
Iron oxide nanoparticles for immune cell labeling and cancer immunotherapy
Seokhwan
Chung
,
Richard A.
Revia
and
Miqin
Zhang
 *
*
Department of Materials Science and Engineering, University of Washington, Seattle, Washington 98195, USA. E-mail: mzhang@uw.edu
First published on 15th July 2021
Abstract
Cancer immunotherapy is a novel approach to cancer treatment that leverages components of the immune system as opposed to chemotherapeutics or radiation. Cell migration is an integral process in a therapeutic immune response, and the ability to track and image the migration of immune cells in vivo allows for better characterization of the disease and monitoring of the therapeutic outcomes. Iron oxide nanoparticles (IONPs) are promising candidates for use in immunotherapy as they are biocompatible, have flexible surface chemistry, and display magnetic properties that may be used in contrast-enhanced magnetic resonance imaging (MRI). In this review, advances in application of IONPs in cell tracking and cancer immunotherapy are presented. Following a brief overview of the cancer immunity cycle, developments in labeling and tracking various immune cells using IONPs are highlighted. We also discuss factors that influence the effectiveness of IONPs as MRI contrast agents. Finally, we outline different approaches for cancer immunotherapy and highlight current efforts that utilize IONPs to stimulate immune cells to enhance their activity and response to cancer.
 Seokhwan Chung | Seokhwan Chung earned his BS in materials and science engineering from the University of Illinois at Urbana-Champaign. He is currently pursuing his PhD degree in materials science and engineering at the University of Washington. His current research interest is in synthesis and application of iron oxide and carbon-based nanomaterials for cancer therapy and diagnostics. |
 Richard A. Revia | Richard A. Revia earned his BS in biomedical engineering from the University of Texas at Austin in 2008 and his MS in electrical engineering from Montana State University in 2013. He received his PhD degree in materials science and engineering at the University of Washington in 2020. He is currently pursuing his MD at University of Central Florida. His research interests include the development of nanoparticle platforms as contrast agents in magnetic resonance imaging and designing devices for use in tissue engineering. |
 Miqin Zhang | Miqin Zhang is Kyocera Chair Professor of Materials Science & Engineering and Neurological Surgery, and adjunct professor in the Departments of Bioengineering, Radiology, and Orthopaedics & Sports Medicine at the University of Washington (UW). She received her PhD from University of California at Berkley in 1999. Her research focuses on nanomedicine for cancer diagnosis and treatment, biomaterials for tissue engineering, and biosensors for detection of chemical and biological agents. |
1 Introduction
Cancer remains as one of the leading causes of mortality worldwide. Current treatment methods include invasive surgery, toxic chemotherapeutics, and low-specificity radiotherapy, all of which have their attendant shortcomings or harmful side effects; furthermore, advanced malignant tumors can develop resistance to drug and radiation treatments over time, diminishing the efficacy of these therapeutic efforts.1–5 Immunotherapy has emerged as an alternative approach to cancer remediation.1,6,7 By harnessing the natural defense mechanisms of the patient, immunotherapy elicits an antitumor response that is systemic and specific to the tumor, thereby overcoming the lack of specificity associated with current chemotherapeutic and radiotherapy approaches. Immunotherapy also creates a prolonged antitumor response through immunological memory.8–11 Immunotherapeutic approaches to cancer therapy include adoptive cell therapy,12–14 cancer vaccines,15–18 monoclonal antibodies for checkpoint blockade,19–21 and modulation of the tumor microenvironment.22,23 Several cancer immunotherapy drugs have either been approved by the United States Food and Drug Administration (FDA) or begun clinical trials, and promising results have brought immunotherapy one step closer to being part of the standard treatment regimen for many forms of cancer.24–27Understanding the role and function of immune cells in response to cancer has led to the development of novel therapeutic approaches. Cell tracking, which includes observation of migration, expansion, and depletion of immune cells, in cell-based immunotherapy allows more informed decision making process in clinical trials, ultimately leading to improved efficacy and safety of the therapy.28 Advances in the field of molecular bioimaging have brought about the development of non-invasive modalities of dynamic in vivo imaging of biologically active immune cells, elucidating factors such as targeting efficiency, pharmacokinetics, spatial heterogeneity in therapeutic delivery, and correlation between therapeutic presence and efficacy.29 The impact of an effective cell tracking strategy extends beyond cancer immunotherapy, and can be applied to various diseases arising from immune disorders by better understanding the role of immune cells in various tissues and pathological conditions. By coupling imaging modalities with effective cell labeling strategies, cell tracking could shed light on the complex cellular and molecular mechanisms utilized by the immune system and lead to the development of novel and sophisticated immunotherapeutic approaches.
Among the various molecular imaging modalities used in clinical settings, single photon emission computed tomography (SPECT) and positron emission tomography (PET) have been applied to image cells labelled with radioactive tracers. Despite their high sensitivity, these methods are limited by the use of radiotracers with short half-life, use of ionizing radiation, low spatial resolution, and cost.30 Fluorescence and bioluminescence-based whole-body imaging has shown promise in animal models, but are inherently limited by their tissue penetration depth and their two-dimensional nature.29,31 In contrast, MRI is a non-invasive imaging modality that provides high-resolution images of the body's soft tissues using the signals generated by protons present throughout the body. With the use of contrast agents, which can alter the signal from the protons, labelled immune cells can be imaged with high contrast against the background from the host tissue.32 Iron oxide nanoparticles (IONPs) have been used as MRI contrast agents due to their superparamagnetic properties, excellent water solubility, and biocompatibility.33–39 Labeling cells with IONPs allows for the monitoring of therapeutic delivery and tracking of cells such as immune cells and stem cells in vivo via contrast-enhanced MRI, and can be used to improve and evaluate therapeutic outcomes.40,41
In addition to their application in MRI-based cell tracking, IONPs have been used to improve the efficacy of current immunotherapeutic approaches in in vivo studies. In addition to biocompatibility, IONPs are also known to be biodegradable; as iron is utilized by various cellular processes and can be found abundantly throughout the human body, IONPs do not pose long-term toxicity concerns regarding their degradation products. This presents an advantage of IONPs over other inorganic nanoparticle systems as novel platforms for improving cancer immunotherapy. Other nanoparticle systems such as liposomes and polymeric nanoparticles suffer from hydrophobicity, poor stability, and large size, whereas IONPs can be tailored to display specific physiochemical properties such as hydrodynamic size and surface charge.42,43
The iron oxide core of IONPs has high surface energy and chemical reactivity and needs to be surrounded by molecules such as polymers, lipids, or proteins in order to lower its chemical potential in biological applications (otherwise, the surfaces of the IONPs will quickly be opsonized by innate proteins in the body to mark the IONPs for removal from circulation before they may impart any beneficial action onto the host). In addition, these coatings may be engineered to allow flexible surface chemistry for conjugation of therapeutics and antibodies. Furthermore, therapeutic efficiency can be improved through active targeting by conjugation of homing ligands onto the surface of IONPs, increasing the selectivity and specificity towards cell types of interest. Due to these properties, IONPs have been incorporated into cancer immunotherapy through applications such as improving the efficiency of therapeutic and regulatory molecules to immune cells, increasing the presence of immune cells at the tumor sites through magnetic-guided cell delivery, and inducing local hyperthermia as a part of combination therapy with delivery of immunostimulants.
In this review, we highlight the versatility of IONPs in immune cell tracking and their application in cancer immunotherapy. After a brief overview of the cancer immunity cycle, we present recent advances in MRI-based immune cell tracking and cell labeling techniques. Then, the effects of various factors on IONP-mediated MRI imaging are discussed. Finally, a review of applications of IONP in various immunotherapeutic approaches will be provided.
2 Cancer immunity cycle
Understanding the response of the immune system to cancer is important in discussing the specific molecular and cellular pathways that are exploited in cancer immunotherapy, and in turn, how these processes can be enhanced through the use of IONPs. The cancer immunity cycle describes the interaction between the immune system and tumor cells.44 Though specific pathways and biomarkers vary amongst different cancer types, the initial step in the cancer immune response begins with the release of cancer antigens by cancer cells (Fig. 1a). Antigens are, by definition, substances (usually proteins or carbohydrates) that elicit an immune response from their host, and are classified according to their source. Although cancer cells are endogenous to the body, cancer antigens are classified as neoantigens, meaning that they are absent from the normal human genome as they arise from mutations in normal cells. Cancer antigens can be either tumor-specific antigens, present only in tumor cells, or tumor-associated antigens, which are aberrantly expressed in tumor cells but also found in normal cells and therefore, can induce central immune tolerance.45 These antigens are captured by antigen-presenting cells (APCs) such as dendritic cells (DCs) (Fig. 1b). The DCs process and present these antigens onto their surface with major histocompatibility complex class I or II molecules and migrate to lymph nodes (Fig. 1c). Naïve T cells residing in lymph nodes can then recognize the antigens present on DCs through T-cell receptors and this interaction subsequently leads to priming and activation of T cells which are then able to migrate away from the lymph node and recognize the antigen on tumor cells (Fig. 1d). Different types of T cells play different roles in cancer immunotherapy: CD8+ T cells are also known as cytotoxic T lymphocytes (CTLs) and can directly recognize antigens and kill tumor cells, whereas CD4+ T cells are helper T cells and play an indirect role by regulating the immune response through release of cytokines that can activate and signal other immune cells including CTLs (Fig. 1e). Activated T cells migrate and infiltrate tumors, and an increased presence of T cells in the tumor microenvironment has been associated with improved prognosis in various cancer types46,47 (Fig. 1f). The release of cancer antigens upon tumor cell death restarts the cancer immunity cycle again.48,49 In addition to the main cancer immunity cycle, other types of immune cells are also involved in the immune response to cancer, such as natural killer (NK) cells that can identify and kill tumor cells by identifying oncogenic transformations, and macrophages in the tumor environment that can regulate the inflammatory response and recruitment of other immune cells.50,51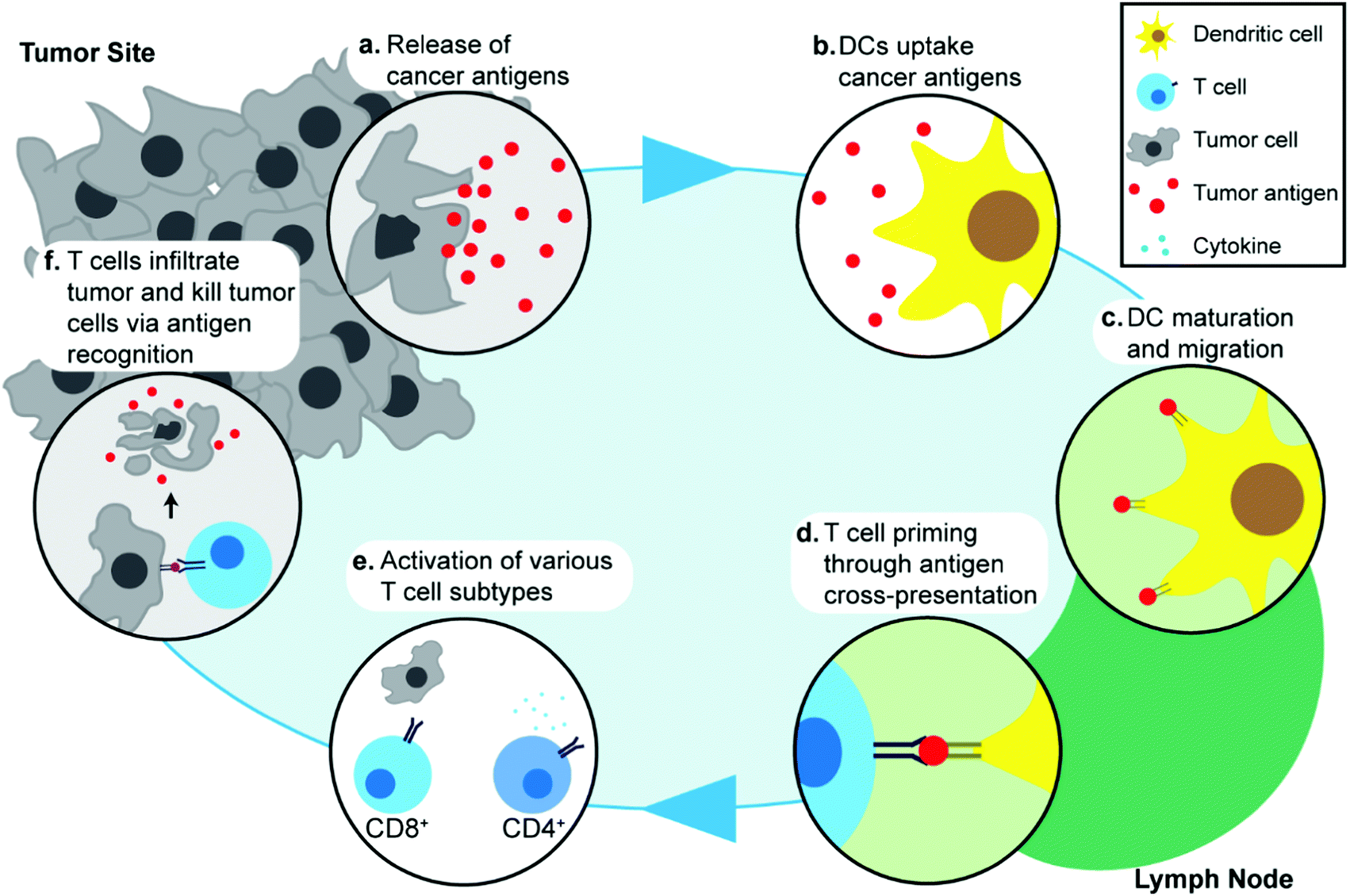 | ||
| Fig. 1 A schematic representation of the immune system's response to cancer. (a) Tumor cells release tumor antigens as they die. (b) These tumor antigens are recognized by antigen-presenting cells such as dendritic cells. (c) Dendritic cells undergo maturation and present the processed antigen on their surface with the major histocompatibility complex. Matured dendritic cells migrate to lymph nodes (d) Dendritic cells present antigens to T cells resident in the lymph node, and T cells can recognize the antigens using T cell receptors. Upon this recognition, T cells are activated into cytotoxic T lymphocytes. (e) The activated T cells then migrate to the tumor site. Various subtypes of T cells are involved in immune response. CD8+ cells are directly involved in killing tumor cells, whereas CD4+ cells release cytokines to regulate the immune response. (f) Activated T cells recognize tumor cells and induce apoptosis, upon which more tumor antigens are released, beginning the cycle again. | ||
As demonstrated through this cycle, migration of specific immune cells to the correct site is critical in eliciting a successful antitumor response. The ability to track and monitor innate or implanted immune cells in therapeutic approaches such as adoptive cell therapy and DC-based vaccines would be invaluable in assessing the efficacy of such treatments. Dynamic tracking of the immune cell migration would also allow early assessment of therapeutic effects, which in turn would help in determination of suitable treatment regimen for personalized therapy. Development of imaging systems capable of tracking immune cells in vivo in clinical settings using nanotechnology would lead to better understanding of the specific pathways in the cancer immunity cycle, and aid in development of novel therapeutic methods.
