
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIs enzyme immobilization a mature discipline? Some critical considerations to capitalize on the benefits of immobilization
Juan M.
Bolivar
 a,
John M.
Woodley
*b and
Roberto
Fernandez-Lafuente
*cd
a,
John M.
Woodley
*b and
Roberto
Fernandez-Lafuente
*cd
aFQPIMA group, Chemical and Materials Engineering Department, Faculty of Chemical Sciences, Complutense University of Madrid, Madrid, 28040, Spain
bDepartment of Chemical and Biochemical Engineering, Technical University of Denmark, 2800 Kgs Lyngby, Denmark. E-mail: jw@kt.dtu.dk; Tel: +45 4525 2885
cDepartamento de Biocatálisis. ICP-CSIC, C/Marie Curie 2, Campus UAM-CSIC Cantoblanco, Madrid 28049, Spain. E-mail: rfl@icp.csic.es; Tel: +34 91 585 4804
dCenter of Excellence in Bionanoscience Research, External Scientific Advisory Academic, King Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 15th July 2022
Abstract
Enzyme immobilization has been developing since the 1960s and although many industrial biocatalytic processes use the technology to improve enzyme performance, still today we are far from full exploitation of the field. One clear reason is that many evaluate immobilization based on only a few experiments that are not always well-designed. In contrast to many other reviews on the subject, here we highlight the pitfalls of using incorrectly designed immobilization protocols and explain why in many cases sub-optimal results are obtained. We also describe solutions to overcome these challenges and come to the conclusion that recent developments in material science, bioprocess engineering and protein science continue to open new opportunities for the future. In this way, enzyme immobilization, far from being a mature discipline, remains as a subject of high interest and where intense research is still necessary to take full advantage of the possibilities.
 Juan M. Bolivar | Dr Juan M. Bolivar is chemical engineer (2004, University of Granada) and PhD in sciences (2009, Complutense University of Madrid). He has been researcher at Institute of Catalysis (2005–2009, ICP-CSIC, Madrid), postdoc and and university assistant at the Graz University of Technology (Austria), 2010–2019. He is currently Senior Research Associate at the department of Chemical and Materials Engineering at the Complutense University of Madrid within excellence program “Atracción de Talento” from Madrid. Current interest lies in industrial biotechnology, design and characterization of heterogeneous biocatalysts catalysis, and bioprocess engineering in the context of integrated biorefineries and sustainable chemistry. |
 John M. Woodley | John Woodley is a Professor at DTU Chemical Engineering in Denmark. He has around 35 years’ experience in scaling-up and implementing new bioprocesses. His current research interests are in bioprocess science, with particular focus on (1) oxygen transfer, (2) selective separation, (3) enzyme stability, and (4) continuous processes. He has published around 300 scientific papers and 30 book chapters. In 2016, he was the elected Chair of the Biocatalysis GRC (USA) and in 2021 he was awarded an ERC Advanced Grant. He is a Fellow of the Institution of Chemical Engineers (UK) and a Fellow of the Royal Academy of Engineering (UK). |
 Roberto Fernandez-Lafuente | Roberto Fernandez-Lafuente is a Research Professor at ICP-CSIC in Madrid, Spain. He has around 35 years’ experience in studying the interactions between bio-macromolecules and supports, applying this to the preparation of biocatalysts and biosensors. His current research interests remain in enzyme immobilization, with particular focus on (1) enzyme co-immobilization, (2) effects of supports surface-enzyme on enzyme function, (3) enzyme stabilization, and (4) design of new immobilization/stabilization strategies. He has published over 500 scientific papers with an H index of 89 (Scopus). He has been recognized as highly cited researcher in the Clarivate lists of years 2019, 2020 and 2021. |
Key learning points(1) Is enzyme immobilization a mature discipline?(2) Should any enzyme immobilization improve the enzyme features? (3) Is the failure of one support and one reactive group using a specific immobilization protocol sufficient to discard the method to immobilize an enzyme? (4) What can go wrong using a random immobilization system? (5) Is enzyme immobilization always recommended? |
1. Introduction
Enzyme immobilization, defined as confining one or more enzymes in a defined space, was originally developed to solve the problems of enzyme recovery and reuse, during the middle of the previous century.1–4 Protein and enzyme immobilization have been used in both the design of biosensors as well as of biocatalysts. Immobilized enzymes and proteins have also been used in a wide variety of industrial applications, such as the analytical, pharmaceutical, commodity chemicals, food and cosmetic industries, as well as energy production and biomedicine.1,5–9 In biosensors, the enzyme immobilization is often essential, because it is necessary to have the enzyme located in a specific place (e.g., on the tip of a transducer).8,10 However, that may not always be the case if an immobilized enzyme is going to be applied as an industrial catalyst. Here, the use of an immobilized enzyme may not always be essential, other than to fulfil economic aspects of the process.1,5,11,12 Initially, the price of enzymes was incompatible with the single cycle use of enzymes, making convenient or even indispensable the use of heterogeneous biocatalysts to facilitate enzyme reuse to make the process economically feasible.1,5There are many different enzyme immobilization strategies. In some of them, an ex novo solid is constituted after the enzyme immobilization. That is the case for co-polymers, where the enzymes are trapped or crosslinked during the polymerization of one or several co-polymers.13–15 This strategy became quite popular in the last century, and even the company Boehringer commercialized a biocatalyst of Penicillin G acylase using this immobilization method,16 but it is currently scarcely utilized, as the reproducibility of the method is not very high and the mechanical properties of the final biocatalyst, determined by the co-polymerization process, cannot be selected in function of the requirements of the reactor. In a broad sense, as a specific case of enzyme immobilization via co-polymerization the immobilization using sol gel strategies become also very popular, mainly in the area of the biosensors because it may be easily located on the tip of the sensors.17–21 Nevertheless, it is still quite a utilized enzyme immobilization strategy today.22–24 Here the enzyme is coated with a silicate that is formed by polymerization of different polymers, that require a curing step usually coupled to UV irritation or heat, the immobilization is trapped and require the formation of small pores, that generate high diffusional restrictions. Due to the hydrophobicity of these systems, results in terms of activity recovery and stability use to be not very high, in fact some authors propose to previously immobilize the enzyme in a porous support before trapping on the sol–gel matrix.25 Another enzyme immobilization strategy that was developed to produce ex novo solids was the production of cross-linked enzyme crystals, commercialized by the company Altus.26–29 This strategy requires the purification of the target enzyme, its purification and the final crosslink with some bi-functional reagent, giving biocatalysts with a very high mass activity, but quite expensive. Prof Sheldon proposed at the beginning of this century a simplification of the strategy; the cross-linked enzyme aggregates.30 Instead of crosslinking enzyme crystals, he proposed the precipitation of the enzymes to get an aggregate, which after its chemical crosslinking could be utilized under any reaction conditions.31–34 This strategy is compatible with the aggregation of the enzyme with other enzymes (to prepare combi-CLEAs),35–37 polymers (to improve crosslinking or generate enzyme nano-environments)38,39 or magnetic nanoparticles (to facilitate the handling).40–43 A company (CLEA technology) was created to commercialize CLEAs from different enzymes. However, the poor mechanical resistance makes that this biocatalysts must be utilized in reactors that do not submit the particles to mechanical stress, such as basket44–46 or vortex reactors.47 In fact, some researchers have proposed the trapping of the cross-linked aggregates in solids with better mechanical features48–51 Other very popular strategy of enzyme immobilization in ex novo solids is the formation of nano-flowers, where the enzyme is incubated in some metal salts and a metal crystal grows using the enzyme surface as nucleation points.52–58 In some instances, enzyme stability or activity may increase.27,52,55 However, again the mechanical fragility makes complex to use these biocatalysts in industrial processes, and many researcher tarp the nano-flowers in materials with better enzyme features.59–63 In fact, very recently has been proposed to make the treatment on previously immobilized enzymes to have the positive effects of nano-flowers and of immobilization in preexisting supports. The enzymes may be also immobilized in the form of crystals coated by proteins. In this strategy, a highly water soluble compound (or an amino acid, a salt or a sugar) is mixed with the enzyme and drop by drop added to a solvent where its solubility is almost null.64,65 This permits the formation of a crystal in whose surface the enzyme is present. This relative old strategy permits a simple preparation of immobilized enzymes that are only useful under low water reaction systems.66 Metal–organic frameworks (MOFs) have an increased popularity as candidates for enzyme immobilization platform, thanks to their remarkable versatility in structural design of the frameworks and simple surface tunability, Although they can be also previously generated, forming a pre-existing support, and utilized to immobilize enzymes, the enzyme immobilization on MOFs via in situ encapsulation or biomineralization belong to the production of ex novo solids after the enzyme immobilization and deserve to be mentioned67,68 The possibility of establish some enzyme–support physical interactions and tailor the pore size prevent enzyme leakage (except if the support particles are broken during operation).
All this immobilization methods producing ex novo solids are very interesting, but their massive industrial implementation may be complex due to the difficulties to produce them in tons/year, moreover, the mechanical resistance problems that they present constitute a very important drawback that can limit their implementation.
The other alternative to immobilize enzymes is the use of preexisting solids as immobilization matrix. There is a huge amount of available different commercial supports with different mechanical features (flexibility, rigidity, pores sizes, etc.) to immobilize enzymes that may be supplied in hundreds of kg without problems. We will mainly focus on the use of preexisting solids in this review, although many of the points may be extrapolated to this kind of biocatalysts.
In any case, although enzyme immobilization has some impact on the final cost of the biocatalyst, this was compensated provided the costly enzymes could be reused.1 Among the immobilization costs using pre-existing supports, we can include the support cost (including transport to the final user and storage until utilization), the immobilization process itself (reagents, reactor and manpower) and the final disposal of the inactivated biocatalysts (if the support is not biodegradable, which may include storage, transportation and final processing of the inactivated biocatalyst).1,2,5,69 In those cases where the enzyme lost some activity after immobilization, this decrease of enzyme activity could also be considered a cost, as more enzyme or longer reaction time would be required in each reaction cycle.
Today, however, the costs of producing enzymes (at large scale) have been much reduced through microbial strain improvement (by genetic engineering and design of better protein overexpression systems), optimization of fermentation conditions and improved enzyme recovery processes.70,71 Although not all enzyme prices have been so reduced, it may be expected that in the future a further decrease of enzyme price can be produced.70,71 Moreover, of particular interest in recent years, is that other approaches have also been developed to address the reuse of enzymes such as the use of two-liquid (aqueous–organic) systems, where the product is in the organic phase and the enzyme in the aqueous phase.72,73 Following the reaction cycle, liquid–liquid separation allows facile separation and thereby recycle and reuse of the soluble enzyme, provided that it remains active, or could even be run continuously.72–76
Thus, other advantages aside from enzyme recovery and reuse might also be needed today to justify enzyme immobilization. One obvious general advantage of using immobilized enzymes is the possibility of a stricter control of the process, which may be critical in some instances.5,77,78 Examples of such processes where a precise control is required include kinetically controlled synthesis (where maximum yields are transient and frequently over the thermodynamic equilibrium)79–81 and reactions where partial modification of a substrate is required (e.g., production of bioactive peptides by controlled partial hydrolysis of proteins).82,83 Another advantage of using an immobilized enzyme is the prevention of product contamination by the enzyme, which (mainly in food technology) may be very undesirable.5,84 Likewise, for pharmaceutical products, the FDA requires all protein be removed prior to the formulation of small molecule APIs.5,85 Immobilized enzymes may also be used in many alternative reactor configurations5,77,86,87 and, on the path towards lower-priced products, the use of continuous reactor technology, with high enzyme loading, is also of great relevance for the introduction of enzyme immobilization.86,88–93 For example, using immobilized enzymes, it is possible to use high concentrations of enzyme to get an accelerated reaction, because now the risks of enzyme aggregation may be avoided (enzyme in a support may be over 100 g l−1 of packed biocatalyst).88 In this way enzyme immobilization is receiving a renewed interest as an enabling technology in the trend of the transition from batch to continuous processes and further process intensification.72,86,88–95 Examples of process intensification associated with enzyme immobilization include separation unit integration (e.g. integration of enzyme purification and catalyst preparation; in-line product isolation facilitated by easier downstream processing) and reaction intensification (e.g. reaction acceleration by enzyme concentration or by the facilitated use of non-conventional reaction media) can be found in the scientific literature.72,86,88–94,96 Moreover, the recent growth of the so-called flow-biocatalysis field rests in many cases on the off-the shelf application of established immobilization techniques.88,97,98 However, the achievement of these goals still requires going beyond ready-to-use immobilization protocols and the careful design of the immobilized enzyme.88,97 Additionally, enzyme proteolysis (if the enzyme is a protease or if the extract containing the enzyme of interest also contains proteases) may be prevented by immobilization (at least in porous supports).84,99,100 However, these requirements and advantages are not general for all enzymatic processes, and they may not be sufficient to justify the enzyme immobilization in a particular case. Fig. 1 summarizes some of the often-claimed advantages of enzyme immobilization.
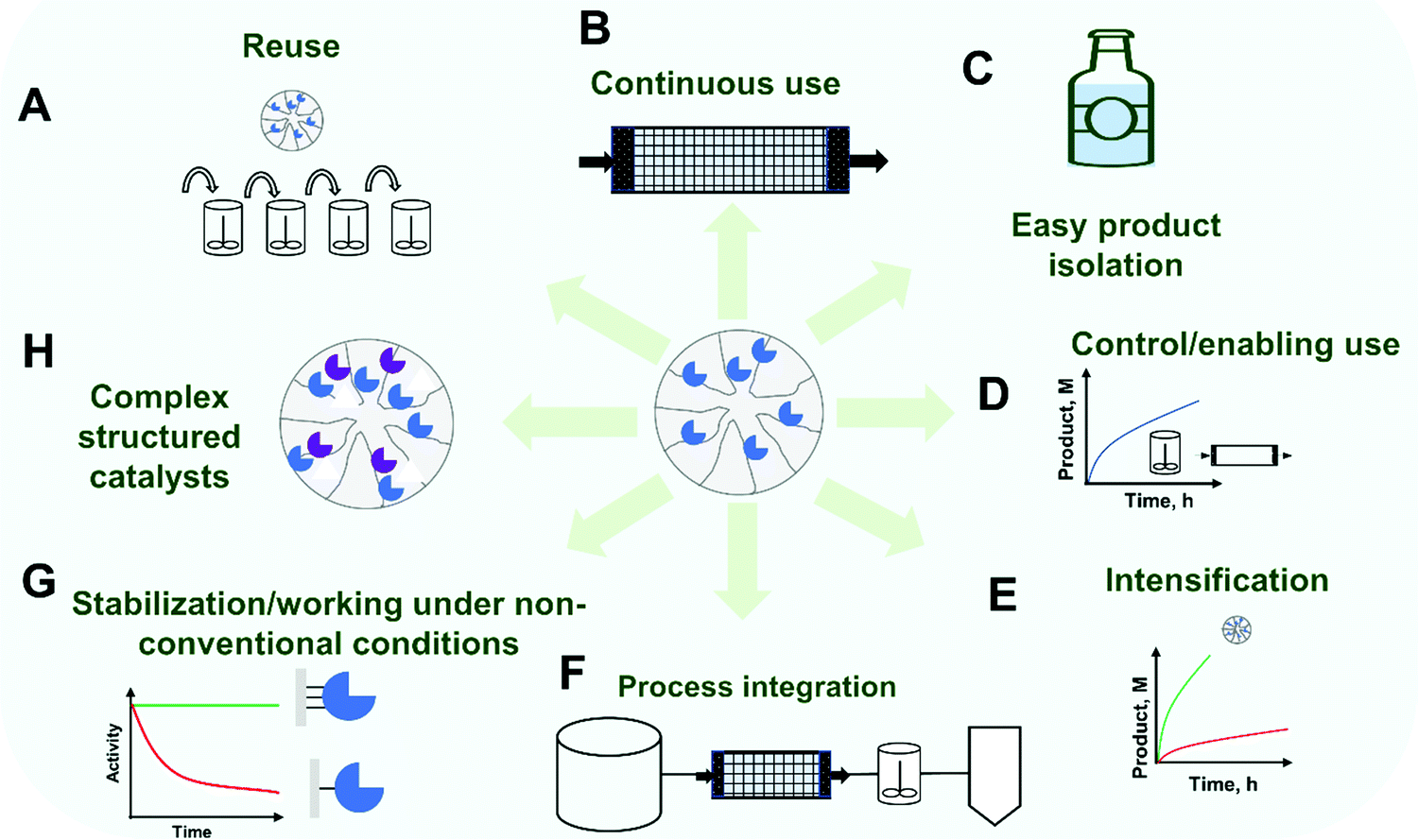 | ||
| Fig. 1 Potential advantages of enzyme immobilization. Enzyme immobilization is often associated with general advantages at different stages of bioprocess and biocatalyst application. Biocatalyst reuse (A), and enabling continuous use (B), are common consequences of supporting the enzyme within a porous support. Downstream processing is also facilitated due to the easier separation of product medium and biocatalyst (C). Other claimed advantages include enhanced design of the bioprocess since immobilized enzymes are usually associated with easier reactor design and application (D), practical intensification of reactions (E), and ease of continuous modular processing (F). Enzyme immobilization is frequently associated with the enhancement of the functional properties compared to soluble, e.g. stabilization (G). Finally, enzyme immobilization is considered an enabling technology for the preparation of multi-catalyst and hybrid chemo-biocatalysts. | ||
Considering the original concept of enzyme reuse to justify enzyme immobilization, many researchers have tried to couple this requirement to the solution of many other enzyme drawbacks (Fig. 2). In this way, it has also been shown that enzyme immobilization, if properly designed, may increase enzyme stability (e.g., via multi-point or multi-subunit immobilization), and thereby not only the enzyme may be used for more reactions cycles, but also the range of conditions where the enzyme can be used may be expanded, leading to better process performance.99–103 In some processes, only the immobilized/improved enzyme is able to catalyze the reaction under conditions (e.g. high temperature, presence of co-solvents, presence of deleterious chemicals) where the product yields, solubility and stability of the substrate/product, or any other reaction parameter are suitable for scale-up and with commercially relevant performance.100,104–106 A recent discussion about the commercial requirements for scale-up highlights not only reaction yield and product concentration as critical, but also productivity and specific yield (which are in a trade-off).107 Immobilization may also be coupled with enzyme purification, as occurs in the case of lipase immobilization on hydrophobic supports and tagged enzymes on their corresponding affinity supports (e.g., using hetero-functional supports).10,108–110 Immobilization may also have other positive effects on certain enzyme features: activity may be increased, selectivity and specificity may be tuned, or inhibition may be decreased.99,101,102,111–113
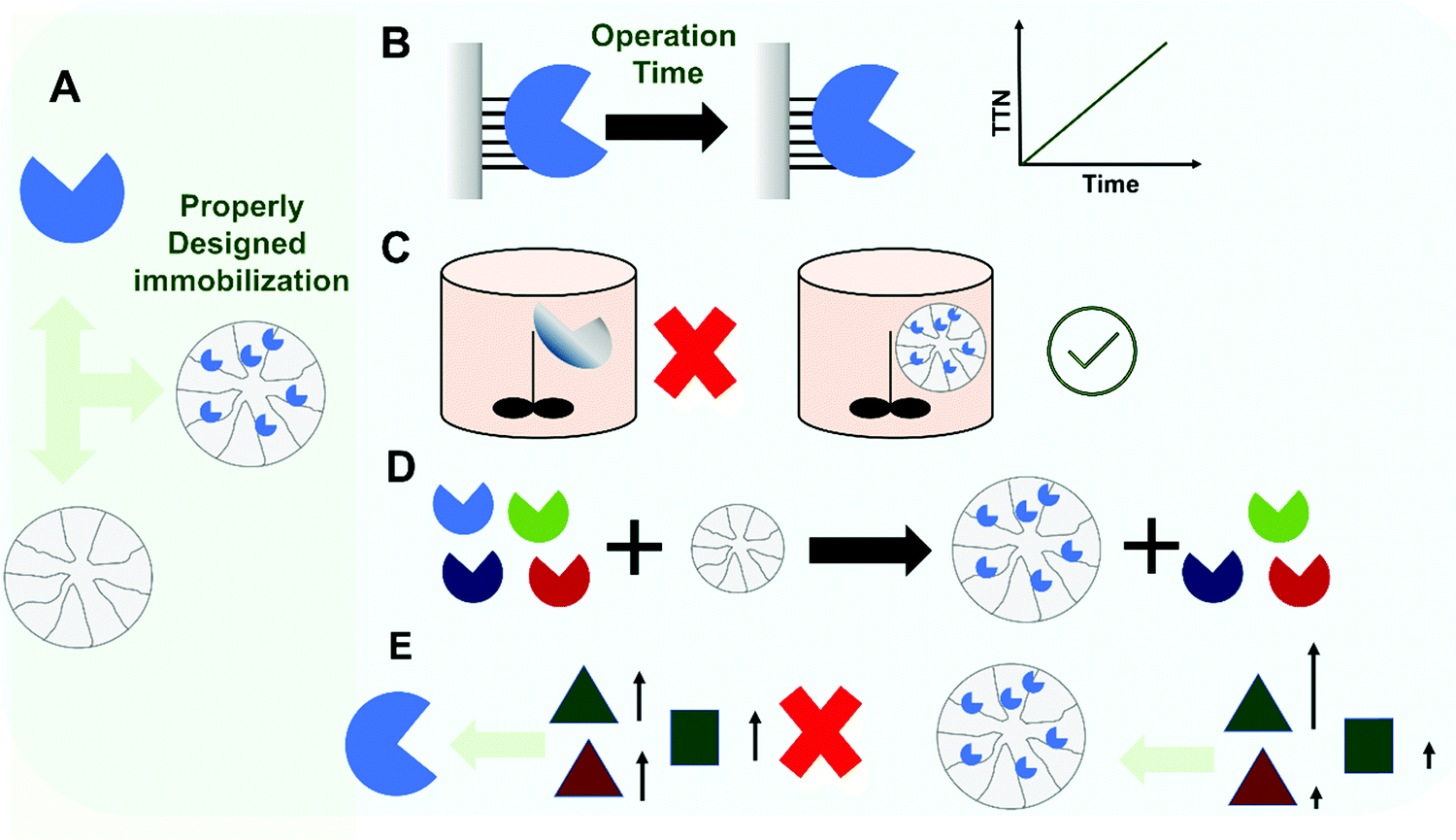 | ||
| Fig. 2 Functional advantages of enzyme immobilization under properly designed methodologies. While enzyme immobilization is often associated with enhancement of the functional properties of the enzyme when compared with the soluble counterpart, this is mainly true in selected cases when the immobilization is properly designed (A). In those cases, different functional advantages can be obtained such as enzyme stabilization enabling the increase of the operational use and enhancing total turnover (B), enabling the use of enzyme under otherwise incompatible reaction conditions (C), enabling one-step purification-immobilization (D), and increasing enzyme reaction specificity (E). | ||
To achieve such results, a library of different biocatalysts using the same enzyme needs to be prepared. The library should be of sufficient size that the possibilities to have a biocatalyst with improved features in a specific process are large enough.101 Furthermore, in many instances, only a properly immobilized enzyme will be able to perform the target reaction in an adequate way.114–118 Hence, immobilization may still be critical to the application of enzymes in many processes.
Moreover, the use of single-enzyme processes has, to some extent, been replaced in recent years by multi-enzyme processes (e.g., for co-factor regeneration and/or cascade reactions).119–125 For instance, it has been demonstrated that the possibility of co-immobilizing enzymes and cofactors enables not only the re-use of enzymes, but also the regeneration and continuous reuse of expensive cofactors.126–130 Many enzymes can be found (or else tuned) to operate under relatively similar reaction conditions (pH, temperature, and pressure) meaning that the linking together of enzymes in this way is much easier than with conventional metal-based catalysts. Indeed, some spectacular industrial examples in the pharmaceutical industry exemplify the power of this approach.125,131–135
Nevertheless, multi-enzyme processes introduce entirely new challenges where controlled spatial and temporal positioning of enzymes becomes of great importance, and dissimilar stabilities of the involved enzymes play a role in biocatalyst re-usability.120 Here, the development of new enzyme immobilization strategies to solve the problem of poorly matched enzyme stability plays a special role and is an area of particular growth.136–140 Enzyme immobilization may also be coupled to other techniques to improve enzyme features, such as site-directed mutagenesis, directed evolution or chemical modification.10,109,141–144 In fact, of special interest are those strategies where site-directed mutagenesis or chemical modification has been developed to improve enzyme immobilization itself.10,141,145–150
However, these techniques may in some cases produce more active soluble enzymes, which may inadvertently give greater challenges for enzyme immobilization, if they are not properly considered.151–153 Thus, an aspect often overlooked is that protein engineering has been used to improve the specific activity of many enzymes.154 This is of course positive, but also means that the risks of diffusional limitations (including substrate and/or pH gradients, see later in this review) are increased when enzymes are immobilized in porous supports.151–153 This represents a new reaction engineering challenge, but one where the design of new immobilization strategies can play a significant role.
Enzyme improvements that can be achieved following immobilization may be a good justification for investment. However, all these advantages, even the most general ones, will only be achieved if using properly designed immobilization protocols. Thus, aside from the challenge of having to design specific immobilization protocols to match the requirements of a given process, there are also several new scientific issues of significant relevance, which today require investigation. Fig. 3 illustrates some potential problems of enzyme immobilization that will be discussed in the following sections.
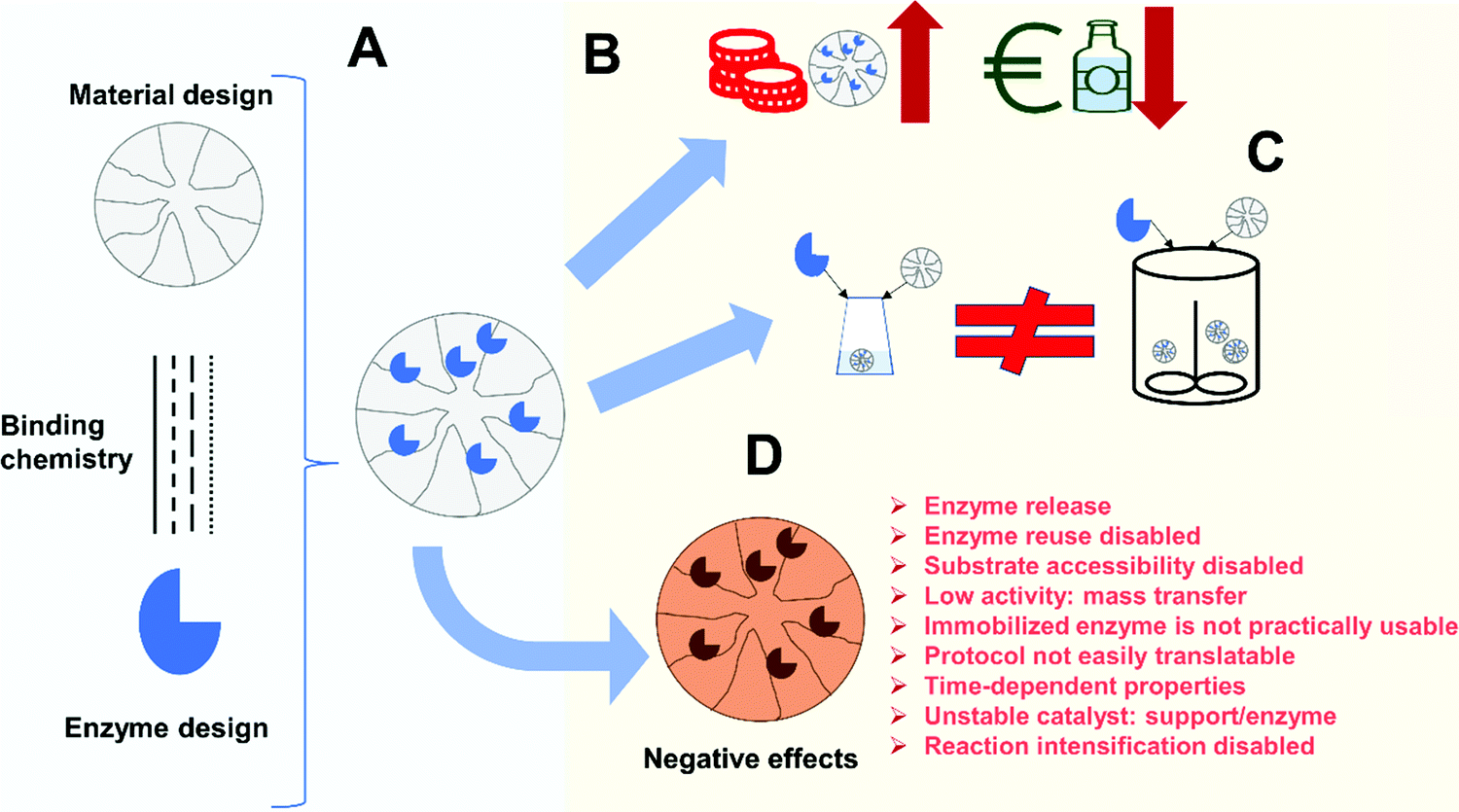 | ||
| Fig. 3 Potential problems of a poorly designed immobilization protocol. (A) Enzyme immobilization design is a multi-parameter process design that involves selection of material, chemical or protein engineering of the enzyme as well as immobilization chemistry. Problems for the implementation can be due to: (B) cost of the biocatalyst; (C) difficulty of scale-up of the immobilization protocol to industrial scale. (D) Negative effects on the biocatalyst performance. | ||
2. Costs of enzyme immobilization
Before starting with the immobilization studies of an enzyme for a specific process, we should consider if the inevitable costs as a result of immobilization justify the potential benefits1,2,77,155–158 (Fig. 3B).As described previously, one of the costs of the enzyme immobilization is the price of the support and of the immobilization process itself.69,159 In this way, one of the common statements in the scientific literature is that the immobilization protocol, and mainly the support, must be cheap, to the point that in some instances this fact is even mentioned in the paper title.160–163 Unfortunately, in many instances this is stated without any real economic balance. Likewise, sometimes important factors are not considered. For example, the loading capacity of the support may be critical to determine the real support costs. With respect to this parameter, a support able to immobilize only 5 mg of enzyme per g of support is really much more expensive than a support that costs 3-fold as much, but is able to immobilize 50 mg enzyme per g of support. For example, agarose beads can immobilize a mass of enzyme even higher than the mass of solid in the bead.164 Preparing a biocatalyst with low enzyme loading will also produce some additional problems for the reactor, since it will require a high mass of support per volume of reactor. For a stirred reactor this may be limited dependent upon the type of mixing (usually to around 10% v/v).155 Similarly, if a proper immobilization system makes the enzyme 5-fold more active than the soluble enzyme in a given process, one can reduce the fraction of solids in the reactor. This increase in enzyme operational activity can occur if the immobilized enzyme is used under extreme conditions and the enzyme stability is improved by the immobilization. In such cases, the immobilized/stabilized enzyme may have more activity than the free soluble enzyme, because it is not disturbed by the severe conditions, unlike the soluble enzyme103,165,166 or if the enzyme adopts a conformation with higher activity than the free soluble enzyme.167 Here the final price will be lower than other cases where an immobilization process (and support) is cheap but did not improve enzyme activity under the reaction conditions. If the enzyme stability is improved by a specific immobilization strategy in the target process and conditions, the increase in the operational stability should also be considered, although this may reach a limit. This limit will be fixed by each specific factory configuration, since at some moment the reactor will need to be stopped, washed and reloaded, even if some enzyme remains active. In fact, enzyme stability may be better considered by mass of product produced per unit of biocatalyst than by considering the traditional temporal half-live.168 Hence, the selection of a proper immobilization system is not a trivial task, even from the economic point of view alone.
