
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of CRBN as a target of thalidomide: a breakthrough for progress in the development of protein degraders
Junichi
Yamamoto
 a,
Takumi
Ito
b,
Yuki
Yamaguchi
a and
Hiroshi
Handa
*c
a,
Takumi
Ito
b,
Yuki
Yamaguchi
a and
Hiroshi
Handa
*c
aSchool of Life Science and Technology, Tokyo Institute of Technology, Yokohama 226-8501, Japan
bInstitute of Medical Science, Tokyo Medical University, Shinjuku, Tokyo 160-8402, Japan
cCenter for Future Medical Research, Tokyo Medical University, Shinjuku, Tokyo 160-8402, Japan. E-mail: hhanda@tokyo-med.ac.jp
First published on 7th July 2022
Abstract
Progress in strategies aimed at breaking down therapeutic target proteins has led to a paradigm shift in drug discovery. Thalidomide and its derivatives are the only protein degraders currently used in clinical practice. Our understanding of the molecular mechanism of action of thalidomide and its derivatives has advanced dramatically since the identification of cereblon (CRBN) as their direct target. The binding of thalidomide derivatives to CRBN, a substrate recognition receptor for Cullin 4 RING E3 ubiquitin ligase (CRL4), induces the recruitment of non-native substrates to CRL4CRBN and their subsequent degradation. This discovery was a breakthrough in the current rapid development of protein-degrading agents because clarification of the mechanism of action of thalidomide derivatives has demonstrated the clinical value of these compounds. This review provides an overview of the mechanism of action of thalidomide and its derivatives and describes perspectives for protein degraders.
 Junichi Yamamoto | Junichi Yamamoto is currently an assistant professor at Tokyo Institute of Technology. He received his PhD degree at Tokyo Institute of Technology in 2014. After postdoctoral training at Tokyo Medical University under the supervision of professor Hiroshi Handa, he joined the faculty at Tokyo Institute of Technology. His current research interest is focused on targeted protein degradation for cancer therapy. |
 Takumi Ito | Takumi Ito is currently an associate professor at Tokyo Medical University. He received his PhD in Engineering from Tokyo Institute of Technology (Supervisor: Hiroshi Handa). From 2008 to 2012, he was a researcher at Tokyo Institute of Technology. From 2012 to 2013, he worked as an Assistant Professor at Tokyo Institute of Technology. Then, he joined the faculty at Tokyo Medical University. His current research interests are small molecule-induced protein degradation and molecular mechanisms of cereblon-binding drugs. |
 Yuki Yamaguchi | Yuki Yamaguchi is currently Professor of Life Science and Technology at Tokyo Institute of Technology. He received his PhD at the same university under then-professor Hiroshi Handa. After postdoctoral training, he was appointed as Principal Investigator of JST's PRESTO program. Then, he joined the faculty at Tokyo Institute of Technology. His research has focused on the mechanisms of chromatin, transcription, and RNA processing and chemical biology aimed at developing new therapeutic strategies. |
 Hiroshi Handa | Hiroshi Handa is a professor at Tokyo Medical University and a professor emeritus at Tokyo Institute of Technology. He graduated from Keio University, School of Medicine in 1972 and got his MD, PhD in 1976 from the University's Graduate School of Medicine. In 1976, he became an assistant professor at the University of Tokyo. He worked as a postdoctoral fellow in the P. Sharp Lab at MIT from 1978 to 1980. He became an associate professor in 1984 at the University of Tokyo, and a full professor from 1991 to 2012 at the Tokyo Institute of Technology. |
Key learning points1. Historical overview of thalidomide and its derivatives as pharmaceuticals.2. Mechanism of action of thalidomide and its derivatives at the molecular level. 3. The structure and function of cereblon, a direct target of thalidomide. 4. Mechanism of action of protein degraders with a mode of action different from conventional small-molecule compounds. 5. Advantages and perspectives of the two classes of protein degraders, molecular glue degraders and PROTACs. |
1. Introduction
Protein degraders are compounds that selectively knock down target proteins via intracellular protein degradation pathways, and are regarded as a novel class of therapeutic agents that employ a different mechanism of action than conventional small molecule compounds, which lead to the breakdown of disease-related proteins. The ubiquitin-proteasome system (UPS) is a major protein degradation pathway that is responsible for the degradation of over 80% of proteins in eukaryotic cells. The UPS degrades target proteins by multiple sequential reactions (Fig. 1). First, the ubiquitin-activating enzyme (E1) forms a thioester bond with ubiquitin in an ATP-dependent manner. The activated ubiquitin moiety is then transferred to the ubiquitin-conjugating enzyme (E2) through a transthioesterification reaction. Next, the E2 enzyme cooperates with a variety of ubiquitin ligases (E3) to transfer ubiquitin to lysine residues on substrate proteins. Polyubiquitinated substrate proteins are recognized, unfolded, and degraded by the proteasome. Protein degraders are small molecule compounds that induce selective degradation of target proteins by hijacking the intracellular UPS. This targeted protein degradation strategy dramatically expands the spectrum of druggable target proteins and is considered as a new promising modality for drug development, as it does not require a clear active site on target proteins.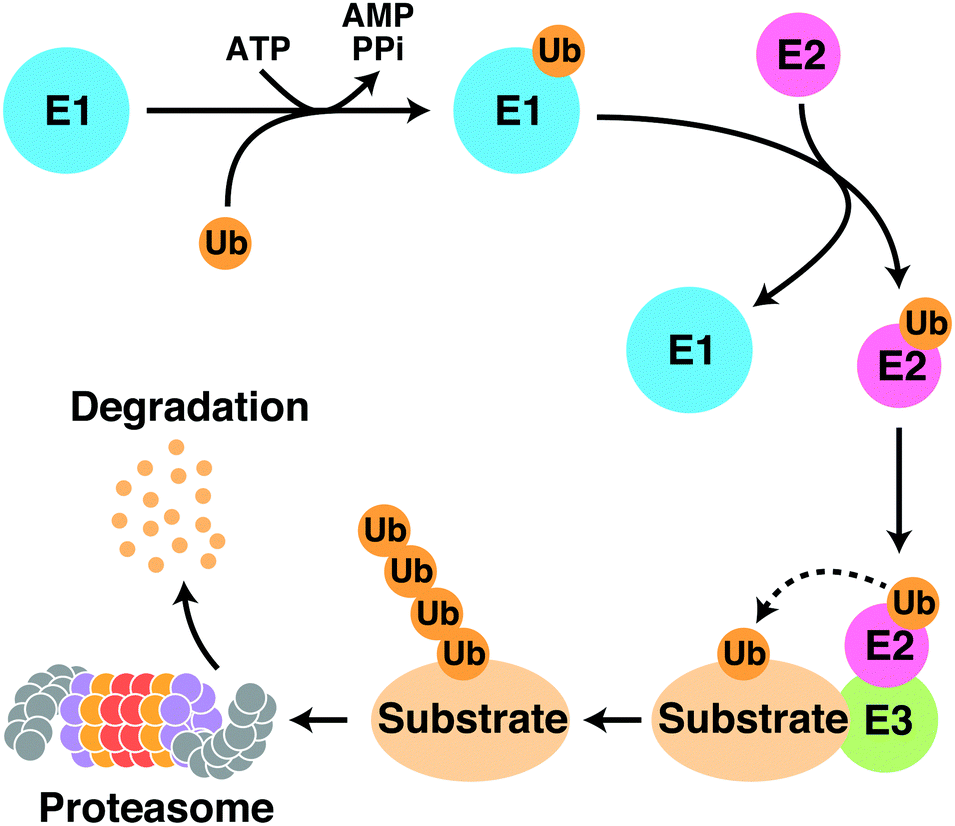 | ||
| Fig. 1 Schematic representation of the ubiquitin proteasome system. Abbreviations: E1, ubiquitin-activating enzyme; E2, ubiquitin-conjugating enzyme; E3, ubiquitin ligase; Ub, ubiquitin; ATP, adenosine triphosphate; AMP, adenosine monophosphate; PPi, pyrophosphate. | ||
Thalidomide derivatives, such as lenalidomide and pomalidomide, collectively referred to as immunomodulatory imide drugs (IMiDs), are mainly used to treat hematological malignancies such as multiple myeloma (Fig. 2). Multiple myeloma is a disease in which plasma cells in the bone marrow become cancerous; the development of IMiDs and other medications have greatly improved its prognosis over the last 20 years. Thalidomide was one of the worst drug disasters in history; however, it has attracted significant attention not only because of its clinical value, but also because of its novel mechanism of action, which has great promise. Our understanding of the mechanism of action of these drugs has advanced rapidly in the last decade with the identification of cereblon (CRBN) as their direct target.1 CRBN is a substrate recognition receptor for Cullin 4 RING E3 ubiquitin ligase (CRL4); the binding of thalidomide derivatives to CRBN triggers the recruitment of non-native substrates to CRL4CRBN and their subsequent proteasomal degradation.2–13 Thalidomide derivatives are the first, and currently the only, protein degraders with demonstrated clinical benefit.
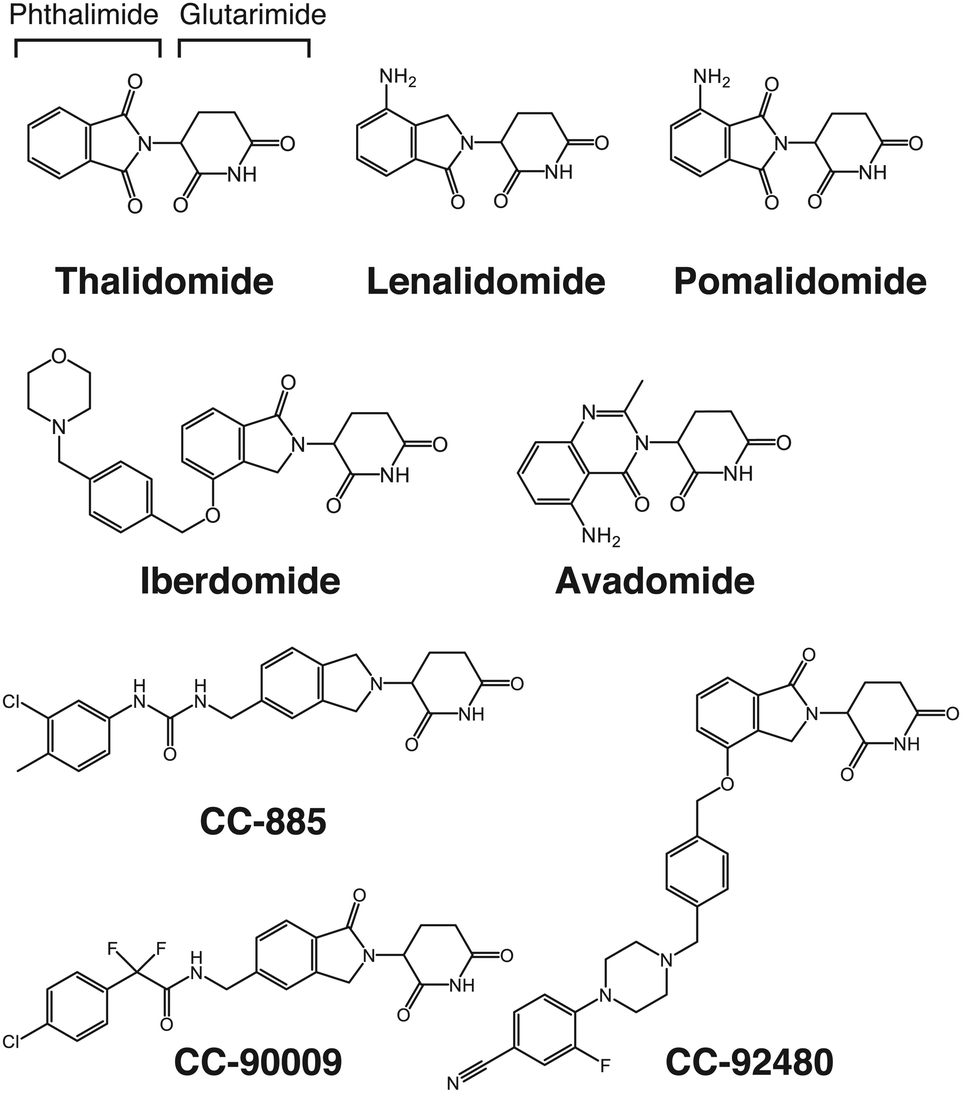 | ||
| Fig. 2 Structure of thalidomide-based glue degraders. Thalidomide is composed of glutarimide and phthalimide. Thalidomide, lenalidomide, and pomalidomide are collectively referred to as IMiDs. All thalidomide-based glue degraders targeting CRBN are collectively referred to as CELMoDs. | ||
Protein degraders are currently under rapid development as a promising modality for drug discovery. The identification of CRBN as a primary target of thalidomide was a breakthrough that is now driving this innovative area of research. In this review, we will provide an overview of the mechanism of action of thalidomide and its derivatives, which will be followed by a discussion of the current status and future perspectives of protein degraders.
2. Pharmacological activities of thalidomide and its derivatives
Thalidomide was first developed in the 1950s by the German pharmaceutical company Grünenthal as a sedative or antiemetic for morning sickness in pregnant women. Thalidomide was marketed in over 40 countries worldwide but was later withdrawn from the market because it causes a wide range of serious teratogenic effects in the fetus, including characteristic limb defects.14,15 By 1962, thalidomide was discontinued in most countries. It is estimated that thalidomide caused serious birth defects in over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 newborns; however, the total number of fetuses actually affected by thalidomide is unknown because the rate of miscarriage increased during this period, and because thalidomide causes damage to a wide variety of tissues that can lead to miscarriage.16
000 newborns; however, the total number of fetuses actually affected by thalidomide is unknown because the rate of miscarriage increased during this period, and because thalidomide causes damage to a wide variety of tissues that can lead to miscarriage.16
Subsequent studies, however, showed that thalidomide has unexpected clinical benefits, allowing it to re-enter the market. Thalidomide is effective for treating erythema nodosum leprosum (ENL), an infectious inflammatory disease characteristic of leprosy.17 Moreover, thalidomide has immunomodulatory properties, modulating the expression of inflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interleukins.18,19 Research on the pharmacological effects of thalidomide has continued, demonstrating its unique properties with diverse pharmacological activities such as inhibition of angiogenesis20 and anti-myeloma21 and immunomodulatory effects. The U.S. Food and Drug Administration (FDA) approved thalidomide for use in ENL and multiple myeloma in 1998 and 2006, respectively. The use of thalidomide is strictly controlled under the program called the Thalidomide Risk Evaluation and Mitigation Strategy. Perhaps, the most important clinical benefit of thalidomide and its derivatives is their anti-cancer activity against hematological malignancies including multiple myeloma. Lenalidomide and pomalidomide are thalidomide derivatives that were developed by Celgene Corporation (currently part of Bristol-Myers Squibb) and collectively referred to as IMiDs because of their potent immunomodulatory effects (Fig. 2).22,23 These derivatives have considerably higher anti-myeloma activity than thalidomide. Lenalidomide is now the first-line treatment for multiple myeloma and is also FDA-approved for treating mantle lymphoma, follicular lymphoma, and myelodysplastic syndromes (MDS) with deletion of chromosome 5q (del(5q)). Pomalidomide is approved for treating patients with relapsed/refractory multiple myeloma because it is also effective in patients with multiple myeloma refractory to lenalidomide. The prognosis of multiple myeloma has been greatly improved with the advent of IMiDs and proteasome inhibitors. Continued research efforts have uncovered many pharmacologically beneficial effects of thalidomide; the molecular mechanisms underlying its clinical benefits and side effects, however, were unclear until we identified CRBN as a direct target of thalidomide.
