
A medium-firm drug-candidate library of cryptand-like structures on T7 phage: design and selection of a strong binder for Hsp90†
Kazuto
Mochizuki‡§
a,
Lisa
Matsukura‡
b,
Yuji
Ito
c,
Naoyuki
Miyashita
 *b and
Masumi
Taki
*a
*b and
Masumi
Taki
*a
aDepartment of Engineering Science, Bioscience and Technology Program, The Graduate School of Informatics and Engineering, The University of Electro-Communications (UEC), 1-5-1 Chofugaoka, Chofu, Tokyo 182-8585, Japan. E-mail: taki@pc.uec.ac.jp; Tel: +81-42-443-5980
bDepartment of Biological Systems Engineering, Graduate School of Biology-Oriented Science and Technology, KINDAI University, 930 Nishimitani, Kinokawa, Wakayama 649-6493, Japan. E-mail: miya@waka.kindai.ac.jp; Tel: +81-736-77-3888
cDepartment of Chemistry and Bioscience, Graduate School of Science and Engineering, Kagoshima University, 1-21-35 Korimoto, Kagoshima, Kagoshima 890-0065, Japan
First published on 14th October 2020
Abstract
We designed and synthesized a medium-firm drug-candidate library of cryptand-like structures possessing a randomized peptide linker on the bacteriophage T7. From the macrocyclic library with a 109 diversity, we obtained a binder toward a cancer-related protein (Hsp90) with an antibody-like strong affinity (KD = 62 nM) and the binding was driven by the enthalpy. The selected supramolecular ligand inhibited Hsp90 activity by site-specific binding outside of the well-known ATP-binding pocket on the N-terminal domain (NTD).
Macrocycles, such as cyclophanes,1,2 calixarenes,3–6 porphyrinoids,7,8 cyclodextrins,9,10 cucurbiturils,11,12 and many other types15 including their hybrids,16–18 have been utilized to recognize specific molecules especially in supramolecular host–guest chemistry19,20 fields. Recently, their applications have been broadened to the fields of polymer/materials chemistry,10,20,21 nanotechnology,22 bio-oriented chemistry,5,10,17,23 optic sensors,24 and more. Among these applications, specific recognition of biopolymers, such as peptides or proteins, by supramolecular ligands6,25–27 as potential targeted-drugs is attracting attention, and yet it is at an early stage.27,28
Meanwhile, we postulate that the limited utilization of such macrocyclic supramolecular ligands for biomolecule recognition arises from a lack of structural diversity. For example, a synthesizable library size of azacrown ether derivatives is usually up to the order of 101.29,30 To expand the diversity, we have previously demonstrated the first example of the construction of a library of azacrown-ether-like structures with vast diversity of 109 on T7 phage via the gp10 based-thioetherification (10BASEd-T![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 31), and successfully found a specific binder toward a cancer-related protein (i.e., Heat shock protein 90 N-terminal domain; Hsp90NTD).14 Nevertheless, one of the major drawbacks of the library is that strong binders as antibody substitutes can never be obtained. Judging from circular dichroism (CD) spectroscopy and isothermal titration calorimetry (ITC) measurements, the whole structure of the obtained binder seems too flexible and an unfavourable entropy loss happens upon the Hsp90 binding. The artificial binder should have possessed more rigidity32 or more enthalpy compensation over the entropy penalty,33 like natural antibodies or cyclic peptides do.
31), and successfully found a specific binder toward a cancer-related protein (i.e., Heat shock protein 90 N-terminal domain; Hsp90NTD).14 Nevertheless, one of the major drawbacks of the library is that strong binders as antibody substitutes can never be obtained. Judging from circular dichroism (CD) spectroscopy and isothermal titration calorimetry (ITC) measurements, the whole structure of the obtained binder seems too flexible and an unfavourable entropy loss happens upon the Hsp90 binding. The artificial binder should have possessed more rigidity32 or more enthalpy compensation over the entropy penalty,33 like natural antibodies or cyclic peptides do.