If enzyme immobilization is essential, or it has been decided to reduce the cost of the process by reusing immobilized enzymes, the balance between product added value and enzyme costs must be evaluated. If the price of the product (or even the substrate) is very high, this may be produced best by soluble enzymes, provided they have sufficient activity (i.e. the enzyme contributes a small fraction to the final product cost). This is often the case in the pharmaceutical industry.1,5 However, if the immobilized/improved enzyme alone is capable of carrying out the desired reaction at the desired yields and reactor productivity, then immobilization becomes a necessity.80,81,104 In this situation, the enzyme engineer has an advantage: the allowable cost of the immobilization may be quite high, as this will be just a small fraction of the price of the product produced. For example, in the case of biosensors, since the amount of immobilized protein required per assay may be in the nanogram scale, even very sophisticated immobilization protocols can be used, provided a good biosensor performance can be justified.169,170 Likewise, in biomedicine, the costs of immobilization become relatively small compared to the final benefits, and the use of sophisticated protocols may be justified if they are the only way of getting the desired result (low toxicity, high bio-stability, low immune response). For example, the use of the expensive genipin as a support activator reagent is justified by its lack of toxicity in these instances,171–174 while the use of genipin in industrial biocatalysis would be much harder to justify.175 In fact, as the price of products which industry could consider producing using sustainable technologies like biocatalysis reduces, the need for effective reuse and recycle of enzymes increases, in order to keep the cost contribution of the enzyme to the final product low enough.12,158,176 In this way, enzyme immobilization maintains its relevance today.
If the added value of the final product is low (e.g., bulk chemicals, biofuels or many food products), all costs are important and the enzyme immobilization, if it permits the reuse of the enzyme for many cycles, may improve the competitiveness of the process.156,158,176 Any concomitant improvements to the enzyme regarding its stability or increased activity via immobilization are also of particular relevance for these low-priced products. However, in this case, the immobilization will be limited to simple (and cheap) protocols, since the allowable cost of the biocatalysts cannot be excessive. In this case, if only a very sophisticated immobilization protocol permits preparation of a biocatalyst with the desired performance, this will prevent the use of that immobilized enzyme, and necessitate the search for alternative enzymes that have intrinsically better properties. If Nature did not provide a suitable enzyme, the properties of the available enzymes may be improved by site-directed mutagenesis or directed evolution to a level where the enzyme features are good-enough for the particular process.11,143,144,177,178 The improved enzyme may then be immobilized, if immobilization is the only way to reuse it. The potential of the new techniques to improve enzyme properties is truly impressive and growing all the time.179–185 This requires an initial investment, but later the improved enzyme may be produced at a low cost. A proof of the potential of these new techniques is the design of artificial plurizymes (bearing two different biologically active centers),186 and how, one of these active centers can be modified using a selective irreversible inhibitor coupled to an organometallic catalyst. This way, an enzyme is generated bearing two active centers with entirely different properties, useful to catalyze cascade reactions.187 Likewise, enzyme stabilization via protein engineering has proven very effective in many cases.144,177,188–190 However, these alternative approaches do not mean that enzyme immobilization is irrelevant, especially considering the possibility of integrating immobilization with other enzyme stabilization techniques.10,141,145–150 In order to ensure reasonable enzyme immobilization costs, deeper investigation into enzyme immobilization is required, often on a case-by-case basis.
3. Enzyme immobilization: large-scale versus small-scale
When defining an enzyme immobilization protocol, the researcher/engineer should consider the ultimate scale of the process (Fig. 3C), always keeping the end use of the biocatalyst in mind. If we need to design an immobilization protocol to prepare 1 kg of a biosensor (that may be valid for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 assays), the jump from the laboratory to industry may be relatively simple. However, when we need to produce many hundreds of kgs of biocatalyst, the change of scale from the laboratory to industry necessitates suitable large-scale immobilization protocols.99,159
000000 assays), the jump from the laboratory to industry may be relatively simple. However, when we need to produce many hundreds of kgs of biocatalyst, the change of scale from the laboratory to industry necessitates suitable large-scale immobilization protocols.99,159
One critical point is the stability of the functional groups in the support. Using small volumes of enzyme and mass of support, the mixing of support and enzyme may take seconds, and even a relatively unstable active group in the support may be suitable to immobilize the enzyme. On the other hand, using thousands of liters of enzyme and hundreds of kilograms of support, the mixing can take a relatively long time, and a support activated with unstable groups will be inappropriate.159
Another problem is the toxicity of some of the more hazardous reagents. If the factory is specialized in enzyme immobilization, this may be a minor problem, as the engineers will be familiar with the management of these kinds of compounds, and they will have all the required licenses and security precautions in place. For example, a factory producing activated agarose beads will be familiar with the management of sodium borohydride and sodium periodate,164,191,192 being able to immobilize enzymes in glyoxyl agarose without serious logistical problems. However, if the factory is devoted to a biotechnological process using the immobilized enzyme, and the only immobilized enzyme that it produces is the one in question, the requirements to use certain reagents may be excessively complex and may prevent the use of this particular immobilization strategy.
The complexity of the immobilization process itself is also something to be considered. Using some few milliliters, steps for the washing and change of the reaction conditions can take seconds, while in a reactor of thousands of liters, this may take many minutes. In this way a protocol that involves many quick changes in experimental conditions may be inadequate for large-scale production, although well-suited for a small-scale laboratory protocol. Moreover, at the small-scale, the handling of solid suspensions is relatively easy and keeping well-mixed protein/support suspensions is readily achieved. In contrast, maintaining well-mixed suspensions of protein and solids at a large-scale requires consideration of the mechanical stability of the support material and the characteristic mixing time, especially in cases of quick immobilization protocols. Hence, there are clear differences between the requirements for large-scale and small-scale immobilization processes. Perhaps for this reason, enzyme immobilization via simple physical adsorption is often preferred at large-scale for immobilized enzymes, even with the inherent risk of enzyme leakage during operation that this may have.193 Among the covalent protocols, epoxide activated supports are preferred due to their simple application (they are very stable).194–199 This popularity is despite the fact that the necessary final blocking step of the remaining epoxy groups in the supports is often ignored. This blocking step can make the final immobilization process still more complex, but may be necessary in some instances, as we will discuss later.200
On this basis, research on enzyme immobilization should be focused on how to convert these “easy-to-use” supports and techniques into potent tools to improve enzyme features (e.g., by separating immobilization and incubation steps),84,90 although this may not have the same academic impact as a sophisticated new immobilization protocol. Sophisticated enzyme immobilization protocols, even if they can greatly improve enzyme features, are nowadays of interest mostly for small-scale implementation (or for very high-priced products) and academia, while awaiting engineering developments that can make feasible their application at a larger scale.
4. Negative effects of an inappropriate enzyme immobilization on biocatalysts performance
Immobilization, considered as the incorporation of an enzyme in, or on, a porous solid support, is a relatively simple process. Most of the solids are not physically inert and therefore most of them will be able to immobilize enzymes.159,201,202 Ironically, a real problem is how to prevent enzyme fixation to solid surfaces when this is not desired.170,191,203–207 In fact, such unspecific and uncontrolled adsorption of proteins to solids becomes a problem in many instances, such as in immuno-biosensors, the use of membrane reactors and also medical implants.159 Although the scientific literature is full of reports about new supports and new protocols to immobilize enzymes, most of them are inadequate for improving enzyme features by immobilization, and many of them are not even suitable for proper enzyme immobilization.Enzyme immobilization inside a porous support may affect the enzyme expressed activity due to different causes;
– Enzyme distortion. In most instances, the interaction between enzyme and support can lead to some enzyme distortion, which usually will produce negative effects on enzyme activity, but in certain cases may produce an enzyme hyper-activation. This may be due to interaction with the support reactive groups (e.g., during multipoint covalent immobilization) or by interactions with the matrix.101,208
– Diffusional matters (substrate, product, pH gradients). The enzymes will be in a confined space and the high activity can produce some diffusion matters.101
– Steric hindrances. If the substrate is large enough, only the active center of properly oriented enzymes will be accessible for the substrate, making that enzyme orientation plays a critical role on the final biocatalyst performance.101
All these facts should be considered on all the topics treated in this review, and, as it will be discussed, may be altered as function of the substrate, enzyme loading or support internal morphology.
Today, few would try to improve enzyme performance solely by performing a single random mutation on an enzyme without proper protein modelling and enzyme dynamics studies to guide the work,187,209,210 but many researchers still expect that any immobilization should lead to a significant improvement of the enzyme features. And when one immobilization protocol fails, the conclusion is that “immobilization” is not adequate for this enzyme, for this reaction or for this process. The actual situation is that an improperly designed immobilized enzyme biocatalyst may have a far worst performance than the soluble enzyme, including lower activity and stability.200,211,212 And it may even be that retention of the immobilized enzyme under the required operational conditions is not achieved, as will be discussed later.
Enzyme immobilization has been extensively used since the 1960s for a range of industrial processes,1,4,5,69,213,214 and some researchers even argue that the majority of work on immobilization has already been done, and there is little left to investigate. In our opinion, the research on enzyme immobilization is still very much necessary, as we are far from understanding many of the phenomena occurring at the level of enzyme–support interactions. Most methods that are acknowledged as valid protocols to improve enzyme features have some limits that prevent their universal application, and there remains a lack of an “ideal” support at a reasonable price. As previously discussed, although the enzyme cost is nowadays lower, still reuse, recycle and retention of enzymes are in many cases relevant and in many cases essential to ensure economic viability of a process. Likewise, in the vast majority of cases, the tuning of enzyme features is also a key to achieve successful enzyme performance. Enzyme immobilization is clearly one way to do this, and thus enzyme immobilization remains of key relevance today.
In this way, this paper, in contrast to many other papers in the scientific literature that review how a proper enzyme immobilization may lead to very good results, will list some of the most common drawbacks that an uncontrolled immobilization can produce (Fig. 3D). That is, how after the effort and extra-cost of enzyme immobilization, the produced biocatalyst may offer a more limited performance than that of the equivalent soluble enzyme, at least in certain aspects. Where possible, solutions to these drawbacks will also be presented. Fig. 4 summarizes the main content of this section.
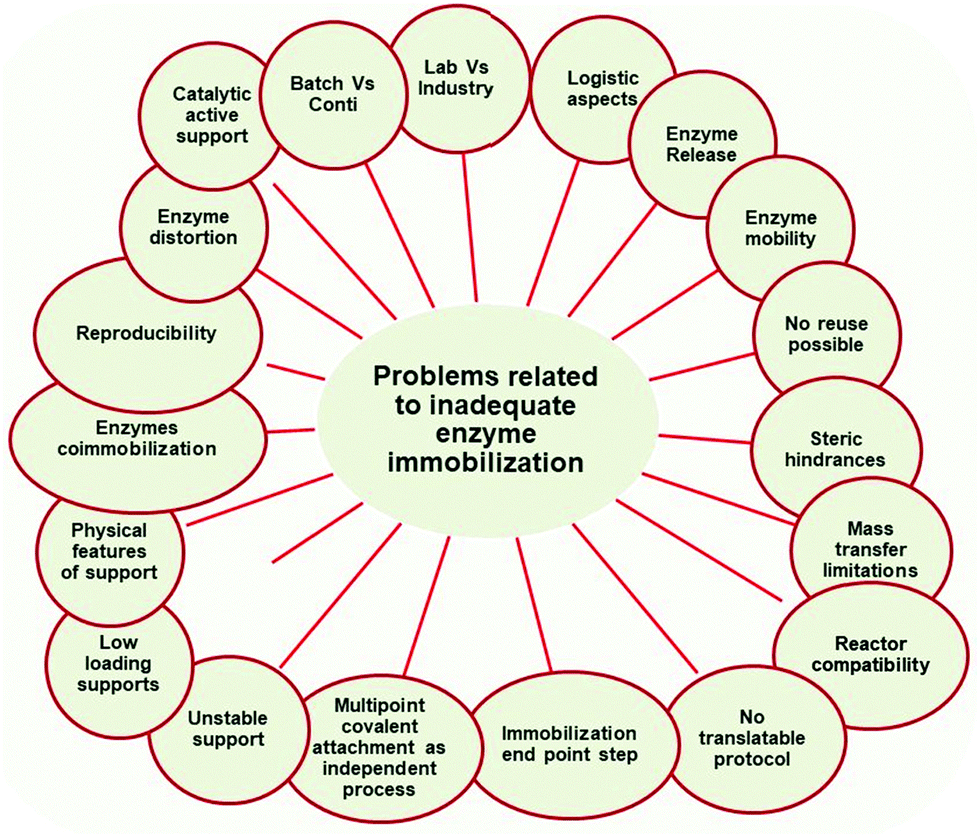 | ||
| Fig. 4 Pitfalls of inadequate enzyme immobilization treated on this review. | ||
4.1. Enzyme release from the support
As stated in the introduction, the first goal of an immobilization process is to solve the problem of enzyme recovery and thereby its reuse in multiple reaction batches or its retention for continuous processing.1–4,72,86,88–95 Hence, a first requisite of immobilization is that there is no risk of enzyme release during operation (Fig. 5). Researchers are often aware of this possibility when using physical immobilization protocols2,69,99,108,110,202 (Fig. 5A). For example, using ion exchange to immobilize an enzyme, changes in the pH value or ionic strength in the medium may facilitate enzyme release. This is very relevant in some cases. For instance, when hydrolyzing an ester, an acid is released and this results in a decrease in pH. In many cases, the pH is not controlled during the reaction because this causes additional problems (e.g., formation of soaps in hydrolysis of oils or fats catalyzed by lipases),215,216 meaning a drop in the final pH value in the reaction medium. This can be problematic if the researcher checked enzyme release under initial conditions alone.202 Even if the pH is controlled during the reaction, if the enzyme volumetric activity is high enough and the support particle is large enough, an internal pH gradient may be formed inside the biocatalyst particle217–223 (this will be specifically discussed later in this review), and this may also facilitate the release of enzyme molecules located in the inner part of the particle.224,225 Furthermore, the ionic strength attributed to the substrate and the product may be very different. That can be the case in the hydrolysis of amides, for example. Amides and their hydrolytic products (acid and amine) are very different. Therefore, while it can be confirmed by the researcher that the enzyme remains immobilized under the initial conditions, the progressive increase of the concentration of the reaction product may favor enzyme release in the final stages of the reaction. Hence, an apparently “optimal” ion exchange immobilized enzyme under initial conditions, may be released later, naturally affecting the operational stability of the biocatalyst.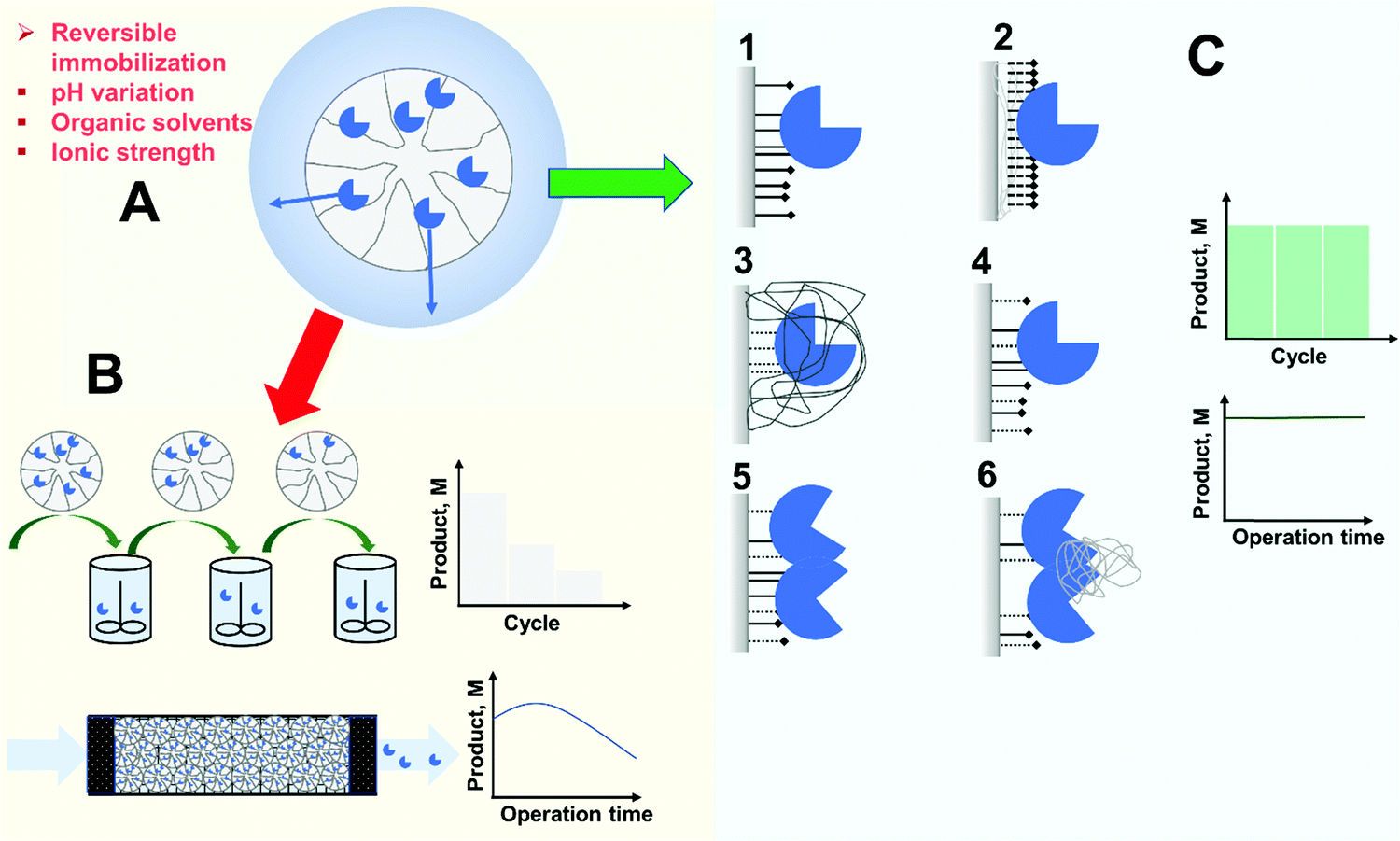 | ||
| Fig. 5 Enzyme release from support: problems and solutions. (A) In reversible immobilization enzyme release can occur under certain circumstances. (B) Enzyme release prevents the reuse or continuous use of the enzyme, resulting in product contamination and a decrease of reactor productivity over time. (C) Immobilization engineering can overcome enzyme release ensuring enzyme reuse and stable productivity. Strengthening of the immobilization can be based on reinforcing enzyme–support interactions (1), using polymeric beds (2), crosslinking of the adsorbed enzyme (3), use of hetero-functional supports (4), ensuring binding of subunits in multimeric enzymes (5), cross-linking of subunits in multimeric enzymes. | ||
Similarly, lipases immobilized on hydrophobic supports via strong interfacial activation adsorption may also be released under some circumstances.108 Such lipase preparations may be used at moderate concentrations of organic solvents and at elevated temperatures, but due to the hydrophobic nature of the enzyme–support interactions, they may be released at high concentrations of some hydrophobic organic solvents or at very high temperatures.226 This is already well known and may be investigated in the development of the biocatalyst. However, even if the enzyme remains fully immobilized under initial conditions (medium, temperature, substrates), the reaction products may favor their release from the support. That can be the case if the product(s) have some detergent-like properties.227 In hydrolysis of oils and fats, mono- and di-glycerides as well as free fatty acids are all likely to generate such problems, but it has been shown that this may be caused also with less obvious molecules, such as dibutyrin.228,229
Released enzyme molecules will contaminate the reaction media (one of the features we try to prevent by enzyme immobilization)5,84 and disqualify the enzyme from reuse (the main feature we try to enable by enzyme immobilization), because the operational stability of the biocatalyst may be poor even if the initial assessment gave promising indications of stability (Fig. 5B). Where the released enzyme remains fully active in the product solution, reaction control is lost, which would otherwise be an advantage of immobilization as commented previously.
Another problem generated by the possibility of enzyme release is that this prevents exploitation of immobilized enzymes in aqueous biphasic systems. Historically, these reaction systems were formed from solutions of moderately hydrophobic polymers mixed with salts or hydrophilic polymer solutions,230–234 and nowadays include the use of ionic liquids235,236 and other compounds.237 They are especially suitable for extraction of hydrophilic compounds, where conventional organic solvent-aqueous biphasic systems cannot be employed.238–243 By extracting the reaction product, the concomitant product modification may be reduced (e.g., hydrolysis of the product in kinetically controlled synthesis) or the reaction equilibrium shifted to the product.237–241,244 In such systems, the soluble enzyme may be in both phases although not at identical concentrations, since it may also suffer partition. In fact, such biphasic systems may even be used for protein fractionation and purification.245,246 However, using immobilized enzymes, the filling of the biocatalyst pores with one of the solutions means that the biocatalyst remains in that phase by capillary action.244 This permits location of the enzyme in one phase while extracting the product in the other. However, this extraction will be not so advantageous if the enzyme is released from the support and some enzyme activity is also in that phase.
In such cases, there are several solutions (Fig. 5C). In the case of ion exchange, the use of an optimized support coated with an ionic polymeric bed has proved to be a way to make the enzyme adsorption stronger, reducing release108,193,211,247 (Fig. 5C). In the case of lipases, a more hydrophobic support may increase the strength of the enzyme adsorption, reducing leakage.108 A more sophisticated strategy may be chemical crosslinking of the immobilized enzyme molecules; using polymers (e.g., dextran aldehyde)203,248–251 or even just glutaraldehyde193 (if the enzyme molecules are close enough to each other, as frequently found when the immobilization rate is very high).252 This also helps to prevent enzyme release (Fig. 5C). The researcher may also utilize some hetero-functional supports (supports with some groups bearing chemical reactivity added to the adsorbent groups) to ensure some enzyme–support covalent bonds110,226,253–256 (Fig. 5C). However, such chemical groups will make the immobilization process more complicated and may prevent the enzyme release when the enzyme becomes inactivated, disqualifying reuse of the support. One alternative is to use ionic polymers to achieve physical intermolecular crosslinking,257 or the use of ionic/hydrophobic groups to immobilize in a mixed way the enzyme molecules.258–260 These strategies make enzyme release more difficult (while not fully preventing it), are simpler and do not preclude the reversibility of the enzyme immobilization, allowing supports to be reused.
However, the main problem of enzyme release is when it is supposed, and assumed, by the researcher that the immobilization is irreversible, but actually it is not, e.g., using stable covalent bonds, when that is not actually the case. A first example is when immobilizing a multimeric enzyme. If the enzyme is immobilized via just one subunit, even using an irreversible bond, those subunits that are not involved in the immobilization may be released into the medium.261,262 If the enzyme subunit is released after enzyme inactivation, the main effect will be a contamination of the final product by the inactivated protein, meaning that the immobilization did not fulfill this objective. However, if the enzyme subunit is released in the context of association/dissociation equilibrium, the problem is more serious. The immobilized enzyme will then be inactivated by washing a fraction of the released enzyme subunit from the support, and the product will be contaminated by active enzyme.261 Even worse, the enzyme subunits may associate in the reaction solution, continuing product modification in an uncontrolled way. To prevent this, first it is necessary to check if all enzyme subunits of multimeric enzymes are, or are not, attached to the support (e.g., by using SDS-PAGE),262 and if not, it is necessary to perform a further chemical (effective although more complex)203,263 or physical (simpler although reversible)193,264,265 step with suitable polymers to promote inter-subunit enzyme crosslinking262,266 (Fig. 5C).
Another problem arises when a covalent immobilization protocol is utilized, but the researchers are unaware of the fact they are using a hetero-functional support. For example, it is possible that the support has a hydrophobic nature, and the reactive chemical groups are over its surface. Using lipases in such a case, the enzyme will very rapidly be immobilized via interfacial activation, even if no covalent bonds are formed.108 Later, after some time, some covalent bonds may be formed (or not). That way, the fraction of enzyme molecules that are only physically immobilized may be released from the support during operation. This may be prevented if the researcher makes a final study on the possibilities of enzyme release under severe conditions, (e.g., by checking the release of enzyme molecules from the support by SDS-PAGE after boiling the biocatalyst in breaking buffer solution226,253–256).
One more general case is if an aminated support activated with glutaraldehyde is used.103,248,267–269 These supports have ionic and hydrophobic features, as well as chemical reactivity.248 Usually, it is assumed that the chemical reactivity is the most important driver of immobilization on this support, but it has been shown that this is not in fact the situation in the majority of cases. Ionic exchange (or interfacial activation in the case of lipases) is far more rapid than covalent immobilization on such supports.103,108,212,248,267–271 This does not mean that after the physical enzyme immobilization some enzyme–support covalent bonds may be not formed, but simply that enzyme immobilization per se does not mean that covalent bonds have been made between the enzyme and the support.198,272,273 Hence a fraction of merely physically immobilized enzymes may be released from the support during operation, with the consequent negative effects discussed previously. A proper study may prevent this by optimizing the covalent immobilization, and if 100% covalent immobilization is not possible, and the release of some enzyme molecules during operation is a real problem, then the researcher should consider the possibility of washing the biocatalysts under conditions where the non-covalently bound enzymes can be deliberately released. The released enzyme molecules could even be used in future preparations of new immobilized biocatalysts, but in any case this will have a cost in terms of volumetric activity of the produced biocatalyst. However, awareness of the multi-functional possibilities of the support will increase its versatility and, that way, the possibilities of designing an optimal immobilization protocol for each enzyme.103,108,212,248,267–271 The real problems arise when hetero-functionality is unknown or is neglected.110
Deliberately using hetero-functional supports, the fact that the first immobilization step is via physical adsorption is already well-known and researchers can easily adopt measures to prevent enzyme release by ensuring that all enzyme molecules are covalently attached to the support.110,198,214 Using hetero-functional supports bearing acyl groups and glyoxyl groups to immobilize lipases, options exist to wash the support with detergent (to eliminate non-covalently bound enzyme),226 or to change glyoxyl groups by the much more reactive vinyl sulfone moieties, able to covalently immobilize in all studied cases 100% of the adsorbed enzyme.256
In other cases, the enzyme–support bonds may be strong but not irreversible. That is the case for immobilization of enzymes using thiol disulfide exchange.110,274–280 The disulfide bonds may be broken under reducing conditions, or if the media is able to oxidize the disulfide, or even if there are compounds in the reaction media bearing thiol groups.53,219–222 Using cyanogen bromide supports, the enzyme is immobilized via several kinds of bond, and some of them are reversible, enabling enzyme release.281–286 Enzyme immobilization via interaction of mainly His groups belonging to the enzyme (but also Cys, Tyr and others) with immobilized metal chelates may also have problems,287–291 since the interaction may be broken at certain values of pH or if the medium contains particular compounds (e.g., His free amino acids or contained in peptides if using immobilized proteases to hydrolyze proteins). This means that these immobilization protocols should be used only after considering all the risks.
One outstanding case is the immobilization of enzymes on glyoxyl supports191,192 (and the problem may be extended to many other aldehyde activated supports, but not to glutaraldehyde, that gives stable and unreactive cycles after some time191,192,214,292,293). The enzymes are immobilized on the glyoxyl support via very weak imine bonds, and the enzyme is only immobilized on the support via several enzyme–support bonds.191,192 Usually, this requires the immobilization to be carried out at pH 10 or above.191,192 However, enzymes containing several terminal amino groups may be immobilized even at pH 7.294 After multi-point immobilization, the enzyme remains in the glyoxyl support even at neutral pH values. However, the enzyme may be released from the support if exposed to high temperatures or high concentrations of compounds containing aminated moieties, making the glyoxyl method apparently unsuitable for enzyme immobilization.295 However, if the weak imino bonds are reduced to highly stable secondary amine bonds using borohydride191 (or some alternative reducing reagent if this is deleterious to the enzyme),296 the enzyme remains immobilized on the support under all experimental conditions, and this support becomes one of the most suitable to get enzyme stabilization by multi-point covalent attachment.100 In this way, not only the immobilization protocol is relevant, but also the reaction end point may be of importance for the suitability of a given immobilization method (as will be discussed later).
4.2. Mobility of immobilized enzymes on the support surface
The enzyme position on the support surface is important for the enzyme expressed activity. The challenges that theoretical and computational methods encounter to achieve accurate descriptions of the physical, chemical and mechanical properties of protein-surface systems makes this a very hard task, The use of multiscale modelling to model these process was reviewed in 2016,297 concluding that the orientation of proteins when they are adsorbed on ionic surfaces or hydrophobic surfaces can be controlled by the electrical dipole and the hydrophobic dipole of proteins, respectively. Tailoring the medium conditions, the properties of the adsorbing surfaces can be altered, and thus, the adsorption/desorption of proteins can be controlled.297 More recently, it has been stated that it should be considered that the protein-surfaces adsorption events can take from nanoseconds to days, and from nanometers to micrometers. And this renders the use of multi-scale approaches unavoidable.298 Alterations in the atomic structure of the adsorbing surfaces can lead to surface reconstruction. Furthermore, changes in the structure of proteins can result in complete denaturation of the adsorbed molecules. These facts can many intermediate structural and energetic states that makes very difficult to make an appropriate sampling. The applicability of techniques such as quantum mechanics through all-atom molecular mechanics, coarse-grained approaches, etc. and the free energy calculations. Additionally, sampling methods can have an important role in the accuracy of the final model.298 This should determine the area of the enzyme that will be in contact with the support, that is, the area of the protein that will have more steric barriers top interact with the medium, the most distorted and the most rigidified (if the enzyme is multipoint covalently immobilized).When the enzyme is covalently immobilized on the support surface, the enzyme molecule will have fixed its position on the support surface. If the enzyme is immobilized via just one point, the enzyme may have some freedom of movement around this point. If the number of bonds is 2, the only possibility of enzyme mobility will be back and forth in the axis formed by the two points. However, when the number of bonds is 3 or more, and they are not aligned linearly, they will form a plane and enzyme movement on the support surface will not be possible.
Nevertheless, this is not the case for reversible immobilization strategies where enzyme may even be released from the support. If enzymes can be released from the support, it is not impossible that reversibly immobilized enzymes can passively migrate on the support surface as a consequence of adsorption/desorption and surface diffusion mechanisms.299–303 These movements of the enzyme on the support surface have been recently confirmed.304 These movements may be relevant when the researcher tries to get some order in the immobilization, e.g., when trying to get concentric crowns of co-immobilized enzymes.305–309 This may be more relevant as some recent reports show that the distribution of some immobilized enzymes on the support surface has a clear effect on their final catalytic performance (Fig. 6). In a first example, advanced single-particle analysis was used to understand the effect of O2 and NADH on the activity of immobilized NADH oxidase (NOX). The determination of the intra-particle O2 showed that the enzyme that was immobilized on the outer surface of the particle has more available O2.130 Another example uses several cofactor-dependent enzymes. The enzymes were co-immobilized with their respective cofactors to give a self-sufficient biocatalyst. In order to check the activity shown by different distributions of the enzyme on the support surface, several techniques were used including image analysis at both intra-particle and single-particle levels, time-lapse fluorescence microscopy and fluorescence recovery after photo-bleaching measurements. The research revealed the relationship between the apparent Michaelis–Menten kinetic parameters of the involved enzyme and the enzyme spatial organization and enzyme density in the confined space, suggesting a negative effect of enzyme molecular crowding on the performance of some of the studied enzymes.310 The possibility of enzyme migration on the support surface may alter these effects.304 Such enzyme migration would be hampered if using fully loaded biocatalysts, although the spaces between the immobilized enzymes may still permit some enzyme mobility, even in this situation. As this research is very recent, it may be expected that some exciting phenomena, with significant implications for designed enzyme features, will be forthcoming in the future.
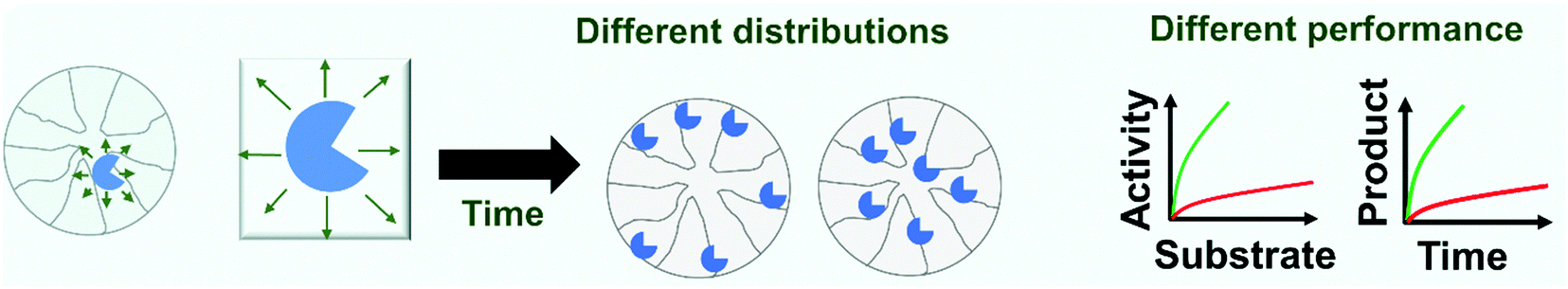 | ||
| Fig. 6 Effects of immobilized enzyme distribution on enzyme performance. When reversible immobilization is used, surface diffusion and intra-pore transport can occur obtaining time-dependent catalyst distribution across the particulate catalyst. From the perspective of performance, different values of activity or operational stability can occur in reactions controlled by substrate-transport into the particle. | ||
4.3. Impossibility of biocatalyst reuse for different reasons
In some instances, the enzyme remains immobilized, even fully active, but the reuse of the biocatalyst may not be straightforward311 (Fig. 7). This is the case when using immobilized enzymes in a medium where the final product is a suspension (even if the substrate is fully soluble).84,312 If the medium contains solids, the recovery of the biocatalyst by filtration will not be possible (e.g., treatment of fruit juices, milk, etc.)41,313 (Fig. 7A). The alternative in these instances is the use of magnetic supports (it may be a porous macro-support or a non-porous nano-particle).41,314 This way, the biocatalyst can be recovered by exposing the outlet of the reactor to a magnetic field.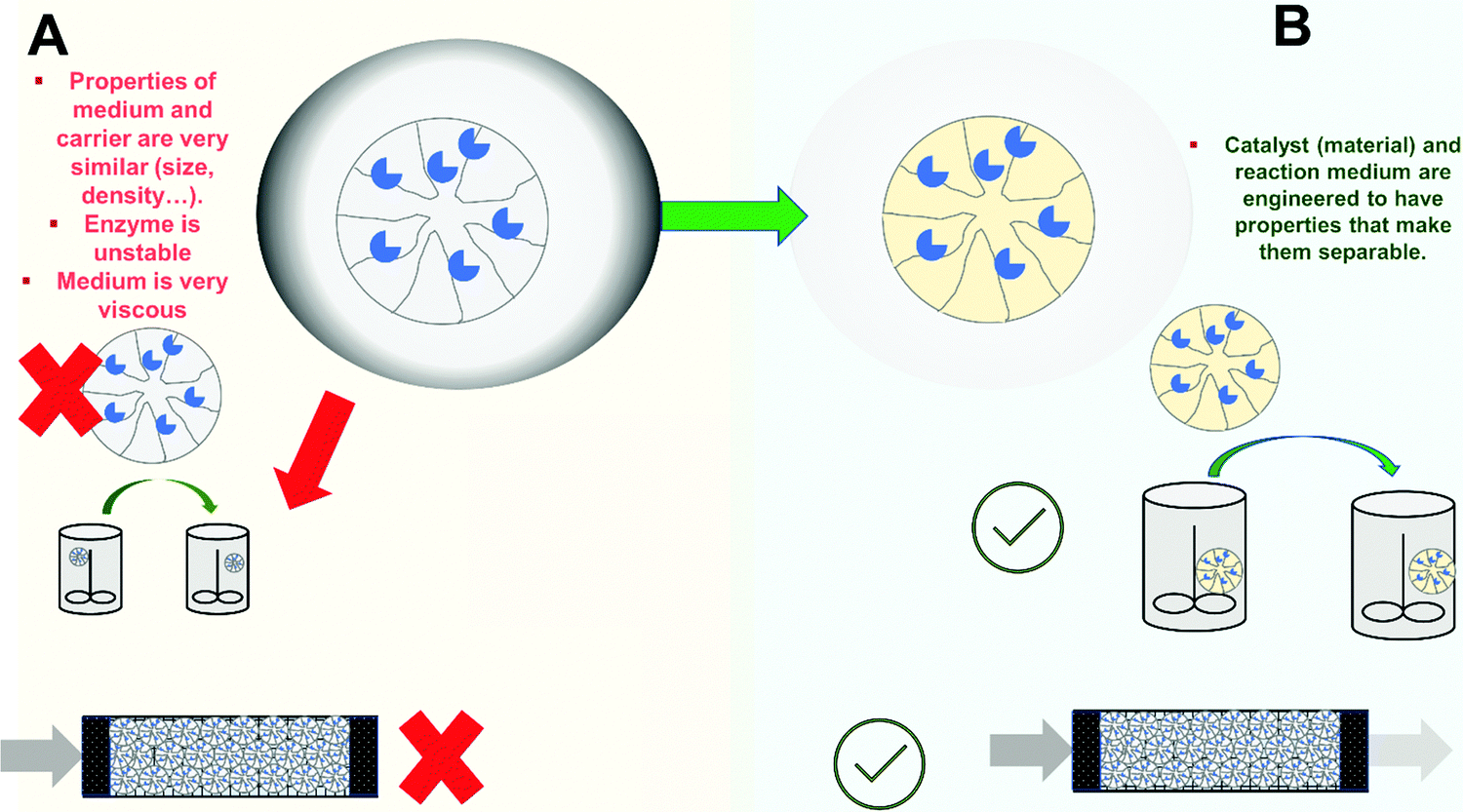 | ||
| Fig. 7 Problems with the enzyme reuse. (A) Characteristics of the reaction medium or physico-chemical similarities between support and medium prevent reuse. (B) Immobilization design and reaction engineering should focus on facilitating separation. | ||
The problem also exists when a liquid–liquid multiphase reaction medium is used, depending on the properties of the support (i.e., its density and size), a partition of the biocatalyst between phases takes place and separation/recovery becomes tedious. In any case immobilization and reaction engineering should focus on creating and exploiting those features that make separation possible (Fig. 7B).
A further problem is if the final reaction medium is highly viscous (Fig. 7A). This can make very complex the use of a batch reactor and recovery of the biocatalyst by filtration. Rigid supports with high density (to facilitate decantation and prevent floating in the reaction media) may be required to avoid high pressure filtration. Likewise, there is the risk of support breakage when using rigid supports (see below). An example of this is the production of bio-lubricants using immobilized lipases.315
A further problem occurs when the reaction conditions lead to enzyme inactivation. In this case, it is possible to recover the immobilized enzyme, but reuse is not possible, reducing the motivation for enzyme immobilization. In such cases, it is necessary to use another immobilization strategy or a more stable enzyme to ensure effective use of an immobilized biocatalyst for the process. Otherwise, the use of the soluble enzyme will be preferred, unless the immobilization is strictly required to produce an enzyme with the desired catalytic properties or to prevent product contamination by the enzyme.
In some instances, the biocatalyst is inactivated because the pores are closed by accumulation of layers of compounds contained in the substrate solution (in many instances contaminants of the actual enzyme substrates, as is the case with oils). This means that even having a perfectly stable and active enzyme, which resides inside the biocatalyst, after some time, all enzyme activity is apparently lost. This may be solved if the researcher investigates the means to eliminate the accumulated substance (e.g., washing under conditions that do not affect the enzyme), so as to prevent its presence in the biocatalyst at levels where the substrate cannot penetrate inside the support pores.
A more complex case can arise if the immobilized enzyme is utilized in processes strictly related with the catalytic features of the enzyme (selectivity or specificity), such as kinetically controlled synthesis,79–81 resolution of racemic mixtures,316,317 asymmetric modification of pro-chiral substrates,318–321 selective modification of multifunctional compounds,322,323etc. In these cases, even very small conformational changes to the enzyme, that perhaps have no effect in simple hydrolytic processes, may dramatically alter the performance of the enzyme in that process, making their reuse unsuitable. This problem cannot be solved adding fresh biocatalyst; the problem is not a decrease in enzyme activity but a radical change in the whole enzyme performance.324,325 For these processes, enzyme operational stability should be evaluated under the real final application conditions and reaction, and decisions on the convenience or not of using immobilized enzymes should be based on this information. However, as stated in many points in this review, it is possible that the optimal properties of the enzyme may only be achieved after its immobilization.101 One possible solution is to use the immobilized enzyme while it keeps the proper catalytic features for the “complex” process, and subsequently, use it for simpler processes where the activity may be maintained (e.g., hydrolysis).
4.4. Generation of steric hindrances preventing substrate access to the enzyme active center
In this section, we consider two problems that can arise when the enzyme is immobilized in a porous support (Fig. 8A). In these cases, most of the specific area of the support will be in the inner part of the support particle (over 99%), and that way, most of the enzyme molecules will be inside the pores, with just a small fraction of the enzyme molecules in the external part.69,99,159,326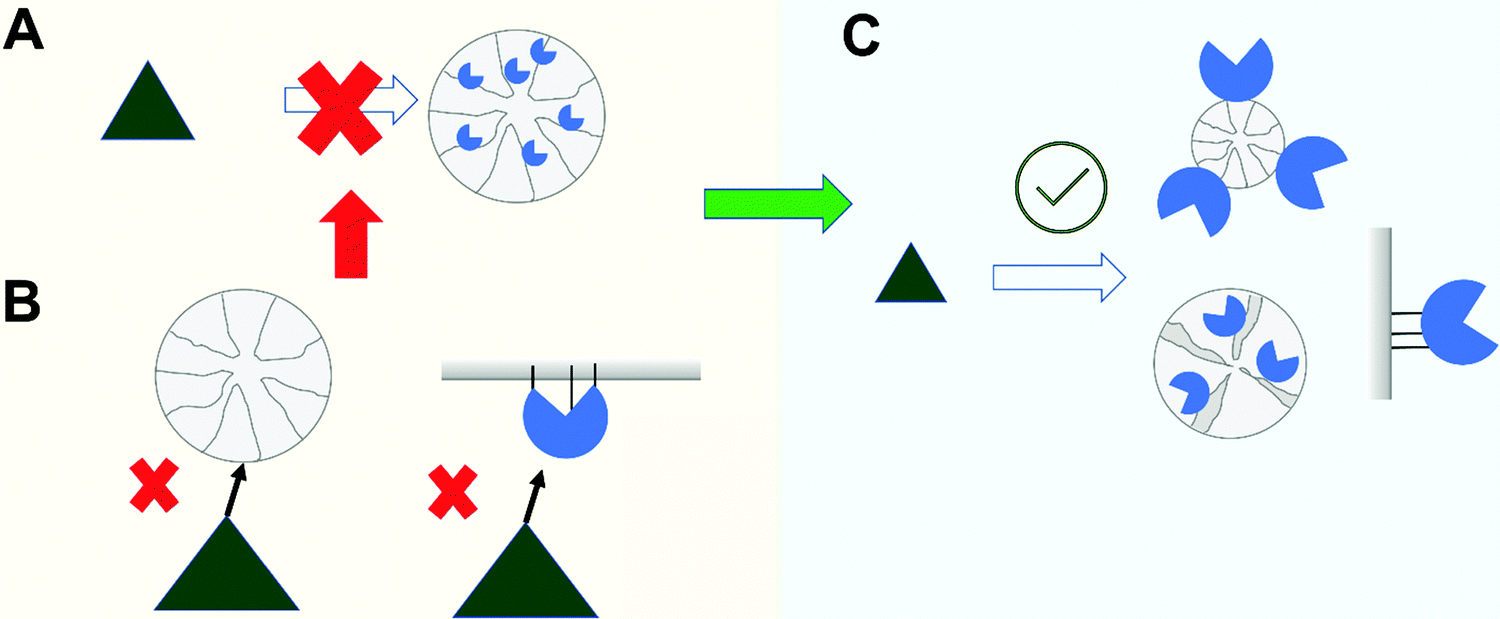 | ||
| Fig. 8 Problems of substrate accessibility to the immobilized enzyme. When enzymes are immobilized into porous solid supports (A) substrate access can be prevented by too large a substrate size to diffuse into the support or due to incorrect enzyme orientation (B). Solutions include use of non-porous particles, use of tailor-made porosity and/or control of enzyme orientation (C). | ||
The most obvious steric problem to an active biocatalyst is when the substrate size exceeds that of the pore size of the support where the enzyme is immobilized (e.g., the substrate is a solid or a large macromolecule, much larger than the enzyme)99 (Fig. 8B). In this case, the enzyme in the external support surface is the only one that can start the modification of the substrate, and when it is fragmented, the substrate can reach a size that permits it access to the inner part of the biocatalyst particle, thereby enabling more enzyme molecules to attack the fragmented substrate. Although this seems a reasonable assumption, we have not found reports in the scientific literature on this matter. In most cases, the activity is evaluated by measuring the fragments (e.g., the free hydroxyls in hydrolysis of polysaccharides or primary amines in the hydrolysis of proteins), and the reaction will be seen to be accelerating over time. In fact, if the time of the first activity measurement is long enough, it should be possible to detect a relatively significant enzyme activity. This apparent activity will be nearer to reality if we determinate the residual substrate, since in these cases the only enzyme acting on it will be that at the surface. This value will be closer to that of the soluble enzyme when very low enzyme loads are employed (as is usual during biocatalyst optimization, to prevent diffusional limitations, as we will comment later). We can assume that the enzyme will be immobilized first in the support external surface and later, in the pores. In this way, the change in the biocatalyst from low to high enzyme loading can give unexpected negative results in terms of observed activity. In these cases, the use of non-porous supports may prove a better alternative (Fig. 8C), even if that means using nano-particles which are not so easily handled, unless they are magnetic.314,327–329 The current price of these supports means that they are hardly competitive, except if the final product has a very high price or the application is in biomedicine or as a biosensor. Moreover, even using magnetic nano-particles, an often overlooked problem may arise: the nano-particles can aggregate, forming an ex novo macro support with a porous structure, and generating again steric hindrances for the action of the enzyme molecules on large substrates.41
The other problem in this category is when the active center of the enzyme is not correctly oriented towards the support surface and the substrate can encounter some steric or partition problems to reach the enzyme activity center84 (Fig. 8B). It is very unlikely that the support active groups can reach the active group of the enzyme active center, as these will be in pockets and unable to interact with the support surface (although they can be rapidly inactivated in presence of similar free reagents). That way, using relatively small substrates and supports that present large flat surfaces for the enzyme–support interaction, this can cause an apparent increase in the KM, although most immobilized enzyme molecules may be accessed by the substrate. For example, lipases that have been immobilized via interfacial activation on hydrophobic flat surfaces are able to recognize large triglycerides, and have their active center perfectly oriented towards the support surface.108,330 However, if the hydrophobic support is formed by fibers of a size similar or smaller than the protein, which can be more suited to the space of the active center pocket, the lipase activity even with small esters will decrease.331
The problem is more significant using large substrates (e.g., polysaccharides, proteins, nucleic acids). In this case, if the active center is perfectly oriented towards the support surface, even if it is a large flat surface, we can have an enzyme with the active center fully preserved, but inactive on large substrates (but not on small ones).84 That way, measuring the activity with large and small substrates is convenient in these cases, to better understand the mechanism that determines the final activity of the biocatalyst.
If the substrate is larger than the enzyme, serious enzyme loading-enzyme activity dependence against the large substrate may be found. Using low loading, enzymes having an active center in the middle of the enzyme (considering the enzyme parts in contact with the support at the start) may have an activity similar to that of the soluble enzyme (if the active center is not distorted) (Fig. 8C). When the loading of the support is increased, then the activity against large and small substrates decreases due to diffusional limitations (see later). However, when approaching the maximum loading of the support, the immobilized enzyme molecules may be so close to each other that there is not enough space for the substrate to reach the active center, producing a sudden decrease in the observed enzyme activity.84 Very high immobilization rates may also result in the appearance of these kinds of problems.130,332,333 If the immobilization rate is much higher than that of the enzyme diffusion rate in the pores, the enzymes will be packed together even at low loading, and then steric problems may arise even using moderate enzyme loadings (perhaps not preventing activity on large substrates, but still decreasing it significantly).332
These kinds of steric problems may be solved using protocols that enable other enzyme orientations99,101,214 (Fig. 8C). However, the comparison of the immobilized enzyme activity using large and small substrates can already give some clues on the real problem.
Finally, as commented above, some recent reports suggest that the spatial distribution of enzymes on the support surface (e.g., enzyme surface density) may greatly affect the kinetic properties of the enzyme.310
4.5. Generation of mass transfer resistances and internal diffusional limitations
Operational activity and stability of immobilized enzymes depends on both chemical phenomena (the reaction rate being given by the intrinsic kinetics of the immobilized enzyme)302,326,334 and physical phenomena.4,224,326,335 The physical phenomena can be due to partition effects or mass transfer resistances. Partition effects are those caused by the different physical properties of the carrier material and reaction medium, as a consequence of the concentration of certain compounds being different at the solid–liquid interface where the enzyme is immobilized.4,224,326,335 In contrast, mass transfer resistances are caused by differences in the rate of the physical processes and the enzyme-catalyzed reaction. There are two basic steps for transfer. The first one is the external mass transfer from the bulk liquid medium to the biocatalyst particle, that can be reduced by using suitable mixing.336–339 The second is the internal mass transfer, that is controlled by molecular diffusion within the porous material340–342 and where conventional mechanical stirring has no effect. However, the use of ultrasound has been reported to permit agitation even inside the biocatalyst particles.343–348When the mass transfer is slower than the reaction rate, substrate and product concentration gradients are developed inside the biocatalyst particle, resulting in the enzyme located in the core of the biocatalyst particle catalyzing the reaction under conditions far from the ones set and monitored in the liquid phase (Fig. 9A). The concentration gradients can lead to an increase in the apparent KM of the enzyme for the substrate, product accumulation (important if it can lead to inhibition or inactivation) and decreases or increases in the pH of the enzyme molecules’ microenvironment.224,326,335,349 Substrate depletion gradients into immobilized enzyme supports decrease the operational activity of the biocatalysts, the efficiency of use, and therefore, affect reaction times and the various metrics of reactor performance224,326,335 (Fig. 9).
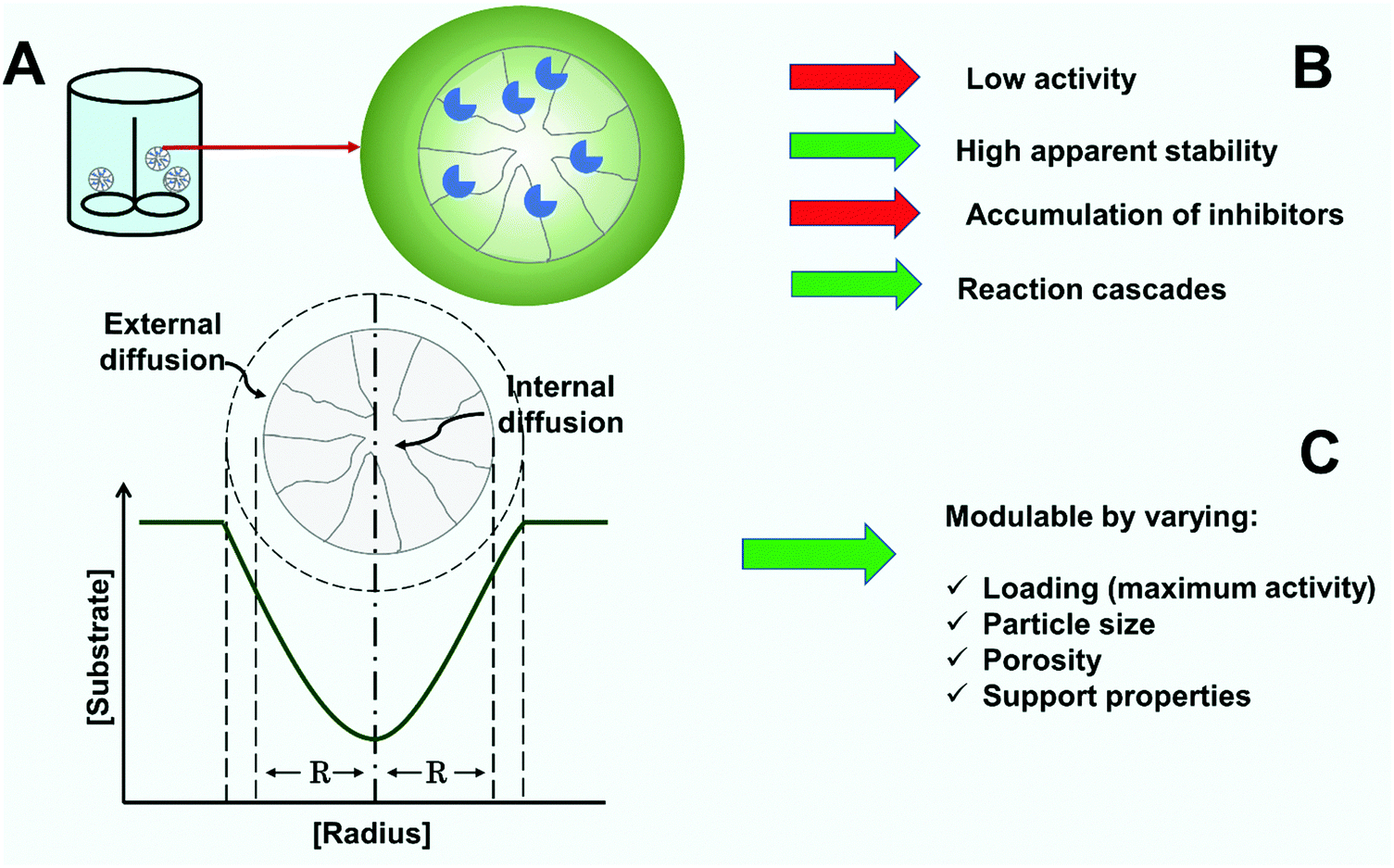 | ||
| Fig. 9 Mass transfer limitations in solid-supported immobilized enzymes. (A) due to the different velocities of chemical reaction and mass transfer, concentration gradients develop in the porous particle (e.g. substrate depletion along the particle radius). (B) Effects of mass transfer resistance on the performance of the immobilized enzyme. (C) Variable designs enable modulation of mass transfer limitations. | ||
One obvious negative effect is the decrease of the catalytic effectiveness of the catalyst. However, the generation of concentration gradients due to mass transfer limitations is not always negative (Fig. 9B). One positive effect is when the substrate has a deleterious effect on the enzyme. The high diffusional limitations reduce exposure of the enzyme molecules located in the inner areas of the particle to this substrate, increasing therefore the apparent stability of the enzyme (under conditions where higher substrate concentrations may be negative for the enzyme stability). This apparent increase in enzyme stability will disappear when the enzyme molecules in the outer positions become inactivated and do not reduce the substrate concentration in the core, but it can be enough to increase the operational stability of the biocatalyst, even allowing for the cost of having an initial activity lower than that expected. This is the case for the decomposition of hydrogen peroxide by those enzymes consuming hydrogen peroxide.350,351 However, in many instances the substrate may present stabilizing effects on the enzyme, and the gradient will give reduced stabilization effects in the inner part of the biocatalyst particle. One further consideration is the possibility of substrate inhibition (e.g., penicillin acylase inhibition by its substrate, penicillin G).352,353 In this instance, the inhibition will be decreased in the inner part of the biocatalyst due to the substrate gradient. As the affinity of the enzyme in the active center will be higher than in the inhibitory location of the substrate, an adequate selection of these gradients may permit optimization of the enzyme activity, although again we have not found reports in the scientific literature using these ideas.
Another positive effect might be the increase of the apparent operational stability of the biocatalyst under conditions of high diffusional limitations and an operationally low effectiveness factor. When the effectiveness factor of an immobilized biocatalyst is low due to substrate diffusional limitation, the reaction rate might remain almost constant even though a significant proportion of the enzyme molecules have been inactivated.
In the case of product gradients, this will be more relevant in the inner areas of the biocatalyst particle. If the product has a stabilizing effect (a relatively common phenomenon),354,355 its accumulation may increase the enzyme stability, and more so for enzyme located in the core of the biocatalyst particle than for enzyme in the outer part. The same may occur if the product exhibits inhibition; the effect will be more relevant for enzyme in the core than for enzymes located in the outer areas of the particle. Moreover, when the product has inactivating effects (e.g., the case of oxidases producing hydrogen peroxide), the enzyme molecules in the core will be exposed to a much higher concentration than those in the bulk.
A positive effect of product accumulation occurs in the case of enzyme cascades and co-immobilized enzymes, where the high concentration of the product of the first enzyme in the pores of the particle enables the second enzyme to act from the start of the reaction at full activity, without the usual lag-times.120,121,356
The promotion of pH gradients can also be positive or negative.225 For example, in the case of immobilized penicillin G acylase, with the formation of a pH gradient in the hydrolysis of penicillin G (e.g., using an external pH of 8 and assuming an internal pH in the core of the particle of 5), this will have a negative effect on the biocatalyst activity (optimal penicillin G acylase activity is at pH 8), but a positive effect on the enzyme stability (optimal at pH 4.5–5).357 In any case, these pH gradients may be controlled by using H+ transporters.358–360
The formation of internal concentration gradients within a support depends on the design of the immobilization4,326 (Fig. 9C). On the one hand, it is determined by the reaction rate as given by the intrinsic enzyme kinetics and enzyme loading.361,362 On the other, it relies on the physico-chemical properties of the carrier support and mass transfer.130,349,362,363 As previously stated,1,4,89,99,326 achieving reaction intensification is based on the immobilization of high protein loads to achieve a high volumetric activity in the carrier. This is the reason why many technically useful catalysts are diffusion limited, meaning activity and stability are thereby both affected. Whereas this is a common feature, the problem can first be observed during the catalyst development and characterization steps. Kinetic models and kinetic constants are dependent on the enzyme loading.4,363,364 Knowledge of the intrinsic KM is important, and in this way, intrinsic and mechanistic aspects can be disentangled.326,362 Characterization of the immobilized biocatalyst should be performed under realistic conditions and the desired load. The modulation of the mass transfer depends also on the physical properties of the support material such as particle and pore radius, as well as porosity. While mass transfer resistances can be minimized (reducing the particle size, the enzyme load, etc.), this may need to be addressed dependent on the biocatalyst format.365
4.6. Use of unstable supports
The use of mechanically unstable supports may generate some “minor” problems at laboratory scale, but can generate far more significant problems at a larger scale99,159 (Fig. 10A). This means that the particle size of the biocatalyst will decrease through the operational use of the biocatalyst. One initial problem, very relevant at industrial scale, is that the filters utilized in the reactor may not be suitable if the biocatalyst particle size decreases, producing the blocking of the pores of the filter and making it necessary to manually remove material from the reactor.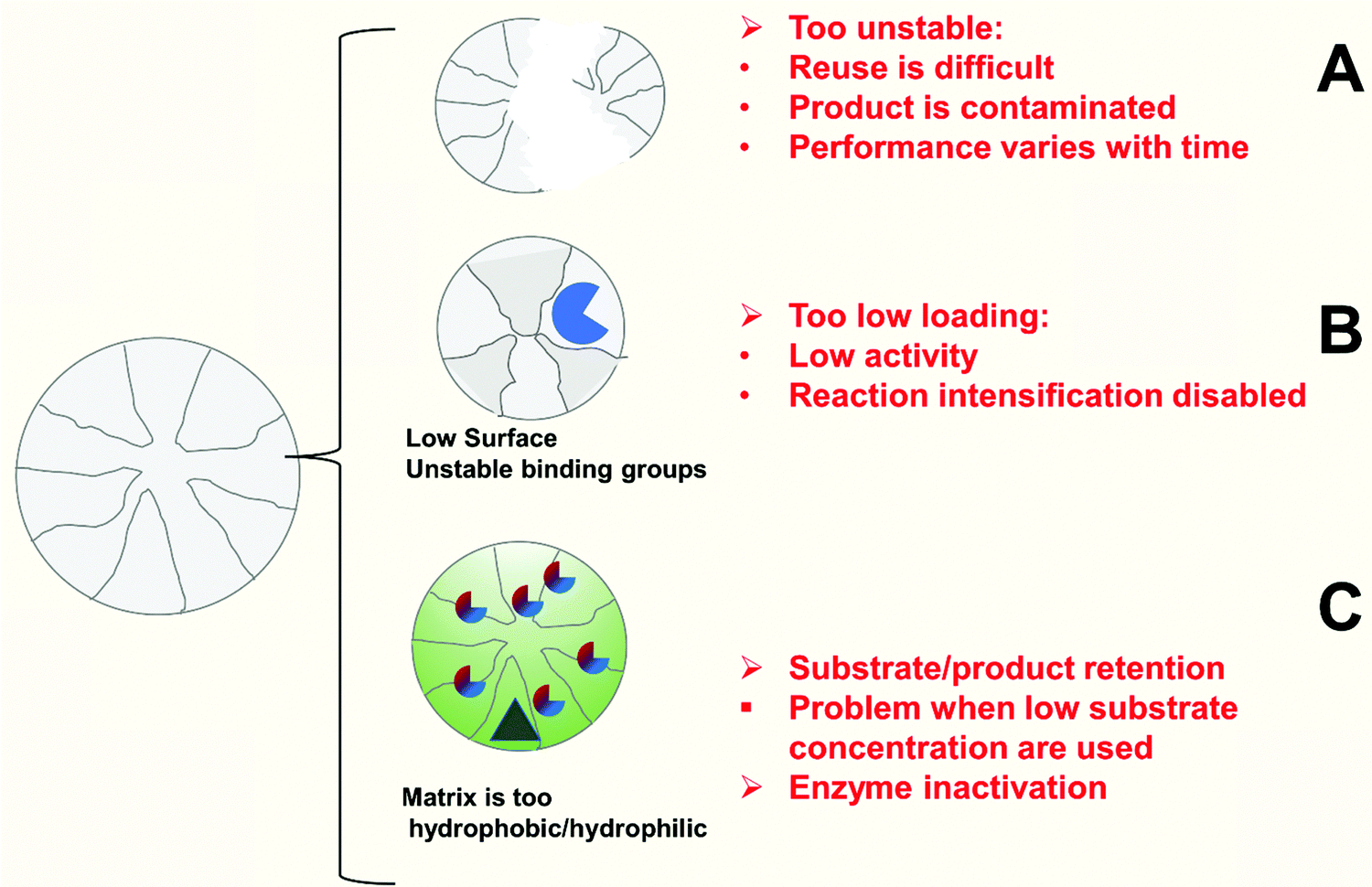 | ||
| Fig. 10 Problems of biocatalyst performance due to support features. (A) Support is unstable. (B) Achievable loading is too low. (C) Matrix/surface properties are not compatible with reaction medium. | ||
However, the decrease in the particle size can produce many more problems. Some of these problems are related to the diffusional and mass transfer issues discussed in the previous section. As discussed previously, they can affect the enzyme activity or even stability.4,89,99,130,159,326,349,362,363 Substrate, product or pH gradients will be much higher when the size of the particle is larger. If the biocatalyst is physically broken and the particle size is reduced, the biocatalyst activity will increase during the reaction cycle and successive reuses, the positive or negative effects of mass transfer (see above section) detected with the original particle size will be altered, and this can produce some unexpected results.365
Even at a laboratory scale such particle breakage can give confusing results. For example, in inactivation experiments, if the particle of the biocatalyst is broken, the researcher will find an “apparent” hyper-activation of the enzyme, or at least an apparent stabilization of the enzyme. Initially, the biocatalyst activity is underestimated, and when the biocatalyst particles are broken, its activity approaches reality. In this way, “false” stabilizations may be reported if particle breakage is not considered.
Moreover, as commented previously, pH gradients may also be generated, with the outer areas of the particle at pH values near to the external pH, and the pH in the core with values quite different. The importance of these problems also depends on the enzyme loading and particle size. In this way, these effects will decrease when the particle size is reduced.151–153
Using co-immobilized enzymes acting in cascade reactions, the support breakage may be particularly problematic. Assuming that these combi-biocatalysts have previously been optimized for the ratio of the enzyme activities to maximize the overall volumetric activity and product yield (in cases where one of the intermediate products is unstable),120,366 then the reduction of particle size may have relevance if one uses co-localized enzymes, and even more if one has produced concentric crowns of each of the enzymes. In the case where we use co-localized enzymes, this particle breakage will reduce the diffusional problems of the initial substrate, making it possible that the activity of the first enzyme is increased.4,89,99,130,159,326,349,362,363,366 Similarly, the potential gradients of the product from the first reaction in the chain will be reduced, decreasing the concentration of the substrate for the second enzyme, and hence, its activity.367–373 Hence, the ratio of enzyme activities may become much altered, decreasing the intrinsic advantage of using co-immobilized enzymes.120 This may be more important if the intermediate product is unstable or if the formed by-product may react with it, as the changed enzyme activity ratio may drive a decrease in the final yield and the presence of the undesired by-product that one intends to prevent using co-immobilized enzymes.303–311,366 Using enzymes co-immobilized in concentric crowns, the problem is much serious, as it is possible that in some of the biocatalyst fragments only one of the co-immobilized enzymes is present, and in any case the “distribution” and “activity optimization” effects will also be missed.