3. CRBN as a direct target of thalidomide
This section briefly outlines how CRBN was identified as a direct molecular target of thalidomide by affinity chromatography using nano-magnetic beads technology.1 Ferrite glycidyl methacrylate (FG) beads used for this identification are nano-sized magnetic particles composed of ferrite nanoparticles coated with a copolymer of glycidyl methacrylate (GMA) and styrene as a core and poly(GMA) as an outer surface (Fig. 3(a) and (b)).24 FG beads have a high binding capacity because of their small diameter of approximately 200 nm; poly(GMA) on the surface reduces non-specific protein binding. Target proteins of a wide variety of compounds, including pharmaceuticals,25 metabolites,26 and natural products,27 have been isolated from cell lysates in a single-step with FG beads. When FR259625, a carboxyl derivative of thalidomide, was immobilized to FG beads and used for affinity purification from HeLa cell extracts or other complex samples, CRBN and damage specific DNA binding protein 1 (DDB1) were specifically purified and identified as thalidomide-binding proteins (Fig. 3(c)). Further analysis revealed that CRBN is the protein that binds directly to thalidomide, and that DDB1 binds indirectly to thalidomide through CRBN.1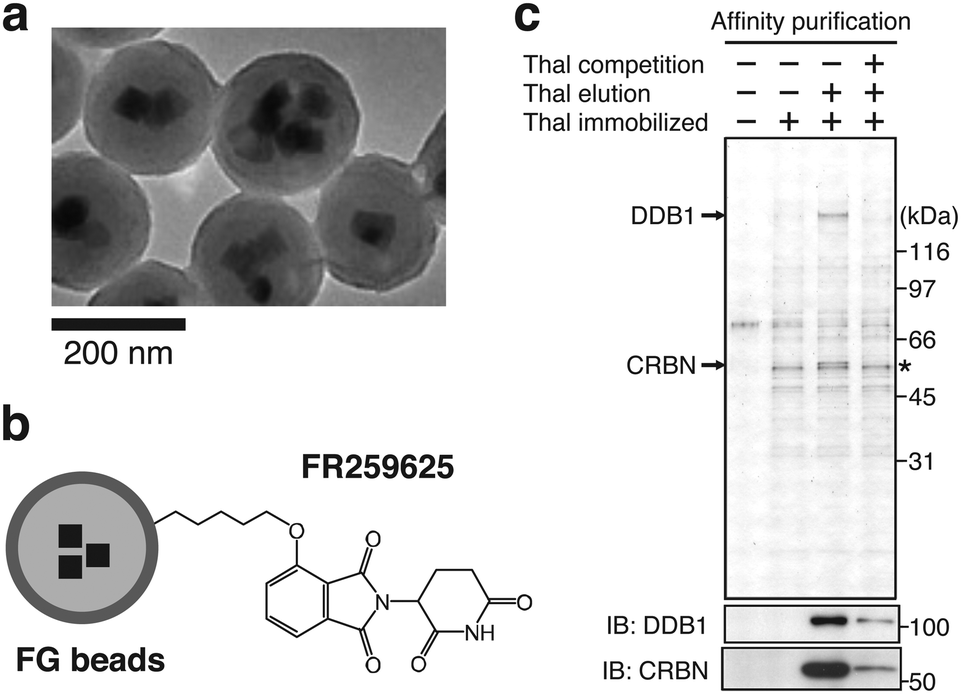 | ||
| Fig. 3 Identification of CRBN as a direct target of CRBN using FG beads. (a) Electron microscope image of FG beads. Reproduced from ref. 24 with author right. (b) Schematic representation of thalidomide-immobilized FG beads. FR259625, a carboxy derivative of thalidomide, was immobilized to the beads. (c) Affinity purification of CRBN and DDB1 from HeLa cell extract using thalidomide-immobilized FG beads. Bound fractions were analyzed by silver staining (top) and immunoblotting (IB) (bottom). The asterisk indicates non-specific signal. From ref. 1. Reprinted with permission from AAAS. | ||
Thalidomide binds to the highly conserved C-terminal region of CRBN; alanine substitutions of the conserved tyrosine residue at position 384 and the tryptophan residue at position 386 of the 442 amino acid protein remove the ability to bind thalidomide while maintaining the ability to form CRL4CRBN. Animal experiments with zebrafish and chickens showed that overexpression of the YW/AA mutant of CRBN conferred resistance to thalidomide-induced limb defects in these embryos (Fig. 4). CRBN is, therefore, a direct target of thalidomide, mediating its teratogenicity.1 CRBN and DDB1 together with Cullin 4 (CUL4A or CUL4B) and Ring-Box 1 (RBX1) form the E3 ubiquitin ligase complex CRL4CRBN. In fact, a large number of proteins such as DDB2, CSA, COP1, VprBP, and CDT2 are known, or suspected, to serve as substrate recognition receptors of CRL4, and CRBN is a recent addition to this growing list. Subsequent studies have revealed that most, if not all, of the diverse pharmacological effects of thalidomide and its derivatives are mediated by their binding to and modulation of CRL4CRBN.
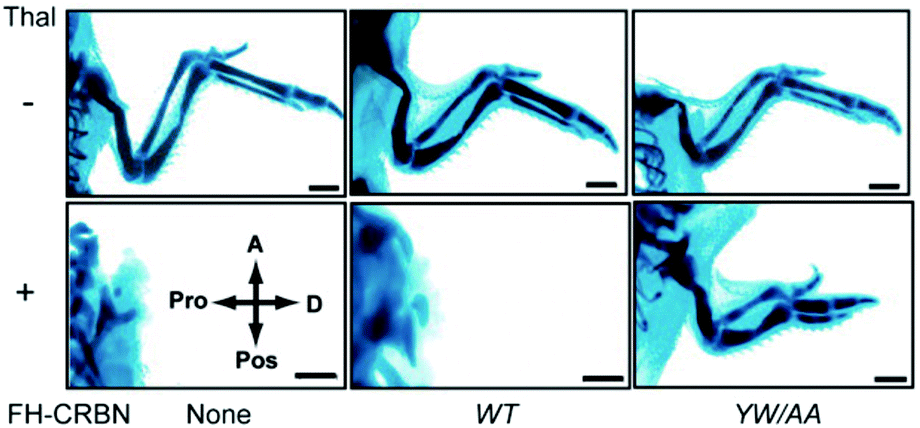 | ||
| Fig. 4 CRBN mediates thalidomide teratogenicity. The plasmids expressing wild-type (WT) CRBN or its thalidomide binding-deficient mutant (YW/AA) were electroporated as indicated into the forelimb field of stage 14 chicken embryos. Forelimbs were then treated with thalidomide or vehicle and stained with Victoria blue at stage 36. Abbreviations: thal, thalidomide; A, anterior; Pos, posterior; Pro, proximal; D, distal; FH, Flag and HA tag. The scale bar indicates 1 mm. From ref. 1. Reprinted with permission from AAAS. | ||
4. Molecular mechanisms of action of thalidomide and its derivatives
CRBN was considered as a substrate recognition receptor for CRL4CRBN, but its actual substrates were unknown. Identification of its substrates, therefore, has been a key focus for researchers trying to elucidate the molecular mechanism of action of thalidomide. To date, a number of substrates that are recognized by CRL4CRBN only in the presence of thalidomide or its derivatives have been identified; these are collectively referred to as neosubstrates (Table 1). Thalidomide and its derivatives largely exert their pharmacological activities by binding to CRBN and altering its substrate specificity, leading to the recognition and degradation of neosubstrates (Fig. 5). In other words, thalidomide and its derivatives act as molecular glues between CRBN and neosubstrates. Protein degraders have attracted interest as a new modality for drug discovery; in general, they are compounds that promote the binding of an E3 ubiquitin ligase to a specific target protein and lead to its degradation. Many thalidomide-based degraders that are clinically used or under development reportedly have multiple neosubstrates, suggesting that their diverse pharmacological effects are caused by the degradation of diverse target proteins by CRL4CRBN.| Genes | Compounds | Binding motifs | Ref. |
|---|---|---|---|
| Ikaros Aiolos | Lenalidomide Pomalidomide Iberdomide CC-885 CC-92480 | C2H2 zinc finger | 2 and 3 |
| CK1α | Lenalidomide | 4 | |
| GSPT1 | CC-885 CC-90009 | 5 | |
| SALL4 | Thalidomide Pomalidomide | C2H2 zinc finger | 6 and 7 |
| TAP63 ΔNP63 | Thalidomide | 8 | |
| ARID2 | Pomalidomide | 9 | |
| PLZF | Pomalidomide | C2H2 zinc finger | 10 and 11 |
| ZMYM2 | Pomalidomide Avadomide | MYM zinc finger | 12 and 13 |
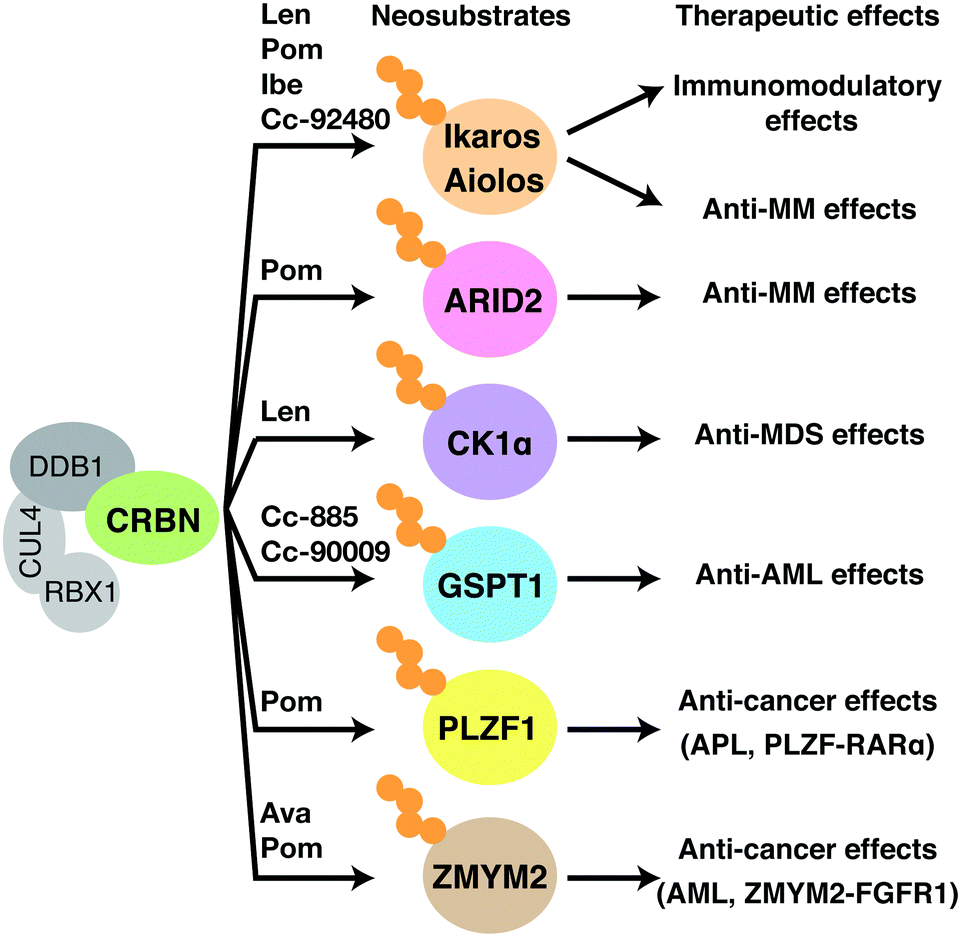 | ||
| Fig. 5 Mechanisms of therapeutic effects of thalidomide-based glue degraders. Binding of thalidomide-based glue degraders to CRBN induces degradation of corresponding neosubstrates, resulting in the therapeutic effects shown in the figure. Abbreviations: len, lenalidomide; Pom, pomalidomide; Ava, avadomide; Ibe, iberdomide; MM, multiple myeloma; MDS, myelodysplastic syndromes; AML, acute myeloid leukemia; APL, acute promyelocytic leukemia. | ||
CRISPR library screens have recently identified two E2 enzymes that are recruited to RBX1 and are involved in the ubiquitination process by CRL4CRBN.28,29 These enzymes play distinct roles in the ubiquitination reaction: UBE2D3 triggers mono-ubiquitination of neosubstrates, whereas UBE2G1 elongates the polyubiquitin chain. In the following section, we describe detailed mechanisms by which thalidomide and its derivatives exert pharmacological and teratogenic effects.
4.1. Anti-myeloma effects
Thalidomide derivatives of the IMiDs class, such as lenalidomide and pomalidomide, are promising anti-myeloma agents. Lenalidomide affects the expression of a variety of cytokines and associated factors.23 Prior to the discovery of CRBN, interferon regulatory factor 4 (IRF4) was identified as an important downstream gene that mediates the anti-myeloma effects of lenalidomide. IRF4 is a hematopoietic cell-specific gene that forms an autoregulatory circuit with another oncogene MYC; IRF4 and MYC play important roles in myeloma cell survival, and their downregulation by lenalidomide causes growth inhibition.30 The loss of CRBN confers lenalidomide and pomalidomide resistance in multiple myeloma cell lines.31 Lenalidomide and pomalidomide bind directly to CRBN more strongly than thalidomide, and their binding to CRBN results in IRF4 downregulation and inhibition of myeloma cell growth.32 In IMiD-treated multiple myeloma patients, CRBN is a favorable prognostic factor,33,34 suggesting that CRBN is an important mediator of the anti-myeloma effects of IMiDs also in a clinical setting.In 2014, two groups independently reported Ikaros (IKZF1) and Aiolos (IKZF3) as neosubstrates that are involved in the anti-myeloma effects of lenalidomide;2,3 these were the first reported CRBN neosubstrates. To identify proteins whose ubiquitination levels are affected by lenalidomide, Ebert and colleagues performed ubiquitin-modified proteome analysis by enriching peptides containing di-glycine (K-ε-GG) remnants, which remain in ubiquitinated lysine residues after trypsin digestion.2 Meanwhile, Kaelin and colleagues used a dual reporter vector carrying an ORF library to investigate proteins whose stability is affected by lenalidomide.3 Ikaros and Aiolos are members of the Ikaros family of transcription factors that contain multiple C2H2 zinc finger motifs. Ikaros and Aiolos are specifically expressed in the hematopoietic lineage and play important roles in regulating lymphocyte differentiation and development. Both research groups found that Ikaros and Aiolos bind to CRBN only in the presence of lenalidomide and undergo ubiquitination and subsequent degradation by the proteasome. Furthermore, depletion of Ikaros and Aiolos downregulates IRF4 expression and reduces growth inhibition of myeloma cell lines. Since other Ikaros family proteins, such as Helios and Eos, are not degraded by lenalidomide treatment, amino acids critical for recognition by CRL4CRBN were narrowed down by sequence comparison of family members. Expression of a CRBN-binding deficient mutant of Ikaros or Aiolos conferred lenalidomide resistance to myeloma cells, indicating that Ikaros and Aiolos are major neosubstrates involved in the anti-myeloma effects of lenalidomide. Subsequently, it was shown that pomalidomide also targets Ikaros and Aiolos via CRL4CRBN, and it has become clear that IMiDs exert their anti-myeloma effects by inducing the degradation of Ikaros and Aiolos and thereby downregulating IRF4 and MYC.
