina and N. C. Turner, Nat. Rev. Cancer, 2017, 17, 318–332 CrossRef CAS PubMed.
- M. Katoh and H. Nakagama, Med. Res. Rev., 2014, 34, 280–300 CrossRef CAS PubMed.
- J. Datta, S. Damodaran, H. Parks, C. Ocrainiciuc, J. Miya, L. Yu, E. P. Gardner, E. Samorodnitsky, M. R. Wing, D. Bhatt, J. Hays, J. W. Reeser and S. Roychowdhury, Mol. Cancer Ther., 2017, 16, 614–624 CrossRef CAS PubMed.
- O. E. Pardo, J. Latigo, R. E. Jeffery, E. Nye, R. Poulsom, B. Spencer-Dene, N. R. Lemoine, G. W. Stamp, E. O. Aboagye and M. J. Seckl, Cancer Res., 2009, 69, 8645–8651 CrossRef CAS PubMed.
- C. Cole, S. Lau, A. Backen, A. Clamp, G. Rushton, C. Dive, C. Hodgkinson, R. McVey, H. Kitchener and G. C. Jayson, Cancer Biol. Ther., 2010, 10, 495–504 CrossRef CAS PubMed.
- F. Haq, Y. N. Sung, I. Park, M. A. Kayani, F. Yousuf, S. M. Hong and S. M. Ahn, J. Transl. Med., 2018, 16, 374–381 CrossRef CAS PubMed.
- M. Y. Quan, Q. Guo, J. Liu, R. Yang, J. Bai, W. Wang, Y. Cai, R. Han, Y. Q. Lv, L. Ding, D. D. Billadeau, Z. Lou, S. Bellusci, X. Li and J. S. Zhang, Front. Cell Dev. Biol., 2020, 8, 287–291 CrossRef PubMed.
- H. Dianat-Moghadam and L. Teimoori-Toolabi, Curr. Drug Targets, 2019, 20, 852–870 CrossRef CAS PubMed.
- S. J. Coleman, A. M. Chioni, M. Ghallab, R. K. Anderson, N. R. Lemoine, H. M. Kocher and R. P. Grose, EMBO Mol. Med., 2014, 6, 467–481 CrossRef CAS PubMed.
- Y. Matsuda, H. Yoshimura, T. Suzuki, E. Uchida, Z. Naito and T. Ishiwata, Cancer Sci., 2014, 105, 1212–1219 CrossRef CAS PubMed.
- F. Koinis, P. Corn, N. Parikh, J. Song, I. Vardaki, I. Mourkioti, S. H. Lin, C. Logothetis, T. Panaretakis and G. Gallick, Cancers, 2020, 12, 244–261 CrossRef CAS PubMed.
- E. Labanca, J. Yang, P. D. A. Shepherd, X. Wan, M. W. Starbuck, L. D. Guerra, N. Anselmino, J. A. Bizzotto, J. Dong, A. M. Chinnaiyan, M. K. Ravoori, V. Kundra, B. M. Broom, P. G. Corn, P. Troncoso, G. Gueron, C. J. Logothethis and N. M. Navone, Eur. Urol. Oncol., 2022, 5, 164–175 CrossRef PubMed.
- X. Wan, P. G. Corn, J. Yang, N. Palanisamy, M. W. Starbuck, E. Efstathiou, E. M. Li Ning Tapia, A. J. Zurita, A. Aparicio, M. K. Ravoori, E. S. Vazquez, D. R. Robinson, Y. M. Wu, X. Cao, M. K. Iyer, W. McKeehan, V. Kundra, F. Wang, P. Troncoso, A. M. Chinnaiyan, C. J. Logothetis and N. M. Navone, Sci. Transl. Med., 2014, 252, 252ra122 Search PubMed.
- J. A. Tucker, T. Klein, J. Breed, A. L. Breeze, R. Overman, C. Phillips and R. A. Norman, Structure, 2014, 22, 1764–1774 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2152740, 2152741, 2152742 and 2152743. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt00793b |
| This journal is © The Royal Society of Chemistry 2022 |