
Resilient hollow fiber nanofiltration membranes fabricated from crosslinkable phase-separated copolymers†
Michael P.
Dugas
a,
Graham
Van Every
b,
Bumjun
Park
a,
John R.
Hoffman
a,
Ryan J.
LaRue
b,
Aaron M.
Bush
a,
Yizhou
Zhang
a,
Jennifer L.
Schaefer
 a,
David R.
Latulippe
b and
William A.
Phillip
*a
a,
David R.
Latulippe
b and
William A.
Phillip
*a
a205 McCourtney Hall, Department of Chemical and Biomolecular Engineering, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: wphillip@nd.edu
b1280 Main Street West, Department of Chemical Engineering, McMaster University, Hamilton, Ontario L8S 4L7, Canada
First published on 26th March 2020
Abstract
As wastewater reclamation and reuse technologies become more critical to meeting the growing demand for water, a need has emerged for separation platforms that can be tailored to accommodate the highly varied feed water compositions and treatment demands of these technologies. Nanofiltration (NF) membranes based on copolymer materials are a promising platform in this regard because they can be engineered at the molecular scale to address an array of separation process needs. Here, for example, a resilient NF membrane is developed through the design of a poly(trifluoroethyl methacrylate-co-oligo(ethylene glycol) methyl ether methacrylate-co-glycidyl methacrylate) [P(TFEMA-OEGMA-GMA)] copolymer that can be dip-coated onto hollow fiber supports. By exploiting the microphase separation of the oligomeric ethylene glycol side chains from the copolymer backbone and by elucidating the processing–structure–property relationships for the dip-coating process, membranes with pores 2 nm-in-diameter that exhibit a hydraulic permeability of 15.6 L m−2 h−1 bar−1 were generated. The GMA repeat units were functionalized post-coating with hexamethylene diamine to incorporate positively-charged moieties along the pore walls. This functionality resulted in membranes that rejected 98% of the MgCl2 from a 1 mM feed solution. Moreover, the reaction with the diamine crosslinked the copolymer such that the membranes operated stably in ethanol, an organic solvent that damaged the unreacted parent membranes irreparably. Finally, the stability of the crosslinked P(TFEMA-OEGMA-GMA) copolymer resulted in membranes that could operate continuously for a 24 hour period in aqueous solutions containing 500 ppm chlorine without exhibiting signs of structural degradation as evidenced by consistent rejection of neutral probe solutes. These results demonstrate how resilient, charge-selective NF membranes can be fabricated from microphase separated copolymers by engineering each of the constituent repeat units for a directed purpose.
Design, System, ApplicationNanofiltration membranes are capable of filtering moderately-sized organic solutes (i.e., those with molar masses ∼500 g mol−1) and divalent ions (e.g., Mg2+) from solution. However, they do not remove monovalent ions (e.g., Na+) effectively. These characteristics will allow nanofiltration membranes to address a crucial need in wastewater reuse processes where the indiscriminate removal of dissolved solutes in order to produce water of a potable quality is not necessarily required. Unfortunately, the performance of state-of-the-art thin film composite nanofiltration membranes declines dramatically in the presence of oxidizing agents such as chlorine. Copolymer-based membranes provide an opportunity to address this shortcoming through the molecular design of membrane precursors. Here, the constituent repeat units of a copolymer are designed such that it can be processed into nanofiltration membranes that possess pores 2 nm-in-diameter, that are capable of rejecting MgCl2 to a high extent, and that do not degrade when exposed to chlorine. While the example detailed here is targeted at resilient membranes for water treatment, the convergence of this versatile materials platform with molecular modeling and process systems engineering will allow for the future design of these membranes to occur in an integrated (i.e., from the molecular to the systems scales) and informed manner. |
1. Introduction
Due to their energy efficiency,1 ease of operation,2 and modular design,3 membrane separations have become an essential component of water treatment processes (e.g., in seawater desalination1,4 and membrane bioreactors2). Given these established applications and the growing demand for secure and sustainable sources of fresh water,5 there is significant interest in enhancing the design of membranes from the molecular-to-systems scales6 such that they can be deployed in wastewater reuse7 and resource recovery processes.8,9 These enhancements may target improvements in membrane properties such as overcoming the permeability-selectivity trade-off10,11 or developing more resilient membranes that can withstand prolonged operation under harsh conditions.12,13 Alternatively, novel membrane designs that exhibit solute-specific transport mechanisms are also of immense interest.14–16 As such, materials platforms that provide more precise control over the nanostructure and chemistry of membranes are needed to address the challenges associated with realizing these performance enhancements and the water treatment paradigms they can help to precipitate.Copolymer materials are a promising platform for the fabrication of membranes with properties tailor-made for target applications.17–19 Using molecular design principles to guide the selection of monomer identity and composition ultimately allows specific properties to be incorporated into the membranes when constructing the parent copolymers. For example, prior studies have shown that copolymer materials consisting of hydrophilic, oligomeric side chains that phase separate from the hydrophobic polymer backbone can be used to create membranes with a percolating network of nanoscale pores that filter solutes from solution based on molecular size.20,21 Copolymers consisting of oligo(ethylene glycol) methyl ether methacrylate (OEGMA) repeat units whose oligo(ethylene glycol) side chains phase separate from acrylonitrile (AN) repeat units in the polymer backbone are one example where this approach has been used to generate size-selective nanofiltration (NF) membranes.20,21 In addition to this control over nanostructure, functional groups that allow the pore wall chemistry to be tailored in the solid-state without compromising the structural integrity of the membrane can be incorporated in the design of the copolymer. This can be achieved, for example, through the introduction of the glycidyl methacrylate (GMA) repeat units.20–22 While one recent effort demonstrated that flat sheet (FS) membranes with thin copolymer active layers can be realized through the inclusion of additives such as ionic liquids in the membrane casting solution,23 to date, most of the FS membranes made from this class of copolymer materials suffer from low throughputs, as quantified by their hydraulic permeability values, due to thick (i.e., above 1 μm) active layers that are deposited using blade coating processes.20,21
Hollow fiber (HF) membranes are attractive technical targets due to the high membrane surface area per module volume (i.e., packing density) values that they provide.24,25 Moreover, HF membranes are of interest here because dip-coating was recently demonstrated as a viable method for depositing a thin layer of self-assembled block polymer material onto the surfaces of commercially-available HF supports.26,27 The resulting composite membranes were fabricated utilizing less block polymer material than traditional blade-casting techniques while exhibiting comparable transport properties. Expanding this dip-coating technique to a broader array of copolymer materials could be instrumental in developing highly-selective, highly-permeable NF membranes that would be an attractive platform for future membrane-based water treatment processes.
In order to translate the copolymer materials from the FS to the HF configuration, their macromolecular design needs to be considered. In particular, the evaporation of solvent from the coated HF surface is crucial to driving copolymer assembly during the dip-coating process.26 Therefore, casting solutions that consist of the copolymer dissolved in a more volatile solvent are needed to allow for an appreciable amount of solvent to evaporate in a relatively short period of time. Unfortunately, this constraint makes utilizing AN as the hydrophobic component difficult because it has limited solubility.28 Therefore, in this study, a parent poly(trifluoroethyl methacrylate-co-oligo(ethylene glycol) methyl ether methacrylate-co-glycidyl methacrylate) [P(TFEMA-OEGMA-GMA)] copolymer was designed to create a material that could be cast as robust and easily functionalized NF membranes using both dip- and blade-coating techniques. The solubility of the TFEMA material in a larger number of solvents is critical to enabling this transition.29 In addition to its solubility, the chemical stability of the TFEMA should result in more resilient membranes.7,30–32 For example, whereas the repeat units of AN hydrolyze at a pH above 10 causing membrane performance to degrade, the TFEMA repeat units do not hydrolyze under basic conditions.33 As in prior studies, the oligomeric side chains of the OEGMA were designed to phase separate from the polymer backbone to create a network of nanostructured pores.20,21
The epoxide rings in the GMA repeat units introduce functional groups along the pore walls that are readily modified post-casting.34 These epoxide rings can be modified with small molecule diamines in order to generate membranes capable of rejecting dissolved salts through a Donnan exclusion mechanism.35 Furthermore, the use of diamines can crosslink the membrane, which in combination with the stability of the TFEMA repeat units, may produce membranes that exhibit stable performance under harsh conditions that challenge state-of-the-art NF membranes. For example, standard thin film composite (TFC) NF membranes are made from crosslinked aromatic polyamide materials that cannot withstand prolonged exposure to the presence of oxidizing agents (e.g., chlorine) at low concentrations. These oxidizing agents result in the irreversible degradation of the aromatic backbone of the material and a concurrent precipitous drop in TFC membrane performance.36–38 Therefore, chlorine, which would help to forestall the deleterious effects of biofouling, must be removed from the feed solution prior to the use of TFC membranes in water treatment. In contrast, the chemical resilience of the crosslinked P(TFEMA-OEGMA-GMA) copolymer may allow it to operate in aqueous solutions containing chlorine. Additionally, the crosslinked material may resist dissolution and irreversible disruption of its physical nanostructure when in contact with a solvent environment, which could present the opportunity to apply these copolymer membranes in organic solvent nanofiltration.39–41
In this study, we report the design and development of a HF NF membrane platform that exhibits stable operation over a range of demanding conditions. The nanostructures within the active layers of the HF NF membranes were controlled by combining dip-coating on HF supports with the microphase-separation of a P(TFEMA-OEGMA-GMA) copolymer. Electron microscopy analysis in concert with hydraulic permeability measurements was utilized to elucidate processing–structure–property relationships such that membranes with thin active layers and high permeabilities could be fabricated reproducibly using the dip-coating process. The relationship between the nanostructure of the phase-separated copolymer and the pores within the active layer of the membranes was interrogated using neutral solute rejection tests as well as small angle X-ray scattering analysis. Subsequently, in order to generate membranes with robust performance profiles, small molecule diamines were introduced through simple chemical reactions associated with the GMA moieties that line the pore walls. A series of salt rejection experiments demonstrated the successful covalent attachment of these charge-selective functional groups along the pore walls and spectroscopic analysis suggested that the diamine reaction crosslinked the copolymer. Finally, a series of experiments where the membranes were exposed to feed solutions containing an organic solvent that dissolves the uncrosslinked copolymer or 500 ppm chlorine were used to demonstrate that the crosslinked NF membranes remained viable following prolonged exposure to these harsh conditions. The successful design and fabrication of these resilient HF NF copolymer membranes with controlled nanostructures and chemical functionality relied on the informed selection of the constituent repeat units within the copolymer. This demonstration, along with the vast number of potential copolymer chemistries, highlights the significant potential that this separations platform has for being implemented in water treatment and reuse applications where tailor-made nanofiltration membranes are needed.
2. Results and discussion
2.1 Copolymer synthesis and characterization
The poly(trifluoroethyl methacrylate-co-oligo(ethylene glycol) methyl ether methacrylate-co-glycidyl methacrylate) [P(TFEMA-OEGMA-GMA)] copolymer was synthesized using a free radical copolymerization. Four batches of copolymer were used to complete this study. The analysis of each batch using proton nuclear magnetic resonance spectroscopy (1H NMR) and gel permeation chromatography (GPC) confirmed the generation of high molecular weight copolymers with consistent compositions across all batches. The weight fractions, determined from 1H NMR spectra, were calculated to be 52.9 ± 0.8% TFEMA, 26.5 ± 0.6% OEGMA, and 20.6 ± 1.5% GMA (ESI,† Fig. S5A). GPC analysis, calibrated with poly(methyl methacrylate) standards, indicated the weight average molar mass was 109 ± 7 kg mol−1 with a dispersity (Đ) of 4.8 ± 1.5 (ESI,† Fig. S5B). The standard deviations represent the variation in composition, molar mass, and dispersity across the four batches of copolymer. In addition, differential scanning calorimetry (DSC) revealed that the oligomeric side chains from the OEGMA phase separate from the copolymer, as suggested by the observation of a glass transition temperature at −45 °C, which is similar to that of the poly(ethylene oxide) homopolymer glass transition (ESI,† Fig. S5C).2.2 Membrane fabrication and hydraulic permeability characterization
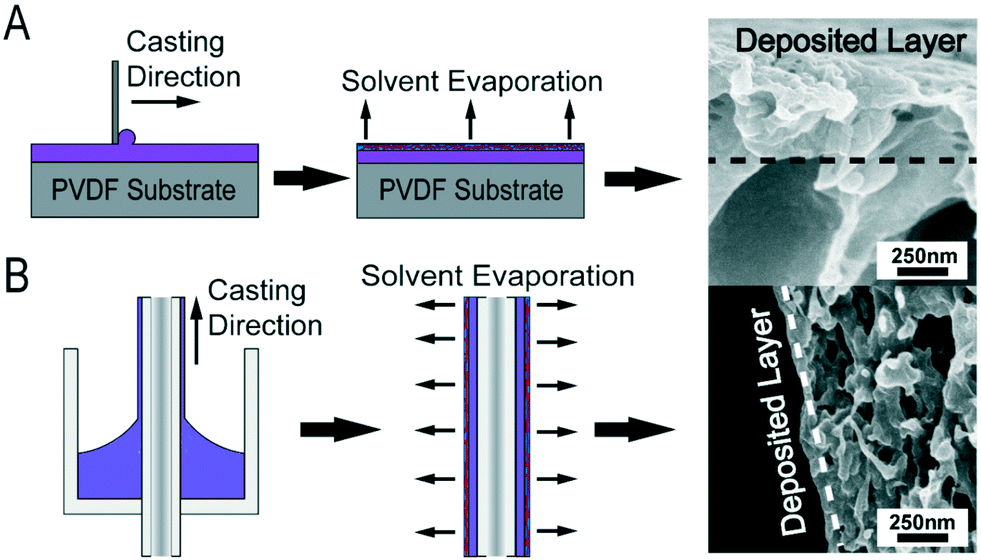 | ||
| Fig. 1 (A) Flat sheet membranes are blade-coated using a doctor's blade fixed at a pre-determined gate height; solvent is allowed to evaporate from the resulting thin film of solution, which drives microphase separation of the copolymer at the solution-air interface. After the evaporation period, the film is plunged into a non-solvent bath to set the membrane structure. (B) Dip-coated hollow fiber membranes are fabricated using a custom die, through which a substrate fiber is pulled. The dip-coating process results in a thin layer of copolymer casting solution being deposited on the substrate. After coating, the procedure is similar to that used in the blade-coated method. A key distinction is the fiber is inverted during the solvent evaporation step and then submerged into the non-solvent bath at the translation velocity used for coating. This consideration ensures a uniform evaporation time over the whole length of the fiber. | ||
Fig. 2 summarizes the relationships between the polymer concentration of the casting solution, the translation velocity of the substrate through the coating die, and the permeability of the HF membranes. The thicknesses of the copolymer layers were measured at three different points around the cross-sectional SEM micrographs (ESI,† Fig. S9) and averaged. The hydraulic permeability of the membranes was measured in a custom-built module (ESI,† Fig. S2). This combination of experiments allowed the desired processing–structure–property relationships to be elucidated.
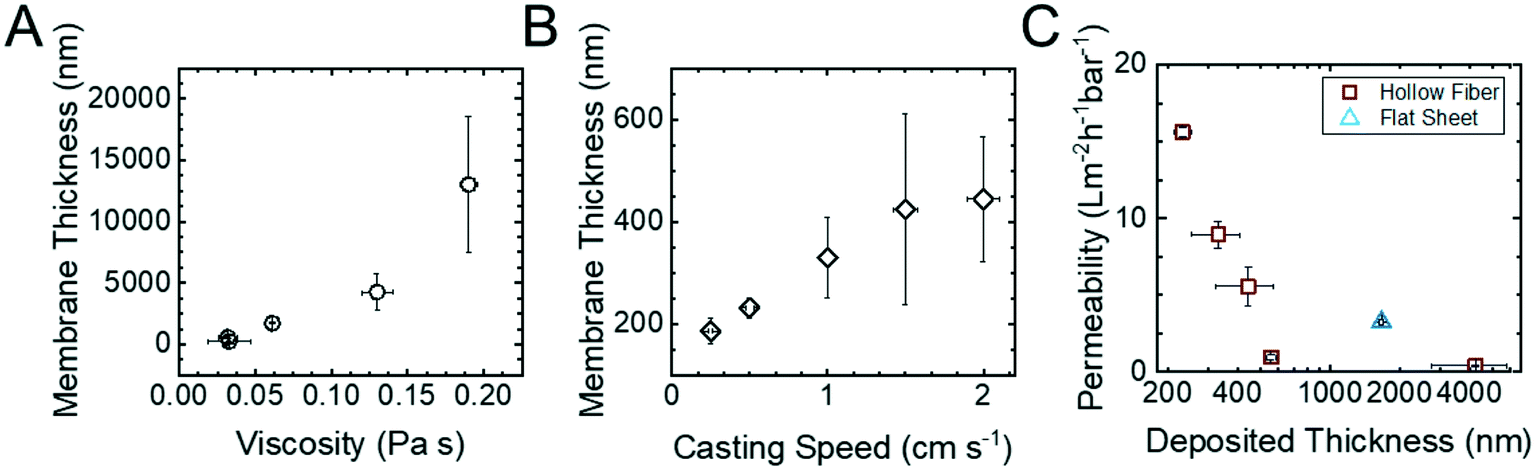 | ||
| Fig. 2 The relationship between the copolymer layer thickness with respect to (A) casting solution viscosity and (B) casting translational velocity. The thickness of the copolymer layer for each sample was determined using cross sectional SEM micrographs of the hollow fibers (ESI,† Fig. S9). (C) The variations in hydraulic permeability as a function of the thickness of the copolymer layer. Experimental permeabilities were determined by measuring the mass of the permeate versus time at varied transmembrane pressures. Error bars represent one standard deviation from n = 3 measurements on the same sample. | ||
In dip coating, the interplay between shear forces, surface tension, and gravity determine the thickness of the solution layer deposited on the translating substrate.26 The shear forces entrain solution on the substrate while surface tension and gravity work to thin the layer. Because a constant evaporation time was utilized in these experiments, this knowledge can be used to rationalize the results in Fig. 2. For example, as the viscosity of the casting solution increases, so does the thickness of the deposited layer (Fig. 2A). The results also show that as the translation velocity increased, the thickness of the deposited layer increased as well (Fig. 2B). These trends are both consistent with increases in the shear force resulting in thicker membranes, which manifest as the reduced permeabilities observed in Fig. 2C. Using a resistances-in-series analysis (ESI,† Fig. S10) demonstrated that for the range of processing conditions examined, the hydraulic permeability of the HF membrane scaled linearly with the inverse of the copolymer thickness. This observation is consistent with the copolymer layer being the dominant resistance to transport even when the copolymer layer is only 200 nm thick and Lp = 15.6 ± 0.3 L m−2 h−1 bar−1. This result highlights the integral role polymer processing plays in producing higher permeability membranes.
A casting speed of 0.5 cm s−1 and a casting solution at a concentration of 2% (by weight) copolymer in trifluoroethanol were used for fabrication of the hollow fibers throughout the rest of the study. Membranes fabricated using these conditions exhibited the highest permeability while being produced reliably by hand. Following fabrication, the membranes were either prepared for transport tests directly or reacted with hexamethylene diamine or spermine and then prepared for transport tests. Attenuated total internal reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) (ESI,† Fig. S11) and X-ray photoelectron spectroscopy (XPS) (ESI,† Fig. S12) were used to characterize the progress of the functionalization reactions. The membranes were classified as either unreacted, amine-functionalized, i.e., the epoxide rings of the GMA repeat units were opened using hexamethylene diamine, or spermine-functionalized, i.e., the epoxide rings were opened using spermine. In regards to preparing the membranes for transport tests, it is important to note that spuriously high permeabilities were sometimes observed while conducting preliminary experiments. This issue was attributed to the membranes drying and cracking while the epoxy used to seal the acrylic modules cured. These effects could be mitigated by limiting the amount of water that evaporated from the membrane surface during the potting process. Ultimately, a new module that used O-rings and washers to secure the membranes (ESI,† Fig. S3 and S4) was utilized to perform the transport studies because it did not require exposing the membrane to the atmosphere for an extended period of time.
2.3 Solute rejection experiments confirm nanostructure and functionalization
The pore sizes of the HF membranes were assessed using neutral solute rejection experiments; the results are reported in Fig. 3. The hydrodynamic diameters of the sucrose and PEO samples used as test solutes were determined from experimental reports of the solute diffusion coefficients in water42 in conjunction with the Stokes–Einstein equation. The observed rejection was calculated based on the measured values of the solute concentration in the feed and permeate solutions. Then, using a mass transfer correlation43 to account for the influence of concentration polarization, the actual rejection was determined.44 The actual rejection values were then correlated with a model for hindered transport through cylindrical pores to determine the ratio of the solute diameter to pore diameter.45 Because the solute diameters are known, this approach allowed the pore diameter to be assessed.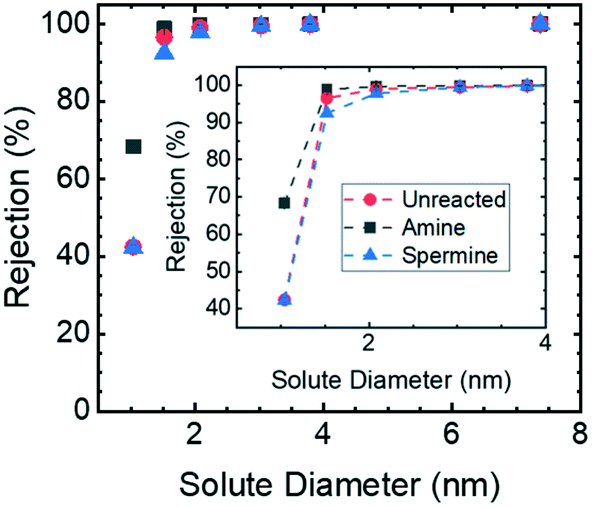 | ||
| Fig. 3 Neutral solute rejection curves for the unreacted, amine-functionalized, and spermine-functionalized membranes. 1 g L−1 solutions of sucrose and PEO with molar masses of 1.1, 2.1, 4.0, 6.0, and 19.0 kg mol−1 dissolved in DI water were used as feed solutions. The solute concentrations in the feed and permeate samples were quantified using TOC analysis. These values were then used to calculate the observed sieving coefficient. Concentration polarization was accounted for using a mass transfer correlation.43 The error bars, which are smaller than the symbols, represent one standard deviation from n = 2 samples taken using the same membrane. | ||
The actual rejection of sucrose by the unreacted, amine-functionalized, and spermine-functionalized membranes were calculated to be 42.4 ± 0.7%, 68.4 ± 0.1%, and 42.2 ± 0.4%, respectively. For both the spermine and unreacted membranes, this sucrose rejection value corresponds to a pore diameter of 2.8 ± 0.1 nm. The sucrose rejection value determined for the amine-functionalized membrane, on the other hand, corresponds to a pore diameter of 1.9 ± 0.1 nm. It is hypothesized that the smaller pore size of the amine-functionalized membrane is the result of both primary amine groups from hexamethylene diamine reacting with GMA repeat units to crosslink the membrane. XPS analysis supports this hypothesis. For the amine-functionalized membrane, the area under the peak associated with secondary amines is greater than the area under the peak associated with primary amines (ESI,† Fig. S12). This inequality is consistent with the higher concentration of secondary amines that would result from diamines reacting with two GMA repeat units. Subsequently, the crosslinked polymer resists swelling in a solvated environment and results in membranes with a smaller pore diameter.
Small angle X-ray spectroscopy (SAXS) measurements on dry membranes (Fig. 4A) and membranes submerged in water (Fig. 4B) were collected to examine this hypothesis in more detail. The samples with or without water were loaded into special glass capillary tubes and sealed with epoxy. SAXS/WAXS measurements were obtained at the Argonne APS synchrotron beamline 12-ID-B, operated by the Chemical and Materials Science group at Argonne National Laboratory. The scatter intensity in relation to the scattering vector, q, from these measurements is shown. The broad nature of the peaks associated with the copolymer structure suggest that it formed a disordered phase-separated network.46 The magnitude of the scattering vector at the maximum peak intensity therefore corresponds to an average characteristic length associated with the network structure. For the dry membranes, the single broad peaks were associated with nanostructures having a characteristic length that ranged from 11 nm for the amine-functionalized membrane to 13 nm for the unreacted and spermine-functionalized membranes. These characteristic lengths likely correspond to the domains formed upon the phase separation of the copolymer backbone and oligo(ethylene glycol) sides chains.
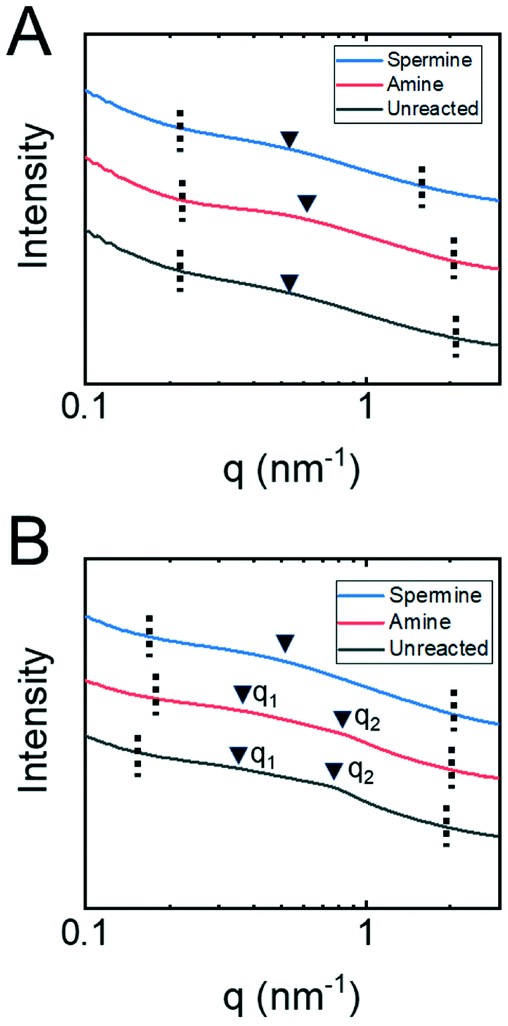 | ||
| Fig. 4 One-dimensional SAXS traces for P(TFEMA-OEGMA-GMA) copolymer membranes (A) in the dry state and (B) submerged in water. The dashed vertical lines indicate the region of interest and the arrows identify maxima in the broad scattering peaks. The scaling and positions of the traces on the y-axis have been altered to facilitate visualizing the peak positions in reference to q-values. | ||
The SAXS traces for the unreacted and amine-functionalized membranes submerged in water exhibit peaks at two distinct ranges of q-values. First, the broad peaks at q2 correspond to characteristic lengths of 7.9 nm and 7.4 nm for the unreacted and amine-functionalized membranes, respectively. Assuming lamellar and hexagonally-packed pore morphologies, a range of pore sizes can be estimated from these lengths.46 For the unreacted membrane, the pore size falls between 2.1–2.8 nm; while for the amine-functionalized membrane, a pore size between 1.9–2.7 nm is estimated. These pore sizes determined from SAXS are comparable to the diameters calculated when analyzing the neutral solute rejection experiments. Prior literature has also demonstrated that the pore diameter inferred from neutral solute rejection curves and the pore diameter determined from other characterization techniques such as SAXS analysis and SEM microscopy compare favorably.47,48 This agreement suggests that the broad peak at q2 is associated with the nanostructural features that control transport through the copolymer active layers. Moreover, these results further suggest that the diamine reaction produces a more constricted pore potentially through crosslinking. The water-swollen membranes also exhibit a peak at q1 ∼ 0.3 nm−1 that corresponds to a larger characteristic length of approximately 21 nm. This larger length scale could potentially be attributed to the water-swollen copolymer forming micellar structures within the membrane. This observation is consistent with other reports in the literature where similar copolymer materials formed micelles in various solvents.49
Unlike the unreacted and amine-functionalized membranes, the SAXS trace for the spermine-functionalized membrane in water exhibited only a single broad peak, which occurred at a q-value comparable to that observed for the dry membrane sample. This result is possibly caused by the capillary tube being poorly sealed thereby allowing the water to evaporate from this sample.
After demonstrating that size-selective nanofiltration membranes with pore diameters in the range of 2–3 nm could be fabricated, ion rejection experiments were conducted to demonstrate the charge-selective nature of the functionalized membranes. Four different 1 mM salt solutions, where the charges of the constituent anions and cations have been varied systematically (i.e., NaCl, MgCl2, MgSO4, and Na2SO4), were used in these experiments. Fig. 5 shows the resulting rejection values. The rejection of all of the salts by the unreacted HF membrane were low, that is, lower than 25%, because the neutral epoxide rings lining the pore wall do not repel the constituent ions of these salts electrostatically. In contrast, the functionalized membranes exhibit behavior that is consistent with the dissolved salts being rejected through a Donnan exclusion mechanism.35 For the Donnan mechanism, when the valences of the dissolved cation and anion are unbalanced, the ion with the higher valence dominates salt transport. In the case of the charge-functionalized membranes reported here, because the diamine and spermine functionality introduce positively-charged moieties on the pore wall, the MgCl2 solution exhibited the highest percent rejection due to the strong electrostatic repulsion of the magnesium ion by the membrane surface. In particular, 98.1 ± 0.1% of the MgCl2 was rejected by amine-functionalized HF membrane, 94.7 ± 0.2% by the spermine-functionalized HF membrane, and 89.3 ± 2.0% by the amine-functionalized flat sheet membrane. Conversely, the Na2SO4 solution demonstrated the lowest percent rejection across all of the functionalized membrane because the sulphate ions are attracted electrostatically to the membrane. Specifically, 13.4 ± 2.6% of the Na2SO4 was rejected by the amine-functionalized HF membrane, 22.8 ± 0.4% was rejected by the spermine-functionalized HF membrane, and 16.1 ± 0.1% was rejected by the amine-functionalized flat sheet membrane. In the Donnan mechanism, salts consisting of ions with balanced valences exhibit intermediate rejection values, which is consistent with the experimentally-measured rejection values for NaCl and MgSO4 across all of the functionalized membranes falling between 40 to 70%.
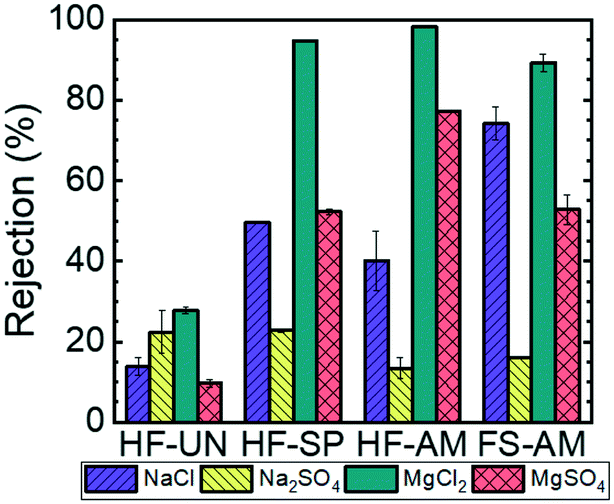 | ||
| Fig. 5 Rejection of salts in a 1 mM feed solution by the unreacted hollow fiber (HF-UN), spermine-functionalized hollow fiber (HF-SP), hexamethylene diamine-functionalized hollow fiber (HF-AM), and hexamethylene diamine-functionalized flat sheet (FS-AM) membranes. The cation concentrations in solution were determined using ICP-OES. The error bars represent one standard deviation from n = 2 measurements. | ||
In addition to salt rejection trends that are consistent with Donnan exclusion, the pH responsive rejection of MgCl2 by the amine-functionalized membranes is consistent with the successful functionalization of the pore walls (ESI,† Fig. S13). The pH of the MgCl2 feed solution was adjusted from 2 to 12 using dilute hydrochloric acid or sodium hydroxide. At a solution pH below the pKa of the amine groups (pKa = 10.7),20 the amine moieties protonate to form ammonium groups that possess a positive charge and reject MgCl2 well. At a solution pH above the pKa of the amine, the deprotonated moieties do not repel the Mg2+ through electrostatic interactions, leading to reduced salt rejection values.
Taken together, the results above demonstrate that the transport properties of the HF membranes are governed by the nanostructure of the copolymer, proving that the copolymer can be transitioned from the flat sheet to the hollow fiber membrane configuration. Moreover, this transition allows for membranes with increased hydraulic permeability values to be fabricated without sacrificing neutral solute rejection capabilities. Finally, the pore wall chemistry can be tailored for targeted performance. Here, for example, charge-functionalized membranes effectively reject dissolved salts that consist of divalent co-ions.
2.4 Amine-functionalized membranes withstand exposure to solvents
Crosslinking of the copolymer active layer during the diamine functionalization process should, by design, generate membranes that are more resilient to harsh conditions (e.g., exposure to solutions containing organic solvents or oxidizing agents). To test this hypothesis, a series of solvent exposure experiments were performed on the unreacted, amine-functionalized, and spermine-functionalized membranes. In this set of experiments, a feed solution containing 90% (by volume) ethanol in water was permeated through the membrane over a 24 hour period. To determine if and how the membrane nanostructures were affected by exposure to the ethanol solution, solvent permeability was quantified before and after exposure to ethanol and at regular intervals throughout the experiments. Additionally, MgCl2 and sucrose rejection experiments were conducted before and after the membranes were exposed to ethanol. Fig. 6 summarizes the results of these experiments, which demonstrate the ability of the amine-functionalized membranes to withstand exposure to an ethanol solution.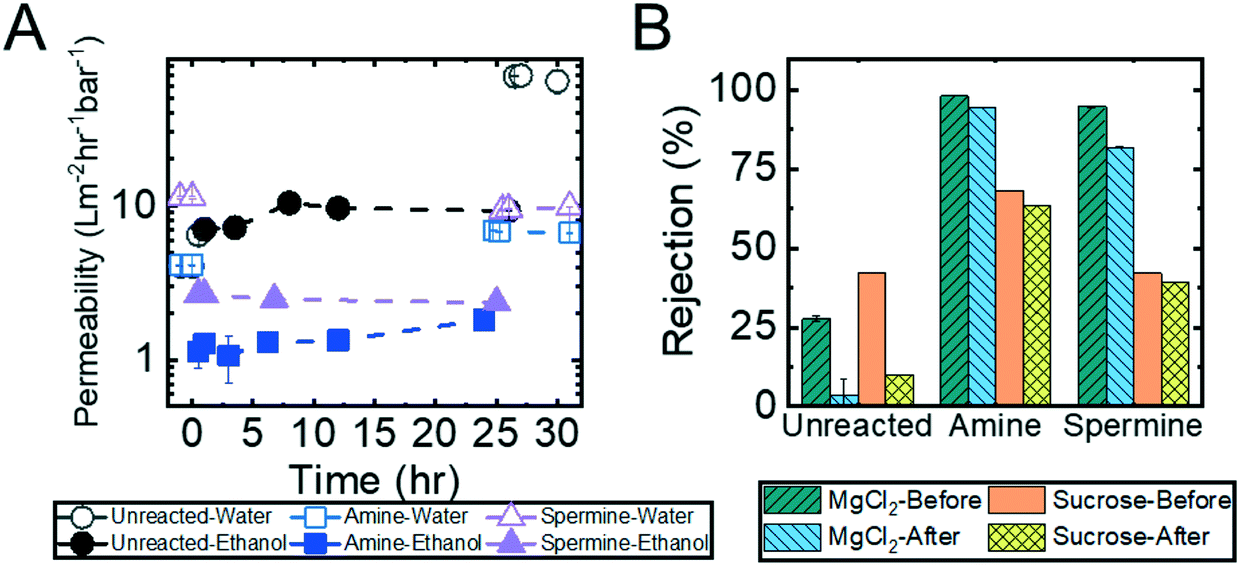 | ||
| Fig. 6 (A) Permeability of unreacted, diamine-functionalized, and spermine-functionalized membranes. These membranes were subjected to ethanol for approximately 24 hours and then changed to water. The time started from the moment the pressure was applied to the membrane in ethanol. The errors are one standard deviation of n = 2 samples taken from one membrane. (B) Rejections of 1 mM MgCl2 and 1 g L−1 sucrose solutions of the unreacted, amine-functionalized, and spermine functionalized membranes before and after subjected to ethanol permeation. The errors are one standard deviation of n = 2 samples from one membrane. | ||
As demonstrated in Fig. 6A, there was a negligible change in permeability when the unreacted membrane was subjected to ethanol. However, the permeability abruptly increased from 9.3 ± 0.1 L m−2 h−1 bar−1 to 68.8 ± 0.6 L m−2 h−1 bar−1 once the membrane was re-exposed to water. The increase in permeability suggests that the membrane nanostructure was disrupted by the exposure to ethanol. This observation that the unreacted, and therefore uncrosslinked, membrane structure degrades upon exposure to solvent is further supported by the dramatic drop in sucrose and MgCl2 rejections that was observed (Fig. 6B). On the other hand, the functionalized membranes exhibit a decrease in permeability (i.e., from 4.1 ± 0.01 L m−2 h−1 bar−1 to 1.1 ± 0.3 L m−2 h−1 bar−1 for the amine-functionalized and from 11.3 ± 0.1 L m−2 h−1 bar−1 to 2.7 ± 0.07 L m−2 h−1 bar−1 for the spermine-functionalized membranes) when they are exposed to the ethanol solution. After the initial decline, the permeability stabilized for the rest of the 24 hour long experiment. When the membranes were transitioned back to a DI water feed solution, the permeability rose to values that were comparable to those observed prior to exposure to ethanol (i.e., 6.7 ± 0.07 L m−2 h−1 bar−1 for the amine-functionalized membrane and 9.4 ± 0.09 L m−2 h−1 bar−1 for the spermine-functionalized membrane). Moreover, as shown in Fig. 6B, in addition to regaining similar permeabilities, the sucrose and MgCl2 rejections values observed before and after ethanol permeation were within 5% of each other. These observations indicated that the crosslinked membranes are not damaged irreparably by exposure to ethanol.
SAXS analysis of membranes submerged in ethanol (Fig. 7) suggests that structural rearrangements driven by polymer swelling lead to the observed changes in membrane performance. The SAXS spectra for all three membranes show two distinct peaks. The peaks at higher q-values, q2, are associated with smaller characteristic lengths of 4.2 nm for the unreacted and spermine-functionalized membranes and 3.7 nm for the amine-functionalized membrane. Using the same arguments that were applied to the water-swollen membranes,46 these characteristic lengths are associated with pore diameters between 1.1–1.5 nm for the unreacted and spermine-functionalized and 1.0–1.3 nm for the amine-reacted membranes. These smaller pore sizes are consistent with the lower permeability values that were observed in the transport experiments. Namely, in the limit of a low Reynolds number, the hydraulic permeability scales with the characteristic length for transport squared. Assuming that the ratio of q2 for the ethanol swollen membrane to q2 for the water swollen membrane is related to the ratio of characteristic lengths, i.e., qethanol/qwater = dwater/dethanol, suggests that the permeability for water should be 3.6 times greater than the permeability for ethanol (1.92 = 3.6). In comparison, the permeability measured in water was 3.9 times larger than that measured in ethanol for the amine-functionalized membrane. The favorable comparison between these two values offers further support for the argument that the smaller characteristic length identified in the SAXS traces are associated with features that control transport through the copolymer active layer.
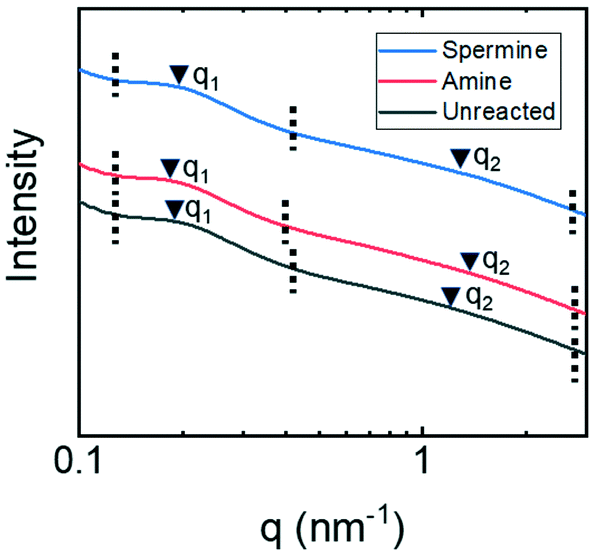 | ||
| Fig. 7 One-dimensional SAXS traces for P(TFEMA-OEGMA-GMA) copolymer membranes submerged in ethanol. The dashed vertical lines indicate the region of interest and the arrows identify maxima in the broad scattering peaks. The scaling and positions of the traces on the y-axis have been altered to facilitate visualizing the peak positions in reference to q-values. | ||
The peaks at q1 that appear in the SAXS traces of membranes submerged in ethanol are consistent with characteristic lengths of 31 nm, 35 nm, and 33 nm for the unreacted membranes, the amine-functionalized, and spermine-functionalized membranes, respectively. These values coincide well with the size of micelles formed by a TFEMA-based copolymer dissolved in methanol that has already been reported.49 AFM analysis of a flat sheet, amine-functionalized P(TFEMA-OEGMA-GMA) membrane in ethanol revealed the formation of nodules about 32 nm in diameter on the membrane surface (ESI,† Fig. S14–S16). In contrast, the same membrane submerged in water did not possess any clear identifying features on the surface. These assessments are further supported by examining the values of Flory–Huggins χ parameters for OEGMA and TFEMA in water29,50 and ethanol.29,51 First, the χ values indicate that the oligo(ethylene glycol) side chains of the OEGMA should swell less in ethanol relative to water, which is a phenomena observed in other experimental systems.52 Additionally, while based on the χ values, ethanol is not a good solvent for either component, it does interact slightly more favorably with the TFEMA component compared to TFEMA in water. This interaction may lead to some swelling that drives the formation of micelles. It is helpful to note that the small structural differences observed on the membrane surface when comparing the AFM and SEM micrographs are likely a result of the AFM analysis being conducted in a liquid environment, which causes the copolymer to swell, while the SEM analysis was conducted under vacuum. This hypothesis is further supported by an AFM micrograph of the dry membrane surface (ESI,† Fig. S14), which consistent with the SEM micrographs, appears smooth and featureless.
2.5 Membrane nanostructure does not degrade upon chlorine exposure
Experiments using feed solutions containing 500 ppm hypochlorite were conducted to assess the resilience of the hollow fiber membranes to chlorine exposure. In addition to measuring permeability, the membranes were tested for MgCl2 and sucrose rejection before hypochlorite exposure, after hypochlorite exposure, and after flushing the membranes with ethanol for 6 hours. Fig. 8 shows the results of these experiments. The permeability, shown in Fig. 8A, remained essentially constant throughout the experiment, ranging from 7.1 L m−2 h−1 bar−1 to 10.2 L m−2 h−1 bar−1. This result suggests that the membrane nanostructure was not affected by the hypochlorite ions permeating the membranes. In comparison, two state-of-the-art thin film composite nanofiltration membranes demonstrated 2× increases in permeability over a similar period of exposure to chlorine (ESI,† Fig. S19).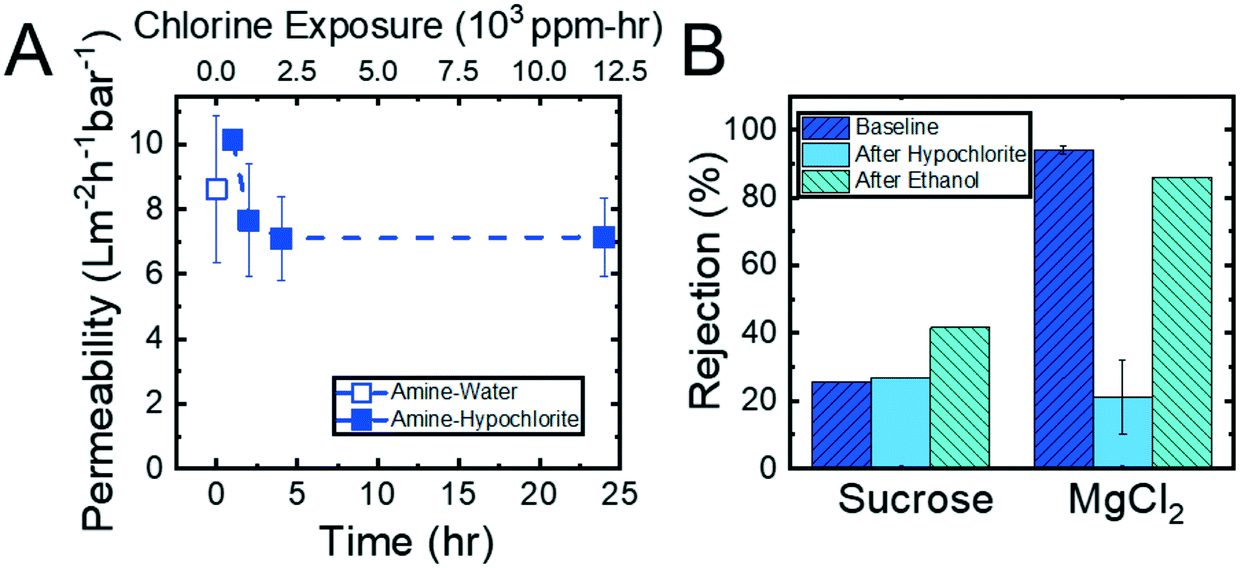 | ||
| Fig. 8 (A) Permeability of the amine-functionalized membranes as a function of chlorine exposure time. The membranes were subjected to an aqueous feed solution containing 500 ppm hypochlorite for 24 hours. The t = 0 time point is determined by the moment when the applied transmembrane pressure stabilized in the module after switching from a DI water feed solution to a chlorine-containing feed solution. The error bars represent one standard deviation of n = 2 samples. (B) 1 mM MgCl2 and 1 g L−1 sucrose rejections of the amine-functionalized membranes before the membranes were exposed to the 500 ppm chlorine feed solution, after the 24 hour experiment, and after the membrane was permeated with ethanol for 6 hours. The error bars represent one standard deviation from n = 2 samples. | ||
Interestingly, it was observed that the physical color of the amine-functionalized membrane changed from a brown to a cream color (ESI,† Fig. S17) upon exposure to the hypochlorite solution. The MgCl2 rejection, seen in Fig. 8B, after the membrane was permeated with chlorine dropped over 70%. This suggested that the loss of color may be the hypochlorite reacting with the primary amine to form chloramine moieties,53 which are neutral, on the membrane surface. This neutralization of the surface charge, in turn, caused the Donnan exclusion mechanism35 to fail, thereby allowing MgCl2 ions to permeate the membrane unhindered. This hypothesis is supported by ATR-FTIR spectra showing the shift of the amine peak after exposure to hypochlorite (ESI,† Fig. S18). Despite this drop in MgCl2 rejection, the copolymer and the membrane nanostructure itself did prove to be resistant to degradation by chlorine as evidenced by the constant hydraulic permeability and sucrose rejection values observed over the course of the experiment. After the amine-reacted membrane was flushed with water it was also permeated with ethanol to ensure no degradation of the reacted amines. The MgCl2 rejection after the ethanol permeation rebounded dramatically to 86%, indicating the chloramine moieties were disrupted to produce the original amine moieties. This assessment is supported by ATR-FTIR spectra showing the amine peak shifting back to its original position (ESI,† Fig. S18). The combination of permeability and rejection results demonstrate that the P(TFEMA-OEGMA-GMA) membrane can withstand prolonged exposure to hypochlorite ions without the structural integrity of the membrane being compromised. In addition, the functional amines remain covalently-bound to the pore walls.
3. Conclusions
The results in the sections above demonstrate that, through the appropriate molecular design and engineering, a resilient, hollow fiber nanofiltration membrane could be fabricated from a P(TFEMA-OEGMA-GMA) copolymer. Each component of the copolymer was incorporated for a targeted purpose that aimed to improve the overall membrane performance. The TFEMA backbone phase separates from the hydrophilic oligo(ethylene glycol) side chains of the OEGMA repeat units to produce well-defined pores about 2 nm in diameter. The solubility of the TFEMA allowed for hollow fiber membranes with controlled active layer thicknesses to be fabricated using dip-coating. In turn, the combination of the phase separated nanostructure and the control provided by dip-coating allowed for highly permeable membranes with sharp size-selective solute rejection curves to be realized. Moreover, by incorporating GMA repeat units, pore walls lined by a high density of epoxide rings were produced. Reacting the epoxide rings postcoating with diamines introduced positive charges along the pore walls such that membranes capable of rejecting divalent co-ions through a Donnan exclusion mechanism were generated. Furthermore, the use of difunctional amine moieties crosslinked the membrane making it more resilient to solvent exposure. Namely, while uncrosslinked membranes degraded irreparably upon exposure to ethanol, the performance of the crosslinked membrane remained consistent. Moreover, the chemical stability of the repeat units resulted in membranes that did not degrade upon exposure to acidic or basic solutions or solutions containing oxidizing agents such as chlorine. As such, this study provides one demonstration of how the copolymer platform allows for membranes with tailored properties to be rationally designed at the molecular scale. Critically, the versatile nature of copolymer materials will allow for this approach to be applied in multiple instances where membrane performance needs to be tailored based on systems-level demands.4. Experimental methods
A detailed description of the materials and methods utilized throughout this study can be found in the ESI.† Briefly, the P(TFEMA-OEGMA-GMA) was synthesized utilizing a free radical copolymerization protocol.20 The copolymer was precipitated and purified then characterized using 1H-NMR and GPC analysis to determine the copolymer composition and molar mass. HF membranes were dip-coated onto a PVDF HF substrate by pulling the support substrate through a Teflon die at a constant translation velocity. Solvent was allowed to evaporate from the coated membrane for 5 minutes before the HF membranes were inverted and plunged into a nonsolvent bath at the same speed as they were pulled through the coating die. This process facilitated a uniform evaporation time over the length of the membrane. After casting, membranes were either left unreacted or functionalized with excess hexamethylene diamine or spermine. ATR-FTIR and XPS analysis was conducted to characterize the functionalization process. FS membranes were subjected to transport experiments in Amicon 8010 stirred cells. HF membranes were either sealed into a custom acrylic module (ESI,† Fig. S2) with epoxy or into a microscale, parallel-structured, crossflow filtration system (ESI,† Fig. S3) for transport tests. SAXS/WAXS measurements were obtained at the Argonne APS synchrotron beamline 12-ID-B, operated by the Chemical and Materials Science group at Argonne National Laboratory, at room temperature with an X-ray beam wavelength of 0.9322 Å (energy of 13.3 keV). Ethanol and chlorine resistance experiments were conducted to determine the ability of the membranes to resist organic solvent and oxidative degradation by permeating unreacted and functionalized membranes with a 90% by volume ethanol solution or a 500 ppm aqueous sodium hypochlorite solution, respectively, for approximately 24 hours.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Portions of this work were made possible with support from the National Science Foundation (NSF) through the Advanced Manufacturing Program (Award Number: 1932206), and we appreciatively acknowledge this support. G. E. thankfully acknowledges support from the Mitacs Globalink Research Award & McMaster Faculty of Engineering. J. R. H. gratefully acknowledges support from the Center of Environmental Science and Technology (CEST)/Bayer Predoctoral Fellowship at the University of Notre Dame. R. J. L. gratefully acknowledges support from a Vanier Canada Graduate Scholarship. We would like to thank the Notre Dame Integrated Imaging Facility (NDIIF), the Center for Environmental Science and Technology (CEST), and the Materials Characterization Facility (MCF) at Notre Dame; portions of this research were performed with instruments at these facilities.References
- O. Lorain, B. Hersant, F. Persin, A. Grasmick, N. Brunard and J. M. Espenan, Desalination, 2007, 203, 277–285 CrossRef CAS.
- S. R. Lewis, S. Datta, M. Gui, E. L. Coker, F. E. Huggins, S. Daunert, L. Bachas and D. Bhattacharyya, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8577–8582 CrossRef PubMed.
- R. W. Baker, Membrane Technology and Applications, John Wiley & Sons, 2nd edn., 2004 Search PubMed.
- Y. Zhang, J. L. Sargent, B. W. Boudouris and W. A. Phillip, J. Appl. Polym. Sci., 2015, 132, 1–17 CAS.
- Advanced Nanomaterials for Water Engineering, Treatment, and Hydraulics, ed. T. A. Saleh, IGI Global, 2017 Search PubMed.
- E. A. Eugene, W. A. Phillip and A. W. Dowling, Curr. Opin. Chem. Eng., 2019, 26, 122–130 CrossRef.
- P. Bengani, Y. Kou and A. Asatekin, J. Membr. Sci., 2015, 493, 755–765 CrossRef CAS.
- J. L. Weidman, R. A. Mulvenna, B. W. Boudouris and W. A. Phillip, Langmuir, 2015, 31, 11113–11123 CrossRef CAS PubMed.
- Y. R. Qiu and L. J. Mao, Desalination, 2013, 329, 78–85 CrossRef CAS.
- X. Kong, Y. Zhang, S. Y. Zeng, B. K. Zhu, L. P. Zhu, L. F. Fang and H. Matsuyama, J. Membr. Sci., 2016, 518, 141–149 CrossRef CAS.
- C. Jiang, L. Tian, Y. Hou and Q. J. Niu, J. Membr. Sci., 2019, 586, 192–201 CrossRef CAS.
- J. Xue, Z. Jiao, R. Bi, R. Zhang, X. You, F. Wang, L. Zhou, Y. Su and Z. Jiang, J. Membr. Sci., 2019, 584, 282–289 CrossRef CAS.
- Q. Li, Z. Liao, X. Fang, D. Wang, J. Xie, X. Sun, L. Wang and J. Li, J. Membr. Sci., 2019, 584, 324–332 CrossRef CAS.
- P. K. Neghlani, M. Rafizadeh and F. A. Taromi, J. Hazard. Mater., 2011, 186, 182–189 CrossRef CAS PubMed.
- S. Bhattacharjee, J. Dong, Y. Ma, S. Hovde, J. H. Geiger, G. L. Baker and M. L. Bruening, Langmuir, 2012, 28, 6885–6892 CrossRef CAS PubMed.
- Y. Jiang and W. C. Wang, Polym. Adv. Technol., 2011, 22, 2509–2516 CrossRef CAS.
- Y. Tang, X. Lin, K. Ito, L. Hong, T. Ishizone, H. Yokoyama and M. Ulbricht, J. Membr. Sci., 2017, 544, 406–415 CrossRef CAS.
- S. P. Nunes, Macromolecules, 2016, 49, 2905–2916 CrossRef CAS.
- Y. Zhang, N. E. Almodovar-Arbelo, J. L. Weidman, D. S. Corti, B. W. Boudouris and W. A. Phillip, npj Clean Water, 2018, 1, 1–14 CrossRef CAS.
- S. Qu, T. Dilenschneider and W. A. Phillip, ACS Appl. Mater. Interfaces, 2015, 7, 19746–19754 CrossRef CAS PubMed.
- S. Qu, Y. Shi, S. Benavides, A. Hunter, H. Gao and W. A. Phillip, Chem. Mater., 2017, 29, 762–772 CrossRef CAS.
- S. Benavides, S. Qu, F. Gao and W. A. Phillip, Langmuir, 2018, 34, 4503–4514 CrossRef CAS PubMed.
- P. Kaner, A. V. Dudchenko, M. S. Mauter and A. Asatekin, J. Mater. Chem. A, 2019, 7, 4829–4846 RSC.
- J. Song, X. M. Li, A. Figoli, H. Huang, C. Pan, T. He and B. Jiang, Water Res., 2013, 47, 2065–2074 CrossRef CAS PubMed.
- S. Darvishmanesh, F. Tasselli, J. C. Jansen, E. Tocci, F. Bazzarelli, P. Bernardo, P. Luis, J. Degrève, E. Drioli and B. Van der Bruggen, J. Membr. Sci., 2011, 384, 89–96 CrossRef CAS.
- Y. Zhang, R. A. Mulvenna, B. W. Boudouris and W. A. Phillip, J. Mater. Chem. A, 2017, 5, 3358–3370 RSC.
- Y. Zhang, R. A. Mulvenna, S. Qu, B. W. Boudouris and W. A. Phillip, ACS Macro Lett., 2017, 6, 726–732 CrossRef CAS.
- Y. Eom and B. C. Kim, Polymer, 2014, 55, 2570–2577 CrossRef CAS.
- S. K. Papadopoulou and C. Panayiotou, J. Chromatogr. A, 2014, 1324, 207–214 CrossRef CAS PubMed.
- I. Sadeghi and A. Asatekin, ACS Appl. Mater. Interfaces, 2019, 11, 12854–12862 CrossRef CAS PubMed.
- P. Bengani-Lutz, R. D. Zaf, P. Z. Culfaz-Emecen and A. Asatekin, J. Membr. Sci., 2017, 543, 184–194 CrossRef CAS.
- P. Bengani-Lutz, E. Converse, P. Cebe and A. Asatekin, ACS Appl. Mater. Interfaces, 2017, 9, 20859–20872 CrossRef CAS PubMed.
- Y. H. Choi, C. M. Choi, D. H. Choi, Y. Paik, B. J. Park, Y. K. Joo and N. J. Kim, J. Membr. Sci., 2011, 371, 84–89 CrossRef CAS.
- R. Barbey, V. Laporte, S. Alnabulsi and H. A. Klok, Macromolecules, 2013, 46, 6151–6158 CrossRef CAS.
- V. S. Rathee, S. Qu, W. A. Phillip and J. K. Whitmer, Mol. Syst. Des. Eng., 2016, 1, 301–312 RSC.
- C. Causserand, B. Pellegrin and J. C. Rouch, Water Res., 2015, 85, 316–326 CrossRef CAS PubMed.
- V. T. Do, C. Y. Tang, M. Reinhard and J. O. Leckie, Environ. Sci. Technol., 2012, 46, 852–859 CrossRef CAS PubMed.
- Y. Hanafi, A. Szymczyk, M. Rabiller-Baudry and K. Baddari, Environ. Sci. Technol., 2014, 48, 13419–13426 CrossRef CAS PubMed.
- M. Cook, P. R. J. Gaffney, L. G. Peeva and A. G. Livingston, J. Membr. Sci., 2018, 558, 52–63 CrossRef CAS.
- C. Li, S. Li, L. Lv, B. Su and M. Z. Hu, J. Membr. Sci., 2018, 564, 10–21 CrossRef CAS.
- I. Soroko, M. P. Lopes and A. Livingston, J. Membr. Sci., 2011, 381, 152–162 CrossRef CAS.
- J. Shao and R. E. Baltus, AIChE J., 2000, 46, 1149–1156 CrossRef CAS.
- M.-C. Yang and E. L. Cussler, AIChE J., 1986, 32, 1910–1916 CrossRef CAS.
- L. J. Zeman and A. L. Zydney, Microfiltration and ultrafiltration: Principles and applications, Marcel Dekker, 1996 Search PubMed.
- L. Z. Michael Wales, Sep. Sci. Technol., 1981, 16, 275–290 CrossRef.
- A. M. Bush, H. O. Ford, F. Gao, M. J. Summe, S. Rouvimov, J. L. Schaefer, W. A. Phillip and R. Guo, J. Membr. Sci., 2018, 567, 104–114 CrossRef CAS.
- R. M. Dorin, W. A. Phillip, H. Sai, J. Werner, M. Elimelech and U. Wiesner, Polymer, 2014, 55, 347–353 CrossRef CAS.
- W. A. Phillip, R. Mika Dorin, J. Werner, E. M. V. Hoek, U. Wiesner and M. Elimelech, Nano Lett., 2011, 11, 2892–2900 CrossRef CAS PubMed.
- I. Sadeghi and A. Asatekin, Macromol. Chem. Phys., 2017, 218, 1–9 CrossRef.
- A. Èliassi, H. Modarress and G. Ali Mansoori, J. Chem. Eng. Data, 1999, 44, 52–55 CrossRef.
- M. Galin, Polymer, 1983, 24, 865–870 CrossRef CAS.
- H. O. Ford, L. C. Merrill, P. He, S. P. Upadhyay and J. L. Schaefer, Macromolecules, 2018, 51, 8629–8636 CrossRef CAS.
- N. R. Ward, R. L. Wolfe and B. H. Olson, Appl. Environ. Microbiol., 1984, 48, 508–514 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI:10.1039/c9me00160c |
| This journal is © The Royal Society of Chemistry 2020 |