Early IMiDs such as lenalidomide and pomalidomide were produced by subtle modifications to thalidomide. The more recently developed thalidomide analogs, however, are structurally more diverse (Fig. 2). These compounds are collectively named cereblon E3 ligase modulators (CELMoDs) because their efficacy is no longer limited to immunomodulation. A number of new CELMoDs have been developed by Celgene Corporation in an effort to identify compounds inducing degradation of Ikaros and Aiolos with greater activity. Iberdomide (Fig. 2), a CELMoD with a higher binding affinity for CRBN than lenalidomide and pomalidomide, induces more efficient degradation of known neosubstrates including Ikaros and Aiolos.35 This compound is thus expected to have a more potent effect against myelomas, and clinical trials for treating relapsed/refractory multiple myeloma with iberdomide are currently underway. CC-92480, another CELMoD, is also in clinical trials for relapsed/refractory multiple myeloma. Lenalidomide-refractory multiple myeloma patients frequently exhibit downregulation of CRBN expression. CC-92480 has a much higher potency for degrading Aiolos than lenalidomide; this compound has high anti-myeloma activity even in lenalidomide-resistant multiple myeloma cell lines with low CRBN expression.36
Ikaros and Aiolos play critical roles in mediating the anti-myeloma effects of lenalidomide and pomalidomide. Given that both compounds induce the degradation of Ikaros and Aiolos, an unresolved question is why pomalidomide shows efficacy in lenalidomide-resistant multiple myeloma. There is conflicting evidence on the association between the expression levels of Ikaros and Aiolos and prognosis.36–39 The idea that Ikaros and Aiolos are major therapeutic targets in patients refractory to lenalidomide is incompatible with multiple reports showing that Ikaros, Aiolos, and IRF4 are not unfavorable but favorable prognostic markers in relapsed multiple myeloma patients treated with lenalidomide.9,36,38 We showed that ARID2 (AT-rich interactive domain-containing protein 2), a subunit of the chromatin remodeling complex PBAF (polybromo-associated BRG1/BRM-associated factors), is a pomalidomide-specific neosubstrate of CRL4CRBN, which is likely to explain its superior efficacy over lenalidomide.9 Pomalidomide induces binding between CRBN and ARID2 more strongly than lenalidomide, resulting in increased ARID2 degradation and growth inhibition in multiple myeloma cell lines. This study also suggested that pomalidomide-induced downregulation of MYC and IRF4 is primarily mediated through the degradation of ARID2 and Ikaros/Aiolos, respectively. ARID2 is an unfavorable prognostic factor that is increased in patients with relapsed and refractory multiple myeloma;9 this is consistent with the idea that ARID2 degradation accounts for the superior anti-myeloma effect of pomalidomide over lenalidomide.
4.2. Anti-MDS effects
Unlike thalidomide and pomalidomide, lenalidomide is approved for del(5q) syndrome. Casein kinase 1α (CK1α) is a lenalidomide-dependent neosubstrate involved in this anti-MDS effect.4 Lenalidomide is much more potent in degrading CK1α than thalidomide or pomalidomide; at clinically relevant concentrations, only lenalidomide promotes its degradation. CK1α is a multifunctional serine/threonine kinase involved in many biological processes, including p53-mediated apoptosis, regulation of the WNT signaling pathway, cell cycle regulation, and the immune response.40–42 The CSNK1A1 gene, which encodes CK1α, is located in the 5q region and is haploinsufficient in del(5q) syndrome. Therefore, del(5q) cells are more sensitive than normal cells to lenalidomide-mediated CK1α degradation; this explains why lenalidomide is particularly effective in MDS with del(5q).43,44 Lenalidomide treatment activates the p53-mediated apoptotic pathway and inhibits proliferation of CSNK1A1 haploinsufficient cells. Concordantly, CK1α overexpression confers lenalidomide resistance to patient-derived MDS del(5q) cells in vitro.44.3. Anti-AML effects
The CELMoD CC-885 exhibits anti-cancer activity against a wide range of cell types via CRL4CRBN; it is particularly effective for acute myeloid leukemia (AML), making it a potential candidate for AML treatment.5 In 2016, G1 to S phase transition 1 (GSPT1) was identified as a CC-885-dependent neosubstrate. GSPT1 is a component of the translation termination complex that recognizes the termination codon and induces translation termination; its deletion causes G1 arrest. GSPT1 specifically associates with CC-885-bound CRBN. Expression of a CRBN-binding deficient mutant of GSPT1 confers resistance to CC-885, demonstrating that the anti-AML effect of CC-885 is caused by the degradation of GSPT1 by CRL4CRBN.5 CC-885 possesses a novel therapeutic property against AML; however, its clinical development is difficult because, in addition to GSPT1, CC-885 also induces degradation of Ikaros, Aiolos, and CK1α and has a complicated mechanism of action. CC-90009 was instead developed as a first-in-class, GSPT1-selective CELMoD and is currently being tested in Phase I and II trials for the treatment of AML. CC-90009 exhibits reduced general cytotoxicity but maintains its anti-AML activity. Global proteomic analysis with AML cell lines suggest that, with the exception of GSPT1, CC-90009 has little effect on the proteome.454.4. Effects on certain types of leukemia with translocations
Chromosomal translocations are characteristic features of many types of cancers including leukemias and lymphomas, and are used as diagnostic markers and therapeutic targets. In a chromosomal translocation, two different chromosomes are broken and fused together, and the resulting chimeric fusion gene drives oncogenic transformation. Thalidomide derivatives lead to the degradation of multiple leukemogenic fusion proteins. The zinc finger MYM-type containing 2 (ZMYM2) gene generates a fusion gene with fibroblast growth factor receptor 1 (FGFR1) through the chromosomal translocation t(8;13)(p11;q12), which is involved in the transformation to AML (Fig. 6(a)). ZMYM2–FGFR1 is a pomalidomide- and avadomide-dependent neosubstrate of CRL4CRBN.12,13 The binding mode of ZMYM2 to CRBN is similar to that of Ikaros and Aiolos, but its drug specificity is different, with ZMYM2 showing strong preference to avadomide. Concordantly, avadomide exhibits antiproliferative activity in vitro in bone marrow samples derived from patients with hematological malignancies harboring the t(8;13)(p11;q12) translocation.12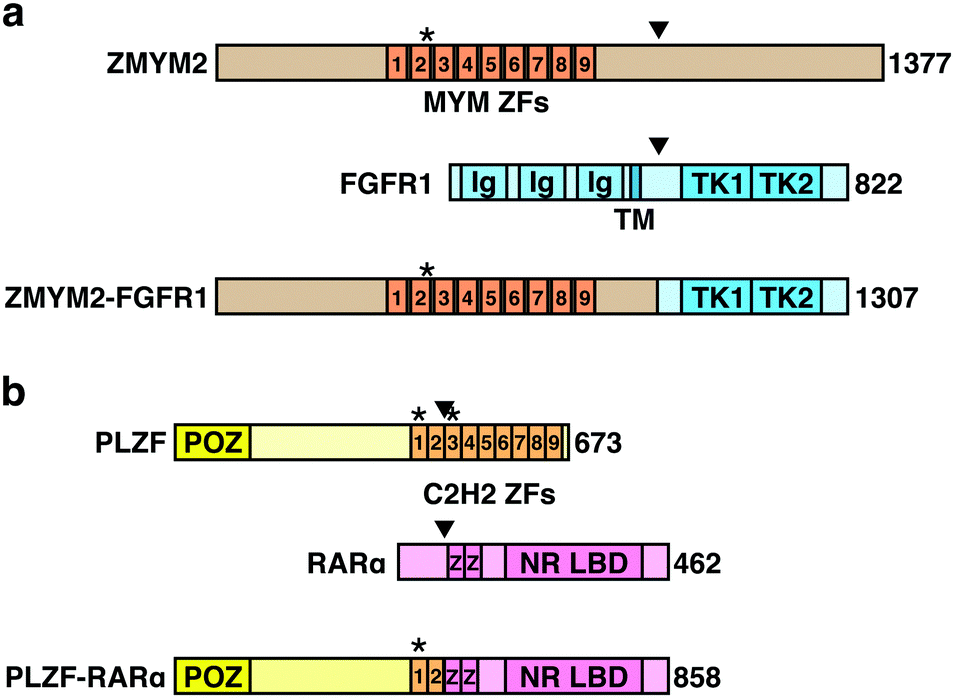 | ||
| Fig. 6 Schematic representation of fusion gene products targeted by thalidomide-based glue degraders. (a) Schematic representation of ZMYM2, FGFR1, and the fusion protein ZMYM2-FGFR1. (b) Schematic representation of PLZF, RARα, and the fusion protein PLZF-RARα. Abbreviations: ZF, zinc finder motif; Ig, immunoglobulin-like domain; TM, transmembrane domain; TK, tyrosine kinase domain; POZ, BTB/POZ domain; NR LBD, nuclear receptor ligand-binding domain. The asterisks indicate degrons recognized by CRBN. The arrowheads indicate breakpoints. | ||
Promyelocytic leukemia zinc finger (PLZF) is another pomalidomide-dependent neosubstrate with leukemogenic potential.11 Chromosomal translocations resulting in the fusions of PLZF with retinoic acid receptor α (RARA) or ABL proto-oncogene 1 (ABL1) are associated with acute promyelocytic leukemia and T cell acute lymphoblastic leukemia (T-ALL), respectively (Fig. 6(b)). PLZF is particularly sensitive to pomalidomide; pomalidomide treatment induces PLZF-RARα degradation and inhibits the growth of PLZF-RARα-expressing model cell lines.11 PLZF is also implicated in the teratogenicity of thalidomide,10 which will be described later. Thus, these findings suggest that pomalidomide and avadomide may be therapeutically beneficial against certain types of leukemias. While many of the translocation-derived fusion genes have been considered undruggable because of the lack of enzymatic activity, thalidomide-based protein degraders are opening up new possibilities.
4.5. Immunomodulatory effects
As the name indicates, IMiDs modulate cytokine production and have multifaceted effects on various immune systems, including activation of T cells, suppression of regulatory T cells (Treg), activation of natural killer cells, and suppression of B cells and monocytes.46–49In vivo immunomodulatory effects of IMiDs are not only cell autonomous, but also involve complex processes with many immune cells. In vitro studies based on cultured cells, such as those derived from multiple myeloma patients, are insufficient for fully understanding the mechanism of immunomodulation. Since, however, Ikaros and Aiolos, key downstream targets of several IMiDs, are important for T cell proliferation and activation, it is likely that the immunostimulatory effects of IMiDs are mediated, at least in part, by their degradation. Indeed, studies using primary T cells and peripheral T cells from healthy volunteers showed that Ikaros and Aiolos degradation contributes to IMiDs-induced production of IL-2, which is associated with co-stimulation of T cells.50 T cell co-stimulation is a secondary, antigen-nonspecific signal that works with a primary, antigen-specific signal to fully activate T cell proliferation and differentiation, increasing IL-2 production in CD4+ and CD8+ T cells and promoting Treg suppression. The immunomodulatory effects of IMiDs are clinically important for treating cancer and inflammatory diseases. For example, iberdomide is being evaluated in clinical trials as a treatment for systemic lupus erythematosus, an autoimmune disease characterized by very low levels of endogenous IL-2 and one that may thus benefit from increased IL-2 production.514.6. Teratogenic effects
Thalidomide has teratogenic effects on embryonic development and induces a broad range of birth defects, including damage to the limbs, face, eyes, ears, genitalia, and internal organs. Prior to the discovery of CRBN as a primary target of thalidomide, its teratogenicity was suggested to be mediated by the inhibition of angiogenesis or DNA damage caused by reactive oxygen species (ROS).52 Subsequent studies, however, illustrated that CRBN is an essential mediator of the teratogenic, immunomodulatory, and anticancer effects of thalidomide. The presence of a chiral carbon in the thalidomide molecule produces (S)- and (R)-enantiomers, which rapidly racemize under physiological conditions. It was suggested, in early studies, that the (S)-enantiomer has teratogenic effects whereas the (R)-enantiomer possesses hypnotic effects. It was therefore expected that the teratogenic side effects may be separated from beneficial effects. However, subsequent studies showed that immunomodulatory and anticancer activities of thalidomide and its derivatives are also substantially stronger in the (S)-enantiomer than in the (R)-enantiomer.53 Concordantly, the (S)-enantiomer exhibits about 10-fold stronger binding to CRBN,54 whereas the hypnotic effect of thalidomide is reportedly CRBN-independent.55Recent findings have improved our understanding of the molecular mechanisms of thalidomide embryopathy. In 2018, two independent groups reported Spalt-like transcription factor 4 (SALL4) as a neosubstrate involved in the teratogenicity of thalidomide.6,7 SALL4 is a C2H2-type zinc finger transcription factor that was identified as the causative gene for Duane-radial ray syndrome, a congenital disorder characterized by malformations of the eyes and limbs.56–58 This syndrome is caused by a loss-of-function mutation in SALL4 and has at times been misdiagnosed as thalidomide embryopathy because of phenotypic similarities.57 To explore proteins whose abundance is altered by thalidomide, lenalidomide, and pomalidomide, Fischer and colleagues conducted a proteomic analysis in human embryonic stem cells and focused on SALL4 as a neosubstrate that is possibly involved in thalidomide embryopathy.6 Chamberlain and colleagues also identified SALL4 from neosubstrate candidates selected by using sequence motifs conserved among known neosubstrates (see below for details) and focused on it because of phenotypic similarities.7 These studies showed that thalidomide induces degradation of SALL4 in humans and rabbits, but not in mice.6 This is consistent with the observation that thalidomide is teratogenic in humans and rabbits, but not in mice. Species specificity of thalidomide will be described later. The hypothesis that SALL4 degradation contributes to thalidomide embryopathy was tested in an in vitro developmental model using human induced pluripotent stem cells. During embryonic development, limb buds are generated from the lateral plate mesoderm (LPM); thalidomide was shown to inhibit the differentiation of stem cells into LPM-like cells via SALL4 degradation.59
Using zebrafish as a model, another group identified tumor protein p63 (TP63) as a neosubstrate possibly involved in thalidomide teratogenicity.8 TP63 belongs to the p53 family of transcription factors with many isoforms with different functions. The major isoforms TAP63 and ΔNP63 are subject to thalidomide-induced degradation. Mutations in human TP63 are associated with congenital defects such as ectrodactyly, ectodermal dysplasia, cleft lip/cleft palate syndrome 3, isolated cleft lip/cleft palate, split-hand/foot malformation type 4, and limb-mammary syndrome.60,61 Moreover, knockout of TP63 in mice and zebrafish causes limb loss resembling thalidomide embryopathy.62,63 Studies using a zebrafish model suggested that TAP63 and ΔNP63 are involved in different teratogenic phenotypes.8 TAP63 is mainly expressed in the ear and heart, and expression of its non-degradable mutant suppresses ear defects and thalidomide-induced downregulation of Atoh1, a gene essential for the development of sensory neurons and cochlea. Meanwhile, ΔNP63 is expressed in the apical ectodermal ridge, the leading edge of the developing limb; expression of its non-degradable mutant suppresses fin defects and thalidomide-induced downregulation of Fgf8, a key regulator of limb development. ΔNP63, furthermore, confers resistance to oxidative stress-induced cell death, which is consistent with the previous observation that thalidomide causes ROS-induced DNA damage. Therefore, recent findings are bridging gaps in information concerning thalidomide embryopathy.
PLZF is another neosubstrate possibly involved in the teratogenicity of thalidomide and its metabolite, 5-hydroxythalidomide.10Plzf knockout mice show defects in the patterning of limb skeletal structures.64 Thalidomide and 5-hydroxythalidomide induce PLZF degradation.10 The authors showed that knockdown of PLZF causes limb defects in chicken embryos, and that PLZF overexpression partially reverses thalidomide-induced phenotypes, such as downregulation of Fgf10/Fgf8. No degradation of SALL4 was observed in chicken embryos, suggesting that, at least in chickens, PLZF serves as a neosubstrate critical for thalidomide embryopathy.
SALL4, TP63, and PLZF are all plausible as neosubstrates involved in thalidomide embryopathy (Fig. 7), but as mentioned above, its actual phenotypes are extremely diverse. It is unclear whether thalidomide-induced degradation of these neosubstrates, alone or in combination, accounts for such a diverse spectrum of defects, particularly given that previous studies used simplified animal models. It is also possible that neosubstrates responsible for the teratogenic effects of thalidomide differ among species. Further validation with primate models may offer more definitive answers as to which neosubstrates are involved in thalidomide embryopathy.
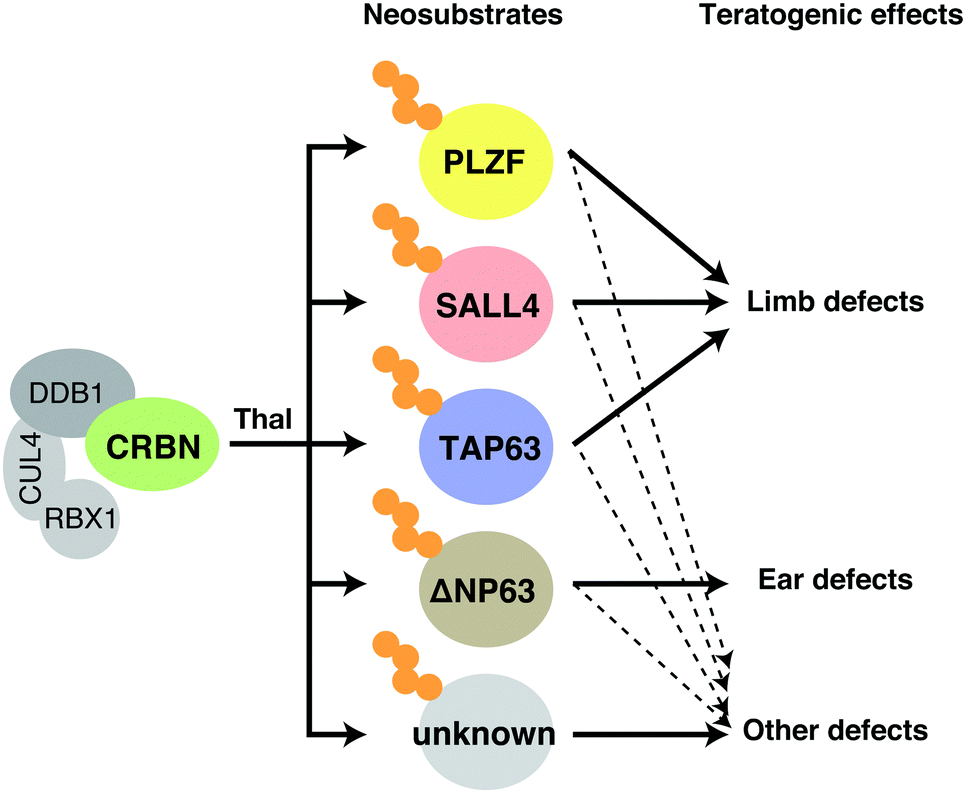 | ||
| Fig. 7 Mechanisms of teratogenic action of thalidomide. Binding of thalidomide to CRBN in fetal development induces degradation of SALL4, TP63, PLZF, and possibly other unknown neosubstrates, resulting in the various defects shown in the figure. Abbreviation: thal, thalidomide. | ||
5. Structural understanding of CRL4CRBN, thalidomide, and its derivatives
The mode of action of thalidomide-based degraders is to physically link CRBN and neosubstrates as molecular glues. Structural knowledge of how CRL4CRBN interacts with a neosubstrate in the presence of a molecular glue is thus important for understanding its specific mechanism of action and developing more effective drugs. Since the discovery of CRBN, determining the structure of complexes that contain CRL4CRBN, thalidomide derivatives, and neosubstrates has been a major focus in this field of research. Human CRBN is a 441- or 442-amino acid protein that is evolutionarily conserved from plants to humans; it was first reported as a gene product associated with people with an intellectual disability.65 CRBN consists of the N-terminal LON-like domain (LLD), which is similar to the bacterial Lon protease, and the C-terminal thalidomide-binding domain (TBD) (Fig. 8(a)). DDB1 binds to the central region of CRBN, and three tryptophan residues (tri-trp) in the TBD are required for thalidomide binding. The glutarimide moiety of thalidomide is critical for binding to CRBN.1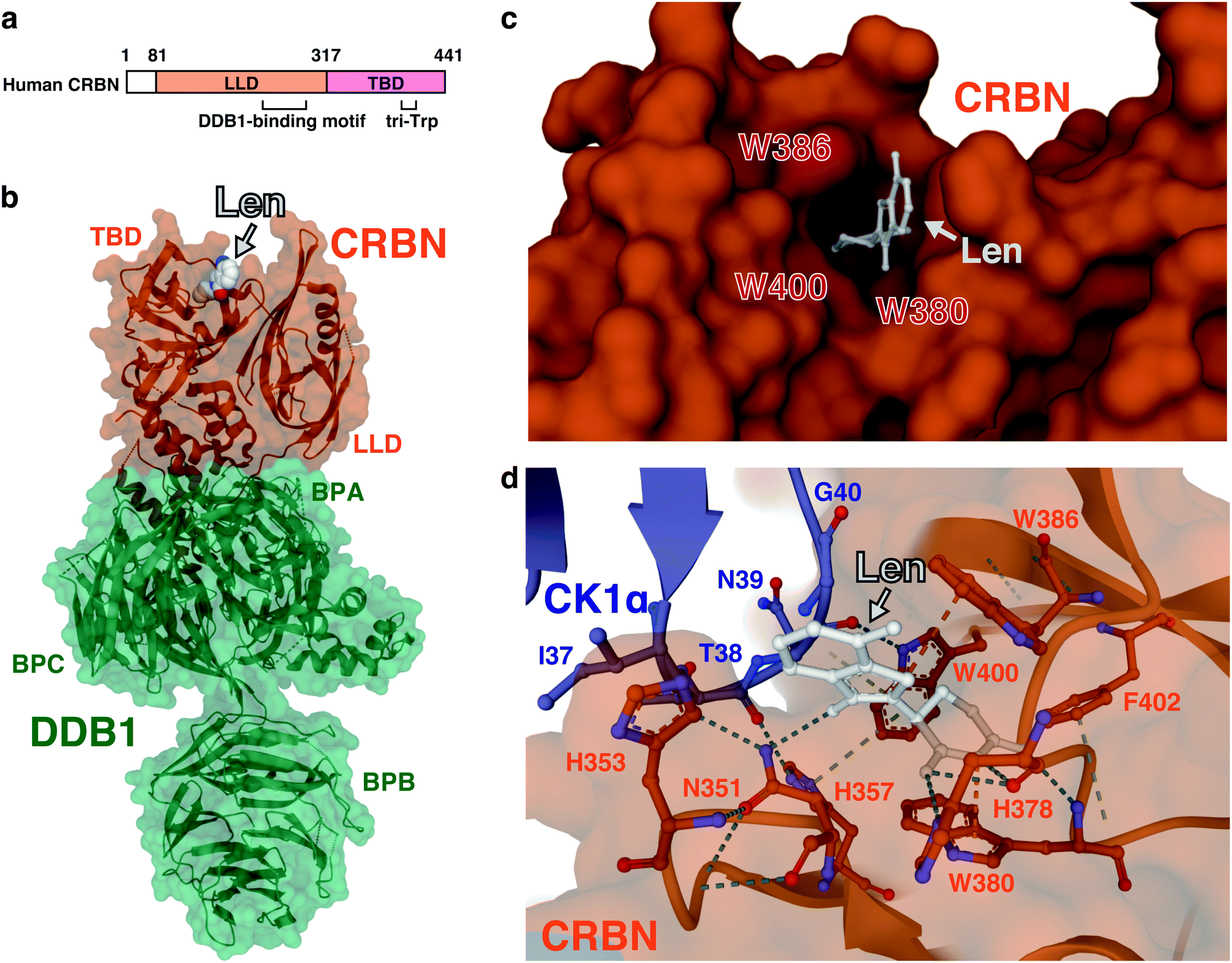 | ||
| Fig. 8 Structure of the ternary complex containing CRBN, a thalidomide-based glue degrader, and a neosubstrate. (a) Schematic representation of CRBN. (b) Three-dimensional structure of the DDB1–CRBN–lenalidomide complex obtained by X-ray crystallography (PDB: 4TZ4). (c) A close-up view of the thalidomide-binding pocket of CRBN. Amino acids comprising tri-Trp are highlighted in red (PDB: 4TZ4). (d) A close-up view of the interface between CRBN and CK1α from the DDB1-CRBN-lenalidomide-CK1α co-crystal structure (PDB: 5FQD). The dash lines indicate non-covalent interactions. Abbreviations: len, lenalidomide; LLD, LON-like domain; TBD, thalidomide-binding domain; BPA, β-propeller A; BPB, β-propeller B; BPC, β-propeller C. | ||
In 2014, two groups independently determined by X-ray crystallography the structure of CRBN–DDB1 bound to IMiDs. Thomä and colleagues reported three-dimensional structures using human DDB1, chicken CRBN, and thalidomide, lenalidomide, or pomalidomide,66 while Cathers and colleagues reported three-dimensional structures using human DDB1, human CRBN, and lenalidomide,67 with consistent results. The DDB1-binding motif of CRBN consists of a series of α-helices, located at residues 188–248 of the LLD. DDB1 is composed of three β-propeller domains (BPA, BPB, and BPC);68 CRBN binds to a cavity between BPA and BPC (Fig. 8(b)).66,67 The glutarimide ring of IMiDs inserts into a hydrophobic pocket consisting of three tryptophan residues (tri-Trp; W380, W386, and W400) and one phenylalanine residue (F402) in the TBD; the phthalimide or isoindolinone ring protrudes outward and is exposed on the surface of CRBN (Fig. 8(c)). Within the tri-Trp pocket, two hydrogen bonds are formed between the glutarimide ring and the backbone of the H378 and W380 residues; a further hydrogen bond is formed with the side chain of H378. This provides a structural explanation as to why the thalidomide-binding deficient mutant of CRBN (YW/AA) discovered in a previous study1 has lost its ability to bind thalidomide. The TBD also has a C4-type zinc finger motif consisting of C323, C326, C391, and C394. Its role is not well understood, but a missense mutation (C391R) in this motif was found in five individuals with a severe intellectual disability from the same Saudi family.69
The tertiary structure of the complex containing a neosubstrate, an IMiD, and CRBN was first reported in 2016. Thomä and colleagues determined the crystal structure of human DDB1–CRBN–lenalidomide–CK1α.70 The β-hairpin loop (residues 35–41) of CK1α was bound to the surface provided by CRBN and lenalidomide (Fig. 8(d)). In the same year, the crystal structure of the DDB1-CRBN–CC-885–GSPT1 complex was also determined.5 CC-885 binds to CRBN in a mode similar to other IMiDs, with the glutarimide moiety inserting into the tri-Trp pocket of CRBN. The non-glutarimide part of CC-885 is exposed on the surface of CRBN, and the isoindolinone ring is in close proximity to the β-hairpin loop (residues 571–575) of GSPT1. In addition, the extended urea and chloromethylphenyl groups of CC-885 also contribute to its interaction with CRBN and GSPT1, respectively. Despite the lack of clear sequence similarity between CK1α and GSPT1, both neosubstrates bind to CRBN via the β-hairpin loop, in which a common glycine residue (G40 in CK1α and G575 in GSPT1) is particularly important for binding. An amino acid at this position must be glycine to avoid steric hindrance. These studies provided a structural basis for the mechanism by which thalidomide-based degraders function as molecular glues. Tertiary structures of other neosubstrates, such as Ikaros and SALL4, bound to CRBN and IMiDs, were subsequently determined, and the common mechanism of neosubstrate recognition involving a β-hairpin containing a key glycine residue has been confirmed and extended.71,72
6. Structure–activity relationships (SARs) of thalidomide-based degraders
The glutarimide moiety of thalidomide-based degraders is mainly involved in the binding to CRBN, while the other part contributes to the recognition of neosubstrates. SARs for CRBN binding have been investigated in detail by a series of studies using the thalidomide-binding domain of cereblon isoform 4 from Magnetospirillum gryphiswaldense (MsCI4).73–76 In an early study, the uracil moiety of uridine was shown to fit in the thalidomide-binding pocket of MsCI4, albeit of lower affinity than thalidomide, suggesting that the minimal structure for CRBN binding is a cyclic imide.73 Subsequently, detailed SARs were studied by fluorescence resonance energy transfer using MsCI4 and various cyclic imides and lactams.75 In order to discuss structural requirements of various, four- to eight-membered cyclic compounds for CRBN binding, a simplified nomenclature shown in Fig. 9(a) will be used below. The carbon linked to the phthalimide group of thalidomide is designated position (a), while positions (b), (c), and (d) correspond to the proximal carbonyl group, the amino group, and the distal carbonyl group, respectively. The SAR study indicated that the carbonyl in position (d) is essential, whereas the other carbonyl in position (b) increases affinity for MsCI4 in both five- and six-membered cyclic compounds; the amino group in position (c) is also required for binding. Four-, five-, and six-membered lactams show measurable affinity, but the seven- and eight-membered lactams do not, suggesting a size limitation of the ligand-binding pocket. These results indicated that the minimal structure for CRBN binding is a lactam with an appropriate size (Fig. 9(b)).74 Interestingly, there are several compounds in existing pharmaceutical products that fulfill this minimal structure. Examples include the anticholinergic drug dexetimide, the aromatase inhibitor rogletimide, the hepatitis C virus RNA polymerase inhibitor dasabuvir, the selective phosphodiesterase inhibitor rolipram, and the anticonvulsant ethosuximide.74 These non-thalidomide compounds might be useful for future development of a new class of CRBN-dependent protein degraders.75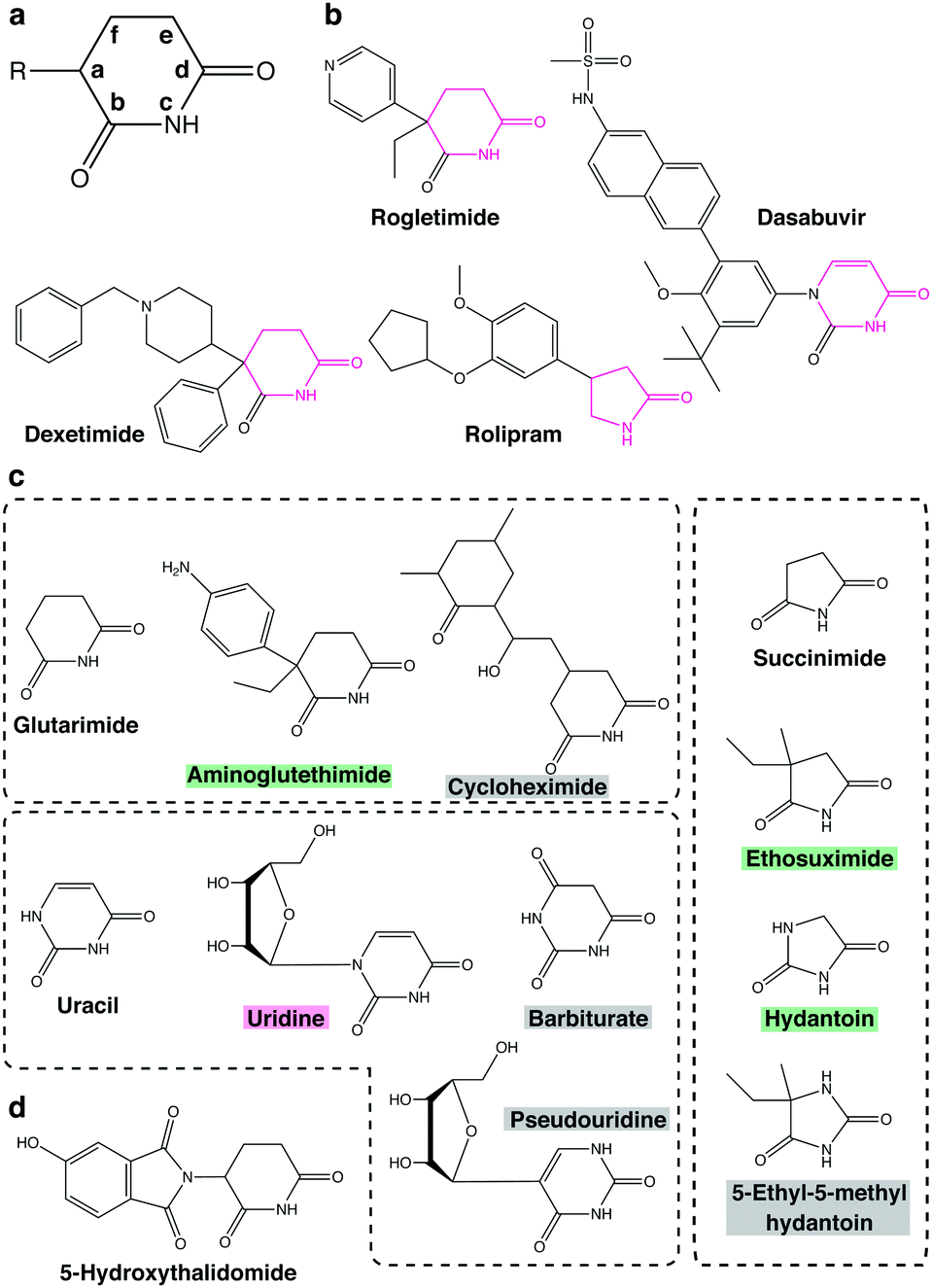 | ||
| Fig. 9 Potential CRBN-binding compounds tested or implicated in SAR studies. (a) A simplified nomenclature for cyclic compounds used in SAR studies. See text for detail. (b) Pharmaceuticals containing the minimal structure for CRBN binding. The minimal structures are highlighted in magenta. (c) Glutarimide-, uracil-, and succinimide-based compounds used in SAR studies. The backgrounds of the compound name are color-coded magenta, green, and gray for higher, lower, or no affinity for MsCI4. (d) Structure of 5-hydroxythalidomide. | ||
Only positions (a) and (b) of imides seem to have potential for structural diversification. Substitutions at position (a) affect the affinity of five- and six-membered cyclic compounds for MsCI4 binding. Thalidomide shows higher affinity than glutarimide; similarly, uridine shows higher affinity than uracil, suggesting that compounds containing a substituent at position (a) bind to MsCI4 more strongly than the corresponding compounds without it. On the other hand, aminoglutethimide, which carries two substituents at position (a), shows slightly lower affinity than glutarimide. Likewise, ethosuximide containing methyl and ethyl groups at position (a) shows much weaker affinity than succinimide (Fig. 9(c)), suggesting that imide compounds containing a quaternary carbon at position (a) may not be good CRBN binders. Meanwhile, the carbonyl at position (b) contributes to the binding but is not essential, and its substitution affects affinity for MsCI4. For example, substitution of the oxygen atom linked to the carbon at position (b) of succinimide with a methyl group decreases affinity for MsCI4, while substitution of the same oxygen atom of hydantoin to sulfur conversely increases the affinity. However, the amino group of position (c) and the carbonyl group of position (d) are critical for MsCI4 binding and cannot be derivatized. Replacement of the carbon atom at position (e) with nitrogen also substantially reduces affinity for MsCI4 in both five- and six-membered rings, suggesting that a polar group at this position disrupts the interaction (Fig. 9(c)). With regard to position (f), cycloheximide, a glutarimide-based six-membered ring with a branched carbon at this position, and barbiturate, a uracil-based six-membered ring with a carbonyl group at the same position, do not bind to MsCI4, suggesting that a large substituent at position (f) causes steric crash with the pocket (Fig. 9(c)).74 Taken together, these results indicate that only positions (a) and (b) can be exploited for structural diversification to modulate affinity for CRBN. According to a recent study using microscale thermophoresis assays, the affinities of these potential CRBN-binders for MsCI4 are generally consistent with those for the TBD of human CRBN with a few exceptions.77
Establishing SARs of thalidomide-based degraders using degradation of intracellular neosubstrates as a measure of activity is more challenging due to concurrent factors, such as membrane permeability and competitive binding to other neosubstrates. Binding affinities of thalidomide-based degraders for both CRBN and neosubstrates are critical for targeted degradation of neosubstrates. The phthalimide moiety is important for ligand-induced degradation of IKZF1, and substitutions on the aromatic ring or deviations from isoindoline structure inhibit its degradation.78 Moreover, the crystal structure of DDB1–CRBN–pomalidomide binding to the zinc finger domain of IKZF1 revealed how lenalidomide and pomalidomide bind to IKZF1 more strongly than thalidomide. The additional amino group present in the phthalimide moiety of lenalidomide and pomalidomide forms a water-mediated hydrogen bond with the Q146 residue of IKZF1, stabilizing the interaction.72
Likewise, an increasing number of studies have provided plausible explanations as to how subtle structural differences among thalidomide-based degraders trigger selective degradation of neosubstrates. While thalidomide, lenalidomide, and pomalidomide have similar structures (Fig. 2), only lenalidomide has the property of efficiently degrading CK1α. Crystal structure analysis of the ternary complex containing CRBN, lenalidomide, and CK1α showed that the carbonyl group only found on the phthalimide ring of thalidomide and pomalidomide causes a steric crash with the CRBN backbone, addressing why CK1α behaves as a lenalidomide-specific neosubstrate.70 Meanwhile, 5-hydroxythalidomide is a major metabolite of thalidomide that induces the degradation of SALL4 via CRL4CRBN more strongly than thalidomide (Fig. 9(d)). Crystal structure analysis revealed that this is because the hydroxyl group forms a water-mediated hydrogen bond with the H353 residue of CRBN, strengthens the affinity for CRBN, and increases the degradation of neosubstrates.79 Similarly, the phenyl and morpholine moieties of iberdomide contribute to binding to CRBN and exhibit stronger CRBN binding capacity than the IMiDs class compounds (Fig. 2).35,76 CC-885 is also structurally extended compared to the IMiDs class. As mentioned above, the urea moiety of CC-885 is located between E377 and H353 of CRBN and contributes to binding by hydrogen bonding to both side chains, while the methylchlorophenyl moiety is located close to GSPT1 domain 3 and probably contributes to the selective binding to GSPT1.7 Thus, modifications to the non-glutarimide part provide the opportunity not only to increase affinity for CRBN, but also to acquire novel neosubstrates; indeed, structural diversification of the non-glutarimide part has been intensively studied in recent years (Fig. 2).
7. C2H2 zinc finger proteins as potential neosubstrates of CRBN
One of the features of thalidomide-based glue degraders is that they can break down a diverse range of neosubstrates. Structural degron is a short sequence that can be recognized as a substrate by a specific E3 ubiquitin ligase; however, there is no apparent consensus sequence in the known neosubstrates of CRL4CRBN, such as Ikaros, Aiolos, CK1α, and GSPT1. The known common interaction motif is a β-hairpin loop that contains a key glycine residue, which in the case of zinc finger proteins such as Ikaros, Aiolos, and SALL4, is located on the C2H2 zinc finger motif.71,72 Since the C2H2 zinc finger domain is necessary and sufficient as a structural degron in the case of Ikaros and Aiolos,2 it was expected that the C2H2 zinc finger protein family would contain new neosubstrates for CRL4CRBN. Thomä and colleagues conducted a comprehensive screen of more than 6500 C2H2 zinc finger motifs predicted from the human proteome.72 The data suggested that 11 zinc finger motifs were degraded in the presence of thalidomide, lenalidomide, or pomalidomide. Detailed analysis revealed that, in addition to Ikaros, Aiolos, and ZFP91, four new zinc finger proteins (ZNF276, ZNF653, ZNF692, and ZNF827) are neosubstrates for CRL4CRBN.80 The 11 zinc finger motifs share the common motif CXXCG, but other than that there is no clear consensus sequence. Despite the large number of zinc fingers containing this motif in the library, only 11 of them actually functioned as structural degrons. These findings point to the complexity of substrate recognition via the CRBN-drug interface. A series of mutagenesis and domain swapping experiments indicated that only a limited number of amino acid combinations are tolerated as degrons.728. Species specificity of thalidomide and its derivatives
Thalidomide is teratogenic in humans, non-human primates, rabbits, chickens, and zebrafish, but not in rodents including mice.81 Animal experiments on mice did not show any serious side effects,82 which is considered to be one of the reasons behind the thalidomide disaster. The anti-myeloma effects of IMiDs are, furthermore, also not observed in mice.83 This species specificity of thalidomide has been a long-standing question, but recent structural studies have now provided important insights. Despite the 94% amino acid sequence identity between human and mouse CRBN, Ikaros and Aiolos were not degraded when mouse Crbn, instead of human CRBN, was expressed in human cell lines.67 The V388 in human CRBN was substituted with isoleucine in mice (I391) (Fig. 10(a)); structural analysis shows that the bulky side chain of isoleucine is sterically hindered to prevent neosubstrate access to mouse Crbn.4 The expression of humanized CRBN, in which I391 of mouse Crbn is substituted with valine (I391V), was sufficient for the degradation of Ikaros and Ck1α in mouse cell lines. Transgenic mice expressing humanized CRBN were then used to test whether the rodent-specific single amino acid substitution in CRBN would explain the species-specific effects of thalidomide and lenalidomide.84,85 Thalidomide- and lenalidomide-induced degradation of Ikaros and CK1α was confirmed in these mice; exposure of pregnant mice to thalidomide or lenalidomide produced a high incidence of fetal lethality. No limb malformation, however, was observed, indicating that the crbnI391V mutation alone may not fully reproduce thalidomide embryopathy in mice.84 SALL4 is a neosubstrate candidate that mediates the teratogenicity of thalidomide; mouse Sall4, however, was resistant to thalidomide-induced degradation in mouse cells expressing humanized CRBN. To degrade mouse Sall4 in mouse cells, it was necessary to humanize not only Crbn but also Sall4 (Fig. 10(b)).6 This suggested that species-specific amino acid substitutions in neosubstrates are also involved in the species specificity of thalidomide teratogenicity. Moreover, V388 is not the only amino acid substitution in CRBN involved in the species-specific effects of thalidomide-based glue degraders. The E377 of human CRBN is replaced by valine in rodents (Fig. 10(a)); this E377 residue is important for CC-885-induced degradation of GSPT1 because the urea moiety of CC-885 interacts with E377.7 Thus, recent structural studies are slowly unraveling the complexity behind the species-specific effects of thalidomide-based glue degraders.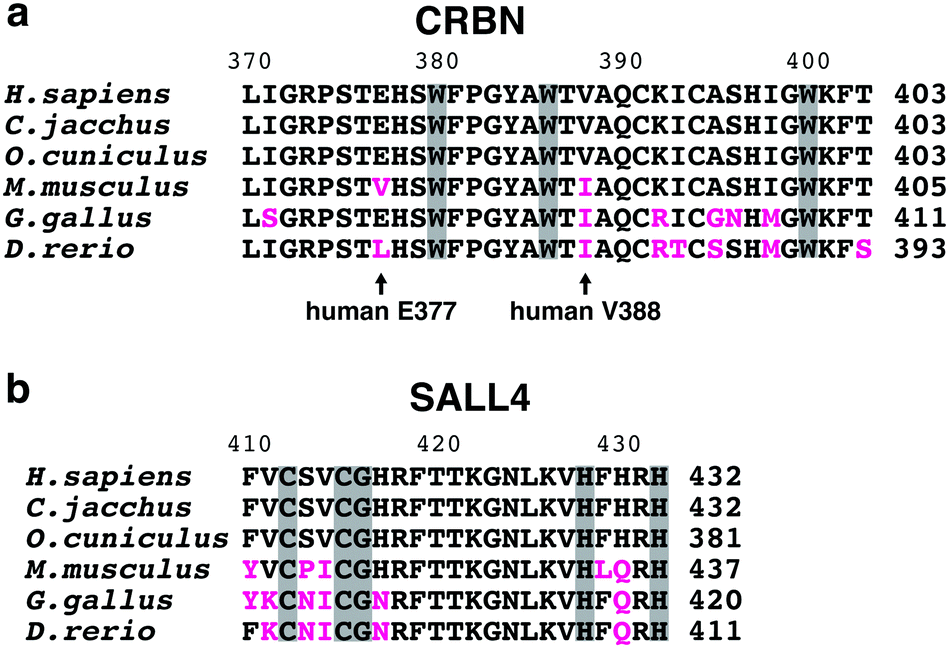 | ||
| Fig. 10 Partial sequences of CRBN TBDs and SALL4 zinc fingers from 6 vertebrate species. Multiple sequence alignment of the CRBN TBD (a) and the SALL4 zinc finger motif containing a degron (b) from 6 vertebrate species. Substitutions from the human orthologs are highlighted in magenta. Arrows denote key residues partially responsible for species specificity of thalidomide and its derivatives. Amino acids comprising the tri-Trp pocket of CRBN (a) and those comprising the C2H2 motif and the degron (CXXCG) of SALL4 (b) are shown in gray background. | ||
9. Protein degraders as a novel pharmacological modality
In past decades, the mainstay of pharmaceuticals has been small molecule compounds that exert their effects by binding to the active site of a target protein, temporarily inhibiting its enzymatic activity. Therefore, proteins that lack a clear active site, such as transcription factors, are considered undruggable; it is estimated that these comprise approximately 80% of the human proteome. Thalidomide-based degraders have a different mode of action, acting as molecular glues that alter the substrate specificity of CRBN, leading to the degradation of new target proteins. Such protein degraders with a new mode of action, i.e. hijacking the UPS, causing the degradation of target proteins, are attracting attention as a new modality with the potential to target undruggable proteins. Protein degraders have a catalytic mechanism of action in which a single compound induces the degradation of multiple target proteins; this is superior to conventional small molecules with the stoichiometric mode of action.One class of protein degraders called “molecular glue degraders” includes thalidomide and its derivatives. The term was first proposed in the study of the plant hormone auxin. Auxin acts by physically linking TIR1, an F-box protein of the Skp, Cullin, F-box containing complex (SCF), to Aux/IAA, leading to its proteasomal degradation.86 The only protein degraders currently used in clinical practice are thalidomide-based glue degraders such as thalidomide, lenalidomide, and pomalidomide. Although many neosubstrates have already been identified, the number is expected to rise substantially in the future. This is because numerous C2H2 zinc finger proteins contain a potential degron(s) that may be recognized by CRL4CRBN; this largest family of transcription factors may be targeted by appropriate modification of the compound.
Recently, new classes of molecular glue degraders have been reported. A series of sulfonamides, such as E7820, Indisulam, and chloroquinoxaline sulfonamides, act as molecular glues; they bind to the DCAF15 subunit of CRL4DCAF15, causing the degradation of the splicing factor RBM39.87,88 The cyclin-dependent kinase (CDK) inhibitors CR8 and HQ461 also function as molecular glue degraders that cause the degradation cyclin K via its binding partner CDK12. This occurs in the absence of substrate recognition receptors because CR8 directly binds to the DDB1 subunit of CRL4 and recruits the CDK-cyclin complex via CDK12.89,90 These findings suggest that molecular glue degraders are more diverse with a broader potential than anticipated as a therapeutic modality.
The other class of protein degraders is the PROteolysis-TArgeting Chimeras (PROTACs), which are composed of two functional groups targeting E3 and protein of interest (POI), respectively, and a spacer linking them. The benefit of this concept is that any desired protein can be targeted for degradation as long as there is a compound that binds to it. On the other hand, due to their tripartite nature, they have larger molecular weights and may have unfavorable pharmacokinetics. Moreover, the so-called hook effect of PROTACs may limit the efficiency of POI degradation. Since two ligands bind to their targets independently, high concentrations of PROTACs favor the formation of two binary complexes over the formation of the ternary complex. PROTACs were first reported in 2001; in that study, methionine aminopeptidase MetAP-2 was successfully recruited to the E3 ubiquitin ligase SCFβ-TRCP for ubiquitination.91 The compound, however, contains a membrane-impermeable phosphopeptide moiety and cannot target intracellular MetAP-2 for degradation. Later, revealing that the mechanism of action of thalidomide is molecular glue fueled the rapid development of PROTACs. A variety of E3s have been targeted by PROTACs, including CRBN, Von Hippel-Lindau (VHL), IAP, and MDM2,92–95 among which CRBN is perhaps most extensively studied as a target of PROTACs. For CRBN-based PROTACs, thalidomide or its derivative is used for the E3 binding site (Fig. 11). Although we will not go into the details in this review, CRBN-based PROTACs targeting BRD4, FKBP12, BCR-ABL, SIRT2, CDK6, and BTK have been reported,92,96–100 and the number of successful PROTACs is steadily increasing. Click-formed proteolysis-targeting chimeras (CLIPTACs) are proposed as a new strategy to improve the poor pharmacokinetics of PROTACs caused by their large molecular weights; in this strategy, PROTACs are generated in cells by click chemistry from two precursors with good pharmacokinetics.101 In addition, light-controllable PROTACs that use light to activate or deactivate PROTACs have also been developed to solve the on-target-off-tissue toxicity, which also limits their clinical application.102,103 PROTAC technology has received particular attention in the field of anti-cancer drugs, and several PROTACs are currently in clinical trials.
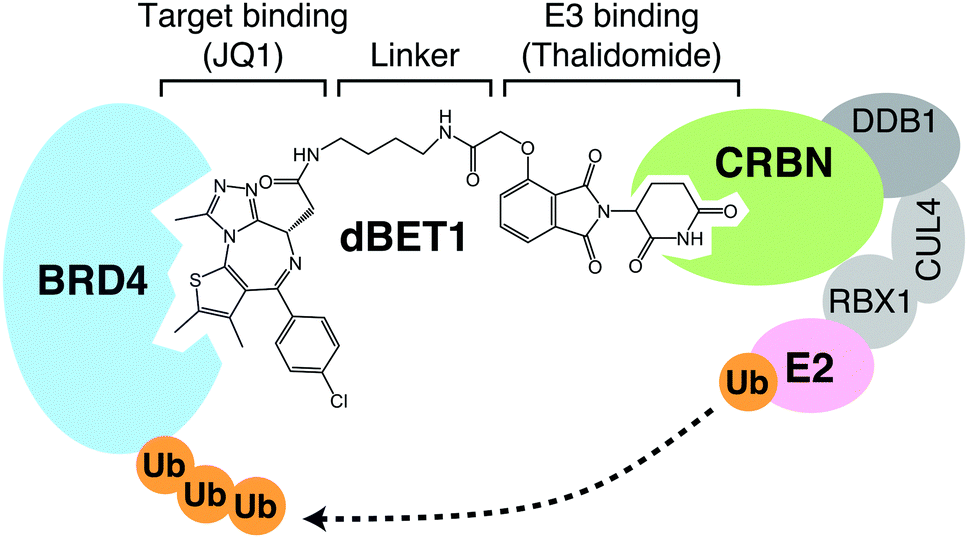 | ||
| Fig. 11 CRBN-based PROTACs. dBET1, one of the CRBN-based PROTACs, consists of a linker connecting thalidomide and JQ1, the latter of which binds to its target BRD4. Formation of the ternary complex shown in the figure leads to the degradation of BRD4 via CRL4CRBN. | ||
10. Conclusions and perspectives
About 70 years have passed since the development of thalidomide. However, a major advance in our understanding of its mechanism of action has been made in the past decade. The breakthrough that drove this advance was the discovery of CRBN as a direct target of thalidomide. A series of studies focusing on the function of CRBN showed that thalidomide and its derivatives act as molecular glues that alter its substrate specificity, leading to the degradation of various neosubstrates. This finding achieved a molecular understanding of the diverse pharmacological effects of thalidomide and its derivatives, including anti-myeloma and teratogenic effects. Furthermore, demonstration of the clinical usefulness of molecular glue degraders has triggered rapid progress in the development of protein degraders, including PROTACs.In the past decade, research on CRBN has focused mainly on the mechanism of action of thalidomide and its derivatives, but there are still unknowns concerning the physiological functions of CRBN. Understanding physiological functions of CRBN will be important in promoting the clinical application of thalidomide-based protein degraders. CRBN was initially identified as a gene associated with people with an intellectual disability, and its function was predicted to be related to brain function. Subsequent studies have shown that it is also involved in the regulation of ion channels and energy metabolism. It seems that the physiological function of CRBN are, at least in part, mediated by the its native substrates, which are targeted for ubiquitination and degradation in the absence of drugs. Previous studies have identified SLO1, MEIS2, glutamine synthetase, AMPKα, and CLC-1 as native substrates of CRL4CRBN.66,104–107 SLO1, a subunit of BK channels, is a protein identified in early studies as an interactor of CRBN,108 and subsequent studies have shown that it is targeted for ubiquitination by CRL4CRBN. Reportedly, polyubiquitination of SLO1 does not cause its degradation, but is important for its retention in the endoplasmic reticulum, which in turn regulates BK channel activity.104 In addition, the nonsense mutation (E419X) of CRBN associated with intellectual disability causes a loss of interaction with BK channels; the resulting destabilization of BK channels affects learning and memory.109 With regard to AMPK, it is a master regulator of energy metabolism, and its dysfunction is responsible for a wide range of metabolic diseases, including obesity, diabetes, and hypertension. Crbn knockout mice were used to show that Crbn is a negative modulator of AMPK; compared to wild-type mice, feeding a normal diet to Crbn-deficient mice produced constitutive activation of AMPK. The knockout mice also showed significantly less weight gain on a high-fat diet and greatly improved insulin sensitivity.110 AMPKα, one of the subunits of AMPK, is ubiquitinated by CRL4CRBN,106 providing a molecular explanation for the regulation of AMPK by CRBN. CRBN is, furthermore, involved in the homeostatic regulation of glutamine. Glutamine synthesis is negatively feedback-regulated by its product, glutamine. The mechanism is that glutamine induces acetylation of glutamine synthetase via p300/CBP, which then induces the degradation of glutamine synthetase via CRL4CRBN, thereby maintaining glutamine homeostasis.105 These findings have provided us with an understanding of the diverse physiological roles of CRBN, but unanswered questions remain. How is CRBN function regulated? How many native substrates are there, in total, for CRL4CRBN? Does any molecule occupy the thalidomide-binding pocket of CRBN when CRL4CRBN catalyzes ubiquitination of a native substrate? With regard to the last question, the observation that thalidomide and its derivatives inhibit the ubiquitination and degradation of MEIS266 and AMPKα,106 which are considered to be native substrates, implies the presence of a native ligand. As mentioned above, common metabolites such as uridine and succinimide can bind to the tri-Trp pocket of CRBN.73,74 These metabolites may serve as native ligands of CRBN, but further research is needed to reach definitive conclusions.
Development of therapeutic agents has not kept pace with the accumulation of basic knowledge, such as the identification of disease-causing genes. Challenges in developing therapeutics are manifold, but one of the difficulties is that most proteins are undruggable as targets of small molecule compounds. Protein degraders have shown great promise as a new modality that may overcome this difficulty. Although molecular glue degraders and PROTACs aim to achieve the same outcome, each strategy is better suited for a different type of targets and requires different considerations in molecular design because of their different target-binding modes and pharmacological properties. Thalidomide-based glue degraders target a wide variety of proteins with similar structural motifs. The design of new molecular glue degraders is limited by the fact that a compound-bound E3 ubiquitin ligase and a neosubstrate must exhibit excellent surface complementarity. However, its better pharmacokinetics and potential to target the large family of zinc finger-containing transcription factors is a major advantage over PROTACs. Since high sequence homology is not required for neosubstrates, the proteome that can be approached by thalidomide-based glue degraders may be broader than expected. Moreover, thalidomide-based glue degraders are already in use in clinical practice. Discovery of novel neosubstrates for these drugs may lead to the discovery of new therapeutic properties of the existing drugs. Thus, “neosubstrate-based” drug development, by which drugs are developed or repurposed based on the function of a novel neosubstrate, is a promising strategy. Considering that omics approaches have led to the identification of multiple neosubstrates for a thalidomide-based glue degrader, however, controlling diverse on-target side effects may be a difficult challenge. On the other hand, the most exciting property of PROTACs is that they have the potential to cause the degradation of any protein with a ligandable site. It allows the use of any small molecule compounds that have sufficiently high affinity and selectivity for POI as ligands on one side, as they do not have to inhibit POI's enzymatic or biological activity per se. For PROTACs to function, two ligands and a linker should work cooperatively to properly align the distance and relative orientation between E3 and POI; this requires a reliable framework for their design and optimization. Thus far, the design of PROTACs has relied largely on trial and error, but recent structural studies of ternary complexes containing PROTACs provide a stepping-stone to rational design.92,111–114
Since formation of stable E3-compound-neosubstrate ternary complexes is critical to the action of both molecular glue degraders and PROTACs, information obtained from structural analysis is critical for their development. Since the reliability of protein structure predictions has been dramatically improved with the advent of AlphaFold,115 computational methods will greatly facilitate future development of protein degraders. Over the past decade, a new modality of protein degraders has been established, and significant resources have been invested in their development, with high expectations. The next decade should be the first time we will see tangible benefits.
Author contributions
All authors conceived and designed the review. J. Y. and Y. Y. mainly wrote the manuscript. J. Y. and T. I. prepared figures and table. H. H. supervised the review.Conflicts of interest
H. H. received research support from Bristol-Meyers Squibb.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 20K08741 (to J. Y.), 20H03182 (to Y. Y.), 18H05502 (to T. I.), and 21H02955 (to T. I.).References
- T. Ito, H. Ando, T. Suzuki, T. Ogura, K. Hotta, Y. Imamura, Y. Yamaguchi and H. Handa, Science, 2010, 327, 1345–1350 CrossRef CAS PubMed.
- J. Krönke, N. Udeshi, A. Narla, P. Grauman, S. Hurst, M. McConkey, T. Svinkina, D. Heckl, E. Comer, X. Li, C. Ciarlo, E. Hartman, N. Munshi, M. Schenone, S. Schreiber, S. Carr and B. Ebert, Science, 2014, 343, 301–305 CrossRef PubMed.