That way, the support fragmentation is not only a trivial operational problem; it can completely alter the biocatalyst performance. For this reason, the researcher should select a support that is physically compatible with the reactor to be used (as discussed elsewhere in this review). For example, to use a very rigid support (e.g., porous glass) in a mechanically stirred reactor is almost certain to lead to problems. For fragile supports, one strategy might be the encapsulation of support particles in a larger structure with better mechanical performance (e.g., LentiKats, alginate beads).48,49,374–377 However, this has an additional cost, and the use of another type of support may be a simpler solution.
Another problem, perhaps even more serious, is if the support becomes dissolved in the reaction medium (e.g., a polymeric support that is not properly cross-linked). This has many negative effects. The first one is that some support components will be incorporated into the final product, leading to a complex downstream product recovery process. Moreover, the released polymer will carry some enzyme, and this will be washed away and incorporated into the product: hence enzyme re-use is not possible, the control of the reaction is lost because there is enzyme in the product, all the filtration systems in the reaction and further processing may be affected, etc. An example of this problem can occur with Novozym 435, one of the most widely used immobilized lipase biocatalysts, where under certain conditions, the support may dissolve.378–381 The solubility of the support under the experimental conditions is a study that not many researchers undertaken and, therefore, the real extent of the problem is not known. The problem itself does not have a ready solution, and if the support may potentially dissolve in the medium, it should be discarded and an alternative support sought.
4.7. Selected support and utilized reactor may be incompatible
Generally, the functional properties of the immobilized enzyme rely on the interplay of the enzyme and the immobilization chemistry with the support, whereas practical applicability depends on the type and properties of the support.69,159,326 The type of support determines the reactor choice5,77,159 (Fig. 11). Indeed, from an engineering perspective, the real catalyst is the support material (particles), not the enzyme itself. Typically, immobilized enzymes in their development phase are extensively characterized from a functional point of view (activity, stability, kinetics),4,88,302,311,326,382 but the consequences of the properties of the selected support material remain more elusive at this early stage. The two design decisions are, however, interdependent, and, as discussed below, the functional properties and practical use are not directly translatable to other supports (Fig. 12). The application of an immobilized enzyme in a reactor with a stirred tank configuration has to consider the stability of the material toward shear stress, and its density in order to ensure a correct mixing throughout the reactor.86,364 Likewise, the method used for biocatalyst retention defines the minimal particle size that can be used. Application in a reactor with a tubular format, e.g. in a fixed bed reactor, implies upper and lower limits. These limits are defined by the maximum pressure drop sustainable and suitable fluid-solid contact which also depends on the superficial velocity and reactor dimensions. Thus, geometrical aspects of reactor, carrier and reaction are interdependent. Rigid materials are more suitable for a fixed-bed compared with elastic materials.164 Material characterization and its consequences for the fluid-dynamics of the reactor (mixing, minimization of external mass transfer resistance etc.) are all key points to be considered. This becomes even more important with the advent of new materials, where there is a lack of material characterization with respect to stability, fluid-dynamics, rheological behavior, chemical resistance, etc. Fortunately, there are some new concepts in reactors that can facilitate the compatibility of the selected material and the bioreactor, where even mechanically fragile biocatalyst supports may find application, such and vortex or basket reactors.47,383–387 Modern biocatalysis has also adopted concepts of reaction engineering where non-conventional media beyond aqueous systems (solvent-free, deep eutectic solvents, biogenic solvents),12,388–394 are taking prominence. It is therefore expected that integrated design of material and reaction medium with comprehensive studies of reactor compatibility will become even more important in the future.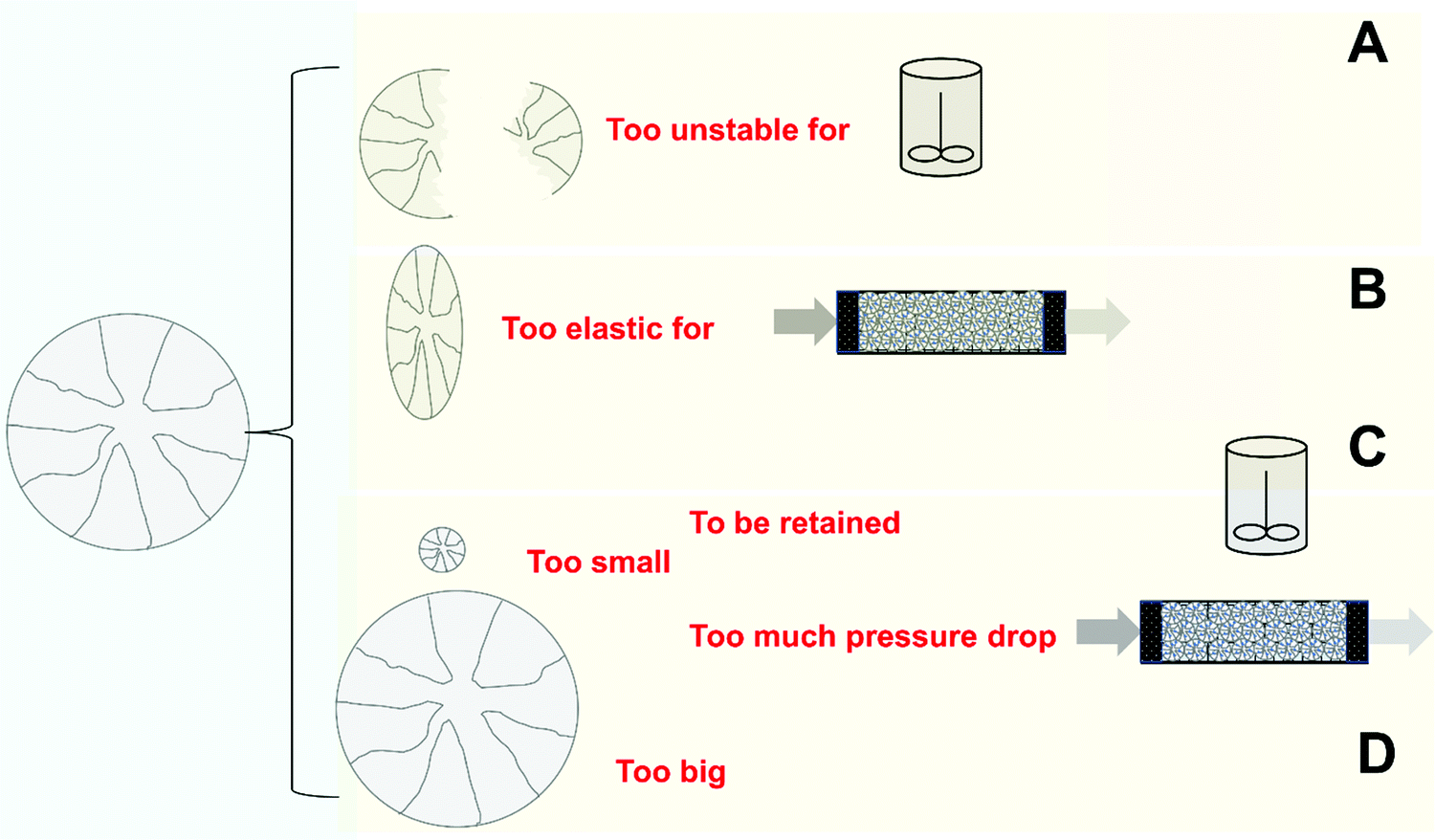 | ||
| Fig. 11 Incompatibilities of the immobilization material support and reactor choice. Immobilization material support is mechanically too unstable under conventional stirring conditions (A). Immobilization material support is not rigid (e.g. supports based on natural organic polymers) which can cause lead to excessive pressure drop across the packed bed (B). Immobilization material support size is too small, which creates problem for retention or excessive pressure drop across fixed bed reactors (C). Immobilization material support size is too big (D). | ||
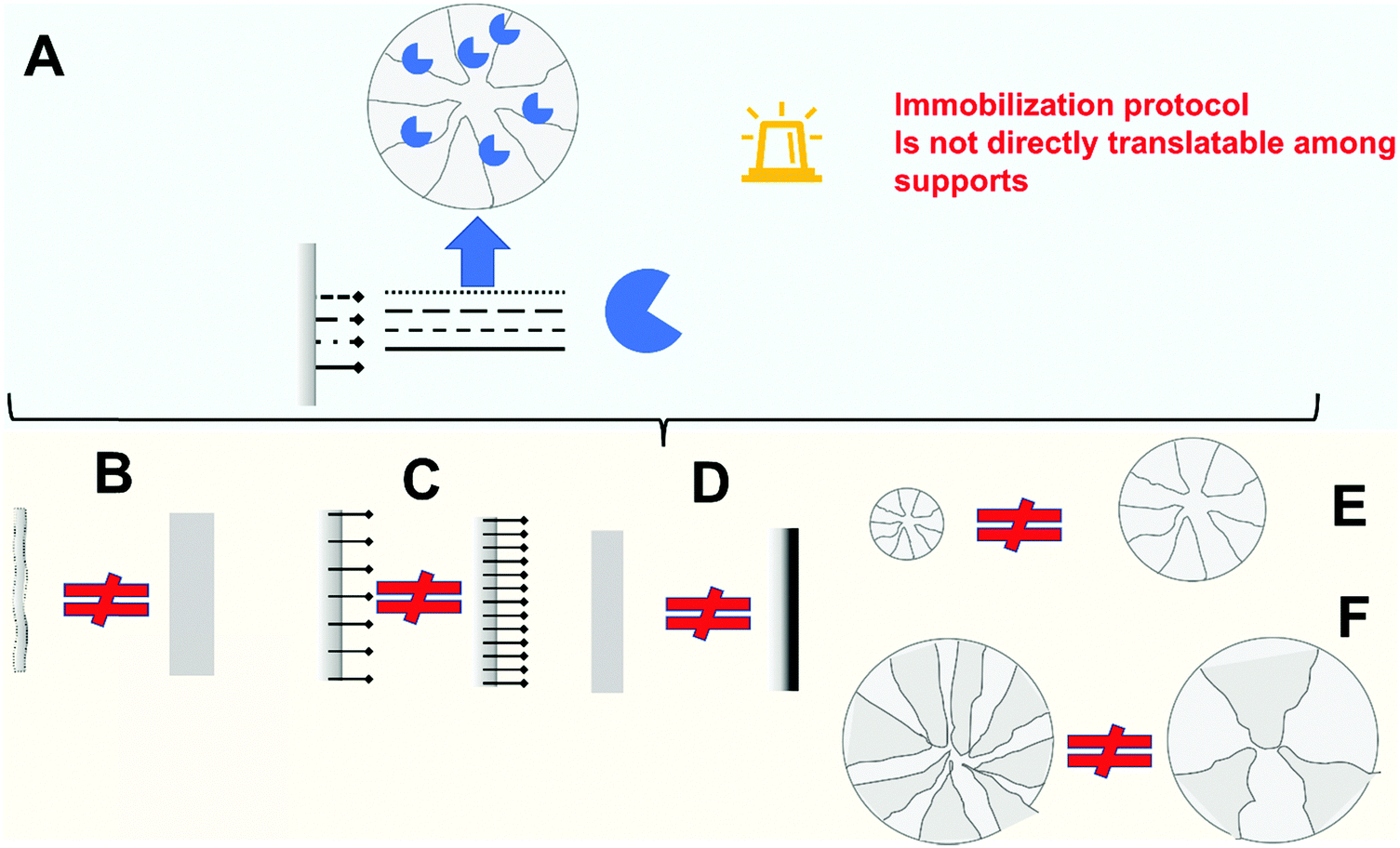 | ||
| Fig. 12 Protocols of immobilization are not directly translatable between carrier materials. The final properties of an immobilized enzyme are a consequence of multiple aspects involving carrier features, immobilization, and enzyme properties (A). Protocols might not be directly translatable among supports due to differences in surface topology (B), the density of surface group activation (C), hydrophobicity of the surface (D), particle size (E), porosity framework (F). | ||
4.8. Extrapolation from one support to another is not always a direct process
In many instances, a frequent confusion occurs between the immobilization support and the active group.99,159,214 That is, many researchers assume that a protocol that works properly for one support, will work similarly when using another support (Fig. 12). And vice versa, if a particular immobilization group does not work with one support, it is discarded as an immobilization method for that specific enzyme.However, several factors should be considered that greatly affect the results related to the use of supports of different natures:
– Not all supports will have similar geometric congruence with the enzyme99,159 (Fig. 12B). The enzyme–support interaction areas will be small if using a support formed by fibers of a size similar or smaller than the enzyme diameter. Even using a support formed by flat surfaces, is not the same if the support pores are convex or concave, so the interaction of the enzyme with the support will be different. This may be for the better (a more intense multi-point attachment may be achieved if the geometrical congruence is increased) or for the worse (a negative enzyme–support interaction may also be increased if the geometrical congruence is increased). An example of this is the results obtained using Eupergit and Sepabead epoxide supports.200,395 Without a proper blocking, the higher geometrical congruence of the moderately hydrophobic Sepabeads gives an enzyme destabilization that is not produced in Eupergit.200,395 However, using a proper blocking agent, the enzyme immobilized in Sepabeads has been found to be much more stable than in Eupergit because of the higher intensity of enzyme–support multi-point covalent attachment.200
– Not all supports will permit a similar extent of activation (Fig. 12C). A higher superficial density of reactive groups (which is the important parameter, not the amount of groups per ml or g) will allow a higher enzyme immobilization rate, a higher intensity of multi-point covalent attachment, and later, a higher effect of the blocking of the support surface.99,159,214 That is, comparing different supports with different superficial densities of groups is not a fair comparison. However, if maximum activation degrees are used in both supports, we can assume that the results obtained with them are the best those supports can offer. Support activation degree can be decreased in case the enzyme–support interaction is excessive, but it cannot go beyond of the maximum activation degree in any case.100
– The physical properties of the different support surfaces may be different (Fig. 12D). After immobilization, the enzyme will be in close contact with the support surface, and even if the soluble enzyme is not able to become adsorbed, even a minimal non-inertness of the support may give rise to undesired enzyme–support interactions after enzyme immobilization (as is the case with Sepabeads epoxy described previously).200 We can minimize these effects by a proper blocking, but a physically active support can never be completely inert. Moreover, it has recently been shown that physical interactions between covalently immobilized enzyme, having exactly the same orientation and number of covalent enzyme–support linkages, and the surface of the support bearing different physically active groups, may greatly affect not only the enzyme activity and stability, but also the mechanism of enzyme inactivation.324 That way, properly utilized, the control of these interactions can become a tool to increase the versatility of the immobilized enzyme.
In some instances, the enzyme is readily inactivated when immobilized in an activated support.139 Only a proper investigation will explain if the problem is the active group or it is the nature of the support itself. For this reason, we recommend characterizing a new immobilization protocol for a specific enzyme using an inert and hydrophilic support (such as agarose),164 and then try to reproduce the protocol with the “target” support. Any discrepancies may be attributed to the support features, and at least the researcher will then know which feature of support is inadequate. That way, it is possible to look for a new support suitable for the reactor, at the desired cost, and suitable for the enzyme immobilization-stabilization.
In some instances, the physical properties able to interact with the enzyme are generated by the active group utilized to immobilize the enzyme and not by the support. For example, activation of an inert surface with vinyl sulfone will give a layer of relatively hydrophobic groups, which can result in immobilization via interfacial activation of lipases. After immobilization, this moderate hydrophobicity may be partially solved by an adequate blocking, but, again, a fully inert surface cannot really be achieved, and these blocking groups may also alter the enzyme properties (for better or worse).324
4.9. No use of enzyme–support interaction end point protocols
In some instances, for example when using physical immobilization protocols, the support must remain fully “active” to avoid enzyme release.99,101,159,311 This means that during enzyme inactivation in operation or storage, new enzyme support interactions may occur, stabilizing inactive forms of the enzyme, until the enzyme is fully unfolded to maximize the enzyme–support interaction (Fig. 13).396,397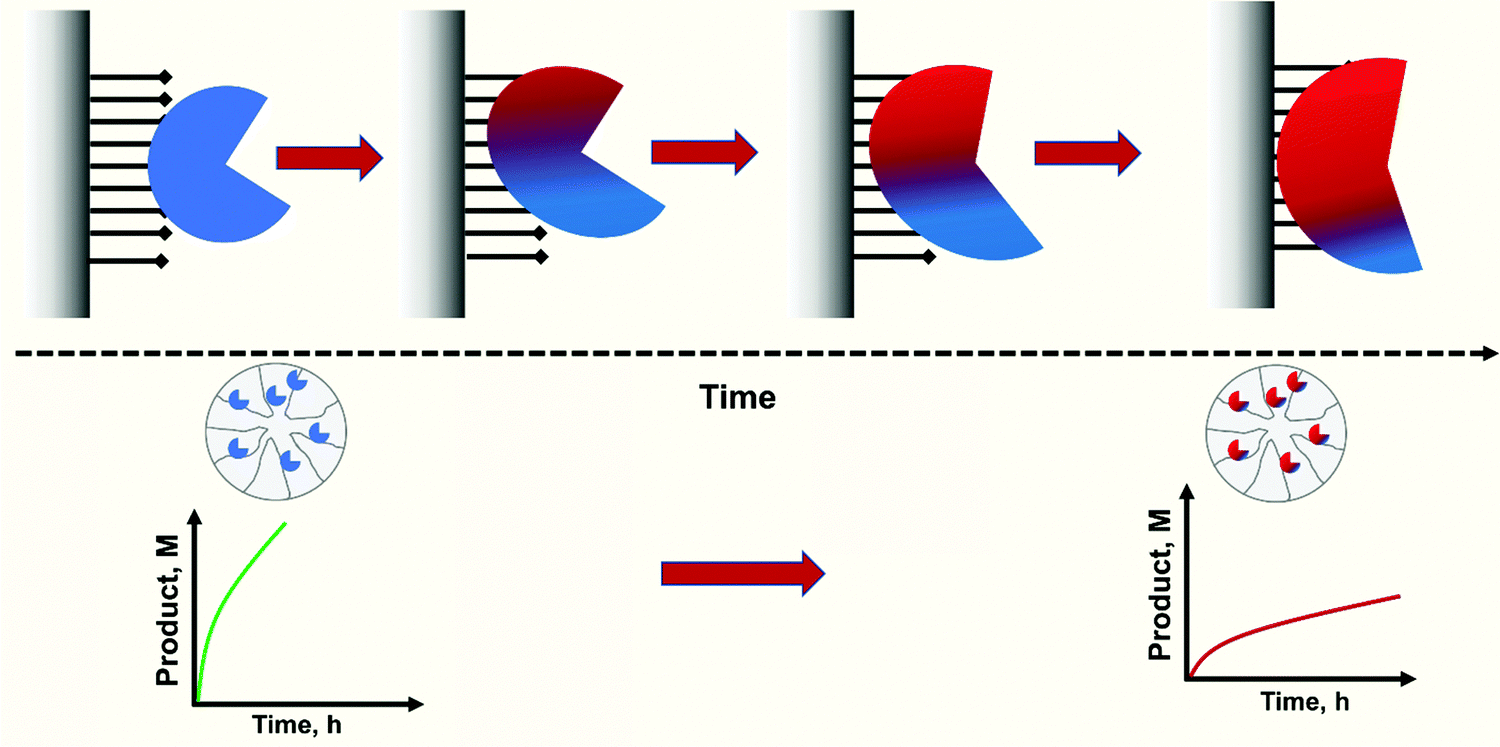 | ||
| Fig. 13 Consequences of a support that remains reactive after immobilization. When enzyme–support interaction end-point protocols are not used, physico-chemical interaction between the support material and the protein can take place over an extended time resulting in structural distortions/modifications that may eventually affect the catalytic properties. | ||
Using covalent immobilization techniques, an end-point strategy is important and frequently available.159 It has many advantages, and can mean that the immobilization protocol becomes a success rather than a failure. First, it prevents the establishment of uncontrolled new enzyme–support covalent bonds during operation, which can stabilize incorrect enzyme structures. Secondly, the reactive support may react with components in the medium, modifying the support surface and perhaps even altering the enzyme properties.324 In some instances, for example when using glyoxyl supports or other aldehydes, the reaction end point (a reduction) simultaneously transforms the remaining aldehydes to inert and hydrophilic hydroxyl groups, and the reversible imino bonds into highly stable secondary amino bonds (as explained above).191 When the end point protocol is via a blocking step, it is an opportunity to reduce enzyme–support surface interactions (important when the support has a hydrophobic nature, as explained previously).200 Finally, it is possible to use this blocking step to tailor the support surface properties, to establish different interactions of the support surface with immobilized enzyme molecules that are in close contact. In this way, it can been used to tune the immobilized enzyme properties: stability, activity, selectivity and specificity.256,398–401 Moreover, as stated above, the enzyme–support interactions may alter the mechanism of enzyme inactivation.324 Hence, if a support–enzyme reaction end point is possible and used to produce a more inert support surface, this opens new possibilities for the design and control of enzyme–support interactions and tailoring enzyme properties. It is an additional step that has a cost, but in many cases, it is essential to take full advantage of the immobilization protocol. For example, it has been shown that this blocking step may be used to perform the co-immobilization of several enzymes, enabling the reuse of the most stable enzyme after the inactivation of the least stable.137,139
4.10. Immobilization protocols are not finished when all the enzyme activity is incorporated in the support
Capitalizing on enzyme immobilization to the full, it is not only necessary to utilize a suitable support, an adequate degree of activation of the support and a convenient reactive group on the support.100 The full immobilization protocol is important in order to maximize the immobilization impact. Previously we have commented on the importance of the enzyme–support reaction end-point and the stability of the reactive groups. However, a conceptual mistake that can be found in many publications is to consider that the immobilization process is finished when the enzyme is fixed to the support. This is not correct using physically active supports (either standard or hetero-functional), since further enzyme–support physical interactions will be developed during storage, and this can alter the enzyme properties.159 This can be partially solved by choosing convenient storage conditions where further interactions may be stopped, but during operation we cannot prevent this. For this reason, it is also convenient to use an inert support to immobilize the enzyme.159However, to have this in mind is even more critical when the researcher intends to get the maximum intensity of enzyme–support multi-point covalent attachment. After the enzyme is incorporated on the support, sometimes by just one point,402 sometimes already by several points,192 many researchers go directly to the end-point step, or in some cases, they finish the immobilization without including the end-point step. In this way, the possibility of having an intense and controlled multi-point covalent attachment using that immobilization protocol may not be properly analyzed.192 In fact, in many instances the conditions for optimal immobilization (determining the enzyme orientation on the support) may differ from the optimal conditions for having an intense enzyme–support multi-point interaction (that are related to the enzyme and support reactivity).402 This conceptual difference between immobilization and multi-point covalent attachment is lost in many publications. The second step should start after enzyme immobilization, and be independently optimized, and usually requires moderately long reaction times. Only in that way, all the possibilities of enzyme stabilization via multi-point covalent immobilization using a specific protocol may be achieved.403 This has been recently reviewed,100 but it is so important in the context of this review that we considered it necessary to emphasize this critical fact.
4.11. Difficulties for the co-immobilization of several enzymes
The order of the enzymes inside the biocatalyst particle may be a key point in the design of a co-immobilized enzymatic biocatalyst.305–309 In some instances, it may be hard to get the desired enzyme distribution. For example, co-localization of the involved enzymes is not granted by their simultaneous immobilization on the support. It is possible that some of the enzymes may be immobilized much faster than others and in that way, will be immobilized mainly in the outer volume of the particle, while the others will be immobilized in the inner zone.252 In order to reach a proper co-localization, the best strategy is to immobilize first the enzyme that immobilizes the slowest, even using conditions where the process may be slowed down, to ensure that the enzyme is distributed along the pores. Then, the enzyme that immobilizes the most rapidly will be immobilized occupying the spaces between the molecules of the other enzyme. This may be even more complicated if using impure enzymes, and still more so considering that the fraction and nature of the contaminants may be different in each batch.Concentric enzyme crowns may also be complex to prepare. Only if some of the involved enzymes immobilize more rapidly than they diffuse, can we guarantee that the immobilization will form a crown.
Moreover, studies should be repeated when the immobilization method changes, or even when we change the support. If the new support can or cannot establish additional interactions with the enzymes, this may affect the relative immobilization rates of the enzymes. The same may be said if the pore diameter is different, even changes in the particle size necessitate a re-optimization of the biocatalyst (in this case because mass transfer will also be affected, as discussed previously).
The localization of the different enzymes on the particle may be analyzed using confocal microscopy with enzymes labelled with fluorophores (that should not interfere with the immobilization rate of the enzyme). In this way, at least we can know the final distribution of the different enzymes in the biocatalyst.332,404–407 Although the optimization may be complex, finally the control of the enzyme order may be possible and confirmed, provided the researcher understands the phenomena occurring in the co-immobilization.
4.12. Use of supports with low loading capacity
We have previously commented that one of the advantages of using immobilized enzymes is the possibility of using very high enzyme concentrations in the reactor without the risk of enzyme aggregation. This is also important for process intensification strategies since the achievable space-time yield scales with the protein loading.88,326 However, these advantages will only be possible using supports that permit high enzyme loadings, or at least a loading suited to the characteristic time of reaction and productivity targets.This may in some cases be a little tricky. The loading capacity is determined by the specific area, provided that the pore size is large enough to immobilize the enzyme. If the specific area is low (e.g., 1 m2 ml−1), to have a low loading is therefore unsurprising (Fig. 10B).
Using supports bearing reactive groups with low stability, results may be confusing (Fig. 10B). The longer the time to make a good mixture of enzyme and support, the smaller the number of reactive groups that are left on the support; and this can alter both immobilization rate and the final enzyme loading on the support. In this case the loading will be determined not only by the specific area of the support, but also by the lifetime of the reactive group on the support.100,110
In some instances, problems may come from the components of the protein sample itself. For example, when the researcher works with unpurified enzyme extracts or with partially purified commercial enzymes (this gives a false sense of security), it is relatively common that the producer neglects contaminant proteins (both, in the production and during the partial purification steps). Indeed, some additional protein contaminants may even confer added stability to an enzyme. However, this may generate a problem for the researcher that intends to immobilize the enzyme. Nevertheless, if the first optimization of the immobilized biocatalyst is performed using an enzyme preparation where the protein of interest is among the one with the highest molecular weight, we can select a support with a pore-size that enables enzyme immobilization and provides a high volumetric activity. However, if some of the future samples of the enzyme contain a much larger molecular weight contaminant protein, or for any reason the aggregation of some of the components of the crude enzyme is favored, even with traces of these proteins (e.g. 1% of the total protein), then serious problems may be ensue for the immobilization. Indeed, aggregation can lead to very large protein forms, such as octamers. First, the larger proteins will have more reactive moieties left to react with the support, becoming immobilized more rapidly than the other proteins, and very likely in the outermost area of the pores in the biocatalyst. Secondly, after its immobilization, the other enzymes cannot be immobilized because the pores of the support are “blocked”. This can mean that the final loading of the biocatalyst is, in this case, much lower than in the first optimization, and most of the target enzyme will remain in the supernatant.99 This may not be discovered so easily, since it is possible that very low concentrations of this large protein may be not detected. Nevertheless, this may be an explanation for many problems observed with the reproducibility of support loading. After discovering the problem, the solution may be just to purify the target enzyme from the problematic protein, or to investigate how to encourage any multimeric protein complexes to dissociate prior to immobilization.
This may have a special relevance for enzymes that can be presented in different oligomerization stages, depending on factors that may be completely unknown to the researcher. This means that in some instances the unpurified enzyme samples are immobilized, but the pure enzyme, which may be more prone to aggregation, requires larger pores. This may be solved if, in the immobilization buffer, some compounds are added that are able to break the “false” oligomer without affecting the active enzyme structure. For example, the addition of 1 M urea was used to immobilize multimeric uridine and purine nucleoside phosphorylases from Bacillus subtilis,408,409 overcoming uncontrolled enzyme aggregation.
4.13. Problems generated by the physical features of the support
Previously, we have mentioned the problems derived from a poor mechanical performance of the support (Fig. 10A). However, in some instances the problems may come from other physical features of the support. One of the main problems comes from the capacity of the support to adsorb particular compounds as a function solely of its hydrophobicity/hydrophilicity99,159 (Fig. 10C). This can generate a partition of some compounds in the enzyme environment or even a decrease of the available substrate. If this partition is made deliberately, it may be used to increase the enzyme resistance to unfavorable components in the reaction media (e.g. components such as organic solvents, deleterious substrates or products such as hydrogen peroxide or phenolic compounds), to reduce the apparent KM or to increase the apparent Ki of the enzyme.39,49,100,350,410–415However, in many cases this partition may occur without having been deliberately designed by the researcher and may produce negative effects for immobilized enzyme activity and stability. For example, if the support is hydrophilic and it has a tendency to capture water and the reaction is in an anhydrous or a solvent free medium, an increase in the amount of biocatalyst will reduce the water molecules available for each enzyme molecule. This can mean that the activity of the biocatalyst is not proportional to the amount of biocatalyst, as the water activity will decrease when the amount of biocatalyst is increased.99,159 This may be solved by measuring the water activity at different enzyme loadings and ensuring that they are similar in all cases. However, such supports can also capture the water that is released in thermodynamically-controlled synthesis (e.g., synthesis of esters, disaccharides or amides), and if the volumetric activity of the enzyme is high enough and the water production rate is higher than the water diffusion from the particle, a water phase may be formed inside the biocatalyst particle, even if the water is captured or eliminated outside. This problem may be extended to other hydrophilic compounds, such as glycerin.416–418 The hydrophilic layer may result in enzyme inactivation (e.g., by concentrating water soluble acids) or inhibition (by preventing access to hydrophobic molecules).416–418 The solution to this problem is the use of ultrasound that permits agitation of the medium inside the pores of the biocatalyst, and prevents a hydrophilic phase from being formed.419–421 Alternatively, one may use a much more hydrophobic support, where hydrophilic compounds are not concentrated.422–425
The support can also partition some of the reactants, concentrating or reducing the accessibility of these to the enzyme, and this may also give a significant alteration of the apparent kinetic parameters of the enzyme (KM, Vmax, Ki).99 At laboratory scale substrate or product adsorption on the support can promote some difficulties in understanding and interpreting results. In the laboratory, usually the used concentration of substrates is not particularly high. The possibility of substrate adsorption to the support can therefore give rise to many problems in determining the real features of the biocatalyst, as the substrate concentration available for the enzyme may be significantly decreased. This may even produce an apparent enzyme inactivation if there is no substrate available for the enzyme. In any case, it can produce an increase in the apparent KM of the immobilized enzyme, un-related to the enzyme conformation, but rather resulting from a decrease of the available substrate. If the product is the one that is adsorbed onto the biocatalyst, we will also under-determine the real enzyme activity because in many cases the researcher only follows the product formation, and if there is no free product, the biocatalyst will be considered inactive and the immobilization protocol discarded. In this way, it is also convenient to study the adsorption of the reaction components onto the support, and if necessary, the standard reaction media may be altered to prevent this adsorption (e.g., by adding some solvent if the problem is a hydrophobic adsorption, or some salts to prevent ion exchange). This can be far from the real practical use of the biocatalysts, but can offer a more accurate visualization of the immobilized enzyme properties. This is a frequent phenomenon when using hydrophobic supports and substrates (e.g., with lipases), as in many cases some solvent is added, even when not required either for its solubilization or any positive effect on enzyme activity. However, at an industrial scale, the high substrate concentration and the high reaction medium/biocatalyst ratio will minimize this problem because the support will be saturated rapidly, and perhaps only in the first reaction cycle. In subsequent cycles, as the support will already be saturated with the substrate (or product), this problem can be discarded.
4.14. Change of the immobilized enzyme features caused by medium composition
The composition of the reaction medium can in some instances have dramatic effects on the final enzyme properties. In some cases, this may be deliberate, such as adding components that can stabilize the enzyme (e.g., polyols, polyethylene glycol, competitive inhibitors, reaction products),426–430 with the objective of reducing enzyme inactivation during the immobilization. These compounds are included in the immobilization protocol and ideally should be added in each immobilization cycle, so as not to alter the reproducibility of the immobilization itself.In other cases, the added components have the objective of preventing or reversing enzyme aggregation, such as the addition of urea to dissociate multi-protein complexes408,409 or the use of detergents when immobilizing lipases to prevent the immobilization of lipase-lipase dimers or break the interaction of lipases with hydrophobic compounds.431–435 Again, these factors may be important to help achieve an optimal immobilized enzyme preparation, but their presence will usually be included in the immobilization protocol.
The problems that we want to discuss in this section arise when the composition of the crude enzyme extract is not fully controlled by the researcher. In most cases, in the laboratory, but even more commonly in industry, the immobilization is performed directly using this crude preparation, whose compositions may vary from batch to batch. One can assume that in these commercial crude enzyme preparations there are additives to stabilize the enzyme, as well as some compounds to inhibit microbial growth, etc. The stabilization may be due to direct enzyme–additive interaction, that can block certain areas of the protein, and that can result even in covalent modification of the enzyme. It has recently been shown that small changes in the immobilization medium, such as the presence of some cations or anions, glycerin, etc., may dramatically alter the functional properties of immobilized lipases.436–438 If this is done deliberately, there is a good chance of producing a library of biocatalysts with different properties and to increase the possibilities of finding some biocatalyst with suitable properties for the target process. However, one can expect that when the enzyme supplier alters the composition of the crude enzyme without informing the users, one can produce biocatalysts with different properties, and this will therefore be in an uncontrolled way. Hence, an immobilization method should be fully reproducible using the same enzyme batch, but this may not be the case using different batches.
A further problem is when some of the components of the crude enzyme, added to alter the enzyme properties (such as enzyme stabilizers), become adsorbed to the support during immobilization. Later, these compounds may be partially released from the support, depending on the conditions, and may even alter the enzyme properties (e.g., stability). The change of the nature or concentration of these compounds among different enzyme batches may greatly alter the apparent enzyme features. The enzyme loading may also play an important role in the extent of this problem. At low enzyme loadings, the concentration of these reagents will be lower, whilst when using high enzyme loadings, this artifact will increase. The effect is important in the characterization of an immobilized enzyme in the laboratory, where small volumes are used in experiments. In industrial operation, this effect will be smaller due to the usually high biocatalyst/reaction media ratio, and in any case, it will decrease over time, number of reuses or reaction time (in continuous reactions). The release of the additive may be higher or lower depending on the reaction conditions, but in any case may produce a discrepancy from the results obtained in the laboratory and in the factory. We have not found any report in the scientific literature analyzing this possibility.
Focusing on the immobilization yield, the interaction of the stabilizers with the enzyme may alter the possibilities of enzyme immobilization, as some of them can interact with the enzyme surface. If the nature or concentration of the additives change, to have fully reproducible results from one batch to the next may not be simple. The problem may be more complex if the enzyme is stored for a long time prior to immobilization, permitting an increase in the enzyme modification or even the production of some covalent adducts. In our laboratory, we have seen such results when immobilizing lipases on octyl agarose.167 In many examples, the initial immobilization yield was almost 100%, but in some cases activity, specificity and even stability of the immobilized enzymes differed from one batch to the next. After long-term storage (for months), even though the lipase activity was maintained, the immobilization yield decreased, as most of the enzyme surface was coated by additives, and after even longer storage (for years), immobilization became impossible (unpublished results). This can suggest that the immobilization protocol is not reproducible, when in reality it is the starting material that is the one that has been changed.
Another possibility is the presence of compounds able to interfere in the immobilization. For example, it has recently been shown that the presence of compounds bearing primary amino groups may alter the immobilization rate and the final stability of enzymes immobilized in glyoxyl agarose.439 The problems were more significant when the aminated compounds were larger and their concentrations significant, but even 1 mM of these compounds may affect the immobilized enzyme properties. The aminated compounds may be peptides, amino acids, or amino saccharides that remain in the crude enzyme mixture or even Tris buffer used to store the enzyme.293 Most of the enzyme suppliers will not advise on their presence nor control their concentrations, which may differ from one batch to the next. In the laboratory, a simple dialysis can avoid this problem, although in industry, this may prove more problematic. The use of different enzyme concentrations during the immobilization will also alter the presence of these unknown compounds, meaning that differences in the immobilization performance may also be found even when using the same enzyme batch, but at different concentrations (e.g., if the researcher wants to increase the enzyme loading).
In this way, even using a very robust immobilization method, if the initial material is not well characterized, we can have quite different results using different enzyme batches.
4.15. Change of results dependent on enzyme loading
Use of immobilized enzyme with different enzyme loadings is one of the cases where the immobilization features of one protocol are not directly translatable to others (Fig. 14). In the laboratory, most of the studies are performed at low enzyme loading, while in industry usually maximal loading is employed, as this is economically advantageous. As discussed in several sections of this paper, high enzyme loading is also interesting for process intensification. However, this can give rise to several problems (Fig. 15) when comparing low and high enzyme loadings.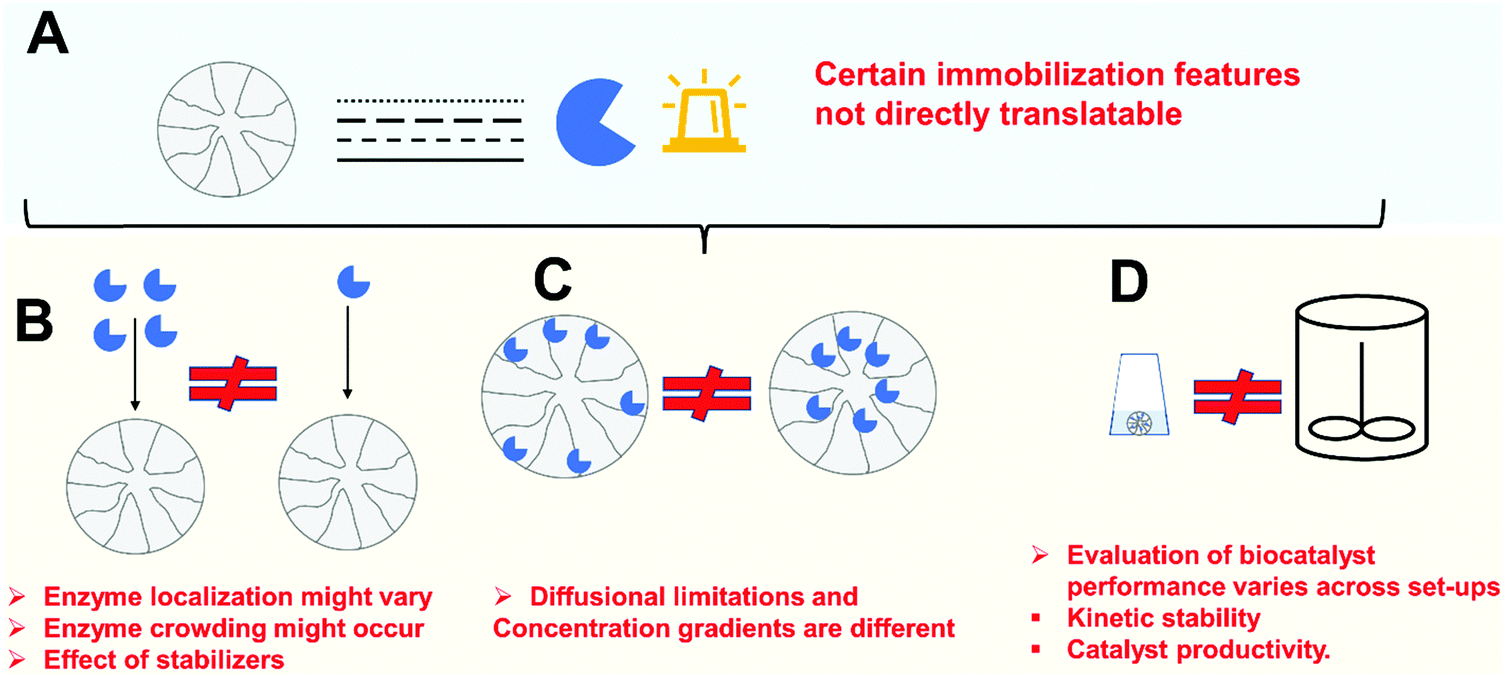 | ||
| Fig. 14 Features of enzyme immobilization that are not easily translatable. Properties of the immobilized enzyme depend on the interplay of material properties, loading, enzyme, chemistry of immobilization (A). Enzyme loading might affect biocatalyst performance beyond volumetric activity (B) enzyme localization might vary, affecting activity/stability (C), properties of the biocatalyst should be evaluated under realistic operating conditions (D). | ||
 | ||
| Fig. 15 Consequences of different enzyme loadings on biocatalyst properties. Use of immobilized enzyme with different enzyme loadings is one of the cases where the immobilization features of one protocol are not directly translatable to another. This can occur when the geometry of the material support is not fully homogeneous and a heterogeneity in the enzyme distribution develops (A); different enzyme loadings lead to very different surface density of immobilized enzymes resulting in steric hindrance for substrate access (B); different enzyme loadings can lead to different enzyme distributions within a porous particle affecting the balance of reaction–diffusion of substrate/product and therefore catalytic properties (C); when the enzyme to be immobilized is not pure different loadings can lead to different apparent enzyme immobilization yields since other (more rapidly immobilized) contaminant proteins can saturate the accessible material support. | ||
The first problem is that the internal geometry of the support may not be fully homogeneous (Fig. 15A). This means that in some areas the geometrical congruence of the enzyme with the support may be higher than in others, even accounting for only 10% of the total surface area (e.g., in knots between fibers). The enzymes will be preferentially immobilized in these areas, where higher enzyme stabilization may be achieved. When using fully loaded biocatalysts, 90% of the enzyme will be immobilized in the area where the enzyme–support interactions are lowest, and it is possible that the immobilized enzyme stability will be decreased compared to studies using low loadings, as the enzyme–support geometric congruence is very important for the positive and negative effects of the support on the resulting immobilized enzyme stability.200,403
The enzyme loading of the immobilized biocatalyst also affects the observed enzyme specificity (but not selectivity), when the enzyme specificity is not absolute (Fig. 15B).365 For example, when using low enzyme loading, as there are no diffusional gradients for any of the substrates, the detection of the real enzyme activity and specificity may be achieved. The use of higher enzyme loadings, that allow higher volumetric activities where the concentration of the better substrate for the enzyme suffers a gradient (see above)4,326,335 in the biocatalyst particle while the concentration of the poorer substrate remains almost constant through the whole particle, may result in an apparent specificity decrease when the enzyme loading is increased.99,365,440–443
A further problem is the possible effects of protein crowding on immobilized enzymes. It has been recently shown that in biocatalysts where the enzymes are rapidly immobilized (much more rapidly than the enzyme diffusion rate) and fully cover the support surface, the protein–protein interactions among the immobilized enzymes may alter the enzyme properties, and produce biocatalysts with very different features, not only activity, but also stability/selectivity or specificity438,444,445 (Fig. 15C).
A final problem that can arise is if the soluble protein solution that is used to prepare the biocatalysts is not pure, but contains contaminant proteins (Fig. 15D). This problem arises when there are other proteins in the solution that can be immobilized on the support more rapidly than the target protein itself. In this case, the immobilization yield/offered protein ratio will reach a maximum and then start to decrease, when the support surface is full and then the target enzyme needs to compete with the other protein. This makes it necessary to control the protein offered to the support in a very strict way in order to build biocatalysts with maximum activity loading. The problem is that most enzyme suppliers only guarantee the enzyme activity per mass or per volume, rather than stating the nature and concentration of the contaminants. This can result in changes when using different batches of soluble enzyme, and therefore the amount of protein that can be immobilized on the support. In turn this may alter the protein loading at which the “maximum” activity is achieved.99
4.16. The catalytic properties of the immobilized biocatalyst are altered because there is more than one enzyme form
Nowadays, most enzymes are produced after cloning them in fungi or yeasts.446–451 That means that the enzymes are glycosylated, although in many cases, the degree of glycosylation is not homogeneous (that is, in one enzyme batch may coexist enzymes bearing different number, size and composition of sugar chains) and the supplier does not guarantee that this is identical in all batches.452 Considering that the glycosylation may affect many enzyme properties452 and that the sugar chains may promote difficulties for the enzyme immobilization via the protein core, these differences in glycosylation may produce different immobilization rates, immobilization yields, and immobilized enzyme properties.100,211,453–458 Again, possible differences in the final biocatalysts may be caused by differences in the used enzyme, not by a lack of reproducibility of the immobilization protocol itself.In some cases, the enzyme solution may contain some other enzymes also able to catalyze the target reaction.459,460 This is an unfortunate situation, but occurs in several instances, mainly when the contaminant is a minority but very active enzyme, sometimes not even detected using standard SDS-PAGE.461 However, its total activity against the target substrate may be very significant, and its properties the opposite to that of the target enzyme of interest. This means that the specificity and selectivity that we observe when using the impure enzyme solution may differ greatly from one batch to the next. As a consequence, the immobilized enzyme preparations may have different features if both enzymes are immobilized and their ratio is altered in different batches.101 In these cases, there are some situations where the immobilization may alter the features of the final biocatalyst, solely by altering the activity ratio between both enzymes.101 Usually, the immobilization of an enzyme is followed using some synthetic substrates, which are easy to measure, and it is possible that the target enzyme is mainly responsible for the activity of the impure enzyme solution on that substrate. However, one of the contaminant proteins may be almost inactive against the synthetic substrate and very active against the target substrate. It is possible that one of the enzymes becomes inactivated after immobilization while the other enzyme is not, or even that only one of the enzymes is immobilized on the support.101 This greatly alters the biocatalyst properties, but not because we are tuning the enzyme features, but because we are eliminating one of the enzyme activities from the final biocatalyst. Following an entire immobilization course with the target substrate will reveal the real reason for these results.
This is not really a problem of the immobilization, but can offer some confusing results unless the researcher considers all the possibilities.
4.17. Change of the enzyme conformation upon immobilization: change of enzyme properties
Enzyme immobilization, except if is via just a single covalent bond using a long spacer arm and a very hydrophilic support, may be expected to alter the enzyme conformation by interactions between the enzyme and the surface of the support.101,297,298 As the catalytic enzyme properties are closely related to the enzyme conformation, it may be assumed that the activity, specificity, selectivity and even inhibition of the immobilized enzyme will be altered. If the enzyme has a flexible active center (e.g., lipases, multimeric enzymes), then these changes may be tolerated without a significant decrease in catalytic activity, and this may be used to tune their properties.331,365,462–470 If the enzyme is to be used in a process where its properties are not completely optimal, generating a large library of very differently immobilized biocatalysts has proved to be a good option to find biocatalysts with the desired features.101 In some cases, even inversion of enzyme specificity has been reported upon immobilization.465 However, this also means that if the soluble enzyme has suitable catalytic properties, its immobilization may alter it and these desirable properties may be lost. It is not unlikely that optimal stability of the enzyme may be achieved using immobilization protocols activity that are different to those that give optimal specificity or selectivity, and a compromise solution may be needed to select the most adequate biocatalyst for the target process.4.18. Use of supports having some catalytic components
In some instances, the support may pose some catalytic activity, sometimes neglected by the researcher. Usually, the catalytic activity of a support in the standard reactions used for enzyme activity determination is assayed, and if the support modifies the substrate, it is discarded. However, this may be not enough, as it is possible that in the final reaction (to be scale-up industrially), the substrate and product may be different, having new groups that can be attacked by the unknown support catalytic components. It should be also considered that the reaction conditions may differ to the standard ones and alter this catalytic activity. In this way, in real operation using the immobilized enzyme, some undesired reaction may be catalyzed by the support, sometimes affecting the reaction substrate or product, but in other cases affecting even the enzyme stability.For example, metals are good catalysts for many reactions471–474 and they are present in many of the currently used enzyme immobilization protocols. That is the case for supports activated with immobilized metal chelates,475,476 magnetic supports,477–479 nano-flowers,52,55,58 or metal organic frameworks,480–483 for example. The metal in the support may be in one stage and not be catalytically active, but it is not unlikely that during operation, it may be released (and this may already alter its catalytic potential) or it can suffer from oxidations or reductions, giving completely new catalytic potential to the metal.484–486 Hence, the use of metals in enzyme immobilization supports should be performed only after careful evaluation of its inertness in the target reaction under operation conditions. In fact, even if the activity follows the direction of interest, they could have other capabilities or selectivities, driving the production of some unexpected by-products. In a similar way, the presence of metals in solution may have negative effects on enzyme stability in the presence of oxidants like hydrogen peroxide, increasing its inactivating power.350
4.19. From batch to continuous reactors and process intensification
Immobilized enzymes are usually assayed and analyzed in small vials or cuvettes submitted to magnetic stirring or orbital shaking. The properties herein analyzed (activity, stability) are therefore targeted to be translated into reaction metrics in a suitable reactor. However these properties might not be directly translatable. This can be due to a lack of standardized procedures for evaluation/characterization311 as commented later or due to the lack of anticipation of implementation bottlenecks based on unknown underlying phenomena. For example, in the state-of-the-art of applied biocatalysis, a lot of attention has been devoted to the transition from batch to continuous reactors, where enzyme is immobilized onto the wall of micro-channel reactors or the particles containing immobilized enzyme are packed in a fixed-bed reactor.86,88,90 The reaction then proceeds with the continuous flow of the reaction medium through the system. The reactor productivity not only depends on the biocatalyst properties (activity, stability) but also on suitable fluid-dynamics of the reaction medium through the bed. From the initial screening and preparation of the immobilized enzyme up to the suitable application in the reactor, a series of issues can generate problems. One is related to the characteristics of the support material. As commented previously, the physico-chemical features of the carrier have a critical influence on the mass-transfer situation and, therefore the effectiveness and catalyst productivity that can be achieved. The particle size and porosity that are suitable to minimize diffusional restrictions in batch might not be suitable for implementation in a fixed bed reactor. In fact, a sub-optimal catalyst, and not the optimal one, prepared and characterized in batch might be the one that fits the operational window of the flow reactor. Something similar can occur with enzyme loading.Different functional consequences as a result of changes in the enzyme loading have been previously discussed in this review. From a practical perspective, the enzyme loading (or better, the volumetric activity of the biocatalysts) defines new constraints in the use of the catalyst in the reactor. Thus, the enzyme loading must not only be suited to switch from kinetic to diffusional control, but also limit the reaction time. Whereas in a stirred tank reactor the enzyme loading in the support and the enzyme loading in the reactor can be set independently (to a certain extent), in a fixed bed reactor the enzyme loading in the packed material is the only variable to define the maximum reactor productivity.487–489 In the extreme, different scenarios can occur that might result in infeasible conditions. For example, if the enzyme loading is very low, and therefore the reactor volumetric productivity is low, the residence time in the reactor might need to be very long to reach the target conversion yield.488 This might cause two undesirable effects. First, when a too low flow rate is used, the fluid velocity through the packed particles is low, which might create external mass transfer limitations and an external concentration gradient of the substrates decreasing the effectiveness factor of the catalyst. Secondly, at low enough superficial velocities, the reaction mixture would not necessarily flow under ideal plug-flow, since back mixing and high axial dispersion in the reactor would take place. Under these conditions, the conversion of the reactor would decrease.86,490 On the contrary, if the enzyme loading is too high, the volumetric activity would also be high, and the contact time (residence time) between the catalyst and the reaction medium might be too short. This might generate high superficial velocities that increase pressure drop over the bed and result in preferential channels through the reactor (so-called channeling), and also decreasing the effectiveness of enzyme use. Additionally, in the case of unstable reaction intermediates or unstable products, it would be necessary to minimize the residence time to prevent decomposition. In conclusion. loading should be adapted to the requirements of the reaction in the flow reactor, and may be quite different to those in batch reactors.491,492
4.20. Differences in the preparation and characterization of immobilized enzymes in the laboratory and under industrial operating conditions. Aspects which are often overlooked
From the previous comments, it is clear that differences between preparation and characterization of an immobilized enzyme at the laboratory scale and those operating under industrial conditions can create problems for implementation, sometimes not easily detectable by routine application of immobilization protocols4,168 (Fig. 16). In the following section we summarize some of these problems. | ||
| Fig. 16 Different scales of preparation and characterization of immobilized enzymes. Differences between preparation and characterization of an immobilized enzyme at the laboratory scale and an industrial scale can create problems for implementation due to the different quantity and quality of information available at the two scales. Routinely applied protocols at a small scale, mean that many immobilization options can be screened and properties analyzed, but operational activity and stability and energy input can be easily misinterpreted. However, preparation and characterization of immobilized enzymes at a large scale requires considerable resources. Identification and selection of intermediate scales where an appropriate number of immobilization variants can be evaluated under a useful range of conditions is of paramount importance. | ||
One of the key issues is the lack of standardization of analysis protocols and activity reporting.311 One classic aspect is the difference in the substrate concentrations used in the quantification of immobilization yield and activity between academia and industry.311,493 For monitoring enzyme immobilization, quick assays based on colorimetric substrates are frequently used. The expression of the activity of the catalyst useful for application however requires a more detailed and broader analysis. At some point in catalyst development high-throughput screening and quantity of information must be substituted by deeper information about the catalyst performance. However, for catalyst implementation, characterization in terms of activity is not sufficient. Instead the variation of the specific rate of the enzyme upon variation of the substrate concentration under realistic conditions of operations should give an insight into the operational window and productivity.4,89,382,490,494,495
Another important aspect is enzyme stability studies. Quick assessment of the suitability of an enzyme immobilization protocol is usually based on studies of stabilization that rely in many cases on measuring the thermodynamic stability of the protein, since it can be performed in automated instruments. However, for process implementation kinetic stability is more relevant.4,89,382,490,494,495 Still the researcher must decide about the amount of information acquired in the context of catalyst development. While analysis under real operational conditions would require extremely long experiments that would slow-down the immobilization development process, a first screening for stabilization consists in the selection of denaturing conditions where the reference catalyst is inactivated in the timeframe of minutes or hours. These conditions are, in many cases far away from the real application. Also, for simplicity, analysis of the stability relies on incubation under resting conditions. However, it has been described in the scientific literature how the substrate and/or product participates in irreversible inactivation/activation mechanisms of enzymes that can alter the operational stability of the biocatalyst.382,496–499
Finally, as in many other biotechnological processes, the mixing and energy input across scales is of major importance. At small scale, the mixing is easily guaranteed, allowing control of the chemical reaction not only during the preparation of the catalyst but also during reaction analysis. At larger scales the mixing time will increase, leading to concentration gradients.87
Suitable scaling-up of enzyme-immobilized catalysts and optimization of the properties of the immobilized enzymes is in many cases limited by the lack of identification of the limiting factor underlying the observable enzyme performance. When immobilized enzymes are designed and properties evaluated with the laboratory, most of the results are based on the measurement of substrate consumption or product formation rates. The analysis can be reduced to an initial reaction rate measurement from where activity is referred and the data are used for kinetic modeling, studies of pH-activity, temperature-activity-profile.311,326,364 Another possibility is the study of complete reaction courses, where data of concentration-time are obtained.89,490,494,500–502 These data can be also used for kinetic modelling, and also identification of different types of inactivation and inhibition.89,490,494,500–502 In any case the enzyme activity or the apparent kinetic constants are the result of a interplay of different factors, namely structural factors of immobilization and the enzyme microenvironment.224,302,326,335,362,503
Structural consequences of immobilization involve multiple phenomena across different scales from enzyme distribution to structural alteration of the immobilized enzyme. The structural alteration of the immobilized enzyme involves a large and long series of potential structural alterations of the enzyme once immobilized, relating from enzyme orientation to structural distortion.302,334 Recent advances in structural characterization of immobilized enzymes using microscopic imaging methods has provided useful insights.302,326 For example, enzyme distribution in porous particles is routinely analyzed by confocal scanning fluorescence microscopy. Different spectroscopic methods are used to analyze protein conformation on solid supports.302,326 Very recent advances in surface-sensitive spectroscopic techniques have provided evidence that push the determination of enzyme structure and orientation at the solid–liquid interface, in particular single-molecule studies showing that analyses sensitive to temporal and spatial heterogeneities in immobilized enzymes are useful to explain the effects of conformational stability and active-site accessibility on activity.302,504–507
In the interplay with the structural aspects of the immobilized enzyme the microenvironment is the other fundamental aspect determining enzyme performance. The microenvironment in which enzymes are acting when they are immobilized in solid materials is usually quite different from conditions in the bulk solution.218,224,326,401,503,508–510 The differences are due to the physical properties of the material and mass transfer effects. As a consequence substrate/product concentrations, pH and ionic strength can be very different and determine the enzyme performance. In a classic biocatalyst characterization, the interpretations are based on observations made in the liquid phase. Elucidation, through direct measurement of differences in the internal as compared to the bulk milieu is, therefore, fundamentally important in the mechanistic characterization of immobilized enzymes. Different approaches have been developed recently based on the direct fluorescent properties of substrate products or the opto-chemical sensing enabled by the labelling of immobilization materials.218,224,326,401,503,508–510
4.21. Some examples where immobilization can hardly improve enzyme stability
Even though the potential of immobilization to solve many enzyme limitations is high, there are some instances where enzyme inactivation cannot be prevented by immobilization, because using an enzyme with greater rigidity or partitioning undesired compounds will not improve the situation. In these cases, the advantages of enzyme immobilization are limited and it should be considered if its other advantages can justify the enzyme immobilization (see Introduction).One example of this situation may be if the enzyme is inactivated via suicide inhibition.511–515 If the enzyme has a certain probability of being inactivated during catalysis, immobilization can hardly improve these chances (perhaps by chance one can generate some better structures, but we have not found reports to support this).
In other cases, the main reason for enzyme inactivation is the loss of an ion, cofactor, or prosthetic group, or the oxidation of an enzyme residue, and this may even be accelerated if the immobilization distorts the enzyme structure and exposes more groups to the medium or decreases the affinity of the enzyme to this component, making easier its release.516,517 In this way, enzyme stability will be not increased even if the protein polymer is more rigid. There are some reports where an increase in the number of enzyme–support bonds reduces enzyme stability, usually when the enzyme has some of the features listed previously.100,166,268,518 Similarly, in some instances a too intense multi-point covalent attachment of multimeric enzymes may produce the weakening of the enzyme assembly, making convenient intermediate levels of multi-point covalent attachment to give the optimal immobilized enzyme stability.519
Hence, to understand the main causes of enzyme inactivation can be a key point to indicate if the immobilization may solve, or not, the stability problems of an enzyme, and help to take decisions regarding the convenience, or not, of using an immobilized enzyme, considering the other advantages discussed in the introduction.
4.22. Logistic problems to be considered before deciding to use an immobilized enzyme at an industrial scale
For an academic laboratory, some logistic problems may not be considered very relevant. However, for a factory, they may be of great importance.The first question is if the user company prefers to buy a commercial biocatalyst or prepare their own biocatalyst. To buy the biocatalysts from a specialist company may be simpler, but that limits possibilities for tuning and developing the biocatalyst, and the user company must rely on the good control of the product that they receive from the supplier.
To make their own biocatalyst gives more opportunities to improve the enzyme features in the direction required by the company. However, this necessitates buying independently supports and enzymes, and the user company must be able to control the reproducible quality of these materials.
In both cases, there are risks of a withdrawal of a specific support from the market, as has been the case of Eupergit by Rohm and Hass. This makes it necessary to look for a similar product in the market and very likely, re-optimize the preparation of the biocatalyst.
This possibility is hard to control, and for many companies, this risk may be excessive and they may prefer the use of soluble enzymes. However, it is very unlikely that an immobilization support, with many costumers, will be withdrawn from the market if it is not by the pressure of some competitor, usually with some advantages over the former.
5. Concluding remarks
This new review has a very different focus compared to most existing reviews on enzyme immobilization. Most reviews to date have focused on the benefits of enzyme immobilization, and how it can improve many enzyme properties (including stability, selectivity, specificity, resistance to inhibition, or how enzyme purification may be coupled to enzyme immobilization), without emphasizing the problems that can be found in immobilization. This has given the general wrong idea that any “enzyme immobilization” can promote these enzyme improvements, and many authors do not make a clear distinction between a properly designed immobilization protocol and a random immobilization. In many cases, if the immobilization of an enzyme has some undesired effects, immobilization as a whole is discarded for this enzyme or process. Even in comparison between different enzyme immobilization techniques or of immobilization techniques with other alternatives (as the use of whole cell biocatalysts), just one support, active group in the support and immobilization protocol (usually not properly optimized) are used, making unfair comparisons in most cases. This review presents some critical considerations to take full advantage of the enzyme immobilization process, commenting on all points that, if not considered, can drive an immobilized enzyme to have properties far from the optimal ones, and in many cases worse than those of the soluble enzyme, without taking full advantage of the enzyme immobilization. We have attempted to describe the different problems that an uncontrolled or non-properly understood enzyme immobilization protocol can present, but also how this may be solved to finally take full advantage of the immobilization process (at least in most instances).In this way, an immobilization system must be conceptually considered to be formed by three different components: an adequate support, a suitable active group in the support and a proper immobilization protocol (including activation of the support, enzyme immobilization conditions, enzyme–support multi-interaction conditions and reaction end-point for covalent immobilization). If one of these aspects is not properly considered in the enzyme immobilization, the results may be far from the best possible, and in some instances give an enzyme with even worse properties than the soluble enzyme, when a proper protocol can give an enzyme much more stable enzyme form (even thousands of fold more stable immobilized enzymes may be produced).
On the other hand, many phenomena are still not fully understood about how enzymes and supports interact and consequently how this can affect the properties of immobilized enzymes. In fact, in some cases an interaction that is positive for an enzyme under specific conditions, is negative for an apparently similar enzyme, or under other conditions.
It cannot be considered that a universal and perfect enzyme immobilization protocol exists to give optimal enzyme features, at least in terms of activity/stability for all enzymes. In fact, there are too few protocols that can be really considered as adequate to give an intense multi-point covalent attachment and each of them has their own advantages and limitations.
Moving to the design of co-immobilized enzymes, the necessity of developing new strategies is even more obvious, as many problems are still without a solution (e.g., how to shortcut the problem of different enzyme stabilities, how this may affect the biocatalyst reusability, how can different enzyme locations be used without discarding immobilization as a tool to improve enzyme features, etc.) or even have not been detected to date. Advances in new immobilization protocols may be key for this situation.
The preparation of an industrial immobilized biocatalyst can be conceptually complex, but to be successfully used, it must be methodologically simple. In this way, we consider that enzyme immobilization is still a discipline where intense research is required. This research should be extended to areas including:
• material sciences, that should design new support materials that are very hydrophilic, physically inert at the end of the immobilization process, mechanical and chemically resistant, with controllable pore and particle size
• reactor engineering, including design of new reactors compatible with fragile supports, methods for biocatalyst recovery, more efficient but not harmful stirring systems, better control of the reaction conditions
• protein chemistry and dynamic simulation, to predict the type and intensity of the enzyme–support interactions
• organic chemistry, including new reactive groups in the support without the limitations of the current ones
• bioprocess engineering, for understanding, modelling and controlling the underlying phenomena: reaction and of the mass transfer phenomena.
Special interest may be required on coupling tools (such as chemical or genetic modifications of enzymes) to develop immobilized enzymes with better properties. Therefore, although enzyme immobilization is considered by many researchers to be a mature discipline where almost all has already been done, considering all these facts, we rather consider that enzyme immobilization requires a yet deeper research to define better immobilization protocols and to explain and avoid some of the undesired effects on enzyme features after immobilization. Moreover, many problems of enzyme immobilization or problems that can be solved by proper immobilization protocols may still be hidden, although we can foresee some and we have advanced some in this review paper (e.g., mobility of the reversibly immobilized enzymes on the support surface). We can consider that we are still far from the borders of this research area and that the future can bring many new and remarkable advances.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully recognize the financial support from Ministerio de Ciencia e Innovación-Spanish Government (project number CTQ2017-86170-R). This work is also part of a project at DTU (JMW) that has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Programme (Grant agreement No. 101021024). JMB acknowledges funding from the Government of Community of Madrid (2018-T1/BIO-10200) and from UCM-Santander (PR108/20).References
- R. DiCosimo, J. McAuliffe, A. J. Poulose and G. Bohlmann, Chem. Soc. Rev., 2013, 42, 6437 RSC.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- U. Hanefeld, L. Gardossi and E. Magner, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- A. Liese and L. Hilterhaus, Chem. Soc. Rev., 2013, 42, 6236–6249 RSC.
- A. Basso and S. Serban, Mol. Catal., 2019, 479, 110607 CrossRef CAS.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- M. B. Ansorge-Schumacher and O. Thum, Chem. Soc. Rev., 2013, 42, 6475–6490 RSC.
- D. Valikhani, J. M. Bolivar and J. N. Pelletier, ACS Catal., 2021, 11, 9418–9434 CrossRef CAS.
- N. Lin and A. Dufresne, Eur. Polym. J., 2014, 59, 302–325 CrossRef CAS.
- K. Hernandez and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 48, 107–122 CrossRef CAS PubMed.
- R. A. Sheldon and P. C. Pereira, Chem. Soc. Rev., 2017, 46, 2678–2691 RSC.
- P. Domínguez de María, Curr. Opin. Green Sustainable Chem., 2021, 31, 100514 CrossRef.
- F. Müller, B. Torger, P. J. Allertz, K. Jähnichen, S. Keßler, M. Müller, F. Simon, K. Salchert, H. Mäurer and D. Pospiech, Eur. Polym. J., 2018, 102, 47–55 CrossRef.
- A. Pollak, H. Blumenfeld, M. Wax, R. L. Baughn and G. M. Whitesides, J. Am. Chem. Soc., 1980, 102, 6324–6336 CrossRef CAS.
- A.-C. Johansson and K. Mosbach, Biochim. Biophys. Acta, Enzymol., 1974, 370, 339–347 CrossRef CAS.
- J. Bryjak and A. Noworyta, Bioprocess Eng., 1993, 9, 37–42 CrossRef CAS.
- S. Vidhya and B. M. Murari, Int. J. Pharma Bio Sci., 2016, 7, P297–P310 Search PubMed.
- P. Tielmann, H. Kierkels, A. Zonta, A. Ilie and M. T. Reetz, Nanoscale, 2014, 6, 6220–6228 RSC.
- M. R. N. Monton, E. M. Forsberg and J. D. Brennan, Chem. Mater., 2012, 24, 796–811 CrossRef CAS.
- K. Sakai-Kato and K. Ishikura, Anal. Sci., 2009, 25, 969–978 CrossRef CAS PubMed.
- J. D. Brennan, Acc. Chem. Res., 2007, 40, 827–835 CrossRef CAS PubMed.
- A. M. M. Ficanha, C. E. D. Oro, E. Franceschi, R. M. Dallago and M. L. Mignoni, Appl. Biochem. Biotechnol., 2021, 193, 2162–2181 CrossRef CAS PubMed.
- A. I. Dudu, M. A. Lăcătuş, L. C. Bencze, C. Paizs and M. I. Toşa, ACS Sustainable Chem. Eng., 2021, 9, 5461–5469 CrossRef CAS.
- N. A. Mohidem, H. Bin Mat, M. Mohamad, F. Hamzah and M. U. Rashid, J. Sol–Gel Sci. Technol., 2021, 98, 462–469 CrossRef CAS.
- L. Betancor, F. López-Gallego, A. Hidalgo, M. Fuentes, O. Podrasky, G. Kuncova, J. M. Guisán and R. Fernández-Lafuente, Biomacromolecules, 2005, 6, 1027–1030 CrossRef CAS PubMed.
- T. E. Abraham, J. R. Joseph, L. B. V. Bindhu and K. K. Jayakumar, Carbohydr. Res., 2004, 339, 1099–1104 CrossRef CAS PubMed.
- M. A. Navia, N. L. St. Clair and J. P. Griffith, Studies in Organic Chemistry, Elsevier, 1993, vol. 47, pp. 63–73 Search PubMed.
- M. Staar, S. Henke, W. Blankenfeldt and A. Schallmey, ChemCatChem, 2022, 14, e202200145 CrossRef CAS.
- J. D. Vaghjiani, T. S. Lee, G. J. Lye and M. K. Turner, Biocatal. Biotransformation, 2000, 18, 151–175 CrossRef CAS.
- L. Cao, F. van Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361–1364 CrossRef CAS PubMed.
- R. A. Sheldon, Appl. Microbiol. Biotechnol., 2011, 92, 467–477 CrossRef CAS PubMed.
- R. A. Sheldon, Org. Process Res. Dev., 2011, 15, 213–223 CrossRef CAS.
- R. Schoevaart, M. W. Wolbers, M. Golubovic, M. Ottens, A. P. G. Kieboom, F. van Rantwijk, L. A. M. van der Wielen and R. A. Sheldon, Biotechnol. Bioeng., 2004, 87, 754–762 CrossRef CAS PubMed.
- R. A. Sheldon, Biochem. Soc. Trans., 2007, 35, 1583–1587 CrossRef CAS PubMed.
- X.-Y. Li, M.-Q. Xu, H. Liu, Q. Zhou, J. Gao and Y.-W. Zhang, Bioprocess Biosyst. Eng., 2022, 45, 353–364 CrossRef CAS PubMed.
- R. Sheldon, Catalysts, 2019, 9, 261 CrossRef CAS.
- N. Gaggero and D. C. M. Albanese, Curr. Org. Chem., 2021, 25, 2666–2675 CrossRef CAS.
- L. Wilson, A. Illanes, O. Abián, B. C. C. Pessela, R. Fernández-Lafuente and J. M. Guisán, Biomacromolecules, 2004, 5, 852–857 CrossRef CAS PubMed.
- C. Mateo, B. Fernandes, F. van Rantwijk, A. Stolz and R. A. Sheldon, J. Mol. Catal. B: Enzym., 2006, 38, 154–157 CrossRef CAS.
- B. Wei, F. Liu, X. Liu, L. Cheng, Q. Yuan, H. Gao and H. Liang, Colloids Surf., B, 2022, 210, 112241 CrossRef CAS PubMed.
- J. Del Arco, A. R. Alcántara, R. Fernández-Lafuente and J. Fernández-Lucas, Bioresour. Technol., 2021, 322, 124547 CrossRef CAS PubMed.
- S. K. Rai, V. Kumar and S. K. Yadav, Catal. Sci. Technol., 2021, 11, 2186–2194 RSC.
- A. A. Mehde, Int. J. Biol. Macromol., 2019, 131, 721–733 CrossRef CAS PubMed.
- J. George, D. S. Rajendran, S. Venkataraman, A. K. Rathankumar, K. Saikia, S. Muthusamy, I. Singh, I. Singh, S. Sinha, S. Ramkumar, H. Cabana and V. K. Vaidyanathan, Environ. Res., 2022, 209, 112882 CrossRef CAS PubMed.
- H. Tirunagari, S. Basetty, H. B. Rode and N. W. Fadnavis, J. Biotechnol., 2018, 282, 67–69 CrossRef CAS PubMed.
- P. Arranz-Martínez, M. Corzo-Martínez, L. Vázquez, G. Reglero and C. F. Torres, Process Biochem., 2018, 64, 38–45 CrossRef.
- M. D. Ramos, L. P. Miranda, R. Fernandez-Lafuente, W. Kopp and P. W. Tardioli, Molecules, 2019, 24, 4392 CrossRef CAS PubMed.
- N. N. Nawawi, Z. Hashim, R. A. Rahman, A. M. A. Murad, F. D. A. Bakar and R. M. Illias, Int. J. Biol. Macromol., 2020, 150, 80–89 CrossRef CAS PubMed.
- L. Wilson, A. Illanes, B. C. C. Pessela, O. Abian, R. Fernández-Lafuente and J. M. Guisán, Biotechnol. Bioeng., 2004, 86, 558–562 CrossRef CAS PubMed.
- J. Cui, S. Jia, L. Liang, Y. Zhao and Y. Feng, Sci. Rep., 2015, 5, 14203 CrossRef PubMed.
- J. D. Cui, L. L. Li and H. J. Bian, PLoS One, 2013, 8, e80581 CrossRef PubMed.
- C. Altinkaynak, S. Tavlasoglu, N. Ÿzdemir and I. Ocsoy, Enzyme Microb. Technol., 2016, 93–94, 105–112 CrossRef CAS PubMed.
- S. Escobar, S. Velasco-Lozano, C.-H. Lu, Y.-F. Lin, M. Mesa, C. Bernal and F. López-Gallego, J. Mater. Chem. B, 2017, 5, 4478–4486 RSC.
- B. Zhang, P. Li, H. Zhang, H. Wang, X. Li, L. Tian, N. Ali, Z. Ali and Q. Zhang, Chem. Eng. J., 2016, 291, 287–297 CrossRef CAS.
- X. Wu, M. Hou and J. Ge, Catal. Sci. Technol., 2015, 5, 5077–5085 RSC.
- Y. Yin, Y. Xiao, G. Lin, Q. Xiao, Z. Lin and Z. Cai, J. Mater. Chem. B, 2015, 3, 2295–2300 RSC.
- F. P. Costa, E. P. Cipolatti, A. Furigo Junior and R. Oliveira Henriques, Chem. Rec., 2022, 22, e202100293 CrossRef PubMed.
- X. Liang, Y. Liu, K. Wen, W. Jiang and Q. Li, J. Mater. Chem. B, 2021, 9, 7597–7607 RSC.
- X. Luo, A. H. Mohammed Al-Antaki, A. Igder, K. A. Stubbs, P. Su, W. Zhang, G. A. Weiss and C. L. Raston, ACS Appl. Mater. Interfaces, 2020, 12, 51999–52007 CrossRef CAS PubMed.
- M. Luo, M. Li, S. Jiang, H. Shao, J. Razal, D. Wang and J. Fang, J. Hazard. Mater., 2020, 381, 120947 CrossRef CAS PubMed.
- T. Sun, M. Fu, J. Xing and Z. Ge, Water Sci. Technol., 2020, 81, 29–39 CrossRef PubMed.
- R. Fotiadou, M. Patila, M. A. Hammami, A. Enotiadis, D. Moschovas, K. Tsirka, K. Spyrou, E. P. Giannelis, A. Avgeropoulos, A. Paipetis, D. Gournis and H. Stamatis, Nanomaterials, 2019, 9, 808 CrossRef CAS PubMed.
- H. Zhang, X. Fei, J. Tian, Y. Li, H. Zhi, K. Wang, L. Xu and Y. Wang, Catal. Commun., 2018, 116, 5–9 CrossRef CAS.
- M. Kreiner, M. C. Parker and B. D. Moore, Chem. Commun., 2001, 1096–1097 RSC.
- M. Kreiner and M.-C. Parker, Biotechnol. Lett., 2005, 27, 1571–1577 CrossRef CAS PubMed.
- R. R. C. Monteiro, J. C. S. dos Santos, A. R. Alcántara and R. Fernandez-Lafuente, Catalysts, 2020, 10, 891 CrossRef CAS.
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C.-K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef CAS PubMed.
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H.-C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC.
- S. Cantone, V. Ferrario, L. Corici, C. Ebert, D. Fattor, P. Spizzo and L. Gardossi, Chem. Soc. Rev., 2013, 42, 6262–6276 RSC.
- A. Fasim, V. S. More and S. S. More, Curr. Opin. Biotechnol., 2021, 69, 68–76 CrossRef CAS PubMed.
- M. Becker, S. Lütz and K. Rosenthal, Molecules, 2021, 26, 573 CrossRef CAS PubMed.
- J. M. Woodley, Comput. Chem. Eng., 2017, 105, 297–307 CrossRef CAS.
- J. M. Bolivar and B. Nidetzky, Green Process. Synth., 2013, 2, 0091 Search PubMed.
- J. Price, M. Nordblad, H. H. Martel, B. Chrabas, H. Wang, P. M. Nielsen and J. M. Woodley, Biotechnol. Bioeng., 2016, 113, 1719–1728 CrossRef CAS PubMed.
- P. M. Nielsen, A. Rancke-Madsen, H. C. Holm and R. Burton, J. Am. Oil Chem. Soc., 2016, 93, 905–910 CrossRef CAS.
- C. Bhatt, P. M. Nielsen, A. Rancke-Madsen and J. M. Woodley, Biotechnol. Appl. Biochem., 2020, 69, 7–19 CrossRef PubMed.
- R. A. Sheldon, A. Basso and D. Brady, Chem. Soc. Rev., 2021, 50, 5850–5862 RSC.
- R. A. Sheldon, Adv. Synth. Catal., 2007, 349, 1289–1307 CrossRef CAS.
- V. Kasche, Enzyme Microb. Technol., 1986, 8, 4–16 CrossRef CAS.
- R. C. Giordano, M. P. A. Ribeiro and R. L. C. Giordano, Biotechnol. Adv., 2006, 24, 27–41 CrossRef CAS PubMed.
- S. R. Marsden, L. Mestrom, D. G. G. McMillan and U. Hanefeld, ChemCatChem, 2020, 12, 426–437 CrossRef CAS.
- V. G. Tacias-Pascacio, R. Morellon-Sterling, E.-H. Siar, O. Tavano, Á. Berenguer-Murcia and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2020, 165, 2143–2196 CrossRef CAS PubMed.
- V. G. Tacias-Pascacio, D. Castañeda-Valbuena, R. Morellon-Sterling, O. Tavano, Á. Berenguer-Murcia, G. Vela-Gutiérrez, I. A. Rather and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2021, 184, 415–428 CrossRef CAS PubMed.
- O. L. Tavano, A. Berenguer-Murcia, F. Secundo and R. Fernandez-Lafuente, Compr. Rev. Food Sci. Food Saf., 2018, 17, 412–436 CrossRef PubMed.
- M. E. Hassan, Q. Yang, Z. Xiao, L. Liu, N. Wang, X. Cui and L. Yang, 3 Biotech, 2019, 9, 440 CrossRef PubMed.
- M. P. Cardoso Marques, A. Lorente-Arevalo and J. M. Bolivar, Advances in Biochemical Engineering/Biotechnology, Springer Berlin Heidelberg, Berlin, Heidelberg, 2021 Search PubMed.
- R. Lindeque and J. Woodley, Catalysts, 2019, 9, 262 CrossRef CAS.
- J. M. Bolivar and F. López-Gallego, Curr. Opin. Green Sustainable Chem., 2020, 25, 100349 CrossRef.
- A. Lorente-Arevalo, M. Ladero and J. M. Bolivar, Curr. Opin. Green Sustainable Chem., 2021, 100544 CrossRef CAS.
- J. M. Bolivar, J. Wiesbauer and B. Nidetzky, Trends Biotechnol., 2011, 29, 333–342 CrossRef CAS PubMed.
- P. De Santis, L.-E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155–2184 RSC.
- A. I. Benítez-Mateos, M. L. Contente, D. Roura Padrosa and F. Paradisi, React. Chem. Eng., 2021, 6, 599–611 RSC.
- R. Wohlgemuth, I. Plazl, P. Žnidaršič-Plazl, K. V. Gernaey and J. M. Woodley, Trends Biotechnol., 2015, 33, 302–314 CrossRef CAS PubMed.
- P. Žnidaršič-Plazl, Curr. Opin. Green Sustainable Chem., 2021, 100546 CrossRef.
- H. M. Salvi and G. D. Yadav, Catal. Sci. Technol., 2021, 11, 1994–2020 RSC.
- F. Meyer, J. Johannsen, A. Liese, G. Fieg, P. Bubenheim and T. Waluga, Chem. Eng. Process. - Process Intensif., 2021, 167, 108506 CrossRef CAS.
- M. Romero-Fernández and F. Paradisi, Curr. Opin. Chem. Biol., 2020, 55, 1–8 CrossRef PubMed.
- M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18 CrossRef CAS.
- C. Garcia-Galan, Á. Berenguer-Murcia, R. Fernandez-Lafuente and R. C. Rodrigues, Adv. Synth. Catal., 2011, 353, 2885–2904 CrossRef CAS.
- R. C. Rodrigues, Á. Berenguer-Murcia, D. Carballares, R. Morellon-Sterling and R. Fernandez-Lafuente, Biotechnol. Adv., 2021, 52, 107821 CrossRef CAS PubMed.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- C. Mateo, J. M. Palomo, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2007, 40, 1451–1463 CrossRef CAS.
- L. Dal Magro, J. F. Kornecki, M. P. Klein, R. C. Rodrigues and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2020, 132, 109397 CrossRef CAS PubMed.
- G. Volpato, R. C. Rodrigues and R. Fernandez-Lafuente, Curr. Med. Chem., 2010, 17, 3855–3873 CrossRef CAS PubMed.
- R. Fernandez-Lafuente, C. M. Rosell and J. M. Guisán, Enzyme Microb. Technol., 1991, 13, 898–905 CrossRef CAS PubMed.
- R. Fernández-Lafuente, O. Hernández-Jústiz, C. Mateo, M. Terreni, G. Fernández-Lorente, M. A. Moreno, J. Alonso, J. L. García-López and J. M. Guisan, Biomacromolecules, 2001, 2, 95–104 CrossRef PubMed.
- M. P. Meissner and J. M. Woodley, Nat. Catal., 2022, 5, 2–4 CrossRef.
- R. C. Rodrigues, J. J. Virgen-Ortíz, J. C. S. dos Santos, Á. Berenguer-Murcia, A. R. Alcantara, O. Barbosa, C. Ortiz and R. Fernandez-Lafuente, Biotechnol. Adv., 2019, 37, 746–770 CrossRef CAS PubMed.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv., 2015, 33, 435–456 CrossRef CAS PubMed.
- O. Barbosa, R. Torres, C. Ortiz, Á. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Biomacromolecules, 2013, 14, 2433–2462 CrossRef CAS PubMed.
- B. C. C. Pessela, C. Mateo, M. Fuentes, A. Vian, J. L. Garcia, A. V. Carrascosa, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2003, 33, 199–205 CrossRef CAS.
- K. Gabrovska, I. Marinov, T. Godjevargova, M. Portaccio, M. Lepore, V. Grano, N. Diano and D. G. Mita, Int. J. Biol. Macromol., 2008, 43, 339–345 CrossRef CAS PubMed.
- C. Mateo, R. Monti, B. C. C. Pessela, M. Fuentes, R. Torres, J. M. Guisan and R. Fernandez-Lafuente, Biotechnol. Prog., 2004, 20, 1259–1262 CrossRef CAS PubMed.
- R. R. C. Monteiro, S. Arana-Peña, T. N. da Rocha, L. P. Miranda, Á. Berenguer-Murcia, P. W. Tardioli, J. C. S. dos Santos and R. Fernandez-Lafuente, Renew. Energy, 2021, 164, 1566–1587 CrossRef CAS.
- W. C. A. Carvalho, J. H. H. Luiz, R. Fernandez-Lafuente, D. B. Hirata and A. A. Mendes, Biomass Bioenergy, 2021, 155, 106302 CrossRef CAS.
- J. R. Guimarães, L. P. Miranda, R. Fernandez-Lafuente and P. W. Tardioli, Molecules, 2021, 26, 193 CrossRef PubMed.
- V. G. Tacias-Pascacio, J. J. Virgen-Ortíz, M. Jiménez-Pérez, M. Yates, B. Torrestiana-Sanchez, A. Rosales-Quintero and R. Fernandez-Lafuente, Fuel, 2017, 200, 1–10 CrossRef CAS.
- C. A. C. G. Neto, N. C. G. e Silva, T. de Oliveira Costa, T. L. de Albuquerque, L. R. B. Gonçalves, R. Fernandez-Lafuente and M. V. P. Rocha, Int. J. Biol. Macromol., 2021, 176, 468–478 CrossRef CAS PubMed.
- N. C. Dubey and B. P. Tripathi, ACS Appl. Bio Mater., 2021, 4, 1077–1114 CrossRef CAS PubMed.
- S. Arana-Peña, D. Carballares, R. Morellon-Sterlling, Á. Berenguer-Murcia, A. R. Alcántara, R. C. Rodrigues and R. Fernandez-Lafuente, Biotechnol. Adv., 2020, 107584 Search PubMed.
- E. T. Hwang and S. Lee, ACS Catal., 2019, 9, 4402–4425 CrossRef CAS.
- K. Xu, X. Chen, R. Zheng and Y. Zheng, Front. Bioeng. Biotechnol., 2020, 8, 660 CrossRef PubMed.
- J. M. Sperl and V. Sieber, ACS Catal., 2018, 8, 2385–2396 CrossRef CAS.
- S. P. France, L. J. Hepworth, N. J. Turner and S. L. Flitsch, ACS Catal., 2017, 7, 710–724 CrossRef CAS.
- P. Fernandes and C. C. C. R. de Carvalho, Processes, 2021, 9, 225 CrossRef CAS.
- A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi and F. López-Gallego, ACS Sustainable Chem. Eng., 2018, 6, 13151–13159 CrossRef.
- A. I. Benítez-Mateos, E. San Sebastian, N. Ríos-Lombardía, F. Morís, J. González-Sabín and F. López-Gallego, Chem. – Eur. J., 2017, 23, 16843–16852 CrossRef PubMed.
- S. Velasco-Lozano, A. I. Benítez-Mateos and F. López-Gallego, Angew. Chem., Int. Ed., 2017, 56, 771–775 CrossRef CAS PubMed.
- A. H. Orrego, D. Andrés-Sanz, S. Velasco-Lozano, M. Sanchez-Costa, J. Berenguer, J. M. Guisan, J. Rocha-Martin and F. López-Gallego, Catal. Sci. Technol., 2021, 11, 3217–3230 RSC.
- A. I. Benítez-Mateos, C. Huber, B. Nidetzky, J. M. Bolivar and F. López-Gallego, ACS Appl. Mater. Interfaces, 2020, 12, 56027–56038 CrossRef PubMed.
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu and H. Yang, Science, 2019, 366, 1255–1259 CrossRef CAS PubMed.
- M. Santi, L. Sancineto, V. Nascimento, J. Braun Azeredo, E. V. M. Orozco, L. H. Andrade, H. Gröger and C. Santi, Int. J. Mol. Sci., 2021, 22, 990 CrossRef CAS PubMed.
- R. A. Sheldon and D. Brady, ACS Sustainable Chem. Eng., 2021, 9, 8032–8052 CrossRef CAS.
- C. J. Hartley, C. C. Williams, J. A. Scoble, Q. I. Churches, A. North, N. G. French, T. Nebl, G. Coia, A. C. Warden, G. Simpson, A. R. Frazer, C. N. Jensen, N. J. Turner and C. Scott, Nat. Catal., 2019, 2, 1006–1015 CrossRef CAS.
- J. A. McIntosh, T. Benkovics, S. M. Silverman, M. A. Huffman, J. Kong, P. E. Maligres, T. Itoh, H. Yang, D. Verma, W. Pan, H.-I. Ho, J. Vroom, A. M. Knight, J. A. Hurtak, A. Klapars, A. Fryszkowska, W. J. Morris, N. A. Strotman, G. S. Murphy, K. M. Maloney and P. S. Fier, ACS Cent. Sci., 2021, 7, 1980–1985 CrossRef CAS PubMed.
- S. Arana-Peña, C. Mendez-Sanchez, N. S. Rios, C. Ortiz, L. R. B. Gonçalves and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2019, 131, 989–997 CrossRef PubMed.
- S. Arana-Peña, D. Carballares, R. Morellon-Sterling, J. Rocha-Martin and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2022, 199, 51–60 CrossRef PubMed.
- S. Peirce, J. J. Virgen-Ortíz, V. G. Tacias-Pascacio, N. Rueda, R. Bartolome-Cabrero, L. Fernandez-Lopez, M. E. Russo, A. Marzocchella and R. Fernandez-Lafuente, RSC Adv., 2016, 6, 61707–61715 RSC.
- R. Morellon-Sterling, D. Carballares, S. Arana-Peña, E.-H. Siar, S. A. Braham and R. Fernandez-Lafuente, ACS Sustainable Chem. Eng., 2021, 9, 7508–7518 CrossRef CAS.
- S. Arana-Peña, D. Carballares, V. Cortés Corberan and R. Fernandez-Lafuente, Catalysts, 2020, 10, 1207 CrossRef.
- R. C. Rodrigues, Á. Berenguer-Murcia and R. Fernandez-Lafuente, Adv. Synth. Catal., 2011, 353, 2216–2238 CrossRef CAS.
- C. Bernal, K. Rodríguez and R. Martínez, Biotechnol. Adv., 2018, 36, 1470–1480 CrossRef CAS PubMed.
- R. Singh, M. Tiwari, R. Singh and J.-K. Lee, Int. J. Mol. Sci., 2013, 14, 1232–1277 CrossRef CAS PubMed.
- J. M. Woodley, Curr. Opin. Chem. Biol., 2013, 17, 310–316 CrossRef CAS PubMed.
- D. A. Cowan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 49, 326–346 CrossRef CAS PubMed.
- N. Rueda, J. C. S. dos Santos, C. Ortiz, R. Torres, O. Barbosa, R. C. Rodrigues, Á. Berenguer-Murcia and R. Fernandez-Lafuente, Chem. Rec., 2016, 16, 1436–1455 CrossRef CAS PubMed.
- V. Grazú, F. López-Gallego, T. Montes, O. Abian, R. González, J. A. Hermoso, J. L. García, C. Mateo and J. M. Guisán, Process Biochem., 2010, 45, 390–398 CrossRef.
- G. Fernandez-Lorente, C. A. Godoy, A. A. Mendes, F. Lopez-Gallego, V. Grazu, B. de las Rivas, J. M. Palomo, J. Hermoso, R. Fernandez-Lafuente and J. M. Guisan, Biomacromolecules, 2008, 9, 2553–2561 CrossRef CAS PubMed.
- R. C. Rodrigues, O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernandez-Lafuente, RSC Adv., 2014, 4, 38350–38374 RSC.
- O. Abian, V. Grazú, J. Hermoso, R. González, J. L. García, R. Fernández-Lafuente and J. M. Guisán, Appl. Environ. Microbiol., 2004, 70, 1249–1251 CrossRef CAS PubMed.
- H. Jaladi, A. Katiyar, S. W. Thiel, V. V. Guliants and N. G. Pinto, Chem. Eng. Sci., 2009, 64, 1474–1479 CrossRef CAS.
- M. I. G. Siso, M. Graber, J.-S. Condoret and D. Combes, J. Chem. Technol. Biotechnol., 2007, 48, 185–200 CrossRef PubMed.
- R. Pang, M. Li and C. Zhang, Talanta, 2015, 131, 38–45 CrossRef CAS PubMed.
- Q. Liu and C. L. Tucker, Curr. Opin. Chem. Biol., 2017, 40, 17–23 CrossRef CAS PubMed.
- P. Tufvesson, W. Fu, J. S. Jensen and J. M. Woodley, Food Bioprod. Process., 2010, 88, 3–11 CrossRef CAS.
- P. Tufvesson, J. Lima-Ramos, M. Nordblad and J. M. Woodley, Org. Process Res. Dev., 2011, 15, 266–274 CrossRef CAS.
- J. M. Woodley, Appl. Microbiol. Biotechnol., 2019, 103, 4733–4739 CrossRef CAS PubMed.
- J. M. Woodley, New Biotechnol., 2020, 59, 59–64 CrossRef CAS PubMed.
- J. C. S. dos Santos, O. Barbosa, C. Ortiz, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, ChemCatChem, 2015, 7, 2413–2432 CrossRef CAS.
- M. Cespugli, S. Lotteria, L. Navarini, V. Lonzarich, L. Del Terra, F. Vita, M. Zweyer, G. Baldini, V. Ferrario, C. Ebert and L. Gardossi, Catalysts, 2018, 8, 471 CrossRef.
- V. S. Nishida, R. F. de Oliveira, T. Brugnari, R. C. G. Correa, R. A. Peralta, R. Castoldi, C. G. M. de Souza, A. Bracht and R. M. Peralta, Int. J. Biol. Macromol., 2018, 111, 1206–1213 CrossRef CAS PubMed.
- L. Corici, V. Ferrario, A. Pellis, C. Ebert, S. Lotteria, S. Cantone, D. Voinovich and L. Gardossi, RSC Adv., 2016, 6, 63256–63270 RSC.
- Y. Kulshrestha and Q. Husain, Biomol. Eng., 2006, 23, 291–297 CrossRef CAS PubMed.
- P. Zucca, R. Fernandez-Lafuente and E. Sanjust, Molecules, 2016, 21, 1577 CrossRef PubMed.
- E.-H. Siar, R. Morellon-Sterling, M. N. Zidoune and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2019, 133, 412–419 CrossRef PubMed.
- E.-H. Siar, H. Zaak, J. F. Kornecki, M. N. Zidoune, O. Barbosa and R. Fernandez-Lafuente, Process Biochem., 2017, 58, 98–104 CrossRef CAS.
- E. A. Manoel, J. C. S. dos Santos, D. M. G. Freire, N. Rueda and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2015, 71, 53–57 CrossRef CAS PubMed.
- M. Dias Gomes and J. M. Woodley, Molecules, 2019, 24, 3573 CrossRef PubMed.
- M. Fuentes, C. Mateo, A. Rodriguez, M. Casqueiro, J. C. Tercero, H. H. Riese, R. Fernández-Lafuente and J. M. Guisán, Biosens. Bioelectron., 2006, 21, 1574–1580 CrossRef CAS PubMed.
- M. Fuentes, C. Mateo, J. M. Guisán and R. Fernández-Lafuente, Biosens. Bioelectron., 2005, 20, 1380–1387 CrossRef CAS PubMed.
- A. Indurkar, A. Pandit, R. Jain and P. Dandekar, J. Biomater. Appl., 2021, 36, 76–94 CrossRef CAS PubMed.
- S. Fujikawa, S. Nakamura and K. Koga, Agric. Biol. Chem., 1988, 52, 869–870 CAS.
- B. Manickam, R. Sreedharan and M. Elumalai, Curr. Drug Delivery, 2014, 11, 139–145 CrossRef CAS PubMed.
- M. Farokhi, M. Aleemardani, A. Solouk, H. Mirzadeh, A. H. Teuschl and H. Redl, Biomed. Mater., 2021, 16, 022004 CrossRef CAS PubMed.
- V. G. Tacias-Pascacio, E. García-Parra, G. Vela-Gutiérrez, J. J. Virgen-Ortiz, Á. Berenguer-Murcia, A. R. Alcántara and R. Fernandez-Lafuente, Catalysts, 2019, 9, 1035 CrossRef CAS.
- N. Guajardo and P. Domínguez de María, Molecules, 2021, 26, 736 CrossRef CAS PubMed.
- U. T. Bornscheuer, Philos. Trans. R. Soc. Math. Phys. Eng. Sci., 2018, 376, 20170063 Search PubMed.
- D. Yi, T. Bayer, C. P. S. Badenhorst, S. Wu, M. Doerr, M. Höhne and U. T. Bornscheuer, Chem. Soc. Rev., 2021, 50, 8003–8049 RSC.
- I. Drienovská and G. Roelfes, Nat. Catal., 2020, 3, 193–202 CrossRef.
- Z. Zhou and G. Roelfes, Nat. Catal., 2020, 3, 289–294 CrossRef CAS.
- A. D. Pagar, M. D. Patil, D. T. Flood, T. H. Yoo, P. E. Dawson and H. Yun, Chem. Rev., 2021, 121, 6173–6245 CrossRef CAS PubMed.
- I. Drienovská, A. Rioz-Martínez, A. Draksharapu and G. Roelfes, Chem. Sci., 2015, 6, 770–776 RSC.
- S. Acebes, E. Fernandez-Fueyo, E. Monza, M. F. Lucas, D. Almendral, F. J. Ruiz-Dueñas, H. Lund, A. T. Martinez and V. Guallar, ACS Catal., 2016, 6, 1624–1629 CrossRef CAS.
- F. H. Arnold, Angew. Chem., Int. Ed., 2018, 57, 4143–4148 CrossRef CAS PubMed.
- M. C. Ebert and J. N. Pelletier, Curr. Opin. Chem. Biol., 2017, 37, 89–96 CrossRef CAS PubMed.
- G. Santiago, M. Martínez-Martínez, S. Alonso, R. Bargiela, C. Coscolín, P. N. Golyshin, V. Guallar and M. Ferrer, Biochemistry, 2018, 57, 2245–2255 CrossRef CAS PubMed.
- S. Alonso, G. Santiago, I. Cea-Rama, L. Fernandez-Lopez, C. Coscolín, J. Modregger, A. K. Ressmann, M. Martínez-Martínez, H. Marrero, R. Bargiela, M. Pita, J. L. Gonzalez-Alfonso, M. L. Briand, D. Rojo, C. Barbas, F. J. Plou, P. N. Golyshin, P. Shahgaldian, J. Sanz-Aparicio, V. Guallar and M. Ferrer, Nat. Catal., 2020, 3, 319–328 CrossRef CAS.
- R. Kazlauskas, Chem. Soc. Rev., 2018, 47, 9026–9045 RSC.
- Q. Liu, G. Xun and Y. Feng, Biotechnol. Adv., 2019, 37, 530–537 CrossRef CAS PubMed.
- A. S. Bommarius and M. F. Paye, Chem. Soc. Rev., 2013, 42, 6534–6565 RSC.
- C. Mateo, J. M. Palomo, M. Fuentes, L. Betancor, V. Grazu, F. López-Gallego, B. C. C. Pessela, A. Hidalgo, G. Fernández-Lorente, R. Fernández-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2006, 39, 274–280 CrossRef CAS.
- C. Mateo, O. Abian, M. Bernedo, E. Cuenca, M. Fuentes, G. Fernandez-Lorente, J. M. Palomo, V. Grazu, B. C. C. Pessela, C. Giacomini, G. Irazoqui, A. Villarino, K. Ovsejevi, F. Batista-Viera, R. Fernandez-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2005, 37, 456–462 CrossRef CAS.
- J. J. Virgen-Ortíz, J. C. S. dos Santos, Á. Berenguer-Murcia, O. Barbosa, R. C. Rodrigues and R. Fernandez-Lafuente, J. Mater. Chem. B, 2017, 5, 7461–7490 RSC.
- T. Boller, C. Meier and S. Menzler, Org. Process Res. Dev., 2002, 6, 509–519 CrossRef CAS.
- C. Mateo, G. Fernández-Lorente, O. Abian, R. Fernández-Lafuente and J. M. Guisán, Biomacromolecules, 2000, 1, 739–745 CrossRef CAS PubMed.
- E. Katchalski-Katzir and D. M. Kraemer, J. Mol. Catal. B: Enzym., 2000, 10, 157–176 CrossRef CAS.
- J. Turková, K. Bláha, M. Malaníková, D. Vančurová, F. Švec and J. Kálal, Biochim. Biophys. Acta, Enzymol., 1978, 524, 162–169 CrossRef.
- C. Mateo, V. Grazu, J. M. Palomo, F. Lopez-Gallego, R. Fernandez-Lafuente and J. M. Guisan, Nat. Protoc., 2007, 2, 1022–1033 CrossRef CAS PubMed.
- C. Mateo, R. Torres, G. Fernández-Lorente, C. Ortiz, M. Fuentes, A. Hidalgo, F. López-Gallego, O. Abian, J. M. Palomo, L. Betancor, B. C. C. Pessela, J. M. Guisan and R. Fernández-Lafuente, Biomacromolecules, 2003, 4, 772–777 CrossRef CAS PubMed.
- C. Mateo, O. Abian, G. Fernandez-Lorente, J. Pedroche, R. Fernandez-Lafuente, J. M. Guisan, A. Tam and M. Daminati, Biotechnol. Prog., 2002, 18, 629–634 CrossRef CAS PubMed.
- M. Hartmann, Chem. Mater., 2005, 17, 4577–4593 CrossRef CAS.
- T. Jesionowski, J. Zdarta and B. Krajewska, Adsorption, 2014, 20, 801–821 CrossRef CAS.
- V. G. Tacias-Pascacio, C. Ortiz, N. Rueda, Á. Berenguer-Murcia, N. Acosta, I. Aranaz, C. Civera, R. Fernandez-Lafuente and A. R. Alcántara, Catalysts, 2019, 9, 622 CrossRef CAS.
- C. Mateo, G. Fernandez-Lorente, B. C. C. Pessela, A. Vian, A. V. Carrascosa, J. L. Garcia, R. Fernandez-Lafuente and J. M. Guisan, J. Chromatogr. A, 2001, 915, 97–106 CrossRef CAS PubMed.
- P. Batalla, C. Mateo, V. Grazu, R. Fernandez-Lafuente and J. M. Guisan, Process Biochem., 2009, 44, 365–368 CrossRef CAS.
- P. Batalla, M. Fuentes, V. Grazu, C. Mateo, R. Fernandez-Lafuente and J. M. Guisan, Biomacromolecules, 2008, 9, 719–723 CrossRef CAS PubMed.
- P. Batalla, M. Fuentes, C. Mateo, V. Grazu, R. Fernandez-Lafuente and J. M. Guisan, Biomacromolecules, 2008, 9, 2230–2236 CrossRef CAS PubMed.
- J. Zhou, J. Zheng and S. Jiang, J. Phys. Chem. B, 2004, 108, 17418–17424 CrossRef CAS.
- E. E. Drufva, N. R. Spengler, E. G. Hix and C. B. Bailey, ChemBioChem, 2021, 22, 1122–1150 CrossRef CAS PubMed.
- S. Roda, L. Fernandez-Lopez, R. Cañadas, G. Santiago, M. Ferrer and V. Guallar, ACS Catal., 2021, 11, 3590–3601 CrossRef CAS.
- D. Carballares, R. Morellon-Sterling, X. Xu, F. Hollmann and R. Fernandez-Lafuente, Catalysts, 2021, 11, 560 CrossRef CAS.
- H. Zaak, S. Peirce, T. de Albuquerque, M. Sassi and R. Fernandez-Lafuente, Catalysts, 2017, 7, 250 CrossRef.
- A. Basso and S. Serban, in Immobilization of Enzymes and Cells, ed. J. M. Guisan, J. M. Bolivar, F. López-Gallego and J. Rocha-Martín, Springer US, New York, NY, 2020, vol. 2100, pp. 27–63 Search PubMed.
- J. M. Guisan, F. López-Gallego, J. M. Bolivar, J. Rocha-Martín and G. Fernandez-Lorente, in Immobilization of Enzymes and Cells, ed. J. M. Guisan, J. M. Bolivar, F. López-Gallego and J. Rocha-Martín, Springer US, New York, NY, 2020, vol. 2100, pp. 1–26 Search PubMed.
- V. R. Murty, J. Bhat and P. K. A. Muniswaran, Biotechnol. Bioprocess Eng., 2002, 7, 57–66 CrossRef CAS.
- D. Goswami, J. K. Basu and S. De, Crit. Rev. Biotechnol., 2013, 33, 81–96 CrossRef CAS PubMed.
- C. Boniello, T. Mayr, J. M. Bolivar and B. Nidetzky, BMC Biotechnol., 2012, 12, 11 CrossRef CAS PubMed.
- H. Luo, L. Zhu, Y. Chang, X. Liu, Z. Liu, H. Sun, X. Li, H. Yu and Z. Shen, Bioresour. Technol., 2017, 223, 157–165 CrossRef CAS PubMed.
- A. C. Spiess and V. Kasche, Biotechnol. Prog., 2001, 17, 294–303 CrossRef CAS PubMed.
- T. Zahel, C. Boniello and B. Nidetzky, Process Biochem., 2013, 48, 593–604 CrossRef CAS.
- C. Boniello, T. Mayr, I. Klimant, B. Koenig, W. Riethorst and B. Nidetzky, Biotechnol. Bioeng., 2010, 106, 528–540 CrossRef CAS PubMed.
- G. Chen, R. L. Fournier and S. Varanasi, Biotechnol. Bioeng., 1998, 57, 394–408 CrossRef CAS PubMed.
- J. P. Byers, M. B. Shah, R. L. Fournier and S. Varanasi, Biotechnol. Bioeng., 1993, 42, 410–420 CrossRef CAS PubMed.
- J. M. Bolivar and B. Nidetzky, Molecules, 2019, 24, 3460 CrossRef CAS PubMed.
- T. Consolati, J. M. Bolivar, Z. Petrasek, J. Berenguer, A. Hidalgo, J. M. Guisán and B. Nidetzky, ACS Appl. Mater. Interfaces, 2018, 10, 6858–6868 CrossRef CAS PubMed.
- N. Rueda, J. C. S. dos Santos, R. Torres, C. Ortiz, O. Barbosa and R. Fernandez-Lafuente, RSC Adv., 2015, 5, 11212–11222 RSC.
- J. J. Virgen-Ortíz, V. G. Tacias-Pascacio, D. B. Hirata, B. Torrestiana-Sanchez, A. Rosales-Quintero and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2017, 96, 30–35 CrossRef PubMed.
- D. B. Hirata, T. L. Albuquerque, N. Rueda, J. M. Sánchez-Montero, E. Garcia-Verdugo, R. Porcar and R. Fernandez-Lafuente, ChemistrySelect, 2016, 1, 3259–3270 CrossRef CAS.
- D. B. Hirata, T. L. Albuquerque, N. Rueda, J. J. Virgen-Ortíz, V. G. Tacias-Pascacio and R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2016, 133, 117–123 CrossRef CAS.
- V. P. Shanbhag and G. Johansson, Biochem. Biophys. Res. Commun., 1974, 61, 1141–1146 CrossRef CAS PubMed.
- G. Johansson, Mol. Cell. Biochem., 1974, 4, 169–180 CrossRef CAS PubMed.
- Z. M. De Martinez Segovia and A. Diaz, Appl. Microbiol., 1968, 16, 1602–1604 CrossRef CAS PubMed.
- A. Schluck, G. Maurer and M.-R. Kula, Biotechnol. Bioeng., 1995, 47, 252–260 CrossRef CAS PubMed.
- B. Y. Zaslavsky, N. M. Mestechkina and S. V. Rogozhin, J. Chromatogr. A, 1983, 260, 329–336 CrossRef PubMed.
- K. E. Gutowski, G. A. Broker, H. D. Willauer, J. G. Huddleston, R. P. Swatloski, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2003, 125, 6632–6633 CrossRef CAS PubMed.
- M. G. Freire, A. F. M. Cláudio, J. M. M. Araújo, J. A. P. Coutinho, I. M. Marrucho, J. N. C. Lopes and L. P. N. Rebelo, Chem. Soc. Rev., 2012, 41, 4966–4995 RSC.
- M. E. Zakrzewska, A. V. M. Nunes, A. R. Barot, A. Fernández-Castané, Z. P. Visak, W. Kiatkittipong and V. Najdanovic-Visak, Fluid Phase Equilib., 2021, 539, 113022 CrossRef CAS.
- C. Aguirre, I. Concha, J. Vergara, R. Riveros and A. Illanes, Process Biochem., 2010, 45, 1163–1167 CrossRef CAS.
- L. Vobecká, A. Romanov, Z. Slouka, P. Hasal and M. Přibyl, New Biotechnol., 2018, 47, 73–79 CrossRef PubMed.
- J. F. B. Pereira, M. G. Freire and J. A. P. Coutinho, Fluid Phase Equilib., 2020, 505, 112341 CrossRef CAS.
- L. Vobecká, L. Tichá, A. Atanasova, Z. Slouka, P. Hasal and M. Přibyl, Chem. Eng. J., 2020, 396, 125236 CrossRef.
- E. Andersson, B. Mattiasson and B. Hahn-Hägerdal, Enzyme Microb. Technol., 1984, 6, 301–306 CrossRef CAS.
- E. Andersson and B. Hahn-Hägerdal, Enzyme Microb. Technol., 1990, 12, 242–254 CrossRef CAS PubMed.
- O. Hernandez-Justiz, R. Fernandez-Lafuente, M. Terreni and J. M. Guisan, Biotechnol. Bioeng., 1998, 59, 73–79 CrossRef CAS PubMed.
- L. H. M. da Silva and W. Loh, Quím. Nova, 2006, 29, 1345–1351 CrossRef.
- J. Benavides, O. Aguilar, B. H. Lapizco-Encinas and M. Rito-Palomares, Chem. Eng. Technol., 2008, 31, 838–845 CrossRef CAS.
- S. Arana-Peña, N. S. Rios, C. Mendez-Sanchez, Y. Lokha, L. R. B. Gonçalves and R. Fernández-Lafuente, Enzyme Microb. Technol., 2020, 137, 109535 CrossRef PubMed.
- O. Barbosa, C. Ortiz, Á. Berenguer-Murcia, R. Torres, R. C. Rodrigues and R. Fernandez-Lafuente, RSC Adv., 2014, 4, 1583–1600 RSC.
- H. Zaak, L. Fernandez-Lopez, C. Otero, M. Sassi and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2017, 106, 67–74 CrossRef CAS PubMed.
- C. Pizarro, M. C. Brañes, A. Markovits, G. Fernández-Lorente, J. M. Guisán, R. Chamy and L. Wilson, J. Mol. Catal. B: Enzym., 2012, 78, 111–118 CrossRef CAS.
- G. Fernandez-Lorente, M. Filice, D. Lopez-Vela, C. Pizarro, L. Wilson, L. Betancor, Y. Avila and J. M. Guisan, J. Am. Oil Chem. Soc., 2011, 88, 801–807 CrossRef CAS.
- O. Barbosa, R. Torres, C. Ortiz and R. Fernandez-Lafuente, Process Biochem., 2012, 47, 766–774 CrossRef CAS.
- C. Bernal, A. Illanes and L. Wilson, J. Agric. Food Chem., 2015, 63, 3716–3724 CrossRef CAS PubMed.
- N. Guajardo, C. Bernal, L. Wilson and Z. Cabrera, Catal. Today, 2015, 255, 21–26 CrossRef CAS.
- C. Bernal, A. Illanes and L. Wilson, Langmuir, 2014, 30, 3557–3566 CrossRef CAS PubMed.
- T. L. de Albuquerque, N. Rueda, J. C. S. dos Santos, O. Barbosa, C. Ortiz, B. Binay, E. Özdemir, L. R. B. Gonçalves and R. Fernandez-Lafuente, Process Biochem., 2016, 51, 865–874 CrossRef CAS.
- L. Fernandez-Lopez, S. G. Pedrero, N. Lopez-Carrobles, J. J. Virgen-Ortíz, B. C. Gorines, C. Otero and R. Fernandez-Lafuente, Process Biochem., 2017, 54, 81–88 CrossRef CAS.
- N. Rueda, T. Albuquerque, R. Bartolome-Cabrero, L. Fernandez-Lopez, R. Torres, C. Ortiz, J. dos Santos, O. Barbosa and R. Fernandez-Lafuente, Molecules, 2016, 21, 646 CrossRef PubMed.
- N. Rueda, C. S. dos Santos, M. D. Rodriguez, T. L. Albuquerque, O. Barbosa, R. Torres, C. Ortiz and R. Fernandez-Lafuente, J. Mol. Catal. B: Enzym., 2016, 128, 10–18 CrossRef CAS.
- N. S. Okura, G. J. Sabi, M. C. Crivellenti, R. A. B. Gomes, R. Fernandez-Lafuente and A. A. Mendes, Int. J. Biol. Macromol., 2020, 163, 550–561 CrossRef CAS PubMed.
- R. Fernandez-Lafuente, V. Rodriguez, C. Mateo, G. Penzol, O. Hernandez-Justiz, G. Irazoqui, A. Villarino, K. Ovsejevi, F. Batista and J. M. Guisan, J. Mol. Catal. B: Enzym., 1999, 7, 181–189 CrossRef CAS.
- R. Fernandez-Lafuente, Enzyme Microb. Technol., 2009, 45, 405–418 CrossRef CAS.
- M. Fuentes, R. L. Segura, O. Abian, L. Betancor, A. Hidalgo, C. Mateo, R. Fernandez-Lafuente and J. M. Guisan, Proteomics, 2004, 4, 2602–2607 CrossRef CAS PubMed.
- C. Garcia-Galan, O. Barbosa and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2013, 52, 211–217 CrossRef CAS PubMed.
- J. M. Bolivar, J. Rocha-Martin, C. Mateo, F. Cava, J. Berenguer, R. Fernandez-Lafuente and J. M. Guisan, Biomacromolecules, 2009, 10, 742–747 CrossRef CAS PubMed.
- C. Mateo, B. C. C. Pessela, M. Fuentes, R. Torres, L. Betancor, A. Hidalgo, G. Fernandez-Lorente, R. Fernandez-Lafuente and J. M. Guisan, in Immobilization of Enzymes and Cells, ed. J. M. Guisan, J. M. Bolivar, F. López-Gallego and J. Rocha-Martín, Springer US, New York, NY, 2020, vol. 2100, pp. 175–187 Search PubMed.
- L. Betancor, F. López-Gallego, A. Hidalgo, N. Alonso-Morales, G. D.-O. C. Mateo, R. Fernández-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2006, 39, 877–882 CrossRef CAS.
- E.-H. Siar, S. Arana-Peña, O. Barbosa, M. Zidoune and R. Fernandez-Lafuente, Catalysts, 2018, 8, 149 CrossRef.
- P. G. Vazquez-Ortega, M. T. Alcaraz-Fructuoso, J. A. Rojas-Contreras, J. López-Miranda and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2018, 110, 38–45 CrossRef CAS PubMed.
- O. Barbosa, R. Torres, C. Ortiz and R. Fernandez-Lafuente, Process Biochem., 2012, 47, 1220–1227 CrossRef CAS.
- D. de Andrades, N. G. Graebin, M. K. Kadowaki, M. A. Z. Ayub, R. Fernandez-Lafuente and R. C. Rodrigues, Int. J. Biol. Macromol., 2019, 129, 672–678 CrossRef CAS PubMed.
- C. Mateo, J. M. Bolivar, C. A. Godoy, J. Rocha-Martin, B. C. Pessela, J. A. Curiel, R. Muñoz, J. M. Guisan and G. Fernández-Lorente, Biomacromolecules, 2010, 11, 3112–3117 CrossRef CAS PubMed.
- H. Zaak, M. Sassi and R. Fernandez-Lafuente, Process Biochem., 2018, 64, 200–205 CrossRef CAS.
- K. Ovsejevi, C. Manta and F. Batista-Viera, in Immobilization of Enzymes and Cells, ed. J. M. Guisan, Humana Press, Totowa, NJ, 2013, vol. 1051, pp. 89–116 Search PubMed.
- F. Batista-Viera, M. Barbieri, K. Ovsejevi, C. Manta and J. Carlsson, Appl. Biochem. Biotechnol., 1991, 31, 175–195 CrossRef CAS.
- E. Pavlovic, S. Oscarsson and A. P. Quist, Nano Lett., 2003, 3, 779–781 CrossRef CAS.
- J. R. Simons, M. Mosisch, A. E. Torda and L. Hilterhaus, J. Biotechnol., 2013, 167, 1–7 CrossRef CAS PubMed.
- L. Gioia, S. Rodríguez-Couto, M. del, P. Menéndez, C. Manta and K. Ovsejevi, Biotechnol. Appl. Biochem., 2015, 62, 502–513 CrossRef CAS PubMed.
- R. Fraas and M. Franzreb, Biocatal. Biotransform., 2017, 35, 337–348 CrossRef CAS.
- J. Carlsson, R. Axen, K. Brocklehurst and E. M. Crook, Eur. J. Biochem., 1974, 44, 189–194 CrossRef CAS PubMed.
- J. Schnapp and Y. Shalitin, Biochem. Biophys. Res. Commun., 1976, 70, 8–14 CrossRef CAS PubMed.
- H. Sato, T. Kidaka and M. Hori, Appl. Biochem. Biotechnol., 1987, 15, 145–158 CrossRef CAS PubMed.
- J. Lasch and R. Koelsch, Eur. J. Biochem., 1978, 82, 181–186 CrossRef CAS PubMed.
- J. Kohn and M. Wilchek, Enzyme Microb. Technol., 1982, 4, 161–163 CrossRef CAS.
- J. F. Kennedy, J. A. Barnes and J. B. Matthews, J. Chromatogr. A, 1980, 196, 379–389 CrossRef CAS.
- J. F. Kennedy, J. A. Barnes and J. B. Matthews, Br. Polym. J., 1983, 15, 133–138 CrossRef CAS.
- J. W. Wong, R. L. Albright and N.-H. L. Wang, Sep. Purif. Methods, 1991, 20, 49–106 CrossRef CAS.
- J. Porath, TrAC, Trends Anal. Chem., 1988, 7, 254–259 CrossRef CAS.
- M. E. Çorman, N. Öztürk, N. Tüzmen, S. Akgöl and A. Denizli, Biochem. Eng. J., 2010, 49, 159–164 CrossRef.
- A. Fb and G. Altmann-Haase, Biotechnol. Appl. Biochem., 1994, 20, 313–322 CrossRef PubMed.
- D. Q. Tang, D. J. Zhang, D. Y. Tang and H. Ai, Chem. Lett., 2006, 35, 1238–1239 CrossRef CAS.
- F. López-Gallego, G. Fernandez-Lorente, J. Rocha-Martín, J. M. Bolivar, C. Mateo and J. M. Guisan, in Immobilization of Enzymes and Cells, ed. J. M. Guisan, J. M. Bolivar, F. López-Gallego and J. Rocha-Martín, Springer US, New York, NY, 2020, vol. 2100, pp. 93–107 Search PubMed.
- S. A. Braham, R. Morellon-Sterling, D. de Andrades, R. C. Rodrigues, E.-H. Siar, A. Aksas, J. Pedroche, M. del, C. Millán and R. Fernandez-Lafuente, Appl. Biochem. Biotechnol., 2021, 193, 2843–2857 CrossRef CAS PubMed.
- J. M. Bolivar, J. Rocha-Martin, C. Mateo, F. Cava, J. Berenguer, D. Vega, R. Fernandez-Lafuente and J. M. Guisan, J. Mol. Catal. B: Enzym., 2009, 58, 158–163 CrossRef CAS.
- V. Grazu, L. Betancor, T. Montes, F. Lopez-Gallego, J. M. Guisan and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2006, 38, 960–966 CrossRef CAS.
- A. H. Orrego, M. Romero-Fernández, M. Millán-Linares, M. Yust, J. Guisán and J. Rocha-Martin, Catalysts, 2018, 8, 333 CrossRef.
- M. Ozboyaci, D. B. Kokh, S. Corni and R. C. Wade, Q. Rev. Biophys., 2016, 49, e4 CrossRef PubMed.
- X. Quan, J. Liu and J. Zhou, Curr. Opin. Colloid Interface Sci., 2019, 41, 74–85 CrossRef CAS.
- J. S. Weltz, D. K. Schwartz and J. L. Kaar, ACS Nano, 2016, 10, 730–738 CrossRef CAS PubMed.
- N. A. Moringo, H. Shen, L. J. Tauzin, W. Wang, L. D. C. Bishop and C. F. Landes, Langmuir, 2017, 33, 10818–10828 CrossRef CAS PubMed.
- M. Kastantin, B. B. Langdon and D. K. Schwartz, Adv. Colloid Interface Sci., 2014, 207, 240–252 CrossRef CAS PubMed.
- J. M. Bolivar and B. Nidetzky, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140333 CrossRef CAS PubMed.
- F. López-Gallego, I. Acebrón, J. M. Mancheño, S. Raja, M. P. Lillo and J. M. Guisán Seijas, Bioconjugate Chem., 2012, 23, 565–573 CrossRef PubMed.
- E. Diamanti, S. Arana-Peña, P. Ramos-Cabrer, N. Comino, D. Carballares, R. Fernandez-Lafuente and F. López-Galledo, Adv. Mater. Interfaces, 2022, 9, 2200263 CrossRef CAS.
- J. Rocha-Martín, B. de las Rivas, R. Muñoz, J. M. Guisán and F. López-Gallego, ChemCatChem, 2012, 4, 1279–1288 CrossRef.
- J. N. Vranish, M. G. Ancona, E. Oh, K. Susumu, G. Lasarte Aragonés, J. C. Breger, S. A. Walper and I. L. Medintz, ACS Nano, 2018, 12, 7911–7926 CrossRef CAS PubMed.
- V. D. Jäger, R. Lamm, R. Kloß, E. Kaganovitch, A. Grünberger, M. Pohl, J. Büchs, K.-E. Jaeger and U. Krauss, ACS Synth. Biol., 2018, 7, 2282–2295 CrossRef PubMed.
- S. Velasco-Lozano, J. Santiago-Arcos, J. A. Mayoral and F. López-Gallego, ChemCatChem, 2020, 12, 3030–3041 CrossRef CAS.
- L. Trobo-Maseda, A. H. Orrego, J. M. Guisan and J. Rocha-Martin, Int. J. Biol. Macromol., 2020, 157, 510–521 CrossRef CAS PubMed.
- E. Diamanti, J. Santiago-Arcos, D. Grajales-Hernández, N. Czarnievicz, N. Comino, I. Llarena, D. Di Silvio, A. L. Cortajarena and F. López-Gallego, ACS Catal., 2021, 11, 15051–15067 CrossRef CAS PubMed.
- J. Boudrant, J. M. Woodley and R. Fernandez-Lafuente, Process Biochem., 2020, 90, 66–80 CrossRef CAS.
- L. Dal Magro, K. S. de Moura, B. E. Backes, E. W. de Menezes, E. V. Benvenutti, S. Nicolodi, M. P. Klein, R. Fernandez-Lafuente and R. C. Rodrigues, Biotechnol. Rep., 2019, 24, e00373 CrossRef PubMed.
- L. Dal Magro, J. P. S. Pessoa, M. P. Klein, R. Fernandez-Lafuente and R. C. Rodrigues, Catal. Today, 2021, 362, 184–191 CrossRef CAS.
- E. P. Cipolatti, A. Valério, R. O. Henriques, D. E. Moritz, J. L. Ninow, D. M. G. Freire, E. A. Manoel, R. Fernandez-Lafuente and D. de Oliveira, RSC Adv., 2016, 6, 104675–104692 RSC.
- I. C. A. Bolina, R. A. B. Gomes and A. A. Mendes, BioEnergy Res, 2021, 14, 1039–1057 CrossRef CAS.
- H. Stecher and K. Faber, Synthesis, 1997, 1–16 CrossRef CAS.
- A. Ghanem and H. Y. Aboul-Enein, Chirality, 2005, 17, 1–15 CrossRef CAS PubMed.
- M. F. P. Ribeiro, K. C. Pais, B. S. M. de Jesus, R. Fernandez-Lafuente, D. M. G. Freire, E. A. Manoel and A. B. C. Simas, Eur. J. Org. Chem., 2018, 386–391 CrossRef CAS.
- C. Nájera, F. Foubelo, J. M. Sansano and M. Yus, Tetrahedron, 2022, 106–107, 132629 CrossRef.
- H. Hu, Q. Wang, D. Wang and Y. Ao, Adv. Synth. Catal., 2021, 363, 4538–4543 CrossRef CAS.
- Y.-F. Ao, H.-J. Hu, C.-X. Zhao, P. Chen, T. Huang, H. Chen, Q.-Q. Wang, D.-X. Wang and M.-X. Wang, ACS Catal., 2021, 11, 6900–6907 CrossRef CAS.
- S. Martínez-Montero, S. Fernández, Y. S. Sanghvi, V. Gotor and M. Ferrero, Eur. J. Org. Chem., 2012, 5483–5490 CrossRef.
- R. Croitoru, F. Fiţigău, L. A. M. van den Broek, A. E. Frissen, C. M. Davidescu, C. G. Boeriu and F. Peter, Process Biochem., 2012, 47, 1894–1902 CrossRef CAS.
- P. M. P. Souza, D. Carballares, N. Lopez-Carrobles, L. R. B. Gonçalves, F. Lopez-Gallego, S. Rodrigues and R. Fernandez-Lafuente, Int. J. Biol. Macromol., 2021, 191, 79–91 CrossRef CAS PubMed.
- O. Romero, E. Araya, A. Illanes and L. Wilson, J. Mol. Catal. B: Enzym., 2014, 104, 70–74 CrossRef CAS.
- J. M. Bolivar, I. Eisl and B. Nidetzky, Catal. Today, 2016, 259, 66–80 CrossRef CAS.
- D.-M. Liu and C. Dong, Process Biochem., 2020, 92, 464–475 CrossRef.
- M. Bilal, S. A. Qamar, S. S. Ashraf, S. Rodríguez-Couto and H. M. N. Iqbal, Adv. Colloid Interface Sci., 2021, 293, 102438 CrossRef CAS PubMed.
- S. A. Qamar, M. Qamar, M. Bilal, R. N. Bharagava, L. F. R. Ferreira, F. Sher and H. M. N. Iqbal, Int. J. Biol. Macromol., 2021, 185, 1–19 CrossRef CAS PubMed.
- A. Bastida, P. Sabuquillo, P. Armisen, R. Fernández-Lafuente, J. Huguet and J. M. Guisán, Biotechnol. Bioeng., 1998, 58, 486–493 CrossRef CAS PubMed.
- G. Fernandez-Lorente, Z. Cabrera, C. Godoy, R. Fernandez-Lafuente, J. M. Palomo and J. M. Guisan, Process Biochem., 2008, 43, 1061–1067 CrossRef CAS.
- J. M. Bolivar, A. Hidalgo, L. Sánchez-Ruiloba, J. Berenguer, J. M. Guisán and F. López-Gallego, J. Biotechnol., 2011, 155, 412–420 CrossRef CAS PubMed.
- A. H. Orrego, F. López-Gallego, G. Fernandez-Lorente, J. M. Guisan and J. Rocha-Martín, in Immobilization of Enzymes and Cells, ed. J. M. Guisan, J. M. Bolivar, F. López-Gallego and J. Rocha-Martín, Springer US, New York, NY, 2020, vol. 2100, pp. 297–308 Search PubMed.
- F. Secundo, Chem. Soc. Rev., 2013, 42, 6250 RSC.
- J. M. Bolivar, T. Consolati, T. Mayr and B. Nidetzky, Trends Biotechnol., 2013, 31, 194–203 CrossRef CAS PubMed.
- O. Tepe and A. Y. Dursun, J. Hazard. Mater., 2008, 151, 9–16 CrossRef CAS PubMed.
- R. K. Sonwani, B. S. Giri, T. Das, R. S. Singh and B. N. Rai, Process Biochem., 2019, 77, 106–112 CrossRef CAS.
- G. Swain, R. K. Sonwani, B. S. Giri, R. S. Singh, R. P. Jaiswal and B. N. Rai, Water Environ. J., 2021, 35, 285–294 CrossRef CAS.
- W. S. Yeo and A. Yuniarto, J. Environ. Chem. Eng., 2019, 7, 103185 CrossRef CAS.
- D. Caşcaval, M. Turnea, A.-I. Galaction and A. C. Blaga, Biochem. Eng. J., 2012, 69, 113–122 CrossRef.
- A. E. AL-Muftah and I. M. Abu-Reesh, Biochem. Eng. J., 2005, 23, 139–153 CrossRef CAS.
- A. Y. Dursun and O. Tepe, J. Hazard. Mater., 2005, 126, 105–111 CrossRef CAS PubMed.
- B. Xu, S. M. R. Azam, M. Feng, B. Wu, W. Yan, C. Zhou and H. Ma, Ultrason. Sonochem., 2021, 81, 105855 CrossRef CAS PubMed.
- N. Priya and P. R. Gogate, Ind. Eng. Chem. Res., 2021, 60, 9650–9668 CrossRef.
- A. Khan, M. R. Beg and P. Waghmare, Biophys. Rev., 2021, 13, 417–423 CrossRef CAS PubMed.
- J. Luo, Z. Fang and R. L. Smith, Prog. Energy Combust. Sci., 2014, 41, 56–93 CrossRef.
- E. V. Rokhina, P. Lens and J. Virkutyte, Trends Biotechnol., 2009, 27, 298–306 CrossRef CAS PubMed.
- Y. Chisti, Trends Biotechnol., 2003, 21, 89–93 CrossRef CAS PubMed.
- J. M. Bolivar, S. Schelch, T. Mayr and B. Nidetzky, ACS Catal., 2015, 5, 5984–5993 CrossRef CAS.
- K. Hernandez, A. Berenguer-Murcia, R. C. Rodrigues and R. Fernandez-Lafuente, Curr. Org. Chem., 2012, 16, 2652–2672 CrossRef CAS.
- J. M. Bolivar, S. Schelch, M. Pfeiffer and B. Nidetzky, J. Mol. Catal. B: Enzym., 2016, 134, 302–309 CrossRef CAS.
- R. Kumar, A. Krishnamurthy Suresh and H. Subbaraman Shankar, J. Chem. Technol. Biotechnol., 1996, 66, 243–250 CrossRef CAS.
- T.-C. Huang, D.-H. Chen and S.-S. Wang, Chem. Eng. Commun., 1996, 147, 99–117 CrossRef CAS.
- C. M. Rosell, R. Fernandez-Lafuente and J. M. Guisan, Biocatal. Biotransform., 1995, 12, 67–76 CrossRef CAS.
- M. L. Greene, J. A. Boyle and J. E. Seegmiller, Science, 1970, 167, 887–889 CrossRef CAS PubMed.
- Y. Zhang and H. Hess, ACS Catal., 2017, 7, 6018–6027 CrossRef CAS.
- G. Jm, G. Alvaro, R. Cm and R. Fernandez-Lafuente, Biotechnol. Appl. Biochem., 1994, 20, 357–369 CrossRef PubMed.
- E. Ruckenstein and P. Rajora, Biotechnol. Bioeng., 1985, 27, 807–817 CrossRef CAS PubMed.
- E. Ruckenstein and V. Sasidhar, Chem. Eng. Sci., 1984, 39, 1185–1200 CrossRef CAS.
- J.-M. Engasser and C. Horvath, Biochim. Biophys. Acta, Enzymol., 1974, 358, 178–192 CrossRef CAS.
- J. M. Bolivar, T. Consolati, T. Mayr and B. Nidetzky, Biotechnol. Bioeng., 2013, 110, 2086–2095 CrossRef CAS PubMed.
- J. M. Bolivar, S. Schelch, T. Mayr and B. Nidetzky, ChemCatChem, 2014, 6, 981–986 CrossRef CAS.
- E. Muñoz-Morales, S. Velasco-Lozano, A. I. Benítez-Mateos, M. J. Marín, P. Ramos-Cabrer and F. López-Gallego, Catalysts, 2019, 9, 896 CrossRef.
- Enzyme Biocatalysis Principles and Applications, ed. A. Illanes, Springer, Netherlands, Dordrecht, 2008 Search PubMed.
- S. Sabbani, E. Hedenström and O. Nordin, J. Mol. Catal. B: Enzym., 2006, 42, 1–9 CrossRef CAS.
- F. van Rantwijk, C. Mateo, A. Chmura, B. C. M. Fernandes and R. A. Sheldon, in Modern Biocatalysis, ed. W.-D. Fessner and T. Anthonsen, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008, pp. 261–272 Search PubMed.
- J. F. Kornecki, D. Carballares, P. W. Tardioli, R. C. Rodrigues, Á. Berenguer-Murcia, A. R. Alcántara and R. Fernandez-Lafuente, Catal. Sci. Technol., 2020, 10, 5740–5771 RSC.
- A. H. Orrego, F. López-Gallego, A. Espaillat, F. Cava, J. M. Guisan and J. Rocha-Martin, ChemCatChem, 2018, 10, 3002–3011 CrossRef CAS.
- A. Lopalco and V. J. Stella, J. Pharm. Sci., 2016, 105, 2879–2885 CrossRef CAS PubMed.
- S. van Pelt, F. van Rantwijk and R. A. Sheldon, Adv. Synth. Catal., 2009, 351, 397–404 CrossRef CAS.
- A. Chmura, S. Rustler, M. Paravidino, F. van Rantwijk, A. Stolz and R. A. Sheldon, Tetrahedron: Asymmetry, 2013, 24, 1225–1232 CrossRef CAS.
- S. Butò, L. Pollegioni, L. D’Angiuro and M. S. Pilone, Biotechnol. Bioeng., 1994, 44, 1288–1294 CrossRef PubMed.
- R. Fernández-Lafuente, V. Rodriguez and J. M. Guisán, Enzyme Microb. Technol., 1998, 23, 28–33 CrossRef.
- S. Bidmanova, E. Hrdlickova, J. Jaros, L. Ilkovics, A. Hampl, J. Damborsky and Z. Prokop, Biotechnol. J., 2014, 9, 852–860 CrossRef CAS PubMed.
- D.-Y. Xu and Z. Yang, Chemosphere, 2013, 92, 391–398 CrossRef CAS PubMed.
- D.-Y. Xu, J.-Y. Chen and Z. Yang, Biochem. Eng. J., 2012, 63, 88–94 CrossRef CAS.
- L. Tavernini, C. Aburto, O. Romero, A. Illanes and L. Wilson, Catalysts, 2021, 11, 866 CrossRef CAS.
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, Á. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC.
- C. José, R. D. Bonetto, L. A. Gambaro, M. del P. G. Torres, M. L. Foresti, M. L. Ferreira and L. E. Briand, J. Mol. Catal. B: Enzym., 2011, 71, 95–107 CrossRef.
- C. José, G. B. Austic, R. D. Bonetto, R. M. Burton and L. E. Briand, Catal. Today, 2013, 213, 73–80 CrossRef.
- C. José and L. E. Briand, React. Kinet., Mech. Catal., 2010, 99, 17–22 Search PubMed.
- A. Illanes and L. Wilson, in Immobilization of Enzymes and Cells, ed. J. M. Guisan, J. M. Bolivar, F. López-Gallego and J. Rocha-Martín, Springer US, New York, NY, 2020, vol. 2100, pp. 65–81 Search PubMed.
- J. Armando Gamarra, C. M. Cuevas and G. Lescano, J. Ferment. Technol., 1986, 64, 25–28 CrossRef.
- H. Cabana, J. P. Jones and S. N. Agathos, Biotechnol. Bioeng., 2009, 102, 1582–1592 CrossRef CAS PubMed.
- R. L. C. Giordano, R. C. Giordano and C. L. Cooney, Process Biochem., 2000, 35, 1093–1101 CrossRef CAS.
- A. V. Paula, G. F. M. Nunes, H. F. de Castro and J. C. Santos, Ind. Eng. Chem. Res., 2015, 54, 1731–1737 CrossRef CAS.
- G. Sheelu, G. Kavitha and N. W. Fadnavis, J. Am. Oil Chem. Soc., 2008, 85, 739–748 CrossRef CAS.
- R. R. Sousa, A. S. Silva, R. Fernandez-Lafuente and V. S. Ferreira-Leitão, Catal. Sci. Technol., 2021, 11, 5696–5711 RSC.
- E. A. Oke and S. P. Ijardar, Biochem. Eng. J., 2021, 176, 108211 CrossRef CAS.
- J. A. Kist, H. Zhao, K. R. Mitchell-Koch and G. A. Baker, J. Mater. Chem. B, 2021, 9, 536–566 RSC.
- F. M. Perna, P. Vitale and V. Capriati, Curr. Opin. Green Sustainable Chem., 2020, 21, 27–33 CrossRef.
- M. Pätzold, S. Siebenhaller, S. Kara, A. Liese, C. Syldatk and D. Holtmann, Trends Biotechnol., 2019, 37, 943–959 CrossRef PubMed.
- M. M. C. H. van Schie, J.-D. Spöring, M. Bocola, P. Domínguez de María and D. Rother, Green Chem., 2021, 23, 3191–3206 RSC.
- N. Guajardo, K. Ahumada and P. Domínguez de María, J. Biotechnol., 2020, 310, 97–102 CrossRef CAS PubMed.
- C. Mateo, O. Abian, R. Fernandez–Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2000, 26, 509–515 CrossRef CAS PubMed.
- J. Virgen-Ortíz, S. Pedrero, L. Fernandez-Lopez, N. Lopez-Carrobles, B. Gorines, C. Otero and R. Fernandez-Lafuente, Molecules, 2017, 22, 91 CrossRef PubMed.
- J. J. Virgen-Ortíz, S. Peirce, V. G. Tacias-Pascacio, V. Cortes-Corberan, A. Marzocchella, M. E. Russo and R. Fernandez-Lafuente, Process Biochem., 2016, 51, 1391–1396 CrossRef.
- J. C. S. dos Santos, N. Rueda, R. Torres, O. Barbosa, L. R. B. Gonçalves and R. Fernandez-Lafuente, Process Biochem., 2015, 50, 918–927 CrossRef CAS.
- J. C. S. dos Santos, N. Rueda, A. Sanchez, R. Villalonga, L. R. B. Gonçalves and R. Fernandez-Lafuente, RSC Adv., 2015, 5, 35801–35810 RSC.
- J. C. S. dos Santos, N. Rueda, L. R. B. Gonçalves and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2015, 77, 1–7 CrossRef PubMed.
- P. Bonomi, T. Bavaro, I. Serra, A. Tagliani, M. Terreni and D. Ubiali, Molecules, 2013, 18, 14349–14365 CrossRef PubMed.
- J. C. S. dos Santos, N. Rueda, O. Barbosa, J. F. Fernández-Sánchez, A. L. Medina-Castillo, T. Ramón-Márquez, M. C. Arias-Martos, M. C. Millán-Linares, J. Pedroche, M. del, M. Yust, L. R. B. Gonçalves and R. Fernandez-Lafuente, RSC Adv., 2015, 5, 20639–20649 RSC.
- J. Pedroche, M. del Mar Yust, C. Mateo, R. Fernández-Lafuente, J. Girón-Calle, M. Alaiz, J. Vioque, J. M. Guisán and F. Millán, Enzyme Microb. Technol., 2007, 40, 1160–1166 CrossRef CAS.
- N. Zeballos, E. Diamanti, A. I. Benítez-Mateos, C. Schmidt-Dannert and F. López-Gallego, Bioconjugate Chem., 2021, 32, 1966–1972 CrossRef CAS PubMed.
- A. Velasco Abadia, K. M. Herbert, V. M. Matavulj, T. J. White, D. K. Schwartz and J. L. Kaar, J. Am. Chem. Soc., 2021, 143, 16740–16749 CrossRef CAS PubMed.
- D. Andrés-Sanz, E. Diamanti, D. Di Silvo, J. Gurauskis and F. López-Gallego, ACS Appl. Mater. Interfaces, 2022, 14, 4285–4296 CrossRef PubMed.
- S. Velasco-Lozano, E. S. da Silva, J. Llop and F. López-Gallego, ChemBioChem, 2018, 19, 395–403 CrossRef CAS PubMed.
- D. Ubiali, S. Rocchietti, F. Scaramozzino, M. Terreni, A. M. Albertini, R. Fernández-Lafuente, J. M. Guisán and M. Pregnolato, Adv. Synth. Catal., 2004, 346, 1361–1366 CrossRef CAS.
- S. Rocchietti, D. Ubiali, M. Terreni, A. M. Albertini, R. Fernández-Lafuente, J. M. Guisán and M. Pregnolato, Biomacromolecules, 2004, 5, 2195–2200 CrossRef CAS PubMed.
- R. Fernandez-Lafuente, C. M. Rosell, L. Caanan-Haden, L. Rodes and J. M. Guisan, Enzyme Microb. Technol., 1999, 24, 96–103 CrossRef CAS.
- R. Fernandez-Lafuente, V. Rodriguez, C. Mateo, G. Fernandez-Lorente, P. Arminsen, P. Sabuquillo and J. M. Guisan, J. Mol. Catal. B: Enzym., 1999, 7, 173–179 CrossRef CAS.
- K. Hernandez and R. Fernandez-Lafuente, Process Biochem., 2011, 46, 873–878 CrossRef CAS.
- A. M. Klibanov, N. O. Kaplan and M. D. Kamen, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 3640–3643 CrossRef CAS PubMed.
- L. Wilson, A. Illanes, O. Abián, B. C. C. Pessela, R. Fernández-Lafuente and J. M. Guisán, Biomacromolecules, 2004, 5, 852–857 CrossRef CAS PubMed.
- L. Wilson, G. Fernández-Lorente, R. Fernández-Lafuente, A. Illanes, J. M. Guisán and J. M. Palomo, Enzyme Microb. Technol., 2006, 39, 750–755 CrossRef CAS.
- A. Marty, V. Dossat and J.-S. Condoret, Biotechnol. Bioeng., 1997, 56, 232–237 CrossRef CAS.
- E. Séverac, O. Galy, F. Turon, C. A. Pantel, J.-S. Condoret, P. Monsan and A. Marty, Enzyme Microb. Technol., 2011, 48, 61–70 CrossRef PubMed.
- V. Dossat, D. Combes and A. Marty, Enzyme Microb. Technol., 1999, 25, 194–200 CrossRef CAS.
- L. P. Fallavena, F. H. F. Antunes, J. S. Alves, N. Paludo, M. A. Z. Ayub, R. Fernandez-Lafuente and R. C. Rodrigues, RSC Adv., 2014, 4, 8675 RSC.
- A. B. Martins, M. F. Schein, J. L. R. Friedrich, R. Fernandez-Lafuente, M. A. Z. Ayub and R. C. Rodrigues, Ultrason. Sonochem., 2013, 20, 1155–1160 CrossRef CAS PubMed.
- N. Paludo, J. S. Alves, C. Altmann, M. A. Z. Ayub, R. Fernandez-Lafuente and R. C. Rodrigues, Ultrason. Sonochem., 2015, 22, 89–94 CrossRef CAS PubMed.
- A. B. Martins, J. L. R. Friedrich, J. C. Cavalheiro, C. Garcia-Galan, O. Barbosa, M. A. Z. Ayub, R. Fernandez-Lafuente and R. C. Rodrigues, Bioresour. Technol., 2013, 134, 417–422 CrossRef CAS PubMed.
- N. G. Graebin, A. B. Martins, A. S. G. Lorenzoni, C. Garcia-Galan, R. Fernandez-Lafuente, M. A. Z. Ayub and R. C. Rodrigues, Biotechnol. Prog., 2012, 28, 406–412 CrossRef CAS PubMed.
- J. L. R. Friedrich, F. P. Peña, C. Garcia-Galan, R. Fernandez-Lafuente, M. A. Z. Ayub and R. C. Rodrigues, J. Chem. Technol. Biotechnol., 2013, 88, 1089–1095 CrossRef CAS.
- J. K. Poppe, C. Garcia-Galan, C. R. Matte, R. Fernandez-Lafuente, R. C. Rodrigues and M. A. Z. Ayub, J. Mol. Catal. B: Enzym., 2013, 94, 51–56 CrossRef CAS.
- C. J. Gray, Biocatalysis, 1988, 1, 187–196 CrossRef CAS.
- J. D. Brennan, D. Benjamin, E. DiBattista and M. D. Gulcev, Chem. Mater., 2003, 15, 737–745 CrossRef CAS.
- D. J. Leibly, T. N. Nguyen, L. T. Kao, S. N. Hewitt, L. K. Barrett and W. C. Van Voorhis, PLoS One, 2012, 7, e52482 CrossRef CAS PubMed.
- G. Alvaro, R. Fernandez-Lafuente, R. M. Blanco and J. M. Guisán, Enzyme Microb. Technol., 1991, 13, 210–214 CrossRef CAS PubMed.
- R. M. Blanco and J. M. Guisán, Enzyme Microb. Technol., 1988, 10, 227–232 CrossRef CAS.
- J. M. Palomo, C. Ortiz, G. Fernández-Lorente, M. Fuentes, J. M. Guisán and R. Fernández-Lafuente, Enzyme Microb. Technol., 2005, 36, 447–454 CrossRef CAS.
- J. M. Palomo, C. Ortiz, M. Fuentes, G. Fernandez-Lorente, J. M. Guisan and R. Fernandez-Lafuente, J. Chromatogr. A, 2004, 1038, 267–273 CrossRef CAS PubMed.
- G. Fernández-Lorente, J. M. Palomo, M. Fuentes, C. Mateo, J. M. Guisán and R. Fernández-Lafuente, Biotechnol. Bioeng., 2003, 82, 232–237 CrossRef PubMed.
- J. M. Palomo, M. Fuentes, G. Fernández-Lorente, C. Mateo, J. M. Guisan and R. Fernández-Lafuente, Biomacromolecules, 2003, 4, 1–6 CrossRef CAS PubMed.
- J. M. Palomo, M. M. Peñas, G. Fernández-Lorente, C. Mateo, A. G. Pisabarro, R. Fernández-Lafuente, L. Ramírez and J. M. Guisán, Biomacromolecules, 2003, 4, 204–210 CrossRef CAS PubMed.
- Y. Lokha, S. Arana-Peña, N. S. Rios, C. Mendez-Sanchez, L. R. B. Gonçalves, F. Lopez-Gallego and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2020, 133, 109461 CrossRef CAS PubMed.
- S. Arana-Peña, N. S. Rios, D. Carballares, L. R. B. Gonçalves and R. Fernandez-Lafuente, Catal. Today, 2021, 362, 130–140 CrossRef.
- S. Arana-Peña, N. S. Rios, D. Carballares, C. Mendez-Sanchez, Y. Lokha, L. R. B. Gonçalves and R. Fernandez-Lafuente, Front. Bioeng. Biotechnol., 2020, 8, 36 CrossRef PubMed.
- R. Morellon-Sterling, E.-H. Siar, S. A. Braham, D. de Andrades, J. Pedroche, M. del, C. Millán and R. Fernandez-Lafuente, J. Biotechnol., 2021, 329, 128–142 CrossRef CAS PubMed.
- A. P. Ison, A. R. Macrae, C. G. Smith and J. Bosley, Biotechnol. Bioeng., 1994, 43, 122–130 CrossRef CAS PubMed.
- R. J. Barros, E. Wehtje and P. Adlercreutz, Biotechnol. Bioeng., 2000, 67, 319–326 CrossRef CAS PubMed.
- H. Baldascini, K. J. Ganzeveld, D. B. Janssen and A. A. C. M. Beenackers, Biotechnol. Bioeng., 2001, 73, 44–54 CrossRef CAS.
- M. J. J. Litjens, K. Q. Le, A. J. J. Straathof, J. A. Jongejan and J. J. Heijnen, Biocatal. Biotransform., 2001, 19, 1–19 CrossRef CAS.
- L. Fernandez-Lopez, S. G. Pedrero, N. Lopez-Carrobles, B. C. Gorines, J. J. Virgen-Ortíz and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2017, 98, 18–25 CrossRef CAS PubMed.
- H. Zaak, E.-H. Siar, J. F. Kornecki, L. Fernandez-Lopez, S. G. Pedrero, J. J. Virgen-Ortíz and R. Fernandez-Lafuente, Process Biochem., 2017, 56, 117–123 CrossRef CAS.
- A. Kumar, Int. J. Microbiol., 2020, 1–11 Search PubMed.
- C. Li, J. Zhou, G. Du, J. Chen, S. Takahashi and S. Liu, Biotechnol. Adv., 2020, 44, 107630 CrossRef CAS PubMed.
- H. A. B. Wösten, Curr. Opin. Biotechnol, 2019, 59, 65–70 CrossRef PubMed.
- R. S. Kun, A. C. S. Gomes, K. S. Hildén, S. Salazar Cerezo, M. R. Mäkelä and R. P. de Vries, Biotechnol. Adv., 2019, 37, 107361 CrossRef CAS PubMed.
- D. A. Teigiserova, J. Bourgine and M. Thomsen, Sustainable Prod. Consum., 2021, 27, 845–857 CrossRef.
- D. B. Archer, Bioprocess Technol, 1994, 19, 373–393 CAS.
- A. Sánchez, P. Ferrer, A. Serrano, F. Valero, C. Solà, M. Pernas, M. L. Rúa, R. Fernández-Lafuente, J. M. Guisán, R. M. de la Casa, J. V. Sinisterra and J. M. Sánchez-Montero, J. Biotechnol., 1999, 69, 169–182 CrossRef.
- A. Natalello, D. Ami, S. Brocca, M. Lotti and S. M. Doglia, Biochem. J., 2005, 385, 511–517 CrossRef CAS PubMed.
- N. G. Jayaprakash and A. Surolia, Biochem. J., 2017, 474, 2333–2347 CrossRef CAS PubMed.
- G. Williamson, N. J. Belshaw, T. R. Noel, S. G. Ring and M. P. Williamson, Eur. J. Biochem., 1992, 207, 661–670 CrossRef CAS PubMed.
- C. Bonzom, S. Hüttner, E. Mirgorodskaya, S.-L. Chong, S. Uthoff, A. Steinbüchel, R. M. D. Verhaert and L. Olsson, AMB Express, 2019, 9, 126 CrossRef PubMed.
- A. Kahn, S. Moraïs, D. Chung, N. S. Sarai, N. N. Hengge, A. Kahn, M. E. Himmel, E. A. Bayer and Y. J. Bomble, FEBS J., 2020, 287, 4370–4388 CrossRef CAS PubMed.
- M. Tian, J. Fu, Z. Wang, C. Miao, P. Lv, D. He, Z. Li, T. Liu, M. Li and W. Luo, Fuel, 2021, 304, 121514 CrossRef CAS.
- C. W. Liria, C. D. Romagna, N. N. Rodovalho, S. R. Marana and M. T. M. Miranda, J. Braz. Chem. Soc., 2008, 19, 1574–1581 CrossRef CAS.
- J. M. Palomo, G. Fernández-Lorente, C. Mateo, M. Fuentes, J. M. Guisan and R. Fernández-Lafuente, Tetrahedron: Asymmetry, 2002, 13, 2653–2659 CrossRef CAS.
- R. L. Segura, L. Betancor, J. M. Palomo, A. Hidalgo, G. Fernández-Lorente, M. Terreni, C. Mateo, A. Cortés, R. Fernández-Lafuente and J. M. Guisán, Enzyme Microb. Technol., 2006, 39, 817–823 CrossRef CAS.
- J. Arsan and K. L. Parkin, Biotechnol. Bioeng., 2000, 69, 222–226 CrossRef CAS PubMed.
- O. Barbosa, M. Ruiz, C. Ortiz, M. Fernández, R. Torres and R. Fernandez-Lafuente, Process Biochem., 2012, 47, 867–876 CrossRef CAS.
- M. Mohammadi, Z. Habibi, S. Dezvarei, M. Yousefi, S. Samadi and M. Ashjari, Process Biochem., 2014, 49, 1314–1323 CrossRef CAS.
- J. M. Palomo, G. Fernandez-Lorente, C. Mateo, C. Ortiz, R. Fernandez-Lafuente and J. M. Guisan, Enzyme Microb. Technol., 2002, 31, 775–783 CrossRef CAS.
- A. Chaubey, R. Parshad, S. Koul, S. C. Taneja and G. N. Qazi, J. Mol. Catal. B: Enzym., 2006, 42, 39–44 CrossRef CAS.
- V. G. Tacias-Pascacio, S. Peirce, B. Torrestiana-Sanchez, M. Yates, A. Rosales-Quintero, J. J. Virgen-Ortíz and R. Fernandez-Lafuente, RSC Adv., 2016, 6, 100281–100294 RSC.
- S. Takaç and M. Bakkal, Process Biochem., 2007, 42, 1021–1027 CrossRef.
- C. Mateo, A. Archelas, R. Fernandez-Lafuente, J. Manuel Guisan and R. Furstoss, Org. Biomol. Chem., 2003, 1, 2739 RSC.
- G. Yang, J. Wu, G. Xu and L. Yang, J. Mol. Catal. B: Enzym., 2009, 57, 96–103 CrossRef CAS.
- D. Farrusseng, S. Aguado and C. Pinel, Angew. Chem., Int. Ed., 2009, 48, 7502–7513 CrossRef CAS PubMed.
- T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC.
- C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- S. M. J. Rogge, A. Bavykina, J. Hajek, H. Garcia, A. I. Olivos-Suarez, A. Sepúlveda-Escribano, A. Vimont, G. Clet, P. Bazin, F. Kapteijn, M. Daturi, E. V. Ramos-Fernandez, F. X. Llabrés i Xamena, V. Van Speybroeck and J. Gascon, Chem. Soc. Rev., 2017, 46, 3134–3184 RSC.
- M. Puri, A. Kaur, R. S. Singh, W. H. Schwarz and A. Kaur, Process Biochem., 2010, 45, 451–456 CrossRef CAS.
- I. Ardao, M. D. Benaiges, G. Caminal and G. Álvaro, Enzyme Microb. Technol., 2006, 39, 22–27 CrossRef CAS.
- M. Cao, Z. Li, J. Wang, W. Ge, T. Yue, R. Li, V. L. Colvin and W. W. Yu, Trends Food Sci. Technol., 2012, 27, 47–56 CrossRef CAS.
- S. S. Nadar and V. K. Rathod, Int. J. Biol. Macromol., 2018, 120, 2293–2302 CrossRef CAS PubMed.
- K. Khoshnevisan, F. Vakhshiteh, M. Barkhi, H. Baharifar, E. Poor-Akbar, N. Zari, H. Stamatis and A.-K. Bordbar, Mol. Catal, 2017, 442, 66–73 CrossRef CAS.
- J. Mehta, N. Bhardwaj, S. K. Bhardwaj, K.-H. Kim and A. Deep, Coord. Chem. Rev., 2016, 322, 30–40 CrossRef CAS.
- S. Liang, X.-L. Wu, J. Xiong, M.-H. Zong and W.-Y. Lou, Coord. Chem. Rev., 2020, 406, 213149 CrossRef CAS.
- J. Cui, S. Ren, B. Sun and S. Jia, Coord. Chem. Rev., 2018, 370, 22–41 CrossRef CAS.
- S. S. Nadar, L. Vaidya and V. K. Rathod, Int. J. Biol. Macromol., 2020, 149, 861–876 CrossRef CAS PubMed.
- F. Lin, V. L. Dagle, A. D. Winkelman, M. Engelhard, L. Kovarik, Y. Wang, Y. Wang, R. Dagle and H. Wang, ChemCatChem, 2021, 13, 999–1008 CrossRef CAS.
- L. V. Mattos, G. Jacobs, B. H. Davis and F. B. Noronha, Chem. Rev., 2012, 112, 4094–4123 CrossRef CAS PubMed.
- A. M. Saib, D. J. Moodley, I. M. Ciobîcă, M. M. Hauman, B. H. Sigwebela, C. J. Weststrate, J. W. Niemantsverdriet and J. van de Loosdrecht, Catal. Today, 2010, 154, 271–282 CrossRef CAS.
- J. M. Bolivar, D. Valikhani and B. Nidetzky, Biotechnol. J., 2019, 14, 1800244 CrossRef PubMed.
- J. M. Bolivar, C. Luley-Goedl, E. Leitner, T. Sawangwan and B. Nidetzky, J. Biotechnol., 2017, 257, 131–138 CrossRef CAS PubMed.
- J. M. Bolivar, M. A. Tribulato, Z. Petrasek and B. Nidetzky, Biotechnol. Bioeng., 2016, 113, 2342–2349 CrossRef CAS PubMed.
- A. Lorente-Arevalo, A. Garcia-Martin, M. Ladero and J. M. Bolivar, in Enzyme Engineering, ed. F. Magnani, C. Marabelli and F. Paradisi, Springer US, New York, NY, 2022, vol. 2397, pp. 277–320 Search PubMed.
- L. Ruzic, J. M. Bolivar and B. Nidetzky, Biotechnol. Bioeng., 2020, 117, 1597–1602 CrossRef CAS PubMed.
- J. M. Bolivar, A. Mannsberger, M. S. Thomsen, G. Tekautz and B. Nidetzky, Biotechnol. Bioeng., 2019, 116, 503–514 CrossRef CAS PubMed.
- P.-Y. Kim, D. J. Pollard and J. M. Woodley, Biotechnol. Prog., 2007, 23, 74–82 CrossRef CAS PubMed.
- A. Lorente-Arevalo, M. Ladero and J. M. Bolivar, React. Chem. Eng., 2021, 6, 2058–2074 RSC.
- A. Illanes, L. Wilson and C. Vera, in Catalysis Series, ed. G. Williams and M. Hall, Royal Society of Chemistry, Cambridge, 2018, pp. 473–515 Search PubMed.
- P. Silva, V. Arancibia, D. Cid, O. Romero, A. Illanes and L. Wilson, Catalysts, 2021, 11, 1167 CrossRef CAS.
- V. Miranda, L. Wilson, C. Cárdenas and A. Illanes, J. Mol. Catal. B: Enzym., 2011, 68, 77–82 CrossRef CAS.
- A. Illanes, L. Wilson and G. Tomasello, J. Mol. Catal. B: Enzym., 2001, 11, 531–540 CrossRef CAS.
- A. Illanes and L. Wilson, Crit. Rev. Biotechnol., 2003, 23, 61–93 CrossRef CAS PubMed.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2005, 44, 4302–4320 CrossRef CAS PubMed.
- N. Al-Haque, P. A. Santacoloma, W. Neto, P. Tufvesson, R. Gani and J. M. Woodley, Biotechnol. Prog., 2012, 28, 1186–1196 CrossRef CAS PubMed.
- M. Nordblad, M. D. Gomes, M. P. Meissner, H. Ramesh and J. M. Woodley, Org. Process Res. Dev., 2018, 22, 1101–1114 CrossRef CAS.
- A. I. Benítez-Mateos, B. Nidetzky, J. M. Bolivar and F. López-Gallego, ChemCatChem, 2018, 10, 654–665 CrossRef.
- H. Sánchez-Morán, J. S. Weltz, D. K. Schwartz and J. L. Kaar, ACS Appl. Mater. Interfaces, 2021, 13, 26694–26703 CrossRef PubMed.
- A. F. Chaparro Sosa, R. M. Bednar, R. A. Mehl, D. K. Schwartz and J. L. Kaar, J. Am. Chem. Soc., 2021, 143, 7154–7163 CrossRef CAS PubMed.
- J. S. Weltz, D. F. Kienle, D. K. Schwartz and J. L. Kaar, J. Am. Chem. Soc., 2020, 142, 3463–3471 CrossRef CAS PubMed.
- A. F. Chaparro Sosa, K. J. Black, D. F. Kienle, J. L. Kaar and D. K. Schwartz, Adv. Mater. Interfaces, 2020, 7, 2000533 CrossRef CAS.
- L. Lancaster, W. Abdallah, S. Banta and I. Wheeldon, Chem. Soc. Rev., 2018, 47, 5177–5186 RSC.
- W. Abdallah, X. Hong, S. Banta and I. Wheeldon, Curr. Opin. Biotechnol, 2022, 73, 233–239 CrossRef CAS PubMed.
- N. R. Mohamad, N. H. C. Marzuki, N. A. Buang, F. Huyop and R. A. Wahab, Biotechnol. Biotechnol. Equip., 2015, 29, 205–220 CrossRef CAS PubMed.
- R. H. Abeles and A. L. Maycock, Acc. Chem. Res., 1976, 9, 313–319 CrossRef CAS.
- C. T. Walsh, Annu. Rev. Biochem., 1984, 53, 493–535 CrossRef CAS PubMed.
- L. Mazzei, M. Cianci, U. Contaldo, F. Musiani and S. Ciurli, Biochemistry, 2017, 56, 5391–5404 CrossRef CAS PubMed.
- X. Zhang, X.-X. Li, Y. Liu and Y. Wang, Front. Chem., 2017, 5, 3 Search PubMed.
- V. Ahmed, Y. Liu and S. D. Taylor, ChemBioChem, 2009, 10, 1457–1461 CrossRef CAS PubMed.
- L. Betancor, A. Hidalgo, G. Fernandez-Lorente, C. Mateo, V. Rodriguez, M. Fuentes, F. Lopez-Gallego, R. Fernandez-Lafuente and J. M. Guisan, Biotechnol. Prog., 2003, 19, 784–788 CrossRef CAS PubMed.
- S. Kaddour, F. López-Gallego, T. Sadoun, R. Fernandez-Lafuente and J. M. Guisan, J. Mol. Catal. B: Enzym., 2008, 55, 142–145 CrossRef CAS.
- E.-H. Siar, S. Arana-Peña, O. Barbosa, M. N. Zidoune and R. Fernandez-Lafuente, Process Biochem., 2018, 73, 109–116 CrossRef CAS.
- P. García-García, J. M. Guisan and G. Fernandez-Lorente, J. Biotechnol., 2020, 318, 39–44 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |