
Current knowledge on the tissue distribution of mRNA nanocarriers for therapeutic protein expression
Matthias
Zadory
,
Elliot
Lopez
 ,
Samuel
Babity
,
Simon-Pierre
Gravel
and
Davide
Brambilla
*
,
Samuel
Babity
,
Simon-Pierre
Gravel
and
Davide
Brambilla
*
Faculté de Pharmacie, Université de Montréal, 2940 Chemin de Polytechnique, Montréal, Québec, Canada H3T 1J4. E-mail: davide.brambilla@umontreal.ca
First published on 13th September 2022
Abstract
Exogenously delivered mRNA-based drugs are emerging as a new class of therapeutics with the potential to treat several diseases. Over the last decade, advancements in the design of non-viral delivery tools have enabled mRNA to be evaluated for several therapeutic purposes including protein replacement therapies, gene editing, and vaccines. However, in vivo delivery of mRNA to targeted organs and cells remains a critical challenge. Evaluation of the biodistribution of mRNA vehicles is of utmost importance for the development of effective pharmaceutical candidates. In this review, we discuss the recent advances in the design of nanoparticles loaded with mRNA and extrapolate the key factors influencing their biodistribution following administration. Finally, we highlight the latest developments in the preclinical and clinical translation of mRNA therapeutics for protein supplementation therapy.
1. Introduction
Messenger RNA (mRNA) therapeutics can be used to replace proteins, produce antigens that raise an immune response, or transiently express components of gene-editing machinery. Because of this versatility, mRNA acts as a promising class of therapeutics, particularly for Mendelian disorders with established deleterious mutations, infectious diseases, immunological disorders, and cancer. Notably, as 85–90% of the protein-encoding portion of the human genome remains undruggable with current small chemical entities, mRNA approaches provide great hope for efficient treatments.1,2 Initial preclinical trials of local in vitro-transcribed (IVT) mRNA injections in the early 1990s demonstrated the potential of this technology for diverse protein replacement and vaccination applications, but a poor pharmacokinetic profile, along with rapid clearance and a short expression profile, hampered its breakthrough into clinics.3,4 The therapeutic use of mRNA is indeed challenging because of its unfavorable physicochemical characteristics, such as high molecular weight, negative charge, and susceptibility to ribonucleases (RNases), which prevent efficient uptake of mRNA into cells and subsequent translation.5 The design of efficient delivery strategies to resolve these shortcomings has thus been a very active research field, with nanoparticles showing promising results. As early as 1989, Malone and co-workers described the use of the cationic lipid N-[1-(2,3-dioleyloxy) propyl]-N,N,N-trimethylammonium chloride (DOTMA) for in vitro transfection, and by 1993 the first liposomal mRNA vaccine was tested in mice, highlighting the potential of nano-formulations for the development of mRNA therapeutics.6–9From a delivery perspective, mRNA can be efficiently translated within the cytoplasm and does not require transport into the nucleus, a key advantage over DNA-based approaches.10 Years of research on the design of delivery tools have focused on both viral and non-viral technologies, with the former demonstrating more rapid success, but carrying potential hurdles such as unwanted genomic integration, immunogenicity, and costly vector production.11,12 Concurrently, the adaptation of biomaterials platforms for the encapsulation of mRNA for safer delivery has invigorated research on synthetic materials, such as polymers and lipids, and has recently led to the development and approval of two COVID-19 vaccines.13 Despite these improvements, there remains a critical challenge facing the therapeutic use of mRNA, namely the difficulty of delivering mRNA molecules to target cells with high efficiency and specificity. This is particularly important in the context of a deficient or defective intracellular protein that is to be replaced through IVT mRNA expression. In such cases, the total proportion of transfected target cells is most critical, rather than the absolute quantity of protein generated.14 Furthermore, the nanocarrier should be capable of shuttling a mRNA payload to specific cells, so that the mRNA can be translated and adequately exert its function in the surrounding tissues or organs. For cell therapy, in which cell transfection occurs in vitro in a controlled and optimized environment, this consideration is less important.14,15 In contrast, the in vivo delivery of mRNA to defined target cell populations in high proportions is challenging, and depends greatly on the accessibility of the target cells and the biodistribution profile of the nanocarrier. By understanding the fate of mRNA nanocarriers upon systemic injection (intravenous, IV) or local administration (e.g., intravitreal, intratracheal, intrathecal), new opportunities can be identified to improve the efficiency of RNA therapeutics and harness their full potential for the treatment of diseases.
To the best of our knowledge, existing reviews primarily focus on therapeutic gene regulation in the liver and provide limited considerations for extrahepatic delivery. Strikingly, no comprehensive overview of mRNA therapeutics for protein expression and the targeted in vivo delivery of mRNA to different organs using non-viral technology has been presented. In this review, we thus seek to examine and critically discuss the development of mRNA nanocarriers and related protein expression in diverse tissues. First, we describe the biochemical features of mRNA, as well as specific challenges associated with its delivery. Second, we review the current literature to isolate the key features of lipid-based nanoparticles (LNPs) and polymer nanocarriers which enable delivery of mRNA to specific organs of therapeutic relevance. To conclude, we highlight recent preclinical and clinical advances in the development of non-viral mRNA delivery strategies for a range of protein-supplementation therapies.
2. mRNA as a drug
2.1. mRNA engineering for optimization in vivo function
As a drug candidate, mRNA possesses two major advantages over DNA-based approaches, namely that it does not require nuclear access and does not need to integrate into the cell genome, thus mitigating the risk of unwanted genomic alteration.16 Within cells, mRNA is physiologically degraded through a succession of molecular modifications including deadenylation and decapping, followed by RNase-mediated hydrolysis.14 These mechanisms ensure that exogenous mRNA therapeutics are expressed transiently, which directly impact their safety profile.17 Nonetheless, the in vivo development of mRNA therapeutics has long been hampered by two major challenges: the poor stability of mRNA (limiting the amount of protein able to be produced by cellular machinery) and the inefficient delivery of mRNA to target cells.Pioneering efforts have focused on modifying structural elements of mRNA to improve intracellular stability and translational efficiency, achieving protein expression levels up to 10-fold higher than initially observed with unmodified mRNA.18,19 In particular, IVT mRNA is designed to mimic mature and processed mRNA in the cytoplasm of eukaryotic cells. Accordingly, IVT mRNA is single-stranded, and has a 5′ cap and a 3′ poly(A) tail with an open reading frame (ORF) region encoding the protein of interest flanked by untranslated regions (UTRs) (Fig. 1A).
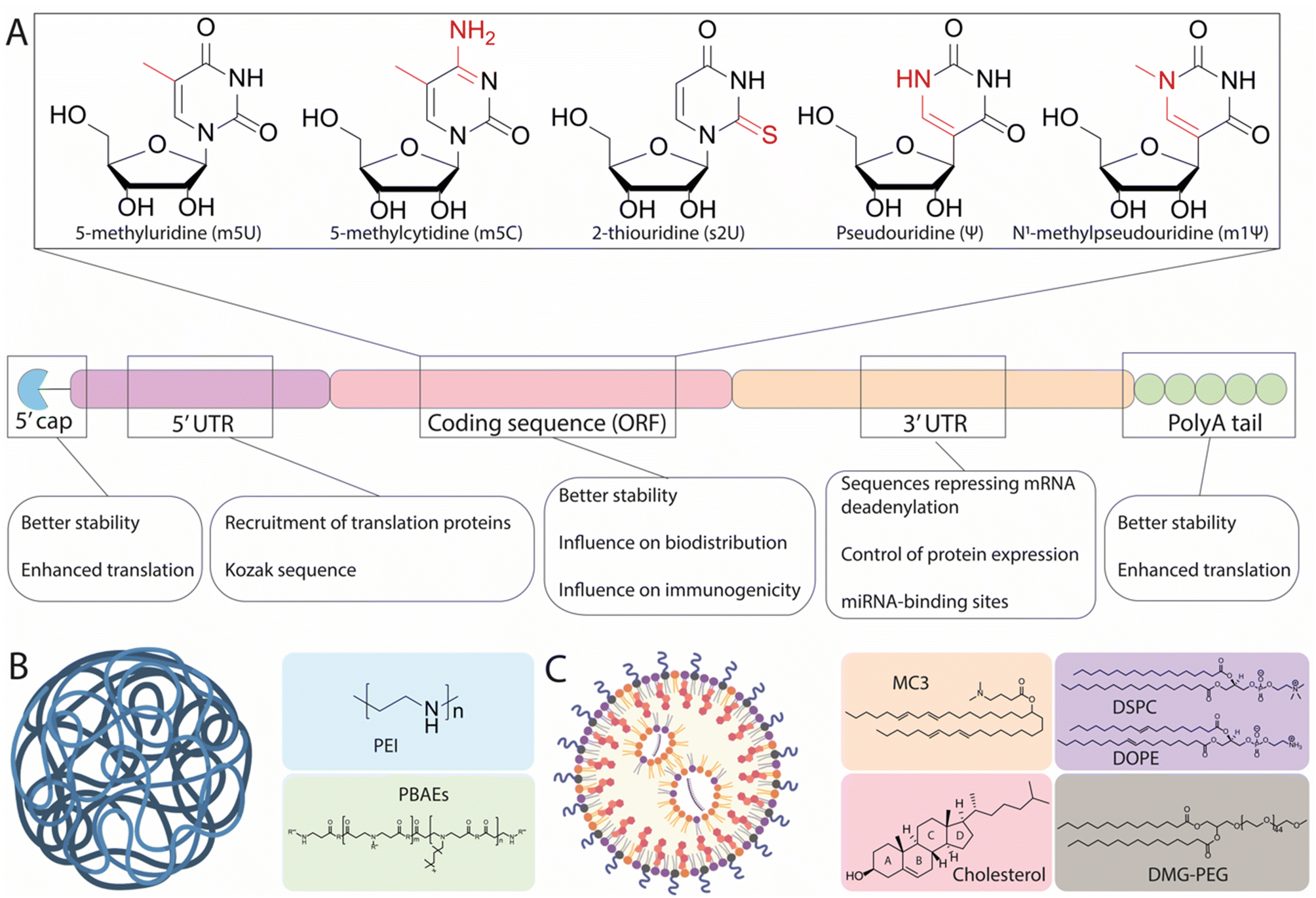 | ||
| Fig. 1 Structural elements of in vitro-transcribed (IVT) mRNA and delivery vehicles. (A) Chemical structure of common modified nucleotides used in the open reading frame (ORF) and scheme of the mRNA structure. Structural elements include a 5′ cap, 5′- and 3′-UTRs, an ORF encoding antigen(s), and a 3′ poly(A) tail. The main function of each region is detailed underneath. (B) and (C) graphical representation of a polymer nanoparticle and a lipid nanoparticle, along with the most common chemical structures associated with each type. Made with Biorender. | ||
The 7-methylguanosine cap (m7G) protects mRNA from exonuclease degradation and promotes translation by recruiting the initiation factor 4E (eIF4E).20 Current approaches to mimic the m7G cap on IVT mRNA utilize synthetic m7G and m7G analogues, including phosphorothioate dinucleotide analogues, boranophosphate analogues, and anti-reverse cap analogues, all of which increase mRNA translation efficiency and extend the cytoplasmic half-life of IVT mRNA.21–24 In synergy, 5′ protection by capping and 3′ polyadenylation also contributes to the stability, translational efficiency, and entry of the mRNA into the ribosomal machinery.25 Many studies have explored the influence of the poly(A) tail on translational efficiency, reporting optimal lengths ranging between 100 and 240 nucleotides.21,26,27 This variation has been shown to depend on the specific modifications of the mRNA sequence, as well as the target cell line used.26
Another strategy to increase mRNA stability and translational efficiency is the integration of 5′- and 3′-UTRs with optimal length and regulatory elements to influence translational efficiency and stability. In a study performing the rational screening of de novo designed UTRs, the authors found that an optimal 5′-UTR length of around 70 nucleotides is required for adequate translation initiation complex assembly and rapid translation, and that the incorporation of positive cis-regulatory elements in the 3′-UTR contributes to a 10-fold increase in protein expression relative to endogenous UTRs.28 Information contained within the UTRs can also regulate the transport and specific subcellular localization of mRNA. Cis-acting RNA elements are specific RNA sequences located in the UTRs that associate with RNA-binding proteins and promote the transport of mRNA to specific regions within targeted cells.29 For example, using the 5′-UTR of a tick-borne encephalitis virus, the inclusion of dendritic targeted cis-acting RNA elements could enable more robust and effective mRNA expression in the neuronal dendrites of mice, in comparison to the control UTR.30 Another feasible way to control the localization of mRNA expression is to insert additional cell-specific microRNA (miRNA) targeting sites (miRTS) within the 3′-UTR of an IVT mRNA scaffold. miRNAs are noncoding RNA sequences ∼20 nucleotides in length that, upon binding to specific regions of the 3′-UTR, can both repress translation and initiate mRNA cleavage, leading to rapid clearance.31 Because miRNAs exhibit cell-type and disease-specific expression profiles, the inclusion of binding sites for miRNAs can be used to suppress protein expression in off-target tissues and has been tested with viral vectors.32 For instance, miR-122 is specifically expressed in healthy hepatocytes and repressed in hepatocellular carcinoma.33 Accordingly, inclusion of a miRTS for miR-122 in the mRNA scaffold resulted in preferential mRNA expression in non-hepatocyte tumor cells in vivo.34 This demonstrates how the appropriate design of UTRs can play an important role in the expression profile and localization of proteins, though this technique has not yet been sufficiently investigated for its capacity to drive expression in specific cells, warranting the use of more sophisticated delivery tools.
Finally, a key development to the understanding of mRNA therapeutics has been the discovery of the immunogenic response elicited by mRNA. It has been shown that exogenous mRNA is immunostimulatory and can be recognized by pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs), which trigger innate immune signaling pathways that restrict protein expression and enhance the clearance of mRNA transcripts within cells.35 Early studies have demonstrated that mRNA recognition by TLRs can be suppressed via the modification of nucleotides used in IVT mRNA.18 The most commonly utilized modifications (Fig. 1A) include 5-methylcytosine (m5C), 5-methyluridine (m5U), 2-thiouridine (s2U), or pseudouridine (ψ) modifications such as 1-methylpseudouridine (m1ψ), which abrogate the immune response by evading the activation of PRRs. The introduction of modified uridines within the mRNA construct is currently the most efficient way to minimize the immune reaction to foreign mRNA. Notably, m1ψ has been used in both the Moderna and BioNTech/Pfizer COVID-19 vaccines.13 Though base modifications have been shown to improve the efficacy of mRNA treatments, other findings have suggested that this effect is not universal. For instance, the presence of pseudouridine modifications demonstrated no significant effect on either protein expression in vivo or mRNA immunogenicity, as compared to unmodified mRNA, when delivered systemically using liver-targeting LNPs.36 As such, it could be hypothesized that the variable advantages offered by base modification may relate to the chemistry and formulation of the mRNA delivery vehicles used, and also to the target cells in which mRNA translation occurs. This notion is supported by a recent study, which systematically screened a series of LNPs incorporating five types of mRNA modifications (ψ, m1ψ, m5U, m5C, m5C/ψ, and m5C/s2U) encoding for luciferase (Luc) and four ionizable amino lipids promoting mRNA delivery to different organs.37 It was demonstrated that the m1ψ modification was the most effective for all vehicles, with an increase of protein expression between 1.5-fold and 11-fold observed, depending on the delivery vehicle. In contrast, the m5U modification was shown to be detrimental to efficacy, inducing up to a 90% reduction in total Luc expression. Together, these findings demonstrate that mRNA modifications ought to be carefully chosen depending on the chemistry of the delivery vehicle and the targeted organ or tissue.
2.2. Delivery of mRNA
Although mRNA sequences can be engineered to limit instability and immunogenicity issues, the in vivo administration of mRNA remains hampered by the effects of extracellular RNase-mediated degradation and by the need to cross the negatively charged cellular membrane.14 Overcoming these effects is where innovations in nanocarrier-based delivery systems are likely to play a crucial role.Naked mRNA is largely unable to cross the cell membrane and achieve efficient availability in the cytoplasm—the primary pharmacodynamic compartment of mRNA. The size of mRNA (300–5000 kDa, 1–15 kb) is significantly larger than that of small interfering RNA (siRNA) and miRNA (13–15 kDa) and antisense oligonucleotides (4–10 kDa).38 Though some cells are capable of spontaneous internalization of naked mRNA through scavenger-receptor-mediated endocytosis, only a small amount of mRNA is delivered to the cytoplasm through this process.39 As an exception, immature dendritic cells have the specialized ability to constantly engulf extracellular fluid, and can take up mRNA more efficiently through macropinocytosis.39,40 Consequently, a suitable carrier is required for the delivery of mRNA into most cell types. Initially, very efficient in vitro delivery tools were developed based on permanent cationic lipids such as 2,3-dioleoyloxy-N-[2(sperminecarboxamido)ethyl]-N,N-dimethyl-1-propaniminium trifluoroacetate (DOSPA), or 1,2-dioleoyl-3-trimethylammonium propane (DOTAP) which, when mixed with phospholipid such as 1,2-dioleoyl-sn-glycerophosphoethanolamine (DOPE), can complex with highly negatively charged mRNA, allowing them to overcome the electrostatic repulsion of the cell membrane. Nevertheless, the major limitation of these lipoplexes (LPXs) for in vivo applications are electrostatic interactions with negatively charged plasma proteins—these interactions lead to fast clearance and recognition by the immune system as well as cytotoxicity, thus hindering in vivo translation.41–43 In light of this, various other classes of materials have been proposed for mRNA delivery, primarily consisting of cationic polymers and LNPs, as illustrated in Fig. 1B and C. Because of their ability to electrostatically condense nucleic acids into nanoparticles, cationic polymers have been presented as potential candidates for intracellular mRNA delivery. Examples of cationic polymers used for in vivo RNA delivery studies include derivatives of polyethyleneimine (PEI), poly(amido-amine) (PAMAM), poly(lactic-co-glycolic acid) (PLGA), and poly(β-amino esters) (PBAEs) (Fig. 1B). Such polymeric structures can be modulated to achieve a high degree of chemical variation in functional groups, allowing for a range of physical properties, which in turn can influence their biodistribution and mRNA delivery.44 However, concerns regarding their limited efficacy and potential toxicity remain, with high net positive charges and their inability to be degraded under physiological conditions potentially leading to hazardous levels of bioaccumulation. In contrast, LNPs have gained popularity in recent years as vehicles for mRNA delivery. It has been determined that four components are generally required for LNP-modulated mRNA delivery: an ionizable lipid, a helper lipid (typically phosphatidylcholine), cholesterol, and a PEG-lipid (as illustrated in Fig. 1C). Ionizable lipids have been widely investigated and display a wide range of structures, yet all share common structural aspects. These include: (i) a headgroup containing tertiary amines with defined pKas, enabling protonation at acidic pH for interaction with the negatively charged mRNA, in addition to preserving a positive charge in the acidic endosomal lumen to promote membrane fusion and delivery to the cytosol, while exhibiting a neutral charge at physiological pH, and (ii) lipid tails enabling incorporation into nanoparticles during formation, facilitating membrane fusion and thereby improving endosomal escape. Additionally, modifications to their structures resulting in altered physicochemical properties can influence the selective delivery of mRNA to different organs.45 Phospholipids (also called helper lipids) and cholesterol are generally used to provide structure to LNPs, promoting formulation stability and potentially aiding in endosomal escape.46–48 Phospholipids such as 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) display strong bilayer-forming properties and high phase-transition temperatures, which help increase membrane rigidity and reduce membrane permeability. As an alternative to DSPC, DOPE has also been widely used as a helper lipid and has been shown to promote superior potency relative to DSPC for mRNA formulations.47 Cholesterol serves to confer stability by preventing the sequestration of endogenous cholesterol within the LNPs while in circulation.49,50 Recent findings have underscored the importance of cholesterol derivatives in the endosomal escape of LNPs, with nanoparticles containing a β-sitosterol substitute exhibiting a 10-fold increase in endosomal retrieval of mRNA in the cytosol when compared to standard cholesterol.51–53 Thus, cholesterol and its derivatives are important for the overall stability of LNPs in circulation and may aid in endosomal escape, thereby enhancing intracellular mRNA delivery. PEG-lipids reside on the surface of LNPs and serve three main functions, namely: (i) tuning particle size by varying their concentration, (ii) improving shelf-stability by preventing particle aggregation, and (iii) increasing the in vivo circulation lifetime of the LNPs. However, it is well established that the shield provided by PEGylation occurs at the expense of transfection efficiency, a problem known as the “PEG dilemma”.54
For potential therapeutic applications, mRNA drugs must generate a sufficient level of encoded protein to achieve their intended effect, while also transfecting a high proportion of target cells. Extensive research into the design of LNPs has resulted in very effective mRNA delivery to the liver, leading to the initiation of multiple clinical investigations.55 Though mRNA therapeutics remain a very promising approach for the treatment of non-liver genetic disorders, effective mRNA delivery to extra-hepatic tissues remains a meaningful challenge. Accordingly, a robust understanding of the behaviour of mRNA nanocarriers throughout the body, as well as the identification of key parameters governing specific delivery to target tissues and cells is required. To this end, tools have been used to assess and quantify particle-based mRNA distribution and expression. For the evaluation of biodistribution, mRNA can be labelled using a fluorescent dye (e.g., Cy5) to allow the tracking of mRNA-loaded nanoparticles throughout the body. As a potential alternative, mRNA transcripts can also be quantified using the reverse transcriptase polymerase chain reaction.56 Another common method of evaluating mRNA translation is the use of mRNA encoding for luciferase (Luc), which exhibits luminescence in transfected cells upon intraperitoneal administration of its substrate, luciferin. As an elegant alternative, detailed evaluation of mRNA expression at the cellular level can also be performed by using Ai14 ‘Cre-reporter’ mice.57 The Ai14 mice contain a Lox-Stop-Lox-tdTomato transgene under the control of a Cre promoter, resulting in conditional expression. As a result, cells in these mice only become tdTomato+ when Cre mRNA has been functionally delivered to the cells or successfully edited. Subsequent labelling with specific cell markers followed by flow cytometry allows for quantification of tdTomato+ cells. These tools have enabled the collection of data on the distribution pattern of mRNA, as well as on transfection performance in targeted cells. In the forthcoming sections, we will discuss properties and examples of particle-based therapeutics which have contributed to increased protein expression in primary organs such as the liver, lungs, and spleen, as well as in organs for which mRNA-based therapies represent a feasible treatment approach.
3. mRNA delivery to the liver, spleen, and lungs
3.1. mRNA delivery to the liver
The liver plays a crucial role in the metabolism of endogenous and exogenous compounds, and alterations in its function underly several metabolic disorders representing a global health burden.58,59 For most inherited metabolic liver disorders, current treatments offer limited efficacy, often leaving liver transplant as the only viable option and raising issues of donor compatibility, surgical risk, and long-term immunosuppression.58,60–62 mRNA therapeutics present an attractive strategy to resolve these issues, owing to their ability to induce efficient gene expression and replace deleterious or missing proteins. The liver is inherently capable of producing secretory proteins, and the administration of mRNA formulations can be used to harness the “liver as a bioreactor”, producing either proteins that are missing in inherited metabolic and hematological disorders, or antibodies for the neutralization of pathogens and tumor cells.63–66Following systemic administration, nanomedicines tend to accumulate in the liver due to discontinuous vasculature, decreased blood flow rates,67 and an abundance of phagocytic cell types lining the hepatic sinusoids.57 Sinusoid blood vessels display a highly fenestrated epithelium with fenestrae ranging from 100–150 nm and lacking basal membrane. This allows for the almost unrestricted passage of blood components and therapeutic carriers into the perisinusoidal space, where the parenchymal cells (hepatocytes) are located.61 Because these cells play a key role in many disorders, their targeting is of great importance. To accomplish this targeting, both active and passive strategies have been proposed. For the active targeting of hepatocytes, the most thoroughly investigated strategy relies on the conjugation of nanocarriers to N-acetylgalactosamine (GalNAc), a ligand for the asialoglycoprotein receptor, which is predominantly expressed on hepatocytes.68 This strategy has been employed in the hepatic delivery of siRNA to silence gene expression, as well as in mRNA delivery for the enhancement of protein expression.69–71 In comparison, passive targeting utilizes the body's existing trafficking systems and endogenous apolipoproteins such as Apolipoprotein E (ApoE), which is a low-density lipoprotein (LDL) and enters cells via binding to LDL receptors (LDLRs).72
For the delivery of mRNA to hepatic tissue, both LNPs and polymer nanocarriers have been investigated. Although not as clinically advanced as lipid-based systems, polymers have shown potential for protein replacement, vaccination, and other applications related to mRNA therapeutics.38,73,74 Herein, we discuss both types of delivery vehicles, with emphasis on LNPs, as this formulation is clearly dominant in hepatic mRNA delivery (as illustrated in Table 1).
| Delivery systems | Formulation (mol% if not specified) | mRNA payload | Administrative route | Tropism features (targeting ligand) | Reported transfected cells or tissue | Clinical indication | Ref. |
|---|---|---|---|---|---|---|---|
| Abbreviations: cholesteryl hemisuccinate (CHEMS), proprotein convertase subtilsin/kexin type 9 (PCSK9), poly[N′-[N(2-aminoethyl)-2-aminoethyl]aspartamide] (PAsp(DET)), SS-cleavable proton-activated lipid-like material (ssPALM). | |||||||
| Liver tissue | |||||||
| Active targeting | |||||||
| mRNA-lipopolyplex | DOTAP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Chol:CHEMS:PEG2000-lipid (+GalNac-targeted diblock polymer) :Chol:CHEMS:PEG2000-lipid (+GalNac-targeted diblock polymer) |
Luc mRNA, OTC mRNA, | IV | GalNAc polymer | Hepatocytes | Ornithine transcarbamylase deficiency | 75 |
| Passive targeting – LNPs | |||||||
| mRNA-LNP | MC3:DSPC:Chol:DMG-PEG |
Cre mRNA | IV | MC3 ionizable lipid with pKa of 6.4 | Hepatocytes (50%), ECs (80%), Kupffer cells (60%) | n.d. | 57 |
| 50:10:38.5:1.5 |
|||||||
| mRNA-LNP | MC3:DSPC:Chol:DMG-PEG |
EPO, modified with m1ψ and m5C | IV | MC3 ionizable lipid with pKa of 6.4 | Liver | Anemia | 81 |
| 50:10:38.5:1.5 |
|||||||
| mRNA-LNP | MC3:DSPC:Chol:DMG-PEG |
EPO mRNA, modified with ψ | IV | MC3 ionizable lipid with pKa of 6.4 | Liver | Anemia | 80 |
| 50:10:38.5:1.5 |
|||||||
| mRNA-LNP | 3060i10:DOPE:Chol:DMG-PEG |
Luc mRNA, EPO mRNA modified with m5U | IV | 3060i10 ionizable lipid, surface pKa of 6–7 | ECs (85%), Kupffer cells (85%), hepatocytes (85%) | Anemia | 93 and 94 |
| 35:16:46.5:2.5 |
|||||||
| mRNA-LNP | cKK-E12-A6:DOPE:Chol:DMG-PEG |
Cre mRNA, Luc mRNA, EPO mRNA | IV | cKK-E12 based LNPs and alkynes, surface pKa of 6–7 | Hepatocytes (73%) | Anemia | 91 |
| 35:16.0:46.5:2.5 |
|||||||
| mRNA-LNP | C12-200:DOPE:Chol:DMG-PEG |
Luc mRNA, EPO mRNA | IV | C12:200 ionizable lipid with pKa of 6.5 | Liver (predominant), spleen | Anemia | 36 and 47 |
| 35:16:46.5:2.5 |
|||||||
| mRNA-LNP | cKK-E12:DOPE:Chol:DMG-PEG |
Cre mRNA | IV | cKK-E12 ionizable lipid, pKa of 6–7 cholesterol variants | Hepatocytes, ECs, Kupffer cells | n.d. | 249 |
| 50:12.5:35:2.5 |
|||||||
| mRNA-LNP | cKK-E12:Chol:DOPE:DMG-PEG |
Cas9 mRNA, sgRNA for PCSK9 | IV | Ionizable lipid cKK-E12, pKa of 6.5 | Liver (>80% edited) | Hypercholesterolemia | 82 |
| 35:46.5:16:2.5 |
|||||||
| mRNA-LNP | Lipid5:DSPC:Chol:DMG-PEG |
Luc or EPO mRNA | IV | Ionizable lipid 5, surface pKa of 6.4 | Liver | Anemia | 89 |
| 50:10:38.5:1.5 |
|||||||
| mRNA LNP | 5A2-SC8:DOPE:Chol:DMG-PEG |
Luc mRNA, FAH mRNA modified with m1ψ | IV | 5A2-SC8 ionizable lipid, surface pKa of 6.5 | Hepatocytes (44%) | Hepatorenal tyrosinemia type I | 48 |
| 23.8:23.6:47.6:4.7 |
|||||||
| Passive targeting polymers | |||||||
| mRNA-lipopolyplex | TarN3C10 poly(tartaratamidoamine):DSPC:Chol:DMG-PEG |
EPO mRNA, Luc mRNA | IV | Polymer structure (73 nm) | Liver, spleen | Anemia | 73 |
| 5:2:2:1 |
|||||||
| N/P ratio, 10:1 (w/w) |
|||||||
| mRNA-polyplex | PEG12000-Pasp(DET):mRNA |
GFP mRNA, Bcl-2 mRNA modified with m5C and ψ | IV | Polymer structure (50 nm) | Liver | Fulminant hepatitis | 110 |
| N/P ratio, 3:1 |
|||||||
| mRNA-lipopolyplex | A-TD3:DOPE:Chol:DMG-PEG |
Luc mRNA | IV | Polymer structure | Liver, spleen | n.d. | 111 |
| 50:25:23.5:1.5 |
|||||||
| N/P ratio, 8:1 |
|||||||
| Splenic tissue | |||||||
| Active targeting | |||||||
| mRNA-LNP | Non-disclosed ionizable lipid:PC:Chol:PEG-lipid |
Luc mRNA, Cre mRNA modified with m1ψ | IV | Affinity ligand towards CD4 | T cells | n.d. | 129 |
| 50:10:38.5:1.5 |
|||||||
| Passive targeting – LNPs | |||||||
| mRNA-LPX | DOTMA:DOPE |
Luc mRNA modified with m1ψ | IV | Negative LPX charge around −30mV | Spleen, dendritic cells | n.d. | 121 |
| RNA-LPX, charge ratio | |||||||
| 1.3:2 |
|||||||
| mRNA-LNP | 5A2-SC8:DOPE:Chol:DMG-PEG:18PA |
Luc mRNA modified with m1ψ | IV | Addition of 30 mol% 18PA | B cells (12%), T cells (10%), macrophages (20%) | n.d. | 122 |
| 16.67:16.67:33.33:3.33:30 |
|||||||
| mRA-LNP | 10A1P16:MDOA:Chol:DMG-PEG |
Luc mRNA | IV | iPhos lipids, alkyl chain linked to the phosphate zwitterionic head | Macrophages (30%), B cells (6%) | n.d. | 126 |
| 29.1:34.9:34.9:1.2 |
|||||||
| mRNA-LNP | 306010:PS:Chol:DMG-PEG |
Luc mRNA | IV | Replacement of DOPE with 40 mol% PS | B cells | n.d. | 125 |
| 35:22.5:40:2.5 |
|||||||
| mRNA-LNP | 4A3-SC8:DOPE:Chol.:DMG-PEG:BMP |
Luc mRNA | IV | Addition of a 20 mol% BMP | Spleen | n.d. | 124 |
| 19.05:19.05:38.10:3.81:20 |
|||||||
| mRNA-LNP | OF-Deg-Lin:DOPE:Chol:DMG-PEG |
Luc mRNA | IV | Ester groups and surface pKa < 6 | B cells (7%), T cells (<1%) | n.d. | 45 |
| 35:16:46.5:2.5 |
|||||||
| Passive targeting – polymers | |||||||
| mRNA-polyplex | PE4K-A17-02SC4:mRNA |
Luc mRNA | IV | Hydrophobic side chain length, surface pKa < 6 | Spleen | n.d. | 132 |
| 30:1 (w/w) |
|||||||
| mRNA-polyplex | CART-D12:A14 poly(carbonate)-b-(α-aminoester) |
eGFP mRNA, fLuc mRNA modified with m5C, ψ | IV | Oleyl CART polymer | CD4+ T cells (1.6%), CD8+ T cells (1.5%) | n.d. | 118 |
| mRNA-polyplex | PA6-4P14 stabilized with 2.5% pluronic F127 | Luc mRNA, Cre mRNA | IV | Phospholipidated polymer | Spleen | n.d. | 133 |
| ZPP:mRNA |
|||||||
| 20:1 (w/w) |
|||||||
| mRNA-lipopolyplex | BDD3 APE:DOPE:Chol:DMG-PEG |
Luc mRNA | IV | Polymer structure (80 nm) | Liver, spleen | n.d. | 111 |
| 50:25:23.5:1.5 |
|||||||
| Lung tissue – local administration | |||||||
| LNPs | |||||||
| mRNA-LNP | 7C1:Chol:DOTAP:DMG-PEG |
Luc mRNA | Nebulization | Local administration | Lung | n.d. | 167 |
| 35:5:5:55 |
|||||||
| Polymers | |||||||
| mRNA-polyplex | Chitosan-coated PLGA:mRNA |
hCFTR mRNA, modified with m1ψ, s2U, m5C | IT | Mucoadhesive chitosan coating, local administration | Epithelial cells | Cystic fibrosis | 152 |
| 25:1 (w/w) |
|||||||
| mRNA-polyplex | Chitosan-coated PLGA:mRNA |
Z3 nuclease-encoding mRNA modified with s2U and m5C | IT | Mucoadhesive chitosan coating, local administration | Epithelial cells, ATII cells | SP-B deficiency | 150 |
| 25:1 (w/w) |
|||||||
| mRNA-polyplex | PBAEs:mRNA |
Luc mRNA | Nebulization | Local administration | Epithelial cells (25%), ECs (4.5%), immune cells (0.4%) | n.d. | 153 |
| 50:1 (w/w) |
|||||||
| mRNA-polyplex | Poloxamine T704-peptide:mRNA |
Luc mRNA, SB100X mRNA | Nebulization | Local administration | Epithelial cells | Cystic fibrosis | 164 |
| mRNA-polyplex | PBAEs:mRNA |
Cas13a mRNA | Nebulization | Local administration | Lung | Influenza and SARS-COV-2 | 165 |
| 50:1 (w/w) |
|||||||
| Lung tissue – systemic administration | |||||||
| Active targeting | |||||||
| Ab-mRNA-LNP | L319:DSPC:Chol:DMG-PEG |
fLuc mRNA, eGFP mRNA modified with m1ψ | IV | Affinity ligand towards PECAM1 | Vascular ECs | n.d. | 171 |
| 50:10:38.5:1.5 |
|||||||
| Passive targeting – LNPs | |||||||
| mRNA-LNP | 5A2-SC8:DOPE:Chol:DMG-PEG:DOTAP |
Luc mRNA, mCherry mRNA, Cre mRNA | IV | Addition of 50 mol% DOTAP | Lung ECs (65%), epithelial cells (40%), immune cells (21%) | n.d. | 122 |
| 11.9:11.9:23.8:2.4:50 |
|||||||
| mRNA-LNP | 9A1P9:DDAB:Chol:DMG-PEG |
Luc mRNA, Cre mRNA | IV | iPhos lipids, alkyl chain linked to the phosphate zwitterionic head | Lung ECs (34%), epithelial cells (20%), immune cells (13%) | n.d. | 126 |
| 46:23:30:0.3 |
|||||||
| mRNA-LNP | ZA3-Ep10:Chol:DMG-PEG |
Luc mRNA, Cas9 mRNA | IV | Zwitterionic amino lipid (ZA3-Ep10) | Lung | n.d. | 173 |
| 50:38.5:1 |
|||||||
| mRNA-LNP | 3060i10:DOTAP:Chol:DMG-PEG |
Luc mRNA | IV | Replacement of DOPE by 40 mol% DOTAP | Lung | n.d. | 125 |
| 35:40:22.5:2.5 |
|||||||
| Passive targeting – polymers | |||||||
| mRNA-polyplex | Polyester PE4K-A17-0.2C8 Polymer:mRNA |
Luc mRNA, Cre mRNA | IV | Surface pKa 7.5–8.0 | Lung | n.d. | 132 |
| 30:1 (w/w) |
|||||||
| mRNA-lipopolyplex | L3 co-formulated with DOPE and C18PEG2000 | Luc mRNA, Cre mRNA | IV | Nanoparticle size of 100 nm, PEG-lipid content of 5%, C18 alkyl PEG-lipid | ECs (75%), immune cells (2%) | n.d. | 176 |
| 20:5, N/P ratio |
|||||||
| 50:1 |
|||||||
| Lymphatics | |||||||
| mRNA-LPX | DOTMA:DOPE |
Luc mRNA modified with m1ψ | IV | Negative charge around −30 mV | Spleen, dendritic cells | n.d. | 121 |
| RNA/LPX ratio | |||||||
| 1.3:2 |
|||||||
| mRNA-LNP | cKK-E12:DOPE:Chol:DMG-PEG:SLS |
fLuc mRNA, Cre mRNA | SC | Administration route, surface charge of −15 mV, size of 50–120 nm | Lymph nodes, dendritic cells (4.6%), macrophages (1.2%), neutrophils (3.3%), B cells (0.06%) | Cancer immunotherapy | 188 |
| 15:26:40.5:2.5:16 |
|||||||
| mRNA-LNP | MC3-analogue:DSPC:Chol:DMG-PEG |
GFP mRNA, VEGFC mRNA modified with m1ψ | ID | Local administration | Lymphatic vessels | Lymphedema | 185 |
| 50:10:38.5:1.5 |
|||||||
| mRNA-Polyplex | PA6–4P14 stabilized with 2.5% F127 | Fluc mRNA, Cre mRNA | IV | Phospholipidated polymer | CD4+ T cells (6%), CD8+ T cells (2%), B cells (<1%), macrophages (6%) | n.d. | 133 |
| ZPP:mRNA |
|||||||
| 20:1 (w/w) |
|||||||
| Brain | |||||||
| Active targeting | |||||||
| mRNA-LNP | A-L01:DSPC:Chol:DSPE-PEG maleimide |
fLuc mRNA, TM mRNA | IV | Active targeting with anti-VCAM antibody | ECs in cerebral vasculature | Brain inflammation | 201 |
| 50:10:38.5:1.5 |
|||||||
| Passive targeting – LNPs | |||||||
| mRNA-LNP | MC3:DSPC:Chol:DMG-PEG |
Luc mRNA, FXN mRNA | Intrathecal | Local administration | Dorsal root ganglia | Friedreich's ataxia | 195 |
| 55:10:32.5:2.5 |
|||||||
| mRNA-LNP | ssPALM:DOPE:Chol:DMG-PEG |
eGFP mRNA, fLuc mRNA modified with ψ, m5C, s2U | Intracerebroventricular injection | Local administration | Neuronal cells, astrocytes | n.d. | 197 |
| 30:30:38:1 |
|||||||
| Passive targeting – polymers | |||||||
| mRNA-polyplex | PEG-PAsp(DET) | GFP mRNA, NEP mRNA | Intracerebroventricular injection | Local administration | Brain ventricle | Alzheimer's | 200 |
| N/P ratio, 3:1 |
|||||||
| Eyes | |||||||
| mRNA-LNP | MC3:DSPC:Chol:DMG-PEG |
fLuc mRNA, eGFP mRNA | SR | Local administration | RPE, Müller glia cells | n.d. | 204 |
| 50:10:38.5:1.5 |
|||||||
| mRNA-LNP | MC3:DSPC:Chol:DMG-PEG |
fLuc mRNA, mCherry mRNA, Cre mRNA | Intravitreal, SR | Local administration | Subretinal: Retinal pigment epithelium | n.d. | 208 |
| 50:10:38.5:0.5 |
Intravitreal: Müller glia, optic nerve and trabecular meshwork | ||||||
| Heart tissues | |||||||
| Naked mRNA | Saline citrate buffer | eGFP mRNA, VEGF mRNA modified with ψ, m5C | Intramyocardial injection | Local administration | Cardiomyocytes | Ischemic injury | 220 |
| mRNA-LNP | C14-113:DSPC:Chol:DMG-PEG |
eGFP mRNA, modified with ψ, m5C | Intramyocardial injection and intraventricular injection | Local administration | Cardiomyocytes | n.d. | 218 |
| 50:10:38.5:1.5 |
|||||||
In recent years, various ionizable lipids have been designed and evaluated for mRNA delivery in vivo (Fig. 2A). The ionizable lipids MC3, 1,1′-((2-(4-(2-((2-(bis(2-hydroxydodecyl)amino)ethyl)(2-hydroxydodecyl)amino)ethyl)piperazin-1-yl)ethyl)azanediyl)bis(dodecan-2-ol) (C12-200) and 3,6-bis(4-(bis(2-hydroxydodecyl)amino)butyl)piperazine2,5-dione (cKK-E12)—previously demonstrated to be effective for siRNA delivery to the liver—were used for initial exploration of mRNA delivery in this organ.77,78 One important physicochemical factor relevant for delivery to the liver has been the discovery that ionizable lipid bearing amino groups exhibiting pKa values between 6 and 7 yield the most effective functional delivery to liver tissue.79 With a pKa between 6.4 and 6.5, MC3 has been the focus of initial liver-targeting mRNA delivery studies. Notably, the IV administration of MC3-based LNPs encapsulating mRNA encoding for erythropoietin (EPO) has demonstrated high levels of hematopoiesis in the liver, resulting in an increased serum concentration of EPO in pigs and cynomolgus monkeys.80,81 Similar to MC3, the lipids cKK-E12 and C12-200 both display pKas around 6.5 and have also been used for effective delivery of mRNA to the liver.36,47,82 A recent study evaluated the biodistribution of cKK-E12 and C12-200-based LNPs using Cy5-labeled mRNA, and reported liver accumulation of around 60% and 70%, respectively. Additionally, an assessment of protein expression six hours after administration showed that 90% of protein expression resulting from the use of these lipids occurred in the liver.37 Related to the pKa of ionizable lipids, another key parameter for mRNA delivery is the surface pKa of the LNP (also termed global or apparent pKa), which has been demonstrated to be a robust predictive value for siRNA activity in vivo.83 The pKa of lipids can differ when incorporated into LNPs; indeed, non-covalent interactions between the ionizable lipid and other constituent lipids of the LNPs, the mRNA payload, and the surrounding medium have all been shown to alter the pKa.84 Supporting this notion, Heyes et al. have reported a fluorescence-based approach using 2-(p-toluidinyl) naphthalene-6-sulfonic acid (TNS), an acidic small molecule showing higher levels of fluorescence when protonated, to assess the pKa of LNPs.85 This method allows the direct determination of the individual pKas of amino lipids in the assembled LNPs, while also serving as an indirect approach to determine the ionization status of the LNPs themselves. Similar to the pKas of ionizable lipids, a surface pKa between 6 and 7—resulting in a neutral surface charge during circulation—has been shown to promote effective gene silencing in the liver, and accordingly this parameter has been used to assess the ability of nanocarriers to deliver mRNA to the liver.86 In one example, surface pKa has been used as a parameter in the development of Moderna's lead ionizable lipid (“Lipid 5”), which has been successfully used for treatment in therapeutic models of acute intermittent porphyria and methylmalonic acidemia.87,88 This lipid was selected based on an in vivo screening of a series of amino lipids, which, when incorporated into LNPs, exhibited surface pKa values ranging from 5.80 to 7.18, as well as distinct protein expression profiles. The optimized “Lipid 5”, with a surface pKa of 6.50, demonstrated a 6-fold increase in protein expression localized in the liver compared to conventional MC3-based LNPs.89
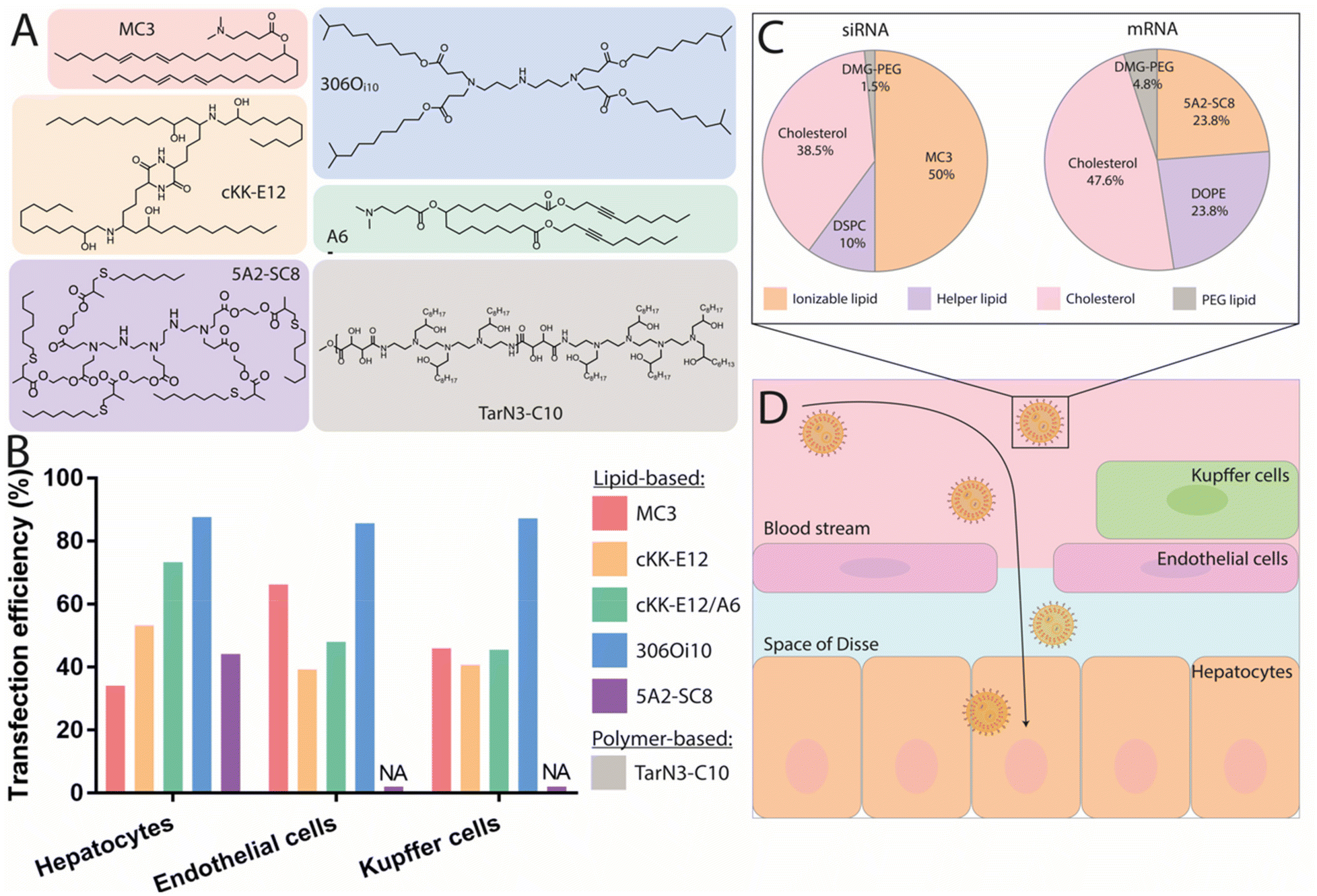 | ||
| Fig. 2 Nanocarriers providing mRNA delivery and protein expression to the liver. (A) Chemical structure of the best performing ionizable lipids and polymers for liver delivery. (B) Transfection efficiency of these compounds for the different liver cell populations. Doses and duration before evaluation: 2 mg kg−1 Cre mRNA after 24 h for MC3,57 0.75 mg kg−1 mCherry mRNA after 48 h for both cKK-E12 and cKK-E12/A6,91 1 mg kg−1 Cre mRNA after 72 h for 306Oi10,94 and 0.5 mg kg−1 mCherry mRNA after 6 h for 5A2-SC8.48 (C) Pie charts of the gold standard LNP formulation used for siRNA delivery, compared to an optimized formulation used to deliver mRNA with the 5A2-SC8 ionizable lipid.48 (D) Graphical representation of the IV delivery and extravasation of LNPs in the liver to reach hepatocytes. | ||
Although it had been assumed that MC3- or cKK-E12-based LNPs targeted hepatocytes, recent findings have shown mRNA expression in the liver's endothelial cells (ECs) and Kupffer cells in Cre reporter mice (see Fig. 2B).57 This suggests that MC3- and cKK-E12-based formulations also enable functional nucleic acid delivery to other cell types within the liver. These findings clearly demonstrate that transfection efficiency must be thoroughly assessed at the cellular level. In addition to targeting the correct cell type, the nanocarrier must also be able to efficiently escape the endosomal lumen to achieve a therapeutically relevant level of protein production. Endosomal escape is a major limitation for mRNA-based drugs, as studies have revealed that only around 1–4% of internalized nanoparticles are able to be retrieved in the cytosol before degradation.90 With this in mind, several works have aimed at identifying strategies to improve the efficiency of this endosomal escape. For instance, a recent study demonstrated that the incorporation of alkyne functionalities, rather than alkenes, in the hydrophobic tails of ionizable lipids could increase the fusogenicity of the resulting particles and facilitate endosomal escape.91 Specifically, the ionizable lipid MC3 was modified by the inclusion of unsaturated alkyne functionalities and ester groups, then formulated into LNPs. Upon injection, superior liver expression was found when using the alkyne lipid, highlighting improved endosomal escape and functional mRNA delivery. Interestingly, analysis of cellular transfection efficiency using Cre-reporter mice revealed that the percentage of Tdtomato+ hepatocytes increased from 50% to 75% when transfected using alkyne-modified lipids. Concurrently, another group developed a series of polyester-based dendrimer ionizable lipids, among which a highly potent ionizable lipid termed 5A2-SC8 (Fig. 2A) with a surface pKa of 6.67 was identified following screening.92 Biodistribution studies showed time-dependent Luc activity with peak expression observed at 6 h post injection followed by a strong decrease with almost no signal detectable after 48 h. Interestingly, increasing the dosage from 0.1 to 0.2 mg kg−1 induced a more disperse expression profile throughout tissues, especially in the spleen, suggesting a potential impact of dose on the biodistribution and functionality of the particles. Flow cytometry quantification of mCherry mRNA delivery to hepatocytes revealed that 45% of the total hepatocyte population strongly expressed the protein at 6 h post injection.48 This lipid was further investigated for the delivery of fumarylacetoacetate hydrolase (FAH) mRNA to hepatocytes and was able to produce a therapeutic level of the protein in a FAH−/− mouse model. Another factor influencing endosomal escape is the surface ionization of LNPs;93 this parameter corresponds to the degree of protonation of the nanoparticle at a given pH and is critical for endosomal escape. Investigation of the relationship between the chemistry of the alkyl tails of lipids and their in vivo efficacy revealed that ionizable lipids bearing branched tail structures are more strongly ionized at the endosomal pH of 5.0 and are consequently more effective at delivering mRNA than their linear counterparts. A biodistribution evaluation of Cy5-labeled mRNA, however, revealed that small changes in lipid chemistry had minimal impact on particle distribution, with LNPs exhibiting accumulation in the spleen, liver, and lungs, though the resulting protein expression was almost entirely observed in the liver. While an explanation for this discrepancy in biodistribution and protein expression profile remains elusive, it can be hypothesized that although the LNPs are able to travel to the sites indicated by the biodistribution profile, they may fail to deliver their mRNA payload for effective translation (e.g., failure of endosomal escape or internalization). Upon in vivo screening, one ionizable lipid termed 3060i10, containing a branched decanonyl alkyl chain and generating a LNP surface pKa of 6.40, demonstrated over 90% of its total protein expression in the liver (Fig. 2A). Single-cell flow cytometry analysis revealed that more than 85% of the total population of Kupffer cells, ECs, and hepatocytes were transfected, making this ionizable lipid one of the most potent agents to mediate functional mRNA delivery to the liver.94
While alterations to LNPs have canonically been focused on the chemistry of ionizable lipids, modifications to the other components of LNPs have been shown to have an influence on the endosomal escape, biodistribution, and circulation of the particles. For example, a strong correlation has been observed between mRNA expression level and the type of helper lipid used, particularly in cases where DSPC was replaced by DOPE, which led to a 7-fold increase in protein production.47 Whereas DSPC contains a quaternary amine headgroup and a fully saturated tail, DOPE contains a primary amine headgroup and a tail with one degree of unsaturation. It has been reported that conical lipids, such as DOPE, tend to adopt the less stable hexagonal phase, while cylindrical lipids, such as DSPC, tend to adopt the more stable lamellar phase.95 Another study has shown that the molar fraction of zwitterionic phospholipids must be increased when switching from siRNA to mRNA delivery, and that this change yields higher levels of protein expression.48 Following these studies, optimizations of siRNA formulations for mRNA delivery have been proposed including: (i) the molar fraction of zwitterionic phospholipids could be increased, while that of ionizable lipids could be decreased, and (ii) the use of DOPE instead of DSPC should result in increased mRNA expression. An optimized formulation is illustrated in Fig. 2C. A third study used an in vivo screening of IV-administered LNPs to demonstrate that those prepared using DOPE preferentially accumulated in the liver, while similar LNPs containing DSPC accumulated primarily in the spleen.96 Using a quartz crystal microbalance with dissipation monitoring, it was found that a DOPE-containing LNP formulation exhibited stronger interactions with ApoE than an identical DSPC-containing formulation, thereby increasing two-fold the extent of mRNA delivery to the liver.
LNPs are commonly formulated with unmodified cholesterol, however, endogenous cholesterol is naturally esterified or oxidized enabling its compact storage and trafficking to several cells including hepatocytes, ECs, and macrophages via lipoproteins.97 Recently, it has been shown that cholesterol derivatives act as an important factor altering the in vivo distribution of LNPs.98 An in vivo screening of LNPs incorporating oxidized, esterified, and unmodified cholesterol showed that esterified cholesterol variants outperformed their unmodified counterparts for mRNA delivery by 1.4-fold. Moreover, the authors reported for the first time enhanced and more specific gene editing in hepatic ECs mediated by a LNP containing a cholesteryl oleate derivative, leading to a 41% increase in editing as compared to unmodified cholesterol. The delivery of this LNP to liver ECs was also three times more efficient than its delivery to hepatocytes, illustrating the higher specificity of cholesterol oleate esters towards hepatic ECs. Targeting these ECs can be of clinical importance given the active role they play in establishing the liver microenvironment and driving fibrosis, inflammation, primary tumor growth, and metastasis.99 In a further study, the same group investigated oxidative cholesterol modifications within the B-ring and the hydrocarbon tail attached to the D-ring (Fig. 1C). Using cKK-E12-based LNP formulations, in vivo screening demonstrated that: (i) LNPs containing oxidized cholesterol deliver mRNA more efficiently to all hepatic cells than those containing unmodified cholesterol, (ii) oxidized cholesterol modifications mediated higher mRNA expression in ECs and Kupffer cells than in hepatocytes compared to unmodified cholesterol, and (iii) modifications of the side chain attached to the D-ring (particularly 20- and 25-hydroxycholesterol) yielded stronger protein expression compared to modifications on the B-ring. Notably, variations containing 20α-OH groups outperformed unmodified cholesterol for all hepatic cells while also demonstrating more robust mRNA delivery to ECs and Kupffer cells. This study highlighted that LNPs containing oxidized cholesterol modifications (especially on the alkyl side chain) can impact mRNA expression in several hepatic cell subtypes, leading to higher mRNA expression in ECs and Kupffer cells than in hepatocytes. Given the role of ECs99 and Kupffer100 cells in the pathogenesis of liver diseases, the use of oxidized and esterified cholesterol in LNP formulations of mRNA could be a feasible strategy to improve targeting to these cells and might be of interest in certain clinical conditions.
Finally, several studies focused on optimizing the PEG-lipid for the enhancement of liver targeting. In particular, it has been shown that diffusible PEG-lipids, which ensure the stability of formulations but dissociate from the LNP upon systemic administration, have been successful for liver delivery.101 Such lipids are generally composed of an acyl chain of 14 carbons conjugated to a PEG polymer such as the predominant DMG-PEG which has been used in almost all reported studies, as well as in the Onpattro© formulation (Table 1).72 These diffusible PEG-lipids rapidly dissociate from the nanoparticle with as little as 20% remaining 2 h after systemic administration.102 In contrast, PEG-lipids with 18-carbon acyl chains are not diffusible and result in LNPs with longer circulation half-lives.102 However, LNPs that target hepatic tissues do not require such extended circulation lifetimes due to the inherent capacity of the liver to rapidly capture nanoparticles. Accordingly, DMG-PEG is an ideal choice of PEG-lipid for delivery to the liver.
Altogether, mRNA-containing LNPs exhibiting surface pKas between 6 and 7 result in high levels of accumulation in the liver, associated with high transfection efficiencies reaching up to 85% for the ionizable lipid 3060i10 (Fig. 2B). These high transfection rates are very promising for therapeutic applications and have helped overcome barriers to the preclinical and clinical development of treatments for rare genetic liver disorders. Passive targeting using ApoE does not only mediate the functional delivery of mRNA to hepatocytes, but also to ECs and Kupffer cells, with which the LNPs come in contact prior to reaching the space of Disse and the hepatocytes (Fig. 2D). Recent findings have also shown that cholesterol and cholesterol derivatives contribute to greater transfection efficiencies and can shift mRNA expression towards different hepatic cell subtypes, indicating their potential—in combination with ionizable lipid and helper lipid chemistry—to achieve improved mRNA delivery to desired cells.
Though not as extensively investigated as LNPs for mRNA delivery to the liver, polymers can deliver functional mRNA to liver cells, resulting in high levels of protein production. Materials such as amino-polyesters synthesized via ring-opening polymerization of readily available lactones represent a viable approach for mRNA delivery, potentially overcoming the biodegradability-related safety concerns raised for cationic polymers.112 Notably, while data on transfection efficiency in various hepatic cells subtypes is now widely reported for LNP formulations, such data are missing from reports of polymer-based nanoparticles.
3.2. mRNA delivery to the spleen
The spleen is the largest secondary lymphoid organ, in which antigen presenting cells (APCs) exist in close proximity to T cells, providing an adequate microenvironment for the efficient priming and amplification of immune responses.113 Leukocytes in the spleen include various subsets of B and T cells, dendritic cells (DCs), and macrophages, and are involved in many functions such as the initiation of innate and adaptive immune responses against pathogens, as well as lymphocyte maturation and recycling (Fig. 3A). Modulation of the activity of immune cells has attracted interest for tumor immunology applications, as highlighted by the rapid growth in the clinical use of immunotherapeutic agents.114 For instance, chimeric antigen receptor T-cell (CAR T) therapy is a technique which relies on the ability of the immune system to fight cancer cells and has achieved clinical approval for the treatment of advanced leukemia and lymphoma.115 While current CAR T cell treatments require the in vitro genetic alteration of T cells prior to their clinical use,116 the development of in vivo T cell-targeted mRNA delivery systems able to induce robust and transient CAR expression could offer an interesting alternative. Such systems could also be valuable for the treatment of several B cell dysfunctions, given the crucial role of B cells in the modulation of immune responses. As an example, the proliferation of dysregulated mature B cells has been shown to be the origin of non-Hodgkin's B cell lymphoma, and effective genetic modulation of B cell function in vivo could have a profound impact on the treatment of this disease.117 Accordingly, improvements to the in vivo delivery methods of mRNA therapeutics targeting both B and T cells would be greatly advantageous. In spite of this, there have been few reports on the effective delivery of mRNA to B and T cells, likely due to their limited capacity for endocytosis and subsequently impaired protein translation.118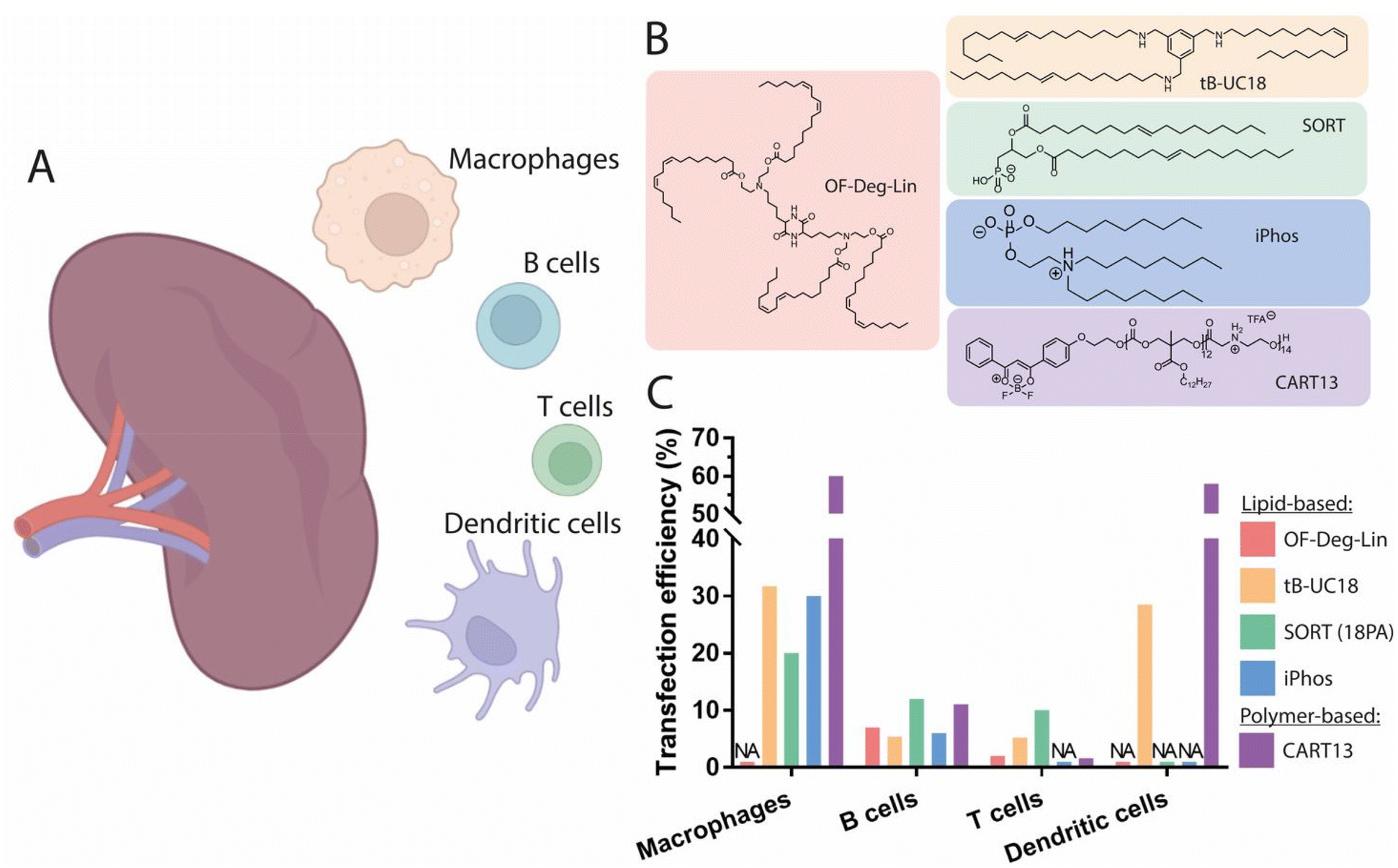 | ||
| Fig. 3 Spleen-targeting nanocarriers. (A) Graphical representation of the spleen cell populations targeted by nanocarriers. (B) Chemical structures of the best performing lipids and polymers for promotion of protein expression in the spleen. (C) Transfection efficiency of these compounds in different splenic cell populations. Doses and duration prior to evaluation: 0.75 mg kg−1 Cy5-labeled mRNA, t = 1 h for OF-Deg-Lin;45 0.5 mg kg−1 mRNA with DiD-labeled LNPs, t = 4 h for tB-UC18;130 0.3 mg kg−1 Cre mRNA, t = 48 h for 18PA;122 0.5 mg kg−1 Cre mRNA, t = 48 h for iPhos,126 non-specified for CART13.118 Made with Biorender. | ||
:1.5 and a zeta potential around −35 mV generated a signal exclusively in the spleen, with no significant expression observed in the liver. Interestingly, the authors revealed that macrophages exhibited the highest rate of mRNA uptake, whereas the largest translation efficiency was observed in dendritic cells, indicating that these cells are more effective at the cytoplasmic translocation and translation of mRNA into protein. The addition of a fifth permanent anionic lipid to a conventional LNP formulation—leading to an altered surface pKa of the resulting nanoparticles—has been identified as a feasible strategy for targeting splenic tissue. This strategy, termed Selective ORgan Targeting (SORT), utilizes the incorporation of 30 mol% of the supplementary anionic lipid 1,2-dioleoyl-sn-glycero-3-phosphate (18PA) into a liver-targeting LNP, allowing the redirection of mRNA expression to splenic tissue.122 Importantly, this formulation demonstrated a remarkable transfection efficiency of ∼12% of all B cells, ∼10% of all T cells, and ∼20% of all macrophages present in the spleen.122 The addition of this supplementary anionic headgroup was found to promote the adsorption of plasma proteins with an isoelectric point above physiological pH, in turn resulting in an altered protein corona. A recent study revealed that the tissue tropism of this formulation is governed by the modulation of surface pKa, with values decreasing from 6.46 to 3.97, far lower than optimal pKa values identified for specific liver targeting.123 Interestingly, zeta potential measurements revealed that the overall net surface charge of the resulting LNPs was not significantly altered, indicating that this strategy for spleen-specific mRNA delivery is feasible using LNPs displaying a near-neutral surface charge—an important feature for in vivo stability and delivery efficiency. Proteomics studies have also demonstrated that, because of this alteration to the surface pKa, spleen-targeting LNPs mostly adsorbed β2-glycoprotein I (β2-GPI, also known as ApoH), in contrast to liver-targeting LNPs which mostly adsorbed ApoE. The administration of spleen-targeting LNPs to ApoE−/− mice did not impair mRNA delivery, but rather enhanced its delivery to splenocytes, confirming an ApoE-independent intracellular mechanism this class of LNPs. Other negatively charged phospholipids have also demonstrated spleen-specific tropism; for instance, the incorporation of bis(monoacylglycerol)phosphate (BMP) allowed for more than 75% mRNA translation efficiency in the spleen.124 When added as a fifth lipid at up to 20% molar ratio in a four-component LNP formulation, spleen-specific mRNA expression was observed, corroborating the results obtained using the aforementioned strategy. A recent study aimed at reproducing this same tissue tropism by maintaining a four-component nanoparticle system and replacing the helper lipid DOPE with the negatively charged helper lipid phosphatidylserine (PS). The incorporation of 40 mol% of PS in a liver-targeting formulation substantially shifted mRNA expression towards the spleen, supporting the previously established notion that the incorporation of negatively charged lipids into LNP formulations represents a feasible approach to promote mRNA expression in the spleen. As previously noted, no significant change in the zeta potential of the LNPs was observed, and only a moderate correlation between zeta potential and spleen-specific mRNA delivery was found.125
The impact of the structure of ionizable lipids and phospholipids on the promotion of spleen selectivity has also been investigated (Fig. 3B). In one example, this was achieved using ionizable phospholipids (iPhos) composed of one pH-switchable zwitterion and three hydrophobic tails, which have been described to adopt a cone shape in acidic endosomal environments, facilitating the release of cargo from endosomes.126 Evaluation of the membrane-disrupting activity of iPhos lipids showed that pH-switchable zwitterions exhibited dramatically higher membrane fusion and rupture than simple tertiary amines in a concentration-dependent manner, validating the superiority of zwitterionic structures in endosomal lumen escape. In vivo structure activity relationships have also identified the length of the alkyl chain bound to the phosphate group as a key moiety for organ selectivity. Indeed, an alkyl chain of 13–16 carbons attached to the phosphate group of the iPhos lipid combined with the helper lipid N-methyldioctadecylamine (MDOA) led to an optimized spleen-specific LNP formulation achieving nearly 30% transfection of all splenic macrophages and 6% transfection of B cells, suggesting that modifications to the chemical structure of iPhos can result in improved organ selectivity towards the spleen (Fig. 3C). Simultaneously, exploration of the rational design of ionizable diketopiperazine lipids for the delivery of mRNA to B cells has resulted in a lead compound—termed OF-Deg-Lin—which exhibits biodegradable ester bonds and, when formulated into mRNA-LNPs, results in particles with a surface pKa of 5.7 (Fig. 3B).45 Upon systemic injection, the biodistribution of Cy5-tagged mRNA encapsulated in OF-Deg-Lin LNPs demonstrated 100-fold preferential accumulation in the liver over spleen; however, over 85% of total protein production from this formulation was found to occur in the spleen, with very little expression observed in the liver. To further investigate this discrepancy between the distribution and expression of mRNA, a non-biodegradable analogue of OF-Deg-Lin was prepared and tested, demonstrating selective distribution and expression in the liver. However, the mechanism by which OF-Deg-Lin mRNA-LNPs promote mRNA expression in the spleen, despite mostly accumulating in the liver, remains elusive. One explanation could be that the electrophilic ester bonds in OF-Deg-Lin are degraded more rapidly in the liver than in other organs, thus preventing either functional delivery of the mRNA to the cytoplasm of hepatic cells (e.g., during cell internalization or trafficking), or successful endosomal escape due to acidic degradation of the lipid.83 In contrast, OF-Deg-Lin might survive enzymatic degradation in the spleen, such that the nanoparticles retain their ability to induce functional protein expression following uptake into the resident splenic cells. This highlights the importance of the ester bonds found in OF-Deg-Lin for the delivery of functional mRNA to the spleen. Flow cytometry analysis of Cy5-mRNA-containing OF-Deg-Lin LNPs demonstrated a transfection efficiency of 7% of the total B cell population and around 1% of the total T lymphocyte population. In another study, the investigation of modifications in tail length and linker spacing in OF-Deg-Lin, resulted in the lipid OF-C4-Deg-Lin, containing a longer linker length of 4 carbons, as well as doubly unsaturated tails. Biodistribution evaluation of nanoparticles formulated using this novel lipid containing Cy5-tagged Luc mRNA demonstrated that over 75% percent of the LNPs were distributed to the liver, while more than 85% of Luc mRNA expression was found to occur in the spleen.127 Moreover, when compared to the lipid cKK-E12, OF-C4-Deg-Lin promoted ∼5-fold higher protein expression in the spleen. Thus, OF-Deg-Lin-based LNPs may be an attractive strategy for targeting splenic B cells in vivo.
Also, the identity of the helper lipid has been shown to influence binding affinity to specific serum proteins, as well as trafficking towards the liver and spleen. Indeed, substituting DOPE with DSPC in LNP formulations has been shown to shift the biodistribution from the liver towards the spleen by reducing the strength of interactions with ApoE.96 Similarly, Dahlman and colleagues observed that changing the composition of helper lipids in a LNP formulation could alter the cell tropism observed upon administration. Notably, they also showed that using DOPE instead of the helper lipid 18:1 Lysophosphatidylcholine (lysoPC) led to an increase in the ratio of transfected splenic ECs to hepatocytes.128 Future combined investigations of charge, surface pKa, and helper lipid composition could serve to greatly increase the potential selectivity of LNP formulations for splenic delivery.
One study evaluated active targeting to enable efficient T-cell transfection. For instance, surface conjugation of anti-CD4 antibodies to LNPs resulted in a 30-fold increase of splenic T-cell uptake in comparison to non-targeted LNPs.129 The localization ratio, calculated as the percentage of initial dose per gram of organ relative to dose per gram of blood, yielded 6-fold higher levels of particles in the spleen compared to conjugated control IgG, confirming that this active targeting platform could outperform the passive uptake usually observed with untargeted mRNA-LNP systems in the spleen. Flow cytometry analysis demonstrated about 60% transfection of CD4+ T cells in the spleen, validating that this formulation can achieve the efficient transfection of T cells.
Overall, most studies have shown that mRNA transfection and translation occur in splenic macrophages and dendritic cells to a greater extent than in B and T cells (Fig. 3C). Typical transfection efficiencies are between 5% and 12% for B cells and around 1% to 10% for T cells.45,122 One feasible strategy for the favorable delivery of functional mRNA to the spleen has been the incorporation of negatively charged phospholipids, which in turn result in alterations to the surface pKa of the LNPs. Optimization of the structure of ionizable lipids—as illustrated with OF-Deg-Lin-derivatives and iPhos lipids—has also proven to be an effective approach for tuning mRNA delivery to the spleen. A recent study reported remarkable protein expression in splenic tissue, with 61% occurring in macrophages and dendritic cells and around 6% occurring in B and T cells, using a structurally optimized lipid (tB-UC18) based on a tri-substituted benzaldehyde derivative containing primary amines (Fig. 3B).130 While the mechanism by which this lipid promotes mRNA expression in the spleen remains unclear, these findings underscore that the exploration of the chemical structures of ionizable lipids might yield potent mRNA delivery vehicles for the spleen. Future investigations of the relationship between helper lipid identity, ionizable lipid structure, surface pKa, and surface charge are thus of prime importance for the development of relevant and efficient solutions for specific mRNA delivery to splenic cells populations in vivo.
3.3. mRNA delivery to the lungs
Lung diseases are a leading cause of death and disability worldwide.136 In particular, genetic disorders leading to functional protein expression deficiencies such as cystic fibrosis (CF), primary ciliary dyskinesia (PCD), α1-antitrypsin deficiency (AATD), and surfactant protein B (SP-B) deficiency, are diseases for which existing treatments can lead to therapeutic resistance, while displaying low overall effectiveness for certain disease variants.16,137,138 In contrast, mRNA-based protein supplementation therapy could act as a viable mutation-independent alternative. The origin of these disorders stems primarily from epithelial cells, which include, among others, goblet cells (which produce mucins and are responsible for regulating the secretion of chloride ions), ciliated cells (which promote mucoclearance), macrophages, and alveolar cells (which monitor gas exchange and the production of lipid surfactants.16 For the delivery of mRNA therapeutics to the lungs, two routes of administration have been investigated: (i) local intratracheal (IT) or nebulization-based administration, and (ii) systemic IV injection. Upon IV administration, nanoparticles circulate directly to the heart, where they reach the right ventricle, and continue into pulmonary circulation.139 Lung capillaries are among the smallest blood vessels in the body, and are comprised mostly of non-fenestrated endothelium, acting as a major barrier for the extravasation of nanoparticles into the alveolar epithelium.140 Generally, the IV administration of nanoparticle-formulated mRNA has the advantage of circumventing the mucus barrier of the lungs, though this route suffers from limited targeting specificity. In contrast, local administration has the advantage of direct application into the airways, but must cross the pulmonary mucus layer, which presents a formidable barrier for nanocarriers. As such, it is of key importance to expand our knowledge of the relevant features and routes of administration for nanocarriers, in order to enable the efficient delivery of mRNA to lung cells.3.3.1.1. Polymer-based nanocarriers. In an attempt to investigate direct IT delivery of mRNA to the lungs, Dewerth and co-workers administered zinc finger nuclease (ZFN)-encoding chemically modified mRNA in chitosan-coated (for mucoadhesive function) PLGA, achieving protein rescue in lasting over 20 days in SP-B deficient mice.150 The study also compared the delivery of ZFN encoded by either plasmid DNA or mRNA, finding that the latter achieved a greater rate homology-directed repair (HDR), approximately 9%. For the treatment of SP-B deficiency in humans, the restoration of SP-B levels by 5–10% in the lungs is sufficient to reduce disease symptoms to mild levels, suggesting a potential therapeutic effect if 9% gene transfection can be achieved.151 Given the transient nature of mRNA-induced ZFN expression, this approach presents a safe and attractive approach. In another independent study, the same group compared the IT and IV delivery of mRNA encoding the transmembrane conductance regulator (CFTR) using chitosan-coated PLGA nanoparticles, observing efficient restoration of CFTR channels in a knockout model. Interestingly, the IV route exhibited greater performance for the delivery of such complexes to the lungs.152 Indeed, to achieve expression at the level reached by delivery of 2 mg kg−1 body weight via IV administration, a doubling of IT dosage was required. These findings are likely due to the presence of mucus in the upper airways, as well as mucociliary movements, which promote the clearance of the nanoparticles. While IT administration allows the direct passage of nanoparticles into the airways, this method usually necessitates invasive procedures such as orotracheal intubation—suitable for testing in animals, but impractical for clinical situations requiring multiple doses. Furthermore, IT delivery is limited to the upper airways of the lungs, unlike non-invasive inhalation technics.153 Evaluation of the IT administration to pigs of Luc mRNA polyplexes consisting of a cationic polymer scaffold of poly(acrylic acid) demonstrated that mRNA was mostly found in cranial lobes of the lungs, as revealed by luminescence measurements of the Luc protein.154 In contrast, inhalation allows deposition throughout the entire bronchiolar and alveolar epithelium, reaching deep lung structure, which could in turn lead to more significant relief of symptoms.155
Nebulization is a non-invasive procedure in which a device transforms a liquid medicine into a mist that can be inhaled into the lungs. For the nebulization of mRNA, the nanocarrier vehicles must withstand shear forces and must be able to be concentrated at high clinical doses, suggesting the suitability of cationic polymers, due to their capacity to complex large quantities of mRNA.156,157 Branched PBAEs (bPBAEs) have been shown to be less toxic than PEI polymers, and polyplexes of this material have been investigated as vehicles for mRNA delivery via nebulization, demonstrating efficient delivery of mRNA to lung tissue. An initial study reported that the transfection percentage of endothelial, epithelial, and immune cells achieved through nebulization was around 25% for the total epithelial cell population, including 4.4% for ECs, and 0.4% for immune cells, as determined by flow cytometry.153 As these data were generated in healthy mice, rather than in a pathological model, it is difficult to evaluate the potential therapeutic effects resulting from this level of transfection efficiency. It has been suggested that for CF, 5–10% CFTR gene correction in epithelial sheets158 could mitigate symptoms, and that 15–30% epithelial transfection would be needed for a 50% restoration of CFTR current.159 To investigate whether PEGylation of the polymer nanocarriers would influence the delivery of mRNA, PBAEs containing an alkyl amine (12 carbons in length) were synthesized to enable coformulation with the PEGylated lipid C18PEG5000, however, it was found that the PEGylated (7.5 mol%) polyplexes did not enhance mRNA delivery in vivo. PEGylation of nanoparticles as a strategy for overcoming the mucus barrier has been widely used for the delivery of payloads other than mRNA and represents a gold-standard for the engineering of mucus-penetrating nanocarriers.160 Additionally, previous studies conducted in pathological and healthy mucus, performed using pDNA-based LPXs and polystyrene beads, reported a positive effect resulting from PEGylation, with the PEG acting as a shield from respiratory fluids and surfactants, thereby facilitating diffusion of mucoinert particles through the mucus.161,162 A recent in vitro study evaluated the mucus penetration rate of PEGylated and non-PEGylated lipopolyplexes in CF sputum from patients and in an artificial model of healthy sputum. While PEGylation improved mucus penetration in healthy sputum, no impact was observed in pathological mucus when compared to non-PEGylated nanoparticles.163 Analysis of sputum composition revealed that the CF mucus contained more mucins (MUC5AC and MUC5B) than the healthy sputum, which strongly interacted with the nanoparticles independent of PEGylation. Nebulization of non-PEGylated mRNA-containing PBAE polyplexes has been successfully achieved in vivo in two studies, which conducted evaluations in animal models of CF. In another example, non-PEGylated peptide-poloxamine nanoparticles have shown encouraging results for the restoration of CFTR activity in mice.164 These vehicles were below 100 nm in size, and the presence of terminal hydrophilic polyethylene oxide blocks from the poloxamine allowed the formation of dense brushes protruding from the nanoparticle surface, resulting in a mucoinert surface that prevented mucoadhesion. Similar results can be seen in a study using polyplexes for the delivery of Cas13 mRNA and guides to target RNA viruses.165 The PBAE-mRNA polyplexes were nebulized in mice challenged with an influenza viral load, and were found to efficiently degrade influenza RNA in lung tissue when delivered after infection, illustrating the potential of non-PEGylated PBAE polymers for the nebulization of mRNA. Results from these studies highlighted that the addition of PEG on the surface of PBAE- or poloxamine-based polyplexes was not a prerequisite for achieving epithelial transfection in the lungs upon local administration.
3.3.1.2. Lipid-based nanocarriers. Though cationic amine polymers have been shown to be efficient carriers for mRNA in the lungs, nebulized therapies using these polymers have not yet been approved by the FDA. In contrast, the clinical implementation of LNPs is more advanced, making these materials an attractive alternative to polymers for delivery of mRNA to the lungs.166 A demonstration that LNPs are capable of delivering clinically relevant doses of mRNA to the lungs via nebulization would present new avenues as a versatile platform for mRNA delivery. A large in vivo screening has yielded interesting insights on (i) the impact of PEG content, and (ii) the impact of helper lipid identity.167 The best-performing formulation for achieving high mRNA expression in the lungs upon nebulization consisted of 55 mol% DMG-PEG and included the cationic helper lipid DOTAP at 5 mol%. While PEG-lipids are generally known to promote stability and prevent clearance in systemic delivery, their function in nebulization remains unclear. One hypothesis is that the large quantity of PEG serves to improve the stability and integrity of LNPs during the harsh conditions applied during the nebulization process. Inclusion of the cationic helper lipid DOTAP also contributed to higher transfection efficiency. Comparison of this strategy with formulations previously optimized for systemic delivery demonstrated superior performance, mainly due to higher stability of the LNPs during nebulization. This study revealed that the structure of PEG, as well as its molar percentage and the charge of the helper lipid can greatly affect mRNA incorporation and subsequent delivery, suggesting the need for further studies to understand the impact of PEG density and charge on pulmonary administration by nebulization.
Altogether, local administration of mRNA to the lungs has been shown to be feasible with both LNPs and polyplexes. The nebulization of mRNA using non-PEGylated biodegradable PBAE polyplexes, which have been shown to be able to complex up to 0.5 mg mL−1 of mRNA, demonstrated 25% transfection of the total lung epithelial cell population in a healthy mouse model (Fig. 4A). Whether PEGylation provides potential benefits for delivery to pulmonary epithelial cells is controversial. Recent studies using poloxamine-based polymers and PBAEs demonstrated successful delivery of mRNA to the lungs without the need of PEGylation.164,165 Further, in a recent study conducted with siRNA, a thorough analysis of comparable PEGylated and non-PEGylated polymer formulations (PEI and PLGA) clearly demonstrated that PEGylation did not enhance gene silencing in CF mucus, corroborating results obtained with non-PEGylated PBAE polyplexes.153,163 This phenomenon could be attributed to lower cellular uptake into epithelial cells resulting from the PEG shield. In contrast, PEG has been shown to be crucial for the stability of LNP formulations for nebulization, by preventing aggregation during storage and promoting structural stability within the vibrating mesh nebulizer.167
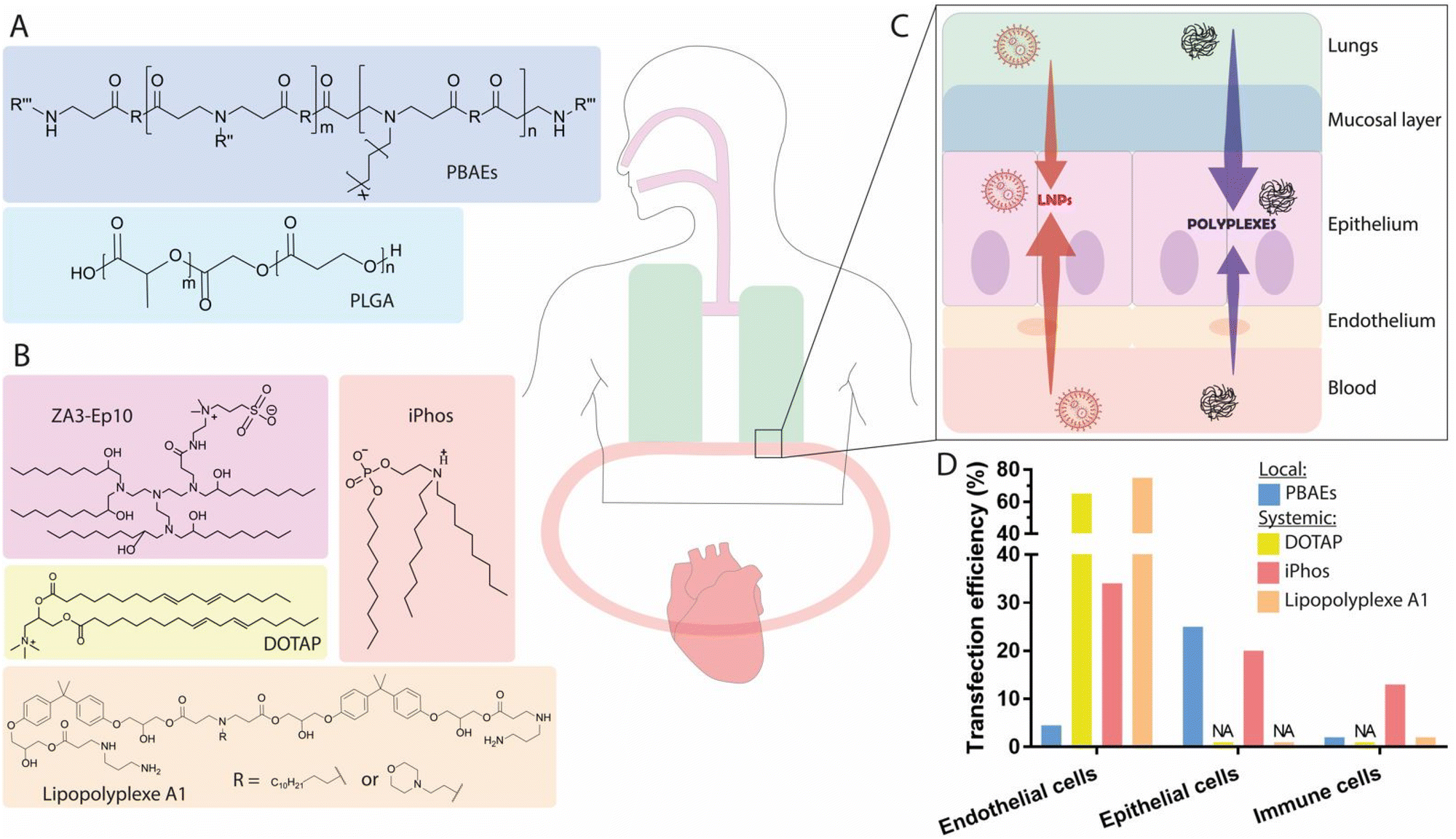 | ||
| Fig. 4 Local and systemic administration of mRNA via nanocarriers to the lungs. (A) Chemical structures of several polymers used for local administration, or (B) ionizable lipids used for systemic delivery. (C) Graphical representation of the two routes of administration, for the principal classes of nanocarriers represented in the literature. (D) Transfection efficiency of the best-performing nanocarriers both for local and systemic delivery. Doses and duration before evaluation: 1 mg kg−1 Cre mRNA, t = 144 h for PBAEs;153 0.3 mg kg−1 Cre mRNA, t = 48 h for DOTAP;122 0.25 mg kg−1 Cre mRNA, t = 48 h for iPhos;126 and unknown dose of Cre mRNA, t = 48 h for lipopolyplex A1.176 | ||
3.3.2.1. Lipid-based nanocarriers. Inducing mRNA expression in the lungs upon systemic administration has mostly been evaluated through passive targeting without the use of targeting ligands. Nonetheless, a recent study has investigated the use of active targeting via conjugation to the LNP surface of antibodies specific for the vascular cell adhesion molecule, PECAM-1. PECAM-1 is highly expressed in vascular cells (including ECs) and early studies have demonstrated that liposomes targeting PECAM-1 are capable of accumulation in the lung endothelium.169,170 Biodistribution evaluation using radiolabeled formulations also revealed that the pulmonary uptake of PECAM-1-targeted-Ab/LNP-mRNA was 16-fold greater than that of control IgG/LNP-mRNA.171 Luc mRNA-induced fluorescent signals showed more specific expression in the lungs, and reduced expression in the liver for PECAM-1-targeted-Ab/LNP-mRNA, but not for the control IgG/LNP-mRNA, with a ∼25-fold elevation of the signal in the lungs. Upon assessment, the lung/liver ratio of targeted LNPs was found to be around 2.5, indicating that this approach might constitute another feasible strategy to achieve specific mRNA delivery to the lungs.
By increasing the molar percentage of the permanent cationic lipid DOTAP in LNPs, mRNA expression was shifted from the liver to the lungs, with an optimal formulation containing 50 mol% DOTAP demonstrating near exclusive mRNA expression in lung tissue. The increased quantity of DOTAP induced a gradual augmentation of the LNP surface pKa from 6.46 to >11 for the optimal lung-targeting formulation, which was capable of transfecting ∼40% of all epithelial cells, ∼65% of all ECs and ∼20% of immune cells in the lungs (Fig. 4B).122 Surprisingly, while the surface pKa of the LNPs was increased, the zeta potential of this formulation showed a surface charge near neutral. A possible explanation might involve the encapsulation of DOTAP within the LNPs alongside the mRNA. Analyses using Cy5-labeled mRNA demonstrated that, despite strong protein expression in the lungs, around 30% of the total mRNA was still delivered to the liver, revealing a discrepancy between the biodistribution and mRNA-induced protein expression profile. The addition of DOTAP and the resulting increase in surface pKa also had the effect of altering the protein corona of the LNPs, as highlighted in a recent proteomics study.123 The composition of the protein corona of lung-targeting LNPs was primarily enriched in vitronectin, in contrast with liver-targeting LNPs, where the corona was mostly enriched in ApoE. In another study, the inclusion of DOTAP at 60% mol in liver-targeting LNPs supported the previous findings, achieving lung-specific gene editing, as indicated by strong fluorescence in the lung tissue of Ai14 Cre-reporter mice upon administration of Cas9/sgTOM mRNA LNPs.172 Replacement of the lipid DOPE with DOTAP in a four-component LNP system was also shown to promote lung-specific mRNA delivery. Specifically, the incorporation of 40 mol% DOTAP substantially shifted mRNA expression to the lung, further supporting that the incorporation of a positively charged lipid into LNP formulations represents a feasible approach to promote selective mRNA expression in lung tissue.125 Interestingly, lung specificity was found to strongly correlate with LNP surface pKa, but correlated poorly with zeta potential. While both of these parameters provide information on the surface charge of the nanoparticles, the absence of a shift in zeta potential is surprising. A complete understanding of this phenomenon remains elusive and future work is required to acquire mechanistic understanding of this discrepancy. The ionizable phospholipid iPhos was also found to be able to promote lung-selective mRNA delivery;126in vivo evaluations revealed that the presence of a 9-carbon alkyl chain attached to the phosphate group was able to mediate lung-specific mRNA localization. Further, the incorporation of the cationic helper lipid dimethyldioctadecylammonium bromide (DDAB) resulted in an optimal lung-targeting LNP formulation with a surface pKa of 7.15, achieving a transfection efficiency of nearly 34% of all ECs, 20% of all epithelial cells, and roughly 13% of immune cells, enabling effective gene editing in the lungs (Fig. 4B and D). This suggests that the inclusion of an optimized ionizable lipid structure, coupled with the insertion of a cationic helper lipid, can promote lung-specific mRNA delivery. Though the surface pKa values for the iPhos formulations were not as high as those exhibited upon the incorporation of 50 mol% DOTAP, both formulations achieved comparable transfection efficiencies in lung cells, suggesting that while the surface pKa of LNPs is relevant, other factors—including the structure of the ionizable lipids—may contribute to the success of lung-specific mRNA expression. Based on the same design principles used for iPhos lipids, zwitterionic amino lipids (ZALs) have been developed, comprising a zwitterionic sulfobetaine headgroup, an amine-rich linker region, and a range of hydrophobic tail structures. These lipids combine the chemical and structural features of zwitterionic and cationic lipids into a single compound, which in turn allows the enhanced delivery of longer RNA sequences, such as Cas9 mRNA. Among the lead ZAL structures prepared, the best performing compound (ZA3-Ep10), resulted in expression of Luc in the lungs, though substantial expression was also observed in the liver and spleen 24 h post-injection (Fig. 4B).173 A further study analysed the biodistribution of the ZA3-Ep10-based LNP formulation and the level of mRNA-induced protein expression 6 h after injection.37 Interestingly, the data showed a wide distribution profile, with mRNA accumulation mostly observed in the liver (66%) and lungs (21%), though protein expression was primarily found to occur in the lungs (>70%).108,111 Thus far, no extensive explanation has been proposed for this discrepancy, and more investigations are required to explore cell-specific differences in nanoparticle internalization, endosomal escape, and translational kinetics, all of which may contribute to better understanding the disparities between biodistribution and mRNA expression.
3.3.2.2. Polymer-based nanocarriers. Systemic mRNA delivery to the lungs has also been investigated using polymeric structures, such as PBAEs and polyester derivatives.132,174,175 In one example, a set of PBAE polymers were synthesized via the Michael addition of three monomers (diacrylate, secondary or tertiary amine, and C12-dodecyl amine), forming random copolymers defined as terpolymers. Nanoparticles were then generated by the incorporation of DMG-PEG (7 mol%) through hydrophobic interactions, promoting particle stability for over 2 hours in phosphate buffered saline (PBS) spiked with 10% fetal bovine serum (FBS). An in vivo evaluation of the best-performing of these polymers resulted in Luc expression primarily in the lungs.108 Similar to previous studies, this work revealed a strong discrepancy between mRNA biodistribution and protein expression. Notably, while the greatest protein expression was detected in the lungs, with no substantial signal recorded in other organs, fluorescently labeled mRNA polyplexes were found to be distributed evenly among the organs, with the strongest signals observed in the spleen and lungs. While the mechanism by which mRNA translation in the lungs is favoured remains unclear, it has been suggested that the surface charge of the lipopolyplexes, ranging from +10 to +15 mV, may result in altered interactions with serum proteins or surface receptors in the lung endothelium, and may account for the level of lung-specific expression observed. Subsequent work on the same terpolymers demonstrated that optimization of the alkyl chain length of the PEG-lipid, alongside the molar % of PEG-lipid, can confer a high degree of lung-specific mRNA expression.176 The in vivo screening of a series of non-covalent polymer formulations made from terpolymers and PEG-lipids containing alkyl chains varying in length between C14 and C18, at proportions ranging from 1–7 mol%, yielded a lipopolyplex formulation comprising PBAE terpolymers + 5% C18PEG2000 complexed with mRNA at a nitrogen to phosphate (N/P) ratio of 50
:1, which outperformed previous formulations containing 5.5 mol% DMG-PEG. As such, optimization of the PEG-lipid structure and mol% could present another feasible approach for improving mRNA expression in the lungs. Furthermore, this mol% of PEG-lipid yielded particles with sizes of ∼100 nm (within the optimal range), a factor which has been strongly correlated with effective lung-specific mRNA delivery in other studies.125 Flow cytometry analysis in Cre-reporter mice revealed that this formulation primarily transfected lung ECs (75% of the total EC population) and, to a lesser extent, immune cells (2% of the total immune cell population). Aside from surface charge and PEGylation, the surface pKa of polyplexes has also been identified as an important parameter for mRNA delivery to the lung. A recent study performed a screening of a series of cationic polyester polymers containing varied amino groups and modified hydrophobic side chains, to evaluate the size, surface charge, and pKa of polyplex nanoparticles for the selective delivery of mRNA to the lungs.132 No correlation was found between the size and charge of polyplexes, as the lead polymer with the strongest signal in lung tissues showed nearly neutral surface charge. Interestingly, surface pKa correlation assessment illustrated a strong relationship between selective mRNA delivery to lung tissue and pKa, with optimal values for lung delivery ranging from 7.5 to 8, in line with the data observed for LNPs. Biodistribution studies also revealed a disperse Cy5-labeled mRNA distribution throughout the body, with strong accumulation in the liver illustrating once more that mRNA distribution does not correlate with mRNA translation.
Altogether, lung-targeted mRNA delivery has been found to be achievable via systemic administration, using both polymeric and lipid-based formulations (Fig. 4C). Interestingly, when comparing transfection efficiency at a cellular level, it appears that transfection in ECs is higher upon systemic administration than upon local administration, suggesting that this administration route could represent a more viable approach for the treatment of acute lung injury/acute respiratory distress syndrome. Transfection efficiency of lung epithelial cells has been reported to range between 20–40% for LNP systems, and up to 75% when using PBAE polymers (Fig. 4D). A common feature of lung-targeting formulations is the introduction of positively-charged lipids into LNPs, resulting in surface pKa values above 7 and consequently altered protein interactions.93,123 In addition, modifications to the side chains of polymers used for mRNA delivery led to similarly altered surface pKa, suggesting that this parameter is critical for achieving lung tissue-specific delivery of mRNA.
3.4. Highlights on the biodistribution of mRNA nanocarriers
As described above for both LNPs and polymeric structures, several parameters must be considered in order to achieve tissue-specific mRNA expression. Here, we underline two key points playing a crucial role in mRNA biodistribution which might provide inspiration for future works aiming to develop more efficient tissue-specific delivery vehicles for mRNA.Thus far, it has been difficult to accurately predict the biodistribution of nanoparticles based on their physicochemical parameters. The surface pKa, corresponding to the pH at which the proportion of charged and uncharged ionizable lipids at the surface of the particle is equal, has been assessed in various studies and is thought to be a key factor. Although this seems persuasive, particularly for liver-targeting formulations, recent findings have revealed that other parameters are also involved. Indeed, surface pKa values above 11 have been reported to promote almost exclusively lung-specific mRNA delivery when using the ionizable lipid 5A2-SC8 and 50 mol% DOTAP, whereas a surface pKa of 7.2–8.0 was sufficient to mediate mRNA delivery to the lungs using iPhos LNPs or the polymer PE4K-A17-0.2C8.122,126,132 This suggests that the surface pKa may depend on the type of ionizable lipids and/or charged helper lipids used in the nanoparticle formulation, and accordingly, the biodistribution profile must be extrapolated from these predictive values keeping in mind the associated formulation. Additionally, only a poor correlation between surface pKa and zeta potential has been reported, which is surprising, given that both parameters provide information regarding the charges present at the surface of LNPs.125 While the underlying mechanism remains unclear, a possible explanation for this lack of alteration of the zeta potential could be due to an internal restructuring of the LNPs when permanently charged lipids are added, resulting in changes to the surface pKa and leaving the zeta potential unaltered. With this in mind, surface pKa appears to have an influence on mRNA biodistribution, but this may need to be thoroughly assessed alongside other parameters, such as the type of ionizable lipids and helper lipids used in the formulation, overall LNP structure, and mRNA-to-lipid ratio, in order to be efficiently used in screening process.
When comparing biodistribution profiles and mRNA-induced protein expression, there is no clear evidence that the massive accumulation of nanocarriers in a given organ will necessarily lead to a high degree of protein expression. Indeed, meaningful discrepancies have been reported for both LNPs and polymeric nanocarriers (Fig. 5).37,94 As an example, the previously described Of-Deg-Lin lipid resulted in over 85% of total protein expression in the spleen, despite the LNPs being primarily distributed to the liver.45 Consequently, biodistribution alone cannot predict the functional delivery of mRNA to a particular tissue. The reasons certain formulations fail to mediate protein expression in certain tissues remain elusive, but possible explanations may lie in endosomal composition, which differs between various cell types, and changes to the formation of the protein corona, which is thought to govern cellular uptake and the fate of nanoparticles in circulation.177,178
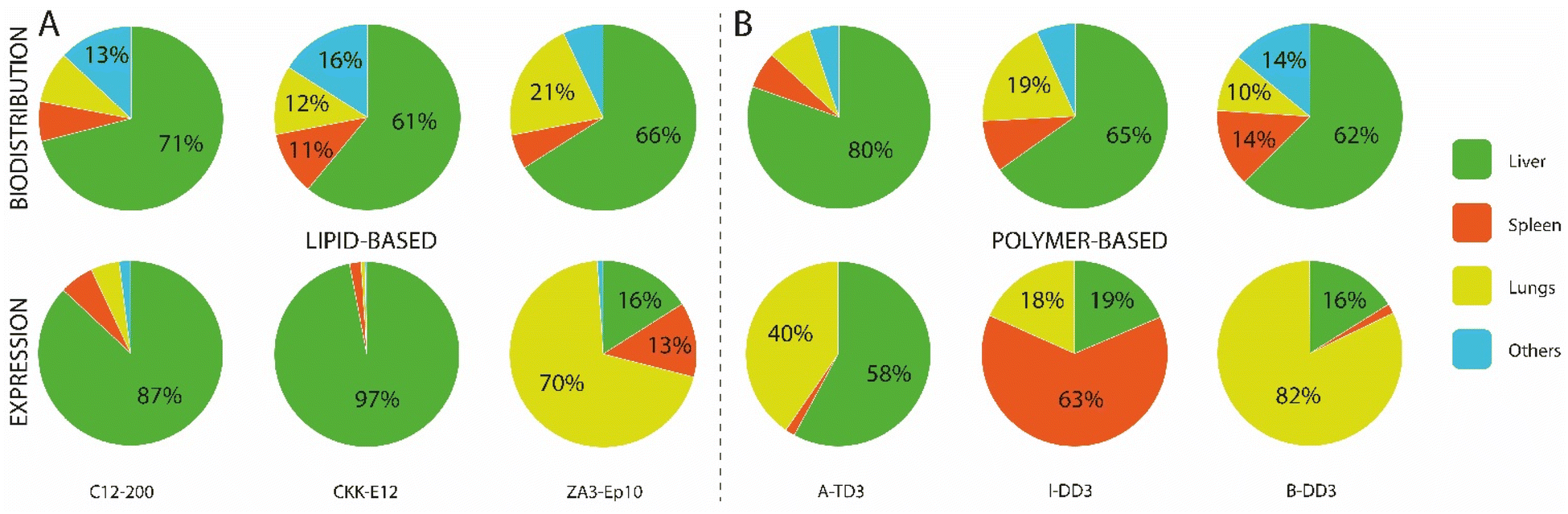 | ||
| Fig. 5 Discrepancies between mRNA biodistribution and resulting expression. (A) mRNA biodistribution and functional delivery for three polymer-based formulations, with a dose of 0.6 mg kg−1 of either Luc mRNA or Cy5-labeled mRNA, after 6 h.111 (B) mRNA biodistribution and functional delivery for three lipid-based formulations, with a dose of 0.75 mg kg−1 of either Luc mRNA or Cy5-labeled mRNA, after 6 h.37 | ||
4. mRNA delivery to other relevant organs
4.1. mRNA delivery to the lymphatic system
Nanoparticle size and charge have both been identified as factors governing drainage through lymphatic vessels. Drainage has been reported for nanoparticles with sizes ranging from 20–200 nm, with optimal lymphatic uptake observed for particles between 20–80 nm.180,186,187 Similarly, it has been reported that following SC injection, mRNA-LNPs with diameters of 150 nm and zeta potentials between −15 and −3 mV could efficiently be drained to the LNs.188 In Cre reporter mice, flow cytometry revealed that 4.6% of DCs, 1.2% of macrophages, 3.3% of neutrophils, and 0.06% of B cells were efficiently transfected. Antitumor function evaluation after a single SC injection of LNPs containing mRNA coding for the tumor-associated antigens gp100 and TRP2 in an aggressive mouse model of melanoma revealed strong CD8+ T cell activation, tumor size reduction, and extended overall survival of the treated group. Direct percutaneous injection into the LNs has also been investigated for immunotherapy applications, specifically for the delivery of mRNA vaccines without the use of nanoparticles.189 Vaccine-induced T cell infiltration and killing of autologous tumor cells was shown in resected post-vaccination metastases. The cumulative rate of metastatic events was highly significantly reduced after the initiation of vaccination, resulting in sustained survival without further disease progression.
4.2. mRNA delivery to the brain
mRNA therapeutics could represent a promising approach for the treatment of brain disorders caused by genetic defects.191,192 However, the effective delivery of large hydrophilic drugs, including mRNA, to the brain following systemic administration is hampered by the blood-brain barrier (BBB).193 Thus, most studies on mRNA delivery to the brain utilize local administration routes, with only one work evaluating the delivery of mRNA to the brain endothelium via IV administration.4.3. mRNA delivery to the eyes
Over the past decade, interest has grown in the development of nucleic acid drugs for the treatment of ocular diseases. The straightforward accessibility and immune-privileged status of the eyes has resulted in a plethora of published works and clinical trials, leading in 2017 to the FDA approval of an adeno-associated virus (AAV2)-based complementary DNA gene augmentation therapy, voretigene neparvovec (Luxturna©).202 LNPs have been used to deliver mRNA to the eyes via two routes of local administration: subretinal (SR) and intravitreal injection. While SR injection delivers the LNPs in close proximity to the retinal pigment epithelium (RPE) in the outer retina, intravitreal administration requires the vehicle to move through the vitreous humor and penetrate a complex network composed of tight junctions and fibronectin, thereby restricting the permeation of nanoparticles prior to reaching the RPE. An initial study demonstrated that the SR injection of Cy5-labeled GFP mRNA encapsulated in Lipofectamine™2000 displayed localization in the RPE layer and specific uptake into photoreceptor cells.203 GFP protein expression was found to be co-localized with the Cy5-mRNA signal in the RPE layer and photoreceptor cells. Similarly, another study, conducted in mice, evaluated the SR injection of a series of LNP formulations containing ionizable lipids with distinct pKa values.204 Examination of Luc expression demonstrated that lipids displaying low pKa values (such as MC3) and containing unsaturated alkyl chains generated the brightest luminescence in the RPE, in comparison to cationic or ionizable lipids with high pKa values such as N,N-dimethyl-2,2-di-(9Z,12Z)-9,12-octadecadien-1-yl-1,3-dioxolane-4-ethanamine and 1,2-dioleyloxy-3-dimethylaminopropane (DODMA) potentially due to the superior ability of MC3 to escape the endosomal lumen. Upon SR injection of MC3 LNPs, mRNA delivery was shown to be cell-type specific with the majority of expression observed in the RPE and in the Müller cells. Owing to the crucial role of RPE cells and Müller cells in the pathological development of several retinal disorders, including macular degeneration,205 the delivery of mRNA to the retinal epithelium might offer new treatment opportunities for these conditions.While SR injection is a very efficient retinal drug delivery route, this technique requires careful manipulation in order to avoid damage to the retinal layer.206 In contrast, intravitreal injection presents a less invasive and safer method of reaching the retina, though the need to travel through the vitreous humor could potentially lower the transfection efficiency of the injected complexes.207 To investigate this approach, a recent work evaluated the influence of particle size and PEG content (0.5–5%) on the penetration and transfection efficiency of LNPs upon intravitreal injection.208 It was found that LNPs with a PEG content of 0.5 mol% and a particle size of 150 nm yielded optimal Luc activity and expression in Müller cells, the optic nerve head, and the trabecular meshwork, but failed to reach the RPE, possibly due to the low quantity of nanoparticles able to reach the retina after diffusion through the vitreous humor. Consequently, the route of administration is a key factor to consider for ocular mRNA delivery, with intravitreal injection enabling transfection of the Müller glia and the optic nerve head in the inner retinal layer, while SR injection is required to reach the RPE in the deeper retinal layer. The FDA-approved gene therapy Luxturna© is administered via SR injection, highlighting the potential of this approach, though the high level of precision required for this procedure should still be emphasized. Future developments enabling transfection of the neural retina through intravitreal delivery, which is less invasive and allows for repeated administration, could broaden the scope of therapeutic applications of mRNA for the treatment of Mendelian eye disorders.
4.4. mRNA delivery to the heart
Myocardial infarction and heart failure are leading causes of death in industrialized nations, with 300000 individuals per year experiencing recurring heart attacks in the United States alone.209,210 Myocardial infarction leads to a loss of cardiomyocytes (CMs), which are replaced with non-CM cells, leading to scarring and, in most cases, heart failure.211 Current therapies such as cell-based treatments using exogenous cells, cardiac progenitor cells, or stem cells have been developed to improve heart function, however, poor clinical improvement has generally been reported.212 With this in mind, there has been a recent focus on the enhancement of angiogenesis and revascularization of the myocardium through direct VEGF administration in both preclinical and clinical studies.213,214 Initial studies have demonstrated that intramyocardial injection of VEGF mRNA complexed with a commercial transfection agent (RNAiMAX©) results in the expansion and directed differentiation of endogenous heart progenitor cells toward cardiovascular cell types, improving heart function in a mouse model of myocardial infarction.215,216 Intramyocardial injection and intracoronary delivery have also been assessed using eGFP-encoding mRNA formulated in LNPs.217,218 Direct intramyocardial injection led to strong eGFP expression in the subepicardial areas of injection and extended to the myocardium, with subsequent biodistribution analyses showing low off-target accumulation (below 10%) in the liver, spleen, and lungs. As a more clinically suitable minimally invasive delivery approach, intracoronary delivery has been investigated in this study. Nonetheless, this approach demonstrated lower levels of protein expression than direct myocardial injection and a more pronounced off-target expression profile, particularly in the spleen. Interestingly, while naked RNA is rapidly degraded in the bloodstream, recent reports have demonstrated that the direct injection of naked mRNA into the myocardium resulted in prolonged VEGF levels in cardiac tissue. In one study, the encapsulation of mRNA with nanoparticles hindered its effective translation, whereas naked m1ψ-modified mRNA in sucrose-citrate buffer was translated very efficiently, with protein expression detected within 10 min of administration in cardiac muscle.219 These findings were supported by another study, which reported efficient intracardiac transfection and protein expression following the delivery of naked Luc mRNA in saline citrate buffer.220 Translation efficiency in mouse hearts was found to be highest with 100 mg of naked mRNA modified with m5C and m1ψ, delivered in a sucrose-citrate buffer. The cause of this prolonged protein expression may be explained by a combination of the interstitial localization of mRNA along the cardiac sarcolemma, potentially acting as a protected reservoir of VEGF mRNA, and the direct local administration into muscle cells, avoiding RNase-mediated degradation in the bloodstream. Building upon these findings, the development of mRNA therapies for cardiac ischemia has been pursued, leading to a first-in-human study of this treatment using VEGF-A mRNA, which has recently been completed (NCT02935712). Overall, these reports indicate that the intramyocardial administration of mRNA-based therapeutics for the induction of cardioprotection and vascular or cardiac regeneration following myocardial infarction is very efficient and shows low off-target biodistribution.
5. Most advanced pre-clinical and clinical nanoparticle-based mRNA therapeutics
The transient nature of mRNA drugs necessitates a chronic administration regimen over an extended period of time. Thus, the ability to achieve long-term efficacy and safety upon repeated dosage is a prerequisite for clinical maturity. To fulfill the task of mRNA delivery, while polymeric nanocarriers have been shown to be effective agents, LNPs have been at the forefront of preclinical and clinical investigations.221 A key component involved in the tolerability and efficacy of these LNP formulations is the ionizable lipid component.222 The insertion of ester functionalities at specific positions on the lipid tail is a feasible strategy for preserving high in vivo transfection efficiency and a favorable metabolic profile, as illustrated by the development of the optimized ionizable lipid “Lipid 5”.89 Evaluation of repeated EPO mRNA dosing in primates showed a consistent expression profile and no increase in liver function biomarkers such as alanine transferase (ALT) or aspartate aminotransferase (AST), illustrating the ability of this lipid to safely deliver mRNA in a chronic dosage regimen. Consequently, recent developments in the design of ionizable lipids and their formulations have enabled preclinical studies and, in some cases, reached the stage of clinical investigations, as highlighted in Tables 2 and 3. Hereafter, we highlight preclinical studies and clinical investigations of mRNA-based protein supplementation therapy explored in the liver, lungs, lymphoid organs, heart, and brain.| Disease | mRNA | Formulation | Route of administration | Ref. |
|---|---|---|---|---|
| Lung | ||||
| Cystic fibrosis | CFTR | Polyplex | IT | 152 and 164 |
| Cystic fibrosis | CFTR | LNP | Nebulization | 234 |
| Cystic fibrosis | CFTR | LNP | Intranasal | 235 |
| SP-B deficiency | ZFN | Polyplex | IT | 150 |
| Influenza, SARS-COV-2 infection | Cas13 and sgRNA | Polyplex | Nebulization | 165 |
| Alpha-1-antitrypsin deficiency | SERPINA | LNP | IV | 250 and 236 |
| Liver | ||||
| Urea cycle disease | OTC | GalNac polymer-conjugated LNP, LUNAR-LNP | IV | 75 |
| Methylmalonic acidemia | MUT | LNP | IV | 88 and 224 |
| Propionic acidemia | PCCA and PCCB | LNP | IV | 251 |
| Glycogen storage disorder | G6Pase-α | LNP | IV | 228 and 252 |
| Acute intermittent porphyria | Human PBGD | LNP | IV | 87 |
| Arginase deficiency | ARG1 | LNP | IV | 226 |
| Hemophilia | hFIX | LNP | IV | 253 and 254 |
| Thrombotic thrombocytopenic purpura | ADAMTS13 | LNP | IV | 255 |
| Anemia | EPO | LNP | IV | 89 |
| Fulminant hepatitis | Bcl-2 | Polyplex | IV | 110 |
| Brain | ||||
| Friedriech's ataxia | FXN | LNP | Intrathecal | 195 |
| Alzheimer disease | NEP | Polyplex | Intracerebroventricular | 200 |
| Inflammatory disorders | TM | Active-targeted LNP | IV | 201 |
| Other organs | ||||
| Cardiovascular diseases | VEGF | Naked | Cardiac injection | 220 |
| Lymphedema | VEGF | LNP | ID | 185 |
| Inflammatory bowel disease | IL-10 | Active-targeted LNP | IV | 238 |
| Product (sponsor) | Gene mRNA and formulation | Indication | Administration route | Clinical stage |
|---|---|---|---|---|
| Heart | ||||
| mRNA AZD-8601 (Astrazeneca/Moderna Tx) | VEGF-A mRNA, naked in citrate buffer | Cardiovascular disease, heart failure | Epicardial injection | Phase I (completed) |
| NCT02935712 | ||||
| Phase II (completed) | ||||
| NCT03370887 | ||||
| Lung | ||||
| MRT5005 (Translate Bio) | CFTR mRNA, LNP | Cystic fibrosis | Nebulization | Phase I/II (recruiting) |
| NCT 03375047 | ||||
| Liver | ||||
| MRT5201 (Translate Bio) | OTC mRNA, LNP | OTC deficiency | IV | Phase I/II (withdrawn) |
| NCT03767270 | ||||
| mRNA-3704 (Moderna Tx) | MUT mRNA, LNP | MUT deficiency | IV | Phase I/II (withdrawn) |
| NCT03810690 | ||||
| mRNA-3927 (Moderna Tx) | PCCA and PCCB mRNA, dispersion | Propionic acidemia | IV | Phase I/II (recruiting) |
| NCT04159103 | ||||
| NCT05130437 (recruiting) | ||||
| ARCT-810 (Arcturus Tx) | OTC mRNA, LNP | OTC deficiency | IV | Phase I (completed) |
| NCT04416126 | ||||
| NCT04442347 (recruiting) | ||||
| mRNA-3745 (ModernaTx) | (G6Pase) mRNA, LNP | Glycogen Storage Disease | IV | Phase I (recruiting) |
| NCT05095727 | ||||
5.1. Genetic liver disorders
mRNA-based protein replacement therapy in the liver has been explored for the treatment of various rare genetic disorders involving the metabolism of amino acids, heme synthesis, and glucose homeostasis for which either no treatment is approved, or where management of the condition restricts quality of life. For example, methylmalonic acidemia is caused by a partial or complete loss of the enzyme methylmalonyl-CoA mutase (MUT) responsible for the catabolism of amino acids, and leads to the accumulation of toxic metabolites such as methylmalonic acid.223 Presently, there is no enzyme-replacement treatment available for this condition, and patients must follow a strict diet, alongside carnitine supplementation. In contrast, a single IV bolus injection of MUT mRNA encapsulated in LNPs (formulated with Lipid 5) demonstrated a 75% decrease in plasma levels of methylmalonic acid in MUT−/− mice.88 A subsequent long-term study evaluated the chronic dosing of this formulation via IV injection every two weeks for 12 weeks at various dosages. The dose-dependent response profile indicated that an mRNA dosage below 0.5 mg kg−1 resulted in fluctuating plasma levels of methylmalonic acid with increases between injections, whereas doses from 0.5 mg kg−1 to 2 mg kg−1 resulted in a more stable profile. Safety evaluations showed no difference in liver toxicity markers, including AST and ALT, nor in other inflammatory markers (e.g., interleukin-6, IFN-γ), indicating that the injection of human MUT mRNA-LNPs did not induce liver toxicity.88,224 Another critical liver condition is arginase deficiency, a disorder resulting in the accumulation of ammonia and arginine in the liver,225 with available treatment options limited to ammonia scavenging and strict dietary regimens. The delivery of arginase-encoding mRNA encapsulated in LNPs every 3 days demonstrated consistent arginine control, without elevation of plasma ammonia, and maintained long-term survival in Arg1−/− mice.226 A safety evaluation in hepatic tissues detected no pathological hepatic abnormalities or lymphocyte infiltration, suggesting the strong tolerability of this approach. Aside from liver inflammation, another safety concern is the development of anti-drug antibodies (ADAs) against the mRNA-encoded protein. In conventional protein replacement therapy, the induction of ADAs upon repeated systemic administration of exogenous recombinant protein is of great concern, as this can activate an immune response, thereby mitigating the efficacy of the treatment.227 In contrast, the repeated dosing of mRNA-LNPs in various studies has shown no substantial increase in induced ADAs in treated mice, indicating that mRNA-based approaches might result in superior long-term efficacy.87,88,228 Indeed, mRNA uses endogenous translation machinery within cells to replace the missing protein, whereas exogenous recombinant proteins injected into the systemic circulation are exposed to immune cells, potentially engaging in complement activation and promoting rapid clearance of the therapeutic protein. Nonetheless, concerns have been raised about the generation of anti-PEG antibodies, which bind to either the backbone or the terminal methoxy group of PEG, as this polymer is commonly used in mRNA nanocarrier formulations and can trigger activation of the complement pathway in humans.229 At present, there are no data available on the repeated dosing of mRNA-LNPs in humans. Clinical evaluation of repeated dosing of Onpattro© over 24 months in 29 patients revealed that, despite the presence of DMG-PEG in the formulation, anti-PEG antibodies were infrequently detected and did not affect the pharmacokinetic exposure or the transthyretin (TTR) reduction effect of the drug.230Though most preclinical and clinical applications of LNPs use passive targeting to traffic particles to the liver, active targeting has also been evaluated, using a hybrid formulation consisting of a combination of LNPs mixed with a GalNAc-targeted polymer. The delivery of ornithine transcarbamylase (OTC) mRNA in hybrid nanoparticles induced a normalization of plasma ammonia levels and urinary orotic acid levels, and led to prolonged survival in a murine model of OTC deficiency, for two different dosing frequencies.75 A twice-per-week dosing regimen led to enhanced survival and normalization of plasma levels of ammonia and orotic acid, whereas for weekly dosing, urinary orotic acid levels became elevated after 20 days, resulting in death shortly thereafter. The twice-per-week dosing regimen also led to a substantial increase in body weight and superior survival, demonstrating the necessity of frequent dosing. In spite of these results, the preclinical study was discontinued in 2018 for unknown reasons. Nonetheless, another clinical study conducted by Arcturus Bio, consisting of an OTC-replacement program using its proprietary lipid formulation platform LUNAR (LUNAR-OTC) without any additional targeting moiety, has successfully completed a Phase I clinical trial (ARCT-810) in healthy adults. The company is currently recruiting participants for a Phase I/II OTC deficiency study, including stable patients with OTC deficiency (Table 3). We note that the first FDA-approved siRNA-LNP-based therapeutic (Onpattro©) did not include a targeting ligand, and a clinical evaluation did not observe any added performance in gene silencing by the GalNac-conjugated formulation over its non-conjugated counterpart.231 Though very promising results have been achieved in animal models for various genetic metabolic liver disorders, clinical investigations remain in early stages, and no clinical data has thus far been provided. Currently, one of the most advanced clinical studies involves the treatment of OTC deficiency, developed by Arcturus Bio, with primary outcomes expected by the end of 2022. Another clinical study conducted by Moderna investigating propionic acidemia is currently recruiting participants, and initial outcomes are expected by 2025 for its first-in-human Phase I/II trial, at which point another study will begin, evaluating the long-term clinical safety of this pharmaceutical candidate (Table 3). Similarly, a study on glycogen storage disorders will begin by the end of 2022. The large number of upcoming clinical trials suggests the great promise of this technology for the development of potential patient treatments, while providing crucial information on clinically suitable dosing regimens.
5.2. Genetic lung disorders
Gene therapy is a promising treatment approach for a range of lung diseases displaying resistance to current treatments. CF, which is caused by mutations in the CFTR gene, has long been a subject at the forefront of lung-disease research. Although several gene therapies have shown potential for treatment of this condition, the correction of CFTR defects using viral or non-viral vectors has thus far remained unsuccessful in clinical trials, largely due to delivery issues.232,233 The local administration of polyplexes containing mRNA encoding for CFTR has been evaluated in a mouse model of CF using chitosan-coated PLGA polyplexes, poloxamine-based polyplexes, and LNPs. It has been found that administration of 40 μg of mRNA complexed with PLGA polyplexes via the IT route significantly improved lung function, specifically enhancing forced expiratory volume (FEV1). Interestingly, the same group also considered IV administration, finding this delivery route to be more efficient for the same dosage, likely due to the poor penetration ability of the polyplexes into CF mucus following IT administration.150 Similarly, the IT administration of peptide-poloxamine nanoparticles encapsulating CFTR gene insertion tools led to the successful knock-in of the CFTR gene in a murine model of CF.164 However, more detailed long-term safety evaluations in larger animal models, such as pigs, as well as in pertinent CF animal models with thicker pulmonary mucus layers, are required before conclusions can be drawn. The nebulization of SORT LNPs including positively charged helper lipid has also been tested for treatment of CF, demonstrating significant rescue of CFTR expression and function in multiple genotypes,234 with immunofluorescent analysis revealing CFTR mRNA expression in several relevant cell types, including basal cells and ionocytes. As an alternative to nebulization and IT administration, the nasal administration of CFTR mRNA-LNPs has also been shown to be capable of mediating chloride secretion restoration in the airway epithelia of CFTR knockout mice for at least 14 days post-treatment. CFTR activity was found to peak on day 3, recovering up to 55% of the net chloride efflux observed in healthy mice.235 The company Translate Bio began the first study on nebulized mRNA-LNP supplementation therapy for CF in 2018 to evaluate the safety and tolerability of single and multiple escalating doses in adult CF patients. Results from the second interim data analysis revealed that, in a single ascending dose study including 16 patients, 20 mg administrations were safe and well tolerated with no marked increases in percent predicted forced expiratory volume (ppFEV1), which would indicate pulmonary exacerbation. Interestingly, patients had no detectable LNP lipid levels in their blood, and neither anti-PEG nor anti-CFTR antibodies were found. In the multiple ascending dose study for doses ranging from 8–16 mg, an analysis of 12 patients found similar safety results, indicating that the nebulization of mRNA was well tolerated among the patients tested. However, the same interim results indicated that no pattern of improvement in lung function was measured, which could be due to the mRNA candidate—which contained no base modifications—used for the clinical investigation.37 Aside from CF treatment, the delivery of mRNA to the lung was also evaluated for the ability to encode a therapeutic antibody with the potential to protect from viral infection. The nebulization of LNPs encapsulating mRNA encoding for membrane-anchored FI6 (aFI6), an antibody binding hemagglutinin, resulted in mice protected from a lethal challenge by the H1N1 subtype of the influenza A virus, whereas control mice progressively died.165 Despite displaying promising transfection efficiencies for lung epithelial cells and ECs, IV injection has not yet been evaluated in a therapeutic model of a lung-specific gene disorder, though one clinical indication affecting both the liver and lungs has been tested, namely alpha-1-antitrypsin deficiency.236 Overall, mRNA-based protein supplementation therapy for the treatment of genetic lung disorders displays clear potential, but future work is required, including a focus on improving delivery tools, which could include pre-treatment with mucolytic agents in order to increase transfection efficiency in epithelial cells in the airways of CF patients.5.3. Cancer immunotherapy and immunomodulation
The delivery of mRNA to immune cells can be used to engage or mitigate the immune response. mRNA has emerged as an attractive cancer vaccine format, as it provides both antigen delivery and innate immune activation.237 This has led to numerous cancer vaccine clinical trials, potentially offering promising alternatives to traditional cancer treatments. Reports have described mRNA-induced tumor-antigen cancer vaccines which can be administered either via the IV route or locally, directly into the lymph nodes or tumor tissue. For example, the SC injection of an optimized mRNA LNP has been shown to efficiently transfect immune cells, eliciting a strong T cell response.188 To evaluate the antitumor function of this approach, the delivery of mRNA coding for tumor-associated antigens in a mouse model of melanoma was investigated. Upon administration, strong T cell activation resulted in a reduction in tumor size and extended the overall survival of the treated mice, which suggests that an mRNA-mediated immune response could act as an effective antitumor strategy. A major limitation of local administration is that only lymph nodes in the vicinity of the injection site can be accessed and participate in T cell activation. In comparison, the IV administration of mRNA to secondary lymphoid organs such as the spleen—as the largest agglomeration of immune cells—could be more potent, potentially holding significant therapeutic potential. As an example, Kranz et al. administered negatively charged mRNA-LPX vaccines in mice resulting in strong T cell stimulation, particularly in the spleen, resulting in tumor shrinkage and improved survival.121 Similarly, in a Phase I clinical trial (NCT02410733), bioimaging studies highlighted the rapid targeting and transient activation of lymphoid-tissue-resident immune cells in the spleen upon IV injection of mRNA-LPXs.190 Melanoma patients were later vaccinated with the same formulation used by Kranz et al., encoding for tumor-associated antigens prevalent in melanoma patients. Interim analysis showed that the vaccine, alone or in combination with PD1 antibody, mediated the induction of strong CD4+ and CD8+ T cell immunity against vaccine antigens, promoting stable disease status. A Phase II study is currently ongoing for the same application (NCT04526899). Similar approaches using other tumor-specific antigens are being evaluated in various Phase I and I/II clinical trials in patients with triple-negative breast cancer (TNBC; NCT02316457), ovarian cancer (NCT04163094), and prostate cancer (NCT04382898).In an opposite approach, mRNA-based therapeutics can be used to downregulate autoimmune responses by expressing anti-inflammatory proteins. Inflammatory bowel disease (IBD) is characterized by elevated and sustained pro-inflammatory expression in the colon and, as such, its treatment and management would benefit from an effective immunosuppressive strategy. To evaluate this, the targeted delivery of mRNA encoding the anti-inflammatory cytokine IL-10 to activated leukocytes (Ly6c+) was conducted in a mouse model of IBD, resulting in a pronounced reduction of proinflammatory cytokines TNFα and IL6 in the colon, when compared to the untreated group.238 Pathological features including inflammatory infiltration of the colon and erythema were significantly reduced, highlighting the therapeutic potential of mRNA as an immunomodulatory approach for inflammation-related diseases.
5.4. Other organs
Lymphedema refers to tissue swelling caused by an accumulation of extracellular fluid normally drained through the body's lymphatic system. Current treatment approaches such as manual lymph drainage and compression therapy have significant limitations and provide only symptomatic relief.239 Studies in a murine model of secondary lymphedema demonstrated that the administration of VEGF mRNA-LNPs effectively induced the growth of lymphatic vessels.185 Consequently, the drainage of extracellular fluid was enhanced, and reduced paw thickness and fibroadipose area were observed. Although more work is needed to investigate the long-term effects of this treatment, mRNA therapy may represent a feasible therapeutic alternative to current approaches.While most mRNA-based therapeutics use nanocarriers to protect the nucleic acids from endonuclease-mediated degradation, an ongoing clinical program is investigating the use of naked mRNA (candidate AZD8601) encoding for VEGF directly injected into the myocardium of patients undergoing elective coronary artery bypass surgery (CABG) surgery to promote the formation of new blood vessels. This study asserts that the direct administration of VEGF-A mRNA to the heart could promote cardioprotective effects and reduce myocardial damage following heart infarction.220 AZD8601 has been investigated for safety and tolerability in patients with impaired systolic function undergoing coronary artery bypass grafting. At present, Phase II trials have been completed, rendering this the most advanced clinical study of mRNA-based therapeutics conducted by Moderna.
5.5. Gene editing
Gene editing technologies could represent an outstanding advancement in the treatment of many genetic disorders through the knock-out or knock-in of a targeted gene with the substantial advantage of an infrequent administration regimen. The mRNA-mediated expression of gene-editing endonucleases is very attractive, as this strategy induces transient and limited expression of endonucleases, thus reducing the probability of off-target genome editing and unwanted effects.240 Various approaches for this have been explored including CRISPR/Cas9-based methodologies, as well as ZFNs. In vivo gene editing using nanoparticles has also been evaluated in different organs including the liver,241–244 the lungs (by local150 and IV administration),122,172,173 the muscles,172 and the spleen.122 An initial study demonstrated successful homology directed-repair (HDR) mediated by an mRNA-encoded ZFN in the lungs of a transgenic mouse model of SP-B deficiency upon IT administration.150 The HDR rate was found to be ∼9%, which in turn prevented the decline of lung function, lung edema, and neutrophilia, all of which were observed in the untreated group. Another study delivering a combination of Cas9 mRNA and sgRNA in a single formulation achieved an editing rate of ∼6% of phosphatase and tensin homolog (PTEN) in lung tissues upon IV administration in wild type C57BL/6 mice. While there are limited works describing gene editing in the lungs, HDR-based gene editing holds great promise for the treatment of genetic lung disorders and will doubtless be explored in future works. Further, HDR-mediated gene editing in vivo has also been shown to be feasible in tumor tissues. The intratumoral injection of LNPs containing Cas9 mRNA, sgRNA, and an ssDNA template was able to achieve HDR editing of up to 23% in tumor tissues.245 Studies investigating gene knockout in the liver are currently under clinical evaluation, showing potent gene silencing of the hepatic protein TTR. Results from a Phase I study in 6 patients demonstrated a dose-dependent response, with the highest dose (0.3 mg kg−1 body weight) achieving a ∼90% reduction of protein, with only mild adverse events.2466. Conclusions
mRNA therapeutics show great potential for the treatment of a multitude of disorders. Nevertheless, important challenges faced in the development of mRNA drugs have been the effective delivery to target cells and the proportion of transfected cells capable of achieving a therapeutic degree of protein expression. mRNA sequences can be engineered to provide increased protein expression and specific localization, but this approach remains insufficiently explored, warranting the further development of safe and efficient nanocarriers for in vivo delivery. In this review, we focused on the biodistribution of both lipid- and polymer-based nanocarriers and their associated parameters which led to preferential biodistribution and expression in the main organs for which mRNA-based drugs hold promise.mRNA delivery has mostly been conducted using passive targeting by modulating the physicochemical properties of the nanocarriers. One predictive physicochemical parameter required for effective in vivo mRNA delivery that has been identified is the surface pKa of nanocarriers. In particular, LNPs with a surface pKas of 6–7 yielded liver-selective mRNA delivery, while achieving high transfection efficiency in a range of hepatic cell subtypes. Transfection amongst these cells can also be modulated using modified cholesterol derivatives. Spleen-specific mRNA delivery has been shown to be feasible through the incorporation of negatively charged phospholipids into LNP formulations, which in turn results in altered surface pKa values below 6. Additionally, optimization of the structure of the ionizable lipids used in LNPs has been demonstrated as an effective approach to favor mRNA delivery to the spleen. In contrast, lung-specific mRNA delivery can be achieved through the introduction of positively charged lipids into LNPs, generating surface pKa values above 7. While surface pKa appears to be a potential predictive parameter for the biodistribution of mRNA nanocarriers, other physicochemical properties of the nanocarriers can influence this value in ways that remain unclear. Future works should attempt to evaluate the relationship between pKa and the surface charge of the nanoparticle, as this is not currently well-understood. Though it seems that liver-targeting mRNA formulations do not require active targeting for efficient delivery, this strategy may prove valuable for cell-specific mRNA delivery in the lungs and spleen. A recent study utilized CD5-targeted LNPs to deliver mRNA encoding a CAR binding to fibroblast activation protein to T cells in vivo.247 The production of transient effective CAR T cells reduced fibrosis and restored cardiac function after injury, indicating that this targeted platform may be a feasible approach for the generation of CAR T cells in vivo. Although these targeting strategies can mediate organ-specific mRNA delivery, future work is required to determine the full relationship between mRNA biodistribution and subsequent mRNA-induced protein expression. A thorough understanding of this phenomenon will provide meaningful insights into the interactions between nanoparticles and cells and may give rise to new opportunities for improved cell-targeted strategies.
Though less clinically advanced, polymer nanocarriers have also been shown to promote substantial mRNA expression in various tissues, but have not yet reached the stage of clinical investigation. On the other hand, several clinical studies have been initiated using LNPs, mainly as a strategy for the treatment of genetic liver diseases. Clinical investigations of mRNA-based protein supplementation therapies in other organs have been less thoroughly explored, with the only studies in progress being limited to local administration to the lungs and heart. A notable disadvantage of mRNA-based treatments is the requirement for repeated dosing which, while demonstrated to be safe in preclinical models, remains an impediment in comparison to DNA-based therapeutics or gene-editing approaches with infrequent administration regimens. Indeed, gene-editing approaches have yielded promising results for gene silencing, though knock-in editing for protein replacement therapy remains far less explored in preclinical models, owing to safety concerns regarding unwanted genetic modifications. Accordingly, mRNA-induced protein replacement approaches yielding prolonged expression over time, while generating fewer safety concerns than gene editing, are a research area of utmost importance. Another strategy for extended protein expression, without the potential risk of insertional mutagenesis, is the delivery of episomal pDNA, which remains in the nucleus for a long period as an extrachromosomal element.248,249 Though this approach has mostly been studied using viral delivery systems, non-viral delivery vectors could also easily be applied.
It is our belief that the foundational works described in this review will continue to be built upon to further enhance the efficacy and tolerability of mRNA nanocarriers, unlocking effective treatments for various Mendelian diseases. Concomitant research on the elucidation of the interactions between surface pKa, surface charge, and their impact on the protein corona formed in circulation, as well as on nanoparticle-cell interactions, can be leveraged to optimize the targeting of diverse cell types and are key factors to be considered in the screening process for nanoformulations. Based on this, we envision that future investigations evaluating the interconnection of these parameters will serve to increase efficacy, while reducing side-effects, for next generation nanoparticle-based mRNA gene therapies.
Author contributions
Matthias Zadory: conceptualization, literature collection and analysis, article writing. Elliot Lopez: figures and article writing. Samuel Babity: manuscript revision and editing. Simon-Pierre Gravel: manuscript revision and editing. Davide Brambilla: conceptualization, supervision, manuscript revision.Conflicts of interest
“There are no conflicts to declare”.Acknowledgements
This work was supported by grants from the Servier foundation of the Faculté de Pharmacie de l'Université de Montréal, the Natural Sciences and Engineering Research Council of Canada, the Canadian Generic Pharmaceutical Association and Biosimilars Canada and funding from the Canada First Research Excellence Fund through the TransMedTech Institute.References
- A. L. Hopkins and C. R. Groom, The druggable genome, Nat. Rev. Drug Discovery, 2002, 1(9), 727–730 CrossRef CAS PubMed
.
- K. D. Warner, C. E. Hajdin and K. M. Weeks, Principles for targeting RNA with drug-like small molecules, Nat. Rev. Drug Discovery, 2018, 17(8), 547–558 CrossRef CAS PubMed
- G. F. Jirikowski, P. P. Sanna, D. Maciejewski-Lenoir and F. E. Bloom, Reversal of diabetes insipidus in Brattleboro rats: intrahypothalamic injection of vasopressin mRNA, Science, 1992, 255(5047), 996–998 CrossRef CAS
- J. A. Wolff, R. W. Malone, P. Williams, W. Chong, G. Acsadi and A. Jani,
et al., Direct Gene-Transfer into Mouse Muscle Invivo, Science, 1990, 247(4949), 1465–1468 CrossRef CAS PubMed
- S. F. Dowdy, Overcoming cellular barriers for RNA therapeutics, Nat. Biotechnol., 2017, 35(3), 222–229 CrossRef CAS
- E. Dolgin, The tangled history of mRNA vaccines, Nature, 2021, 597(7876), 318–324 CrossRef CAS PubMed
- X. Hou, T. Zaks, R. Langer and Y. Dong, Lipid nanoparticles for mRNA delivery, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed
- R. W. Malone, P. L. Felgner and I. M. Verma, Cationic liposome-mediated RNA transfection, Proc. Natl. Acad. Sci. U. S. A., 1989, 86(16), 6077–6081 CrossRef CAS PubMed
- F. Martinon, S. Krishnan, G. Lenzen, R. Magne, E. Gomard and J. G. Guillet,
et al., Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA, Eur. J. Immunol., 1993, 23(7), 1719–1722 CrossRef CAS PubMed
- K. A. Hajj and K. A. Whitehead, Tools for translation: non-viral materials for therapeutic mRNA delivery, Nat. Rev. Mater., 2017, 2(10), 17056 CrossRef CAS
- K. Paunovska, D. Loughrey and J. E. Dahlman, Drug delivery systems for RNA therapeutics, Nat. Rev. Genet., 2022, 23, 265–280 CrossRef CAS PubMed
- A. Wadhwa, A. Aljabbari, A. Lokras, C. Foged and A. Thakur, Opportunities and Challenges in the Delivery of mRNA-based Vaccines, Pharmaceutics, 2020, 12(2) DOI:10.3390/pharmaceutics12020102
- L. Schoenmaker, D. Witzigmann, J. A. Kulkarni, R. Verbeke, G. Kersten and W. Jiskoot,
et al., mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability, Int. J. Pharm., 2021, 601, 120586 CrossRef CAS
- U. Sahin, K. Kariko and O. Tureci, mRNA-based therapeutics–developing a new class of drugs, Nat. Rev. Drug Discovery, 2014, 13(10), 759–780 CrossRef CAS PubMed
- C. H. June, R. S. O'Connor, O. U. Kawalekar, S. Ghassemi and M. C. Milone, CAR T cell immunotherapy for human cancer, Science, 2018, 359(6382), 1361–1365 CrossRef CAS PubMed
- I. Sahu, A. Haque, B. Weidensee, P. Weinmann and M. S. D. Kormann, Recent Developments in mRNA-Based Protein Supplementation Therapy to Target Lung Diseases, Mol. Ther., 2019, 27(4), 803–823 CrossRef CAS PubMed
- H. Tourriere, K. Chebli and J. Tazi, mRNA degradation machines in eukaryotic cells, Biochimie, 2002, 84(8), 821–837 CrossRef CAS PubMed
- K. Kariko, H. Muramatsu, F. A. Welsh, J. Ludwig, H. Kato and S. Akira,
et al., Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector With Increased Translational Capacity and Biological Stability, Mol. Ther., 2008, 16(11), 1833–1840 CrossRef CAS PubMed
- K. Kariko, M. Buckstein, H. Ni and D. Weissman, Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA, Immunity, 2005, 23(2), 165–175 CrossRef CAS PubMed
- Y. Li and M. Kiledjian, Regulation of mRNA decapping, Wiley Interdiscip. Rev.: RNA, 2010, 1(2), 253–265 CrossRef CAS
- M. Mockey, C. Goncalves, F. P. Dupuy, F. M. Lemoine, C. Pichon and P. Midoux, mRNA transfection of dendritic cells: synergistic effect of ARCA mRNA capping with Poly(A) chains in cis and in trans for a high protein expression level, Biochem. Biophys. Res. Commun., 2006, 340(4), 1062–1068 CrossRef CAS
- P. M. Rabinovich, M. E. Komarovskaya, Z. J. Ye, C. Imai, D. Campana and E. Bahceci,
et al., Synthetic messenger RNA as a tool for gene therapy, Hum. Gene Ther., 2006, 17(10), 1027–1035 CrossRef CAS PubMed
- E. Grudzien-Nogalska, J. Jemielity, J. Kowalska, E. Darzynkiewicz and R. E. Rhoads, Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells, RNA, 2007, 13(10), 1745–1755 CrossRef CAS PubMed
- J. Kowalska, M. Lewdorowicz, J. Zuberek, E. Grudzien-Nogalska, E. Bojarska and J. Stepinski,
et al., Synthesis and characterization of mRNA cap analogs containing phosphorothioate substitutions that bind tightly to eIF4E and are resistant to the decapping pyrophosphatase DcpS, RNA, 2008, 14(6), 1119–1131 CrossRef CAS PubMed
- D. R. Gallie, The cap and poly(A) tail function synergistically to regulate mRNA translational efficiency, Genes Dev., 1991, 5(11), 2108–2116 CrossRef CAS PubMed
- M. Ikegami, S. Uchida, H. Uchida, K. Nagata, K. Kataoka and K. Itaka, Enhanced Transfection of mRNA Transcribed From Elongated-poly(A/T) Tail-Containing Vector, Mol. Ther., 2014, 22, S43 Search PubMed
- P. Arbuthnot, A. Ely and K. Bloom, A convenient method to generate and maintain poly(A)-encoding DNA sequences required for in vitro transcription of mRNA, BioTechniques, 2019, 66(1), 37–39 CrossRef CAS
- C. Zeng, X. Hou, J. Yan, C. Zhang, W. Li and W. Zhao,
et al., Leveraging mRNA Sequences and Nanoparticles to Deliver SARS-CoV-2 Antigens In Vivo, Adv. Mater., 2020, 32(40), e2004452 CrossRef PubMed
- G. Tushev, C. Glock, M. Heumuller, A. Biever, M. Jovanovic and E. M. Schuman, Alternative 3′ UTRs Modify the Localization, Regulatory Potential, Stability, and Plasticity of mRNAs in Neuronal Compartments, Neuron, 2018, 98(3), 495–511 CrossRef CAS PubMed
- L. Lu, F. Zhang, Y. Li, A. Yang, C. Guan and X. Ding,
et al., Dendritic targeted mRNA expression via a cis-acting RNA UTR element, Biochem. Biophys. Res. Commun., 2019, 509(2), 402–406 CrossRef CAS PubMed
- M. A. Valencia-Sanchez, J. Liu, G. J. Hannon and R. Parker, Control of translation and mRNA degradation by miRNAs and siRNAs, Genes Dev., 2006, 20(5), 515–524 CrossRef CAS PubMed
- B. D. Brown, B. Gentner, A. Cantore, S. Colleoni, M. Amendola and A. Zingale,
et al., Endogenous microRNA can be broadly exploited to regulate transgene expression according to tissue, lineage and differentiation state, Nat. Biotechnol., 2007, 25(12), 1457–1467 CrossRef CAS PubMed
- C. Coulouarn, V. M. Factor, J. B. Andersen, M. E. Durkin and S. S. Thorgeirsson, Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties, Oncogene, 2009, 28(40), 3526–3536 CrossRef CAS PubMed
- R. Jain, J. P. Frederick, E. Y. Huang, K. E. Burke, D. M. Mauger and E. A. Andrianova,
et al., MicroRNAs Enable mRNA Therapeutics to Selectively Program Cancer Cells to Self-Destruct, Nucleic Acid Ther., 2018, 28(5), 285–296 CrossRef CAS PubMed
- X. Mu and S. Hur, Immunogenicity of In Vitro-Transcribed RNA, Acc. Chem. Res., 2021, 54(21), 4012–4023 CrossRef CAS PubMed
- K. J. Kauffman, F. F. Mir, S. Jhunjhunwala, J. C. Kaczmarek, J. E. Hurtado and J. H. Yang,
et al., Efficacy and immunogenicity of unmodified and pseudouridine-modified mRNA delivered systemically with lipid nanoparticles in vivo, Biomaterials, 2016, 109, 78–87 CrossRef CAS PubMed
- J. R. Melamed, K. A. Hajj, N. Chaudhary, D. Strelkova, M. L. Arral and N. Pardi,
et al., Lipid nanoparticle chemistry determines how nucleoside base modifications alter mRNA delivery, J. Controlled Release, 2022, 341, 206–214 CrossRef CAS PubMed
- P. S. Kowalski, A. Rudra, L. Miao and D. G. Anderson, Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery, Mol. Ther., 2019, 27(4), 710–728 CrossRef CAS PubMed
- C. Lorenz, M. Fotin-Mleczek, G. Roth, C. Becker, T. C. Dam and W. P. Verdurmen,
et al., Protein expression from exogenous mRNA: uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway, RNA Biol., 2011, 8(4), 627–636 CrossRef CAS PubMed
- M. Diken, S. Kreiter, A. Selmi, C. M. Britten, C. Huber and O. Tureci,
et al., Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation, Gene Ther., 2011, 18(7), 702–708 CrossRef CAS PubMed
- B. Dalby, S. Cates, A. Harris, E. C. Ohki, M. L. Tilkins and P. J. Price,
et al., Advanced transfection with Lipofectamine 2000 reagent: primary neurons, siRNA, and high-throughput applications, Methods, 2004, 33(2), 95–103 CrossRef CAS
- Y. Sato, H. Hatakeyama, Y. Sakurai, M. Hyodo, H. Akita and H. Harashima, A pH-sensitive cationic lipid facilitates the delivery of liposomal siRNA and gene silencing activity in vitro and in vivo, J. Controlled Release, 2012, 163(3), 267–276 CrossRef CAS
- Y. Zhang, C. Sun, C. Wang, K. E. Jankovic and Y. Dong, Lipids and Lipid Derivatives for RNA Delivery, Chem. Rev., 2021, 121(20), 12181–12277 CrossRef CAS
- H. Li, T. Lee, T. Dziubla, F. M. Pi, S. J. Guo and J. Xu,
et al., RNA as a stable polymer to build controllable and defined nanostructures for material and biomedical applications, Nano Today, 2015, 10(5), 631–655 CrossRef CAS PubMed
- O. S. Fenton, K. J. Kauffman, J. C. Kaczmarek, R. L. McClellan, S. Jhunjhunwala and M. W. Tibbitt,
et al., Synthesis and Biological Evaluation of Ionizable Lipid Materials for the In Vivo Delivery of Messenger RNA to B Lymphocytes, Adv. Mater., 2017, 29(33), 1606944 CrossRef
- X. Cheng and R. J. Lee, The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery, Adv. Drug Delivery Rev., 2016, 99(Pt A), 129–137 CrossRef CAS PubMed
- K. J. Kauffman, J. R. Dorkin, J. H. Yang, M. W. Heartlein, F. DeRosa and F. F. Mir,
et al., Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs, Nano Lett., 2015, 15(11), 7300–7306 CrossRef CAS PubMed
- Q. Cheng, T. Wei, Y. Jia, L. Farbiak, K. Zhou and S. Zhang,
et al., Dendrimer-Based Lipid Nanoparticles Deliver Therapeutic FAH mRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I, Adv. Mater., 2018, 30(52), e1805308 CrossRef PubMed
- W. V. Rodrigueza, J. J. Wheeler, S. K. Klimuk, C. N. Kitson and M. J. Hope, Transbilayer Movement and Net Flux of Cholesterol and Cholesterol Sulfate between Liposomal Membranes, Biochemistry, 1995, 34(18), 6208–6217 CrossRef CAS
- Y. Sato, N. Okabe, Y. Note, K. Hashiba, M. Maeki and M. Tokeshi,
et al., Hydrophobic scaffolds of pH-sensitive cationic lipids contribute to miscibility with phospholipids and improve the efficiency of delivering short interfering RNA by small-sized lipid nanoparticles, Acta Biomater., 2020, 102, 341–350 CrossRef CAS PubMed
- M. Herrera, J. Kim, Y. Eygeris, A. Jozic and G. Sahay, Illuminating endosomal escape of polymorphic lipid nanoparticles that boost mRNA delivery, Biomater. Sci., 2021, 9(12), 4289–4300 RSC
- S. Patel, N. Ashwanikumar, E. Robinson, Y. Xia, C. Mihai and J.3 Griffith,
et al., Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA, Nat. Commun., 2020, 11(1), 983 CrossRef CAS PubMed
- Y. Eygeris, S. Patel, A. Jozic and G. Sahay, Deconvoluting Lipid Nanoparticle Structure for Messenger RNA Delivery, Nano Lett., 2020, 20(6), 4543–4549 CrossRef CAS
- H. Hatakeyama, H. Akita and H. Harashima, The polyethyleneglycol dilemma: advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors, Biol. Pharm. Bull., 2013, 36(6), 892–899 CrossRef CAS
- I. Vlatkovic, Non-Immunotherapy Application of LNP-mRNA: Maximizing Efficacy and Safety, Biomedicines, 2021, 9(5), 530 CrossRef CAS
- C. D. Sago, M. P. Lokugamage, G. N. Lando, N. Djeddar, N. N. Shah and C. Syed,
et al., Modifying a Commonly Expressed Endocytic Receptor Retargets Nanoparticles in Vivo, Nano Lett., 2018, 18(12), 7590–7600 CrossRef CAS PubMed
- C. D. Sago, B. R. Krupczak, M. P. Lokugamage, Z. Gan and J. E. Dahlman, Cell Subtypes Within the Liver Microenvironment Differentially Interact with Lipid Nanoparticles, Cell. Mol. Bioeng., 2019, 12(5), 389–397 CrossRef PubMed
- S. K. Asrani, H. Devarbhavi, J. Eaton and P. S. Kamath, Burden of liver diseases in the world, J. Hepatol., 2019, 70(1), 151–171 CrossRef PubMed
- O. A. Almazroo, M. K. Miah and R. Venkataramanan, Drug Metabolism in the Liver, Clin. Liver Dis., 2017, 21(1), 1–20 CrossRef PubMed
- F. S. Wang, J. G. Fan, Z. Zhang, B. Gao and H. Y. Wang, The Global Burden of Liver Disease: The Major Impact of China, Hepatology, 2014, 60(6), 2099–2108 CrossRef
- D. Witzigmann, J. A. Kulkarni, J. Leung, S. Chen, P. R. Cullis and R. van der Meel, Lipid nanoparticle technology for therapeutic gene regulation in the liver, Adv. Drug Delivery Rev., 2020, 159, 344–363 CrossRef CAS PubMed
- G. Gonzalez-Aseguinolaza and J. Prieto, Gene therapy of liver diseases: a 2011 perspective, Clin. Res. Hepatol. Gastroenterol., 2011, 35(11), 699–708 CrossRef CAS PubMed
- N. Pardi, A. J. Secreto, X. Shan, F. Debonera, J. Glover and Y. Yi,
et al., Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge, Nat. Commun., 2017, 8, 14630 CrossRef PubMed
- Y. Rybakova, P. S. Kowalski, Y. Huang, J. T. Gonzalez, M. W. Heartlein and F. DeRosa,
et al., mRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo, Mol. Ther., 2019, 27(8), 1415–1423 CrossRef CAS PubMed
- M. Thran, J. Mukherjee, M. Ponisch, K. Fiedler, A. Thess and B. L. Mui,
et al., mRNA mediates passive vaccination against infectious agents, toxins, and tumors, EMBO Mol. Med., 2017, 9(10), 1434–1447 CrossRef CAS
- C. R. Stadler, H. Bahr-Mahmud, L. Celik, B. Hebich, A. S. Roth and R. P. Roth,
et al., Elimination of large tumors in mice by mRNA-encoded bispecific antibodies, Nat. Med., 2017, 23(7), 815–817 CrossRef CAS
- K. M. Tsoi, S. A. MacParland, X. Z. Ma, V. N. Spetzler, J. Echeverri and B. Ouyang,
et al., Mechanism of hard-nanomaterial clearance by the liver, Nat. Mater., 2016, 15(11), 1212–1221 CrossRef CAS PubMed
- A. J. Debacker, J. Voutila, M. Catley, D. Blakey and N. Habib, Delivery of Oligonucleotides to the Liver with GalNAc: From Research to Registered Therapeutic Drug, Mol. Ther., 2020, 28(8), 1759–1771 CrossRef CAS PubMed
- J. K. Nair, J. L. Willoughby, A. Chan, K. Charisse, M. R. Alam and Q. Wang,
et al., Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing, J. Am. Chem. Soc., 2014, 136(49), 16958–16961 CrossRef CAS
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt and P. R. Cullis,
et al., The current landscape of nucleic acid therapeutics, Nat. Nanotechnol., 2021, 16(6), 630–643 CrossRef CAS PubMed
- Y. Sato, H. Matsui, N. Yamamoto, R. Sato, T. Munakata and M. Kohara,
et al., Highly specific delivery of siRNA to hepatocytes circumvents endothelial cell-mediated lipid nanoparticle-associated toxicity leading to the safe and efficacious decrease in the hepatitis B virus, J. Controlled Release, 2017, 266, 216–225 CrossRef CAS PubMed
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman and S. Barros,
et al., The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs, Nat. Nanotechnol., 2019, 14(12), 1084–1087 CrossRef CAS
- Y. Dong, J. R. Dorkin, W. Wang, P. H. Chang, M. J. Webber and B. C. Tang,
et al., Poly(glycoamidoamine) Brushes Formulated Nanomaterials for Systemic siRNA and mRNA Delivery in Vivo, Nano Lett., 2016, 16(2), 842–848 CrossRef CAS
- S. T. Crowley, J. A. Poliskey, N. J. Baumhover and K. G. Rice, Efficient expression of stabilized mRNA PEG-peptide polyplexes in liver, Gene Ther., 2015, 22(12), 993–999 CrossRef CAS PubMed
- M. G. Prieve, P. Harvie, S. D. Monahan, D. Roy, A. G. Li and T. L. Blevins,
et al., Targeted mRNA Therapy for Ornithine Transcarbamylase Deficiency, Mol. Ther., 2018, 26(3), 801–813 CrossRef CAS
- A. Akinc, W. Querbes, S. M. De, J. Qin, M. Frank-Kamenetsky and K. N. Jayaprakash,
et al., Targeted Delivery of RNAi Therapeutics With Endogenous and Exogenous Ligand-Based Mechanisms, Mol. Ther., 2010, 18(7), 1357–1364 CrossRef CAS PubMed
- Y. Z. Dong, K. T. Love, J. R. Dorkin, S. Sirirungruang, Y. L. Zhang and D. L. Chen,
et al., Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates (vol 111, pg 3955, 2014), Proc. Natl. Acad. Sci. U. S. A., 2014, 111(15), 5753 Search PubMed
- K. T. Love, K. P. Mahon, C. G. Levins, K. A. Whitehead, W. Querbes and J. R. Dorkin,
et al., Lipid-like materials for low-dose, in vivo gene silencing, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(5), 1864–1869 CrossRef CAS
- P. R. Cullis and M. J. Hope, Lipid Nanoparticle Systems for Enabling Gene Therapies, Mol. Ther., 2017, 25(7), 1467–1475 CrossRef CAS PubMed
- A. Thess, S. Grund, B. L. Mui, M. J. Hope, P. Baumhof and M. Fotin-Mleczek,
et al., Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals, Mol. Ther., 2015, 23(9), 1456–1464 CrossRef CAS PubMed
- M. Sedic, J. J. Senn, A. Lynn, M. Laska, M. Smith and S. J. Platz,
et al., Safety Evaluation of Lipid Nanoparticle-Formulated Modified mRNA in the Sprague-Dawley Rat and Cynomolgus Monkey, Vet. Pathol., 2018, 55(2), 341–354 CrossRef CAS
- H. Yin, C. Q. Song, S. Suresh, Q. Wu, S. Walsh and L. H. Rhym,
et al., Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing, Nat. Biotechnol., 2017, 35(12), 1179–1187 CrossRef CAS PubMed
- K. A. Whitehead, J. R. Dorkin, A. J. Vegas, P. H. Chang, O. Veiseh and J. Matthews,
et al., Degradable lipid nanoparticles with predictable in vivo siRNA delivery activity, Nat. Commun., 2014, 5, 4277 CrossRef CAS PubMed
- J. Zhang, H. Fan, D. A. Levorse and L. S. Crocker, Ionization behavior of amino lipids for siRNA delivery: determination of ionization constants, SAR, and the impact of lipid pKa on cationic lipid-biomembrane interactions, Langmuir, 2011, 27(5), 1907–1914 CrossRef CAS PubMed
- J. Heyes, L. Palmer, K. Bremner and I. MacLachlan, Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids, J. Controlled Release, 2005, 107(2), 276–287 CrossRef CAS PubMed
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen and X. Du,
et al., Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo, Angew. Chem., Int. Ed., 2012, 51(34), 8529–8533 CrossRef CAS PubMed
- L. Jiang, P. Berraondo, D. Jerico, L. T. Guey, A. Sampedro and A. Frassetto,
et al., Systemic messenger RNA as an etiological treatment for acute intermittent porphyria, Nat. Med., 2018, 24(12), 1899–1909 CrossRef CAS PubMed
- D. An, J. L. Schneller, A. Frassetto, S. Liang, X. L. Zhu and J. S. Park,
et al., Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia, Cell Rep., 2017, 21(12), 3548–3558 CrossRef CAS PubMed
- S. Sabnis, E. S. Kumarasinghe, T. Salerno, C. Mihai, T. Ketova and J. J. Senn,
et al., A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates, Mol. Ther., 2018, 26(6), 1509–1519 CrossRef CAS PubMed
- C. Delehedde, L. Even, P. Midoux, C. Pichon and F. Perche, Intracellular Routing and Recognition of Lipid-Based mRNA Nanoparticles, Pharmaceutics, 2021, 13(7) DOI:10.3390/pharmaceutics13070945
- L. Miao, J. Lin, Y. Huang, L. Li, D. Delcassian and Y. Ge,
et al., Synergistic lipid compositions for albumin receptor mediated delivery of mRNA to the liver, Nat. Commun., 2020, 11(1), 2424 CrossRef CAS PubMed
- K. Zhou, L. H. Nguyen, J. B. Miller, Y. Yan, P. Kos and H. Xiong,
et al., Modular degradable dendrimers enable small RNAs to extend survival in an aggressive liver cancer model, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(3), 520–525 CrossRef CAS PubMed
- K. A. Hajj, R. L. Ball, S. B. Deluty, S. R. Singh, D. Strelkova and C. M. Knapp,
et al., Branched-Tail Lipid Nanoparticles Potently Deliver mRNA In Vivo due to Enhanced Ionization at Endosomal pH, Small, 2019, 15(6), 1805097 CrossRef PubMed
- K. A. Hajj, J. R. Melamed, N. Chaudhary, N. G. Lamson, R. L. Ball and S. S. Yerneni,
et al., A Potent Branched-Tail Lipid Nanoparticle Enables Multiplexed mRNA Delivery and Gene Editing In Vivo, Nano Lett., 2020, 20(7), 5167–5175 CrossRef CAS PubMed
- R. D. Harvey, N. Ara, R. K. Heenan, D. J. Barlow, P. J. Quinn and M. J. Lawrence, Stabilization of distearoylphosphatidylcholine lamellar phases in propylene glycol using cholesterol, Mol. Pharm., 2013, 10(12), 4408–4417 CrossRef CAS PubMed
- R. Zhang, R. El-Mayta, T. J. Murdoch, C. C. Warzecha, M. M. Billingsley and S. J. Shepherd,
et al., Helper lipid structure influences protein adsorption and delivery of lipid nanoparticles to spleen and liver, Biomater. Sci., 2021, 9(4), 1449–1463 RSC
- E. Ikonen, Cellular cholesterol trafficking and compartmentalization, Nat. Rev. Mol. Cell Biol., 2008, 9(2), 125–138 CrossRef CAS PubMed
- K. Paunovska, C. J. Gil, M. P. Lokugamage, C. D. Sago, M. Sato and G. N. Lando,
et al., Analyzing 2000 in Vivo Drug Delivery Data Points Reveals Cholesterol Structure Impacts Nanoparticle Delivery, ACS Nano, 2018, 12(8), 8341–8349 CrossRef CAS PubMed
- J. Poisson, S. Lemoinne, C. Boulanger, F. Durand, R. Moreau and D. Valla,
et al., Liver sinusoidal endothelial cells: Physiology and role in liver diseases, J. Hepatol., 2017, 66(1), 212–227 CrossRef CAS PubMed
- G. Kolios, V. Valatas and E. Kouroumalis, Role of Kupffer cells in the pathogenesis of liver disease, World J. Gastroenterol., 2006, 12(46), 7413–7420 CrossRef CAS PubMed
- P. Harvie, F. M. Wong and M. B. Bally, Use of poly(ethylene glycol)-lipid conjugates to regulate the surface attributes and transfection activity of lipid-DNA particles, J. Pharm. Sci., 2000, 89(5), 652–663 CrossRef CAS
- S. Chen, Y. Y. C. Tam, P. J. C. Lin, M. M. H. Sung, Y. K. Tam and P. R. Cullis, Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA, J. Controlled Release, 2016, 235, 236–244 CrossRef CAS PubMed
- M. Zhao, M. Li, Z. Zhang, T. Gong and X. Sun, Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA, Drug Delivery, 2016, 23(7), 2596–2607 CrossRef CAS PubMed
- M. Li, M. Zhao, Y. Fu, Y. Li, T. Gong and Z. Zhang,
et al., Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra- and paracellular pathways, J. Controlled Release, 2016, 228, 9–19 CrossRef CAS PubMed
- A. A. Eltoukhy, D. Chen, C. A. Alabi, R. Langer and D. G. Anderson, Degradable terpolymers with alkyl side chains demonstrate enhanced gene delivery potency and nanoparticle stability, Adv. Mater., 2013, 25(10), 1487–1493 CrossRef CAS
- K. Hadinoto, A. Sundaresan and W. S. Cheow, Lipid-polymer hybrid nanoparticles as a new generation therapeutic delivery platform: a review, Eur. J. Pharm. Biopharm., 2013, 85(3 Pt A), 427–443 CrossRef CAS PubMed
- J. C. Kaczmarek, A. K. Patel, L. H. Rhym, U. C. Palmiero, B. Bhat and M. W. Heartlein,
et al., Systemic delivery of mRNA and DNA to the lung using polymer-lipid nanoparticles, Biomaterials, 2021, 275, 120966 CrossRef CAS PubMed
- J. C. Kaczmarek, A. K. Patel, K. J. Kauffman, O. S. Fenton, M. J. Webber and M. W. Heartlein,
et al., Polymer-Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs, Angew. Chem., Int. Ed., 2016, 55(44), 13808–13812 CrossRef CAS PubMed
- C. Schumann, D. X. Nguyen, M. Norgard, Y. Bortnyak, T. Korzun and S. Chan,
et al., Increasing lean muscle mass in mice via nanoparticle-mediated hepatic delivery of follistatin mRNA, Theranostics, 2018, 8(19), 5276–5288 CrossRef CAS PubMed
- A. Matsui, S. Uchida, T. Ishii, K. Itaka and K. Kataoka, Messenger RNA-based therapeutics for the treatment of apoptosis-associated diseases, Sci. Rep., 2015, 5, 15810 CrossRef CAS
- P. S. Kowalski, U. Capasso Palmiero, Y. Huang, A. Rudra, R. Langer and D. G. Anderson, Ionizable Amino-Polyesters Synthesized via Ring Opening Polymerization of Tertiary Amino-Alcohols for Tissue Selective mRNA Delivery, Adv. Mater., 2018, 1801151 CrossRef
- J. Zhou, J. Liu, C. J. Cheng, T. R. Patel, C. E. Weller and J. M. Piepmeier,
et al., Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery, Nat. Mater., 2011, 11(1), 82–90 CrossRef PubMed
- S. M. Lewis, A. Williams and S. C. Eisenbarth, Structure and function of the immune system in the spleen, Sci. Immunol., 2019, 4(33), 1–12 Search PubMed
- Y. Zhang and Z. Zhang, The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications, Cell. Mol. Immunol., 2020, 17(8), 807–821 CrossRef CAS
- P. Serra and P. Santamaria, Antigen-specific therapeutic approaches for autoimmunity, Nat. Biotechnol., 2019, 37(3), 238–251 CrossRef CAS PubMed
- M. M. Billingsley, N. Singh, P. Ravikumar, R. Zhang, C. H. June and M. J. Mitchell, Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering, Nano Lett., 2020, 20(3), 1578–1589 CrossRef CAS PubMed
- P. Castiglioni, M. Gerloni and M. Zanetti, Genetically programmed B lymphocytes are highly efficient in inducing anti-virus protective immunity mediated by central memory CD8 T cells, Vaccine, 2004, 23(5), 699–708 CrossRef PubMed
- C. J. McKinlay, N. L. Benner, O. A. Haabeth, R. M. Waymouth and P. A. Wender, Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(26), E5859–E5866 CrossRef PubMed
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nanoparticle Uptake: The Phagocyte Problem, Nano Today, 2015, 10(4), 487–510 CrossRef CAS PubMed
- P. W. Bisso, S. Gaglione, P. P. G. Guimaraes, M. J. Mitchell and R. Langer, Nanomaterial Interactions with Human Neutrophils, ACS Biomater. Sci. Eng., 2018, 4(12), 4255–4265 CrossRef CAS
- L. M. Kranz, M. Diken, H. Haas, S. Kreiter, C. Loquai and K. C. Reuter,
et al., Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy, Nature, 2016, 534(7607), 396–401 CrossRef
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing, Nat. Nanotechnol., 2020, 15(4), 313–320 CrossRef CAS
- S. A. Dilliard, Q. Cheng and D. J. Siegwart, On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(52), e2109256118 CrossRef CAS
- E. Alvarez-Benedicto, L. Farbiak, M. Márquez Ramírez, X. Wang, L. T. Johnson and O. Mian,
et al., Optimization of phospholipid chemistry for improved lipid nanoparticle (LNP) delivery of messenger RNA (mRNA), Biomater. Sci., 2022, 10(2), 549–559 RSC
- S. T. LoPresti, M. L. Arral, N. Chaudhary and K. A. Whitehead, The replacement of helper lipids with charged alternatives in lipid nanoparticles facilitates targeted mRNA delivery to the spleen and lungs, J. Controlled Release, 2022, 345, 819–831 CrossRef CAS PubMed
- S. Liu, Q. Cheng, T. Wei, X. Yu, L. T. Johnson and L. Farbiak,
et al., Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR-Cas gene editing, Nat. Mater., 2021, 20(5), 701–710 CrossRef CAS PubMed
- O. S. Fenton, K. J. Kauffman, R. L. McClellan, J. C. Kaczmarek, M. D. Zeng and J. L. Andresen,
et al., Customizable Lipid Nanoparticle Materials for the Delivery of siRNAs and mRNAs, Angew. Chem., Int. Ed., 2018, 57(41), 13582–13586 CrossRef CAS PubMed
- C. D. Sago, M. P. Lokugamage, K. Paunovska, D. A. Vanover, C. M. Monaco and N. N. Shah,
et al., High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(42), E9944–E9952 CrossRef CAS PubMed
- I. Tombacz, D. Laczko, H. Shahnawaz, H. Muramatsu, A. Natesan and A. Yadegari,
et al., Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4 + cell-homing mRNA-LNPs, Mol. Ther., 2021, 29(11), 3293–3304 CrossRef CAS PubMed
- M. Li, S. Li, Y. Huang, H. Chen, S. Zhang and Z. Zhang,
et al., Secreted Expression of mRNA-Encoded Truncated ACE2 Variants for SARS-CoV-2 via Lipid-Like Nanoassemblies, Adv. Mater., 2021, 33(34), e2101707 CrossRef PubMed
- Z. H. Liu, Z. Y. Zhang, C. R. Zhou and Y. P. Jiao, Hydrophobic modifications of cationic polymers for gene delivery, Prog. Polym. Sci., 2010, 35(9), 1144–1162 CrossRef CAS
- X. Yu, S. Liu, Q. Cheng, S. M. Lee, T. Wei and D. Zhang,
et al., Hydrophobic Optimization of Functional Poly(TPAE-co-suberoyl chloride) for Extrahepatic mRNA Delivery following Intravenous Administration, Pharmaceutics, 2021, 13(11), 1–13 CrossRef PubMed
- S. Liu, X. Wang, X. Yu, Q. Cheng, L. T. Johnson and S. Chatterjee,
et al., Zwitterionic Phospholipidation of Cationic Polymers Facilitates Systemic mRNA Delivery to Spleen and Lymph Nodes, J. Am. Chem. Soc., 2021, 143(50), 21321–21330 CrossRef CAS PubMed
- L. M. Caffrey, B. M. deRonde, L. M. Minter and G. N. Tew, Mapping Optimal Charge Density and Length of ROMP-Based PTDMs for siRNA Internalization, Biomacromolecules, 2016, 17(10), 3205–3212 CrossRef CAS PubMed
- B. M. deRonde, J. A. Torres, L. M. Minter and G. N. Tew, Development of Guanidinium-Rich Protein Mimics for Efficient siRNA Delivery into Human T Cells, Biomacromolecules, 2015, 16(10), 3172–3179 CrossRef CAS PubMed
- Collaborators GBDCRD, Prevalence and attributable health burden of chronic respiratory diseases, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017, Lancet Respir. Med., 2020, 8(6), 585–596 CrossRef
- A. A. Aslam, C. Higgins, I. P. Sinha and K. W. Southern, Ataluren and similar compounds (specific therapies for premature termination codon class I mutations) for cystic fibrosis, Cochrane Database Syst. Rev., 2017, 1, CD012040 Search PubMed
- R. H. Pfister and R. F. Soll, New synthetic surfactants: the next generation?, Biol. Neonate, 2005, 87(4), 338–344 CrossRef CAS PubMed
- N. Bertrand and J. C. Leroux, The journey of a drug-carrier in the body: an anatomo-physiological perspective, J. Controlled Release, 2012, 161(2), 152–163 CrossRef CAS PubMed
- T. Wang, J. R. Upponi and V. P. Torchilin, Design of multifunctional non-viral gene vectors to overcome physiological barriers: dilemmas and strategies, Int. J. Pharm., 2012, 427(1), 3–20 CrossRef CAS PubMed
- S. P. Newman, Aerosol deposition considerations in inhalation therapy, Chest, 1985, 88(2 Suppl), 152S–160S CrossRef CAS
- J. J. Wine, The genesis of cystic fibrosis lung disease, J. Clin. Invest., 1999, 103(3), 309–312 CrossRef CAS PubMed
- J. G. Widdicombe, Airway liquid: a barrier to drug diffusion?, Eur. Respir. J., 1997, 10(10), 2194–2197 CrossRef CAS PubMed
- M. Dawson, D. Wirtz and J. Hanes, Enhanced viscoelasticity of human cystic fibrotic sputum correlates with increasing microheterogeneity in particle transport, J. Biol. Chem., 2003, 278(50), 50393–50401 CrossRef CAS PubMed
- N. N. Sanders, S. C. De Smedt, E. Van Rompaey, P. Simoens, F. De Baets and J. Demeester, Cystic fibrosis sputum - A barrier to the transport of nanospheres, Am. J. Respir. Crit. Care, 2000, 162(5), 1905–1911 CrossRef CAS
- J. S. Suk, S. K. Lai, Y. Y. Wang, L. M. Ensign, P. L. Zeitlin and M. P. Boyle,
et al., The penetration of fresh undiluted sputum expectorated by cystic fibrosis patients by non-adhesive polymer nanoparticles, Biomaterials, 2009, 30(13), 2591–2597 CrossRef CAS PubMed
- S. Ferrari, C. Kitson, R. Farley, R. Steel, C. Marriott and D. A. Parkins,
et al., Mucus altering agents as adjuncts for nonviral gene transfer to airway epithelium, Gene Ther., 2001, 8(18), 1380–1386 CrossRef CAS
- N. Sanders, C. Rudolph, K. Braeckmans, S. C. De Smedt and J. Demeester, Extracellular barriers in respiratory gene therapy, Adv. Drug Delivery Rev., 2009, 61(2), 115–127 CrossRef CAS PubMed
- N. Ernst, S. Ulrichskotter, W. A. Schmalix, J. Radler, R. Galneder and E. Mayer,
et al., Interaction of liposomal and polycationic transfection complexes with pulmonary surfactant, J. Gene Med., 1999, 1(5), 331–340 CrossRef CAS
- A. Dewerth, A. Mahiny, L. E. Mays, M. Alkahled, B. Mothes and E. Malaeksefat,
et al., In vivo genome editing using nuclease-encoding mRNA corrects SP-B deficiency, Eur. Respir. J., 2016, 48, 584–586 Search PubMed
- L. M. Nogee, Alterations in SP-B and SP-C expression in neonatal lung disease, Annu. Rev. Physiol., 2004, 66, 601–623 CrossRef CAS
- A. Haque, A. Dewerth, J. S. Antony, J. Riethmuller, G. R. Schweizer and P. Weinmann,
et al., Chemically modified hCFTR mRNAs recuperate lung function in a mouse model of cystic fibrosis, Sci. Rep., 2018, 8(1), 16776 CrossRef PubMed
- A. K. Patel, J. C. Kaczmarek, S. Bose, K. J. Kauffman, F. Mir and M. W. Heartlein,
et al., Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium, Adv. Mater., 2019, 31(8), e1805116 CrossRef
- A. Jarzebinska, T. Pasewald, J. Lambrecht, O. Mykhaylyk, L. Kummerling and P. Beck,
et al., A Single Methylene Group in Oligoalkylamine-Based Cationic Polymers and Lipids Promotes Enhanced mRNA Delivery, Angew. Chem., Int. Ed., 2016, 55(33), 9591–9595 CrossRef CAS PubMed
- S. E. Beck, B. L. Laube, C. I. Barberena, A. C. Fischer, R. J. Adams and K. Chesnut,
et al., Deposition and expression of aerosolized rAAV vectors in the lungs of Rhesus macaques, Mol. Ther., 2002, 6(4), 546–554 CrossRef CAS PubMed
- C. L. Densmore, F. M. Orson, B. Xu, B. M. Kinsey, J. C. Waldrep and P. Hua,
et al., Aerosol delivery of robust polyethyleneimine-DNA complexes for gene therapy and genetic immunization, Mol. Ther., 2000, 1(2), 180–188 CrossRef CAS PubMed
- G. McLachlan, H. Davidson, E. Holder, L. A. Davies, I. A. Pringle and S. G. Sumner-Jones,
et al., Pre-clinical evaluation of three non-viral gene transfer agents for cystic fibrosis after aerosol delivery to the ovine lung, Gene Ther., 2011, 18(10), 996–1005 CrossRef CAS
- L. G. Johnson, J. C. Olsen, B. Sarkadi, K. L. Moore, R. Swanstrom and R. C. Boucher, Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis, Nat. Genet., 1992, 2(1), 21–25 CrossRef CAS PubMed
- A. E. Potash, T. J. Wallen, P. H. Karp, S. Ernst, T. O. Moninger and N. D. Gansemer,
et al., Adenoviral gene transfer corrects the ion transport defect in the sinus epithelia of a porcine CF model, Mol. Ther., 2013, 21(5), 947–953 CrossRef CAS PubMed
- J. T. Huckaby and S. K. Lai, PEGylation for enhancing nanoparticle diffusion in mucus, Adv. Drug Delivery Rev., 2018, 124, 125–139 CrossRef CAS PubMed
- N. N. Sanders, S. C. De Smedt, S. H. Cheng and J. Demeester, Pegylated GL67 lipoplexes retain their gene transfection activity after exposure to components of CF mucus, Gene Ther., 2002, 9(6), 363–371 CrossRef CAS PubMed
- K. Maisel, M. Reddy, Q. Xu, S. Chattopadhyay, R. Cone and L. M. Ensign,
et al., Nanoparticles coated with high molecular weight PEG penetrate mucus and provide uniform vaginal and colorectal distribution in vivo, Nanomedicine, 2016, 11(11), 1337–1343 CrossRef CAS PubMed
- G. Conte, G. Costabile, D. Baldassi, V. Rondelli, R. Bassi and D. Colombo,
et al., Hybrid
Lipid/Polymer Nanoparticles to Tackle the Cystic Fibrosis Mucus Barrier in siRNA Delivery to the Lungs: Does PEGylation Make the Difference?, ACS Appl. Mater. Interfaces, 2022, 7565–7578 CrossRef CAS PubMed
- S. Guan, A. Munder, S. Hedtfeld, P. Braubach, S. Glage and L. Zhang,
et al., Self-assembled peptide-poloxamine nanoparticles enable in vitro and in vivo genome restoration for cystic fibrosis, Nat. Nanotechnol., 2019, 14(3), 287–297 CrossRef CAS PubMed
- E. L. Blanchard, D. Vanover, S. S. Bawage, P. M. Tiwari, L. Rotolo and J. Beyersdorf,
et al., Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents, Nat. Biotechnol., 2021, 39(6), 717–700 CrossRef CAS PubMed
- R. Y. K. Chang and H. K. Chan, Lipid nanoparticles for the inhalation of mRNA, Nat. Biomed. Eng., 2021, 5(9), 949–950 CrossRef CAS PubMed
- M. P. Lokugamage, D. Vanover, J. Beyersdorf, M. Z. C. Hatit, L. Rotolo and E. S. Echeverri,
et al., Optimization of lipid nanoparticles for the delivery of nebulized therapeutic mRNA to the lungs, Nat. Biomed. Eng., 2021, 5(9), 1059–1068 CrossRef CAS PubMed
- E. Schrom, M. Huber, M. Aneja, C. Dohmen, D. Emrich and J. Geiger,
et al., Translation of Angiotensin-Converting Enzyme 2 upon Liver- and Lung-Targeted Delivery of Optimized Chemically Modified mRNA, Mol. Ther.–Nucleic Acids, 2017, 7, 350–365 CrossRef CAS PubMed
- D. A. Chistiakov, A. N. Orekhov and Y. V. Bobryshev, Endothelial PECAM-1 and its function in vascular physiology and atherogenic pathology, Exp. Mol. Pathol., 2016, 100(3), 409–415 CrossRef CAS
- J. Han, V. V. Shuvaev and V. R. Muzykantov, Targeted interception of signaling reactive oxygen species in the vascular endothelium, Ther. Delivery, 2012, 3(2), 263–276 CrossRef CAS
- H. Parhiz, V. V. Shuvaev, N. Pardi, M. Khoshnejad, R. Y. Kiseleva and J. S. Brenner,
et al., PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake, J. Controlled Release, 2018, 291, 106–115 CrossRef CAS PubMed
- T. Wei, Q. Cheng, Y. L. Min, E. N. Olson and D. J. Siegwart, Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing, Nat. Commun., 2020, 11(1), 3232 CrossRef CAS PubMed
- J. B. Miller, S. Zhang, P. Kos, H. Xiong, K. Zhou and S. S. Perelman,
et al., Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA, Angew. Chem., Int. Ed., 2017, 56(4), 1059–1063 CrossRef CAS PubMed
- Y. Rui, D. R. Wilson, S. Y. Tzeng, H. M. Yamagata, D. Sudhakar and M. Conge,
et al., High-throughput and high-content bioassay enables tuning of polyester nanoparticles for cellular uptake, endosomal escape, and systemic in vivo delivery of mRNA, Sci. Adv., 2022, 8(1), eabk2855 CrossRef CAS PubMed
- Y. Yan, H. Xiong, X. Zhang, Q. Cheng and D. J. Siegwart, Systemic mRNA Delivery to the Lungs by Functional Polyester-based Carriers, Biomacromolecules, 2017, 18(12), 4307–4315 CrossRef CAS PubMed
- J. C. Kaczmarek, K. J. Kauffman, O. S. Fenton, K. Sadtler, A. K. Patel and M. W. Heartlein,
et al., Optimization of a Degradable Polymer-Lipid Nanoparticle for Potent Systemic Delivery of mRNA to the Lung Endothelium and Immune Cells, Nano Lett., 2018, 18(10), 6449–6454 CrossRef CAS PubMed
- C. D. Walkey, J. B. Olsen, F. Song, R. Liu, H. Guo and D. W. Olsen,
et al., Protein corona fingerprinting predicts the cellular interaction of gold and silver nanoparticles, ACS Nano, 2014, 8(3), 2439–2455 CrossRef CAS PubMed
- J. Huotari and A. Helenius, Endosome maturation, EMBO J., 2011, 30(17), 3481–3500 CrossRef CAS PubMed
- F. Morfoisse and A. Noel, Lymphatic and blood systems: Identical or fraternal twins?, Int. J. Biochem. Cell Biol., 2019, 114, 105562 CrossRef CAS PubMed
- Y. Xie, T. R. Bagby, M. S. Cohen and M. L. Forrest, Drug delivery to the lymphatic system: importance in future cancer diagnosis and therapies, Expert Opin. Drug Delivery, 2009, 6(8), 785–792 CrossRef CAS PubMed
- P. Y. von der Weid, S. Rehal and J. G. Ferraz, Role of the lymphatic system in the pathogenesis of Crohn's disease, Curr. Opin. Gastroenterol., 2011, 27(4), 335–341 CrossRef CAS PubMed
- W. F. Richter, S. G. Bhansali and M. E. Morris, Mechanistic determinants of biotherapeutics absorption following SC administration, AAPS J., 2012, 14(3), 559–570 CrossRef CAS PubMed
- K. E. Lindsay, S. M. Bhosle, C. Zurla, J. Beyersdorf, K. A. Rogers and D. Vanover,
et al., Visualization of early events in mRNA vaccine delivery in non-human primates via PET-CT and near-infrared imaging, Nat. Biomed. Eng., 2019, 3(5), 371–380 CrossRef CAS PubMed
- S. N. Thomas, N. A. Rohner and E. E. Edwards, Implications of Lymphatic Transport to Lymph Nodes in Immunity and Immunotherapy, Annu. Rev. Biomed. Eng., 2016, 18, 207–233 CrossRef CAS PubMed
- D. Szoke, G. Kovacs, E. Kemecsei, L. Balint, K. Szotak-Ajtay and P. Aradi,
et al., Nucleoside-modified VEGFC mRNA induces organ-specific lymphatic growth and reverses experimental lymphedema, Nat. Commun., 2021, 12(1), 3460 CrossRef CAS PubMed
- V. Manolova, A. Flace, M. Bauer, K. Schwarz, P. Saudan and M. F. Bachmann, Nanoparticles target distinct dendritic cell populations according to their size, Eur. J. Immunol., 2008, 38(5), 1404–1413 CrossRef CAS
- S. T. Reddy, A. Rehor, H. G. Schmoekel, J. A. Hubbell and M. A. Swartz, In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles, J. Controlled Release, 2006, 112(1), 26–34 CrossRef CAS PubMed
- M. A. Oberli, A. M. Reichmuth, J. R. Dorkin, M. J. Mitchell, O. S. Fenton and A. Jaklenec,
et al., Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy, Nano Lett., 2017, 17(3), 1326–1335 CrossRef CAS PubMed
- U. Sahin, E. Derhovanessian, M. Miller, B. P. Kloke, P. Simon and M. Lower,
et al., Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer, Nature, 2017, 547(7662), 222–226 CrossRef CAS PubMed
- U. Sahin, P. Oehm, E. Derhovanessian, R. A. Jabulowsky, M. Vormehr and M. Gold,
et al., An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma, Nature, 2020, 585(7823), 107–112 CrossRef CAS
- L. F. Wong, L. Goodhead, C. Prat, K. A. Mitrophanous, S. M. Kingsman and N. D. Mazarakis, Lentivirus-mediated gene transfer to the central nervous system: therapeutic and research applications, Hum. Gene Ther., 2006, 17(1), 1–9 CrossRef CAS PubMed
- S. Yang, R. Chang, H. Yang, T. Zhao, Y. Hong and H. E. Kong,
et al., CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington's disease, J. Clin. Invest., 2017, 127(7), 2719–2724 CrossRef PubMed
- S. Ohta, E. Kikuchi, A. Ishijima, T. Azuma, I. Sakuma and T. Ito, Investigating the optimum size of nanoparticles for their delivery into the brain assisted by focused ultrasound-induced blood-brain barrier opening, Sci. Rep., 2020, 10(1), 18220 CrossRef CAS PubMed
- K. T. Householder, S. Dharmaraj, D. I. Sandberg, R. J. Wechsler-Reya and R. W. Sirianni, Fate of nanoparticles in the central nervous system after intrathecal injection in healthy mice, Sci. Rep., 2019, 9(1), 12587 CrossRef CAS PubMed
- J. F. Nabhan, K. M. Wood, V.
P. Rao, J. Morin, S. Bhamidipaty and T. P. LaBranche,
et al., Intrathecal delivery of frataxin mRNA encapsulated in lipid nanoparticles to dorsal root ganglia as a potential therapeutic for Friedreich's ataxia, Sci. Rep., 2016, 6, 20019 CrossRef CAS PubMed
- A.J Atkinson Jr., Intracerebroventricular drug administration, Transl. Clin. Pharmacol., 2017, 25(3), 117–124 CrossRef
- H. Tanaka, T. Nakatani, T. Furihata, K. Tange, Y. Nakai and H. Yoshioka,
et al., In Vivo Introduction of mRNA Encapsulated in Lipid Nanoparticles to Brain Neuronal Cells and Astrocytes via Intracerebroventricular Administration, Mol. Pharm., 2018, 15(5), 2060–2067 CrossRef CAS PubMed
- R. E. Pitas, J. K. Boyles, S. H. Lee, D. Hui and K. H. Weisgraber, Lipoproteins and Their Receptors in the Central-Nervous-System - Characterization of the Lipoproteins in Cerebrospinal-Fluid and Identification of Apolipoprotein-B,E(Ldl) Receptors in the Brain, J. Biol. Chem., 1987, 262(29), 14352–14360 CrossRef CAS
- R. L. Rungta, H. B. Choi, P. J. Lin, R. W. Ko, D. Ashby and J. Nair,
et al., Lipid Nanoparticle Delivery of siRNA to Silence Neuronal Gene Expression in the Brain, Mol. Ther.–Nucleic Acids, 2013, 2, e136 CrossRef CAS PubMed
- C. Y. Lin, F. Perche, M. Ikegami, S. Uchida, K. Kataoka and K. Itaka, Messenger RNA-based therapeutics for brain diseases: An animal study for augmenting clearance of beta-amyloid by intracerebral administration of neprilysin mRNA loaded in polyplex nanomicelles, J. Controlled Release, 2016, 235, 268–275 CrossRef CAS PubMed
- O. A. Marcos-Contreras, C. F. Greineder, R. Y. Kiseleva, H. Parhiz, L. R. Walsh and V. Zuluaga-Ramirez,
et al., Selective targeting of nanomedicine to inflamed cerebral vasculature to enhance the blood-brain barrier, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(7), 3405–3414 CrossRef CAS PubMed
- D. A. Prado, M. Acosta-Acero and R. S. Maldonado, Gene therapy beyond luxturna: a new horizon of the treatment for inherited retinal disease, Curr. Opin. Ophthalmol., 2020, 31(3), 147–154 CrossRef PubMed
- J. Devoldere, K. Peynshaert, H. Dewitte, C. Vanhove, L. De Groef and L. Moons,
et al., Non-viral delivery of chemically modified mRNA to the retina: Subretinal versus intravitreal administration, J. Controlled Release, 2019, 307, 315–330 CrossRef CAS
- S. Patel, R. C. Ryals, K. K. Weller, M. E. Pennesi and G. Sahay, Lipid nanoparticles for delivery of messenger RNA to the back of the eye, J. Controlled Release, 2019, 303, 91–100 CrossRef CAS PubMed
- S. Veleri, C. H. Lazar, B. Chang, P. A. Sieving, E. Banin and A. Swaroop, Biology and therapy of inherited retinal degenerative disease: insights from mouse models, Dis. Models Mech., 2015, 8(2), 109–129 CrossRef PubMed
- R. Ladha, L. E. Caspers, F. Willermain and M. D. de Smet, Subretinal Therapy: Technological Solutions to Surgical and Immunological Challenges, Front. Med., 2022, 9, 846782 CrossRef PubMed
- J. Mains and C. G. Wilson, The vitreous humor as a barrier to nanoparticle distribution, J. Ocul. Pharmacol. Ther., 2013, 29(2), 143–150 CrossRef CAS PubMed
- R. C. Ryals, S. Patel, C. Acosta, M. McKinney, M. E. Pennesi and G. Sahay, The effects of PEGylation on LNP based mRNA delivery to the eye, PLoS One, 2020, 15(10), e0241006 CrossRef CAS
- H. Dargie, Heart failure post-myocardial infarction: a review of the issues, Heart, 2005, 91(Suppl 2), ii3–ii6 Search PubMed
- A. S. Go, D. Mozaffarian, V. L. Roger, E. J. Benjamin, J. D. Berry and M. J. Blaha,
et al., Heart disease and stroke statistics–2014 update: a report from the American Heart Association, Circulation, 2014, 129(3), e28–e292 Search PubMed
- H. Hashimoto, E. N. Olson and R. Bassel-Duby, Therapeutic approaches for cardiac regeneration and repair, Nat.
Rev. Cardiol., 2018, 15(10), 585–600 CrossRef
- A. Magadum, K. Kaur and L. Zangi, mRNA-Based Protein Replacement Therapy for the Heart, Mol. Ther., 2019, 27(4), 785–793 CrossRef CAS
- K. Sato, T. Wu, R. J. Laham, R. B. Johnson, P. Douglas and J. Li,
et al., Efficacy of intracoronary or intravenous VEGF165 in a pig model of chronic myocardial ischemia, J. Am. Coll. Cardiol., 2001, 37(2), 616–623 CrossRef CAS PubMed
- D. J. Stewart, M. J. Kutryk, D. Fitchett, M. Freeman, N. Camack and Y. Su,
et al., VEGF gene therapy fails to improve perfusion of ischemic myocardium in patients with advanced coronary disease: results of the NORTHERN trial, Mol. Ther., 2009, 17(6), 1109–1115 CrossRef CAS PubMed
- K. O. Lui, L. Zangi, E. A. Silva, L. Bu, M. Sahara and R. A. Li,
et al., Driving vascular endothelial cell fate of human multipotent Isl1 + heart progenitors with VEGF modified mRNA, Cell Res., 2013, 23(10), 1172–1186 CrossRef CAS PubMed
- L. Zangi, K. O. Lui, A. von Gise, Q. Ma, W. Ebina and L. M. Ptaszek,
et al., Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction, Nat. Biotechnol., 2013, 31(10), 898–907 CrossRef CAS PubMed
- I. C. Turnbull, A. A. Eltoukhy, D. G. Anderson and K. D. Costa, Lipidoid mRNA Nanoparticles for Myocardial Delivery in Rodents, Methods Mol. Biol., 2017, 1521, 153–166 CrossRef CAS PubMed
- I. C. Turnbull, A. A. Eltoukhy, K. M. Fish, M. Nonnenmacher, K. Ishikawa and J. Chen,
et al., Myocardial Delivery of Lipidoid Nanoparticle Carrying modRNA Induces Rapid and Transient Expression, Mol. Ther., 2016, 24(1), 66–75 CrossRef CAS PubMed
- N. Sultana, A. Magadum, Y. Hadas, J. Kondrat, N. Singh and E. Youssef,
et al., Optimizing Cardiac Delivery of Modified mRNA, Mol. Ther., 2017, 25(6), 1306–1315 CrossRef CAS PubMed
- L. Carlsson, J. C. Clarke, C. Yen, F. Gregoire, T. Albery and M. Billger,
et al., Biocompatible, Purified VEGF-A mRNA Improves Cardiac Function after Intracardiac Injection 1 Week Post-myocardial Infarction in Swine, Mol. Ther.–Methods Clin. Dev., 2018, 9, 330–346 CrossRef CAS PubMed
- T. T. H. Thi, E. J. A. Suys, J. S. Lee, D. H. Nguyen, K. D. Park and N. P. Truong, Lipid-Based Nanoparticles in the Clinic and Clinical Trials: From Cancer Nanomedicine to COVID-19 Vaccines, Vaccines, 2021, 9(4), 1–29 Search PubMed
- M. A. Maier, M. Jayaraman, S. Matsuda, J. Liu, S. Barros and W. Querbes,
et al., Biodegradable Lipids Enabling Rapidly Eliminated Lipid Nanoparticles for Systemic Delivery of RNAi Therapeutics, Mol. Ther., 2013, 21(8), 1570–1578 CrossRef CAS PubMed
- X. Zhou, Y. Cui and J. Han, Methylmalonic acidemia: Current status and research priorities, Intractable Rare Dis. Res., 2018, 7(2), 73–78 CrossRef
- D. An, A. Frassetto, E. Jacquinet, M. Eybye, J. Milano and C. DeAntonis,
et al., Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia, EBioMedicine, 2019, 45, 519–528 CrossRef
- T. Uchino, S. E. Snyderman, M. Lambert, I. A. Qureshi, S. K. Shapira and C. Sansaricq,
et al., Molecular basis of phenotypic variation in patients with argininemia, Hum. Genet., 1995, 96(3), 255–260 CrossRef CAS PubMed
- B. Truong, G. Allegri, X. B. Liu, K. E. Burke, X. L. Zhu and S. D. Cederbaum,
et al., Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(42), 21150–21159 CrossRef CAS
- S. Song, L. Yang, W. L. Trepicchio and T. Wyant, Understanding the Supersensitive Anti-Drug Antibody Assay: Unexpected High Anti-Drug Antibody Incidence and Its Clinical Relevance, J. Immunol. Res., 2016, 2016, 3072586 Search PubMed
- J. Cao, M. Choi, E. Guadagnin, M. Soty, M. Silva and V. Verzieux,
et al., mRNA therapy restores euglycemia and prevents liver tumors in murine model of glycogen storage disease, Nat. Commun., 2021, 12(1), 3090 CrossRef CAS PubMed
- J. Szebeni, G. Storm, J. Y. Ljubimova, M. Castells, E. J. Phillips and K. Turjeman,
et al., Applying lessons learned from nanomedicines to understand rare hypersensitivity reactions to mRNA-based SARS-CoV-2 vaccines, Nat. Nanotechnol., 2022, 17(4), 337–346 CrossRef CAS
- X. Zhang, V. Goel and G. J. Robbie, Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients With Hereditary Transthyretin-Mediated Amyloidosis, J. Clin. Pharmacol., 2019, 60(5), 573–585 CrossRef
- J. S. Butler, A. Chan, S. Costelha, S. Fishman, J. L. S. Willoughby and T. D. Borland,
et al., Preclinical evaluation of RNAi as a treatment for transthyretin-mediated amyloidosis, Amyloid, 2016, 23(2), 109–118 CrossRef CAS
- K. M. Miah, S. C. Hyde and D. R. Gill, Emerging gene therapies for cystic fibrosis, Expert Rev. Respir. Med., 2019, 13(8), 709–725 CrossRef CAS PubMed
- J. A. Lee, A. Cho, E. N. Huang, Y. Xu, H. Quach and J. Hu,
et al., Gene therapy for cystic fibrosis: new tools for precision medicine, J. Transl. Med., 2021, 19(1), 452 CrossRef PubMed
- M. Torres, D. Boudko, E. Meleshkevitch, M. Coquelin, X. Yu and J. Eby,
et al., Variant-agnostic CFTR rescue using aerosolized delivery of CFTR mRNAusing the SORT-LNP in primary human bronchial epithelial cellsderived from patients with cystic fibrosis, J. Cystic Fibrosis, 2021, 20, S278 CrossRef
- E. Robinson, K. D. MacDonald, K. Slaughter, M. McKinney, S. Patel and C. Sun,
et al., Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis, Mol. Ther., 2018, 26(8), 2034–2046 CrossRef CAS PubMed
- A. Karadagi, A. G. Cavedon, H. Zemack, G. Nowak, M. E. Eybye and X. L. Zhu,
et al., Systemic modified messenger RNA for replacement therapy in alpha 1-antitrypsin deficiency, Sci. Rep., 2020, 10(1), 7052 CrossRef CAS
- J. D. Beck, D. Reidenbach, N. Salomon, U. Sahin, O. Tureci and M. Vormehr,
et al., mRNA therapeutics in cancer immunotherapy, Mol. Cancer, 2021, 20(1), 69 CrossRef CAS
- N. Veiga, M. Goldsmith, Y. Granot, D. Rosenblum, N. Dammes and R. Kedmi,
et al., Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes, Nat. Commun., 2018, 9(1), 4493 CrossRef PubMed
- K. Radhakrishnan and S. G. Rockson, The clinical spectrum of lymphatic disease, Ann. N. Y. Acad. Sci., 2008, 1131, 155–184 CrossRef PubMed
- A. Da Silva Sanchez, K. Paunovska, A. Cristian and J. E. Dahlman, Treating Cystic Fibrosis with mRNA and CRISPR, Hum. Gene Ther., 2020, 31(17–18), 940–955 CrossRef CAS
- X. Zhang, W. Zhao, G. N. Nguyen, C. Zhang, C. Zeng and J. Yan,
et al., Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing, Sci. Adv., 2020, 6(34), eabc2315 CrossRef CAS PubMed
- C. Jiang, M. Mei, B. Li, X. Zhu, W. Zu and Y. Tian,
et al., A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo, Cell Res., 2017, 27(3), 440–443 CrossRef CAS PubMed
- M. Qiu, Z. Glass, J. Chen, M. Haas, X. Jin and X. Zhao,
et al., Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3, Proc. Natl. Acad. Sci. U. S. A., 2021, 118(10), e20204011 CrossRef
- J. D. Finn, A. R. Smith, M. C. Patel, L. Shaw, M. R. Youniss and J. van Heteren,
et al., A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing, Cell Rep., 2018, 22(9), 2227–2235 CrossRef CAS PubMed
- L. Farbiak, Q. Cheng, T. Wei, E. Alvarez-Benedicto, L. T. Johnson and S. Lee,
et al., All-In-One Dendrimer-Based Lipid Nanoparticles Enable Precise HDR-Mediated Gene Editing In Vivo, Adv. Mater., 2021, 33(30), e2006619 CrossRef PubMed
- J. D. Gillmore, E. Gane, J. Taubel, J. Kao, M. Fontana and M. L. Maitland,
et al., CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis, N. Engl. J. Med., 2021, 385(6), 493–502 CrossRef CAS PubMed
- J. G. Rurik, I. Tombacz, A. Yadegari, P. O. Méndez Fernández, S. V. Shewale and L. Li,
et al., CAR T cells produced in vivo to treat cardiac injury, Science, 2022, 375(6576), 91–96 CrossRef CAS PubMed
- A. Ehrhardt, R. Haase, A. Schepers, M. J. Deutsch, H. J. Lipps and A. Baiker, Episomal vectors for gene therapy, Curr. Gene Ther., 2008, 8(3), 147–161 CrossRef CAS PubMed
- K. Paunovska, A. J. Da Silva Sanchez, C. D. Sago, Z. Gan, M. P. Lokugamage and F. Z. Islam,
et al., Nanoparticles Containing Oxidized Cholesterol Deliver mRNA to the Liver Microenvironment at Clinically Relevant Doses, Adv. Mater., 2019, 31(14), e1807748 CrossRef PubMed
- B. Connolly, C. Isaacs, L. Cheng, K. H. Asrani and R. R. Subramanian, SERPINA1 mRNA as a Treatment for Alpha-1 Antitrypsin Deficiency, J. Nucleic Acids, 2018, 2018, 8247935 Search PubMed
- L. Jiang, J. S. Park, L. Yin, R. Laureano, E. Jacquinet and J. Yang,
et al., Dual mRNA therapy restores metabolic function in long-term studies in mice with propionic acidemia, Nat. Commun., 2020, 11(1), 5339 CrossRef CAS PubMed
- D. S. Roseman, T. Khan, F. Rajas, L. S. Jun, K. H. Asrani and C. Isaacs,
et al., G6PC mRNA Therapy Positively Regulates Fasting Blood Glucose and Decreases Liver Abnormalities in a Mouse Model of Glycogen Storage Disease 1a, Mol. Ther., 2018, 26(3), 814–821 CrossRef CAS PubMed
- S. Ramaswamy, N. Tonnu, K. Tachikawa, P. Limphong, J. B. Vega and P. P. Karmali,
et al., Systemic delivery of factor IX messenger RNA for protein replacement therapy, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(10), E1941–E1950 CrossRef CAS PubMed
- F. DeRosa, B. Guild, S. Karve, L. Smith, K. Love and J. R. Dorkin,
et al., Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system, Gene Ther., 2016, 23(10), 699–707 CrossRef CAS
- S. Liu-Chen, B. Connolly, L. Cheng, R. R. Subramanian and Z. Han, mRNA treatment produces sustained expression of enzymatically active human ADAMTS13 in mice, Sci. Rep., 2018, 8(1), 7859 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2022 |