
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective electrosynthesis of tetra- and hexa-functionalized [60]fullerene derivatives with unprecedented addition patterns†
Kai-Qing
Liu‡
 a,
Jun-Jie
Wang‡
a,
Xing-Xing
Yan
a,
Chuang
Niu
a and
Guan-Wu
Wang
*ab
a,
Jun-Jie
Wang‡
a,
Xing-Xing
Yan
a,
Chuang
Niu
a and
Guan-Wu
Wang
*ab
aHefei National Laboratory for Physical Sciences at Microscale, CAS Key Laboratory of Soft Matter Chemistry, iChEM (Collaborative Innovation Center of Chemistry for Energy Materials), Center for Excellence in Molecular Synthesis of CAS, Department of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, P. R. China. E-mail: gwang@.ustc.edu.cn
bState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, Gansu 730000, P. R. China
First published on 20th November 2019
Abstract
The efficient and regioselective electrosynthesis of tetra- and hexa-functionalized [60]fullerene derivatives with unprecedented addition patterns has been achieved. The tetra-functionalized [60]fullerene derivative with an intriguing 1,2,4,17-addition pattern is regioselectively obtained by cyclization reaction of the dianionic species generated electrochemically from a [60]fulleroindoline with 1,2-bis(bromomethyl)benzene at 0 °C, and can be converted to the more stable 1,2,3,4-adduct at 25 °C. Furthermore, the hexa-functionalized [60]fullerene derivative with the 1,2,3,4,9,10-addition pattern displaying a unique “S”-shaped configuration can be synthesized by protonation of the electrochemically generated dianion of the obtained tetra-functionalized 1,2,4,17-adduct. The structures of the tetra- and hexa-functionalized products have been determined by spectroscopic data and single-crystal X-ray analysis.
Introduction
Multiply functionalized fullerene derivatives have captured increasing attention for their potential applications in materials and biological science over the past two decades.1 Various methods have been established for the synthesis of multi-functionalized [60]fullerene (C60) derivatives.2 For a [2 + 1]-, [2 + 2]-, [2 + 3]- or [2 + 4]-cycloaddition reaction of C60, the cyclization usually gives one monoadduct fused to a [6,6]-junction of C60, and affords a mixture of 8 regioisomers for bis-cycloadducts, that is, cis-1, cis-2, cis-3, e, trans-4, trans-3, trans-2 and trans-1 isomers (Fig. 1b).3 Theoretically, there are 46 and 262 possible regioisomers for tris-cycloadducts and tetrakis-cycloadducts, respectively.2,3 For non-cyclized or partially cyclized tetra-functionalized derivatives of C60, the most commonly reported regioisomers are 1,2,3,4-adducts,4 1,4,11,15-adducts,5 1,2,4,15-adducts6 and 1,2,3,16-adducts;7 meanwhile for the hexa-functionalized derivatives of C60, the most frequently reported regioisomers are 1,2,3,4,5,6-adducts8 and 1,2,4,11,15,30-adducts5,9 (Fig. 1a). However, most of the previously reported methods suffer from the problem of poor regioselectivity and difficulty in separation of individual regioisomers. Even though the elegant templated multifunctionalizations of fullerenes have been developed to achieve high regioselectivity for bis-cycloadditions,3,10 the regioselective formation of a specific isomer of bis-adducts or multi-adducts with new addition patterns is still quite challenging.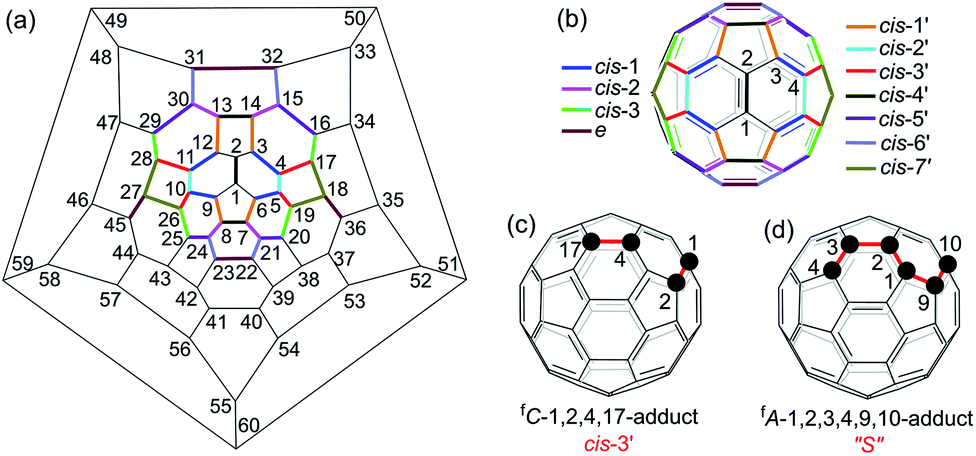 | ||
| Fig. 1 (a) Schlegel diagram of C60 with numbering of the C-atoms. (b) The cis and e isomers fused to two [6,6]-junctions and cis′ isomers fixed to a [6,6]-junction and a [5,6]-junction for bis-cycloadducts of C60. (c) Structure of the 1,2,4,17-adduct (cis-3′). (d) Structure of the “S”-shaped 1,2,3,4,9,10-adduct. | ||