- G. Lu, R. Middleton, H. Sun, M. Naniong, C. Ott, C. Mitsiades, K. Wong, J. Bradner and W. Kaelin, Science, 2014, 343, 305–309 CrossRef CAS PubMed.
- J. Krönke, E. Fink, P. Hollenbach, K. MacBeth, S. Hurst, N. Udeshi, P. Chamberlain, D. Mani, H. Man, A. Gandhi, T. Svinkina, R. Schneider, M. McConkey, M. Järås, E. Griffiths, M. Wetzler, L. Bullinger, B. Cathers, S. Carr, R. Chopra and B. Ebert, Nature, 2015, 523, 183–188 CrossRef PubMed.
- M. Matyskiela, G. Lu, T. Ito, B. Pagarigan, C. Lu, K. Miller, W. Fang, N. Wang, D. Nguyen, J. Houston, G. Carmel, T. Tran, M. Riley, L. Nosaka, G. Lander, S. Gaidarova, S. Xu, A. Ruchelman, H. Handa, J. Carmichael, T. Daniel, B. Cathers, A. Lopez-Girona and P. Chamberlain, Nature, 2016, 535, 252–257 CrossRef CAS PubMed.
- K. Donovan, J. An, R. Nowak, J. Yuan, E. Fink, B. Berry, B. Ebert and E. Fischer, eLife, 2018, 7, e38430 CrossRef PubMed.
- M. Matyskiela, S. Couto, X. Zheng, G. Lu, J. Hui, K. Stamp, C. Drew, Y. Ren, M. Wang, A. Carpenter, C. Lee, T. Clayton, W. Fang, C. Lu, M. Riley, P. Abdubek, K. Blease, J. Hartke, G. Kumar, R. Vessey, M. Rolfe, L. Hamann and P. Chamberlain, Nat. Chem. Biol., 2018, 14, 981–987 CrossRef CAS PubMed.
- T. Asatsuma-Okumura, H. Ando, M. De Simone, J. Yamamoto, T. Sato, N. Shimizu, K. Asakawa, Y. Yamaguchi, T. Ito, L. Guerrini and H. Handa, Nat. Chem. Biol., 2019, 15, 1077–1084 CrossRef CAS PubMed.
- J. Yamamoto, T. Suwa, Y. Murase, S. Tateno, H. Mizutome, T. Asatsuma-Okumura, N. Shimizu, T. Kishi, S. Momose, M. Kizaki, T. Ito, Y. Yamaguchi and H. Handa, Nat. Chem. Biol., 2020, 16, 1208–1217 CrossRef CAS PubMed.
- S. Yamanaka, H. Murai, D. Saito, G. Abe, E. Tokunaga, T. Iwasaki, H. Takahashi, H. Takeda, T. Suzuki, N. Shibata, K. Tamura and T. Sawasaki, EMBO J., 2021, 40, e105375 CrossRef CAS PubMed.
- N. Shimizu, T. Asatsuma-Okumura, J. Yamamoto, Y. Yamaguchi, H. Handa and T. Ito, Commun. Biol., 2021, 4, 277 CrossRef PubMed.
- A. Renneville, J. Gasser, D. Grinshpun, P. Jean Beltran, N. Udeshi, M. Matyskiela, T. Clayton, M. McConkey, K. Viswanathan, A. Tepper, A. Guirguis, R. Sellar, S. Cotteret, C. Marzac, V. Saada, S. De Botton, J. Kiladjian, J. Cayuela, M. Rolfe, P. Chamberlain, S. Carr and B. Ebert, Blood Cancer Discovery, 2021, 2, 250–265 CrossRef PubMed.
- S. Yamanaka, Y. Horiuchi, S. Matsuoka, K. Kido, K. Nishino, M. Maeno, N. Shibata, H. Kosako and T. Sawasaki, Nat. Commun., 2022, 13, 183 CrossRef CAS PubMed.
- W. Mcbride, Lancet, 1961, 278, 1358 CrossRef.
- D. Burley and W. Lenz, Lancet, 1962, 279, 271–272 CrossRef.
- W. Lenz, Teratology, 1988, 38, 203–215 CrossRef CAS PubMed.
- J. Sheskin, Clin. Pharm. Ther., 1965, 6, 303–306 CrossRef CAS PubMed.
- E. Sampaio, E. Sarno, R. Galilly, Z. Cohn and G. Kaplan, J. Exp. Med., 1991, 173, 699–703 CrossRef CAS PubMed.
- S. Mchugh, I. Rifkin, J. Deighton, A. Wilson, P. Lachmann, C. Lockwood and P. Ewan, Clin. Exp. Immunol., 1995, 99, 160–167 CrossRef CAS PubMed.
- R. D’Amato, M. Loughnan, E. Flynn and J. Folkman, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4082–4085 CrossRef PubMed.
- S. Singhal, J. Mehta, R. Desikan, D. Ayers, P. Roberson, P. Eddlemon, N. Munshi, E. Anaissie, C. Wilson, M. Dhodapkar, J. Zeldis, D. Siegel, J. Crowley and B. Barlogie, N. Engl. J. Med., 1999, 341, 1565–1571 CrossRef CAS PubMed.
- K. Dredge, J. Marriott, S. Todryk, G. Muller, R. Chen, D. Stirling and A. Dalgleish, J. Immunol., 2002, 168, 4914–4919 CrossRef CAS PubMed.
- J. Marriott, I. Clarke, K. Dredge, G. Muller, D. Stirling and A. Dalgleish, Clin. Exp. Immunol., 2002, 130, 75–84 CrossRef CAS PubMed.
- K. Nishio, Y. Masaike, M. Ikeda, H. Narimatsu, N. Gokon, S. Tsubouchi, M. Hatakeyama, S. Sakamoto, N. Hanyu, A. Sandhu, H. Kawaguchi, M. Abe and H. Handa, Colloids Surf., B, 2008, 64, 162–169 CrossRef CAS PubMed.
- Y. Masaike, T. Takagi, M. Hirota, J. Yamada, S. Ishihara, T. Yung, T. Inoue, C. Sawa, H. Sagara, S. Sakamoto, Y. Kabe, Y. Takahashi, Y. Yamaguchi and H. Handa, Mol. Pharmacol., 2009, 77, 262–269 CrossRef PubMed.
- M. Azuma, Y. Kabe, C. Kuramori, M. Kondo, Y. Yamaguchi and H. Handa, PLoS One, 2008, 3, e3070 CrossRef PubMed.
- C. Kuramori, M. Azuma, K. Kume, Y. Kaneko, A. Inoue, Y. Yamaguchi, Y. Kabe, T. Hosoya, M. Kizaki, M. Suematsu and H. Handa, Biochem. Biophys. Res. Commun., 2009, 379, 519–525 CrossRef CAS PubMed.
- Q. Sievers, J. Gasser, G. Cowley, E. Fischer and B. Ebert, Blood, 2018, 132, 1293–1303 CrossRef CAS PubMed.
- G. Lu, S. Weng, M. Matyskiela, X. Zheng, W. Fang, S. Wood, C. Surka, R. Mizukoshi, C. Lu, D. Mendy, I. Jang, K. Wang, M. Marella, S. Couto, B. Cathers, J. Carmichael, P. Chamberlain and M. Rolfe, eLife, 2018, 7, e40958 CrossRef PubMed.
- A. Shaffer, N. Emre, L. Lamy, V. Ngo, G. Wright, W. Xiao, J. Powell, S. Dave, X. Yu, H. Zhao, Y. Zeng, B. Chen, J. Epstein and L. Staudt, Nature, 2008, 454, 226–231 CrossRef CAS PubMed.
- Y. Zhu, E. Braggio, C. Shi, L. Bruins, J. Schmidt, S. Van Wier, X. Chang, C. Bjorklund, R. Fonseca, P. Bergsagel, R. Orlowski and A. Stewart, Blood, 2011, 118, 4771–4779 CrossRef CAS PubMed.
- A. Lopez-Girona, D. Mendy, T. Ito, K. Miller, A. Gandhi, J. Kang, S. Karasawa, G. Carmel, P. Jackson, M. Abbasian, A. Mahmoudi, B. Cathers, E. Rychak, S. Gaidarova, R. Chen, P. Schafer, H. Handa, T. Daniel, J. Evans and R. Chopra, Leukemia, 2012, 26, 2326–2335 CrossRef CAS PubMed.
- A. Broyl, R. Kuiper, M. van Duin, B. van der Holt, L. el Jarari, U. Bertsch, S. Zweegman, A. Buijs, D. Hose, H. Lokhorst, H. Goldschmidt and P. Sonneveld, Blood, 2013, 121, 624–627 CrossRef CAS PubMed.
- D. Heintel, A. Rocci, H. Ludwig, A. Bolomsky, S. Caltagirone, M. Schreder, S. Pfeifer, H. Gisslinger, N. Zojer, U. Jäger and A. Palumbo, Br. J. Haematol., 2013, 161, 695–700 CrossRef CAS PubMed.
- M. Matyskiela, W. Zhang, H. Man, G. Muller, G. Khambatta, F. Baculi, M. Hickman, L. LeBrun, B. Pagarigan, G. Carmel, C. Lu, G. Lu, M. Riley, Y. Satoh, P. Schafer, T. Daniel, J. Carmichael, B. Cathers and P. Chamberlain, J. Med. Chem., 2017, 61, 535–542 CrossRef PubMed.
- Y. Zhu, E. Braggio, C. Shi, K. Kortuem, L. Bruins, J. Schmidt, X. Chang, P. Langlais, M. Luo, P. Jedlowski, B. LaPlant, K. Laumann, R. Fonseca, P. Bergsagel, J. Mikhael, M. Lacy, M. Champion and A. Stewart, Blood, 2014, 124, 536–545 CrossRef CAS PubMed.
- J. Krönke, F. Kuchenbauer, M. Kull, V. Teleanu, L. Bullinger, D. Bunjes, A. Greiner, S. Kolmus, S. Köpff, M. Schreder, L. Mügge, C. Straka, M. Engelhardt, H. Döhner, H. Einsele, F. Bassermann, R. Bargou, S. Knop and C. Langer, Leukemia, 2016, 31, 1363–1367 CrossRef PubMed.
- M. Pourabdollah, M. Bahmanyar, E. Atenafu, D. Reece, J. Hou and H. Chang, J. Hematol. Oncol., 2016, 9, 123 CrossRef PubMed.
- I. Misiewicz-Krzeminska, C. de Ramón, L. Corchete, P. Krzeminski, E. Rojas, I. Isidro, R. García-Sanz, J. Martínez-López, A. Oriol, J. Bladé, J. Lahuerta, J. San Miguel, L. Rosiñol, M. Mateos and N. Gutiérrez, Blood Adv., 2020, 4, 6023–6033 CrossRef PubMed.
- A. Huart, N. MacLaine, D. Meek and T. Hupp, J. Biol. Chem., 2009, 284, 32384–32394 CrossRef CAS PubMed.
- S. Amit, A. Hatzubai, Y. Birman, J. Andersen, E. Ben-Shushan, M. Mann, Y. Ben-Neriah and I. Alkalay, Genes Dev., 2002, 16, 1066–1076 CrossRef CAS PubMed.
- N. Bidère, V. Ngo, J. Lee, C. Collins, L. Zheng, F. Wan, R. Davis, G. Lenz, D. Anderson, D. Arnoult, A. Vazquez, K. Sakai, J. Zhang, Z. Meng, T. Veenstra, L. Staudt and M. Lenardo, Nature, 2008, 458, 92–96 Search PubMed.
- A. List, S. Kurtin, D. Roe, A. Buresh, D. Mahadevan, D. Fuchs, L. Rimsza, R. Heaton, R. Knight and J. Zeldis, N. Engl. J. Med., 2005, 352, 549–557 CrossRef CAS PubMed.
- A. List, G. Dewald, J. Bennett, A. Giagounidis, A. Raza, E. Feldman, B. Powell, P. Greenberg, D. Thomas, R. Stone, C. Reeder, K. Wride, J. Patin, M. Schmidt, J. Zeldis and R. Knight, N. Engl. J. Med., 2006, 355, 1456–1465 CrossRef CAS PubMed.
- C. Surka, L. Jin, N. Mbong, C. Lu, I. Jang, E. Rychak, D. Mendy, T. Clayton, E. Tindall, C. Hsu, C. Fontanillo, E. Tran, A. Contreras, S. Ng, M. Matyskiela, K. Wang, P. Chamberlain, B. Cathers, J. Carmichael, J. Hansen, J. Wang, M. Minden, J. Fan, D. Pierce, M. Pourdehnad, M. Rolfe, A. Lopez-Girona, J. Dick and G. Lu, Blood, 2021, 137, 661–677 CrossRef CAS PubMed.
- L. G. Corral, P. A. J. Haslett, G. W. Muller, R. Chen, L. M. Wong, C. J. Ocampo, R. T. Patterson, D. I. Stirling and G. Kaplan, J. Immunol., 1999, 163, 380–386 CAS.
- P. Schafer, A. Gandhi, M. Loveland, R. Chen, H. Man, P. Schnetkamp, G. Wolbring, S. Govinda, L. Corral, F. Payvandi, G. Muller and D. Stirling, J. Pharmacol. Exp. Ther., 2003, 305, 1222–1232 CrossRef CAS PubMed.
- (a) K. Dredge, J. Marriott, S. Todryk, G. Muller, R. Chen, D. Stirling and A. Dalgleish, J. Immunol., 2002, 168, 4914–4919 CrossRef CAS PubMed; (b) A. Gandhi, J. Kang, C. Havens, T. Conklin, Y. Ning, L. Wu, T. Ito, H. Ando, M. Waldman, A. Thakurta, A. Klippel, H. Handa, T. Daniel, P. Schafer and R. Chopra, Br. J. Haematol., 2013, 164, 811–821 CrossRef PubMed.
- L. Wu, M. Adams, T. Carter, R. Chen, G. Muller, D. Stirling, P. Schafer and J. Bartlett, Clin. Cancer Res., 2008, 14, 4650–4657 CrossRef CAS PubMed.
- A. Gandhi, J. Kang, C. Havens, T. Conklin, Y. Ning, L. Wu, T. Ito, H. Ando, M. Waldman, A. Thakurta, A. Klippel, H. Handa, T. Daniel, P. Schafer and R. Chopra, Br. J. Haematol., 2013, 164, 811–821 CrossRef PubMed.
- P. Schafer, Y. Ye, L. Wu, J. Kosek, G. Ringheim, Z. Yang, L. Liu, M. Thomas, M. Palmisano and R. Chopra, Ann. Rheum. Dis., 2018, 77, 1516–1523 CrossRef CAS PubMed.
- T. Parman, M. Wiley and P. Wells, Nat. Med., 1999, 5, 582–585 CrossRef CAS PubMed.
- V. Jacques, A. Czarnik, T. Judge, L. Van der Ploeg and S. DeWitt, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1471–E1479 CrossRef CAS PubMed.
- T. Mori, T. Ito, S. Liu, H. Ando, S. Sakamoto, Y. Yamaguchi, E. Tokunaga, N. Shibata, H. Handa and T. Hakoshima, Sci. Rep., 2018, 8, 1294 CrossRef PubMed.
- Y. Hirose, T. Kitazono, M. Sezaki, M. Abe, K. Sakimura, H. Funato, H. Handa, K. Vogt and M. Yanagisawa, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 23106–23112 CrossRef CAS PubMed.
- R. Al-Baradie, K. Yamada, C. Hilaire, W. Chan, C. Andrews, N. McIntosh, M. Nakano, E. Martonyi, W. Raymond, S. Okumura, M. Okihiro and E. Engle, Am. J. Hum. Genet., 2002, 71, 1195–1199 CrossRef CAS PubMed.
- J. Kohlhase, Hum. Mol. Genet., 2002, 11, 2979–2987 CrossRef CAS PubMed.
- J. Kohlhase and L. Holmes, Birth Defects Res., Part A, 2004, 70, 550–551 CrossRef PubMed.
- D. Belair, G. Lu, L. Waller, J. Gustin, N. Collins and K. Kolaja, Sci. Rep., 2020, 10, 2864 CrossRef CAS PubMed.
- A. Mills, B. Zheng, X. Wang, H. Vogel, D. Roop and A. Bradley, Nature, 1999, 398, 708–713 CrossRef CAS PubMed.
- A. Yang, M. Kaghad, D. Caput and F. McKeon, Trends Genet., 2002, 18, 90–95 CrossRef CAS PubMed.
- A. Yang, R. Schweitzer, D. Sun, M. Kaghad, N. Walker, R. Bronson, C. Tabin, A. Sharpe, D. Caput, C. Crum and F. McKeon, Nature, 1999, 398, 714–718 CrossRef CAS PubMed.
- H. Chen, A. Beasley, Y. Hu and X. Chen, PLoS One, 2015, 10, e0143878 CrossRef PubMed.
- M. Barna, N. Hawe, L. Niswander and P. Pandolfi, Nat. Genet., 2000, 25, 166–172 CrossRef CAS PubMed.
- J. Higgins, J. Pucilowska, R. Lombardi and J. Rooney, Neurology, 2004, 63, 1927–1931 CrossRef CAS PubMed.
- E. Fischer, K. Böhm, J. Lydeard, H. Yang, M. Stadler, S. Cavadini, J. Nagel, F. Serluca, V. Acker, G. Lingaraju, R. Tichkule, M. Schebesta, W. Forrester, M. Schirle, U. Hassiepen, J. Ottl, M. Hild, R. Beckwith, J. Harper, J. Jenkins and N. Thomä, Nature, 2014, 512, 49–53 CrossRef CAS PubMed.
- P. Chamberlain, A. Lopez-Girona, K. Miller, G. Carmel, B. Pagarigan, B. Chie-Leon, E. Rychak, L. Corral, Y. Ren, M. Wang, M. Riley, S. Delker, T. Ito, H. Ando, T. Mori, Y. Hirano, H. Handa, T. Hakoshima, T. Daniel and B. Cathers, Nat. Struct. Mol. Biol., 2014, 21, 803–809 CrossRef CAS PubMed.
- T. Li, X. Chen, K. Garbutt, P. Zhou and N. Zheng, Cell, 2006, 124, 105–117 CrossRef CAS PubMed.
- A. Sheereen, M. Alaamery, S. Bawazeer, Y. Al Yafee, S. Massadeh and W. Eyaid, J. Med. Genet., 2017, 54, 236–240 CrossRef CAS PubMed.
- G. Petzold, E. Fischer and N. Thomä, Nature, 2016, 532, 127–130 CrossRef CAS PubMed.
- M. Matyskiela, T. Clayton, X. Zheng, C. Mayne, E. Tran, A. Carpenter, B. Pagarigan, J. McDonald, M. Rolfe, L. Hamann, G. Lu and P. Chamberlain, Nat. Struct. Mol. Biol., 2020, 27, 319–322 CrossRef CAS PubMed.
- Q. Sievers, G. Petzold, R. Bunker, A. Renneville, M. Słabicki, B. Liddicoat, W. Abdulrahman, T. Mikkelsen, B. Ebert and N. Thomä, Science, 2018, 362, eaat0572 CrossRef PubMed.
- M. Hartmann, I. Boichenko, M. Coles, F. Zanini, A. Lupas and B. Hernandez Alvarez, J. Struct. Biol., 2014, 188, 225–232 CrossRef CAS PubMed.
- I. Boichenko, K. Bär, S. Deiss, C. Heim, R. Albrecht, A. Lupas, B. Hernandez Alvarez and M. Hartmann, ACS Omega, 2018, 3, 11163–11171 CrossRef CAS PubMed.
- C. Heim, D. Pliatsika, F. Mousavizadeh, K. Bär, B. Hernandez Alvarez, A. Giannis and M. Hartmann, J. Med. Chem., 2019, 62, 6615–6629 CrossRef CAS PubMed.
- C. Heim and M. Hartmann, Acta Crystallogr., Sect. D: Struct. Biol., 2022, 78, 290–298 CrossRef CAS PubMed.
- S. Maiwald, C. Heim, B. Hernandez Alvarez and M. Hartmann, ACS Med. Chem. Lett., 2020, 12, 74–81 CrossRef PubMed.
- G. Burslem, P. Ottis, S. Jaime-Figueroa, A. Morgan, P. Cromm, M. Toure and C. Crews, ChemMedChem, 2018, 13, 1508–1512 CrossRef CAS PubMed.
- H. Furihata, S. Yamanaka, T. Honda, Y. Miyauchi, A. Asano, N. Shibata, M. Tanokura, T. Sawasaki and T. Miyakawa, Nat. Commun., 2020, 11, 4578 CrossRef CAS PubMed.
- J. An, C. Ponthier, R. Sack, J. Seebacher, M. Stadler, K. Donovan and E. Fischer, Nat. Commun., 2017, 8, 15398 CrossRef CAS PubMed.
- N. Vargesson, Birth Defects Res., Part C, 2015, 105, 140–156 CrossRef CAS PubMed.
- R. Brent, J. Pediatr., 1964, 64, 762–770 CrossRef CAS PubMed.
- M. Chesi, G. Matthews, V. Garbitt, S. Palmer, J. Shortt, M. Lefebure, A. Stewart, R. Johnstone and P. Bergsagel, Blood, 2012, 120, 376–385 CrossRef CAS PubMed.
- E. Fink, M. McConkey, D. Adams, S. Haldar, J. Kennedy, A. Guirguis, N. Udeshi, D. Mani, M. Chen, B. Liddicoat, T. Svinkina, A. Nguyen, S. Carr and B. Ebert, Blood, 2018, 132, 1535–1544 CrossRef CAS PubMed.
- Y. Gemechu, D. Millrine, S. Hashimoto, J. Prakash, K. Sanchenkova, H. Metwally, P. Gyanu, S. Kang and T. Kishimoto, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 11802–11807 CrossRef CAS PubMed.
- X. Tan, L. Calderon-Villalobos, M. Sharon, C. Zheng, C. Robinson, M. Estelle and N. Zheng, Nature, 2007, 446, 640–645 CrossRef CAS PubMed.
- T. Uehara, Y. Minoshima, K. Sagane, N. Sugi, K. Mitsuhashi, N. Yamamoto, H. Kamiyama, K. Takahashi, Y. Kotake, M. Uesugi, A. Yokoi, A. Inoue, T. Yoshida, M. Mabuchi, A. Tanaka and T. Owa, Nat. Chem. Biol., 2017, 13, 675–680 CrossRef CAS PubMed.
- T. Han, M. Goralski, N. Gaskill, E. Capota, J. Kim, T. Ting, Y. Xie, N. Williams and D. Nijhawan, Science, 2017, 356, eaal3755 CrossRef PubMed.
- M. Słabicki, Z. Kozicka, G. Petzold, Y. Li, M. Manojkumar, R. Bunker, K. Donovan, Q. Sievers, J. Koeppel, D. Suchyta, A. Sperling, E. Fink, J. Gasser, L. Wang, S. Corsello, R. Sellar, M. Jan, D. Gillingham, C. Scholl, S. Fröhling, T. Golub, E. Fischer, N. Thomä and B. Ebert, Nature, 2020, 585, 293–297 CrossRef PubMed.
- L. Lv, P. Chen, L. Cao, Y. Li, Z. Zeng, Y. Cui, Q. Wu, J. Li, J. Wang, M. Dong, X. Qi and T. Han, eLife, 2020, 9, e59994 CrossRef CAS PubMed.
- K. Sakamoto, K. Kim, A. Kumagai, F. Mercurio, C. Crews and R. Deshaies, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 8554–8559 CrossRef CAS PubMed.
- G. Winter, D. Buckley, J. Paulk, J. Roberts, A. Souza, S. Dhe-Paganon and J. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- W. Hon, M. Wilson, K. Harlos, T. Claridge, C. Schofield, C. Pugh, P. Maxwell, P. Ratcliffe, D. Stuart and E. Jones, Nature, 2002, 417, 975–978 CrossRef CAS PubMed.
- Y. Itoh, M. Ishikawa, M. Naito and Y. Hashimoto, J. Am. Chem. Soc., 2010, 132, 5820–5826 CrossRef CAS PubMed.
- A. Schneekloth, M. Pucheault, H. Tae and C. Crews, Bioorg. Med. Chem. Lett., 2008, 18, 5904–5908 CrossRef CAS PubMed.
- G. Winter, D. Buckley, J. Paulk, J. Roberts, A. Souza, S. Dhe-Paganon and J. Bradner, Science, 2015, 348, 1376–1381 CrossRef CAS PubMed.
- A. Lai, M. Toure, D. Hellerschmied, J. Salami, S. Jaime-Figueroa, E. Ko, J. Hines and C. Crews, Angew. Chem., Int. Ed., 2015, 55, 807–810 CrossRef PubMed.
- M. Schiedel, D. Herp, S. Hammelmann, S. Swyter, A. Lehotzky, D. Robaa, J. Oláh, J. Ovádi, W. Sippl and M. Jung, J. Med. Chem., 2017, 61, 482–491 CrossRef PubMed.
- S. Su, Z. Yang, H. Gao, H. Yang, S. Zhu, Z. An, J. Wang, Q. Li, S. Chandarlapaty, H. Deng, W. Wu and Y. Rao, J. Med. Chem., 2019, 62, 7575–7582 CrossRef CAS PubMed.
- A. Zorba, C. Nguyen, Y. Xu, J. Starr, K. Borzilleri, J. Smith, H. Zhu, K. Farley, W. Ding, J. Schiemer, X. Feng, J. Chang, D. Uccello, J. Young, C. Garcia-Irrizary, L. Czabaniuk, B. Schuff, R. Oliver, J. Montgomery, M. Hayward, J. Coe, J. Chen, M. Niosi, S. Luthra, J. Shah, A. El-Kattan, X. Qiu, G. West, M. Noe, V. Shanmugasundaram, A. Gilbert, M. Brown and M. Calabrese, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E7285–E7292 CrossRef PubMed.
- H. Lebraud, D. Wright, C. Johnson and T. Heightman, ACS Cent. Sci., 2016, 2, 927–934 CrossRef CAS PubMed.
- J. Liu, H. Chen, L. Ma, Z. He, D. Wang, Y. Liu, Q. Lin, T. Zhang, N. Gray, H. Kaniskan, J. Jin and W. Wei, Sci. Adv., 2020, 6, eaay5154 CrossRef CAS PubMed.
- M. Reynders, B. Matsuura, M. Bérouti, D. Simoneschi, A. Marzio, M. Pagano and D. Trauner, Sci. Adv., 2020, 6, eaay5064 CrossRef CAS PubMed.
- J. Liu, J. Ye, X. Zou, Z. Xu, Y. Feng, X. Zou, Z. Chen, Y. Li and Y. Cang, Nat. Commun., 2014, 5, 3924 CrossRef CAS PubMed.
- T. Van Nguyen, J. Lee, M. Sweredoski, S. Yang, S. Jeon, J. Harrison, J. Yim, S. Lee, H. Handa, B. Kuhlman, J. Jeong, J. Reitsma, C. Park, S. Hess and R. Deshaies, Mol. Cell, 2016, 61, 809–820 CrossRef PubMed.
- E. Kwon, X. Li, Y. Deng, H. Chang and D. Kim, FASEB J., 2019, 33, 6539–6550 CrossRef CAS PubMed.
- Y. Chen, Y. Peng, M. Hu, J. Huang, Y. Chien, J. Wu, T. Chen and C. Tang, Sci. Rep., 2015, 5, 10667 CrossRef PubMed.
- S. Jo, K. Lee, S. Song, Y. Jung and C. Park, J. Neurochem., 2005, 94, 1212–1224 CrossRef CAS PubMed.
- T. Song, S. Liang, J. Liu, T. Zhang, Y. Yin, C. Geng, S. Gao, Y. Feng, H. Xu, D. Guo, A. Roberts, Y. Gu and Y. Cang, PLoS Genet., 2018, 14, e1007165 CrossRef PubMed.
- K. Lee, S. Yang, Y. Kim, Y. Choi, J. Nam, C. Choi, H. Choi and C. Park, Diabetes, 2013, 62, 1855–1864 CrossRef CAS PubMed.
- M. Gadd, A. Testa, X. Lucas, K. Chan, W. Chen, D. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
- R. Nowak, S. DeAngelo, D. Buckley, Z. He, K. Donovan, J. An, N. Safaee, M. Jedrychowski, C. Ponthier, M. Ishoey, T. Zhang, J. Mancias, N. Gray, J. Bradner and E. Fischer, Nat. Chem. Biol., 2018, 14, 706–714 CrossRef CAS PubMed.
- B. Smith, S. Wang, S. Jaime-Figueroa, A. Harbin, J. Wang, B. Hamman and C. Crews, Nat. Commun., 2019, 10, 131 CrossRef PubMed.
- W. Farnaby, M. Koegl, M. Roy, C. Whitworth, E. Diers, N. Trainor, D. Zollman, S. Steurer, J. Karolyi-Oezguer, C. Riedmueller, T. Gmaschitz, J. Wachter, C. Dank, M. Galant, B. Sharps, K. Rumpel, E. Traxler, T. Gerstberger, R. Schnitzer, O. Petermann, P. Greb, H. Weinstabl, G. Bader, A. Zoephel, A. Weiss-Puxbaum, K. Ehrenhöfer-Wölfer, S. Wöhrle, G. Boehmelt, J. Rinnenthal, H. Arnhof, N. Wiechens, M. Wu, T. Owen-Hughes, P. Ettmayer, M. Pearson, D. McConnell and A. Ciulli, Nat. Chem. Biol., 2019, 15, 672–680 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgland, C. Meyer, S. Kohl, A. Ballard, A. Cowie, B. Romera-Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |