
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of sulfur-doped porous carbon from heavy coker gas oil and its application in CO2 capture†
Yi
Du
 *,
Bradley
Wooler
,
Stuart E.
Smith
,
Brenda
Raich
,
Clarence E.
Chase
,
Lesheng
Wang
,
Chris
Kliewer
and
Kanmi
Mao
*,
Bradley
Wooler
,
Stuart E.
Smith
,
Brenda
Raich
,
Clarence E.
Chase
,
Lesheng
Wang
,
Chris
Kliewer
and
Kanmi
Mao
Corporate Strategic Research, ExxonMobil Research and Engineering Company, 1545 US22, Annandale, NJ, USA. E-mail: yi.du@exxonmobil.com
First published on 17th December 2020
Abstract
Elemental sulfur is used to cross-link heavy coker gas oil (HKGO) with only 0.44 wt% olefinic hydrogen content in the absence of solvent; subsequent pyrolysis yields sulfur-doped porous carbon with Brunauer–Emmett–Tell (BET) surface area (s.a.) around 1714–1785 m2 g−1. The heavy coker gas oil does not need demetallation or hydrotreating to enable this. The usual problems (residue metal and sulfur/nitrogen) in HKGO became an opportunity in product carbon to anchor CO2 resulting in high sorption capacity.
To decrease atmosphere CO2, one should consider both capturing and reducing its emission. Separating CO2 from process streams is an important and most widely studied step in carbon capture and storage, natural gas purification, and enhanced oil recovery (EOR). To practice this commercially, it is important that the potential materials be effective, low-cost, and amenable to mass production. In the other end of the spectrum, eliminating the steps that generate CO2 ought to be examined. Among refineries, energy consumption to perform demetallation and hydrotreating contributes to CO2 generation significantly. The heavier the feedstock is, the more energy required to convert. Products such as asphaltene, coal, coker gas oil will require very severe demetallation to remove metal residue, and high H2 consumption for hydrotreating and hydrocracking to turn into products. Because of the energy intensity and low margin, these heavy feedstock are often opted to apply to a low value utilization such as road construction. This is essentially a form of carbon rejection benefiting CO2 life cycle efficiency.1,2 A more beneficial form of carbon rejection is to produce porous carbon which can then be used for CO2 capture.3,4
To produce porous carbon for CO2 capture, the carbon precursor has often been a subject of research. Biomass5 and MOF precursor,6–8 or oxidation and exfoliation of graphene9 have all been subjects of interests, offering attractive physical properties such as high surface area and high pore volume. Some research groups took a different approach in designing the low cost carbon precursors prior to porous carbon formation, using low value feedstock4 or petroleum redundant product sulfur as the modifier.10–18 This is rather intriguing because redundant elemental sulfur has become an issue at refinery due to an imbalance in the rate of production vs. demand.10,11 In the field of incorporating elemental sulfur into carbon rich precursors, Pyun pioneer studies on inverse vulcanization using elemental sulfur to make >90% S containing functional polymers, application of which lies in IR optical materials,12–14 Chalker reacted sulfur and limonene to form polysulfide material15 which firstly reveals a complete consumption of both exocyclic and endocyclic alkene, and Hasell's later demonstrated16–18 that reacting industrial by-product dicyclopentadienes (DCPA) with elemental sulfur, without any added solvent, can yield an excellent CO2 sorbent after pyrolysis with a proper chemical activator such as KOH. Hasell also demonstrated his approach can readily extend to other olefin-rich precursors such as myrcene, farnesene, etc., indicating its versatile utilization with olefins.16–19
These advances inspired us to click these three seemingly separate but highly linkable challenges – abundance sulfur production, energy intensive process to treat heavy feedstocks, and the challenge to produce large scale, economical, but still effective CO2 sorbents.
In this study, we provide a unique solution for CO2 capture to match the scale economically. Porous carbons were made from low-value hydrocarbons feedstock (heavy coker gas oil/HKGO or steam cracked tar/STC) that contains residue metal and untreated S. Refinery abundance by-product elemental sulfur was added as the first step to crosslink to aid final carbon yield, and activator KOH to generate porosity in product carbon. The residue metal from the heavy streams were speculated to act as anchoring sites for CO2 capture providing a means to turn waste into value.
CO2 global emission is in the scale of 30 billion tons per year, which translates into 25 kilotons of sorbent materials assuming an optimum adsorption of 30 wt% of its own weight, and recycle ∼5000 times a year. On the other hand, refinery produce a large volume of heavy streams such as heavy coker gas oil. Vacuum resid production alone is in the range of 12.5 kilotons per year. The only consumable chemical used in this study is elemental sulfur, which is also produced in abundance from our refinery (60million tons globally). The two raw reactants’ scale and cost will match CO2 sorbent's need beautifully.
Although the concept is desired, it was unexpected that this will actually work. Heavy feedstock has ∼100 times less olefin content per H as compared to DCPA or other olefins used in Pyun12–14 and Hasell's16–18 system. The feed has a large portion of saturates or aromatics that is olefin free, and heteroatom free, so there is no known reactive site to crosslink with elemental sulfur. Without adding solvent, the feedstock is also highly viscous and is challenging to mix well, a step of which was emphasized to be key by both Pyun and Hasell. Details of the synthesis and characterization of pore size, pore volume by varying S content or mixing conditions are given in the experimental section in ESI,† as well as summary in Tables S1–S6.
Surprisingly, Fig. 1 (Fig. S1–S10 in ESI†) shows that it not only works but outperforms the best materials reported by Hasell18,19 using a similar sulfur crosslinked chemistry. The best sorption capacity is at 2.6 mmol g−1 at 298 K, 800 Torr, vs. ∼1.9 mmol g−1 for Hasell's carbon at the same condition (Fig. S1, ESI†). This capacity is also comparable to other best porous carbon's reported capacity (2.1–3.9 mmol g−1) for dry CO2 sorption in the same condition,3 even though our carbon's surface area is only around 1714–1785 m2 g−1, which is considered moderate or even low for the fast advancing porous carbon community.3–9 The heat of adsorption is 20 kJ mol−1 (Fig. S1b, ESI†) based on Clausius–Clapeyron equation,20 and is slightly decreasing as CO2 loading increasing. That implies a moderate exothermic physical adsorption of CO2, on a slightly energetically heterogeneous surfaces.
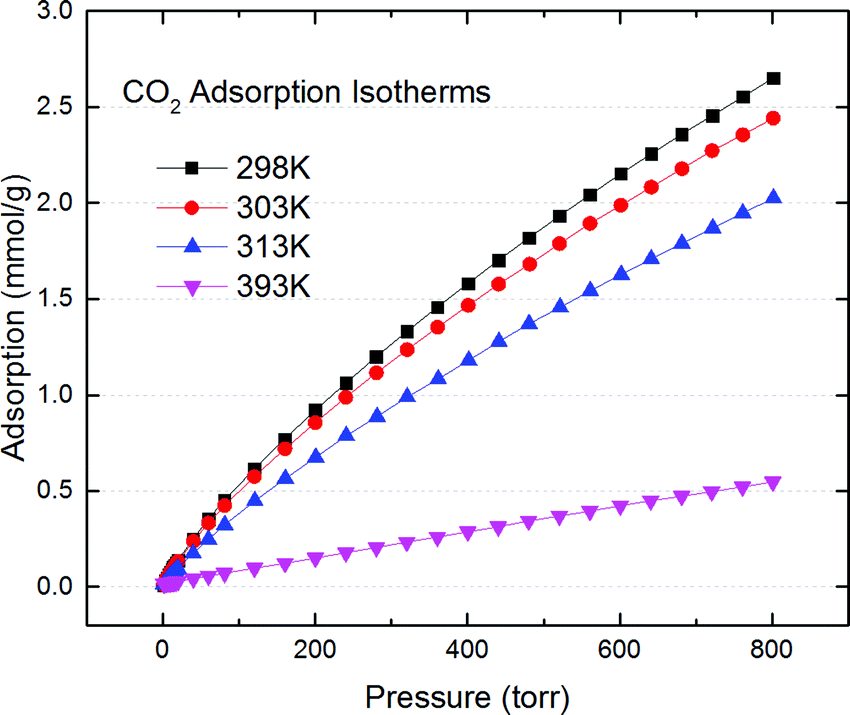 | ||
| Fig. 1 CO2 adsorption isotherm of S doped porous carbon from HKGO (SD-Carbon 1) at varies temperatures up to 800 Torr. | ||
The elemental analysis of the HKGO feed and the carbon product are summarized in Table 1. With the carbonization process, it is expected concentrations of metals will rise with two exceptions, V disappearance and significant rise of K. The disappearance of V is due to its instability in basic solution, which happens during the post water treatment of the carbon product. An increased amount of potassium is residue from the KOH activator, unavoidably even though multiple washes were involved. All the other metal species, such as Ni, Ca, Fe and Cr, concentrate in the carbon product carbon, all originated fromthe HKGO. With a moderate s.a., limited micropore, the high CO2 sorption capacity also cannot be explained by its heat of adsorption and lack of amine like chemical functions. The presence of the metal species offers a good explanation. Although it is challenging to pin point the exact trace metal's effect in CO2 sorption, the function of metal has been reported in prior art with Zhu et al.21a and Wolfgang et al.21b reported K and Fe for enhanced CO2 sorption especially at low pressure. On the other hand, interestingly, although we introduce a large amount of sulfur in the precursor (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 sulfur:HKGO weight ratio), the product sulfur is always in the range of 3–5 wt%, meaning that the majority of sulfur has sublimed during the high temperature thermal treatment, and/or lost as H2S during reaction. This is consistent with Hasell's observation. Even though most of the sulfur added was absent in the product, sulfur's presence is essential to crosslink the small amount of olefins in HKGO to increase both yield, and porosity of the carbon products. A control study shows that without sulfur, the carbon residue will decrease by more than 50% with only HKGO even with KOH as activator. Product carbon exhibit amorphous graphitic nature as evidenced by 13C Solid State NMR and TEM furthering verifying the absence of molecule sieve nature, hence lack of micropore volume as CO2 sorption sites (Fig. S11 and S12, Table S7 in ESI†).
:2 sulfur:HKGO weight ratio), the product sulfur is always in the range of 3–5 wt%, meaning that the majority of sulfur has sublimed during the high temperature thermal treatment, and/or lost as H2S during reaction. This is consistent with Hasell's observation. Even though most of the sulfur added was absent in the product, sulfur's presence is essential to crosslink the small amount of olefins in HKGO to increase both yield, and porosity of the carbon products. A control study shows that without sulfur, the carbon residue will decrease by more than 50% with only HKGO even with KOH as activator. Product carbon exhibit amorphous graphitic nature as evidenced by 13C Solid State NMR and TEM furthering verifying the absence of molecule sieve nature, hence lack of micropore volume as CO2 sorption sites (Fig. S11 and S12, Table S7 in ESI†).
| S | C | N | H | V | K | Ni | Ca | Fe | Cr | |
|---|---|---|---|---|---|---|---|---|---|---|
| HKGO feed | 2.79% | 86.3% | 0.67% | 9.86% | 2.3 ppm | 0.7 ppm | 1 ppm | <0.1 | −<0.1 | <0.1 |
| Carbon product | 3.19% | 88.9% | na | na | <0.1 | 0.19% | 30 ppm | 175 ppm | 496 ppm | 55 ppm |
The nature of the feed composition, the crosslinked intermediate, and the graphitic product lead us to derive the reaction mechanism shown in Fig. 2 and Fig. S2 (ESI†), where the potential olefin reacts with elemental sulfur which further fuses the aromatic rings to enable a rigid network. The latter will reject H2 and form S embedded graphite sheets upon pyrolysis condition. This mechanism is supported by Chalk's observation of endocycle alkene reaction with elemental sulfur,15 and is based on a model derived internally (data not included). The overall representative model structure in HKGO is included also in Fig. S2 (ESI†) for readers of interests. The total sulfur content in HKGO and in carbon do not differentiate too much meaning the S embedded in the 3 + Ring structure retains its position vs. the added elemental sulfur's crucial function is to facilitate cross linkage through the limited olefinic double bonds.
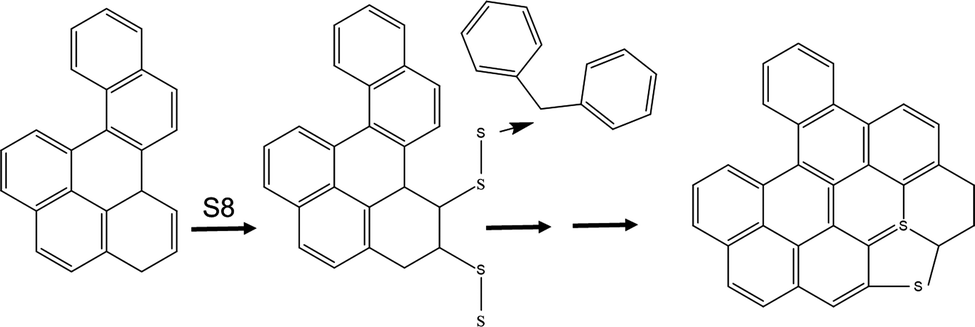 | ||
| Fig. 2 Reaction scheme of HKGO and elemental sulfur to form carbon. | ||
Although our carbon product show good CO2 uptake with a moderate surface area, H2 uptake at various temperatures (Fig. 3) indicated a max of 8.9 mmol g−1 at 77 K and 800 Torr; roughly 1.78 wt% uptake. That is only slightly larger than previously reported inverse-vulcanized polymer,22 but noticeably smaller than the optimized S-DCPD.18 This is expected since H2 sorption is largely due to physisorption, and hence is not affected by the trace metals present in HKGO, and is a mere reflection its surface area and pore volume.
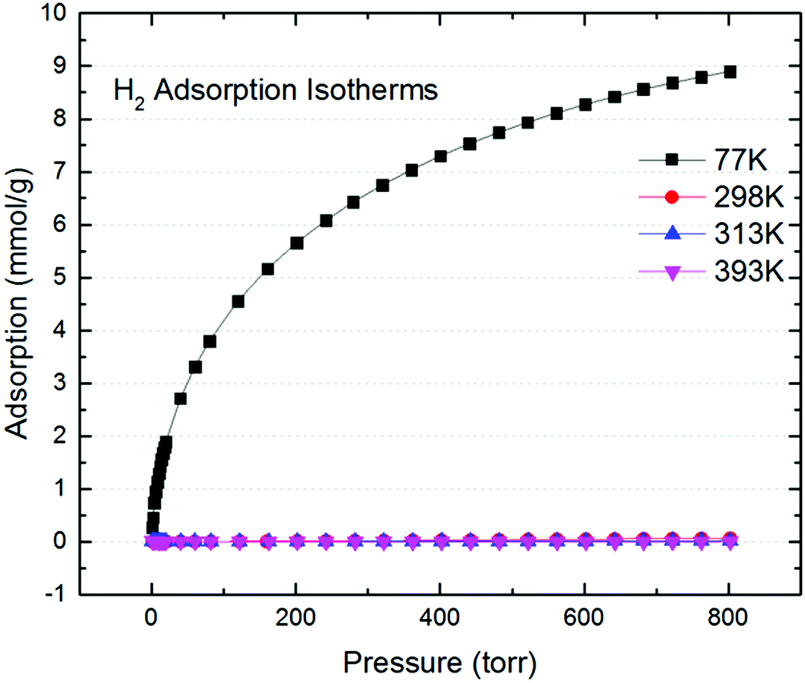 | ||
| Fig. 3 The H2 adsorption isotherms of SD-Carbon 1 at 77 K, 298 K, 313 K, 393 K. | ||
The HKGO is a viscous black liquid at room temperature and its basic physical properties summarized below in Table 2. Without the S-crosslinker, the carbon yield is only 5 wt%, with a s.a. <10 m2 g−1.23 the complexity of the feed and the crosslinked intermediate presents challenges to derive mechanism. A key analytical breakthrough came from 1H NMR and GPC.
| Feed properties HKGO | |
|---|---|
| Feed S, wt% | 2.79 |
| N, WPPM | 6700 |
| 10 PCT OFF – SIMDIS | 620.4 |
| 30 PCT OFF – SIMDIS | 670.9 |
| 50 PCT OFF – SIMDIS | 718.2 |
| 70 PCT OFF – SIMDIS | 776.5 |
| 90 PCT OFF – SIMDIS | 859.8 |
| 99 PCT OFF – SIMDIS | 994.3 |
1H NMR analysis reveals the presence of olefins and represents 0.44 wt% total H in Fig. 4. The quantitative olefinic H is determined from signals 4.5–6 ppm for heavy hydrocarbons (HC). 5.65–5.95 ppm is the vinyl H (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 5.55–5.31 ppm is for vinylenes H (–CHCH–), 5.30–4.95 ppm is for trisubstituted olefins (
CH2), 5.55–5.31 ppm is for vinylenes H (–CHCH–), 5.30–4.95 ppm is for trisubstituted olefins (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CCH) and the other vinyl H (–CHCH2), and 4.84–4.5 ppm is for vinylidenes H (CCH2). It is also worth noting that the 0.44 wt% olefin content is referring to the H directly linked to the CC and is not a reflection of the total weight of the olefin containing molecules. After the cross-linking reaction, the olefin content in HKGO dropped to 0.0%, strongly indicating the reaction is between elemental sulfur and the olefins, following a reaction mechanism similar to S and DCPD reported by Hasell.18
CCH) and the other vinyl H (–CHCH2), and 4.84–4.5 ppm is for vinylidenes H (CCH2). It is also worth noting that the 0.44 wt% olefin content is referring to the H directly linked to the CC and is not a reflection of the total weight of the olefin containing molecules. After the cross-linking reaction, the olefin content in HKGO dropped to 0.0%, strongly indicating the reaction is between elemental sulfur and the olefins, following a reaction mechanism similar to S and DCPD reported by Hasell.18
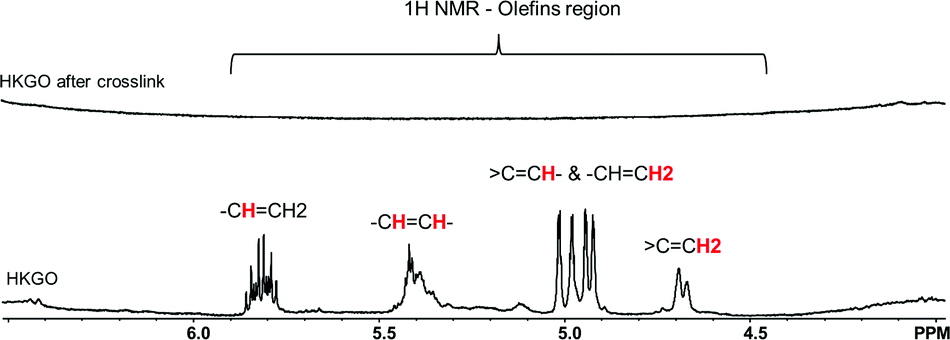 | ||
| Fig. 4 Liquid 1H NMR of HKGO before and after reaction with sulfur. | ||
We have also examined the liquid 13C NMR and separated the changes in the aliphatic and aromatic regions below in Fig. 5. The 13C aromatic changed from 39.5–40.0 wt% to 41.8–47.1 wt%, suggesting dehydrogenation of naphthenic rings from reaction with sulfur, and formation of new aromatic rings and H2S. The ranges listed exist due to a slight variation in HKGO origin and aging period. In general, fresh HKGO will have less change in aromatic carbon after crosslinking, but will have very similar olefin change and resulting porous carbon product. This observation, together with a separate study using an alternative low-value feedstock steam cracked tar, in which we can observe a noticeable aromatic increase after the heat treatment with sulfur, it is believed the NMR changes in aromatic region is associated with dehydrogenation during the heat treatment and is actually not useful in the formation of sulfur-crosslinks. Only the decrease of olefinic H correlated well with the degree of crosslinking which will then affect the product sulfur. Details of this study is included in Table S7 (ESI†).
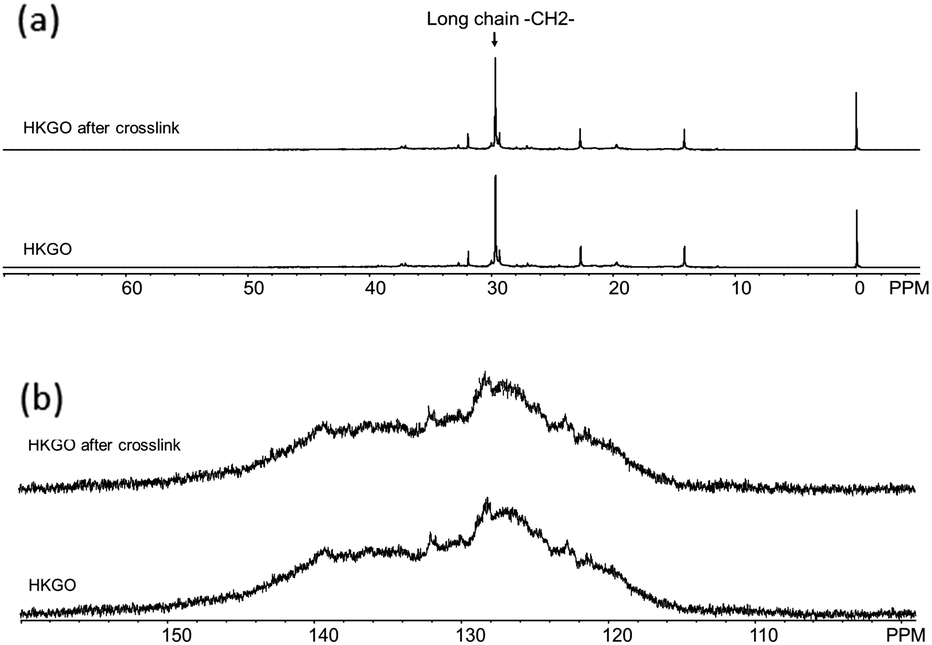 | ||
| Fig. 5 Liquid 13C NMR of HKGO, and HKGO reacted with sulfur (a) aliphatic region, and (b) aromatic region. | ||
The change in molecular weight distribution is revealed by Gel Permeation Chromatography (GPC). GPC results in Fig. 6 indicate the molecular weight growth of cross-linked product shifted, with an upper limit around 19379 vs. the unreacted precursor's upper limit of 4107. The peak molecular weight (Mp) of the cross-linked product was around 629 vs. the unreacted precursor with the Mp value of 188. This numbers are based on polystyrene calibration curve from GPC. By changing HKGO origin, which seemed to change NMR aromatic H%, the molecular weight distribution remain constant in GPC. Elemental sulfur appeared with an elution time around 46.6 min, the content of which gradually decreases with increasing reaction time, mainly due to potential sublimation of elemental sulfur at the reaction temperature (160 °C). Olefin concentrations in HKGO decrease from 0.44 wt% to 0 wt% when large amounts of elemental sulfur persist, as shown in Fig. 4. GPC analysis for samples made with higher olefin content was included in Fig. S13 and Table S8 (ESI†) for readers of interest.
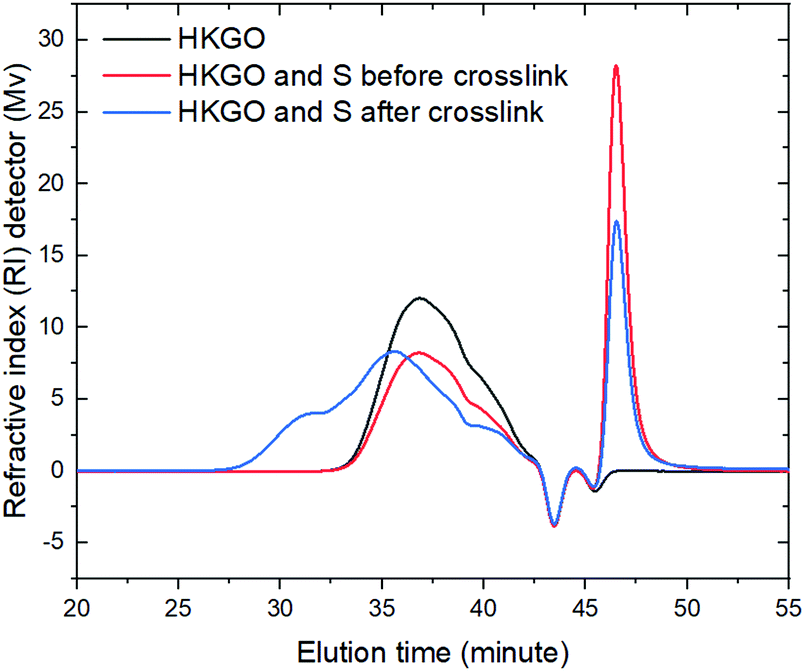 | ||
| Fig. 6 GPC overlay plot for RI detector for control S, S/HKGO before heat treatment and after heat treatment. | ||
Although a lot of elemental sulfur still remains after the crosslinking reaction (Fig. 6), the olefinic H was completely converted (Fig. 4). When we further increase or decrease the S to HKGO ratio from 1:2, to either 1.5 to 2, or 0.6:2, the product carbon's surface area will both decrease from 1714–1785 m2 g−1 to 1092 m2 g−1 and 1625 m2 g−1, respectively (Fig. S10–S14 in ESI†). We conclude the presence of S is critical in enable higher s.a. carbon product but the exact function of sulfur in CO2 capture and the explicit detail of how it is retained in the final structure is not clear.
Another critical step to achieve high s.a. in the carbon product relates to addition and mixing of the chemical activator KOH. Chemical activation is often employed to lower the pyrolysis temperature, increase yield, and create microporosity in activated carbon.24–29 The typical chemicals used are phosphoric acid, zinc chloride, potassium or sodium hydroxide, and they are the subject of interest for activated carbon synthesis. In our study we focus on KOH with controlled N2 atmosphere, which is believed to result in the formation of CO2 when hydroxides and carbon precursors react, and to create microporosity in the carbon product. Solano et al.25 traced CO2 formation with IR at various NaOH or KOH to carbon ratios, and concluded that the reaction ratio should be 1:1 to 1.7:1. Hence, we chose to vary KOH to S-HKGO weight ratio from 0.5:1 to 2:1. With a high hydroxide to carbon ratio, more CO2 could be formed resulting in higher porosity, especially in the micropore region; however, more carbon is also lost resulting in a lower yield (Fig. S14, ESI†). Some papers indicate that a further increase in the KOH to carbon ratio will result in lowering its surface area due to micropore collapse. Since the yield severely drops when the KOH:S-HKGO ratio increases from 1:1 to 2:1 (Fig. S15, ESI†), the KOH:S-HKGO ratio is kept at 1:1 in this study. The role of KOH is also interestingly. We observed a notable amount of K2SO4 (Fig. S15, ESI†). Although K2SO4 is soluble in water, its solubility is 10 times less than K2CO3 and so one can intercept washed carbon product with only K2SO4 impurity but no K2CO3. Most other papers we found reported the activation mechanism to be 6KOH + 2C → 2K + 3H2 + 2K2CO3. Here we proposed a modified mechanism 4KOH + S + CH2– → K2SO4 + 3H2 + 2K + C to reflect the sulfur rich system due to the evidence of K2SO4.
Last but not least, we have studied a different heavy feed steam cracked tar (STC) and successfully demonstrated the applicability of S crosslink chemistry to ultimately produce porous carbon. Details of its synthesis (Tables S1–S6, ESI†) and NMR analysis (Fig. S16–18 and Table S7, ESI†) are including for readers of interest. The main difference between STC and HKGO is S-crosslinked STC appears to be much heavier in molecular weight from both NMR and GPC, as well as enhanced insolubility.
In summary, porous sulfur-doped carbon can be made with surface areas approaching ∼1714–1785 m2 g−1 from elemental sulfur and heavy coker gas oil through a crosslinked intermediate. KOH:S-HKGO and sulfur:HKGO ratios are the two key parameters to tune the product carbon's porosity. The presence of olefins in HKGO and the disappearance of it after inverse vulcanization are the critical steps for success. The sulfur-doped porous carbon has good CO2 sorption capacity. This is possibly due to the trace metals left from the feedstock, providing an excellent way to turn undesired contaminants to designed features for sorption anchoring site.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors would like to thank ExxonMobil Research and Engineering for funding, Yu Wang for heat of adsorption calculation, Steve Rucker for 1H NMR analysis Joe Gatt and Christine Elia for their management support.Notes and references
- M. Siskin, S. Kelemen, C. Eppig, D. Brown and M. Afeworki, Energy Fuels, 2006, 20(3), 1227–1234 CrossRef CAS.
- J. Artz, T. Mueller, K. Thenert, J. Kleinekorte, R. Meys, A. Sternberg, A. Bardow and W. Leitner, Chem. Rev., 2018, 118, 434–504 CrossRef CAS.
- M. Sevilla, P. Valle-Vigon and A. Fuertes, Adv. Funct. Mater., 2011, 21(14), 2781–2787 CrossRef CAS.
- E. Jang, S. Choi, S. Hong, S. Shin and K. Lee, Appl. Surf. Sci., 2018, 429, 62–71 CrossRef CAS.
- W. Travis, S. Gadipelli and Z. Guo, RSC Adv., 2015, 5, 29558–29562 RSC.
- S. Gadipelli and Z. Guo, ChemSusChem, 2015, 8, 2123–2132 CrossRef CAS.
- S. Gadipelli, Z. Li, Y. Lu, J. Li, J. Guo, N. Skipper, P. Shearing and D. Brett, Adv. Sci., 2019, 6, 1901517 CrossRef CAS.
- S. Gadipelli, C. Howard, J. Guo, N. Skipper, H. Zhang, P. Shearing and D. Brett, Adv. Energy Mater., 2020, 10, 1903649 CrossRef CAS.
- S. Gaduoekkum, Y. Lu, N. Skipper, T. Yildirim and Z. Guo, J. Mater. Chem. A, 2017, 5, 17833–17840 RSC.
- Y. Sun and R. Prins, Angew. Chem., Int. Ed., 2008, 47(44), 8478–8481 CrossRef CAS.
- S. Eijsbouts, N. Van Gestel, J. Van Veen, V. de Beer and R. Prins, J. Catal., 1991, 131, 412–432 CrossRef CAS.
- W. Chung, J. Griebel, E. Kim, H. Yoon, A. Simmonds, H. Ji, P. Dirlam, R. Glass, J. Wie, N. Nguyen, B. Guralnick, J. Park, A. Somogyi, P. Theato, M. Mackay, Y. Sung, K. Char and J. Pyun, Nat. Chem., 2013, 5, 518 CrossRef CAS.
- J. J. Griebel, N. A. Nguyen, S. Namnabat, L. E. Anderson, R. S. Glass, R. A. Norwood, M. E. Mackay, K. Char and J. Pyun, ACS Macro Lett., 2014, 3, 1258 CrossRef CAS.
- J. Lim, J. Pyun and K. Char, Angew. Chem., Int. Ed., 2015, 54, 3249 CrossRef CAS.
- M. Crockett, A. Evans, M. Worthington, I. Albuquerque, A. Slattery, C. Gibson, J. Campbell, D. Lewis, G. Bernardes and J. Chalker, Angew. Chem., Int. Ed., 2016, 55, 1714–1718 CrossRef CAS.
- T. Hasell, D. J. Parker, H. A. Jones, T. McAllisterb and S. M. Howdle, Chem. Commun., 2016, 52, 538 RSC.
- D. J. Parker, H. A. Jones, S. Petcher, L. Cervini, J. M. Griffin, R. Akhtarb and T. Hasell, J. Mater. Chem. A, 2017, 5, 11682–11692 RSC.
- J. S. Lee, D. J. Parker, A. I. Cooper and T. Hasell, J. Mater. Chem. A, 2017, 5, 18603 RSC.
- H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 8875–8883 CrossRef CAS.
- Y. Wang, Ind. Eng. Chem. Res., 2020, 59(17), 8304–8314 CrossRef CAS.
- (a) A. Liu, Q. Zhang, Q. Ye, D. Sun and Z. Zhu, AIChE Annual Meeting, 2012, 69QWRU/1-69QWRU/8; (b) G. Cornelius and H. Wolfgang, Chem. Ing. Tech., 2004, 76, 1649–1652 CrossRef.
- J. C. Bear, J. D. McGetterick, I. P. Parkin, C. W. Dunnill and T. Hasell, Microporous Mesoporous Mater., 2016, 232, 189–195 CrossRef CAS.
- N. P. Zambrano, L. J. Daurte, J. C. Poveda-Jaramillo, H. J. Picon, F. M. Ortega and M. E. Nino-Gomez, Energy Fuels, 2018, 32(3), 2722–2732 CrossRef CAS.
- W. Kicinski, M. Szala and M. Bystrzejewski, Carbon, 2014, 68, 1–32 CrossRef CAS.
- M. A. Lillo-Ródenas, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2003, 41(2), 267–275 CrossRef.
- B. Kueh, M. Kapsi, C. M. Veziri, C. Athanasekou, G. Pilatos, K. S. Reddy, A. Raj and G. Karanikolos, Energy Fuels, 2018, 32(11), 11718–11730 CrossRef CAS.
- H. C. Lee, N. Byamba-Ochir, W. G. Shim, M. S. Balathanigaimani and H. Moon, J. Power Sources, 2015, 275, 668–674 CrossRef CAS.
- X. Ma, Y. Yang, Q. Wu, B. Liu, D. Li, R. Chen, C. Wang, H. Li, Z. Zeng and L. Li, Fuel, 2020, 282, 118727 CrossRef CAS.
- X. Ma, C. Su, B. Liu, Q. Wu, K. Zhou, Z. Zeng and L. Li, Sep. Purif. Technol., 2020, 118065 CrossRef , in press.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental detail, separation detail, NMR and GPCs. See DOI: 10.1039/d0ma00859a |
| This journal is © The Royal Society of Chemistry 2021 |