To increase the affinity while retaining the balance, here we generated a medium-firm34,35 drug-candidate library of cryptand36,37-like structures instead of the library of crown-like structures as shown in Fig. 1, and successfully selected a Hsp90NTD binder with an antibody-like38 strong affinity. The advantages of using the cryptand library over the crown one are: (1) the more constrained cryptand scaffold would minimize the conformational entropy loss upon binding.39 (2) A favourable enthalpy contribution would be expected by increasing the numbers of hydrogen-bond acceptors (i.e., ether oxygen atoms on the macrocycle) when their geometries are optimized32 to fit to the basic protein (i.e., Hsp90NTD, which possesses many hydrogen-bond donors). To design the core structure of the artificial macrocycle, we estimated the flexibility of several core candidates from long-time molecular dynamics (MD) simulations (Fig. 2).40,41 Two out of the three candidates of cryptand-precursors possessed well-balanced flexibility (i.e., cr2 and cr4 in Fig. 2C), and the average distances between the nitrogen atoms of these cryptand-precursors were similar to those of the corresponding crown-precursors (Fig. 2D). In addition to the flexibility and geometric homogeneity balance, cr2 has been known to recognize the side-chain of the basic amino acids (i.e., lysine and arginine).25,42 These points indicate that the cr2 precursor is the most appropriate to the Hsp90 binder.
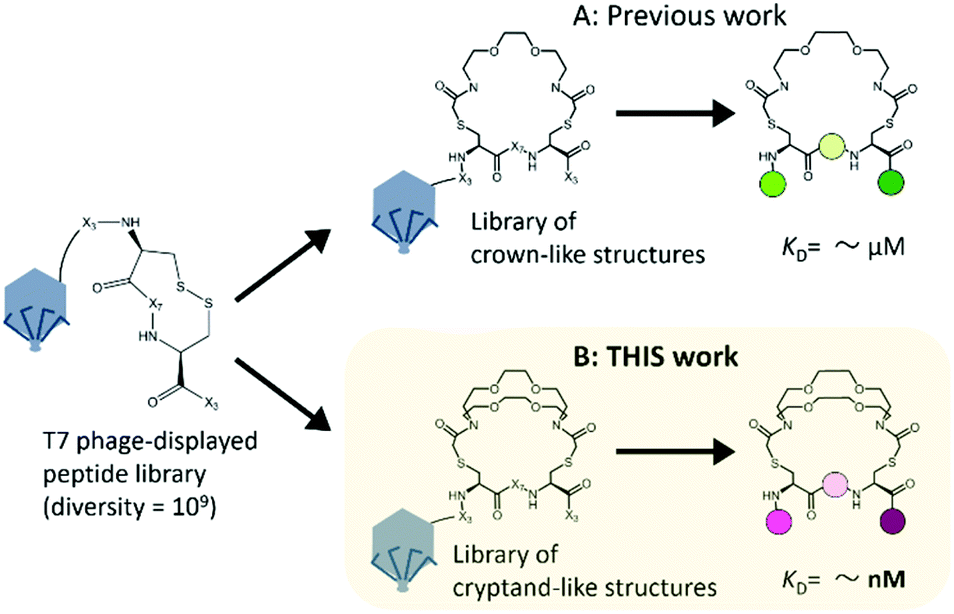 | ||
| Fig. 1 Two types of macrocyclic libraries on bacteriophage T7 for finding Hsp90 binders. The starting peptide library on the phage was the very same; X represents the randomized amino acid. (A) Previous work: a selected crown-like binder possesses a weak affinity for Hsp90NTD.14 (B) This work: a cryptand-like binder possessed a high affinity. | ||
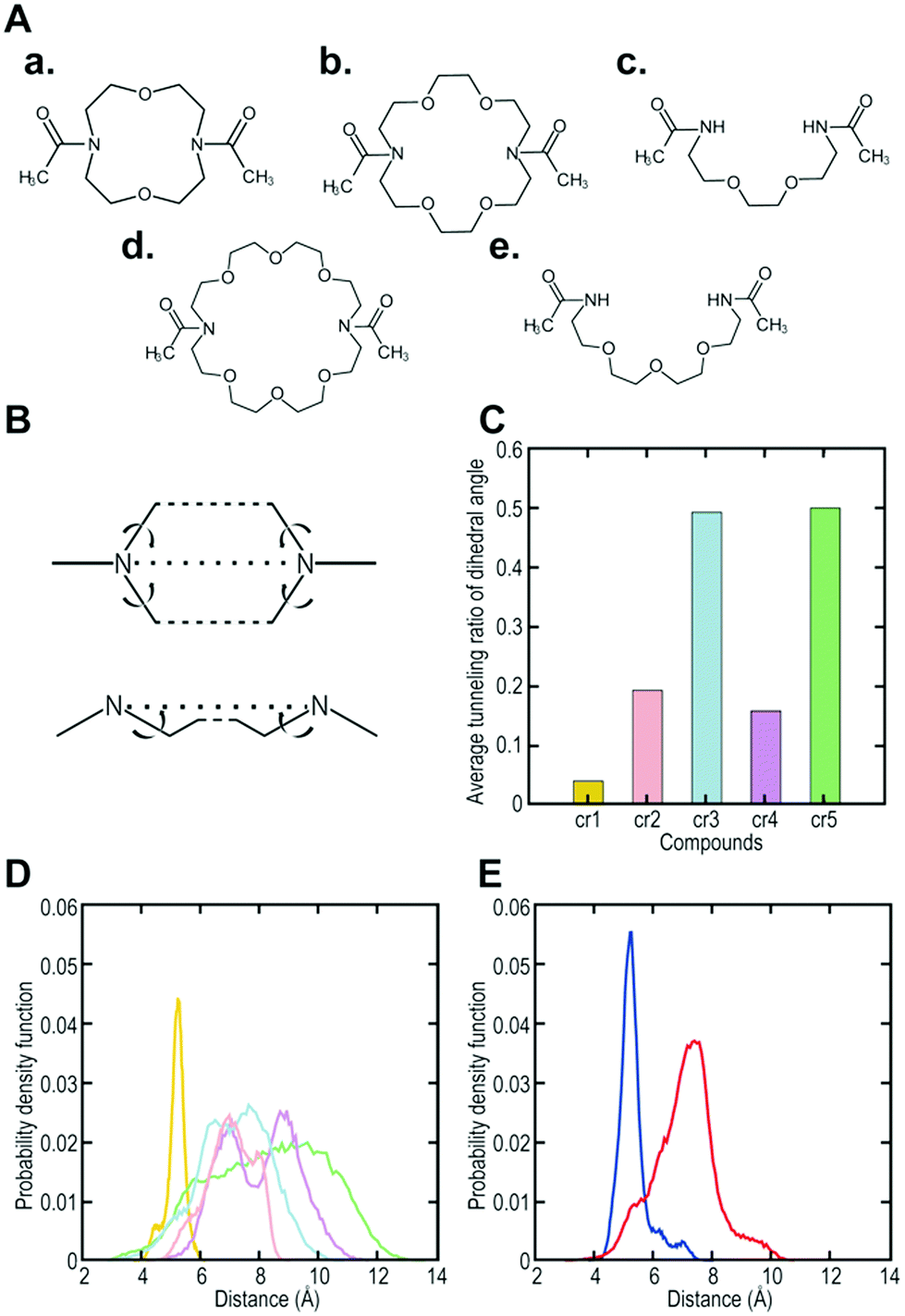 | ||
| Fig. 2 Computer designing of a core structure possessing well-balanced flexibility. (A) (a) to (e) are core candidates (i.e., precursors; cr1, cr2, cr3, cr4, and cr5, respectively) for synthesizing cryptand/crown-like macrocyclic libraries. (B) A schematic view of the dihedral angles to evaluate the fluctuation of the candidate compounds. (C) Average tunneling ratio of the dihedral angle for the candidate compounds. (D) Probability density function of the distance between nitrogen-nitrogen atoms (N–N) in the core candidates. Yellow, pink, cyan, magenta, and green indicate cr1, cr2, cr3, cr4, and cr5, respectively. (E) Probability density function of the distance between N–N in the selected cryptand-like Hsp90NTD binder (blue) or crown-like binder14 (red). | ||
In advance of performing the library construction, we confirmed the expected cyclization using a T7 phage displaying a model peptide (-GSRVS-C-GGRDRPG-C-LSV); we conjugated both amino ends of 1,10-diaza-18-crown-6 ether and designated two cysteines (Cys) on the peptide. After the cyclization against the model phage, the total proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie Brilliant Blue (CBB) staining. The model-peptide-fused T7 capsid protein (i.e., gp10) was excised from the gel and then digested with trypsin. The resulting peptide fragments were analysed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).14 The m/z values of the parent ion from MS and those of the cyclized peptide from MS/MS indicate that Cys in the model phage-displayed peptide were position-specifically conjugated with the diazacrown derivative to form the macrocyclic cryptand structure (Fig. S1, ESI†).
Next, we constructed a library of cryptand-like structures and performed a selection against Hsp90NTD. From a phage-displayed peptide library (-SGGG-X3-C-X7-C-X3; where X represents any amino acids),31,43 the library of cryptand-like structures was constructed in the same way via 10BASEd-T (Fig. 1B). Seven rounds of biopanning were performed against biotinylated-Hsp90NTD fused with glutathione-S-transferase (GST), and enrichment of the Hsp90NTD binders was assessed by enzyme-linked immunosorbent assay (ELISA) as shown in Fig. 3B. After the biopanning, the selected cryptand on the T7 phage polyclone showed the strongest binding to Hsp90NTD, whereas ones lacking the cryptand structure did not (Fig. S2A, ESI†). Among the 9 monoclones of the randomly chosen T7 phage from the polyclone, 6 clones had the same peptide sequence, QWV-C-LNPWLSI-C-RA (Cys was cyclized with the diazacrown derivative) (Fig. S2B, ESI†). The sequence was different from what is discovered from the library of crown-like structures,14 although the starting phage-displayed peptide library is the very same (Fig. 1A). This suggests that the different oligoethylene glycol moieties may affect the geometries of the whole macrocycle structures constructed from the starting library, to generate independent libraries.
 | ||
| Fig. 3 (A) Structure of the selected cryptand-like binder. The azacrown moiety and N-terminal lariat (i.e., QW) were essential for the Hsp90NTD-specific strong binding. (B) The cryptand-binding domain on Hsp90 was identified by enzyme-linked immunosorbent assay (ELISA). The N-terminal (NTD), middle (MD), and C-terminal (CTD) domains of Hsp90 fused with glutathione-S-transferase (GST) were independently assayed against the selected cryptand on phage. Bovine serum albumin (BSA; target-unrelated protein) served as a negative control. (C) The isothermal titration calorimetry (ITC) profiles of the titrations of the selected cryptand against GST-Hsp90NTD. N: number of binding sites, KD: dissociation constant, ΔH: enthalpy change, ΔS: entropy change, T: temperature (i.e., 298 K), and ΔG: free energy change. (D) Identification of the cryptand binding site on Hsp90NTD by mass spectrometry (MS). A crosslinked cryptand-Hsp90NTD was digested with trypsin, and the conjugated fragment (i.e., Hsp90 + cryptand; green line) was analysed by MALDI−TOF MS. A mock crosslinking was also performed in the absence of cryptand (i.e., Hsp90; blue line). The new fragment was observed only when both GST-Hsp90NTD and the selected cryptand were present. | ||
To determine the affinity of the Hsp90NTD-binding cryptand, we chemically synthesized the selected peptide sequence followed by cyclization with the azacrown derivative. ITC measurement was performed to obtain the dissociation constant (KD) as well as the thermodynamic parameters (Fig. 3C). The selected cryptand possessed a high affinity toward GST-Hsp90NTD with the KD value of 62 ± 10 nM, whereas the selected peptide lacking the cryptand structure did not (Fig. S6, ESI†). This indicates that both the structures of the azacrown moiety and the rest of the peptide are essential for the Hsp90-specific strong binding. Also, an expected larger negative enthalpy change (ΔH = −16.9 ± 0.3 kcal mol−1) compared to our previous crown-like binder (−13.7 ± 1.6 kcal mol−1),14 was observed, suggesting that the numbers of hydrogen bonds and van der Waals interactions may contribute to the favourable binding between the selected cryptand and Hsp90NTD. To investigate the secondary structure of the macrocycle, we measured the circular dichroism (CD) spectra before and after the cyclization of the selected peptide (Fig. S3, ESI†). They were almost superimposable on each other, which indicated that the selected peptide possessed a β-turn structure regardless of the azacrown moiety. Taking into account the rigidity of the β-turn, we tentatively supposed that a slight unfavourable entropy change (−TΔS = +7.0 kcal mol−1) could arise from the enthalpy penalty associated with desolvation of the polar groups (i.e., the entropy-enthalpy compensation),32 rather than from the conformational entropy loss deduced from our previous crown-like binder,14 which possesses a more flexible core structure. In the MD simulations, it was shown that the fluctuation in the cryptand-like binder was less than that in the crown-like binder (Fig. 2E). Moreover, the interaction between Trp2 (W2) and Arg13 (R13) was frequently found during a long-time MD simulation of the cryptand-like binder (Fig. S12D†). The importance of W2 at the N-terminal lariat for the strong binding was also indicated experimentally; a Q1W2-deleted cryptand at the same molar concentration showed less intense saturation transfer difference (STD) signals44 in the proton nuclear magnetic resonance (1H NMR) spectroscopy (Fig. S4B, ESI†), which increased the dissociation constant in the ITC measurements by an order of about 103 (Fig. S6, ESI†). To sum up, appropriate electrostatic/hydrophobic effects arose from whole regions (i.e., azacrown and peptide moieties) of the cryptand to afford a strong supramolecular interaction toward Hsp90NTD.
Using the selected cryptand, we performed more detailed structural analysis including binding-site determination on Hsp90NTD. A fluorescence polarization (FP) competition assay using fluorescein-5-isothiocyanate labelled geldanamycin (GA-FITC), which binds to the ATP-binding pocket on Hsp90NTD, indicated that the selected cryptand did not compete with GA-FITC (Fig. S5, ESI†). This means that the cryptand bound to the outside of the ATP-binding pocket unlike most of the reported Hsp90NTD inhibitors.45,46 To determine the binding site, the cryptand on Hsp90NTD was covalently conjugated with a bifunctional crosslinker (succinimidyl-ester diazirine; SDA). After the hetero-crosslinking, it was digested with trypsin and the crosslinked fragment was analysed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS; Fig. 3D). From the m/z value of a newly appeared fragment, the binding site was deduced to be the β-sheets region in Hsp90NTD, which was consistent with the FP competition assay above. Also, the binding site was determined by a long-time MD simulation between the selected cryptand and Hsp90NTD,47 followed by a docking simulation by AutoDock Vina.48 As shown in Fig. 4, a most reasonable structure was obtained, which was again consistent with all of the experimental results. The β-sheets region of Hsp90NTD, in which the cryptand bound, could possibly be a novel drug targeting site, because Hsp90 chaperone machinery was inhibited upon binding (Fig. S7†).49
 | ||
| Fig. 4 The docking model of the cryptand and Hsp90NTD deduced from both experimental and computational results. Yellow and purple indicate the β-sheets and α-helices.13 The cryptand-like binder, which is indicated by a red colour, bound to the β-sheets region. | ||
Conclusions
In conclusion, we demonstrated the first example of the design and construction of a library of an unclosed-cryptand36 with two lariat arms using the T7-phage display system, and successfully selected a strong Hsp90NTD-specific binder. It is emphasized that such a discovery of a functional supramolecular ligand was attained by an ensemble of MD-simulation-assisted rational design of the artificial macrocyclic moiety and genetically-encoded random peptide structure.Since there are sophisticated alternative library-construction systems for obtaining macrocyclic targeted drugs50,51 with strong affinities,52–55 we think that using such an artificial moiety just for the target recognition is not the final achievement. More advanced artificial functionalization on the moiety, such as the addition of catalytic activity (e.g., proteolytic actions by complexation with metals56,57), fluorescent chemosensors,58 optochemical properties,59 or cell permeability,60–62 could be one of the future directions of bio-oriented supramolecules possessing expanded diversities.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers 17K05925, 17J10365 and 17K00421 to M.T., K.M., and N.M., respectively. It was also supported by UEC internal research grant. Simulation studies used the computational resources of MOMO provided by BOST Computer Centre, KINDAI University (N.M., L.M.), and TSUBAME3.0 provided by Tokyo Institute of Technology through TSUBAME Encouragement Program for Young/Female Users (Project ID: 19IJ0037) (L.M.). We are grateful to Dr F.U. Hartl for providing plasmids, Dr Y. Minami for providing proteins, Dr K. Ikebukuro and Dr J. Lee for use of ITC instrument installed by the Low-Carbon Research Network Japan (LCnet), Dr S. Hosoya (TMDU), and Mr Ordan for technical assistance and valuable suggestions.Notes and references
- Z. Hassan, E. Spuling, D. M. Knoll and S. Brase, Angew. Chem., Int. Ed., 2020, 59, 2156–2170 CrossRef CAS.
- J. Nishimura, Y. Nakamura, Y. Hayashida and T. Kudo, Acc. Chem. Res., 2000, 33, 679–686 CrossRef CAS.
- L. Troian-Gautier, A. Mattiuzzi, O. Reinaud, C. Lagrost and I. Jabin, Org. Biomol. Chem., 2020, 18, 3624–3637 RSC.
- M. X. Wang, Acc. Chem. Res., 2012, 45, 182–195 CrossRef CAS.
- S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366–386 RSC.
- A. M. Doolan, M. L. Rennie and P. B. Crowley, Chem. – Eur. J., 2018, 24, 984–991 CrossRef CAS.
- W. Stawski, M. Kijewska and M. Pawlicki, Chem. – Asian J., 2020, 15, 8–20 CrossRef CAS.
- R. S. Czernuszewicz, V. Mody, A. Czader, M. Galezowski and D. T. Gryko, J. Am. Chem. Soc., 2009, 131, 14214–14215 CrossRef CAS.
- Y. M. Zhang, Y. H. Liu and Y. Liu, Adv. Mater., 2020, 32, e1806158 CrossRef.
- B. Kost, M. Brzezinski, M. Socka, M. Basko and T. Biela, Molecules, 2020, 25, 3404 CrossRef CAS.
- K. M. Park, M. Y. Hur, S. K. Ghosh, D. R. Boraste, S. Kim and K. Kim, Chem. Commun., 2019, 55, 10654–10664 RSC.
- L. Isaacs, Acc. Chem. Res., 2014, 47, 2052–2062 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics Modell., 1996, 14, 33–38 CrossRef CAS.
- K. Fukunaga, T. Hatanaka, Y. Ito, M. Minami and M. Taki, Chem. Commun., 2014, 50, 3921–3923 RSC.
- L. P. Yang and W. Jiang, Angew. Chem., 2020, 59, 15794–15796 CrossRef CAS.
- E. Pazos, P. Novo, C. Peinador, A. E. Kaifer and M. D. Garcia, Angew. Chem., Int. Ed., 2019, 58, 403–416 CrossRef CAS.
- I. M. Mavridis and K. Yannakopoulou, J. Med. Chem., 2020, 63, 3391–3424 CrossRef CAS.
- X. Wu, Y. Chen, Q. Yu, F. Q. Li and Y. Liu, Chem. Commun., 2019, 55, 4343–4346 RSC.
- A. Blanco-Gomez, P. Corton, L. Barravecchia, I. Neira, E. Pazos, C. Peinador and M. D. Garcia, Chem. Soc. Rev., 2020, 49, 3834–3862 RSC.
- S. Dong, B. Zheng, F. Wang and F. Huang, Acc. Chem. Res., 2014, 47, 1982–1994 CrossRef CAS.
- K. Jie, Y. Zhou, E. Li and F. Huang, Acc. Chem. Res., 2018, 51, 2064–2072 CrossRef CAS.
- V. Montes-Garcia, J. Perez-Juste, I. Pastoriza-Santos and L. M. Liz-Marzan, Chemistry, 2014, 20, 10874–10883 CrossRef CAS.
- C. C. Lee, M. Maestre-Reyna, K. C. Hsu, H. C. Wang, C. I. Liu, W. Y. Jeng, L. L. Lin, R. Wood, C. C. Chou, J. M. Yang and A. H. J. Wang, Angew. Chem., Int. Ed., 2014, 53, 13054–13058 CrossRef CAS.
- M. Qi, N. M. Y. Zhang, K. Li, S. C. Tjin and L. Wei, Sensors, 2020, 20, 3266 CrossRef CAS.
- S. X. Fa and Y. Zhao, Chem. Mater., 2019, 31, 4889–4896 CrossRef CAS.
- Y. Zhao, Chem. – Eur. J., 2018, 24, 14001–14009 CrossRef CAS.
- S. van Dun, C. Ottmann, L. G. Milroy and L. Brunsveld, J. Am. Chem. Soc., 2017, 139, 13960–13968 CrossRef CAS.
- T. Yokoyama and M. Mizuguchi, J. Med. Chem., 2019, 62, 2076–2082 CrossRef CAS.
- Y. Luo, G. Ouyang, Y. Tang, Y. M. He and Q. H. Fan, J. Org. Chem., 2020, 85, 8176–8184 CrossRef CAS.
- G. Farruggia, S. Iotti, M. Lombardo, C. Marraccini, D. Petruzziello, L. Prodi, M. Sgarzi, C. Trombini and N. Zaccheroni, J. Org. Chem., 2010, 75, 6275–6278 CrossRef CAS.
- K. Fukunaga, T. Hatanaka, Y. Ito and M. Taki, Mol. BioSyst., 2013, 9, 2988–2991 RSC.
- E. Freire, Drug Discovery Today, 2008, 13, 869–874 CrossRef CAS.
- B. Zhao, P. Xu, L. Jiang, B. Paaske, T. Kromann-Hansen, J. K. Jensen, H. P. Sorensen, Z. Liu, J. T. Nielsen, A. Christensen, M. Hosseini, K. K. Sorensen, N. C. Nielsen, K. J. Jensen, M. Huang and P. A. Andreasen, PLoS One, 2014, 9, e115872 CrossRef.
- B. C. Doak, J. Zheng, D. Dobritzsch and J. Kihlberg, J. Med. Chem., 2016, 59, 2312–2327 CrossRef CAS.
- F. Giordanetto and J. Kihlberg, J. Med. Chem., 2014, 57, 278–295 CrossRef CAS.
- K. Dabrowa, M. Pawlak, P. Duszewski and J. Jurczak, Org. Lett., 2012, 14, 6298–6301 CrossRef CAS.
- Y. Han, Y. Jiang and C. F. Chen, Tetrahedron, 2015, 71, 503–522 CrossRef CAS.
- R. Ahirwar, S. Nahar, S. Aggarwal, S. Ramachandran, S. Maiti and P. Nahar, Sci. Rep., 2016, 6, 21285 CrossRef CAS.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608–624 CrossRef CAS.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS.
- S. Fa and Y. Zhao, Chem. Mater., 2017, 29, 9284–9291 CrossRef CAS.
- T. Hatanaka, S. Ohzono, M. Park, K. Sakamoto, S. Tsukamoto, R. Sugita, H. Ishitobi, T. Mori, O. Ito, K. Sorajo, K. Sugimura, S. Ham and Y. Ito, J. Biol. Chem., 2012, 287, 43126–43136 CrossRef CAS.
- A. Viegas, J. Manso, F. L. Nobrega and E. J. Cabrita, J. Chem. Educ., 2011, 88, 990–994 CrossRef CAS.
- L. Li, L. Wang, Q. D. You and X. L. Xu, J. Med. Chem., 2020, 63, 1798–1822 CrossRef CAS.
- T. Ueda, T. Tamura and I. Hamachi, Biochemistry, 2020, 59, 179–182 CrossRef CAS.
- R. Galindo-Murillo, J. C. Robertson, M. Zgarbova, J. Sponer, M. Otyepka, P. Jurecka and T. E. Cheatham, J. Chem. Theory Comput., 2016, 12, 4114–4127 CrossRef CAS.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- J. Davenport, M. Balch, L. Galam, A. Girgis, J. Hall, B. S. Blagg and R. L. Matts, Biology, 2014, 3, 101–138 CrossRef CAS.
- P. G. Dougherty, Z. Q. Qian and D. H. Pei, Biochem. J., 2017, 474, 1109–1125 CrossRef CAS.
- T. Passioura, Biochemistry, 2020, 59, 139–145 CrossRef CAS.
- T. Anananuchatkul, I. V. Chang, T. Miki, H. Tsutsumi and H. Mihara, ACS Omega, 2020, 5, 5666–5674 CrossRef CAS.
- S. Chen, I. R. Rebollo, S. A. Buth, J. Morales-Sanfrutos, J. Touati, P. G. Leiman and C. Heinis, J. Am. Chem. Soc., 2013, 135, 6562–6569 CrossRef CAS.
- A. A. Vinogradov, Y. Z. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS.
- C. Heinis and G. Winter, Curr. Opin. Chem. Biol., 2015, 26, 89–98 CrossRef CAS.
- S. Mahesh, K. C. Tang and M. Raj, Molecules, 2018, 23, 2615 CrossRef.
- J. Suh, Asian J. Org. Chem., 2014, 3, 18–32 CrossRef CAS.
- M. A. Beatty, A. J. Selinger, Y. Li and F. Hof, J. Am. Chem. Soc., 2019, 141, 16763–16771 CrossRef CAS.
- M. R. Jafari, H. Yu, J. M. Wickware, Y. S. Lin and R. Derda, Org. Biomol. Chem., 2018, 16, 7588–7594 RSC.
- J. Mosquera, I. Garcia and L. M. Liz-Marzan, Acc. Chem. Res., 2018, 51, 2305–2313 CrossRef CAS.
- J. Rodriguez, J. Mosquera, J. R. Couceiro, J. R. Nitschke, M. E. Vazquez and J. L. Mascarenas, J. Am. Chem. Soc., 2017, 139, 55–58 CrossRef CAS.
- H. Fernandez-Caro, I. Lostale-Seijo, M. Martinez-Calvo, J. Mosquera, J. L. Mascarenas and J. Montenegro, Chem. Sci., 2019, 10, 8930–8938 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01855d |
| ‡ These authors equally contributed to the work. |
| § Current affiliation: Tokyo Metropolitan Industrial Technology Research Institute, 2-4-10 Aomi, Koto-ku, Tokyo 135-0064, Japan. |
| This journal is © The Royal Society of Chemistry 2021 |