
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh performance LiMnFePO4/Li4Ti5O12 full cells by functionalized polymeric additives†
Jean-Christophe
Daigle
 *a,
Sylviane
Rochon
a,
Yuichiro
Asakawa
b,
Benoît
Fleutot
a,
Charlotte
Mallet
a,
Kamyab
Amouzegar
a and
Karim
Zaghib
*c
*a,
Sylviane
Rochon
a,
Yuichiro
Asakawa
b,
Benoît
Fleutot
a,
Charlotte
Mallet
a,
Kamyab
Amouzegar
a and
Karim
Zaghib
*c
aCenter of Excellence in Transportation Electrification and Energy Storage (CETEES), Hydro-Québec, 1806, Lionel-Boulet Blvd., Varennes, Québec J3X 1S1, Canada. E-mail: daigle.jean-christophe@hydroquebec.com
bMurata Munufacturing, 10-1 Higashikotari 1-chrome, Nagaokakyo-shi, Kyoto 617-8555, Japan
cDepartment of Materials Engineering, McGill University, 3610 University Street, Montréal, Québec H3A 0C5, Canada. E-mail: karim.zaghib@mcgill.ca
First published on 15th October 2020
Abstract
Herein, we report a novel class of additives based on functional polymers for electrode fabrication. A small quantity of a 2,3-dimethoxybenzene (DMB)-based polymer was mixed with a state-of-the-art binder, poly(vinylene difluoride) (PVDF), to greatly improve the first fast charge–discharge and high-voltage LiMnFePO4/Li4Ti5O12 batteries operating at 3.0 V. Dual effects of the polymer were reported, i.e., trapping of soluble Mn2+ of positive electrodes and the in situ formation of a passivation layer on the negative electrode (AlF3). Both effects are possible because a functional polymer was used; this synergic effect enhances the cell performance by 16.7% after more than 300 cycles at 45 °C and 1C.
Introduction
At present, considerable industrial research effort is devoted to extending the cycle life of commercial Li-ion battery technologies that can be synthesized safely and consequently employed in EVs. Moreover, a high C-rate, an increase in power, and a high energy density of battery packs lead to a higher internal operation temperature that can decrease the lifetime and induce thermal runaway.1–5 Therefore, management of temperature by complex cooling systems is essential for preventing any side chemical reactions during cell operation, which can cause degradation, and, by extension, battery fires.6,7From a chemical perspective point of view, extending the cycle life of batteries despite extreme operating conditions functions as a powerful catalyst to promote innovation.8,9 One of the fundamental approaches that has garnered significant research attention is the mitigation of degradation during cycling by adding organic molecules in the battery electrolyte. For example, additives can be used with a redox shuttle effect to protect cells against overcharging, which could otherwise cause severe safety issues. Methoxybenzene derivatives, which were initially introduced by Adachi et al.10,11 and described in numerous reports,12–14 were effectively added to the electrolyte to achieve this purpose. A reverse reaction occurs by oxidation at the positive electrode, followed by diffusion to the negative electrode. The molecule exhibits a shuttle effect and carries the current.
Additives are also applied to induce the formation of a protective solid electrolyte interface (SEI) on the negative electrode surface by decomposition or polymerization. Vinylene carbonate (VC) is extensively used in academia and industries to protect graphite or lithium titanium oxide (LTO) anodes. The decomposition of VC instigates the formation of a passive nanolayer that creates a barrier between the negative electrode and the electrolyte and improves the interfacial stability.8,15 In our group, we introduced a Schiff base as an additive in LiFePO4/Li4Ti5O12 batteries; 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) initiates ring-opening polymerization of propylene carbonate to form a layer of ionic conductive polymer on the LTO surface. Furthermore, we reported effective implementation in cylindrical cells.16 The formation of a protective layer of LTO is a well-known strategy to prevent the degradation (e.g., solvents and salts) that is induced by the LTO reactive surface. Interaction between LTO and the electrolytes can result in a myriad of side reactions, resulting in accelerated degradation and gas evolution.17–20
Degradation may also be prevented by limiting the diffusion of undesirable species, such as HF, water, or metal ions, into the electrolyte, thereby leading to a series of side reactions. Undesirable species such as metal ions commonly occur in the presence of high-voltage manganese-based and cobalt-free cathodes. Moreover, dissolution of Mn2+ in the electrolyte during cell operation is caused by water and acid, and can induce the deposition of Mn0 on the negative electrode surface, resulting in cell polarization and capacity deterioration. This thin layer of metal can function as a catalyst and accelerate the degradation of electrolytes, compromising the sustainability of cells.4,21–23
Silane-based additives are effective for reactions with HF and water, and for preventing Mn dissolution.9 Cyclic aminosilane (0.5%) recently has enhanced the stability of NMC-graphite cells by 28% at 45 °C. A dual effect is reported; the additive captures HF and improves the interfacial layer on the positive electrode.24 Cyclic ether-based and acid-based molecules that can interact with bivalent ions have been used to restrict the mobility of bivalent ions in the electrolyte, and their subsequent plating at the negative electrode. For example, separators have been chemically designed to trap metal ions (M2+) by incorporating aza-crown and crown-ether groups in the polymer structure to improve the cell durability.25–28 Lee et al. reported an alginate-based polymer that is used as an alternative binder for LMO-based batteries instead of poly(vinylene difluoride) (PVDF).29 The chemical interaction between dissolved Mn2+ and the polymer prevents dissolution in the electrolyte. Degradation of cells is substantially reduced at 55 °C, particularly with lithium bis(oxalato)borate (LiBOB) as the salt in the electrolyte composition. LiBOB suppresses Mn2+ dissolution by coordination; the resultant complex forms a stable interface at the positive electrode surface.9
Herein, we report a new strategy for preventing the dissolution of multivalent metal ions. This novel strategy entails a mixture of a functional polymer, poly(pentafluorostyrene-co-2,3-dimethoxystyrene), with a state-of-art binder, PVDF. 2,3-Dimethoxystyrene enables the polymer to coordinate metal ions30–33 and prevent dissolution. Therefore, the metal is trapped in the positive electrode.
Moreover, we implemented this strategy in a positive electrode with LiMn0.75Fe0.20Mg0.05PO4 (LMFP) as the active material and tested its validity in Li-ion coin cells paired with Li4Ti5O12 (LTO) as the negative electrode. These active materials were selected to evaluate our polymer on a challenging full cell, i.e., an Mn-based electrode and a reactive negative electrode (LTO), which undergo several side reactions (gas evolution). Among the olivine-type LiMPO4 positive electrode materials, LiFePO4 nanomaterials with carbon coating are predominantly used due to their excellent battery performance. However, it is important to increase their redox potential reaction to improve the energy density. LiMnPO4 is a promising material due to a redox potential around 4.1 V vs. Li+/Li, which is still within the stability window of typical carbonate electrolytes.34,35 Moreover, the material comprises an abundance of relevant elements35,36 (neither Ni nor Co is used) but suffers from low ionic and electronic conductivity. Due to the partial substitution of Mn2+ with Fe2+, LMFP demonstrates better cycling capacity and energy density than LMP and LiFePO4, respectively. The design of the LMFP–LTO cell detailed in this study facilitates a voltage operation of 3.0 V because in our configuration the negative electrode is the limiting material in the balancing when compared with other materials versus LTO (Scheme 1). At the same time, this balancing limits the oxidation of the liquid electrolyte at high potential. Due to the redox potential reaction of the LTO material, aluminum current collectors can be used as positive and negative electrodes, thus reducing the cost. Therefore, investigation was made to understand the impact on the full-cell performance and the synergic effects of both electrodes.
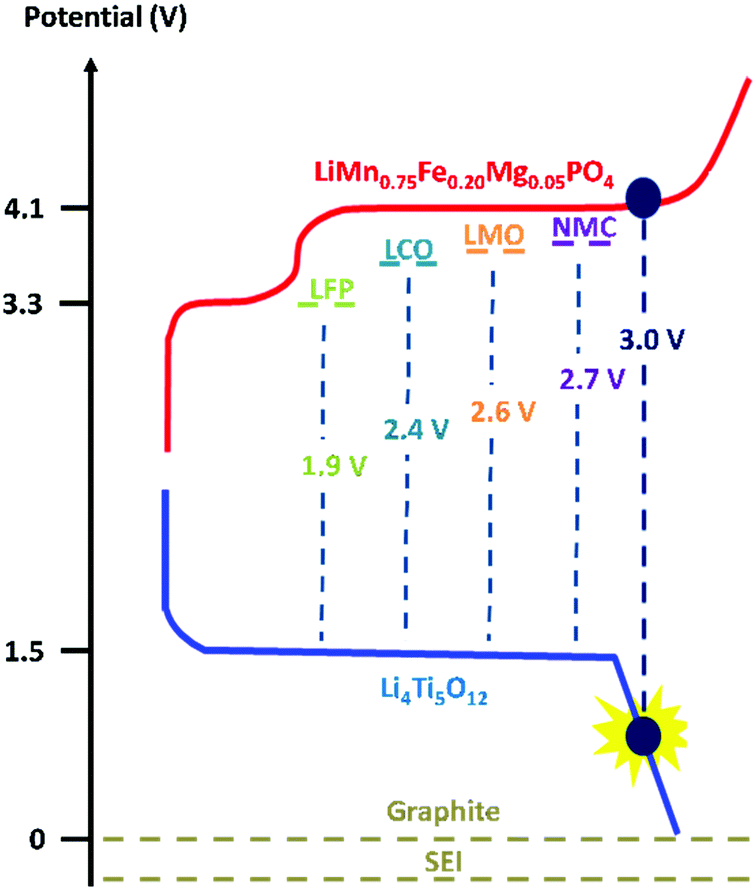 | ||
| Scheme 1 Potential of the electrode materials used for constructing a full cell. | ||
Experimental
Materials
All chemicals were purchased from Sigma Aldrich and VWR chemicals and were used without further purification, unless otherwise stated. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was crystallized in hot methanol.Random co-polymerization of 3,4-dimethoxystyrene (DMSt) and pentafluorostyrene (PFSt)
DMSt and PFSt were first passed through basic aluminum oxide (alumina, Al2O3). Polymerization was achieved by introducing dioxane (50.0 mL), DMSt (3.2 g, 19.5 mmol), and PFSt (3.7 g, 19.1 mmol) into a round-bottom flask (100 mL). The solution was then stirred with a magnetic bar for 30 min and bubbled with nitrogen gas. AIBN (80 mg, 0.488 mmol) was then added to the obtained solution. A condenser was then attached to the round-bottom flask and the reaction mixture was heated at 80 °C for 12 h under N2. The resulting solution was then cooled to 22 °C and poured in 10% (v/v) methanol. The supernatant was separated from the precipitated polymer, which was then dried under vacuum at 60 °C for 12 h. Yield = 4.1 g, 48 mol% in DMSt determined by 1H NMR in CDCl3, Mn = 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, PDI = 1.7.
000 g mol−1, PDI = 1.7.
Demethylation of the co-polymers was performed according to a method reported by Westwood et al.30 by using the polymer (0.50 g) previously described and BBr3 (4.3 mL) in 1.0 M DCM.
Physical and surface characterization
Liquid 1H NMR analysis was performed on a Bruker 300 instrument operating at 300 MHz with an acquisition time of 1.9 s. The samples were dissolved in CDCl3 and the residual solvent peak was used for calibration. Molecular weight distribution (Mn, Mw, and PDI) was determined using gel permeation chromatography (GPC, 1260 Infinity II Multi-detector from Agilent) equipped with triple detection (LS at 690.0 nm, viscosity, and RI) and operating at 25 °C with THF as the eluent. The columns used were Agilent (Varian) PL gel mixed-B 10 mm. The samples were treated in a glovebox and transferred to a TOF-SIMS instrument using a transfer vessel to prevent the samples from being exposed to air. TOF-SIMS measurements were performed using a time-of-flight secondary ion mass spectrometer (TOF-SIMS V, ION-TOF GmbH, Germany). The vacuum pressure in the main chamber during the measurements was <4 × 10−7 Pa. During the analysis, the targets were bombarded with Bi3++ cluster ions (30 kV) with a primary ion current of 0.05 pA. The analysis area was 200 × 200 μm2 and the total ion dose was 6 × 1010 ions cm−2.Convergence of the primary ion beam <1 μm enabled microanalysis and high spatial resolution imaging (observation of distribution). The detection depth of this analytical method was less than several nanometers (<3 nm). The chemical composition of the cathode surface was investigated by XPS using a PHI 5600-ci spectrometer (Physical Electronics, Eden Prairie, MN). The main XPS chamber was maintained at a base pressure of <8 × 10−9 Torr. A standard aluminum X-ray source (Al kα = 1486.6 eV) was used to record the survey spectra (1400–0 eV, 10 min), whereas pure magnesium was used for the high-resolution spectra, both without charge neutralization. The detection angle was set at 45° with respect to the surface normal and the analyzed area was 0.05 cm2 (aperture 4). High-resolution spectra were obtained for C 1s with 40 sweeps. The spectrometer work function was adjusted to achieve 285.0 eV for the main C (1s) peak.
Cell assembly and electrochemical measurements
Carbon coated LMFP and LTO were supplied by Sumitomo Osaka Cement and Posco, respectively. LiPF6, solvents, and PVDF were purchased from BASF. Electronically conductive materials such as acetylene black and VGCF were purchased from Denka and Showa Denko, respectively.The positive electrode contained LMFP (90 wt%), acetylene black (4.0 wt%), VGCF (1.0 wt%), and PVDF or poly(pentafluorostyrene-co-3,4-dimethoxystyrene) as the binder (4.0 wt% and 1.0 wt% or 3.0 wt% and 2.0 wt%, respectively). The slurry was mixed using a Thinky mixer and coated on a 15 μm aluminum collector using the Doctor Blade method. The negative electrodes contained LTO (90 wt%), acetylene black (5.0 wt%), and the PVDF binder (5.0 wt%). The slurry, was mixed using a Thinky mixer, and was coated on a 15 μm aluminum collector using the Doctor Blade method. The poly(ethylene) separator was 16 μm thick. The electrolyte used was LiPF6 1.0 mol kg−1 with carbonate solvents,8,28 and 1.0 wt% VC was added as the additive. The electrodes were fabricated in a dried room with a maximum dew point of −58 °C. The cells were assembled in a glovebox under Argon.
Prior to cycling, the cells were cycled (formation) at 0.05, 0.1, and 0.2C at 25 °C, and the capacities were recorded. Retention capacities at different C-rates were calculated based on an average of the three cells and four cycles per cell at 25 °C; load tests were performed by charging or discharging at 0.2C before applying each C-rate. Static capacities were recorded at 0.05, 0.1, and 0.2C and 25 °C during the cycle-life test. All measurements were done on a Bio Logic VMP3 workstation. The cycle life was performed at 45 °C for continuous charge–discharge at 1C and recorded on a Toscat-3000 battery tester using a Toyo-system.
Results and discussion
Binder additive
A functional binder additive was developed to enhance the physical or electrochemical properties of the state-of-the-art binder (PVDF). The concept of an additive binder is based on introducing a small fraction of a specific binder and adding it to the state-of-the-art binder, i.e., PVDF. Hence, PVDF is the dominant binder. We reported, in a previous study, the good compatibility of poly(pentafluorostyrene) grafted on LTO particles with a PVDF binder.17 Moreover, styrene-butadiene rubber (SBR) was used as a stable binder for the positive electrode; however, the voltage stability was limited to 4.3 V.37,38 In parallel, the methylbenzene-based compound exhibited promising applications in the battery caused by the shuttle effect.10,39 Moreover, polymers based on quinone and catechol moieties were reported as active electrode materials, and oxidation occurred to form stable anions that could coordinate ions.40,41 Hence, the 2,3-dimethoxystyrene-based polymer could act as functional monomers upon the coordination of metal ions, thereby preventing the dissolution of bivalent ions in the electrolyte, with Mn2+ being the main target. We presumed that the combination of the 2,3-dimethoxystyrene-based polymer and pentafluorostyrene could have a dual effect.Free-radical polymerization of pentafluorostyrene and 2,3 dimethoxystyrene (DMSt) was performed (Scheme S1 in the ESI†), as described in the Experimental section. The polymer obtained had a backbone comprising 48 mol% DMSt, as calculated by 1H NMR (Fig. S1 in the ESI†) with a molecular weight of 26000 g mol−1 (GPC trace in the ESI†).
Electrochemical effects of the polymer on the LMFP/LTO cell
The DMB-based polymer was mixed with PVDF in proportions of 1.0 wt% and 2.0 wt% to facilitate use as a binder. Initial electrochemical evaluation demonstrated that the binder containing 2.0 wt% DMB-based polymer significantly hindered the performances at 2C and 4C. Therefore, we fabricated an LMFP-based positive electrode and used it to assess the impact of our functional binder additive in a proportion of 1.0 wt% for 4.0 wt% PVDF. To facilitate a comparison with a reference battery, the standard LMFP positive electrode was exclusively fabricated with PVDF as the binder; 5.0 wt% PVDF was used to achieve the same total composition of the binder in the positive electrode, thereby enabling an easier comparison of the variation in performances and phenomena. The initial evaluation was performed in a coin cell configuration, as depicted in Fig. 1. The same LTO negative electrode was used and prepared as described in the experimental section. Hence, we were able to isolate and extract the effect of our functional binder additive. As expected, the initial charge/discharge cycle (Fig. 1a) exhibits a slightly lower initial capacity at low C-rates (0.05C, 0.1C and 0.2C) with 1.0 wt% additive polymer compared to the corresponding pure PVDF binder, which is still acceptable. Indeed, as reported by us previously, the introduction of polar groups in the polymer impeded the diffusion of lithium in the electrode.17 This effect is confirmed and is considerably more visible at high C rates because the efficiency diminishes as the C-rate increases. Fig. 1b and c shows that this trend is more significant with our additive binder. However, further experiments are required because the loss of capacity retention remains largely unchanged at 1, 2, and 4C, regardless of whether our functional polymer additive was used.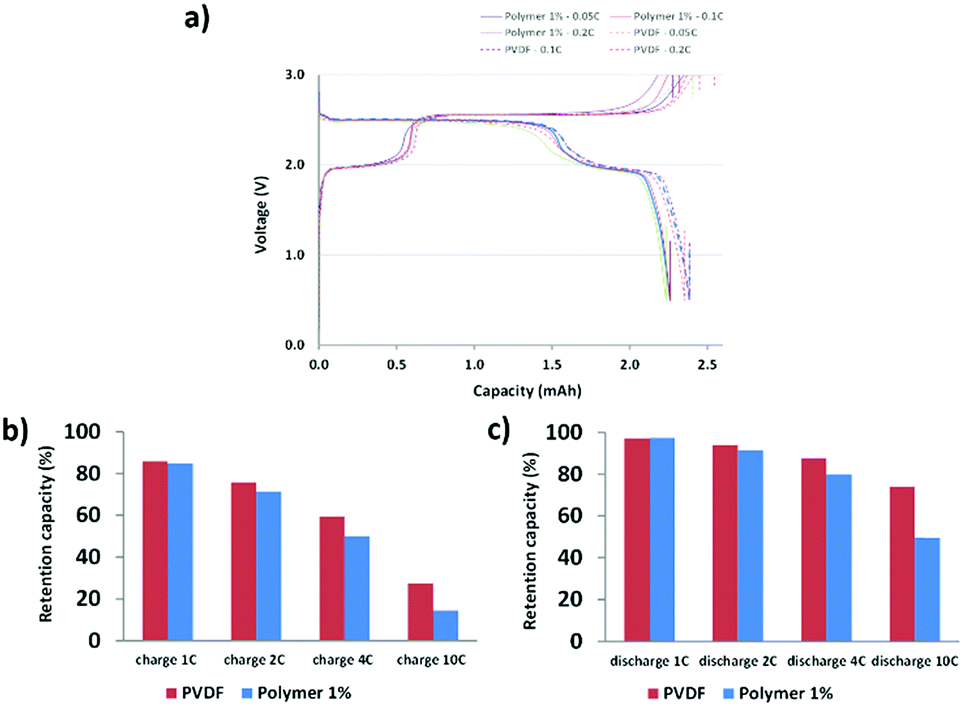 | ||
| Fig. 1 Initial electrochemical evaluation of cells. (a) Charge–discharge curves at different C rates. (b) Bar graphs of the charging cells at different C rates. (c) Bar graphs of the discharging cells at different C rates. | ||
Plating of metals on the LTO negative electrode in a full-cell configuration can induce side reactions that lead to cell degradation.17 Hence, the primary objective of using a functional polymer as an additive is to prevent Mn2+ and other metal ions from interacting with the electrolyte. Scheme 2 shows a reaction that can occur in the positive electrode (LMFP in our case) during the battery cycle.
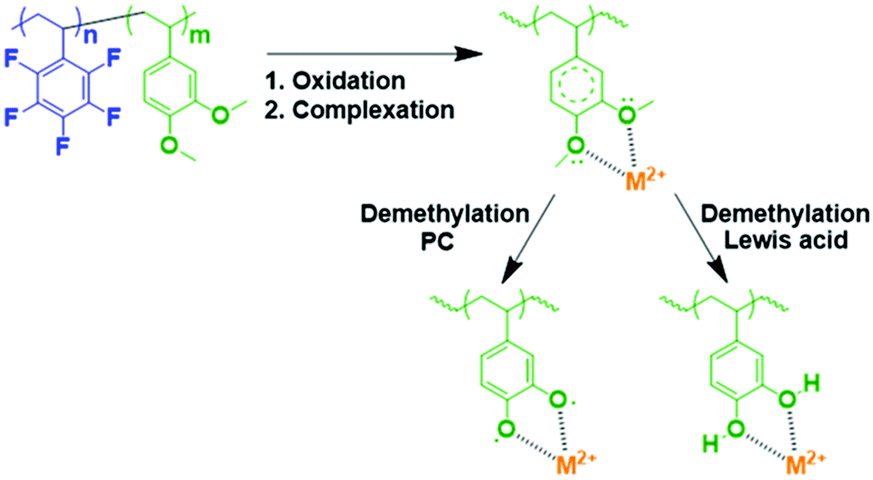 | ||
| Scheme 2 Mechanism of reaction for the polymer additive with the metal ions in the cathode. | ||
The coordination of multivalent metal ions is extremely feasible with methoxy groups, and the subsequent in situ demethylation can proceed via two mechanisms; demethylation did not alter the coordination ability.
Demethylation using a Lewis acid (BBr3) is extensively used in organic chemistry to form adhesive catechol-based polymers with high efficiency; thanks to PF5− (Lewis acid), the degradation product in the electrolyte, this reaction is possible via this pathway in the cell.30 Another pathway was theoretically reported and confirmed by calculations.39 This alternative pathway involved the formation of a stable radical and a reaction with propylene carbonate, which was a constituent of the electrolyte.
In both cases, M+, M2+, and M3+ coordination is expected. This method could prevent the dissolution of undesirable multivalent ion metals (e.g., Fe2+, Mn2+, and Fe3+) because a highly stable complex is formed with the catechol-based polymer and the reaction with Li+ occurs at 3.2 V vs. Li+/Li0. Therefore, this reaction will be reversible in our proposed cell (LMFP–LTO).33,40,41
Briefly, the functional polymer additive could trap multivalent metal ions and improve the aging of the cell based on the LMFP positive electrode. Hence, we performed continuous cycling at 1C (charge–discharge) and 45 °C for over 300 cycles (Fig. 2a) to accelerate the dissolution of ions and aging of the cells and to verify our hypotheses.
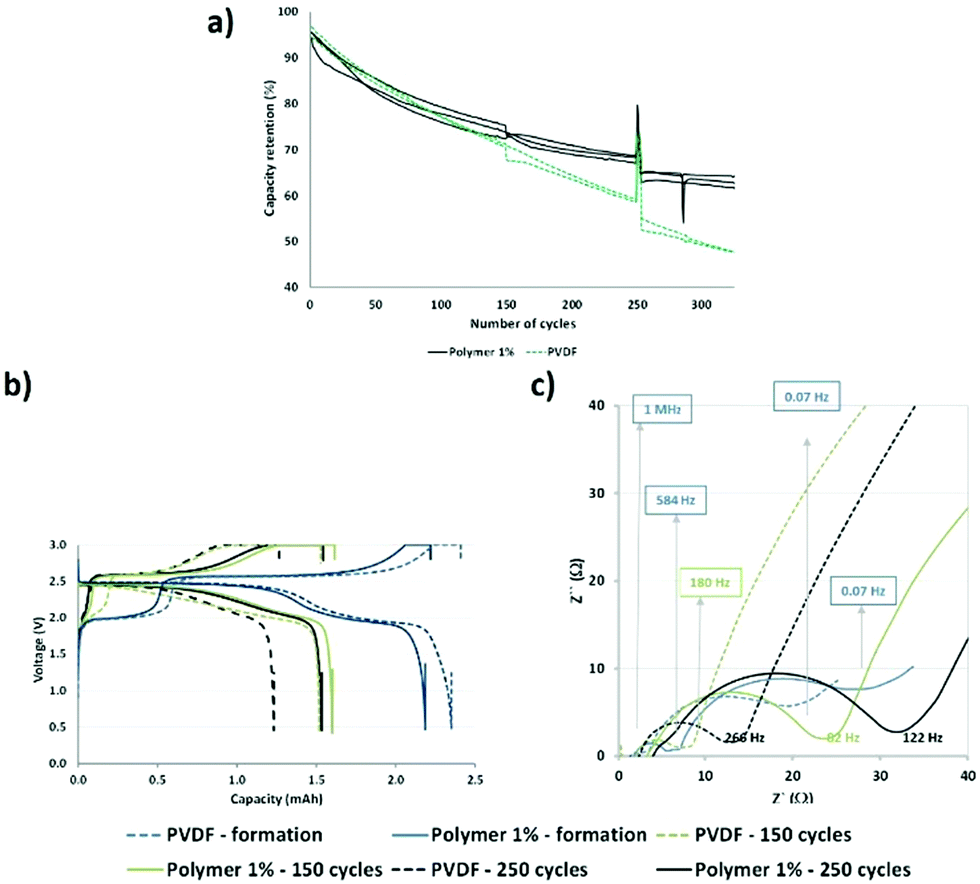 | ||
| Fig. 2 (a) Cycle-life curves of the coin cells at 1C and 45 °C. (N.B.: The break at 250 cycles is due to the recorded static capacities at 0.05, 0.1, and 0.2C). (b) Charge–discharge curves during the cycle-life experiment. (c) Nyquist plots at different cycle times. | ||
From Fig. 2a, a good reproducibility is observed for the three cells, which enables us to extract reliable data. Our batteries demonstrated better capacity retention, with a larger improvement in aging with our functional polymer over 300 cycles, compared to those reported by Borgel et al.42 Therefore, we confirmed that our polymer additive is effective for preventing degradation and extending the cycle life because the only difference between the cells is the presence or absence of a functional polymer. After 80 cycles, a deviation in the capacity retention curves (PVDF vs. polymer 1%) is observed and this may be ascribed to several phenomena occurring in the cells. Static capacities (0.2C and 25 °C) and impedances were measured at the 1st, 150th, and 250th cycles to enable a more comprehensive understanding. The capacity values are similar for the reference (PVDF) and the polymer additive (polymer 1%) after 150 and 250 cycles, respectively (Fig. 2b). We observed stabilization of capacity retention between 150 and 250 cycles in the presence of the polymer additive. However, in the absence of this additive, a large decrease was observed. Therefore, our polymer additive improves the capacity retention at long cycles.
Moreover, we observed a shape modification of the discharge curve (green dashed line) for the reference cells at the 150th cycle. This may be associated with the dissolution of Mn ions in the electrolyte because the biphasic phenomenon related to the reduction of Mn3+ to Mn2+ in LMFP is not easily observable and does not occur at a fixed potential. Fig. 2c confirms that there is no added resistance during cycling. We previously reported that the polymer additive revealed a high resistance at the first cycle and above 0.07 Hz, probably due to the presence of polar groups which hinder diffusion.14
After 150 and 250 cycles, there is a higher impedance increase for the cell above 100 Hz containing the functional additive polymer, even if the impedance increases with aging regardless of the type of positive electrode (with and without our polymer). Table S1 in the ESI† reports the resistance and capacitance values extracted from Fig. 2c. The aforementioned frequency (100 Hz) corresponds to the frequency domains of the interface and charge transfer. Hence, this phenomenon could be ascribed to the formation of a specific interface at the positive or negative electrode or both related to a chemical or electrochemical reaction or both with our functional polymer additive, as discussed in the subsection Post-mortem analysis of electrodes. However, this interface is advantageous in that it helps to improve the capacity retention during aging (Fig. 2a (after 150 and 250 cycles)).
To better understand the phenomena occurring during aging, the electrochemical data were analyzed more thoroughly by deriving the capacities after every ten cycles and plotting the dQ/dV curves (Fig. S3 in the ESI†). All the batteries comprised of the same loading electrode, balancing, and surfaces. Hence it is possible to directly compare the different results. From these curves, the maximum derivatives for each metal insertion were extracted, corresponding to Mn3+/Mn2+ at ∼2.6 V and Fe3+/Fe2+ at ∼2.0 V versus LTO, and plotted as a function of the cycle number (Fig. 3). The LTO active material was stable during aging.
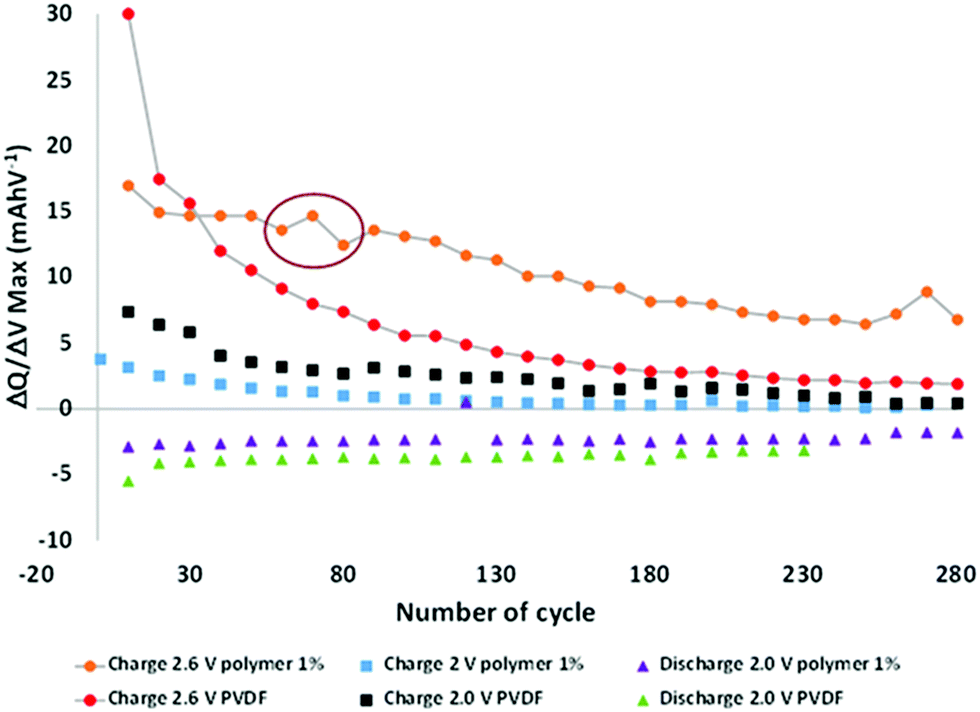 | ||
| Fig. 3 Maximum derivatives as a function of the number of cycles at different voltages. | ||
Hence, all observable variations may be attributed to the degradation of the positive electrode, and potentially to the formation of a specific interface at the negative electrode surface.
The charge curves at 2.6 V versus LTO demonstrate two different behaviors as a function of the modified binder, i.e., in the presence or absence of the functional polymer additive. The PVDF-based cells exhibit a rapid decrease in the number of coulombs consumed relative to the oxidation of Mn2+ to Mn3+ as cycling progresses, thereby indicating a decrease in the reversible Mn3+/Mn2+ redox reaction, which can be related to a rapid loss of Mn. After approximately 60 cycles, the change in the slope indicates a decline in the Mn dissolution with a continuous decrease in the rate of dissolution. After 120 cycles, the slope becomes relatively flat (changed rates). An entirely different behavior is observed when the polymer additive is used. A plateau is observed between 20 and 50 cycles, which indicates that, contrary to the pure PVDF binder, the redox reaction is reversible, hence there is no Mn2+ dissolution. As expected, the polymer additive had coordinated some ions. However, after ∼60–80 cycles, a fluctuation was observed (red circle in Fig. 3), which we believe is related to a side reaction in the cell; depletion subsequently occurred at a constant rate that is lower than that of the pure PVDF without our functional polymer additive. This variation validated the hypothesis that a continuous phenomenon (reaction) played a role in the remainder of thecycles. Also, this fluctuation correlates with what is observed in Fig. 2a—the change in the slope for the cycle-life curves is attributed to the additive polymers and the enhanced capacity retention.
Based on these observations, we postulate that our polymer additive may be involved in the coordination of Mn2+ (before 60 cycles); subsequently, an internal reaction occurred, followed by the release of metal ions. The reactions or other phenomena that occurred after 80 cycles were directly associated with the decreased degradation and Mn2+ dissolution, which were prevented during the initial stage of aging and limited in the latter stage. No major effects were detected at other voltages during charging and discharging.
To more accurately verify the behavioral variation at the negative electrode or in the domain of the Fe3+/Fe2+ redox reaction at the positive electrode, the maximum derivate dQ/dV for the Fe3+/Fe2+ couple (∼2.0 V vs. LTO) was plotted as a function of the potential at this maximum (Fig. S4 in the ESI†).
A decrease in the number of coulombs consumed relative to the oxidation of Fe2+ to Fe3+ as cycling progresses is observed, with no large modification of the reaction potential. Hence, a decrease in the rate of the reversible reaction can be observed with aging, but without the formation of an interface with huge impedance. If an impeded interface is generated during cycling, the resistance will increase and so the reaction potential during charging (decrease in the reaction potential during discharging). A variation of 0.05 V is extracted with the pure PVDF binder, thereby confirming the small increase in impedance during cycling, as shown in the Nyquist plot (Fig. 2c) during aging. The reaction voltage varies more extensively with variation of the order of 0.2 V during cycling, even if the same decrease in the rate of the reversible reaction is observed with our functional polymer additive. A more resistive interface is created during cycling with our polymer, which is confirmed by the higher impedance measured by electrochemical impedance spectroscopy (Fig. 2c). Since the same decrease in capacity is observed in the domain of Fe2+/Fe3+, regardless of the binder used, we assumed that the modification of the interface occurred at the negative electrode, i.e., at the surface of the LTO electrode.
Post-mortem analysis of electrodes
Consequently, there must be a reaction in the positive electrode that triggers PL formation on the negative surface electrode (LTO). To validate this hypothesis, the surface of the negative electrode was analyzed by time-of-flight secondary ion mass spectrometry (TOF-SIMS) under an inert atmosphere after cycling. Table 1 outlines the main compounds detected and the corresponding normalized intensities. Fig. 4 depicts the negative electrode (polymer 1%) after disassembly. Two surfaces were observed: a light-colored center (major) and a dark-colored edge (minor); the spectra of both were analyzed. Reduced amounts of Ca and Mg were detected in the center for the sample containing the polymer additive compared to a similar intensity observed on the edge for the negative electrode with the pure PVDF binder. This observation allows us to hypothesize that the polymer effectively coordinates Mn2+ and Fe2+ throughout the cycling, as observed for the catechol-based polymer applied as organic electrodes.33Table 1 and Fig. 4 show that a negligible amount of Mn was detected in the center of the LTO negative electrode with 1.0 wt% polymer (black color, 15 times less vs. anode PVDF).| Positive spectrum | |||
|---|---|---|---|
| LTO PVDF | LTO Center | LTO Edge | |
| ID | Normalized intensity (×10−5) | ||
| Mg | 222 | 6.12 | 91.9 |
| Al | 30.0 | 262 | 106 |
| Li | 2500 | 3000 | 2160 |
| Li2OH | 71.2 | 394 | 354 |
| Li3O | 137 | 97.4 | 13.9 |
| Ca | 169 | 5.96 | 7.18 |
| Mn | 209 | 13.5 | 847 |
| Fe | 50.8 | 2.84 | 48.0 |
| Li3F2 | 4150 | 8030 | 1900 |
| C3HF4 | 787 | 253 | 286 |
| Negative spectrum | |||
|---|---|---|---|
| LTO PVDF | LTO Center | LTO Edge | |
| ID | Normalized intensity (×10−5) | ||
| CN | 631 | 62.1 | 79.6 |
| HF2 | 562 | 120 | 45.9 |
| CHO2 | 209 | 68 | 97.3 |
| C2H3O | 190 | 103 | 94.6 |
| LiCO3 | 31.1 | 14.5 | 17.1 |
| Li2F3 | 626 | 607 | 54.9 |
| AlF4 | 431 | 13800 |
2380 |
| PF6 | 10500 |
6820 | 11300 |
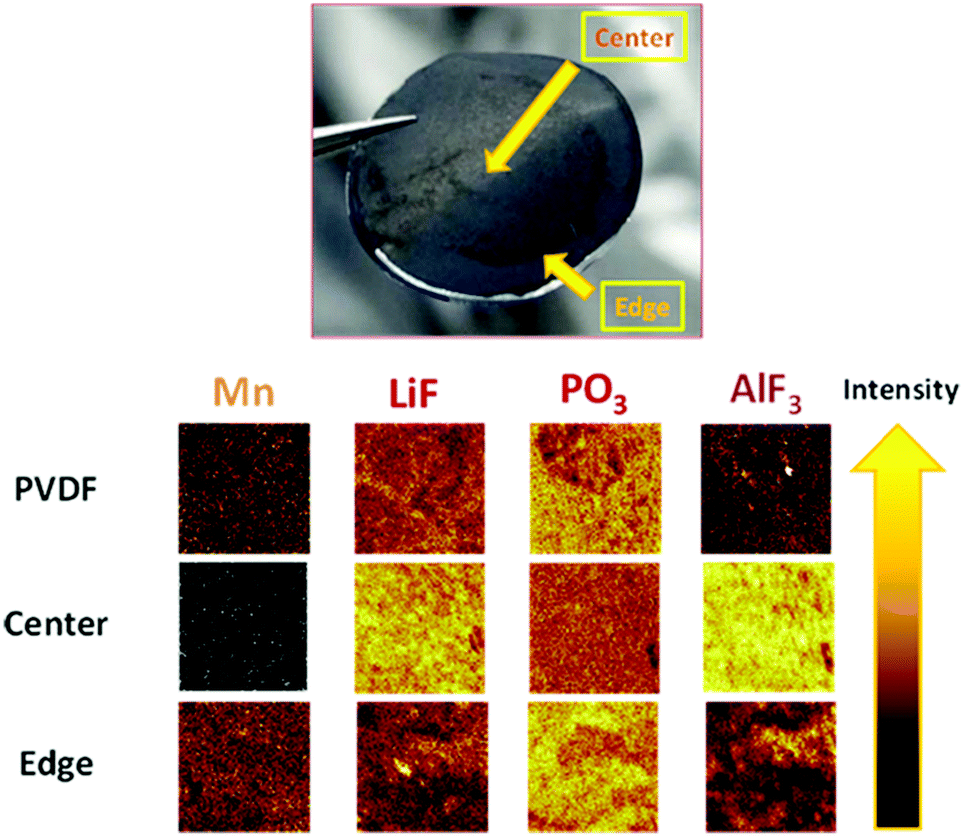 | ||
| Fig. 4 Photograph of the negative electrode with 1 wt% polymer and TOF-SIMS images with and without 1 wt% polymer as a function of the analyze point position and the material detected. | ||
A strong intensity associated with Mn was identified on the edge, which is superior to that of the negative electrode with PVDF. The amount of Mn in the anode with the polymer determined by ICP-MS was twice as superior comparing with PVDF. We believe that this was related to the continuous exponential release observed in Fig. 3. However, we cannot speculate on the valence of Mn detected, but the deposition of MnF2 was feasible.43
This lack of deposition of metals in the center of the anode directly correlates with the presence of AlF3 on the surface (Fig. 4) and the detection of Al+. Therefore, the PL of AlF3 was formed, LiF formation was induced, and deposition was prevented. Moreover, fewer derivatives of HF were detected (5 times less, see Table 1); therefore, we presumed that AlF3 was possibly formed upon the reaction with HF. This consumption of HF to form PL also justifies why less solvents degraded based on the fragments (organic fragments, Table 1) detected and the PO3− (linked with LiPF6) observed, which is ascribed to the electrolyte decomposition (Fig. 4). The utility of the functional polymer enabled the in situ formation of a thin layer of AlF3 on the LTO surface and the prevention of degradation.18 The degradation caused by Mn and the decomposition of the solvents can be mitigated effectively by PL formation. Aurbach et al.42 attributed the capacity fading to the Li+ consumption of positive electrodes to form a passivation layer on the LTO surface; Li2CO3 was detected and ascribed to solvent degradation. They suggested that pre-treatment of the LTO electrode was an effective strategy for protecting the negative electrode to assemble a highly stable LMFP–LTO-based Li-ion battery. By using our additive, the detection of lithium carbonate was approximately two times less. Therefore, the PL formed by AlF3 prevented the electrolyte decomposition and Li+ consumption more effectively. Our strategy allows implementation of a stable Li-ion battery without pre-treatment of the LTO electrode. Moreover, the AlF3-based PL was more effective than the VC-based PL (the cell contained 1.0 wt% VC); VC was used to form the PL on the LTO surface in the reference cells.
We hypothesized that Al3+ originated from the cathode collector and Mn plays a role in the mechanism. XPS analysis of the cathode revealed a peak corresponding to aluminum oxide (75.5–76.0 eV), and a shift of the Mn peak associated with coordination to oxygen (Mn–O). Only one organic species, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (ketones, aldehydes), differed from that of the reference (Fig. S5 in the ESI†).
O (ketones, aldehydes), differed from that of the reference (Fig. S5 in the ESI†).
Proposed mechanism for the formation of the passivation layer
All these observations enabled us to address a putative mechanism for the coordination of metal ions to the polymer and the in situ formation of AlF3 on the anode surface by the reaction between the aluminum derivative and HF. Scheme 3 shows our hypothesis.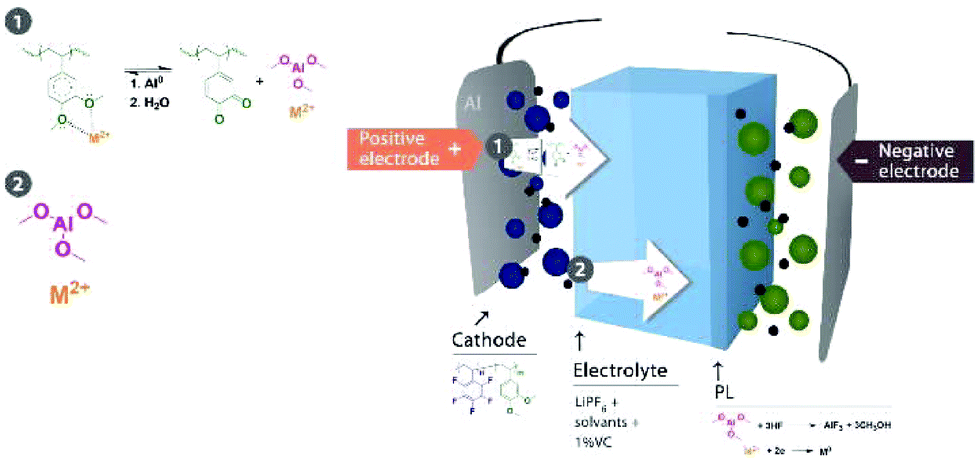 | ||
| Scheme 3 Proposed mechanism for PL formation. | ||
The detection of AlF3 on the anode suggests that Al3+ was coordinated to the polymer, forming a stable complex.33 Therefore, we presume that Al originated from the current collector and the release of Mn2+ and Al3+ was possible due to a synergic chemical reaction induced by electrons. Al2O3, which was detected on the cathode surface, and AlF3 were insoluble in the electrolyte. The organometallic complex of aluminum was more susceptible to diffusion in organic solvents. We presume that Al from the current collector reacted with the dimethoxy group in the polymer to form Al(OCH3)3 and simultaneously released the trapped Mn2+; this reaction between Al and an alcohol is a common reaction in chemical engineering.44 The reaction is usually catalyzed by HgCl2, but we hypothesized that Mn2+ can feasibly function as a catalyst in our cell when the concentration reaches a specified level after 60 cycles (Fig. 3). This hypothesis was also supported by the detection of oxygen linked to Mn in the cathode as well as the detection of ketones (quinone), i.e., the equilibrium reaction between quinone and alkoxide in the presence of protons and electrons.41 The cells fabricated with 1.0 wt% of the catechol-based polymer did not experience an improvement in the retention capacity after cycle-life testing, which confirmed the role of methoxy groups in the formation of the AlF3 PL (Fig. S6 in the ESI†). Therefore, the demethylation reaction of the polymer during cell operation was a determining factor in the formation of the AlF3 PL on the negative electrode. Aluminum trimethoxide diffused in the electrolyte and reacted with HF to form AlF3 on the LTO surface because Mn2+ is simultaneously released, which may explain the detection of Mn on the edges. Moreover, an insoluble AlF3 PL was reported on the Al current collector surface of the LMFP-based electrode; therefore, the diffusion of AlF3 in solvents is not plausible.42 Using 4.0 wt% of PVDF the integrity of the positive electrode could be maintained as visualized by the eyes after disassembling the cells, and the additive polymer (1.0 wt%) was sacrificed to form a PL.
Conclusions
We have shown for an LMFP/LTO cell how to improve the capacity retention by 16.7% after more than 300 cycles at 45 °C and 1C. We achieved this by the first-time implementation of a DMB-based additive polymer as a binder in the positive electrode (LMFP). This functional polymer trapped metal ions and prevented diffusion to the negative electrode. Moreover, a reaction of the aluminum current collector with the polymer to form a soluble organometallic derivative was observed. Migration of this species through the liquid electrolyte enabled the formation of an AlF3 PL on the LTO surface by consuming the HF. However, we believe that further investigations are probably necessary to confirm the mechanism. The PL formed using this in situ strategy is more efficient than the traditional SEI formed by VC to reduce solvent degradation. All these reactions assisted in improving the performance of the battery. Moreover, we report the first example of the LMFP/LTO cell at 3.0 V having a good cycle-life. There are limited articles on LMFP/LTO cells42,45–47 and only one LTO-based Li-ion battery has been commercialized.48 Hence, this discovery can pave the way for novel research on additive polymers that can interact positively with active materials or electrolytes during battery operation in an effort to enhance the performance.Conflicts of interest
Two patent applications covering this work have been filed by Hydro-Québec and Murata (International patent application no. WO 2020065831 A1 20200402 and no. WO 2020034031 A1 20200220.)Acknowledgements
This work was supported by Hydro-Québec and Murata. The authors thank Mr Kojima Sachiko and Mrs Mami Oda of Toray Research Center for TOF-SIMS analysis and Dr Pascale Chevalier of Laval University for XPS analysis. We would like to thank Dr Andrea Paolella for his illuminating writing advice.Notes and references
- J. Duan, X. Tang, H. Dai, Y. Yang, W. Wu, X. Wei and Y. Huang, Electrochem. Energy Rev., 2020, 3, 1–42 CrossRef CAS.
- X. Feng, M. Ouyang, X. Liu, L. Lu, Y. Xia and X. He, Energy Storage Mater., 2018, 10, 246–267 CrossRef.
- S. J. Harris, D. J. Harris and C. Li, J. Power Sources, 2017, 342, 589–597 CrossRef CAS.
- Z. Mao, M. Farkhondeh, M. Pritzker, M. Fowler and Z. Chen, J. Electrochem. Soc., 2017, 164, A3469–A3483 CrossRef CAS.
- R. Zhao, J. Gu and J. Liu, J. Power Sources, 2015, 273, 1089–1097 CrossRef CAS.
- D. Chen, J. Jiang, G.-H. Kim, C. Yang and A. Pesaran, Appl. Therm. Eng., 2016, 94, 846–854 CrossRef CAS.
- K. Li, J. Yan, H. Chen and Q. Wang, Appl. Therm. Eng., 2018, 132, 575–585 CrossRef CAS.
- N.-S. Choi, J.-G. Han, S.-Y. Ha, I. Park and C.-K. Back, RSC Adv., 2015, 5, 2732–2748 RSC.
- G. Xu, Z. Liu, C. Zhang, G. Cui and L. Chen, J. Mater. Chem. A, 2015, 3, 4092–4123 RSC.
- M. Adachi, K. Tanaka and K. Sekai, J. Electrochem. Soc., 1999, 146, 1256–1261 CrossRef CAS.
- EP740359A1, 1996.
- C. Buhrmester, J. Chen, L. Moshurchak, J. Jiang, R. L. Wang and J. R. Dahn, J. Electrochem. Soc., 2005, 152, A2390 CrossRef CAS.
- J. Chen, C. Buhrmester and J. R. Dahn, Electrochem. Solid-State Lett., 2005, 8, A59 CrossRef CAS.
- J. Huang, N. Azimi, L. Cheng, I. A. Shkrob, Z. Xue, J. Zhang, N. L. Dietz Rago, L. A. Curtiss, K. Amine, Z. Zhang and L. Zhang, J. Mater. Chem. A, 2015, 3, 10710–10714 RSC.
- J.-B. Gieu, C. Courrèges, L. El Ouatani, C. Tessier and H. Martinez, J. Electrochem. Soc., 2017, 164, A1314–A1320 CrossRef CAS.
- J.-C. Daigle, Y. Asakawa, P. Hovington and K. Zaghib, ACS Appl. Mater. Interfaces, 2017, 9, 41371–41377 CrossRef CAS.
- J.-C. Daigle, Y. Asakawa, M. Beaupré, A. A. Arnold, D. Laul, M. Trudeau and K. Zaghib, J. Power Sources, 2019, 421, 116–123 CrossRef CAS.
- W. Li, X. Li, M. Chen, Z. Xie, J. Zhang, S. Dong and M. Qu, Electrochim. Acta, 2014, 139, 104–110 CrossRef CAS.
- M. Milien, J. Hoffmann, M. Payne and B. Lucht, J. Electrochem. Soc., 2018, 165, A3925–A3931 CrossRef CAS.
- C. Han, Y.-B. He, M. Liu, B. Li, Q.-H. Yang, C.-P. Wong and F. Kang, J. Mater. Chem. A, 2017, 5, 6368–6381 RSC.
- B. Michalak, B. B. Berkes, H. Sommer, T. Bergfeldt, T. Brezesinski and J. Janek, Anal. Chem., 2016, 88, 2877–2883 CrossRef CAS.
- F. Diaz, Y. Wang, R. Weyhe and B. Friedrich, Waste Manage., 2019, 84, 102–111 CrossRef CAS.
- C. Zhan, T. Wu, J. Lu and K. Amine, Energy Environ. Sci., 2018, 11, 243–257 RSC.
- K. Kim, D. Hwang, S. Kim, S. O. Park, H. Cha, Y.-S. Lee, J. Cho, S. K. Kwak and N.-S. Choi, Adv. Energy Mater., 2020, 10, 2000012 CrossRef CAS.
- L. Danis, S. M. Gateman, M. E. Snowden, I. C. Halalay, J. Y. Howe and J. Mauzeroll, Electrochim. Acta, 2015, 162, 169–175 CrossRef CAS.
- Z. Li, A. D. Pauric, G. R. Goward, T. J. Fuller, J. M. Ziegelbauer, M. P. Balogh and I. C. Halalay, J. Power Sources, 2014, 272, 1134–1141 CrossRef CAS.
- H. Saneifar, K. Zaghib and D. Bélanger, ACS Appl. Energy Mater., 2020, 3, 647–657 CrossRef CAS.
- B. Ziv, N. Levy, V. Borgel, Z. Li, M. D. Levi, D. Aurbach, A. D. Pauric, G. R. Goward, T. J. Fuller, M. P. Balogh and I. C. Halalay, J. Electrochem. Soc., 2014, 161, A1213–A1217 CrossRef CAS.
- M.-H. Ryou, S. Hong, M. Winter, H. Lee and J. Choi, J. Mater. Chem. A, 2013, 1 Search PubMed.
- G. Westwood, T. N. Horton and J. J. Wilker, Macromolecules, 2007, 40, 3960–3964 CrossRef CAS.
- M. A. North, C. A. Del Grosso and J. J. Wilker, ACS Appl. Mater. Interfaces, 2017, 9, 7866–7872 CrossRef CAS.
- N. Patil, C. Jérôme and C. Detrembleur, Prog. Polym. Sci., 2018, 82, 34–91 CrossRef CAS.
- N. Patil, A. Mavrandonakis, C. Jérôme, C. Detrembleur, J. Palma and R. Marcilla, ACS Appl. Energy Mater., 2019, 2, 3035–3041 CrossRef CAS.
- M. Pivko, M. Bele, E. Tchernychova, N. Z. Logar, R. Dominko and M. Gaberscek, Chem. Mater., 2012, 24, 1041–1047 CrossRef CAS.
- G. Li, H. Azuma and M. Tohda, Electrochem. Solid-State Lett., 2002, 5, A135 CrossRef CAS.
- C. Delacourt, P. Poizot, M. Morcrette, J. M. Tarascon and C. Masquelier, Chem. Mater., 2004, 16, 93–99 CrossRef CAS.
- H. Isozumi, K. Kubota, R. Tatara, T. Horiba, K. Hida, T. Matsuyama, S. Yasuno and S. Komaba, ACS Appl. Energy Mater., 2020, 3, 7978–7987 CrossRef CAS.
- N. Yabuuchi, Y. Kinoshita, K. Misaki, T. Matsuyama and S. Komaba, J. Electrochem. Soc., 2015, 162, A538–A544 CrossRef CAS.
- R. S. Assary, L. Zhang, J. Huang and L. A. Curtiss, J. Phys. Chem. C, 2016, 120, 14531–14538 CrossRef CAS.
- N. Patil, A. Aqil, F. Ouhib, S. Admassie, O. Inganäs, C. Jérôme and C. Detrembleur, Adv. Mater., 2017, 29, 1703373 CrossRef.
- Y. Liang, Y. Jing, S. Gheytani, K.-Y. Lee, P. Liu, A. Facchetti and Y. Yao, Nat. Mater., 2017, 16, 841–848 CrossRef CAS.
- V. Borgel, G. Gershinsky, T. Hu, M. G. Theivanayagam and D. Aurbach, J. Electrochem. Soc., 2013, 160, A650–A657 CrossRef CAS.
- N. Gauthier, C. Courrèges, J. Demeaux, C. Tessier and H. Martinez, Appl. Surf. Sci., 2020, 501, 144266 CrossRef CAS.
- M. Khosravi, M. B. Andrus, S. R. Burt and B. F. Woodfield, Polyhedron, 2013, 62, 18–25 CrossRef.
- N. Takami, H. Inagaki, Y. Tatebayashi, H. Saruwatari, K. Honda and S. Egusa, J. Power Sources, 2013, 244, 469–475 CrossRef CAS.
- N. Takami, K. Yoshima and Y. Harada, J. Electrochem. Soc., 2017, 164, A6254–A6259 CrossRef CAS.
- J.-C. Daigle, Y. Asakawa, A. Perea, M. Dontigny and K. Zaghib,, Sci. Rep., 2020, 10, 10305 CrossRef CAS.
- https://www.scib.jp/en/, accessed August 2020.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00679c |
| This journal is © The Royal Society of Chemistry 2021 |