3 IONP-mediated immune cell tracking
As proper migration of immune cells is necessary for activation of antitumor immune responses, immune cell tracking provides a method to assess the efficacy of immunotherapy treatments. The ability to visualize the distribution and migration of specific immune cells throughout the body would lead to not only a better understanding of the role of immune cells in cancer therapy, but also identification of biological targets for more effective immunotherapeutic strategies. Non-invasive, real-time monitoring of immune cells would allow rapid evaluation of patient response to the therapeutic approach, and modification of the therapy for personalized treatment regimen. In order to visualize the immune cells of interest through MRI, the cells must be labelled with contrast agents in order to distinguish the cells from surrounding tissues and other cells. While labeling of certain cells such as macrophages is possible through systemic injection of IONPs, development of cell-based immunotherapeutic approaches, such as adoptive cell therapy where target cells are isolated and modified ex vivo and then transplanted into the patient, suggest ex vivo labeling of immune cells is more viable.52,53 Furthermore, ex vivo labeling not only leads to greater signal contrast, but also eliminates the need to inject high concentrations of IONP systemically to achieve similar signal intensity in the cells of interest. In this section, applications of IONPs in tracking different types of immune cells will be presented, along with insights to the approaches to labeling the immune cells.3.1 DCs
DCs play an important role in the cancer immunity cycle by first recognizing and internalizing cancer antigens and then processing and presenting the antigens on the surface of the DCs. The migration of DCs into regional draining lymph nodes after uptake of antigens in peripheral organs allows DCs to activate CTLs and regulate adaptive immune responses. Several studies have demonstrated the capability of tracking DCs with MRI by transplanting IONP-labeled DCs into the flanks or footpads of mice and imaging nearby lymph nodes for changes in MRI signal.18,54,55 In addition to altering the properties of the IONPs, studies using clinical 3 T scanners and optimized MRI pulse sequences demonstrate the clinical relevance of utilizing IONPs for DC tracking.56 Phagocytic cells, including DCs, macrophages, and monocytes (which can differentiate into the other two cell types), can internalize IONPs via endocytosis for IONPs with diameters between 20 and 200 nm. In studies comparing cellular uptake of IONPs in various immune cell types, DCs showed higher intracellular iron concentration compared to non-phagocytic cells, such as T cells.15 The utility of IONPs as immunotherapeutic imaging probes can be improved by conferring additional functionalities onto the nanoparticle. Nanoparticles consisting of Fe3O4-cores and ZnO-shells were synthesized as MRI/fluorescence multimodal imaging agents. ZnO-binding peptides were also utilized to deliver tumor antigens to DCs so that the nanoparticles could stimulate and monitor the migration of DCs simultaneously. DCs labelled with either ZnO-IONPs or ZnO nanoparticles were injected into the hind footpads of C57BL/6 mice, and popliteal lymph nodes were observed for changes in MRI signal to monitor the migration of DCs. T2-weighted signal reduction was seen in the central region of the lymph node on the side with ZnO-IONP-labelled DCs as a darker region, whereas no T2-weighted signal reduction was observed in the lymph nodes corresponding to the injection site where DCs were labelled with ZnO nanoparticles. Furthermore, injection of free ZnO-IONPs instead of nanoparticle-labelled DCs also did not result in significant reduction in T2, showing that the signal change observed in lymph nodes was due to migration of DCs labelled with the ZnO-IONPs.573.2 T cells
T cells are a type of lymphocyte that originate from the thymus and reside in lymph nodes. Upon interaction with cancer antigens on APCs, naïve T cells can differentiate into various types of T cells that can contribute to eliminating tumor cells.58 While the ultimate evaluation of therapeutic efficacy is dependent on the observation of tumor reduction post-treatment, visualization of T cell migration could reveal the activity and function of T cells in the therapeutic approach in a timely manner. Coupled with growing interest in adoptive T cell therapy, MRI-mediated T cell tracking would be an invaluable tool in understanding the role and behavior of adoptive T cells in vivo.59,60 Recently, an IONP-based dual modality (MRI/fluorescence) cellular imaging probe was developed, consisting of an iron oxide core coated with PEG conjugated with fluorescent dyes. These IONPs did not alter the cellular function of human and murine T cells and were also effective labels for T cell tracking in vivo in animal models. In this study, labelled T cells were injected intravenously and migration of T cells to transplanted allograft heart and lung as a result of immune rejection was observed. Regions of hypointense MRI signal were observed in both allograft heart and lung 24 h and 48 h after administration of IONPs, corresponding to increased presence of T cells in the organs as part of an acute rejection of the transplants.61 Similar results were also observed in another study that utilized IONPs conjugated with rhodamine B as dual MRI/fluorescence probes.62However, the non-phagocytic nature of T cells is a barrier to overcome for efficient labeling of T cells. Internalization of IONPs by T cells was observed to be an order of magnitude lower than that of monocytes and other phagocytic immune cells.15,34 Various methods such as electroporation and surface modification with human immunodeficiency virus 1 (HIV-1) transactivator peptides, to improve the cellular uptake of IONPs by T cells via transfection, have been explored.63–65 Transfection agents aid the crossing of the cell membrane and can be used to facilitate the internalization of IONPs for cell tracking applications. IONPs and transfection agents were mixed to form complexes, which were incubated with T cells extracted from Lewis rats. The transfection agents tested include lipofectamine, poly-L-lysine, polyethyleneimine (PEI), and FuGene6, a commercially available lipidic multicomponent transfection agent. The labeling efficiency was analyzed through magnetic separation and X-ray fluorescence spectroscopy. IONP:PEI complexes, yielded the highest labeling efficiency (60%), followed by poly-L-lysine and lipofectamine, and FuGene6. The trend in the labeling efficiency was correlated with the zeta potential of IONP:PEI as PEI had the highest zeta potential. IONP:PEI was also shown to induce the greatest cytotoxicity, indicating a trade-off between labeling efficiency and cytotoxicity.66 As advances in T cell tracking are made, efficacy of T cell-based cancer immunotherapy such as CAR T-cell therapy could be assessed through correlation of T-cell migration and antitumor activity of the T cells.
3.3 NK cells
NK cells are another type of lymphocyte, and they play an important role in innate immunity by eliminating aberrant cells ranging from virally infected cells to tumor cells. NK cells can recognize and lyse tumor cells without differentiation or maturation, and this phenomenon has led to efforts in using NK cells in adoptive cell therapy. Monitoring the migration of NK cells to the target tumor, which is crucial in elimination of tumor cells, allows tracking of the biodistribution of transplanted NK cells, and could reveal underlying biological and biochemical mechanism behind NK cell-based therapy.67 Ferumoxytol, an FDA-approved formulation of IONPs, was complexed with heparin and protamine (HPF) to label NK cells for treatment and monitoring of liver tumors. The cellular uptake of HPF by NK-92MI cells increased when incubated with cell culture media containing increasing concentration of HPF, and up to 3.47 pg per cell intracellular iron content was observed. A rat hepatoma model was used to evaluate the viability of MRI-based NK cell tracking, and systemic delivery of labelled NK cells via intravenous injection and catheter-assisted intraportal vein delivery was compared. Migration of NK cells into the tumor from surrounding liver tissue was observed with the transcatheter transfusion, and intraportal vein delivery of NK cells allowed more efficient targeting of tumors than intravenous systemic delivery. To assess the tumor-targeting capability of the NK cells,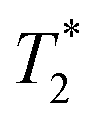 -weighted images and
-weighted images and 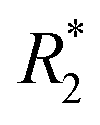 values in the tumor and surrounding healthy liver tissues pre-infusion, 0.5 h post-infusion, and 12 h post-infusion were evaluated. While
values in the tumor and surrounding healthy liver tissues pre-infusion, 0.5 h post-infusion, and 12 h post-infusion were evaluated. While 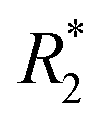 values increased in the initial 0.5 h post-infusion in healthy liver tissues, the values dropped significantly 12 h post-infusion; simultaneously, in tumor nodules, no significant changes in
values increased in the initial 0.5 h post-infusion in healthy liver tissues, the values dropped significantly 12 h post-infusion; simultaneously, in tumor nodules, no significant changes in 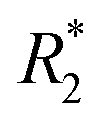 values were observed in the first 0.5 h, which was then followed by a significant increase 12 h post-infusion. Representative images of the liver before and after infusion of IONP-labelled NK cells show increasing heterogeneity in the signal in the tumor nodules 12 h post-infusion, as well as the T2 reduction caused by the infiltration of the NK cells into the tumor.68 Further investigation of the biodistribution of IONP-labelled NK cells and their efficacy in treating other malignancies would highlight the utility and efficiency NK-based adoptive cell therapy.
values were observed in the first 0.5 h, which was then followed by a significant increase 12 h post-infusion. Representative images of the liver before and after infusion of IONP-labelled NK cells show increasing heterogeneity in the signal in the tumor nodules 12 h post-infusion, as well as the T2 reduction caused by the infiltration of the NK cells into the tumor.68 Further investigation of the biodistribution of IONP-labelled NK cells and their efficacy in treating other malignancies would highlight the utility and efficiency NK-based adoptive cell therapy.
Approaches to efficient labeling NK cells have also been explored with increased interest in NK cell-based immunotherapy. Using the magnetic properties of IONPs, magnetic field-assisted labeling methods have been developed. Silica-coated IONPs (silica-IONPs) were used to label NK-92MI cells for magnetic-guided delivery of these cells into tumor sites. The silica-IONP was conjugated with a Cy5.5 fluorescent dye and incubated with NK cells. An external magnetic field was produced by a magnet placed underneath the cell culture plate; the magnetic field drew the silica-IONP towards the surface of NK cells at the bottom of the plate and increased the chances of internalization of silica-IONP through endocytosis. Fluorescence activated cell sorting (FACS) was used to assess the uptake of the nanoparticles, and showed greater degree of internalization of silica-IONP in cells placed above the magnet.69 In another cell labeling approach, a “biohybrid” composed of NK cells and nanoparticles was synthesized by attaching streptavidin-modified IONPs on the exterior of biotin-coated NK cells. The labeling efficiency of this design was assessed by quantifying the IONP bound to the biotinylated NK cell and the IONP that did not, and was found to be greater than 50%. Furthermore, the labeled NK cells continued to remain viable.70 While the reported labeling efficiency of the biohybrid approach is lower than other NK labeling methods, the stabilization of IONPs on cell surface rather than internalization of IONPs preserves the intrinsic composition of NK cells. Strategies to label immune cells with IONP for both MRI and magnetic guided-delivery applications have been developed. These approaches utilize the property of IONPs, as well as material properties of cationic polymers and liposomes to enhance the internalization of IONPs for greater extent of cell labeling.
In these approaches, the intended destination of the transplanted immune cells already identified prior to observation of MRI signal in different regions of the animal model, and injection sites of the immune cells was in close proximity to the intended target. The visualized migration and accumulation of immune cells to intended target sites would be indicative of proper priming and activation of immune cells in response to tumor antigens; however, as a diagnostic tool, cell tracking via MRI should indicate migration of activated immune cells to tumor sites without prior confirmation bias, and there is a lack of evidence that this could be used to reveal tumors throughout the body. More in-depth studies of systemic migration of immune cells, and its visualization through MRI mediated cell tracking would allow this technology to be more widely used in cancer diagnostic applications, in addition to monitoring the treatment of cell-based immunotherapy.
4 IONPs as MRI probes
With the development of cancer immunotherapy, visualization of immune cells in vivo has become more important in assessing the success of the therapeutic approach. The presence of cells of interest at specific sites implies increased stimulation of immune cells, and enhanced antitumor activity, leading to tumor size reduction and increased survival.18 MRI is a non-invasive, radiation-free imaging modality that can be used to track and monitor the migration of IONP-laden immune cells. The MRI signal is generated by disrupting the aligned net magnetization of hydrogen nuclei placed in a strong external static magnetic field using a radio frequency electromagnetic pulse and measuring the signal returned by the hydrogen nuclei in the sample under study as they reform their net magnetization vector aligned with the external magnetic field; such measurements can be made in the same plane as the external magnetic-field in so-called T1-weighted imaging or in the plane perpendicular to the direction of the external magnetic field in T2-weighted imaging. In MRI, the time it takes for the magnetic moment associated with a hydrogen atom to realign with the external magnetic field can be estimated as the time constant of an exponential decay curve. Further, smaller values of the time constant T1 result in brighter spots in the resulting image, whereas smaller values of T2 result in darker loci in an image. IONPs have been shown to shorten both T1 and T2 when they are near hydrogen atoms under measurement, but IONPs more strongly affect the T2 time constant as opposed to T1 and thereby act as contrast agents in T2-weighted scans (note that the degree to which IONPs perturb the values of T1 or T2 may be altered by adjusting the size of the iron oxide core; in fact, IONPs with core sizes less than 4 nm have been shown to act as T1 contrast agents as opposed to T2-weighted contrast agents). As T2 relaxation depends on the inhomogeneity in the local magnetic field created by the magnetic nanostructure, IONPs for T2 contrast-enhanced MRI should exhibit high saturation magnetization. This can be achieved through altering the properties of IONPs such as size and morphology.71,72 Hydrophilic surface coating of the IONP also contributes to the relaxivity mechanism by facilitating the diffusion of water molecules in the outer sphere of the field inhomogeneity induced by the IONP. Furthermore, the surface coating also provides stability in aqueous environments as well as retard the degradation of the iron oxide core.73–75 By labeling immune cells with IONPs, strong contrast can be achieved between the labelled cells and the host tissue.76Properties of the iron oxide core such as size, composition, and morphology influence the magnetic properties of IONPs, and can affect the performance of IONPs as MRI contrast agents. Additionally, a greater concentration of contrast agent correlates with greater MRI signal; cells must be sufficiently labelled with IONPs in order to be distinguished from the background signal from host tissues. Insights into factors that influence the effectiveness of IONPs as MRI contrast agents for immune cell tracking such as size and surface charge of IONPs, and the magnetic field strength of the MRI machine in use will be highlighted in this section, followed by recent advances in tracking immune cells through IONP-mediated contrast-enhanced MRI (Fig. 2).
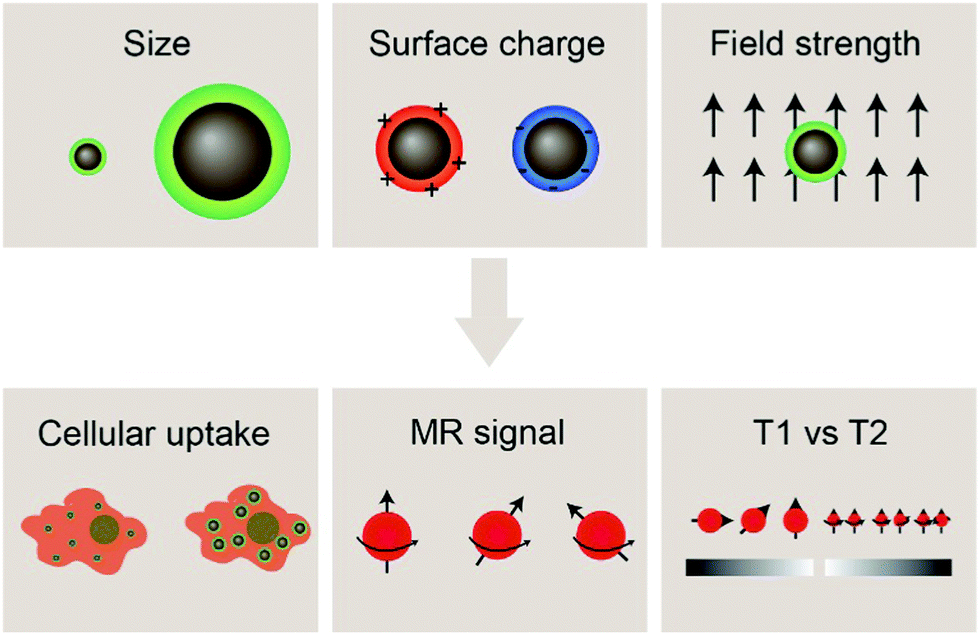 | ||
| Fig. 2 Upper row: Nanoparticle properties such as size and surface charge, as well as the MRI field strength are practical design parameters for IONP-based cell labeling. Bottom row: Cellular uptake, MRI signal, and T1versus T2 imaging are some of the factors to be considered in tracking IONP-labelled immune cells. | ||
4.1 Effect of nanoparticle size
The size of IONPs is an important factor affecting their interaction with biological systems in terms of cellular uptake, pharmacokinetics, and toxicity. The size of the iron oxide core has been shown to alter the magnetic properties of IONPs and can be tuned to achieve the desired relaxivity for MRI applications. In terms of interaction with cells and tissues, the hydrodynamic size of IONPs should be considered, as it reflects the interaction of the nanoparticle with the aqueous medium. The hydrodynamic size is larger than the core size of the IONPs as the hydrodynamic size accounts for the solid core, polymer coating, the solvent layer associated with surface of the nanoparticle, and any nanoparticle aggregation occurring in aqueous environments. In most in vivo applications, IONPs are systemically administered via intravenous injection and are delivered to their targets by either passive or active targeting. In systemic delivery of IONPs, size constraints posed by the clearance mechanisms of the body must be considered – IONPs smaller than 10 nm in diameter are removed by the kidneys, while those greater than 200 nm have been shown to accumulate in the liver.77 Hence, the size of the IONPs plays an important role in their blood circulation half-life, biodistribution, and tumor permeation. Furthermore, in ex vivo cell labeling applications, immune cells of interest are extracted and expanded, followed by incubation in IONP-containing growth medium, and then injected back into the body. In vitro interactions between immune cells and IONPs such as cytotoxicity, cellular uptake, and any alteration of the function of the immune cells must also be considered. Effective cell tracking relies on sufficient uptake of IONPs in order to produce a specific and selective MRI signal, while not disrupting the inherent function of the immune cells.IONPs with core sizes greater than 5 nm are well-documented T2 contrast agents for MRI. This means that if regions of interest containing IONPs are imaged by MRI with a T2-weighted imaging sequence, those regions will appear darker than if IONPs are not present. Further, increasing the concentration of IONPs within a region of interest will cause more intense darkening of the T2-weighted MRI image until a saturation limit is reached (i.e., the image cannot get blacker than pure black).
The idea of loading cells with IONPs, introducing those cells into an in vivo environment, and using MRI to track the migration of those cells in vivo by following the movement of the IONPs (which are visible due to their MRI contrast enhancing properties) requires a sufficient amount of IONPs taken up by the cells to be tracked. Otherwise, the cells will not be visible in MRI because the signal provided from IONPs will be below the detection limit set by the MRI hardware.
The size of IONPs influences cellular uptake and function of immune cells as the physical interaction of nanoparticles and cellular membranes will employ a multitude of internalization pathways. While the size of IONPs, in particular the size of the iron oxide core, influences the magnetic properties of the IONPs, and hence the contrast enhancement provided by the IONPs, greater contrast in T2-weighted MRI also correlates with a higher concentration of IONPs, and thus, ensuring sufficient uptake of IONPs allows immune cells to be more clearly visualized in vivo. In macrophages treated with IONPs of different sizes (20 and 100 nm), the intracellular content was ten times greater in macrophages treated with 100 nm particles than in macrophages treated with 20 nm particles (7.4 versus 73.46 pg per cell).78 In DCs incubated with IONPs of various hydrodynamic diameters, larger IONPs (145.5 nm) were taken up more than smaller IONPs (44.4 and 125.6 nm) after 2 h; however, no significant difference in intracellular concentration was observed after 24 h.79 Size dependent cellular uptake of IONPs was also observed in T cells, where dextran-coated IONPs of 107 nm in hydrodynamic size had an uptake of near tenfold greater than smaller IONPs (53.5 and 33.4 nm) by. However, particles larger than 289 nm were taken up less, possibly due to sedimentation.80 In these studies, size dependent cellular uptake was observed, and cellular uptake of IONP is optimized at a certain hydrodynamic size range. The specific size range at which cellular uptake is optimal differs from study to study, likely due to the different cell types involved as well as the different materials used to coat IONPs. The trend between size and nanoparticle uptake was also observed in other studies on non-immune cells and other nanoparticle formulations.81,82 It is known that cells utilize several mechanisms for endocytosis such as caveolae-mediated and clathrin-mediated endocytosis, as well as phagocytosis for large particles (250 nm to 3 μm).83 Other internalization mechanisms have been identified to be mediated by neither caveolin nor clathrin for particles smaller than 25 nm.84 As the nanoparticle size increases, more endocytosis mechanisms could be involved in the cellular internalization of nanoparticles, which may explain the increased IONP uptake with increased size, up to a range that can employ the clathrin- and caveolae-mediated pathways.83 By utilizing IONPs with optimal size for favorable cellular uptake, clearer MRI images can be acquired with increased contrast.
In addition to cellular uptake, the effect of IONP size on the cellular function of immune cells and their potential cytotoxicity must also be considered for in vivo applications. An imaging probe should not disrupt the regular function of the target cell and should not cause cytotoxicity at applicable concentrations. In a study, mouse bone marrow-derived macrophages were treated with IONPs of various sizes to assess the effect of size on macrophage function. Secretion of cytokines such as interleukin 6 (IL-6) or tumor necrosis factor alpha (TNF-α) is indicative of macrophage activity. Lipopolysaccharides, which activate macrophages, have been used to prime the macrophages prior to incubation with IONPs. The production of IL-6 and TNF-α in macrophages incubated with 30, 80, and 120 nm IONPs did not differ significantly, indicating that IONP uptake did not significantly alter macrophage function.85 In another study, silica-coated IONPs of various sizes were synthesized, and RW264.7 macrophages were incubated with the IONPs to assess cytotoxicity and TNF- α secretion. Similarly, TNF-α secretion levels did not vary significantly with incubation with IONPs of different diameters: 20, 40, and 100 nm. However, IONPs larger than 200 nm did induce significantly increased expression of TNF-α in macrophages.86 Macrophage release TNF-α in response to injury or infection, eliciting a pro-inflammatory response.87 Phagocytosis mediated by certain phagocytic receptors have shown to directly induce the pro-inflammatory response, and as larger nanoparticles tend to interact with more receptors due to their surface area, it is possible that they would elicit stronger immune response from macrophages.88 To label macrophages without disrupting their innate cellular functions, IONPs under 200 nm in size have shown to be more effective as they are internalized without eliciting pro-inflammatory responses. These results suggest that increasing the IONP size would result in more efficient phagocytosis, leading to increased cellular uptake; however, this effect is limited to a certain size range, beyond which cellular uptake is less efficient. In addition, larger IONPs have also shown to induce pro-inflammatory response from macrophages, whereas smaller IONPs did not cause significant alteration in cell functions.
The hydrodynamic size, which is influenced largely by the material used to coat the iron oxide core, is an important design factor for regulating their interaction with cells, such as cellular uptake and cytotoxicity, which in turn affects their utility as cell labeling tools. The MRI signal of IONPs is also affected by their size, more specifically, the size the iron oxide core. IONPs act as T2 MRI contrast agents by shortening the T2 time for decay of transverse magnetization, making an IONP-rich region in tissue appears darker on the MRI image. The relationship between IONP core size and T2 relaxation can be classified into three size regimes. For small core sizes (<15–20 nm), reduction of the core size of IONPs suppresses the magnetic moment of the IONP and, in turn, reduces the transverse relaxivity (r2), which is a measure T2 shortening effect as a function of concentration, of IONPs.41,80,89 A reduction in core size increases the surface area-to-volume ratio of the IONP. As more atoms are proportionately present at the particle surface, they exhibit greater surface effects such as noncollinear electronic spins, spin canting, and spin-glass-like behavior, leading to the suppression of saturation magnetization of IONPs which would become less potent as T2 shortening contrast agents.90 The size range in which this behavior is observed is termed the motional average regime, where water protons can diffuse quickly, and experience a changing magnetic field from the IONP. As the core size increases, the T2 relaxation behavior enters the static dephasing regime, where r2 is maximized.91 The IONP cores are sufficiently large compared to the diffusion of water molecules so that the water protons would feel a constant magnetic field. The static dephasing regime encompasses a small size range in which r2 does not change with core size.92 Beyond the static dephasing region, for larger IONP core sizes, r2 decreases with increasing size. This behavior occurs in the echo-limiting regime, or the “Luz-Meiboom” regime. As the nanoparticle size further increases, the nanoparticles start to exhibit ferromagnetism as opposed to superparamagnetism, and generate strong magnetic fields that completely dephase nearby water protons which in turn are unable to contribute to the MR signal.93 Furthermore, other factors such as aggregation due to lower stability, as well as presence of multiple domains also lead to decrease in r2 for IONP in the echo-limiting regime.92,94 While this implies that increasing the core size of IONPs will increase r2, effects from other factors such as water exchange rate and mean residence time become more prominent and may lead to reduction of r2 as the core size is greater than a size limit; above this limit, an increase in particle size reduces surface accessibility and water exchange rate between the bulk solvent and inner sphere layer of IONPs, limiting the number of hydrogen nuclei accessible to the paramagnetic core.95 This trend of r2 dependence on IONP core size was demonstrated by measurement of the relaxivity of the solutions containing IONPs of various core sizes. By comparing the r2 measurements in solutions containing IONPs with different core sizes, r2 was shown to increase with increased core size from 7.7 nm to 13.1 nm, then decrease as the core size continued to increase to 17.2 nm96 (Fig. 3a–c). Both uptake and MRI signal were shown to increase with size up to a certain point, and then start to become less efficient as cell labeling tools. These results suggest that there is an optimal range of both hydrodynamic size and core size that would lead to the greatest contrast in MRI through maximizing cellular uptake and MRI contrast-enhancing ability. Many studies utilize commercially available IONPs that fall within this range of optimized core size.97–99 While optimization of IONP size to maximize the contrast in T2-weighted imaging would be beneficial to visualization of migration of IONP-labelled cells, IONPs within a certain size range are adequately taken up by immune cells and can provide MRI contrast.
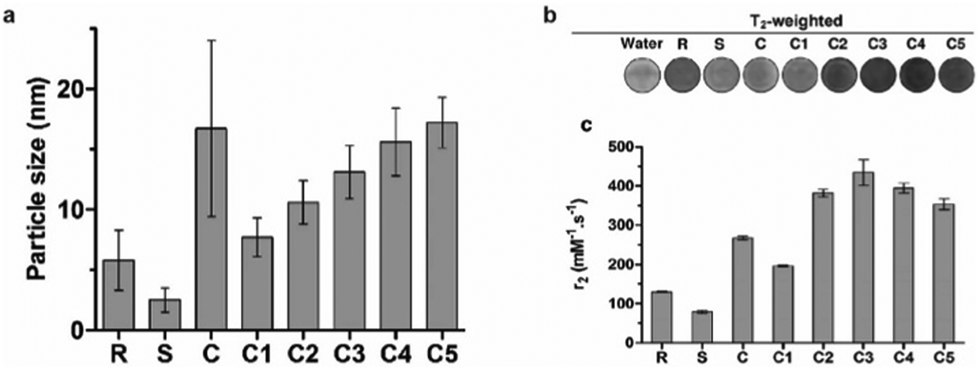 | ||
| Fig. 3 Effect of iron oxide core size on MRI relaxivity. (a) Crude IONPs were divided by centrifugation, and size distributions were measured using transmission electron microscopy. (b) T2-weighted images of each size-separated batch was taken and (c) transverse relaxivity (r2) was measured. Reproduced with permission.96 Copyright 2020, Springer Nature. | ||
4.2 Effect of surface charge
The surface charge of nanoparticles plays a vital role in their interaction with cellular components once internalized into the cell. Positively charged nanoparticles, such as cationic liposomes and cationic polymer-coated nanoparticles, have shown to be internalized to a greater extent than neutral or negatively charged nanoparticles, as they can associate with the negatively charged phospholipids in the plasma membrane via electrostatic interaction, and are subsequently internalized through endocytosis.83 These nanoparticles are used notably in drug delivery and gene delivery applications.100,101 Nanoparticles with high surface charge may also form a protein corona when the biomolecules are adsorbed onto the surface of the nanoparticle to lower the surface energy, leading to significantly different surface charge and capacity to interact with cellular components in biological systems.102The effect of IONP surface charge when IONPs serve as adjuvants was investigated by delivering ovalbumin (OVA) which can induce an observable immune response, as a model antigen to murine DCs using IONPs with different zeta potentials (NP-OVA). The zeta potential is the electrokinetic potential at the interface between the bulk fluid and stationary layer on the nanoparticle caused by distribution of counterions to the surface of the nanoparticle, and is a more relevant quantity in colloidal solutions of IONPs than surface charge of the bare nanoparticles.103 DCs incubated with positively charged NP-OVA were able to activate T cells, whereas negatively charged NP-OVA with the same dose of OVA did not result in antigen cross-presentation (Fig. 4a and b). The lack of antigen cross-presentation in DCs incubated with negatively charged nanoparticles was attributed to the sequestering of the antigen in intracellular compartments, where it could not be recognized by the proteasome, which processes the antigens, and not participate in the cross-presentation pathway.104 In order to evaluate the potential of charged IONPs as immunoadjuvants, the relationship between surface charge of IONPs and function of DCs was further investigated by observing the production of the pro-inflammatory cytokine IL-1β in human and murine DCs. Negatively charged IONPs induced much greater levels of IL-1β in murine DCs in a dose dependent manner, compared to positively charged and unmodified IONPs. However, DCs loaded with negatively charged NPs were not able to activate T cells, while DCs loaded with positively charged NPs could. The inability of DCs loaded with negatively charged NPs to activate T cells was attributed to cellular dysfunction of DCs in antigen cross-presentation due to the aberrant secretion of IL-1β.105
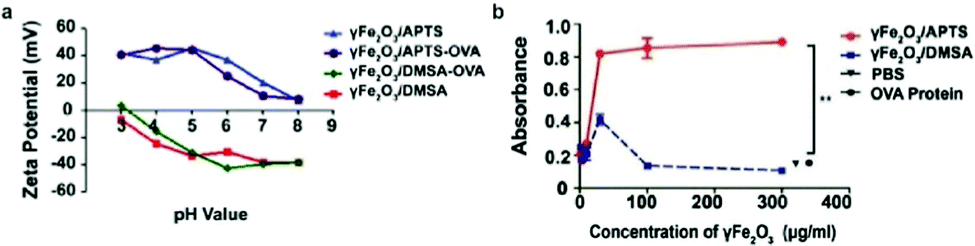 | ||
| Fig. 4 Effect of surface charge on properties of IONPs. (a) IONPs were modified to display different surface charges as measured by zeta potential. (b) β-Galactosidase assay measured the activation of T cells by murine DCs incubated with IONPs of different surface charge. β-Galactosidase upregulated in activated T cells can cleave reporter molecule in the assay, leading to increased absorbance at 595 nm. OVA protein and phosphate buffered saline (PBS) were administered as controls. Reproduced with permission.104 Copyright 2017, Springer Nature. | ||
The effect of surface charge on nanoparticle uptake was also investigated in T cells. Less cellular uptake was observed with decreasing number of amine groups on the IONPs, similar to the trend observed with DCs.80 As shown through these studies, positive surface charge leads to increased cellular uptake in immune cells through electrostatic interaction with the cell membrane. The high capacity of positively charged IONPs to be internalized in immune cells not only makes them efficient cell labeling probes but also highlights their utility in applications in delivery of antigens or immunotherapeutics.
4.3 Effect of field strength
The field strength of an MRI apparatus also strongly influences the relaxivity of the contrast agent, as well as the resolution and contrast in the obtained image. In clinical settings, 1.5 T is commonly used, whereas higher field strengths up to 14 T are used in research settings. The r1 and r2 relaxivities are also dependent on field strength, and the ratio between the two relaxivities determines the extent of contrast in MRI – low r2/r1 ratios results in bright, positive contrast in T1-weighted imaging, while high r2/r1 ratios leads to dark, negative contrast in T2-weighted imaging. Commercially available IONPs coated with polyethylene glycol (PEG) with varying molecular weights were imaged under different field strengths. The value of r1 decreased with increasing field strength for all IONP formulations; on the other hand, while r2 increased as the field strength increased from 0.35 to 1.5 T across all samples, some of the samples showed continued increase in r2 from 1.5 T to 3 T, and others showed a decrease. However, there was an overall increase in r2/r1 with increasing field strength for all samples.99 In another study, similar effects were seen with synthesized IONPs, where r1 had decreased with increasing field strength, but r2 had initially increased with increasing field strength, then changed depending on the type of surface modification as field strength was increased to 9.4 T (Fig. 5a and b).106 As these results suggest, higher magnetic field strength MRI would further enhance the contrast in IONP-mediated cell tracking. However, MRI scanners with field strengths greater than 3 T are rarely seen in clinical settings due to the small regions over which they can acquire an image, and high static magnetic fields have been reported to induce nausea, dizziness, magnetophosphene, and metallic taste.107 Altering the magnetic properties of IONPs by controlling the core size of the IONPs to optimize the r1 and r2 values at lower field strengths would be a more practical approach to bring IONP-mediated cell tracking closer to widespread clinical application.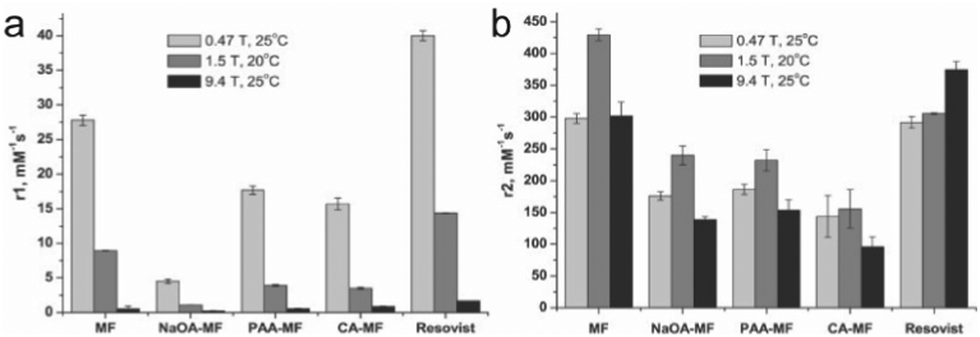 | ||
| Fig. 5 Effect of magnetic field strength on the relaxivity of IONP. (a) r1 values of naked magnetite nanoparticles (MF), oleic acid coated nanoparticles (NaOA-MF), polyacrylic acid coated nanoparticles (PAA-MF), citric acid coated nanoparticles (CA-MF), and Resovist, a commercially available iron oxide nanoparticle formulation. Relaxivity values were measured at different field strengths between 0.47 T and 9.4 T. (b) The r2 values of the nanoparticle formulations measured at various field strengths. Reproduced with permission.106 Copyright 2012, Elsevier. | ||
4.4 IONPs as T1 MRI contrast agents
Up until this point, the discussion of IONPs as MRI contrast agent has pertained to their role as T2 contrast agent, which means that regions with high concentrations of IONPs will appear darker due to the faster decay of transverse magnetization. On the other hand, T1-weighted MRI offers advantages that address some limitations of T2-weighted imaging. As the intrinsic nature of T2 relaxation is the decay of transverse magnetization, the resulting signal leads to regions of low contrast which could be misidentified as other hypointense areas such as bleeding and calcification. In contrast, T1-weighted imaging is dependent on the recovery of longitudinal magnetization, leading to greater contrast in cells or tissues with high concentrations of contrast agents.108 However, commercially available T1 contrast agents are limited to gadolinium-based chelates, which have been associated with unfavorable side effects such as nephrogenic systemic fibrosis.109,110 Furthermore, T1 contrast agents have low sensitivity, and need to be administered at high dosages, further limiting their usage in clinical settings. While available IONP formulations for MRI contrast can provide high T2 contrast, they have not been clinically used for T1 contrast-enhanced MRI.111,112Recent developments have demonstrated that ultrasmall IONPs with core sizes smaller than 5 nm are capable of providing T1 contrast enhancement as an alternative to gadolinium-based contrast agents.113–115 In general, contrast agents for T1 imaging tend to have lower r2/r1 ratio (<5) while those for T2 imaging have a larger value of r2/r1.116 By reducing the core size of the IONP to 5 nm or smaller, the saturation magnetization responsible for high r2 values in IONP can be suppressed, and decrease the r2/r1 ratio for T1 contrast. To produce such small nanoparticles with high control over size, Mn-IONPs were synthesized via dynamic simultaneous thermal decomposition. The resulting particles had an average size of 3 nm, and the r2/r1 value was as low as 2.49 in a 3 T scanner, which is close to the value for Omniscan, a commercially available gadolinium-based contrast agent, whose r2/r1 value was reported to be 1.04. In vivo MRI after intravenous administration showed much brighter vasculature in the mouse treated with the Mn-IONP than the mouse treated with Omniscan.114 Altering the composition of the iron oxide core from magnetite (Fe3O4) to maghemite (Fe2O3) also reduces the saturation magnetization;, bulk magnetite shows greater magnetization than bulk maghemite. Zwitterion-coated IONPs with maghemite core and average diameter of 3 nm were synthesized via thermal decomposition. These IONPs showed greater magnetization than Magnevist, a commercially available Gd-based T1 contrast agent, but smaller magnetization than ferumoxytol. The r2/r1 ratio of the zwitterion-coated IONPs was 2.0 in 1.5 T, close to that of Magnevist (1.1).117
The development of IONP-based T1 contrast agents provides a safe alternative to gadolinium-based formulations. As T1 contrast provides signals that are clearly distinguished from the background and other biological effects, T1 contrast-based immune cell tracking would be invaluable to cancer immunotherapy for real-time visualization of migration and distribution of immune cells. However, barriers still remain for IONP to be widely utilized as T1 contrast agent for MRI. As T1 contrast agents have low sensitivity, in vivo cell labeling applications would require injection of high dosages of these agents. Furthermore, upon cellular uptake of the nanoparticles, they would be confined to endosomes. The shortening of T1 relaxation could be restricted only to the protons inside the endosomal membrane, and increased concentration of IONP in endosomes would lead to increased field inhomogeneities and increased r2. Finally, the nanoparticles would be exposed to the harsh environment inside the endosome compared to the cytoplasm or extra cellular matrix. Additional design consideration of facilitating endosomal escape of the IONP would further enhance their T1 contrast capabilities.118–120 Further investigation of the interaction between these ultrasmall nanoparticles and immune cells could address some of the current limitations of T2-weighted MRI-based cell tracking.
5 Applications of IONPs for cancer immunotherapy
Nanoparticles can be used to target immune cells that partake in the immunotherapy mechanism. Among these nanoparticle-immune cell interactions are stimulation of APCs, activation of T cells, and reprogramming of macrophages. The flexible surface chemistry of IONPs allows delivery of antigens, adjuvants, and therapeutics to immune cells and conjugation of antigens onto IONPs protects the antigens from in vivo degradation. The interaction between iron oxide particles and APCs, especially DCs, has led to the development of cancer vaccines highlighting the potential of IONP-based technology in cancer immunotherapy. Cancer vaccines prime the immune system against cancer through administration of tumor antigens which are recognized and taken up by DCs, which in turn activate T cells to specifically attack tumor cells associated with the antigen (Fig. 6a). Furthermore, the magnetic properties of IONPs have also been used in adoptive cell therapy and T-cell enrichment, and magnetization of cytotoxic T cells and NK cells allows guided delivery of these cells to specific tumor sites (Fig. 6b). The polarization of tumor associated macrophages (TAMs) in the tumor microenvironment into pro-inflammatory M1 macrophages using IONPs can elicit anti-tumor immune response (Fig. 6c). The iron oxide cores of IONPs can also be used in photothermal therapy to induce immune responses in tumors and deliver immunostimulants to enhance the immune response (Fig. 6d). Finally, IONPs can be used to deliver checkpoint inhibitory molecules in immune checkpoint blockade therapy (Fig. 6e). This section will highlight the various methods in which IONPs have been used to stimulate immune cells to enhance their activity and response to cancer.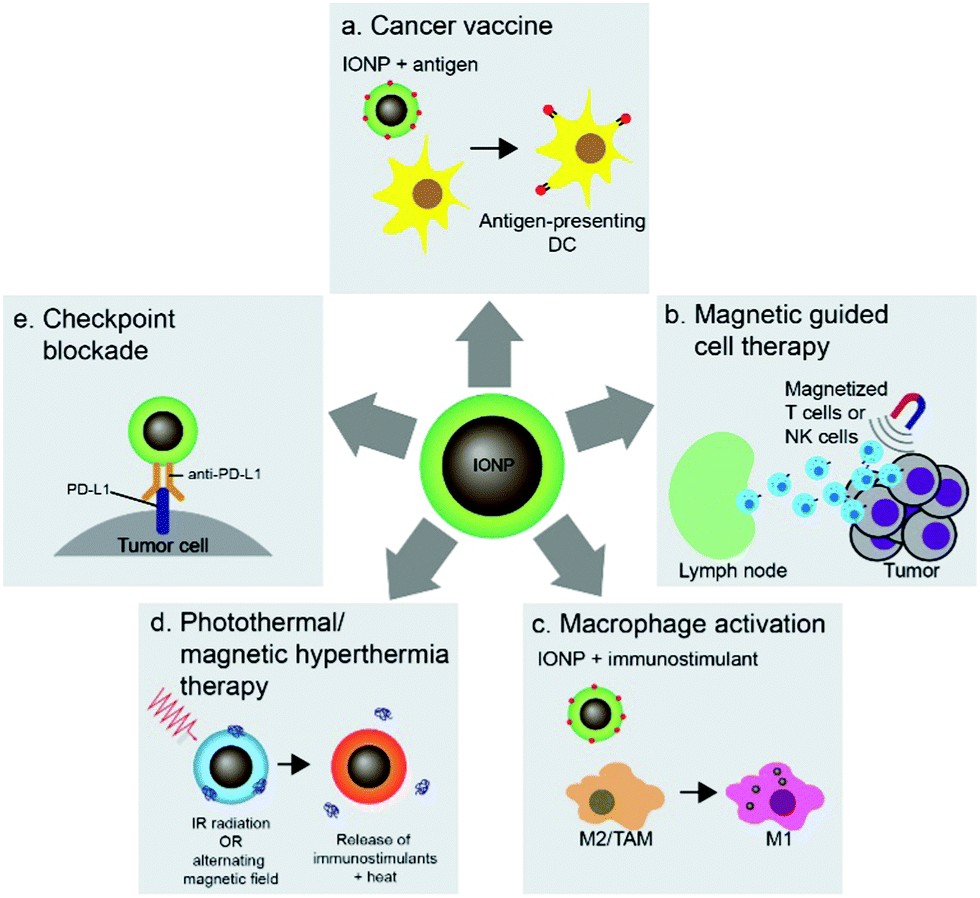 | ||
| Fig. 6 Applications of IONPs in cancer immunotherapy. (a) Cancer vaccines can be designed to enhance the delivery of antigens using IONPs. (b) Magnetized cytotoxic cells can be guided to tumor sites by pretreatment with IONPs. (c) Activation of macrophages to induce tumor cell death can be achieved by delivering IONPs with immunostimulants to macrophages in the tumor environment. (d) Immunotherapy can be combined with photothermal/magnetic hyperthermia therapy using IONPs to trigger local hyperthermia and deliver immunostimulants. (e) IONPs can be used to deliver inhibitory molecules for checkpoint blockade therapy. | ||
5.1 DC-based cancer vaccines
In the cancer immunity cycle, APCs capture cancer antigens, process them, and present the resulting peptides onto their surface with major histocompatibility complex class I or II molecules. The antigen is then recognized by naïve T cells, which differentiate into CTLs or helper T cells that can carry out the immune response in the body. Regulation of antigen uptake and migration of APCs can play a critical role in enhancing the immune system's response to cancer.121 DCs are a class of APC that can capture and process various types of cancer antigens and activate naive T cells into CTLs after migrating to lymph nodes. Hence, as a critical initiator of the pathway to elicit an antitumor response, DCs have been a major target for cancer vaccine development.122IONP-based formulations of cancer vaccines have shown to improve antitumor response by efficient delivery of antigens to APCs through enhancing the solubility and availability of the antigens. A study using IONP-OVA nanocomposites showed a markedly improved stimulation of DCs and tumor reduction in vivo. Increased expression of pro-inflammatory cytokines IL-6, TNF-α, and interferon-γ (IFN-γ) was observed in DC2.4 murine DCs treated with the IONP-OVA nanocomposites as compared to cells treated with free OVA. To further test the immunotherapeutic capability of IONP-OVA in vivo, mice with subcutaneously grown tumors were administered with saline, free OVA, free IONP, or IONP-OVA. A dramatic tumor reduction was observed in the group treated with IONP-OVA, whereas no tumor growth inhibition occurred in the group treated with free OVA.15 Similar results were observed in other studies that utilized IONP-OVA complexes.19
These nanoparticle formulations were also shown to stimulate macrophages as well, and thus they were investigated to act as prophylactic vaccines against solid tumors and metastasis. Mice were administered PBS, free OVA, IONP, and two formulations of IONP-OVA consisting of different ratios of IONP to OVA near lymph nodes, and after an interval of 1 week, mice were injected with B16-OVA melanoma cells. No tumor growth was observed in mice administered with IONP-OVA, whereas free OVA was not able to prevent tumor growth.123 The marked improvement of in vivo antitumor immune response resulted from the administration of IONP-OVA complexes as compared to administration of free soluble OVA was attributed to protection of cancer antigens from intracellular degradation and inactivation. These studies also showed that the IONP-based vaccines stimulated macrophages, as shown through increased TNF-α expression, which accelerated tumor immunotherapy. The immunostimulatory capability of free IONPs was observed through tumor reduction in some experiments, meaning that IONPs could potentially serve as adjuvants on their own; however, the trend has not been consistently observed in other studies.19 Release of signaling molecules by the macrophage as a response to interaction with IONP could potentially be a reason for the observed immunostimulatory effect of IONPs.
Antigens may be delivered to DCs by transfection with RNA coding for a specific tumor antigen. Cationic liposomes were used to encapsulate IONPs and therapeutic mRNA (IO-RNA-NP), and IONPs were used as MRI contrast agents to track DC migration in vivo. Transfection efficiency was enhanced when IONP was incorporated into the liposomal vector compared to when IONP and the liposomal vector were not initially incorporated as the cationic lipid molecules were able to interact with the cell membrane via electrostatic interactions and the lipid nature of the vector allowed fusion of the vector with the membrane; DC activation and function was also increased. Compared to the transfection of RNA through electroporation, the conventional method for ex vivo transfection of DCs through application of an electric field in cells in order to enhance the permeability of the cell membrane, the transfection through IO-RNA-NP exhibited increased secretion of co-stimulatory molecules, which are proteins that can amplify the activating signals to T cells, and IFN-α, which is needed to initiate an antitumor immune response. DCs transfected with either IO-RNA-NP or through electroporation were injected intradermally to subcutaneous B16F10-OVA tumor bearing mice. The IO-RNA-NP treatment group showed significant tumor growth suppression compared to the electroporation treatment group. Finally, the migration of DCs loaded with IO-RNA-NP was observed using MRI, and the MRI intensity resulted from the IONPs in lymph nodes was evaluated as a biomarker of antitumor response (Fig. 7a). Mice were separated into groups depending on the observed MRI intensity on day 2 after the treatment, and their survival outcomes were correlated to initial DC migration (Fig. 7b). MRI-predicted responders, or mice with DC migration in the top 75th percentile on day 2 had a 39% increase in median survival compared to those with DC migration in the bottom 25th percentile when the tumor cells and vaccines were administered on the same day. The similar results were observed in models with an already established tumor18 (Fig. 7c and d). A potent cancer vaccine design requires selection of the right tumor antigen to sufficiently activate CTLs and must achieve an optimal level of antigen concentration in DCs so that they can be cross presented to prime T cells.17 Cancer vaccines based on IONPs have demonstrated to activate DCs by efficiently delivering tumor antigens while protecting free antigens from degradation and allow monitoring of DC migration to evaluate the therapeutic response to the vaccine.
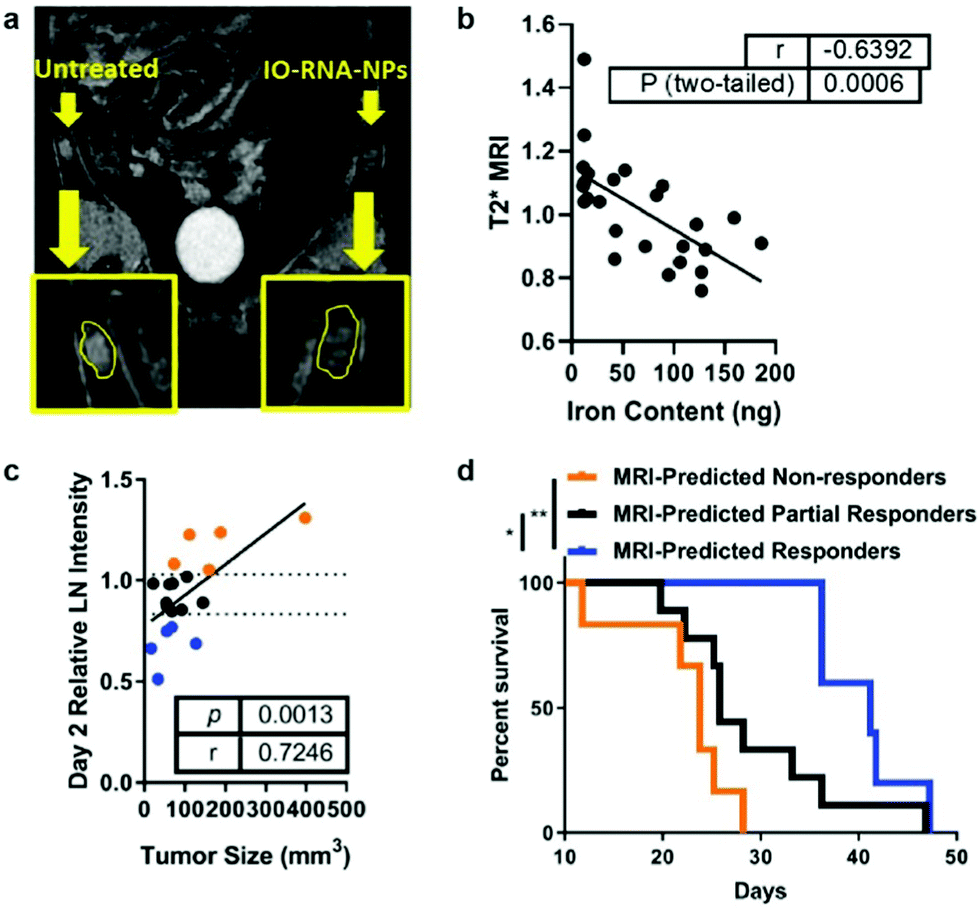 | ||
| Fig. 7 IONP-based cancer vaccine as a predictive biomarker for cancer treatment. (a) DCs incubated with IONPs complexed with RNA were tracked using MRI, as shown in the difference in signal intensity in each lymph node. (b) Comparison between iron content in the lymph node and T2 MRI signal shows that IONPs can be used to quantitatively track DC migration. (c) Correlation between tumor size 17 days after treatment and MRI intensity in the lymph node at day 2. (d) Kaplan–Meier survival curves for all treated mice, divided into groups that responded to vaccine treatment depending on MRI intensity in the lymph node at day 2. Reproduced with permission.18 Copyright 2019, American Chemical Society. | ||
5.2 Magnetic field-assisted cell migration
T cells are distinguished from other lymphocytes by the T-cell receptor on their surface, which can recognize antigens that can lead to maturation of naïve T cells into various types of T cells. The antitumor effects of immunotherapy begin only when cytotoxic T cells are activated and successfully kill tumor cells. In order to increase the presence of CTLs at tumor sites, T cells can be functionalized with IONPs and stimulated to move toward the tumor site with an external magnetic field. This magnetic field-directed migration of T cells may mitigate the side effects in the body from T cell exposure and requires only a small amount of T cells to achieve therapeutic efficacy. Preliminary studies have shown that IONPs taken up by T cells can make the cells to move along an external magnetic field, and in vitro migration due to the external magnetic fields has been demonstrated.124,125 In an in vivo experiment, migration of IONP-labeled T cells guided by an external magnetic field was explored by applying a magnetic field near the popliteal lymph node and observing T cell retention. More of CD4+ and CD8+ cells labelled with IONPs were found to be retained than the T cells free of IONP (Fig. 8).126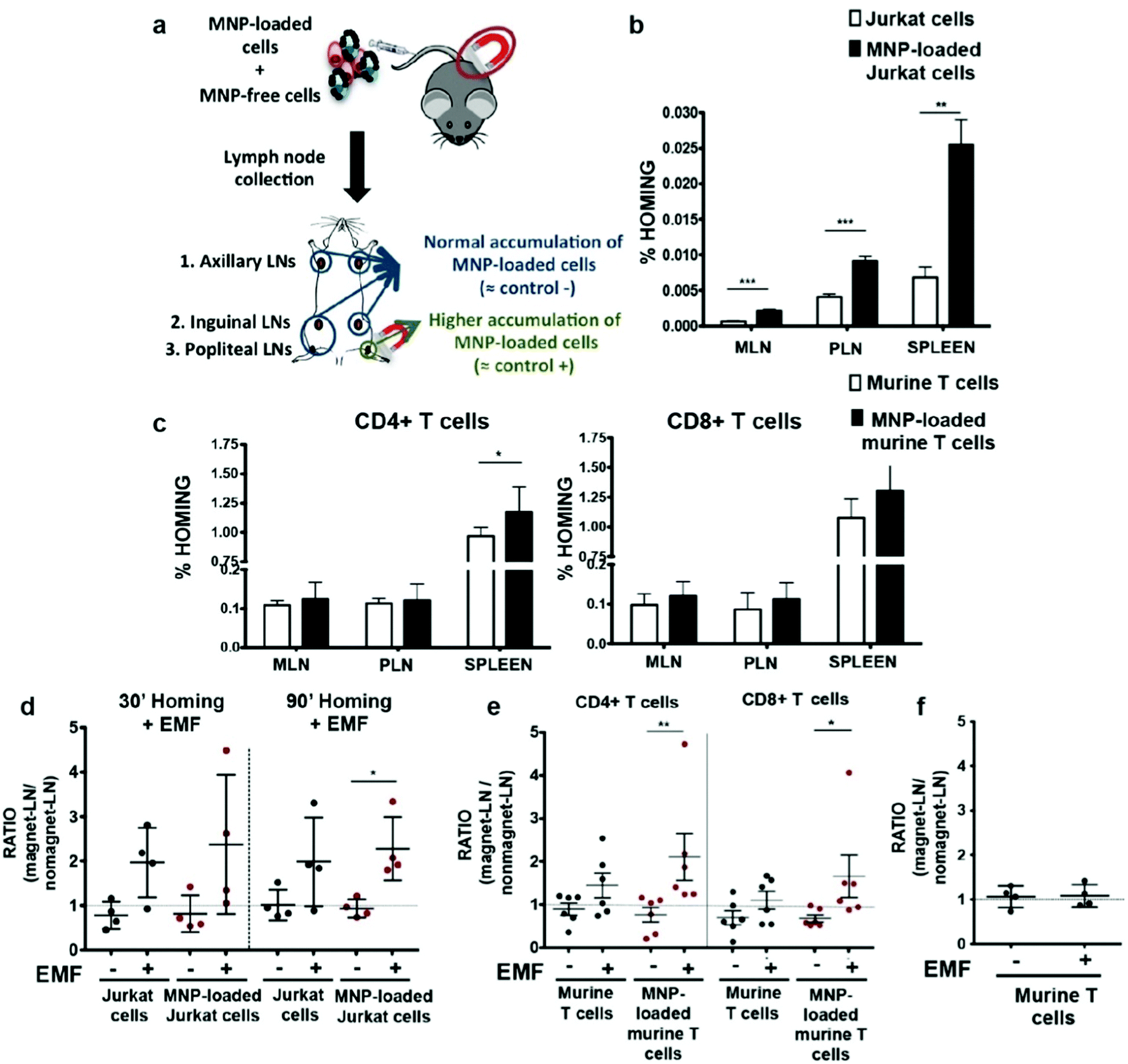 | ||
| Fig. 8 In vivo homing capacity of Jurkat human T cells and murine primary T cells after magnetic iron oxide nanoparticle (MNP) treatment with and without an external magnetic field (EMF). (a) Experimental set-up for determining the homing capacity of MNP-loaded cells compared to MNP-free cells. A mixture of differentially fluorescence-labelled MNP-free and MNP-loaded Jurkat or murine T cells was prepared and intravenously injected into nude (Jurkat) or C57BL/6J (murine T cells) recipient mice. After 1 h, peripheral (PLN) and mesenteric (MLN) lymph nodes (LN) and spleen were collected, processed, and analyzed by flow cytometry. Homing capacity of MNP-free and MNP-loaded (b) Jurkat and (c) murine T cells in the absence of an EMF, 1 h after cell injection. Ratio of MNP-free and MNP-loaded (d) Jurkat and (e) murine T cells in the LNs exposed to an EMF to control LN (no EMF), 20 min after intravenous injection of the cell mixture into recipient mice, normalized to the input ratio. (f) Ratio of MNP-free murine T cells, administered alone as control, in the LN exposed to an EMF to control LN (no EMF) after intravenous injection. Reproduced with permission.126 Copyright 2019, Springer Nature. | ||
NK cells are another subpopulation of lymphocytes that can induce cell death. In contrast with CTLs, NK cells are able to kill tumor cells without prior sensitization.127 Evidence has also shown that NK cells are able to prevent metastasis128 and can eliminate tumor cells through various methods including releasing cytolytic granules that contain cytotoxic proteins and activation of target-cell apoptosis. However, NK cell-based therapy has many barriers to overcome, including the dispersion of NK cells upon in vivo administration13 and limited infiltration in tumor environments.129–131 Functionalizing NK cells with magnetic properties and guiding them to the intended tumor site would greatly enhance the therapeutic effects of NK cell-based adoptive cell therapy. The effect of IONPs on NK cell function and cytotoxicity was evaluated using IONPs coated with (3-aminopropyl)triethoxysilane. No significant differences in various NK cell activities such as pro-inflammatory cytokine production or cytolytic activity were observed with increasing IONP concentration. NK cells incubated with IONPs were placed in an external magnetic field, and movement of the NK cells in response to the magnetic field was observed. NK cells incubated with greater concentration of IONPs were shown to migrate further.14 In an in vivo experiment, mice with A549 cancer cells subcutaneously transplanted were divided into four groups that were treated with PBS, NK cells, NK + IONP, and NK + IONP + magnetic field, respectively. All groups injected with NK cells had their tumor growth significantly reduced compared to the group treated with PBS; however, the greatest tumor growth inhibition was observed in the NK + IONP + magnetic field group due to the local retention of NK cells contributing to a greater antitumor response.132 Magnetic-guided delivery of IONP-labelled NK cells overcomes the barrier of NK cell-based immunotherapy by allowing more NK cells to migrate into the tumor site without affecting the function of the NK cells.
5.3 Activation of tumor associated macrophages
Macrophages play an important role in immune responses by eliminating foreign materials and cellular debris via phagocytosis and orchestrating inflammatory responses by secreting cytokines. In tumor microenvironments, TAMs are polarized into either pro-inflammatory M1 or cell proliferation-promoting M2 states by anti-inflammatory cytokines in the tumor microenvironment and promote tumor growth as well as tumor angiogenesis.97,133 By polarizing TAMs to the M1 state, these macrophages could induce apoptosis of tumor cells while already residing in the tumor microenvironment, overcoming the difficulty of tumor penetration by M1 macrophages outside of the tumor. Hence, reprogramming of macrophages, or macrophage activation, has attracted much interest as a form of cancer immunotherapy.IONPs can serve to deliver biomolecules such as OVA123 and toll-like receptor agonist97 that could trigger the polarization of macrophages. Even on their own, IONPs have shown to suppress tumor growth by macrophage activation, as metabolic degradation of IONPs leads to increased iron content in the macrophages, stimulating their polarization to an M1 state. Iron levels in macrophages can regulate their polarization – M2 macrophages have high expression of ferroportin, which transports iron out of cells, and M1 macrophages have high expression of ferritin, which helps store iron inside cells. Administration and subsequent lysosomal degradation of liposomes release iron which are then ingested by macrophages. The accumulation of iron in macrophages leads to their M1 polarization.134 Administration of IONPs following injection of cancer cells in mice showed a significant tumor growth suppression compared to ones that did not receive IONP treatment. The potential of IONPs to address metastasis was also demonstrated by IONP administration prior to injection of cancer cells, which led to a tumor size that was six times smaller than the control.135 In another study, porous hollow IONPs were used to deliver 3-methyladenine, an inhibitor of phosphoinositide 3-kinase (PI3K) γ that could promote an immune response and trigger repolarization of TAMs to M1-type macrophages. This system was able to successfully inhibit expression of PI3K γ and upregulate the production of NF-κB p65, a protein in the complex NF-κB whose expression is associated with promoting immune responses. The synergistic effect of macrophage polarization by IONP and 3-metyladenine suppressed the tumor growth in mouse models.136
In a different approach, macrophages were artificially reprogrammed using hyaluronic acid-coated IONPs to enhance the effect of macrophage polarization. In this study, RAW 264.7 mouse macrophages were incubated with IONPs, and then transplanted into 4T1 tumor-bearing mice. A schematic representation of the approach is outlined in Fig. 9a. The artificially programmed macrophages were able to specifically target and kill cancer cells, induce polarization of resident TAMs to M1-type macrophages, amplify the anticancer effect, and remain insusceptible to cytokines in the tumor microenvironment that would have suppressed other pro-inflammatory macrophages. BALB/c mice with subcutaneously inoculated with 4T1 tumors were injected with various treatments, and transplantation of macrophages programmed with IONPs resulted in much greater tumor growth suppression than treatment with macrophages or IONPs alone. In addition to chemotaxis of macrophages along the cytokine gradient, IONP labeling enabled magnetic guidance of the transplanted macrophages, resulting in further tumor growth suppression (Fig. 9b and c).137 These results showed that IONPs can serve as delivery vessels of immunostimulatory molecules and innately induce polarization of M1 macrophage through their degradation into iron. Macrophage activation is an important process in anticancer immune responses as it could amplify the therapeutic effects of other immune cells involved in the cancer immunity cycle. Further investigation of concurrent administration of IONP-based macrophages and other therapeutic modalities would further demonstrate the utility of IONP in cancer immunotherapy.
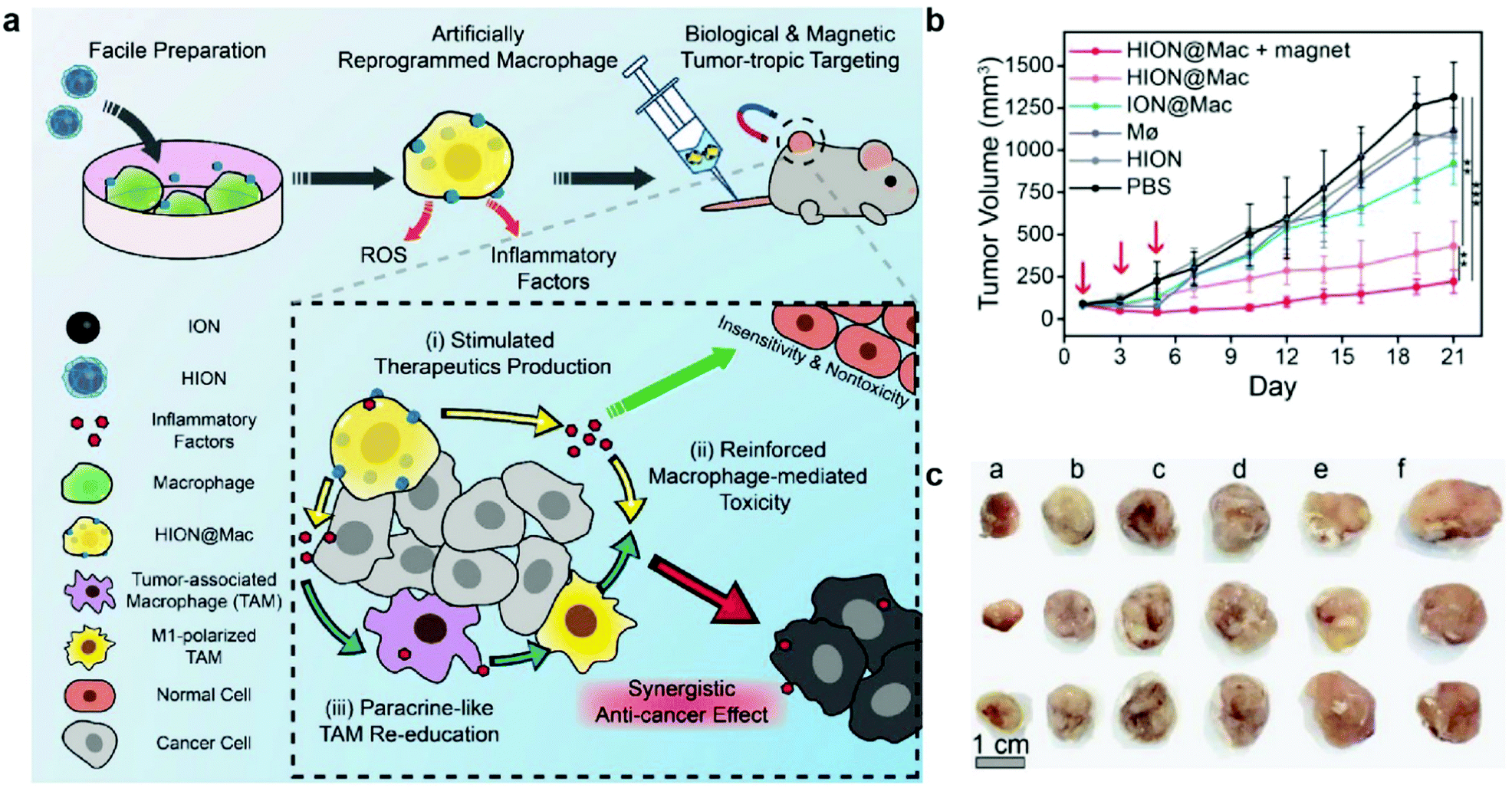 | ||
| Fig. 9 Artificial reprogramming of macrophages. (a) Schematic illustration of the role of reprogrammed macrophages by IONPs (HION@Mac) in anticancer immune response. (b) Tumor growth profiles of 4T1 inoculated mice over 21 days. (c) Representative images of tumor tissues from each group: (a) HION@Macs + magnet, (b) HION@Macs, (c) macrophages reprogrammed with IONPs without hyaluronic acid coating, (d) native macrophage, (e) HION, and (f) PBS injection. Reproduced with permission.137 Copyright 2019, Wiley. | ||
5.4 Photothermal therapy and magnetic hyperthermia therapy
Synergistic effects of combining different therapeutic modalities have been widely investigated. In traditional cancer therapy, chemotherapy is sometimes accompanied by radiotherapy to enhance the therapeutic response.138 Similarly, immunotherapeutic applications of IONPs can be combined with other cancer therapy modalities to induce a greater antitumor response. Photothermal therapy (PTT) is an approach that utilizes the conversion of light, usually infrared, to heat in order to ablate the target tissue; nanoparticles can amplify this effect, and constrain the ablation to a localized region, minimizing damage to healthy tissue. Furthermore, local hyperthermia elicits an immune response, which can lead to recruitment of immune cells to develop anticancer immune responses.139 While gold nanoparticles have been used in clinical trials of PTT as photosensitizers, IONPs provide a safe and biocompatible alternative to gold nanoparticles for PTT, and have shown positive therapeutic outcomes in animal models.140,141An IONP-based immunostimulatory and magnetic-responsive nanoagent was developed to target tumors through magnetic and photoacoustic guidance through MRI and ultrasound imaging, while triggering immunostimulatory effects by amplifying photothermal effects. This design also incorporated cytosine-phosphate-guanine (CpG) oligodeoxynucleotides as potent immunoadjuvants to be delivered to the tumor site. CpG is a motif that is prevalent in bacterial DNA, and is known to induce strong immunostimulatory effects.142 This design was initially tested in vitro, where 4T1 breast cancer cells were treated with IONPs with and without incorporation of CpG, free CpG, and saline in the upper chamber of a transwell system, and bone marrow-derived DCs were cultured on the bottom chamber. Immunostimulatory effects from the treatment on 4T1 cells were evaluated through maturation of DCs by measuring the upregulation of typical co-stimulatory molecules. The treatment with free CpG led to greater extent of maturation of DCs, compared to the treatment with CpG-incorporated IONP; however, upon laser irradiation onto the cell culture, DC maturation was significantly accelerated, demonstrating the immunostimulatory effects of PTT mediated by IONPs. In the in vivo treatment model, mice inoculated with two 4T1 tumors on, one on each flank were treated with various combinations of IONP, CpG, infrared laser, and an external magnetic field (Fig. 10a). Only one of the tumors was treated to model the primary tumor response, and the untreated tumor was used to model metastatic tumor. In the primary tumor, treatment with IONPs with and without the incorporation of CpG, infrared laser, and an external magnetic field had greatly suppressed tumor growth, with or without CpG. In the metastatic model, decrease in tumor size was also observed in treatment groups where the primary tumor was treated with IONPs, infrared laser, and external magnetic field. The metastatic tumor size decreased significantly more when CpG-incorporated IONPs were utilized in the treatment of the primary tumor, compared to the use of CpG-free IONPs, indicating the release of CpG from the IONP had elicited an immune response to treat distant tumors143 (Fig. 10b and c). The treatment of actual metastatic cancer is much more complicated due to the heterogeneity of the tumors, and detection of early metastasis, rather than treatment of advanced metastatic tumors, remains a greater challenge; however, these results indicate that stimulation of immune response using IONP-mediated PTT can effectively treat distant tumors away from the localized treatment region.144,145
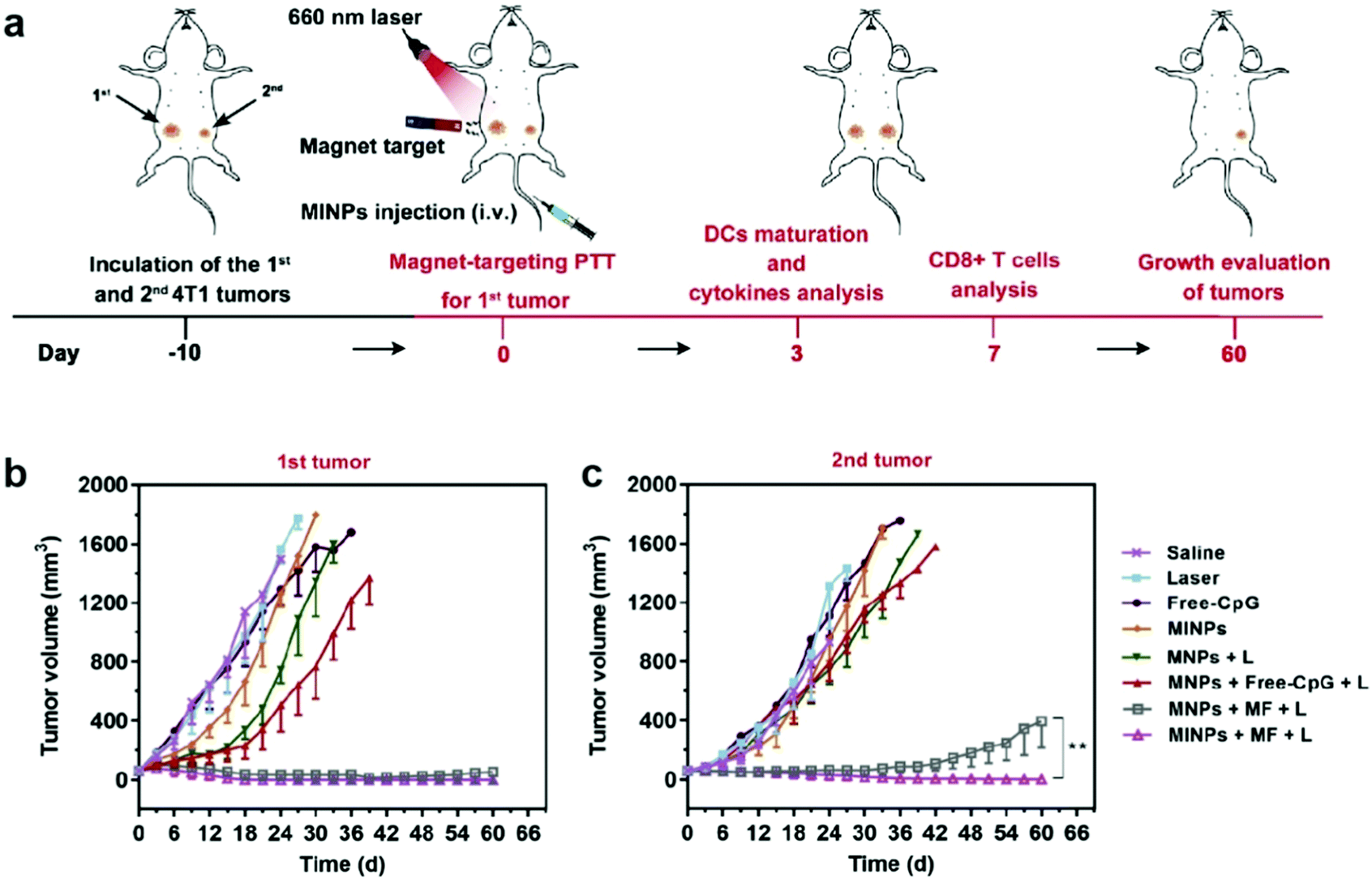 | ||
| Fig. 10 IONPs for delivery of immunostimulants and PTT. (a) Schematic illustration of the in vivo experimental design. Growth curves of (b) primary and (c) distant tumors in different treatment groups. MINP refers to nanoparticles with CpG incorporated, MNP refers to the CpG-free formulation, MF refers to presence of external magnetic field, and L refers to laser irradiation. Reproduced with permission.143 Copyright 2019, Elsevier. | ||
In another approach, IONP-induced macrophage polarization was combined with PTT mediated by IONPs. Here, a biomimetic approach to the IONP design employed the membranes of myeloid-derived suppressor cells to coat the IONPs in order to increase tumor targeting and facilitate immune escape. Mice bearing B16/F10 melanoma tumors were treated with the IONPs and irradiated with an infrared laser, which resulted in a temporary increase of tumor temperature from 34.4 to 54.7 °C. Proteins such as high-mobility group protein B1 was detected near the irradiated site, indicating PTT-induced immunogenic cell death. These IONPs were also able to reprogram macrophages from M2 to M1 to induce a greater immune response.146 Compared to other photosensitizers such as gold nanoparticles, nanorods, and carbon nanotubes, however, IONPs exhibit limited absorbance in the near infrared (NIR) region, and require irradiation levels greater than the safe limit for cutaneous tissues (0.33 W cm−2 for 808 nm laser). This precludes the use of IONPs photosensitizers by themselves in clinical settings.141 Hence, PTT methods that utilize IONPs have been accompanied by delivery of immunostimulatory molecules and surface coatings to modulate immune response. Recently, it has been shown that clustering of IONPs can improve their NIR conversion, compared to individual IONPs. Individual IONPs were synthesized via thermal decomposition, and clustered IONP were synthesized via solvothermal method. The individual nanoparticles were 15 nm in diameter, whereas the clustered IONP were around 225 nm in size and comprised of nanocrystals 5–10 nm in diameter. The clustered IONPs displayed NIR absorbance 3.6 times greater than that of individual IONPs.147 Other approaches to improving the photothermal effect of IONPs have also been investigated. Highly crystallized IONPs synthesized via thermal decomposition exhibited greater photothermal efficiency compared to commercially available IONPs of lower crystallinity. The correlation between improved photothermal efficiency and crystallinity was attributed to preferred lattice orientations in the highly crystalline core.148
Magnetic hyperthermia therapy (MHT) is another method of utilizing IONP to induce hyperthermia. In MHT, an alternating magnetic field is applied to IONPs, where the alternating magnetic moments dissipate heat approximately equal to the area of the hysteresis loop. Intrinsic magnetic properties of IONPs, as well as their biocompatibility and stability make them an ideal candidate for MHT for cancer treatment. Compared to PTT, where the use of NIR light limits their tissue penetration, MHT presents a significant advantage as alternating magnetic field has no penetration depth limitation.149 Efforts have been made to improve the heating efficiency of the IONPs in MHT applications. The morphology of iron oxide nanostructures has been shown to affect the heating efficiency in alternating magnetic fields. Ellipsoidal IONPs, obtained from growth of ellipsoidal hematite nanoparticle and subsequent conversion to magnetite, were shown to induce greater magnetic heating than Resovist, a commercially available formulation of IONPs with spherical morphology. Combined with exposure to an alternating magnetic field, administration of ellipsoidal nanoparticles was also able to inhibit tumor growth more efficiently compared to treatment with Resovist in murine 4T1 breast cancer model.150 In a similar manner, iron oxide nanorods were also shown to induce efficient magnetic heating. These iron oxide nanorods were synthesized via reduction of nonmagnetic precursor and treatment in a microwave reactor. By altering the concentration of hydrazine hydrate in the initial solution, the morphology was changed from rod-like to polyhedral plates. The iron oxide nanorods were more effective in magnetic hyperthermia than their plate-like counterparts.151 In these traditional iron oxide nanoparticle configurations, the magnetic moments exist as a pole in the axis through the center of the nanoparticle. Recently, a class of nanoparticles with a magnetic vortex have been explored as efficient nano-heaters. In these nanoparticles, the magnetic moment curls in concentric circles, and contains magnetic flux within the particle, effectively creating a particle with no magnetic pole. These vortex particles exhibited morphologies such as nanoring, or nanotube, and were able to induce hyperthermia inside the cells. In fact, these nanostructures were able to heat only the cells with high nanoparticle uptake in the first 100 s of exposure to alternating magnetic field. With improved design and specific targeting, these nanoparticles would be able to induce magnetic heating specifically in cancer cells.152
Similar to its effect in photothermal therapy, clustering of nanoparticles also influences the efficiency of magnetic heating. Citric acid-coated IONPs were encapsulated in silica matrix to explore the effect of clustering IONPs on heating efficiency in an alternating magnetic field. The IONPs were individually encapsulated in silica, encapsulated as a cluster in silica, or not encapsulated in silica at all. While no significant differences were observed in temperature elevation profile of uncoated and individually encapsulated IONPs, the IONP clusters encapsulated in silica was the least effective in magnetic heating. This effect was attributed to increased interaction between adjacent nanoparticles, which in turn impairs their Néel relaxation, which is the mean time between two flips of the magnetization of a magnetic moment.153 To prevent clustering, nanoparticles must be stabilized with binding ligands such as citric acid. In comparison of uncoated IONP and citric acid-coated IONP, the coated IONP showed not only much smaller hydrodynamic size, but also greater heating efficiency.154 Novel techniques that increase the targeting and infiltration of tumors have highlighted the potential of PTT- and MHT-based immunotherapy.
5.5 Checkpoint blockade
One of the obstacles to T cell-mediated immunotherapeutic approaches is the ability of tumor cells to evade immune surveillance. Programmed cell death protein 1 (PD-1) is a protein expressed on the surface of T cells that can bind to PD-1 ligand 1 (PD-L1) which can be found in a variety of healthy cell types, and inhibits the activity of T cells upon binding. PD-L1 is also known to be overexpressed in certain tumor cells, which allows them to evade immune surveillance and PD-L1 in tumor cells was linked to tumor aggressiveness and poor clinical prognosis.155,156 Such molecular interactions between immune cells and cancer cells is known as immune checkpoint, and by targeting inhibitory checkpoint molecules such as PD-1 and PD-L1, the immune response can be regulated so that T cells can recognize and eradicate tumor cells more efficiently.1,21 Currently, several antibodies against immune checkpoints such as nivolumab against PD-1 and ipilimumab against cytotoxic T-lymphocyte-associated protein 4 have been approved by the FDA, and many more checkpoint inhibitor molecule-based immunotherapeutic drugs are undergoing clinical trials, signifying a growing interest in checkpoint inhibition as a standard regimen in cancer therapy.25,157–159However, checkpoint inhibitors such as proteins and antibodies are prone to degradation in physiological conditions, and require administration of high dosages which is a concern for both safety and financial reasons.160 Nanoparticle systems allow efficient delivery of therapeutics to target sites, while protecting their cargo by shielding it from degradation mechanisms, and IONPs provide a layer of multifunctionality in addition to being a robust drug delivery system. Multifunctional nanoparticles consisting of lactic-co-glycolic acid, IONPs, PEG, and GRGDS peptide which is a ligand that targets membrane receptors highly expressed in melanoma cells, were synthesized to deliver anti-PD-1 antibody (aPD-1). The targeting efficiency of this nanoparticle system was assessed 24 h after systemic injection into mice bearing melanoma tumors; the incorporation of GRGDS into the nanoparticle led to a 6.7-fold increase of nanoparticle concentration in the tumors compared to the administration of free aPD-1. The nanoparticles were able to deliver more aPD-1 to tumors and also led to significant decrease in uptake and accumulation of aPD-1 by the kidneys, which could minimize drug side effects such as renal failure and pneumonitis.161 Antibodies against immune checkpoints can also be fixed onto the surface of IONPs, as demonstrated by an “immunoswitch” design consisting of dextran-coated IONPs with antibodies against multiple checkpoints conjugated on their surface. In this design, antibodies against PD-L1 served to inhibit the immunosuppressive PD-L1/PD-1 pathway, and antibodies against 4-1BB, which is an immunostimulatory molecule expressed on T cells, were utilized to stimulate T cells (Fig. 11a). The effect of incorporating these antibodies on the IONP surface was investigated by treating tumor-bearing C57BL/6 mice with adoptively transferred T cells, then administering the “immunoswitch” nanoparticles or free individual antibodies. The treatment with the antibody-conjugated IONPs resulted in greater tumor growth suppression and enhanced survival rate, as shown in Fig. 11b and c. To investigate the effect of applying both types of antibodies on a single nanoparticle, the tumor-being mice were treated with nanoparticles conjugated with both anti PD-L1 and anti 4-1BB antibodies or nanoparticles conjugated with only one type of antibody, this time without adoptive transfer of T cells. The results showed that administration of nanoparticles with only one type of antibody did not result in significant extension of survival or reduction of tumor size, whereas these effects were seen in mice treated with the “immunoswitch” nanoparticles162 (Fig. 11d and e). These results not only suggest the potential of targeting multiple checkpoint pathways for cancer immunotherapy, but also highlight the utility of IONPs as multifunctional delivery systems for checkpoint inhibition.
 | ||
| Fig. 11 IONP conjugated with anti-4-1BB and anti-PD-L1 antibodies as an “immunoswitch”. (a) Schematic representation of the role of the “immunoswitch” nanoparticle in regulating the immune checkpoints between a CD8 T cell and a tumor cell. (b) Tumor growth curves and (c) survival curves of mice treated with various combinations of adoptively transferred CD8 cells, nanoparticle, and antibodies. (d) Tumor growth curves and (e) survival curves of mice treated with the “immunoswitch” design and concurrent administration of IONP conjugated with individual antibody types. Reproduced with permission.162 Copyright 2017, American Chemical Society. | ||
While antibodies can physically target and inhibit immune checkpoints through ligand-receptor interaction, knockdown of genes that express the immunosuppressive checkpoint proteins has been investigated as another means of checkpoint inhibition. IONPs coated with PEG, polyethylenimine, and folic acid were used to deliver short interfering RNA (siRNA) to silence PD-L1 in T cells. Folic acid was incorporated to target folate-receptors that are overexpressed in gastric cancer cells, and polyethylenimine was used to enhance transfection efficiency by imparting positive surface charge on the IONPs. The IONP design demonstrated transfection efficiencies similar to that exhibited by the commercially available transfection agent Lipofectamine, and the extent of PD-L1 knockdown, measured as PD-L1 mRNA levels in treated cells, was shown to be 90.93%.163
These studies demonstrate the utility of IONPs in checkpoint inhibition therapy, as the use of IONPs as a means of delivery of inhibitor antibodies significantly increased the targeting efficiency and improved the subsequent treatment outcomes in mouse models. However, the concept of utilizing IONPs to deliver cancer therapeutic drugs has already long been studied, and only few studies have incorporated IONPs into delivery of checkpoint inhibitors. This suggests that more in-depth investigations of interactions between the IONP and the inhibitor molecules, and the conjugated IONPs and immune cells in vitro and in vivo, are needed to comprehensively evaluate the utility of IONP-mediated checkpoint blockade as a viable immunotherapeutic strategy.
6 Conclusion
IONPs provide an innovative pathway to improve immunotherapeutic approaches to cancer. Control of IONP size through adjustment of synthesis parameters and flexible surface chemistry allow modification of not only the innate properties of the nanoparticles, but also their interaction with immune cells. As IONPs do not induce substantial changes in the activity or viability of immune cells, IONP-labelled immune cells can be tracked using modalities such as MRI that leverage the unique properties of the iron oxide core. Migration of immune cells to lymph nodes and tumor tissues is vital in triggering antitumor responses, and IONPs present a non-invasive method of tracking transplanted cells to assess therapeutic outcomes. In addition to cell tracking, IONPs have also been applied to cancer vaccines targeting DCs. Antigens can be more efficiently recognized by DCs as IONPs prevent them from degradation in circulation, which allows more antigen cross-presentation to prime T cells to attack tumor cells. IONPs have also been used to increase the migration and retention of immune cells that can induce tumor cell death. Guided delivery of T cells and NK cells incorporated with IONPs to tumors via an external magnetic field has shown increased presence of these cells at tumor sites and subsequent reduction in tumor sizes mediated by the immunogenic activity these cells. By reprogramming and polarizing macrophages to their pro-inflammatory state, IONPs have been shown to suppress tumor growth through macrophage activation. IONP-based systems have also been developed to induce local hyperthermia either by NIR irradiation or with an alternating magnetic field to ablate tumors as well as trigger immune responses by delivering immunostimulants to the tumor site. The efficacy of checkpoint blockade was enhanced by IONPs by delivering and monitoring the response to inhibitory molecules.Some obstacles remain to be overcome for cancer immunotherapy to be more prevalent in clinical use. Current cancer immunotherapy has shown to induce systemic side effects.164,165 Solid tumors have shown to be less responsive to immunotherapy compared to lymphoma, as the immunotherapeutic drugs must penetrate the abnormal extracellular matrix and the immune-suppressive tumor microenvironment reduces the efficacy of the immunotherapeutics.166 Economic barriers also prevent more widespread application of immunotherapy; for example, sipuleucel-T, an immunostimulant for prostate cancer approved by the FDA in 2010, cost $93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for three injections, while the median overall survival benefit was 4.1 months.167 As novel cancer immunotherapy drugs undergo clinical trials and become more practical, this presents an opportunity for IONPs to enhance the efficiency and safety of these approaches. As a potent delivery vesicle, IONPs can more efficiently deliver immunotherapeutic molecules to the intended target while protecting them from degradation in the extracellular matrix; this could lower the initial dosage needed to achieve the same therapeutic index and potentially alleviate some of the economic burdens. Furthermore, by programming cells via adoptive cell therapy, immune cells can be engineered to more efficiently diffuse through the tumor and be protected from the immune-suppressive tumor microenvironment.
000 for three injections, while the median overall survival benefit was 4.1 months.167 As novel cancer immunotherapy drugs undergo clinical trials and become more practical, this presents an opportunity for IONPs to enhance the efficiency and safety of these approaches. As a potent delivery vesicle, IONPs can more efficiently deliver immunotherapeutic molecules to the intended target while protecting them from degradation in the extracellular matrix; this could lower the initial dosage needed to achieve the same therapeutic index and potentially alleviate some of the economic burdens. Furthermore, by programming cells via adoptive cell therapy, immune cells can be engineered to more efficiently diffuse through the tumor and be protected from the immune-suppressive tumor microenvironment.
Labeling and tracking T cells presents a challenge as their non-phagocytic nature prevents high loading of imaging probes. As evidenced by the studies presented in this review, IONPs can efficiently label and magnetize T cells so they may respond to externally applied magnetic fields. This presents the potential for IONPs to be used in conjunction with novel T cell-based immunotherapies such as CAR T cell therapy to monitor and guide the migration of T cells in vivo. Furthermore, studies have shown IONP-based vaccines to induce therapeutic effects that could potentially target metastatic cells. IONPs could be used to develop more potent vaccine designs that could effectively regulate and monitor the activation and migration of T cells to target metastasis. Advances in NK-based immunotherapy such as engineered CAR NK cells and the use of allogeneic versus autologous NK cells has led to increases in their anti-tumor activity, opening a window for IONPs to be used in conjunction with these novel approaches.168–170
A thorough study of the interaction between biomaterials and the host tissue is vital in assessing the biocompatibility of the material and improving the therapeutic effects of the material. While some studies have delved into the interaction between IONPs and immune cells, a systematic overview of the role of IONPs in the immune response and immune cell activity could highlight the utility of IONPs as a cancer immunotherapeutic agent. Clinical applications of FDA approved IONP formulations have already demonstrated IONPs to be safe and biocompatible, which is unparalleled by other metal-based nanoparticle systems for clinical use. This presents an advantage in utilizing IONPs to enhance therapeutic outcomes as further developments in cancer immunotherapy are made.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by National Institutes of Health Grant (NIH/NIBIB R01EB026890). R. A. R. acknowledges the support provided by the National Cancer Institute of the National Institutes of Health (NIH) under Award Number F31CA232546 and M. Z. acknowledges the support of Kyocera Chair Professor Endowment.Notes and references
- M.-J. Gorbet and A. Ranjan, Pharmacol. Ther., 2020, 207, 107456 CrossRef CAS PubMed.
- A. Lasfar, M. Balan, K. A. Cohen-Solal and A. Zloza, Front. Oncol., 2019, 9 DOI:10.3389/fonc.2019.01131.
- M. Riesco-Martinez, K. Parra, R. Saluja, G. Francia and U. Emmenegger, Cancer Lett., 2017, 400, 311–318 CrossRef CAS PubMed.
- K. Gong, Z.-J. Gong, P.-X. Lu, X. Ni, S. Shen, H. Liu, J.-W. Wang, D.-X. Zhang, H.-B. Liu and T. Suo, Biochem. Biophys. Res. Commun., 2019, 516, 983–990 CrossRef CAS PubMed.
- Q. Wang, Y. Shi, K. Zhou, L. Wang, Z. Yan, Y. Liu, L. Xu, S. Zhao, H. Chu, T. Shi, Q. Ma and J. Bi, Cell Death Dis., 2018, 9, 1–11 CrossRef PubMed.
- M. Abbott and Y. Ustoyev, Semin. Oncol. Nurs., 2019, 35, 150923 CrossRef PubMed.
- J. M. Caster, C. Callaghan, S. N. Seyedin, K. Henderson, B. Sun and A. Z. Wang, Adv. Drug Delivery Rev., 2019, 144, 3–15 CrossRef CAS PubMed.
- A. Byrne, P. Savas, S. Sant, R. Li, B. Virassamy, S. J. Luen, P. A. Beavis, L. K. Mackay, P. J. Neeson and S. Loi, Nat. Rev. Clin. Oncol., 2020, 17, 341–348 CrossRef PubMed.
- I. Sarkar, S. Pati, A. Dutta, U. Basak and G. Sa, Cell. Immunol., 2019, 338, 27–31 CrossRef CAS PubMed.
- Y. Gao and I. Bergman, Anticancer Res., 2018, 38, 6621–6629 CrossRef CAS PubMed.
- J. L. Reading, F. Gálvez-Cancino, C. Swanton, A. Lladser, K. S. Peggs and S. A. Quezada, Immunol. Rev., 2018, 283, 194–212 CrossRef CAS PubMed.
- A. Rivera-Rodriguez, L. B. Hoang-Minh, A. Chiu-Lam, N. Sarna, L. Marrero-Morales, D. A. Mitchell and C. Rinaldi, Nanotheranostics, 2021, 5(4), 431–444, DOI:10.1101/2020.06.02.128587.
- M. R. Parkhurst, J. P. Riley, M. E. Dudley and S. A. Rosenberg, Clin. Cancer Res., 2011, 17, 6287–6297 CrossRef CAS PubMed.
- L. Sanz-Ortega, J. M. Rojas, Y. Portilla, S. Pérez-Yagüe and D. F. Barber, Front. Immunol., 2019, 10 DOI:10.3389/fimmu.2019.02073.
- Y. Zhao, X. Zhao, Y. Cheng, X. Guo and W. Yuan, Mol. Pharmaceutics, 2018, 15, 1791–1799 CrossRef CAS PubMed.
- A. Alshamsan, in Cancer Nanotechnology: Methods and Protocols, ed. R. Zeineldin, Springer, New York, NY, 2017, pp. 257–270 Search PubMed.
- J. Liu, L. Miao, J. Sui, Y. Hao and G. Huang, Asian J. Pharm. Sci., 2020, 15(5), 576–590, DOI:10.1016/j.ajps.2019.10.006.
- A. J. Grippin, B. Wummer, T. Wildes, K. Dyson, V. Trivedi, C. Yang, M. Sebastian, H. R. Mendez-Gomez, S. Padala, M. Grubb, M. Fillingim, A. Monsalve, E. J. Sayour, J. Dobson and D. A. Mitchell, ACS Nano, 2019, 13, 13884–13898 CrossRef CAS PubMed.
- G. Traini, A. Ruiz-de-Angulo, J. B. Blanco-Canosa, K. Z. Bascarán, A. Molinaro, A. Silipo, D. Escors and J. C. Mareque-Rivas, Small, 2019, 15, 1803993 CrossRef PubMed.
- E. N. Mojarad and P. J. Kuppen, Immunotherapy, 2013, 5, 1267–1269 CrossRef CAS PubMed.
- N. Asmar, T. Ibrahim and J.-F. Rey, Dig. Dis. Sci., 2018, 63, 2177–2179 CrossRef PubMed.
- Y. Liu, J. Guo and L. Huang, Theranostics, 2020, 10, 3099–3117 CrossRef CAS PubMed.
- M. Saeed, J. Gao, Y. Shi, T. Lammers and H. Yu, Theranostics, 2019, 9, 7981–8000 CrossRef CAS PubMed.
- W. Park, Y.-J. Heo and D. K. Han, Biomater. Res., 2018, 22, 24, DOI:10.1186/s40824-018-0133-y.
- D. Zamarin, R. A. Burger, M. W. Sill, D. J. Powell, H. A. Lankes, M. D. Feldman, O. Zivanovic, C. Gunderson, E. Ko, C. Mathews, S. Sharma, A. R. Hagemann, S. Khleif and C. Aghajanian, JCO, 2020, 38, 1814–1823 CrossRef CAS PubMed.
- Bayer, A Phase III, Randomized, Double-blind, Controlled Multicenter Study of Intravenous PI3K Inhibitor Copanlisib in Combination With Standard Immunochemotherapy Versus Standard Immunochemotherapy in Patients With Relapsed Indolent Non-Hodgkin's Lymphoma (iNHL), clinicaltrials.gov, 2020.
- L. Qian, Treatment of Primary CNS Lymphoma With Systemic R-IDARAM Chemotherapy and Intrathecal Immunochemotherapy, http://clinicaltrials.gov, 2019.
- H. W. Lee, P. Gangadaran, S. Kalimuthu and B.-C. Ahn, BioMed Res. Int., 2016, e1946585 Search PubMed.
- M. Iafrate and G. O. Fruhwirth, Front. Physiol., 2020, 11 DOI:10.3389/fphys.2020.00154.
- C. Villa, S. Erratico, P. Razini, F. Fiori, F. Rustichelli, Y. Torrente and M. Belicchi, Int. J. Mol. Sci., 2010, 11, 1070–1081 CrossRef CAS PubMed.
- M. M. Welling, M. Duijvestein, A. Signore and L. van der Weerd, J. Cell. Physiol., 2011, 226, 1444–1452 CrossRef CAS PubMed.
- U. Himmelreich and T. Dresselaers, Methods, 2009, 48, 112–124 CrossRef CAS PubMed.
- K. R. Hurley, H. L. Ring, M. Etheridge, J. Zhang, Z. Gao, Q. Shao, N. D. Klein, V. M. Szlag, C. Chung, T. M. Reineke, M. Garwood, J. C. Bischof and C. L. Haynes, Mol. Pharmaceutics, 2016, 13, 2172–2183 CrossRef CAS PubMed.
- S. J. Kim, B. Lewis, M.-S. Steiner, U. V. Bissa, C. Dose and J. A. Frank, Contrast Media Mol. Imaging, 2016, 11, 55–64 CrossRef CAS PubMed.
- C. Sun, C. Fang, Z. Stephen, O. Veiseh, S. Hansen, D. Lee, R. G. Ellenbogen, J. Olson and M. Zhang, Nanomedicine, 2008, 3, 495–505 CrossRef CAS PubMed.
- C. Janko, M. Pöttler, J. Matuszak, H. Unterweger, A. Hornung, R. P. Friedrich and C. Alexiou, Chem. Ing. Tech., 2017, 89, 244–251 CrossRef CAS.
- V. Kalidasan, X. L. Liu, T. S. Herng, Y. Yang and J. Ding, Nano-Micro Lett., 2016, 8, 80–93 CrossRef PubMed.
- H. Wang, Q. Mu, R. Revia, K. Wang, B. Tian, G. Lin, W. Lee, Y.-K. Hong and M. Zhang, J. Controlled Release, 2018, 289, 70–78 CrossRef CAS PubMed.
- O. Veiseh, F. M. Kievit, C. Fang, N. Mu, S. Jana, M. C. Leung, H. Mok, R. G. Ellenbogen, J. O. Park and M. Zhang, Biomaterials, 2010, 31, 8032–8042 CrossRef CAS PubMed.
- N. Guldris, B. Argibay, J. Gallo, R. Iglesias-Rey, E. Carbó-Argibay, Y. V. Kolen’ko, F. Campos, T. Sobrino, L. M. Salonen, M. Bañobre-López, J. Castillo and J. Rivas, Bioconjugate Chem., 2017, 28, 362–370 CrossRef CAS PubMed.
- L. Li, W. Jiang, K. Luo, H. Song, F. Lan, Y. Wu and Z. Gu, Theranostics, 2013, 3, 595–615 CrossRef PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed.
- J. Han, D. Zhao, D. Li, X. Wang, Z. Jin and K. Zhao, Polymers, 2018, 10(1), 31, DOI:10.3390/polym10010031.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1–10 CrossRef CAS PubMed.
- V. Janelle, C. Rulleau, S. Del Testa, C. Carli and J.-S. Delisle, Front. Immunol., 2020, 11 DOI:10.3389/fimmu.2020.00276.
- W. H. Fridman, F. Pagès, C. Sautès-Fridman and J. Galon, Nat. Rev. Cancer, 2012, 12, 298–306 CrossRef CAS PubMed.
- J. Reiser and A. Banerjee, J. Immunol. Res., 2016, 2016, 8941260 Search PubMed.
- F. Yang and X.-F. Yang, Cell. Mol. Immunol., 2005, 2, 331–341 CAS.
- C. Fu and A. Jiang, Front. Immunol., 2018, 9 DOI:10.3389/fimmu.2018.03059.
- N. Shimasaki, A. Jain and D. Campana, Nat. Rev. Drug Discovery, 2020, 19, 200–218 CrossRef CAS PubMed.
- J. Kowal, M. Kornete and J. A. Joyce, Immunotherapy, 2019, 11, 677–689 CrossRef CAS PubMed.
- J. Perrin, M. Capitao, M. Mougin-Degraef, F. Guérard, A. Faivre-Chauvet, L. Rbah-Vidal, J. Gaschet, Y. Guilloux, F. Kraeber-Bodéré, M. Chérel and J. Barbet, Front. Med., 2020, 7 DOI:10.3389/fmed.2020.00034.
- E. T. Ahrens and J. W. M. Bulte, Nat. Rev. Immunol., 2013, 13(10) DOI:10.1038/nri3531.
- C. Wu, Y. Xu, L. Yang, J. Wu, W. Zhu, D. Li, Z. Cheng, C. Xia, Y. Guo, Q. Gong, B. Song and H. Ai, Adv. Funct. Mater., 2015, 25, 3581–3591 CrossRef CAS.
- S. Schwarz, F. Fernandes, L. Sanroman, M. Hodenius, C. Lang, U. Himmelreich, T. Schmitz-Rode, D. Schueler, M. Hoehn, M. Zenke and T. Hieronymus, J. Magn. Magn. Mater., 2009, 321, 1533–1538 CrossRef CAS.
- Y. Xu, C. Wu, W. Zhu, C. Xia, D. Wang, H. Zhang, J. Wu, G. Lin, B. Wu, Q. Gong, B. Song and H. Ai, Biomaterials, 2015, 58, 63–71 CrossRef CAS PubMed.
- N.-H. Cho, T.-C. Cheong, J. H. Min, J. H. Wu, S. J. Lee, D. Kim, J.-S. Yang, S. Kim, Y. K. Kim and S.-Y. Seong, Nat. Nanotechnol., 2011, 6, 675–682 CrossRef CAS PubMed.
- C. A. Janeway, P. Travers, M. Walport and M. J. Shlomchik, Immunobiology: The Immune System, 2001 Search PubMed.
- J. Gunn, H. Wallen, O. Veiseh, C. Sun, C. Fang, J. Cao, C. Yee and M. Zhang, Small, 2008, 4, 712–715 CrossRef CAS PubMed.
- O. A. Garden, P. R. Reynolds, J. Yates, D. J. Larkman, F. M. Marelli-Berg, D. O. Haskard, A. D. Edwards and A. J. T. George, J. Immunol. Methods, 2006, 314, 123–133 CrossRef CAS PubMed.
- L. Liu, Q. Ye, Y. Wu, W.-Y. Hsieh, C.-L. Chen, H.-H. Shen, S.-J. Wang, H. Zhang, T. K. Hitchens and C. Ho, Nanomedicine, 2012, 8, 1345–1354 CrossRef CAS PubMed.
- W.-N. Jin, X. Yang, Z. Li, M. Li, S. X.-Y. Shi, K. Wood, Q. Liu, Y. Fu, W. Han, Y. Xu, F.-D. Shi and Q. Liu, J. Cereb. Blood Flow Metab., 2016, 36, 1464–1476 CrossRef CAS PubMed.
- L. Wang, Z. Wang, T. G. Frank, S. I. Brown, S. A. Chudek and A. Cuschieri, Nanomedicine, 2009, 4, 305–315 CrossRef CAS PubMed.
- M. F. Kircher, J. R. Allport, E. E. Graves, V. Love, L. Josephson, A. H. Lichtman and R. Weissleder, Cancer Res., 2003, 63, 6838–6846 CAS.
- M. Lewin, N. Carlesso, C.-H. Tung, X.-W. Tang, D. Cory, D. T. Scadden and R. Weissleder, Nat. Biotechnol., 2000, 18, 410–414 CrossRef CAS PubMed.
- C. Berger, M. Rausch, P. Schmidt and M. Rudin, Mol. Imaging, 2006, 5(00010), 7290.2006 Search PubMed.
- P. Gangadaran, R. L. Rajendran and B.-C. Ahn, Cancers, 2020, 12(5) DOI:10.3390/cancers12051318.
- K. Li, A. C. Gordon, L. Zheng, W. Li, Y. Guo, J. Sun, G. Zhang, G. Han, A. C. Larson and Z. Zhang, Nanomedicine, 2015, 10, 1761–1774 CrossRef CAS PubMed.
- E.-S. Jang, J.-H. Shin, G. Ren, M.-J. Park, K. Cheng, X. Chen, J. C. Wu, J. B. Sunwoo and Z. Cheng, Biomaterials, 2012, 33, 5584–5592 CrossRef CAS PubMed.
- R. A. Burga, D. H. Khan, N. Agrawal, C. M. Bollard and R. Fernandes, Bioconjugate Chem., 2019, 30, 552–560 CrossRef CAS PubMed.
- Z. Zhao, Z. Zhou, J. Bao, Z. Wang, J. Hu, X. Chi, K. Ni, R. Wang, X. Chen, Z. Chen and J. Gao, Nat. Commun., 2013, 4, 2266 CrossRef PubMed.
- M. W. Marashdeh, B. Ababneh, O. M. Lemine, A. Alsadig, K. Omri, L. El Mir, A. Sulieman and E. Mattar, Results Phys., 2019, 15, 102651 CrossRef.
- W. Zhang, L. Liu, H. Chen, K. Hu, I. Delahunty, S. Gao and J. Xie, Theranostics, 2018, 8, 2521–2548 CrossRef CAS PubMed.
- F. Hu, K. W. MacRenaris, E. A. Waters, T. Liang, E. A. Schultz-Sikma, A. L. Eckermann and T. J. Meade, J. Phys. Chem. C, 2009, 113, 20855–20860 CrossRef CAS PubMed.
- M. Barrow, A. Taylor, P. Murray, M. J. Rosseinsky and D. J. Adams, Chem. Soc. Rev., 2015, 44, 6733–6748 RSC.
- M. Hoehn, D. Wiedermann, C. Justicia, P. Ramos-Cabrer, K. Kruttwig, T. Farr and U. Himmelreich, J. Physiol., 2007, 584, 25–30 CrossRef CAS PubMed.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicine, 2016, 11, 673–692 CrossRef CAS PubMed.
- B. Dalzon, A. Torres, S. Reymond, B. Gallet, F. Saint-Antonin, V. Collin-Faure, C. Moriscot, D. Fenel, G. Schoehn, C. Aude-Garcia and T. Rabilloud, Nanomaterials, 2020, 10, 266 CrossRef CAS PubMed.
- A. Kunzmann, B. Andersson, C. Vogt, N. Feliu, F. Ye, S. Gabrielsson, M. S. Toprak, T. Buerki-Thurnherr, S. Laurent, M. Vahter, H. Krug, M. Muhammed, A. Scheynius and B. Fadeel, Toxicol. Appl. Pharmacol., 2011, 253, 81–93 CrossRef CAS PubMed.
- D. L. J. Thorek and A. Tsourkas, Biomaterials, 2008, 29, 3583–3590 CrossRef CAS PubMed.
- M. Wu, H. Guo, L. Liu, Y. Liu and L. Xie, Int. J. Nanomed., 2019, 14, 4247–4259 CrossRef CAS PubMed.
- L. Li, W.-S. Xi, Q. Su, Y. Li, G.-H. Yan, Y. Liu, H. Wang and A. Cao, Small, 2019, 15, 1901687 CrossRef PubMed.
- P. Foroozandeh and A. A. Aziz, Nanoscale Res. Lett., 2018, 13(1) DOI:10.1186/s11671-018-2728-6.
- S. K. Lai, K. Hida, S. T. Man, C. Chen, C. Machamer, T. A. Schroer and J. Hanes, Biomaterials, 2007, 28, 2876–2884 CrossRef CAS PubMed.
- S. Chen, S. Chen, Y. Zeng, L. Lin, C. Wu, Y. Ke and G. Liu, J. Appl. Toxicol., 2018, 38, 978–986 CrossRef CAS PubMed.
- W. Injumpa, P. Ritprajak and N. Insin, J. Magn. Magn. Mater., 2017, 427, 60–66 CrossRef CAS.
- M. Baer, A. Dillner, R. C. Schwartz, C. Sedon, S. Nedospasov and P. F. Johnson, Mol. Cell. Biol., 1998, 18, 5678–5689 CrossRef CAS PubMed.
- D. M. Underhill and A. Ozinsky, Annu. Rev. Immunol., 2002, 20, 825–852 CrossRef CAS PubMed.
- M. Jeon, M. V. Halbert, Z. R. Stephen and M. Zhang, Adv. Mater., 2021, 33(23), 1906539 CrossRef CAS PubMed.
- Z. R. Stephen, F. M. Kievit and M. Zhang, Mater. Today, 2011, 14, 330–338 CrossRef CAS PubMed.
- E. Pöselt, H. Kloust, U. Tromsdorf, M. Janschel, C. Hahn, C. Maßlo and H. Weller, ACS Nano, 2012, 6, 1619–1624 CrossRef PubMed.
- R. A. Brooks, Magn. Reson. Med., 2002, 47, 388–391 CrossRef CAS PubMed.
- N. Lee, Y. Choi, Y. Lee, M. Park, W. K. Moon, S. H. Choi and T. Hyeon, Nano Lett., 2012, 12, 3127–3131 CrossRef CAS PubMed.
- V. E. Orel, M. Tselepi, T. Mitrelias, M. Zabolotny, A. Shevchenko, A. Rykhalskyi, A. Romanov, V. B. Orel, A. Burlaka, S. Lukin, V. Kyiashko and C. H. W. Barnes, Nanotechnology, 2019, 30, 415701 CrossRef CAS PubMed.
- A. D. Sherry and Y. Wu, Curr. Opin. Chem. Biol., 2013, 17, 167–174 CrossRef CAS PubMed.
- S. M. Dadfar, D. Camozzi, M. Darguzyte, K. Roemhild, P. Varvarà, J. Metselaar, S. Banala, M. Straub, N. Güvener, U. Engelmann, I. Slabu, M. Buhl, J. van Leusen, P. Kögerler, B. Hermanns-Sachweh, V. Schulz, F. Kiessling and T. Lammers, J. Nanobiotechnol., 2020, 18, 22 CrossRef CAS PubMed.
- J. Zhao, Z. Zhang, Y. Xue, G. Wang, Y. Cheng, Y. Pan, S. Zhao and Y. Hou, Theranostics, 2018, 8, 6307–6321 CrossRef CAS PubMed.
- A. Y. Sheu, Z. Zhang, R. A. Omary and A. C. Larson, Invest. Radiol., 2013, 48, 492–499 CrossRef PubMed.
- A. Farzadniya, F. Faeghi and S. Shanehsazzadeh, Appl. Magn. Reson., 2017, 48, 597–607 CrossRef CAS.
- Z. R. Stephen, C. J. Dayringer, J. J. Lim, R. A. Revia, M. V. Halbert, M. Jeon, A. Bakthavatsalam, R. G. Ellenbogen and M. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 6320–6328 CrossRef CAS PubMed.
- F. M. Kievit, O. Veiseh, N. Bhattarai, C. Fang, J. W. Gunn, D. Lee, R. G. Ellenbogen, J. M. Olson and M. Zhang, Adv. Funct. Mater., 2009, 19, 2244–2251 CrossRef CAS PubMed.
- J. Y. Oh, H. S. Kim, L. Palanikumar, E. M. Go, B. Jana, S. A. Park, H. Y. Kim, K. Kim, J. K. Seo, S. K. Kwak, C. Kim, S. Kang and J.-H. Ryu, Nat. Commun., 2018, 9, 4548 CrossRef PubMed.
- S. Ferraris, M. Cazzola, V. Peretti, B. Stella and S. Spriano, Front. Bioeng. Biotechnol., 2018, 6 DOI:10.3389/fbioe.2018.00060.
- Y. Mou, Y. Xing, H. Ren, Z. Cui, Y. Zhang, G. Yu, W. J. Urba, Q. Hu and H. Hu, Nanoscale Res. Lett., 2017, 12, 52 CrossRef PubMed.
- H. Liu, H. Dong, N. Zhou, S. Dong, L. Chen, Y. Zhu, H. Hu and Y. Mou, Nanoscale Res. Lett., 2018, 13, 409 CrossRef PubMed.
- A. Jedlovszky-Hajdú, E. Tombácz, I. Bányai, M. Babos and A. Palkó, J. Magn. Magn. Mater., 2012, 324, 3173–3180 CrossRef.
- M. E. Ladd, P. Bachert, M. Meyerspeer, E. Moser, A. M. Nagel, D. G. Norris, S. Schmitter, O. Speck, S. Straub and M. Zaiss, Prog. Nucl. Magn. Reson. Spectrosc., 2018, 109, 1–50 CrossRef CAS PubMed.
- Y.-K. Peng, S. C. E. Tsang and P.-T. Chou, Mater. Today, 2016, 19, 336–348 CrossRef CAS.
- T. J. Fraum, D. R. Ludwig, M. R. Bashir and K. J. Fowler, J. Magn. Reson. Imaging, 2017, 46, 338–353 CrossRef PubMed.
- H. Wang, R. Revia, K. Wang, R. J. Kant, Q. Mu, Z. Gai, K. Hong and M. Zhang, Adv. Mater., 2017, 29, 1605416 CrossRef PubMed.
- X. Song, Z. Liu, X. Xu and Q. Tang, New J. Chem., 2014, 38, 3813–3818 RSC.
- P. Caravan, Chem. Soc. Rev., 2006, 35, 512–523 RSC.
- Y. Li, X. Zhao, X. Liu, K. Cheng, X. Han, Y. Zhang, H. Min, G. Liu, J. Xu, J. Shi, H. Qin, H. Fan, L. Ren and G. Nie, Adv. Mater., 2020, 32, 1906799 CrossRef CAS PubMed.
- H. Zhang, L. Li, X. L. Liu, J. Jiao, C.-T. Ng, J. B. Yi, Y. E. Luo, B.-H. Bay, L. Y. Zhao, M. L. Peng, N. Gu and H. M. Fan, ACS Nano, 2017, 11, 3614–3631 CrossRef CAS PubMed.
- A. P. Khandhar, G. J. Wilson, M. G. Kaul, J. Salamon, C. Jung and K. M. Krishnan, J. Biomed. Mater. Res., Part A, 2018, 106, 2440–2447 CrossRef CAS PubMed.
- S. Caspani, R. Magalhães, J. P. Araújo and C. T. Sousa, Materials, 2020, 13(11) DOI:10.3390/ma13112586.
- H. Wei, O. T. Bruns, M. G. Kaul, E. C. Hansen, M. Barch, A. Wiśniowska, O. Chen, Y. Chen, N. Li, S. Okada, J. M. Cordero, M. Heine, C. T. Farrar, D. M. Montana, G. Adam, H. Ittrich, A. Jasanoff, P. Nielsen and M. G. Bawendi, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 2325–2330 CrossRef CAS PubMed.
- S. A. Anderson, K. K. Lee and J. A. Frank, Invest. Radiol., 2006, 41, 332–338 CrossRef PubMed.
- B. Gustafsson, S. Youens and A. Y. Louie, Bioconjugate Chem., 2006, 17, 538–547 CrossRef CAS PubMed.
- I. E. Haedicke, S. Loai and H.-L. M. Cheng, Contrast Media Mol. Imaging, 2019, e3475786 Search PubMed.
- A. Faith and C. M. Hawrylowicz, Clin. Exp. Immunol., 2005, 139, 395–397 CrossRef CAS PubMed.
- T. H. Tran, T. T. P. Tran, H. T. Nguyen, C. D. Phung, J.-H. Jeong, M. H. Stenzel, S. G. Jin, C. S. Yong, D. H. Truong and J. O. Kim, Int. J. Pharm., 2018, 542, 253–265 CrossRef CAS PubMed.
- L. Luo, M. Z. Iqbal, C. Liu, J. Xing, O. U. Akakuru, Q. Fang, Z. Li, Y. Dai, A. Li, Y. Guan and A. Wu, Biomaterials, 2019, 223, 119464 CrossRef CAS PubMed.
- M. Mühlberger, C. Janko, H. Unterweger, E. Schreiber, J. Band, C. Lehmann, D. Dudziak, G. Lee, C. Alexiou and R. Tietze, J. Magn. Magn. Mater., 2019, 473, 61–67 CrossRef.
- M. Mühlberger, C. Janko, H. Unterweger, R. P. Friedrich, B. Friedrich, J. Band, N. Cebulla, C. Alexiou, D. Dudziak, G. Lee and R. Tietze, Int. J. Nanomed., 2019, 14, 8421–8432 CrossRef PubMed.
- L. Sanz-Ortega, J. M. Rojas, A. Marcos, Y. Portilla, J. V. Stein and D. F. Barber, J. Nanobiotechnol., 2019, 17, 14 CrossRef PubMed.
- A. D. McLellan, Oncoimmunology, 2014, 3 DOI:10.4161/onci.28582.
- S. Lorenzo-Herrero, A. López-Soto, C. Sordo-Bahamonde, A. P. Gonzalez-Rodriguez, M. Vitale and S. Gonzalez, Cancers, 2018, 11(1) DOI:10.3390/cancers11010029.
- M. Balsamo, W. Vermi, M. Parodi, G. Pietra, C. Manzini, P. Queirolo, S. Lonardi, R. Augugliaro, A. Moretta, F. Facchetti, L. Moretta, M. C. Mingari and M. Vitale, Eur. J. Immunol., 2012, 42, 1833–1842 CrossRef CAS PubMed.
- G. Sconocchia, R. Arriga, L. Tornillo, L. Terracciano, S. Ferrone and G. C. Spagnoli, Cancer Res., 2012, 72, 5428–5429 CrossRef CAS PubMed.
- G. Sconocchia, G. C. Spagnoli, D. Del Principe, S. Ferrone, M. Anselmi, W. Wongsena, V. Cervelli, E. Schultz-Thater, S. Wyler, V. Carafa, H. Moch, L. Terracciano and L. Tornillo, Neoplasia, 2009, 11, 662–671 CrossRef CAS PubMed.
- L. Wu, F. Zhang, Z. Wei, X. Li, H. Zhao, H. Lv, R. Ge, H. Ma, H. Zhang, B. Yang, J. Li and J. Jiang, Biomater. Sci., 2018, 6, 2714–2725 RSC.
- M. Orecchioni, Y. Ghosheh, A. B. Pramod and K. Ley, Front. Immunol., 2019, 10 DOI:10.3389/fimmu.2019.01084.
- A. Laskar, J. Eilertsen, W. Li and X.-M. Yuan, Biochem. Biophys. Res. Commun., 2013, 441, 737–742 CrossRef CAS PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS PubMed.
- K. Li, L. Lu, C. Xue, J. Liu, Y. He, J. Zhou, Z. Xia, L. Dai, Z. Luo, Y. Mao and K. Cai, Nanoscale, 2019, 12, 130–144 RSC.
- C.-X. Li, Y. Zhang, X. Dong, L. Zhang, M.-D. Liu, B. Li, M.-K. Zhang, J. Feng and X.-Z. Zhang, Adv. Mater., 2019, 31, 1807211 CrossRef PubMed.
- E. C. Saputra, L. Huang, Y. Chen and L. Tucker-Kellogg, Cancer Res., 2018, 78, 2419–2431 CrossRef CAS PubMed.
- A. C. V. Doughty, A. R. Hoover, E. Layton, C. K. Murray, E. W. Howard and W. R. Chen, Materials, 2019, 12(5) DOI:10.3390/ma12050779.
- K. Bilici, A. Muti, F. D. Duman, A. Sennaroğlu and H. Y. Acar, Photochem. Photobiol. Sci., 2018, 17, 1787–1793 CrossRef CAS PubMed.
- J. Estelrich and M. A. Busquets, Molecules, 2018, 23(7) DOI:10.3390/molecules23071567.
- B. Scheicher, A.-L. Schachner-Nedherer and A. Zimmer, Eur. J. Pharm. Sci., 2015, 75, 54–59 CrossRef CAS PubMed.
- Y. Guo, Y. Ran, Z. Wang, J. Cheng, Y. Cao, C. Yang, F. Liu and H. Ran, Biomaterials, 2019, 219, 119370 CrossRef CAS PubMed.
- C. Liang, L. Xu, G. Song and Z. Liu, Chem. Soc. Rev., 2016, 45, 6250–6269 RSC.
- C.-N. Qian, Y. Mei and J. Zhang, Chin. J. Cancer, 2017, 36 DOI:10.1186/s40880-017-0206-7.
- G.-T. Yu, L. Rao, H. Wu, L.-L. Yang, L.-L. Bu, W.-W. Deng, L. Wu, X. Nan, W.-F. Zhang, X.-Z. Zhao, W. Liu and Z.-J. Sun, Adv. Funct. Mater., 2018, 28, 1801389 CrossRef.
- S. Shen, S. Wang, R. Zheng, X. Zhu, X. Jiang, D. Fu and W. Yang, Biomaterials, 2015, 39, 67–74 CrossRef CAS PubMed.
- H. Chen, J. Burnett, F. Zhang, J. Zhang, H. Paholak and D. Sun, J. Mater. Chem. B, 2014, 2, 757–765 RSC.
- Z. R. Stephen and M. Zhang, Adv. Healthcare Mater., 2021, 10, 2001415 CrossRef CAS PubMed.
- H. Gao, T. Zhang, Y. Zhang, Y. Chen, B. Liu, J. Wu, X. Liu, Y. Li, M. Peng, Y. Zhang, G. Xie, F. Zhao and H. M. Fan, J. Mater. Chem. B, 2020, 8, 515–522 RSC.
- A. Nikitin, M. Khramtsov, A. Garanina, P. Mogilnikov, N. Sviridenkova, I. Shchetinin, A. Savchenko, M. Abakumov and A. Majouga, J. Magn. Magn. Mater., 2019, 469, 443–449 CrossRef CAS.
- C. S. B. Dias, T. D. M. Hanchuk, H. Wender, W. T. Shigeyosi, J. Kobarg, A. L. Rossi, M. N. Tanaka, M. B. Cardoso and F. Garcia, Sci. Rep., 2017, 7, 14843 CrossRef PubMed.
- S. Nemec, S. Kralj, C. Wilhelm, A. Abou-Hassan, M.-P. Rols and J. Kolosnjaj-Tabi, Appl. Sci., 2020, 10, 7322 CrossRef CAS.
- M. Hammad, S. Hardt, B. Mues, S. Salamon, J. Landers, I. Slabu, H. Wende, C. Schulz and H. Wiggers, J. Alloys Compd., 2020, 824, 153814 CrossRef CAS.
- S. Inaguma, Z. Wang, J. Lasota, M. Sarlomo-Rikala, P. A. McCue, H. Ikeda and M. Miettinen, Am. J. Surg. Pathol., 2016, 40, 1133–1142 CrossRef PubMed.
- A. A. Davis and V. G. Patel, Journal for ImmunoTherapy of Cancer, 2019, 7, 278 CrossRef PubMed.
- H. Zhang and J. Chen, J. Cancer, 2018, 9, 1773–1781 CrossRef PubMed.
- K. Diao, S. X. Bian, D. M. Routman, C. Yu, J. C. Ye, N. A. Wagle, M. K. Wong, G. Zada and E. L. Chang, J. Neurooncol., 2018, 139, 421–429 CrossRef CAS PubMed.
- M. T. Wan and M. E. Ming, Br. J. Dermatol., 2018, 179, 296–300 CAS.
- Z. Yang, Y. Ma, H. Zhao, Y. Yuan and B. Y. S. Kim, WIREs Nanomedicine and Nanobiotechnology, 2020, 12, e1590 Search PubMed.
- N. Zhang, J. Song, Y. Liu, M. Liu, L. Zhang, D. Sheng, L. Deng, H. Yi, M. Wu, Y. Zheng, Z. Wang and Z. Yang, J. Controlled Release, 2019, 306, 15–28 CrossRef CAS PubMed.
- A. K. Kosmides, J.-W. Sidhom, A. Fraser, C. A. Bessell and J. P. Schneck, ACS Nano, 2017, 11, 5417–5429 CrossRef CAS PubMed.
- X. Luo, X. Peng, J. Hou, S. Wu, J. Shen and L. Wang, Int. J. Nanomed., 2017, 12, 5331–5343 CrossRef CAS PubMed.
- P. W. Kantoff, C. S. Higano, N. D. Shore, E. R. Berger, E. J. Small, D. F. Penson, C. H. Redfern, A. C. Ferrari, R. Dreicer, R. B. Sims, Y. Xu, M. W. Frohlich and P. F. Schellhammer, N. Engl. J. Med., 2010, 363, 411–422 CrossRef CAS PubMed.
- S. L. Maude, D. Barrett, D. T. Teachey and S. A. Grupp, Cancer J., 2014, 20, 119–122 CrossRef CAS PubMed.
- S. Yan, P. Zhao, T. Yu and N. Gu, Cancer Biol. Med., 2019, 16, 486–497 CAS.
- S. Simoens, Hum. Vaccines Immunother., 2012, 8, 506–508 CrossRef PubMed.
- J. P. Veluchamy, N. Kok, H. J. van der Vliet, H. M. W. Verheul, T. D. de Gruijl and J. Spanholtz, Front. Immunol., 2017, 8 DOI:10.3389/fimmu.2017.00631.
- C. Quintarelli, S. Sivori, S. Caruso, S. Carlomagno, M. Falco, I. Boffa, D. Orlando, M. Guercio, Z. Abbaszadeh, M. Sinibaldi, S. Di Cecca, A. Camera, B. Cembrola, A. Pitisci, M. Andreani, L. Vinti, S. Gattari, F. Del Bufalo, M. Algeri, G. Li Pira, A. Moseley, B. De Angelis, L. Moretta and F. Locatelli, Leukemia, 2020, 34, 1102–1115 CrossRef CAS PubMed.
- W. Hu, G. Wang, D. Huang, M. Sui and Y. Xu, Front. Immunol., 2019, 10 DOI:10.3389/fimmu.2019.01205.
| This journal is © The Royal Society of Chemistry 2021 |