
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBreaking the bottleneck: stilbene as a model compound for optimizing 6π e− photocyclization efficiency†
Joshua Seylar a,
Dmytro Stasioukb,
Davide L. Simonec,
Vikas Varshneyc,
James E. Hecklercd and
Ruel McKenzie*a
a,
Dmytro Stasioukb,
Davide L. Simonec,
Vikas Varshneyc,
James E. Hecklercd and
Ruel McKenzie*a
aSchool of Polymer Science and Polymer Engineering, The University of Akron, Akron, OH 44325, USA. E-mail: rmckenzie@uakron.edu
bDepartment of Chemistry, The University of Akron, Akron, OH 44325, USA
cMaterials and Manufacturing Directorate, Air Force Research Laboratory, WPAFB, OH 45433, USA
dARCTOS Technology Solutions, Dayton, OH 45432, USA
First published on 5th February 2021
Abstract
TEMPO was more suitable at photocyclizing stilbene than iodine. As stilbene concentration increased, TEMPO produced a higher yield of phenanthrene at shorter times and significantly reduced the potential for undesired [2+2] cycloadditions. Iodine retarded phenanthrene formation because it promoted isomerization to (E)-stilbene which encouraged [2+2] cycloaddition.
Phenanthrenes are primarily synthesized via a photocyclodehydrogenation reaction1,2 where stilbenes or other di-aryl alkenes undergo a photochemically driven, conrotatory electrocyclic3–5 reaction to form dihydrophenanthrenes (DHPs)6 which are generally unstable and have a short lifetime. DHPs are conventionally oxidized by an oxygen7,8 or iodine9,10 radical to form phenanthrene.
The photooxidative cyclization of stilbenes to form phenanthrenes is often referred to as the Mallory reaction.11 Mallory significantly advanced photochemical synthesis of phenanthrene and other polycyclic aromatic compounds (PACs) by suggesting the use of a catalytic amount of iodine in conjunction with environmental oxygen as an oxidizing agent.7 This reaction was originally performed by irradiation with unfiltered, high-pressure mercury vapor lamps under ambient conditions with 3–5 mol% iodine.12 These reaction conditions were widely adopted and went unmodified until Liu et al.10 suggested the use of a stoichiometric amount of iodine under an inert atmosphere in conjunction with a scavenger to remove the resulting hydrogen iodide that forms. This approach enhanced the yield of the desired product but was limited to low concentrations for efficient conversion. Increasing the reactant concentration results in the formation of undesired cycloalkanes via [2+2] photocycloaddition.13–17 Scale-up using this approach is largely inefficient and may inadvertently discourage industrial production and applications of PACs for emerging technologies. Alternative approaches must be considered to overcome this production bottleneck. One approach utilizes flow reactors18,19 but these systems also operate at relatively dilute conditions.
The photochemical reaction pathway of stilbene is illustrated in Fig. 1. It has been proposed that phenanthrenes are formed through the excited state of (Z)-stilbene which makes DHP that can be subsequently oxidized.20–22 However, there have been limited empirical studies confirming the proposed pathway of phenanthrene formation.23–29 This is partially due to the difficulty in isolating DHP as a stable product, and also due to the predictive limitations of modelling techniques for formation of DHP via centroid carbon interactions in excited-state (Z)-stilbene.25
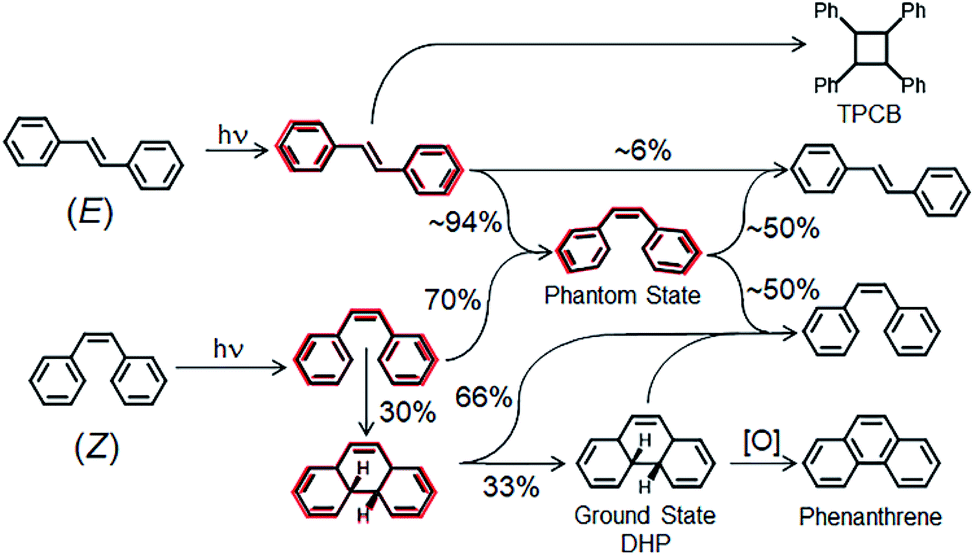 | ||
| Fig. 1 Simplified stilbene photochemical reaction pathway. Red outlines indicate excited states. | ||
UV light excites (Z)-stilbene to the singlet (S1) state which has a lifetime of 0.7–1.4 ps.5 S1 (Z)-stilbene then proceeds through two possible pathways: roughly 70% becomes a twisted pyramidal state known as the phantom state; and the remaining 30% becomes planar S1 DHP via centroid carbon interactions.5,27,30 The phantom state will then form ground state (S0) (Z)- and (E)-stilbene in roughly equal amounts, depending on the reaction conditions. Excited state DHP will form roughly 66% S0 (Z)-stilbene and 33% S0 DHP which can form phenanthrene in the presence of an oxidizing agent.4,23,26,29 When (E)-stilbene is photochemically excited to the S1 state, it has a lifetime of 80–140 ps5 and follows three possible pathways. It can directly reform S0 (E)-stilbene (∼6%),26 interact with an S0 (E)-stilbene to form cyclobutane through a [2+2] photocycloaddition, or form the same twisted phantom state as (Z)-stilbene which then reforms S0 (Z)- or (E)-stilbene.
It is theorized that intermolecular [2+2] cycloaddition of stilbenes is more likely to occur through the (E)-stilbene excited state than through (Z)-stilbene.30,31 This is due to the shorter (Z)-stilbene S1 lifetime, along with the much faster timescale for (Z)-(E) isomerization (2 ps) compared to the (E)-(Z) isomerization (10–200 ps).23 This reduces the probability for intermolecular dimerization to occur through (Z)-stilbene and indicates a preference for the (Z) conformation for the increased effectiveness of phenanthrene synthesis.30 A goal of this study is to assess the impact of stilbene stereoisomers on the synthesis of the desired phenanthrene and the undesired tetraphenylcyclobutane (TPCB).
Iodine is known to catalyse the formation of (E)-stilbene as well as reduce the absorption of light into the reaction volume.21 These facts, along with the much longer lifetime of the (E)-stilbene S1 compared to the (Z)-stilbene S1 and the longer time to convert (E) to (Z) may lead to a significant decrease in the efficiency of phenanthrene formation. Iodine has served as the conventional oxidizing agent in the photochemical synthesis of PACs, but the propensity to promote (E)-stilbene formation limits its utility. It is desired to replace iodine with an oxidizing agent that circumvents this limitation. Ideally, the replacement for iodine should utilize mild reaction conditions, produce easy to remove side products, and should not interfere with the reaction pathway. (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) was recently demonstrated to be a potentially suitable alternative from a study that hypothesized that the photooxidative cyclization of stilbenes is free-radical mediated.32 Even though TEMPO has a lower and pH-dependent redox potential (+0.25 V at pH 7)33 than iodine (+0.54 V)34 in aqueous solutions, TEMPO may be an ideal alternative to iodine because it is a commercially available free-radical source that does not require photochemical or thermal activation.
A series of photochemical reactions was performed on solutions initially containing only (Z)- or (E)-stilbene at concentrations ranging from 1–100 mM to compare and contrast the impact of iodine and TEMPO on the formation of the desired phenanthrene and undesired TPCB. 1 mM to 20 mM reactions were performed in 10 mL of cyclohexane in borosilicate glass vials with rubber septa unless otherwise stated. 100 mM concentrations of stilbene were reacted in 5 mL of cyclohexane. The borosilicate glass vials act as short-wavelength UV filters below 300 nm. Samples were prepared in a dark room. All samples were sparged with argon before irradiation with a 450 W medium pressure mercury vapor lamp equipped with a quartz water jacket for discrete periods of time. Reactions with iodine were performed at 1 eq. with 100 eq. propylene oxide (PPO). Reactions containing TEMPO were performed using 5 eq. Stoichiometrically, a minimum of 2 eq. of TEMPO is required to completely oxidize DHP but it was found that using an excess of TEMPO was more beneficial. The dependency of the reaction dynamics on concentration of oxidizing agent was not assessed in this study. Excess TEMPO and iodine were removed under vacuum. Higher (20 mM) concentrations of iodine were removed by washing with sodium thiosulphate. Samples were analysed by 1H NMR. Extent of reaction and yields were analysed by monitoring the 2-proton singlet peaks of (Z)-stilbene (6.60 ppm), (E)-stilbene (7.11 ppm), phenanthrene (7.73 ppm), and TPCB (4.47, 3.69 ppm) which were well-separated in 1H NMR (Fig. S2–S5†).
The equilibrium conformation of stilbene is light sensitive. The wavelength range and intensity of the light source remained constant. The impact of concentration and initial conformation of stilbene was assessed as a function of exposure time to the light source. Temperature of the system was not controlled, and any temperature rise in the reaction medium due to the light source was assumed to be negligible because of cooling from the water jacket. Fig. 2 shows the ratio of (Z)-stilbene to the total amount of stilbene over time in the presence of no oxidizing agent (Fig. 2A), iodine (Fig. 2B) and TEMPO (Fig. 2C). These figures represent the relative amount of (Z)-stilbene available over the course of the reaction. There is negligible consumption of (Z)-stilbene in the absence of an oxidizing agent because oxygen was thoroughly removed from the sample by sparging with argon. Fig. 2A shows that the reaction approached an equilibrium of approximately 85% (Z)-stilbene with the light source used irrespective of whether the system initially started with 100% (Z) or 100% (E) conformation. Increasing the concentration of (E)-stilbene increased the time to reach the equilibrium conformation.
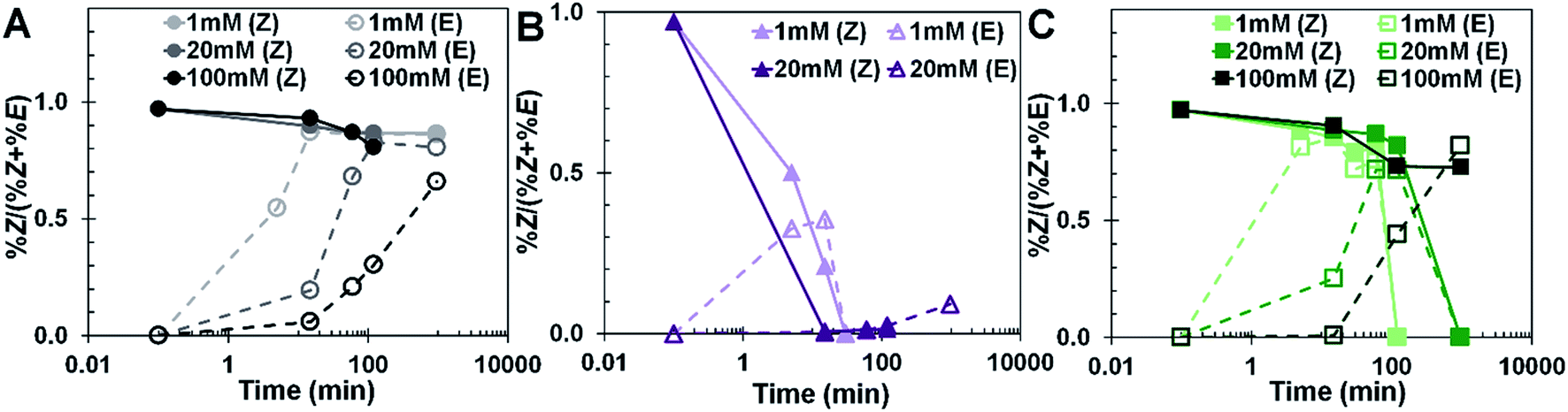 | ||
| Fig. 2 Relative amount of (Z)-stilbene available for (A) non-oxidizing, (B) iodine- and (C) TEMPO-oxidized reactions. | ||
The effect of initial conformer concentration on the timescale to attain equilibrium concentrations of (Z)- and (E)-stilbene conformers was most apparent with (E)-stilbene, while there was no noticeable effect with (Z)-stilbene. This implies that intermolecular interactions may be causing a decrease in the rate of conversion of (E)-stilbene to (Z)-stilbene. The oxidizing agents (iodine (Fig. 2B) and TEMPO (Fig. 2C)) had a distinguishable effect on the amount of (Z)-stilbene available which was either due to oxidation to form phenanthrene or isomerization to form (E)-stilbene. As seen in Fig. 2B, the addition of iodine drastically reduced the amount of (Z)-stilbene available for the reaction, particularly as concentration increased. The presence of TEMPO (Fig. 2C) did not appear to have a noticeable impact on the (Z) isomer availability because of the similarity in trajectory to when there was no oxidizing agent in the system (Fig. 2A).
Starting with higher (Z) content should lead to a greater probability of phenanthrene formation. The relative impact of oxidizing agent on the yield of phenanthrene is displayed in Fig. 3. Iodine (Fig. 3A) and TEMPO (Fig. 3B) are equally effective in the formation of phenanthrene at low concentrations. The transient data (Fig. 3A and B) displayed a progressive lag in the reaction as concentration of the reactant increased. (E)-stilbene produced phenanthrene at a lower rate than (Z)-stilbene as the concentration increased. Utilizing TEMPO revealed that the reaction became more sensitive to stereoisomers as concentration increased. The reaction did not display a sensitivity to stereoisomers in the presence of iodine because iodine demonstrated a tendency to promote isomerization to the (E) conformer. The results of Fig. 2B indicate that the effect of iodine on promoting the formation of the (E) isomer occurred at early times in the reaction. Therefore, the iodine systems were primarily reacting through (E)-stilbene as an intermediate. The low availability of (Z)-stilbene in the presence of iodine limited the amount of phenanthrene that could be synthesized. This implies (E)-stilbene needs to convert to (Z)-stilbene for phenanthrene formation to occur. This activity in the presence of iodine effectively suppressed the rate of phenanthrene formation irrespective of the initial conformer used.
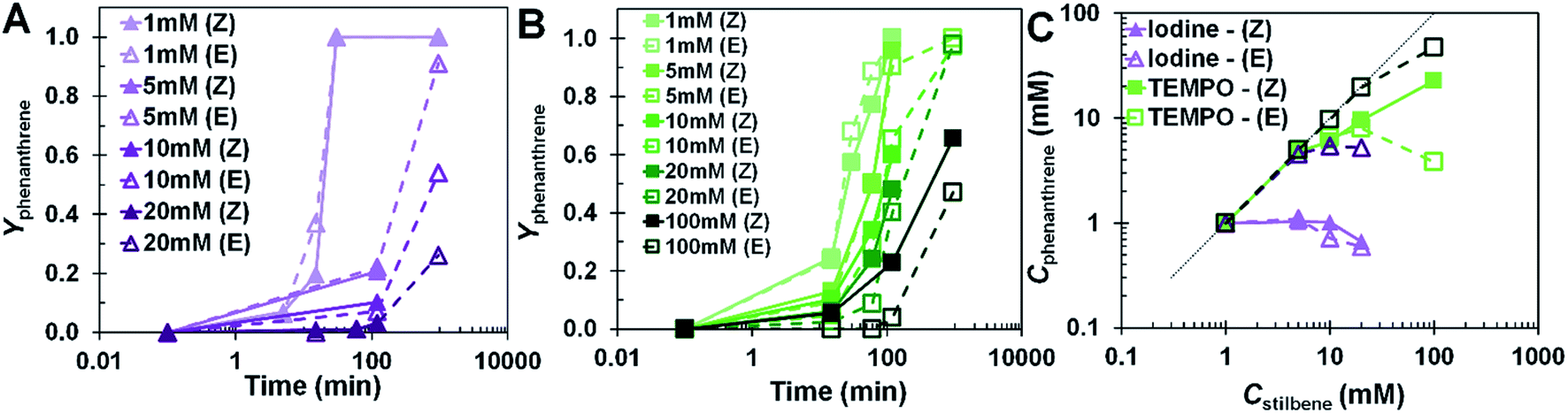 | ||
| Fig. 3 NMR yield of phenanthrene evolution for (A) iodine- and (B) TEMPO-oxidized systems; (C) concentration dependence of phenanthrene formation at 2 h (light lines and symbols) and 16 h (dark lines and symbols). Dotted line represents complete conversion. | ||
Fig. 3C shows that TEMPO became more effective at producing phenanthrene than iodine as the concentration of stilbene increased from as low as 5 mM. TEMPO had a much higher yield at higher concentrations than that of iodine. When comparing the impact of concentration at 2 and 16 hours of reaction time, the concentration of phenanthrene formed remained approximately constant and was low for iodine while there was a systematic increase in the case of TEMPO. For example, at 20 mM, there was 14× more phenanthrene after 2 h when TEMPO was used. The reaction sensitivity to stereoisomers in the presence of TEMPO was most apparent at concentrations above 20 mM.
From the results thus far, it has been demonstrated that TEMPO may be a suitable alternative to iodine, particularly at high reactant concentrations where iodine was shown to be limited. TEMPO also showed particular effectiveness in phenanthrene formation when used with (Z)-stilbene, but still outperformed iodine when (E)-stilbene was used. However, the formation of the undesired product, TPCB, must be analysed to effectively conclude on the overall potential of TEMPO in photocyclization reactions.
It has been suggested that [2+2] photocycloadditions occur primarily through the (E) isomer of stilbene.13,30,31 The results of this study suggest the same. There was a steady evolution of TPCB with increasing concentration of stilbene with and without oxidizing agent. The formation of TPCB was sensitive to the conformation of stilbene and primarily formed in the presence of (E)-stilbene. Samples with higher overall (E)-stilbene content yielded a higher quantity of TPCB (Fig. 4A). In the presence of iodine, concentration was the main factor influencing the formation of TPCB because of the tendency for iodine to promote formation of the (E) isomer which occurred at early times in the reaction. Therefore, in the case of iodine, the yield of TPCB was similar irrespective of the stereoisomer used. TEMPO-oxidized samples had a lower overall yield of TPCB when compared to iodine and non-oxidized systems. The impact of the starting conformer in the presence of TEMPO was also evident in the formation of TPCB. Reactions that started with (E)-stilbene evolved TPCB at lower rates than iodine and in the case where no oxidant was present. Reactions that started with (Z)-stilbene produced negligible amounts of TPCB.
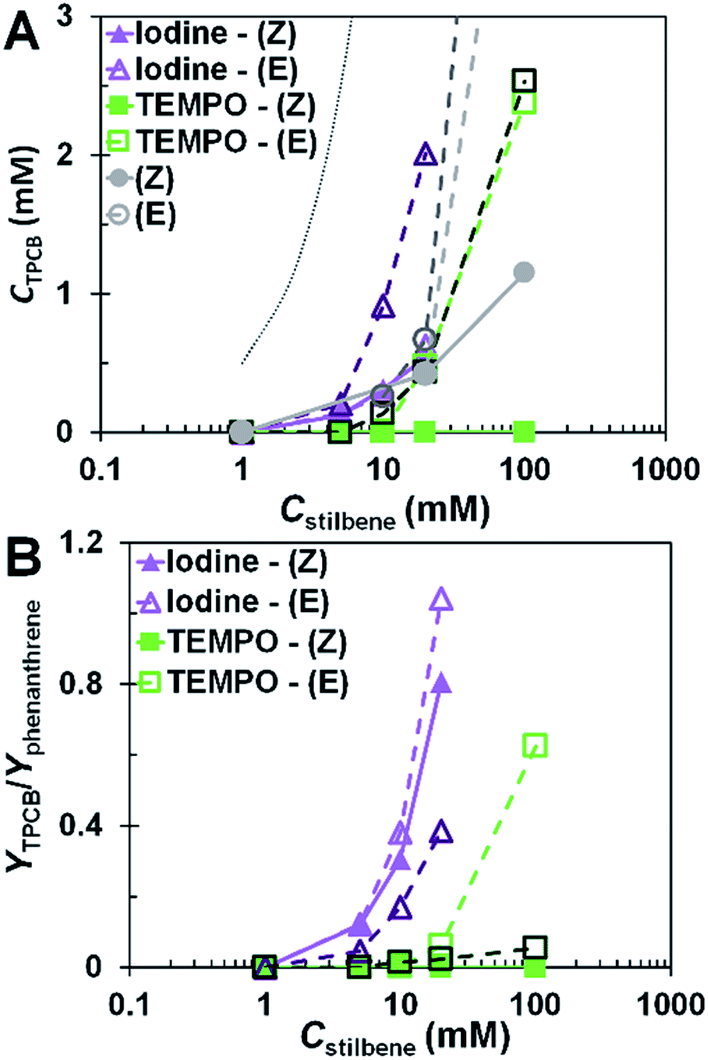 | ||
| Fig. 4 (A) Concentration dependence of TPCB formation. Dotted line represents complete conversion. (B) Relative yield of TPCB to phenanthrene. Data is at 2 h (light lines and symbols) and 16 h (dark lines and symbols). | ||
TPCB was primarily formed when a preponderance of (E)-stilbene was present. The lower yield of TPCB using TEMPO compared to having no oxidizing agent present was attributed to the consumption of (Z)-stilbene for the formation of phenanthrene. This minimized the probability of (Z)-(E) isomerization, thus decreasing the probability for [2+2] photocycloaddition side products to form. Starting with (E)-stilbene enhanced the probability for [2+2] photocycloaddition to occur. Since using TEMPO does not promote (E) isomerization, the main competing reaction was consumption of the as-formed (Z)-stilbene. Iodine-containing samples continued to produce TPCB throughout the reaction which confirmed that iodine promoted (E)-isomerization of stilbene. It can be seen in Fig. 4B that at 20 mM, the ratio of the yield of TPCB to phenanthrene was as high as unity in the presence of iodine after 2 h and only fell to about 0.4 after 16 h. Overall, the ratio of TPCB to phenanthrene formed was much higher with iodine than with TEMPO. This indicated that TEMPO was better suited for mitigating the production of TPCB as concentration increased. The impact of (E)-stilbene on the formation of TPCB along with the (Z) isomer not forming TPCB demonstrated that [2+2] photocycloaddition predominantly occurred through the (E) isomer.
Understanding the photocyclodehydrogenation reaction pathway is relevant to the synthesis of PACs in large quantities for cost-effective commercialization of next-generation applications. Iodine is the conventional oxidizing agent. Although iodine is a stronger oxidizing agent than TEMPO, the UV absorptivity of iodine affected the stereo-conformation of stilbene, causing transformation of (Z)-stilbene to the less productive (E)-stilbene. This inadvertently allowed the undesired [2+2] reaction to occur. In addition, the use of iodine generates a strong acid (HI) that requires neutralization by an excess amount of weak base. The evolution of the acidic by-product stimulates the formation of non-aromatic products.10 This study demonstrated the limitations of using iodine at elevated concentrations. The impact of concentration and stilbene conformation on the photochemical reaction pathway in the presence of select oxidizing agents was studied. Critical aspects of the reaction pathway that limited the scalability of this photochemical reaction were revealed. Phenanthrene formation primarily occurred through (Z)-stilbene and [2+2] cycloaddition primarily occurred through (E)-stilbene. Iodine promoted formation of (E)-stilbene which drastically reduced the phenanthrene formation rate and encouraged the undesired [2+2] cycloaddition reaction. TEMPO did not produce a strongly acidic by-product nor did it encourage (E)-stilbene formation. These advantages, aside from TEMPO being the weaker oxidizing agent, demonstrated that TEMPO was more suitable than iodine at oxidative photocyclodehydrogenation of stilbene, particularly at elevated concentrations. TEMPO facilitated photocyclodehydrogenation of stilbenes at concentrations as high as 100 mM without the occurrence of unwanted side reactions. Using TEMPO revealed that photocyclodehydrogenation reactions were isomer dependent. The impact of (Z)- or (E)-stilbene conformers became more pronounced with increasing concentration.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Air Force Research Laboratory Scholars Program.Notes and references
- F. B. Mallory and C. W. Mallory, in Organic Reactions, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1984, pp. 1–456 Search PubMed.
- R. B. Woodward and R. Hoffman, Angew. Chem., Int. Ed., 1969, 8, 781–853 CrossRef CAS.
- P. Celani, S. Ottani, M. Olivucci, F. Bernardi and M. A. Robb, J. Am. Chem. Soc., 1994, 116, 10141–10151 CrossRef CAS.
- H. Petek, K. Yoshihara, Y. Fujiwara, Z. Lin, J. H. Penn and J. H. Frederick, J. Phys. Chem., 1990, 94, 7539–7543 CrossRef CAS.
- H. Meier, Angew. Chem., Int. Ed. Engl., 1992, 31, 1399–1420 CrossRef.
- K. A. Muszkat, in Organic Chemistry Syntheses and Reactivity, Springer-Verlag, Berlin/Heidelberg, 1980, vol. 88, pp. 89–143 Search PubMed.
- F. B. Mallory, C. S. Wood, J. T. Gordon, L. C. Lindquist and M. L. Savitz, J. Am. Chem. Soc., 1962, 84, 4361–4362 CrossRef CAS.
- A. Bromberg and K. A. Muszkat, J. Am. Chem. Soc., 1969, 91, 2860–2866 CrossRef CAS.
- W. H. Laarhoven, Recl. Trav. Chim. Pays-Bas, 1983, 102, 241–254 CrossRef CAS.
- L. Liu, B. Yang, T. J. Katz and M. K. Poindexter, J. Org. Chem., 1991, 56, 3769–3775 CrossRef CAS.
- K. B. Jørgensen, Molecules, 2010, 15, 4334–4358 CrossRef.
- W. H. Laarhoven and W. J. C. Prinsen, in Stereochemistry, ed. F. Vögtle and E. Weber, Springer Berlin Heidelberg, Berlin, Heidelberg, 1984, vol. 125, pp. 63–130 Search PubMed.
- S. Poplata, A. Tröster, Y.-Q. Zou and T. Bach, Chem. Rev., 2016, 116, 9748–9815 CrossRef CAS.
- A. G. Griesbeck and J. Mattay, Synthetic Organic Photochemistry, Marcel Dekker, New York, 2005 Search PubMed.
- Y. Nakamura, T. Tsuihiji, T. Mita, T. Minowa, S. Tobita, H. Shizuka and J. Nishimura, J. Am. Chem. Soc., 1996, 118, 1006–1012 CrossRef CAS.
- B. S. Green, M. Rejto, D. E. Johnson, C. E. Hoyle, J. T. Simpson, P. E. Correa, T. I. Ho, F. McCoy and F. D. Lewis, J. Am. Chem. Soc., 1979, 101, 3325–3331 CrossRef CAS.
- D. G. Amirsakis, A. M. Elizarov, M. A. Garcia-Garibay, P. T. Glink, J. F. Stoddart, A. J. White and D. J. Williams, Angew. Chem., Int. Ed., 2003, 42, 1126–1132 CrossRef CAS.
- H. Okamoto, T. Takane, S. Gohda, Y. Kubozono, K. Sato, M. Yamaji and K. Satake, Chem. Lett., 2014, 43, 994–996 CrossRef CAS.
- Q. Lefebvre, M. Jentsch and M. Rueping, Beilstein J. Org. Chem., 2013, 9, 1883–1890 CrossRef.
- W. M. Moore, D. D. Morgan and F. R. Stermitz, J. Am. Chem. Soc., 1963, 85(6), 829–830 CrossRef CAS.
- F. B. Mallory, C. S. Wood and J. T. Gordon, J. Am. Chem. Soc., 1964, 86, 3094–3102 CrossRef CAS.
- W. H. Laarhoven, T. J. H. M. Cuppen and R. J. F. Nivard, Recl. Trav. Chim. Pays-Bas, 1968, 87, 687–698 CrossRef CAS.
- N. Minezawa and M. S. Gordon, J. Phys. Chem. A, 2011, 115, 7901–7911 CrossRef CAS.
- C.-M. Chung, F. Nakamura, Y. Hashimoto and M. Hasegawa, Chem. Lett., 1991, 20, 779–782 CrossRef.
- R. Improta and F. Santoro, J. Phys. Chem. A, 2005, 109, 10058–10067 CrossRef CAS.
- D. C. Todd and G. R. Fleming, J. Chem. Phys., 1998, 98, 269 CrossRef.
- I. N. Ioffe, M. Quick, M. T. Quick, A. L. Dobryakov, C. Richter, A. A. Granovsky, F. Berndt, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, J. Am. Chem. Soc., 2017, 139, 15265–15274 CrossRef CAS.
- J. Saltiel, J. Am. Chem. Soc., 1967, 89, 1036–1037 CrossRef CAS.
- M. J. Bearpark, F. Bernardi, S. Clifford, M. Olivucci, M. A. Robb and T. Vreven, J. Phys. Chem. A, 1997, 101, 3841–3847 CrossRef CAS.
- M. Schraub, H. Gray and N. Hampp, Macromolecules, 2011, 44, 8755–8762 CrossRef CAS.
- F. D. Lewis, T. Wu, E. L. Burch, D. M. Bassani, J.-S. Yang, S. Schneider, W. Jaeger and R. L. Letsinger, J. Am. Chem. Soc., 1995, 117, 8785–8792 CrossRef CAS.
- T. Matsushima, S. Kobayashi and S. Watanabe, J. Org. Chem., 2016, 81, 7799–7806 CrossRef CAS.
- J. B. Gerken, Y. Q. Pang, M. B. Lauber and S. S. Stahl, J. Org. Chem., 2018, 83, 7323–7330 CrossRef CAS.
- P. Atkins and J. De Paula, Physical Chemistry, 8th edn, W. H. Freeman and Company, New York, NY, 2006 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and NMR spectra for all compounds. See DOI: 10.1039/d0ra10619d |
| This journal is © The Royal Society of Chemistry 2021 |