Electrosynthesis turns out to be an efficient protocol in organic synthesis due to its mild reaction conditions, good chemoselectivity and regioselectivity, and relatively high yield.11,12 Furthermore, electrosynthesis has been increasingly applied to the synthesis of multi-functionalized fullerene derivatives including the above-mentioned tetra-functionalized 1,2,3,4-, 1,2,4,15- and 1,2,3,16-adducts in excellent regioselectivity. However, the electrosynthesis has not been applied to the synthesis of hexa-functionalized fullerene derivatives until now. In continuation of our interest in electrochemical functionalizations of C60 derivatives,7b,e,f,13 particularly the successful synthesis of 1,2,3,16-adducts from the reaction of an electroreduced dianionic [60]fulleroindoline with alkyl bromides7b prompted us to explore the reaction of the dianionic species with a bis-electrophile in order to produce cyclized products with novel addition patterns. Herein, we disclose an efficient and straightforward electrosynthesis of tetra-functionalized derivative of C60 with an unprecedented 1,2,4,17-addition pattern, also named as cis-3′ adduct (Fig. 1b and c),14 by the reaction of the dianionic [60]fulleroindoline with 1,2-bis(bromomethyl)benzene. A unique “S”-shaped hexa-functionalized derivative with the 1,2,3,4,9,10-addition pattern (Fig. 1d) can be obtained by further protonation of the electrochemically generated dianion of the synthesized 1,2,4,17-adduct.
Results and discussion
A heterolytic cleavage of the C60–N bond would occur after [60]fulleroindoline 1 accepts two electrons, and the dianionic 12− has its most electronegative carbon atom at the para position of the carbon linked with the aryl group, which is more prone to react with an electrophile.7b,e,f We surmise that when 12− is allowed to react with a bis-electrophile such as 1,2-bis(bromomethyl)benzene, the most electronegative para carbon on the fullerene skeleton is expected to attack one of the two –CH2Br groups of 1,2-bis(bromomethyl)benzene, followed by a ring-closure process via the C60–N bond formation to afford the fullerenyl monoanion INT-1 bearing a heterocycle fused with a [5,6]-junction, and final intramolecular cyclization of the nearby anionic fullerenyl carbon with another –CH2Br group to provide product 2 with an unprecedented 1,2,4,17-addition pattern. Product 2 has a carbocycle fused to a [6,6]-junction and a heterocycle fixed to a [5,6]-junction at the cis-3′ site (Fig. 1c). Density functional theory (DFT) calculations at the B3LYP/6-31G(d) level15 indicate that INT-1 is preferably formed and that the desired product 2 is most likely generated because it is more stable than other two possible isomers 2′ and 2′′ by at least 18.9 kcal mol−1 (Fig. 2) (see ESI† for details).[60]Fulleroindoline 1 was synthesized according to our previous procedure.16 To testify our assumption, we performed the reaction of 12−, which was generated from neutral 1 in ortho-dichlorobenzene (o-DCB) solution containing 0.1 M tetra-n-butylammonium perchlorate (TBAP) by controlled potential electrolysis (CPE) at −1.1 V,7b with 10 equiv. of 1,2-bis(bromomethyl)benzene and 10 equiv. of NaH solution under an argon atmosphere. NaH was added to the reaction system in order to remove the trace amount of residual water and further reduce byproduct formation.17 We found that when the reaction was allowed to proceed at 25 °C for 3 min and then immediately quenched with 2 equiv. of trifluoroacetic acid (TFA), 1,2,3,16-adduct 3 was obtained in 48% yield. The yield of 3 could be further increased to 58% if the reaction was conducted at 0 °C for 10 min under otherwise same conditions (Scheme 1).
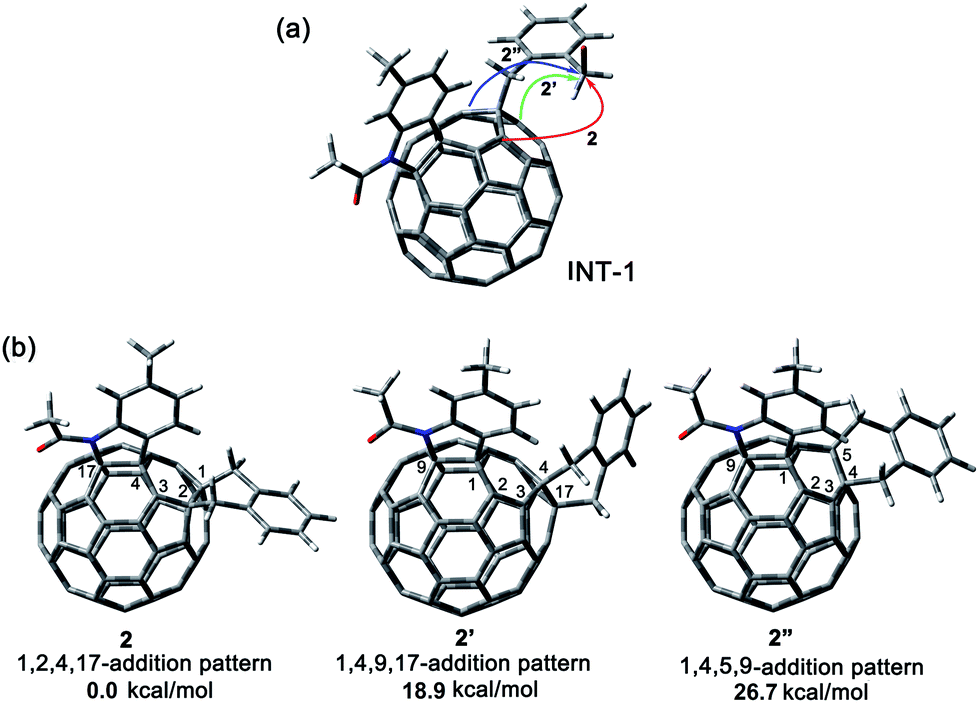 | ||
| Fig. 2 (a) Possible ring-closure pathways of the monoanionic INT-1. (b) Relative energies of the possible three ring-closure products 2, 2′ and 2′′. | ||
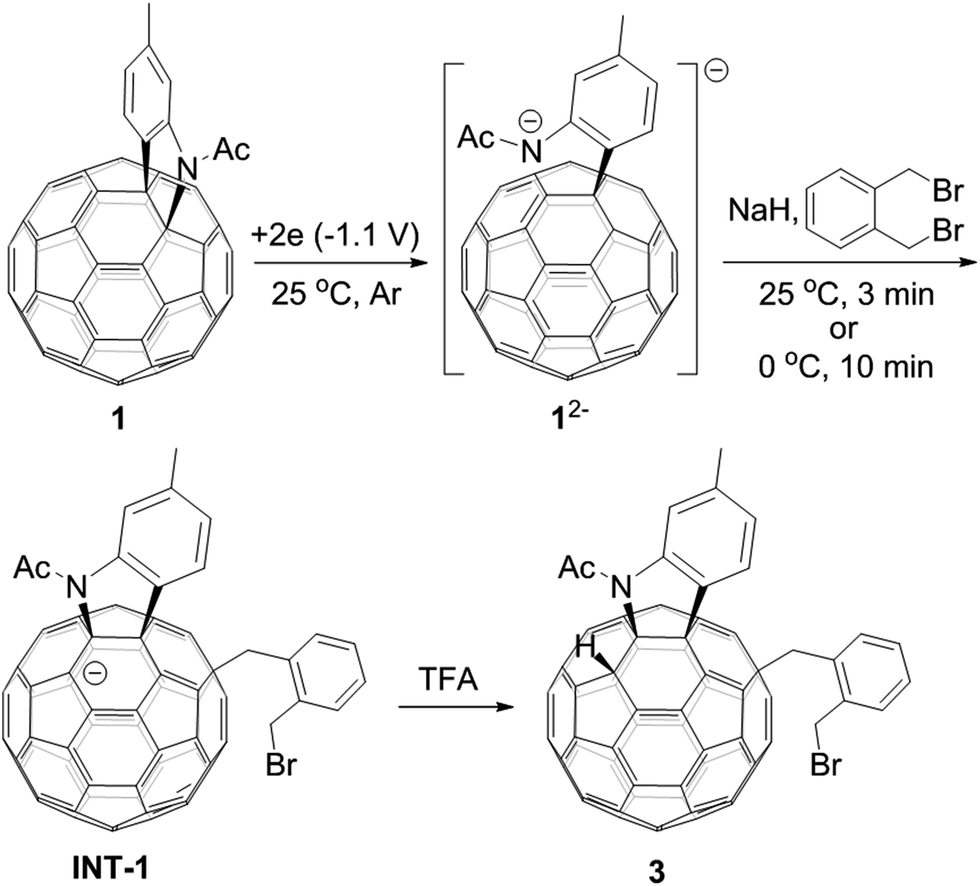 | ||
| Scheme 1 Reaction of 12− with 1,2-bis(bromomethyl)benzene at 25 °C for 3 min or 0 °C for 10 min and subsequent quenching with TFA. | ||
The isolation of 3 indicated that the proposed intermediate INT-1 was indeed formed. Surprisingly, it was found that further reaction at 25 °C and 0 °C for 5 h afforded totally different products, exhibiting temperature-controlled regioselectivity (Scheme 2). When the reaction was performed at 0 °C, the desired product 2 could be generated in 49% yield. Product 2 has an unprecedented 1,2,4,17-addition pattern, which can also be named as cis-3′ isomer and possesses a carbocycle fused to a [6,6]-junction and a heterocycle connected to a [5,6]-junction (see Fig. 1c). For a bis-cycloadduct, the two cycles are usually attached to two [6,6]-junctions of C60.2,3 To the best of our knowledge, there has been no precedent with the two cycles bonded to a [6,6]-junction and a [5,6]-junction, respectively, for a non-tethered bis-cycloadduct.18 In sharp contrast, another regioisomer 4 with the 1,2,3,4-addition pattern, also named as cis-1 isomer where both the carbocycle and heterocycle are fused to two neighboring [6,6]-junctions of C60 (see Fig. 1b), could be generated in 33% yield at 25 °C. It should be mentioned that part of 2 decomposed when it was purified on a silica gel column at 25 °C, and the isomerized product 4 could be isolated. This phenomenon was not observed when the purification process was performed at 0 °C.
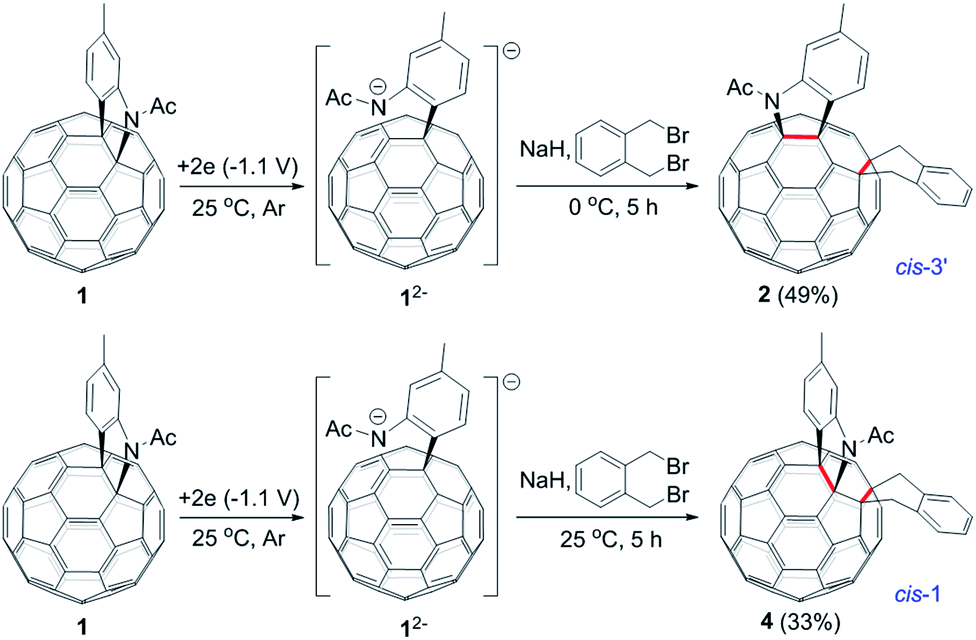 | ||
| Scheme 2 Reaction of 12− with 1,2-bis(bromomethyl)benzene at 0 °C and 25 °C to afford 2 and 4, respectively. | ||
Theoretical calculations showed that cis-3′ isomer 2 was less stable than cis-1 isomer 4 by 14.2 kcal mol−1 at the B3LYP/6-31G(d) level (see ESI† for details). Although 2 was not very stable, it remained nearly unchanged after being stirred in pure o-DCB at ambient temperature for 5 h. However, 2 tended to decompose to generate isomeric 4 in a low yield of 13% and some unidentified residue along with 21% of recovered 2 when stirred in o-DCB containing NaH and 0.1 M TBAP at room temperature for 5 h.
Monitoring of the reaction of 12− with 1,2-bis(bromomethyl)benzene at 25 °C showed that cis-3′ isomer 2 was initially formed and then gradually converted to cis-1 isomer 4, indicating that 2 was the precursor of 4 under our electrochemical conditions at 25 °C. Control experiments precluded the thermal rearrangement of 2 to 4 at 25 °C as the predominant process due to the low efficiency and poor yield (vide supra). The cyclic voltammogram (CV) of 2 exhibited irreversible first redox process of 2 (Fig. S3†) and more positively shifted than that of 1 (Table S1†).7b This CV result suggested that the formed 2 could be reduced in situ by the unreacted 12− to produce radical anions 1˙− and 2˙− during the reaction. On the other hand, after completion of the reaction, 2 could also be reduced to 2˙− by the negatively charged system generated by CPE. Radical anion 2˙− might undergo rearrangement to provide 4˙−, which was subsequently oxidized to neutral 4 by an oxidizing species such as oxygen during the workup process. This assumption was supported by a control experiment, which showed that the electrochemically generated radical monoanion 2˙− by CPE from 2 indeed underwent rearrangement within 1 h to afford 4 in 78% yield at 25 °C (Scheme 3).
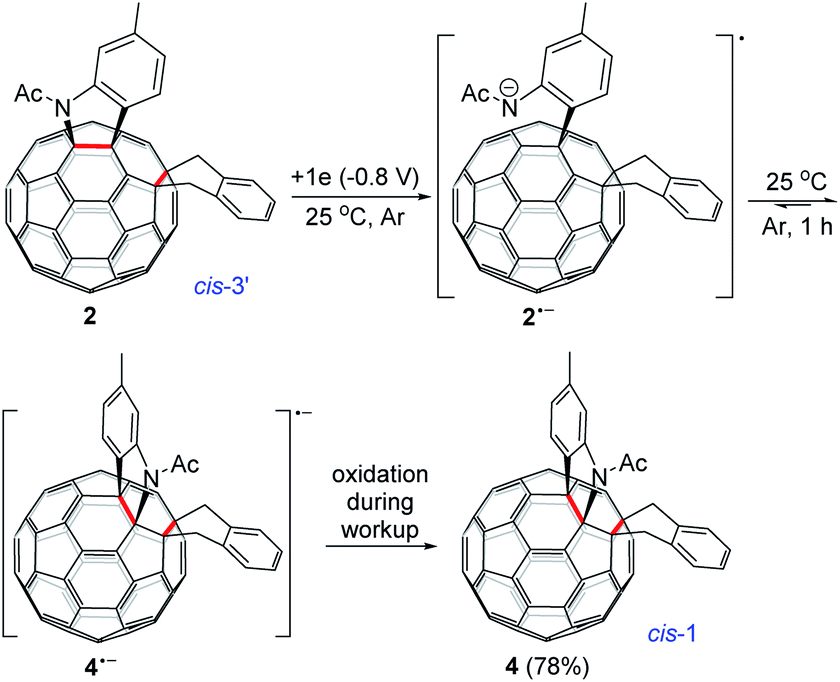 | ||
| Scheme 3 Electrochemical rearrangement of 2 to 4 at 25 °C. | ||
As seen in the CV of 2 (Fig. S3†), the first redox process of 2 was irreversible and its second one was quasi-reversible. This result indicated that the dianionic species 22− should also have a ring-opened structure and could be employed for further functionalization. Pleasingly, it was found that the protonation of the dianionic 22− with 2 equiv. of TFA at 0 °C for 5 min afforded 1,2,3,4,9,10-adduct 5 in 59% yield (Scheme 4). The preferred formation of 5 from the protonation of 22− was also supported by theoretical calculations (see ESI† for details).
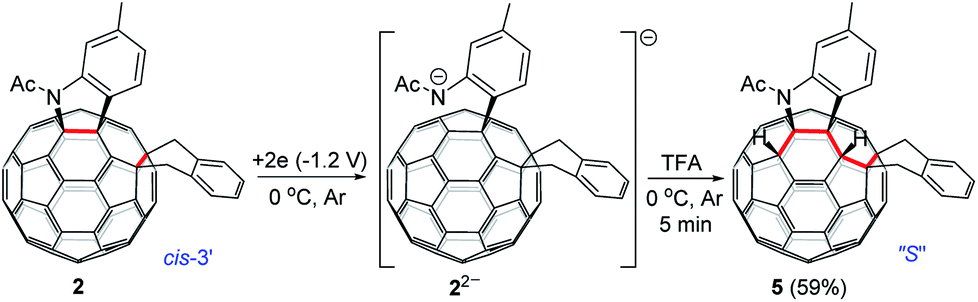 | ||
| Scheme 4 Electrochemical transformation of 2 into 5. | ||
Products 2, 3, 4 and 5 were characterized by HRMS, 1H NMR, 13C NMR, FT-IR, UV-vis and fluorescence spectra as well as CV and DPV. Particularly, the UV-vis spectra exhibit characteristic absorption peaks and features for each type of fullerene derivatives.2 Products 2 and 5 are new types of fullerene derivatives with unprecedented addition patterns, thus displaying new absorption features. However, the UV-vis absorption feature of 3 was very similar to those of other 1,2,3,16-adducts,7 while that of 4 showed a diagnostic spike at 431 nm, pretty close to those of other 1,2,3,4-adducts.4 The observed similar absorption features of 3 and 4 to those reported in the literature confirmed their structural assignments. The fluorescence spectra of bis-cycloadducts 2 and 4 in chloroform solution with an excitation wavelength of 550 nm resembled each other and exhibited two peaks at ∼720 and ∼800 nm, while those of 3 and 5 showed two peaks at 711 and 790 nm for the former and 696 and 766 nm for the latter, respectively. Two similar fluorescence peaks have also been observed for bis-cycloadducts of C60 in the literature.19 The CVs and DPVs of tetra-functionalized products 2–4 in the range of 0 to −2.0 V showed predominantly four redox processes, whereas those of hexa-functionalized product 5 exhibited only three redoxes in the same range probably due to its decreased electron-accepting ability. The first and second redox waves of 4 were reversible, while the first redox of 3 was quasi-reversible. In contrast, both 2 and 5 showed an irreversible wave for the first redox process. These results hint that compounds 2, 3 and 5 may be further functionalized by the electrochemical reduction to their monoanionic and/or dianionic stages.
In addition, the assigned structure of 5 was unequivocally confirmed by its single-crystal X-ray crystallography (Fig. 3).20 As a consequence of the inherent chirality possessed by 5, the crystal structure contains a pair of enantiomers, and they are in equal amounts, which is encountered in other fullerene crystals.6f,7c The structure of 5 reveals clearly that a heterocycle is bonded to C60 through a Caryl atom and a N atom at C2 and C3 sites, respectively, and that a carbocycle is bonded to C60 through two benzyl groups at the C9 and C10 sites, respectively. Two hydrogen atoms are attached to C1 and C4, neighboring the heterocycle. The six-functionalized carbon atoms of the fullerene cage are uplifted from the spherical surface notably because of their sp3 characters. The bond lengths for C1–C2, C2–C3, C3–C4, C1–C9, C9–C10 and C10–C11 are 1.580(2) Å, 1.642(2) Å, 1.557(2) Å, 1.622(3) Å, 1.596(2) Å, and 1.522(2) Å, respectively, and are within the range of typical C–C single bond lengths. In comparison, C5–C6 has a bond length of 1.351(2) Å, thus possessing double bond character.7b The single-crystal structure of 5 clearly demonstrates that it has a unique “S”-shaped configuration.
 | ||
| Fig. 3 ORTEP diagram of two enantiomers of 5 with 30% thermal ellipsoids. Solvent molecules are omitted for clarity. | ||
Conclusions
In summary, we have achieved the efficient and regioselective synthesis of the tetra-functionalized 1,2,4,17-adduct and the hexa-functionalized 1,2,3,4,9,10-adduct of C60. The 1,2,4,17-adduct has an unprecedented cis-3′ addition pattern, meanwhile the 1,2,3,4,9,10-adduct exhibits a unique “S”-shaped addition pattern. Both of them bear a carbocycle fused to a [6,6]-junction and a heterocycle fixed to a [5,6]-junction. The reaction of the electrochemically generated dianonic [60]fulleroindoline with 1,2-bis(bromomethyl)benzene for a short time and subsequent acid quenching afford the expected 1,2,3,16-adduct, proving our assumed addition site in the first step. Further reaction without acid quenching provides products with different addition patterns depending critically on the reaction temperature. The product obtained at 0 °C for 5 h is the desired unprecedented cis-3′ adduct. In contrast, the same reaction at 25 °C for 5 h selectively affords the more stable cis-1 isomer, which turns out to be generated by the rearrangement of the cis-3′ isomer induced by the negatively charged system. Intriguingly, the obtained cis-3′ adduct can be further regioselectively transformed into the “S”-shaped hexa-functionalized product. The observed high regioselectivities are controlled by charge distribution, steric effect and reaction temperature. It is anticipated that further chemical or electrochemical manipulations of the tetra- and hexa-functionalized [60]fullerene derivatives would provide new fullerene derivatives with novel addition patterns and physical properties.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are thankful for financial support from the National Natural Science Foundation of China (21572211) and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB20000000). The calculations in this paper have been done at the Supercomputing Center of University of Science and Technology of China.Notes and references
- For selected reviews, see: (a) F. Diederich and M. Gómez-López, Chem. Soc. Rev., 1999, 28, 263–277 RSC; (b) E. Nakamura and H. Isobe, Acc. Chem. Res., 2003, 36, 807–815 CrossRef CAS PubMed; (c) C.-Z. Li, H.-L. Yip and A. K. Y. Jen, J. Mater. Chem., 2012, 22, 4161–4177 RSC. For selected examples, see: (d) R. Tao, T. Umeyama, K. Kurotobi and H. Imahori, ACS Appl. Mater. Interfaces, 2014, 6, 17313–17322 CrossRef CAS PubMed; (e) R. Tao, T. Umeyama, T. Higashino, T. Koganezawa and H. Imahori, Chem. Commun., 2015, 51, 8233–8236 RSC.
- A. Hirsch and M. Brettreich, Fullerenes: Chemistry and Reactions, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2005 Search PubMed.
- For a review, see: Y. Nakamura, K. O-kawa and J. Nishimura, Bull. Chem. Soc. Jpn., 2003, 76, 865–882 CrossRef CAS.
- For selected examples on 1,2,3,4-adducts, see: (a) A. W. Jensen, A. Khong, M. Saunders, S. R. Wilson and D. I. Schuster, J. Am. Chem. Soc., 1997, 119, 7303–7307 CrossRef CAS; (b) M. Yamada, W. B. Schweizer, F. Schoenebeck and F. Diederich, Chem. Commun., 2010, 46, 5334–5336 RSC; (c) W.-W. Yang, Z.-J. Li, F.-F. Li and X. Gao, J. Org. Chem., 2011, 76, 1384–1389 CrossRef CAS PubMed; (d) C.-L. He, R. Liu, D.-D. Li, S.-E. Zhu and G.-W. Wang, Org. Lett., 2013, 15, 1532–1535 CrossRef CAS PubMed; (e) M. Chen, L. Bao, P. Peng, S. Zheng, Y. Xie and X. Lu, Angew. Chem., Int. Ed., 2016, 55, 11887–11891 CrossRef CAS PubMed; (f) S.-P. Jiang, M. Zhang, C.-Y. Wang, S. Yang and G.-W. Wang, Org. Lett., 2017, 19, 5110–5113 CrossRef CAS PubMed.
- For selected examples on 1,4,11,15-adducts, see: (a) G. Schick, K.-D. Kampe and A. Hirsch, J. Chem. Soc., Chem. Commun., 1995, 2023–2024 RSC; (b) Y. Murata, M. Shiro and K. Komatsu, J. Am. Chem. Soc., 1997, 119, 8117–8118 CrossRef CAS; (c) L.-L. Deng, S.-L. Xie, C. Yuan, R.-F. Liu, J. Feng, L.-C. Sun, X. Lu, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, Sol. Energy Mater. Sol. Cells, 2013, 111, 193–199 CrossRef CAS; (d) T. T. Clikeman, S. H. M. Deng, S. Avdoshenko, X.-B. Wang, A. A. Popov, S. H. Strauss and O. V. Boltalina, Chem.–Eur. J., 2013, 19, 15404–15409 CrossRef CAS PubMed.
- For selected examples on 1,2,4,15-adducts, see: (a) K. M. Kadish, X. Gao, E. V. Caemelbecke, T. Suenobu and S. Fukuzumi, J. Am. Chem. Soc., 2000, 122, 563–570 CrossRef CAS; (b) Y. Matsuo, A. Iwashita, Y. Abe, C.-Z. Li, K. Matsuo, M. Hashiguchi and E. Nakamura, J. Am. Chem. Soc., 2008, 130, 15429–15436 CrossRef CAS PubMed; (c) M. Nambo, A. Wakamiya, S. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2009, 131, 15112–15113 CrossRef CAS PubMed; (d) I. V. Kuvychko, A. V. Streletskii, N. B. Shustova, K. Seppelt, T. Drewello, A. A. Popov, S. H. Strauss and O. V. Boltalina, J. Am. Chem. Soc., 2010, 132, 6443–6462 CrossRef CAS PubMed; (e) W.-W. Chang, Z.-J. Li, W.-W. Yang and X. Gao, Org. Lett., 2012, 14, 2386–2389 CrossRef CAS PubMed; (f) M. Nambo, A. Wakamiya and K. Itami, Chem. Sci., 2012, 3, 3474–3481 RSC.
- For selected examples on 1,2,3,16-adducts, see: (a) Y. Rubin, P. S. Ganapathi, A. Franz, Y.-Z. An, W. Qian and R. Neier, Chem.–Eur. J., 1999, 5, 3162–3184 CrossRef CAS; (b) Y. Xiao, S.-E. Zhu, D.-J. Liu, M. Suzuki, X. Lu and G.-W. Wang, Angew. Chem., Int. Ed., 2014, 53, 3006–3010 CrossRef CAS PubMed; (c) H.-L. Hou, Z.-J. Li and X. Gao, Org. Lett., 2014, 16, 712–715 CrossRef CAS PubMed; (d) Z.-J. Li, S.-H. Li, T. Sun, H.-L. Hou and X. Gao, J. Org. Chem., 2015, 80, 3566–3571 CrossRef CAS PubMed; (e) H.-S. Lin, Y. Matsuo, J.-J. Wang and G.-W. Wang, Org. Chem. Front., 2017, 4, 603–607 RSC; (f) F. Li, J.-J. Wang and G.-W. Wang, Chem. Commun., 2017, 53, 1852–1855 RSC.
- For selected examples on 1,2,3,4,5,6-adducts, see: (a) H.-F. Hsu and J. R. Shapley, J. Am. Chem. Soc., 1996, 118, 9192–9193 CrossRef CAS; (b) Y. Tajima and K. Takeuchi, J. Org. Chem., 2002, 67, 1696–1698 CrossRef CAS PubMed; (c) S.-C. Chuang, F. R. Clemente, S. I. Khan, K. N. Houk and Y. Rubin, Org. Lett., 2006, 8, 4525–4528 CrossRef CAS PubMed.
- For selected examples on 1,2,4,11,15,30-adducts, see: (a) P. R. Birkett, P. B. Hitchcock, H. W. Kroto, R. Taylor and D. R. M. Walton, Nature, 1992, 357, 479–481 CrossRef CAS; (b) L. Gan, S. Huang, X. Zhang, A. Zhang, B. Cheng, H. Cheng, X. Li and G. Shang, J. Am. Chem. Soc., 2002, 124, 13384–13385 CrossRef CAS PubMed; (c) Y. Matsuo and E. Nakamura, Chem. Rev., 2008, 108, 3016–3028 CrossRef CAS PubMed.
- For a review, see: F. Diederich and R. Kessinger, Acc. Chem. Res., 1999, 32, 537–545 CrossRef CAS.
- For selected reviews on organic electrosysnthesis, see: (a) J. Utley, Chem. Soc. Rev., 1997, 26, 157–167 RSC; (b) B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC; (c) M. D. Kärkäs, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- For a review on electrochemistry of fullerenes, see: L. Echegoyen and L. E. Echegoyen, Acc. Chem. Res., 1998, 31, 593–601 CrossRef CAS.
- C. Niu, D.-B. Zhou, Y. Yang, Z.-C. Yin and G.-W. Wang, Chem. Sci., 2019, 10, 3012–3017 RSC.
- The isomers of bis-cycloadducts fused to a [6,6]-junction and a [5,6]-junction are named as cis′ isomers in order to differentiate them from those (cis isomers) fixed to two [6,6]-junctions. The order of precedence for the cis′ isomers is decided by the priority in locant numbers. The nomenclature of fC or fA for an enantiomer is dependent on the clockwise or anticlockwise numbering of the fullerene carbon atoms, see ref. 2.
- M. J. Frisch, et al., Gaussian 09 (RevisionB.01), Gaussian Inc., Wallingford, CT, 2010 Search PubMed.
- B. Zhu and G.-W. Wang, Org. Lett., 2009, 11, 4334–4337 CrossRef CAS PubMed.
- W.-W. Yang, Z.-J. Li and X. Gao, J. Org. Chem., 2010, 75, 4086–4094 CrossRef CAS PubMed.
- N. Martín, M. Altable, S. Filippone, Á. Martín-Domenech, A. Poater and M. Solà, Chem.–Eur. J., 2005, 11, 2716–2729 CrossRef PubMed.
- G. Accorsi and N. Armaroli, J. Phys. Chem. C, 2010, 114, 1385–1403 CrossRef CAS.
- The CCDC number of compound 5 is 1900924. For more details, see the ESI.†.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data, calculation results, NMR, UV-vis and fluorescence spectra, CVs and DPVs of 2–5 (PDF). X-ray crystallographic data for 5 (CIF). CCDC 1900924. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc02131k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |