
Complexation behaviour of LiCl and LiPF6 – model studies in the solid-state and in solution using a bidentate picolyl-based ligand†
Noel Angel
Espinosa-Jalapa
 a,
Nele
Berg
b,
Michael
Seidl
a,
Ilya G.
Shenderovich
b,
Ruth M.
Gschwind
b and
Jonathan O.
Bauer
*a
a,
Nele
Berg
b,
Michael
Seidl
a,
Ilya G.
Shenderovich
b,
Ruth M.
Gschwind
b and
Jonathan O.
Bauer
*a
aInstitut für Anorganische Chemie, Fakultät für Chemie und Pharmazie, Universität Regensburg, Universitätsstraße 31, D-93053 Regensburg, Germany. E-mail: jonathan.bauer@ur.de
bInstitut für Organische Chemie, Fakultät für Chemie und Pharmazie, Universität Regensburg, Universitätsstraße 31, D-93053 Regensburg, Germany
First published on 15th October 2020
Abstract
Structural knowledge on ubiquitous lithium salts in solution and in the crystalline state is of paramount importance for our understanding of many chemical reactions and of the electrolyte behaviour in lithium ion batteries. A bulky bidentate Si-based ligand (6) was used to create simplified model systems suitable for correlating structures of LiCl and LiPF6 complexes in the solid-state and in solution by combining various experimental, spectroscopic, and computational methods. Solution studies were performed using 1H DOSY, multinuclear variable temperature NMR spectroscopy, and quantum chemical calculations. [Ph2Si(2-CH2Py)2·LiCl]2 (3) dissociates into a monomeric species (9) in THF. For [Ph2Si(2-CH2Py)2·LiPF6]2 (11), low temperature NMR studies revealed an unprecedented chiral coordination mode (12) in non-coordinating solvents.
Structural knowledge on synthetically and technically relevant lithium salts like LiX (X = halogenide) and LiPF6 in the solid-state and in solution in the presence of coordinating solvents or ligands is particularly important for a better understanding of salt effects both in chemical reactions and technical applications such as in lithium ion batteries.1–4 LiCl is very often a by-product in chemical reactions with lithiated species involved and has long been known to be a potential key to reactivity and selectivity in reactions mediated by organometallic reagents.5 Combinations of inorganic salts or donor ligands with certain organometallic reagents (e.g. organolithium, -magnesium, and -zinc compounds) have gained broad applications in preparative chemistry.6,7 Significant progress has recently also been made in the field of LiCl catalysis.8 Understanding cation–anion coordination modes between Li+ and PF6− in LiPF6 electrolytes is of great importance and has contributed significantly to the improvement of lithium ion batteries.2 The development of defined [ligand·metal salt] combinations can open up entirely new paths in synthetic chemistry and provide hitherto unknown information on cation–anion interactions. However, the structural behaviour in solution often remains a black-box and correlations between solid-state and solution are only possible to a limited extent.9 Structural investigations on lithium compounds in solution using diffusion-ordered NMR spectroscopic methods have therefore become an increasingly important field, but have been reported mainly for lithium amide, mixed-metal, and organolithium reagents.10 Simplified model systems that use bulky and specially designed ligands have proven to be helpful tools for obtaining in-depth information on structure-forming principles of fundamentally important metal compounds, which are either used in preparative chemistry or found in nature.11 Especially in solution, the structural space often shows great versatility and flexibility and can be efficiently restricted using bulky ligands with diagnostic molecular patterns in order to achieve information on coordination modes that are otherwise difficult to identify.12
This gave us the idea to introduce diphenylbis(2-picolyl)-silane [Ph2Si(2-CH2Py)2] (6) (Scheme 1) because we expected this to be a promising backbone for the design of new bulky bidentate ligands that would enable detailed model studies of widely used synthetically and technically important lithium salts by showing defined structural patterns in solution. Different to phosphorus-based bis(2-picolyl) NPN ligands,13 tetravalent bis(2-picolyl)-substituted group 14 elements do not have an additional free donor site capable of coordination, but sterically demanding substituents can easily be incorporated.14 This makes them proper target molecules for use as bulky bidentate ligand systems that can limit structural flexibility in coordination modes. With this ligand in hands, we shed light on the structural behaviour of an LiCl and an LiPF6 complex both in the solid-state and in solution by combining various NMR spectroscopic methods with quantum chemical calculations.
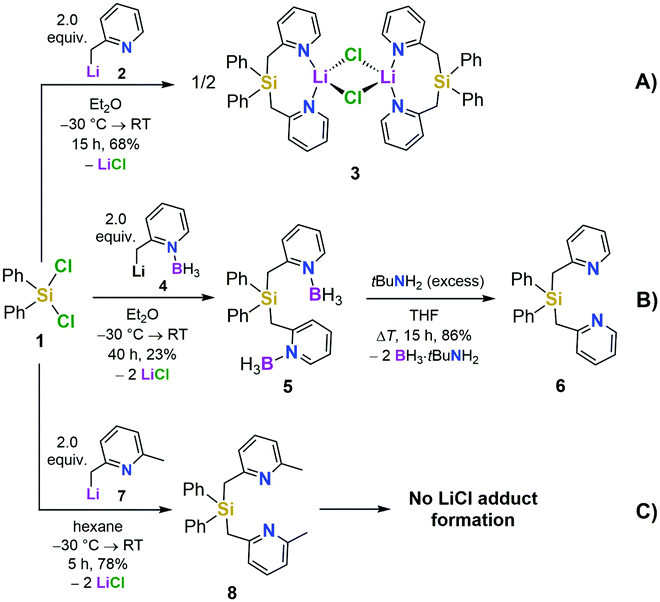 | ||
| Scheme 1 Synthesis of [Ph2Si(2-CH2Py)2·LiCl]2 (3) (route A), the free ligand 6via the borane-protected intermediates 4 and 5 (route B), and the lutidine derivative 8 (route C). | ||
We first set out to investigate a straightforward synthetic route to the new bidentate ligand (6) (Scheme 1). 2-Methyl-pyridines (2-picolines) that are functionalized by main group elements at the methyl position have long been the subject of intensive research due to their interesting electronic properties and coordinating abilities.15 However, only little is known about the coordination abilities of multidentate, unsubstituted (2-picolyl)silanes.16 Direct reaction of dichlorodiphenylsilane (1) with two equiv. of 2-picolyllithium17 (2) resulted in the formation of a dimeric LiCl complex (3) coordinated by the new bidentate ligand diphenylbis(2-picolyl)silane (6). Compound 3 could easily be isolated by extraction of the formed solids with DCM in 68% yield (Scheme 1A) and was characterized by single-crystal X-ray diffraction analysis (Fig. 1, top).
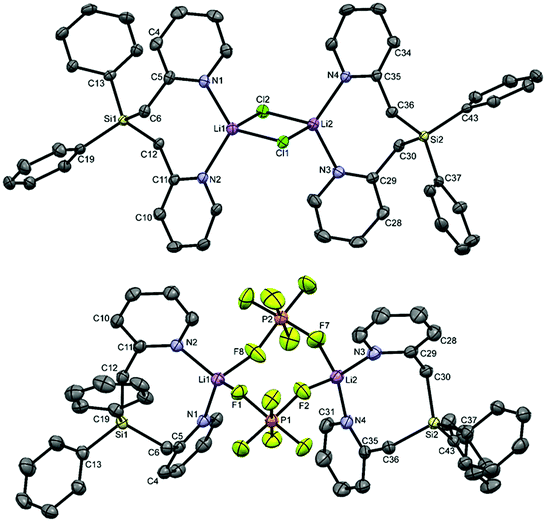 | ||
| Fig. 1 Molecular structures of complexes 3 (top) and 11 (bottom) in the crystal (displacement ellipsoids set at the 50% probability level, hydrogen atoms and included solvent molecules are omitted for clarity). | ||
Interestingly, Ph2Si(2-CH2Py)2 (6) is quite similar to the phosphorus analog18 PhP(2-CH2Py)2 concerning its coordination behaviour to LiCl.18c In the molecular structure of the latter, only the two pyridyl nitrogen atoms are involved in the coordination to LiCl forming a similar dimer as reported herein.18c
This prompted us to investigate the structure of [Ph2Si(2-CH2Py)2·LiCl]2 (3) in more detail in organic solvents. For this purpose, we developed a two-step synthesis to obtain Ph2Si(2-CH2Py)2 (6) in a LiCl-free fashion using BH3 as protecting agent (Scheme 1B). In addition, we made the lutidine derivative 8 available to study the influence of a methyl group in the 6-position of the pyridyl ring on the coordination behaviour (Scheme 1C). In contrast to the picoline derivative 6, the bis(2,6-lutidinyl)silane 8 could easily be obtained without complexation of LiCl when carrying out the direct reaction of two equiv. 2-picolyllithium with dichlorosilane 1 (Scheme 1C). Both compounds 6 and 8 were obtained in crystalline form, suitable for single-crystal X-ray diffraction analysis (for details, see the ESI†).
Solid-state 7Li NMR spectroscopy of dimer 3 shows a signal at δ = 2.6 ppm. In DCM, the 7Li NMR chemical shift of complex 3 (δ = 2.5 ppm) fits perfectly with the results of the solid-state NMR spectroscopy, which led us to the conclusion that the dimeric structure is retained in this weakly coordinating solvent. However, in a THF solution of 3, the 7Li NMR signal is highfield-shifted to δ = 0.8 ppm while the 1H NMR chemical shift of the pyridinyl-H atom in 6-position (δ = 8.50 ppm) still shows an interaction of the ligand with the lithium salt when compared with the 1H NMR spectrum of the free ligand Ph2Si(2-CH2Py)2 (6) (δ = 8.34 ppm). LiCl, dissolved in THF without any ligand, shows a 7Li NMR signal at δ = 0.5 ppm. We therefore assumed that in THF, dimer 3 breaks up into a monomeric species (9) as a result of the good coordination properties of the solvent (Scheme 2). This was further supported by quantum chemical calculations at the B3LYP/6-31+G(d) level of theory using the polarizable continuum model (PCM) (solvent: THF),19 which showed that this deaggregation is indeed a slightly exothermic process in THF (ΔH = −5 kJ mol−1), whereas the complete dissociation into the free ligand 6 and a plausible LiCl·THF species1b,20 (10) is a thermodynamically unfavorable process (ΔH = +10 kJ mol−1 with respect to monomer 9). Compound 10 was previously identified as a crucial species in equilibrium processes of organometallic reagents in the presence of LiCl in THF solutions.20 The same calculations for the lutidine derivative show a highly exothermic process (ΔH = −63 kJ mol−1 in total) for the dissociation of a hypothetical dimer into the free ligand 8 and [LiCl·2THF]2 (10) (for details, see the ESI†), so that even the formation of a monomeric LiCl species can be excluded if there is a methyl group in the 6-position of the pyridyl ring. 7Li NMR spectroscopy of 8 with one equiv. LiCl in THF shows one signal at δ = 0.5 ppm in accordance with free LiCl in THF. DFT calculations of the 7Li NMR chemical shift at the B3LYP/6-311+G(2d,p)//B3LYP/6-31+G(d) level21 strengthen the dissociation of dimer 3 into monomer 9 in THF. For the monomeric LiCl complex 9, a 7Li NMR chemical shift of δ = 1.3 ppm was calculated, which matches well with the experimentally observed 7Li NMR signal (δ = 0.8 ppm) of the LiCl adduct in THF. The calculated 7Li NMR chemical shift of dimer 3 was clearly downfield-shifted both in THF (δ = 1.9 ppm) and in the gas phase (δ = 2.3 ppm). Although the differences in the 7Li NMR chemical shifts are only marginal, the trend agrees well with the proposed model.
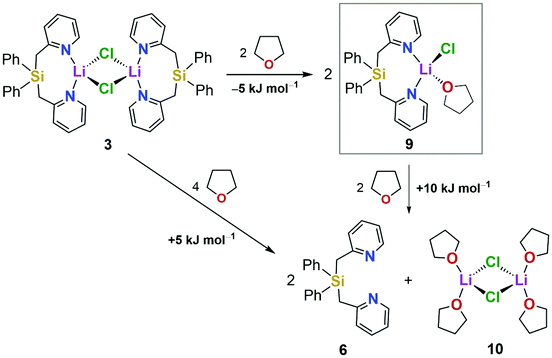 | ||
| Scheme 2 Calculated reaction enthalpies (ΔH) for the proposed dissociation of dimer 3 into a monomeric LiCl complex (9) and for the unlikely dissociation into the free ligand 6 and a dissolved LiCl·THF complex (10) in THF [B3LYP/6-31+G(d)] (PCM calculations; solvent: THF). | ||
We finally performed 1H-diffusion-ordered NMR spectroscopic (1H DOSY)22 measurements to get additional support for the formation of a monomeric Ph2Si(2-CH2Py)2·LiCl adduct (9) in THF solution. The hydrodynamic radius (rH) of ligand 6 in THF was determined to be 4.70 Å. For a solution of complex 3 in THF, the 1H DOSY experiment gave a radius of rH = 6.12 Å. This radius corresponds much more to a monomeric LiCl complex such as 9, as assumed in THF (see Scheme 2), than to the intact dimer 3 (for details on the measurement, see the ESI†). For comparison, 1H-diffusion measurements of the lutidine derivative 8 with and without addition of LiCl gave hydrodynamic radii of rH = 4.08 Å (without LiCl) and rH = 4.88 Å (addition of one equiv. LiCl), respectively, which indicates interactions between compound 8 and LiCl in THF, but the formation of a stable complex can be excluded.
In order to get more insight into the coordinating abilities of ligand 6 towards technically important lithium salts, we performed a ligand exchange reaction on adduct 3. The LiCl complex 3 could easily be converted into the dinuclear LiPF6 complex 11 by anion exchange with TlPF6 (Scheme 3). The molecular structure of compound 11 was determined by single-crystal X-ray crystallography (Fig. 1, bottom) and is another example for the rare bridging coordination of two lithium cations by two PF6− anions, each of which providing two fluorine atoms.23 Solid-state 7Li NMR spectroscopy of 11 shows a signal at δ = 0.7 ppm. In DCM at room temperature, dimer 11 can still be detected in very small amounts [δ(7Li) = 0.6 ppm], but is mainly converted into a new species [δ(7Li) = 1.7 ppm]. In view of the high structural flexibility of Li+⋯PF6− interactions,24 we carried out preliminary and for the first time variable temperature multinuclear NMR studies on a LiPF6 complex, which gave interesting information on the structural behaviour in solution (Fig. 2). In DCM, a defined major species [δ(7Li) = 1.2 ppm] becomes more and more evident starting from −30 to −80 °C, while at the same time formation of a minor species [δ(7Li) = 3.3 ppm] occurs. Remarkably, as the temperature decreases, 2JHH coupling (13.2 Hz) within the CH2 groups of the main compound becomes increasingly clear in the 1H NMR spectrum. The diastereotopicity of the methylene hydrogen atoms at −80 °C is a stereochemical probe that gives a clear hint to the formation of a defined and unprecedented complex exhibiting a chiral coordination sphere. A suggestion of a plausible structure (12) of the LiPF6 complex in DCM in accordance with the solution NMR studies is also shown in Fig. 2. Two lithium cations are each chelated by two fluorine atoms of one PF6− anion in a chiral complexation mode.25 A flipping coordination of the lithium centers and a rapid exchange between the coordinating and non-coordinating counteranion can be expected and matches with the 19F and 31P NMR spectra at ambient and low temperature. The formation of such a species can open new possibilities for generating polar chiral environments in solution. Furthermore, knowledge on ionic association in the presence of coordinating additives on a molecular level can provide helpful information concerning the conductivity behaviour of LiPF6-containing non-aqueous electrolyte solutions.26
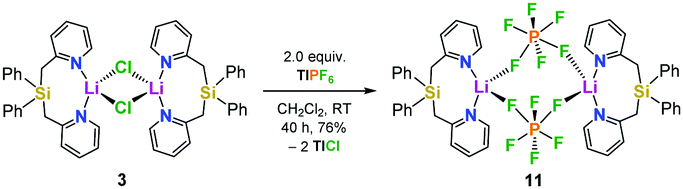 | ||
| Scheme 3 Synthesis of [Ph2Si(2-CH2Py)2·LiPF6]2 (11) from the LiCl complex 3via anion exchange with TlPF6. | ||
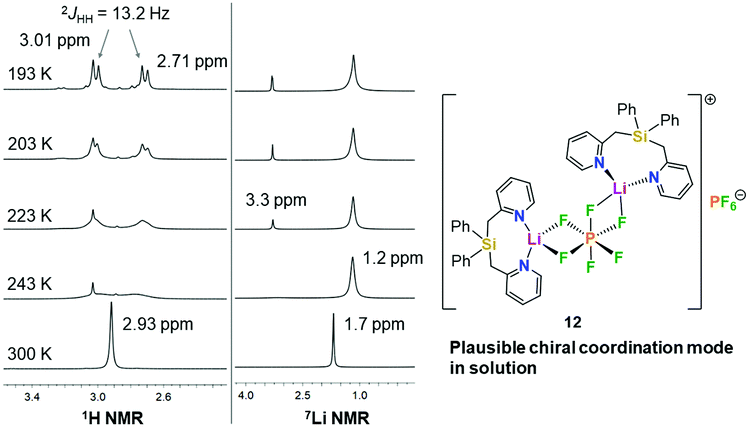 | ||
| Fig. 2 Left: Variable temperature NMR measurement (400 MHz) of complex 11 in DCM-d2 showing the diastereotopic splitting of the CH2 groups in the 1H NMR spectrum and the 7Li NMR signals. Right: Suggestion of a plausible structure (12) of the LiPF6 complex in solution. | ||
In summary, we presented a combined experimental, spectroscopic, and computational model study concerning structures of fundamentally important lithium salts in the solid-state and in solution in the presence of a new bidentate silicon-based ligand (6). We have gained detailed insights into fundamental dissociation and association processes in coordinating and non-coordinating solvents. It was shown that complex 3 breaks up into a monomeric LiCl adduct (9) in THF solution, but remains a dimer in less-coordinating DCM. The chloride ions could easily be exchanged by PF6− ions, while maintaining the dinuclear structure in the solid-state. [Ph2Si(2-CH2Py)2·LiPF6]2 (11) shows a rare bridging coordination of the lithium centers in the solid-state and forms a unique chiral species in solution, which was elucidated by variable temperature NMR measurements. In our ongoing studies we will address the targeted functionalization of ligand 6 at the silicon α-position. This might lead to new prototypes of multifunctional bulky bidentate C2-symmetric ligands,27 which can be used as stereochemical probes to reveal unusual coordination spheres in solution.
The Elite Network of Bavaria (ENB), the Bavarian State Ministry of Science and the Arts (StMWK), and the University of Regensburg are gratefully acknowledged for financial support (N-LW-NW-2016-366). We also thank Prof. Manfred Scheer and Prof. Jörg Heilmann for generous and continuous support.
Conflicts of interest
There are no conflicts to declare.References
- Structural investigations on LiCl solvates and complexes: (a) D. Barr, W. Clegg, R. E. Mulvey and R. Snaith, J. Chem. Soc., Chem. Commun., 1984, 79 RSC; (b) F. E. Hahn and S. Rupprecht, Z. Naturforsch., 1991, 46b, 143 Search PubMed; (c) F. S. Mair, J. Am. Chem. Soc., 1993, 115, 3388 CrossRef CAS; (d) R. Snaith and D. W. Wright, in Lithium Chemistry: A Theoretical and Experimental Overview, ed. A.-M. Sapse and P. v. R. Schleyer, John Wiley & Sons, New York, 1995 Search PubMed; (e) D. Hoffmann, A. Dorigo, P. v. R. Schleyer, H. Reif, D. Stalke, G. M. Sheldrick, E. Weiss and M. Geissler, Inorg. Chem., 1995, 34, 262 CrossRef CAS; (f) N. W. Mitzel and C. Lustig, Z. Naturforsch., 2001, 56b, 443 Search PubMed; (g) P. García-Álvarez, D. V. Graham, E. Hevia, A. R. Kennedy, J. Klett, R. E. Mulvey, C. T. ÒHara and S. Weatherstone, Angew. Chem., Int. Ed., 2008, 47, 8079 CrossRef.
- Structural investigations on LiPF6 electrolytes: (a) D. M. Seo, P. D. Boyle, O. Borodin and W. A. Henderson, RSC Adv., 2012, 2, 8014 RSC; (b) C.-C. Su, M. He, R. Amine, Z. Chen and K. Amine, J. Phys. Chem. Lett., 2018, 9, 3714 CrossRef CAS; (c) S. Hwang, D.-H. Kim, J. H. Shin, J. E. Jang, K. H. Ahn, C. Lee and H. Lee, J. Phys. Chem. C, 2018, 122, 19438 CrossRef CAS; (d) C.-C. Su, M. He, J. Shi, R. Amine, J. Zhang and K. Amine, Angew. Chem., Int. Ed., 2020, 59, 18229 CrossRef CAS.
- Reviews on lithium ion batteries: (a) K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS; (b) M. Winter, B. Barnett and K. Xu, Chem. Rev., 2018, 118, 11433 CrossRef CAS; (c) A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS; (d) J. Xie and Y.-C. Lu, Nat. Commun., 2020, 11, 2499 CrossRef CAS; (e) H. Zhang, C. Li, G. G. Eshetu, S. Laruelle, S. Grugeon, K. Zaghib, C. Julien, A. Mauger, D. Guyomard, T. Rojo, N. Gisbert-Trejo, S. Passerini, X. Huang, Z. Zhou, P. Johansson and M. Forsyth, Angew. Chem., Int. Ed., 2020, 59, 534 CrossRef CAS.
- Nobel Prize in Chemistry 2019: P. V. Kamat, ACS Energy Lett., 2019, 4, 2757 CrossRef.
- (a) D. Seebach, Angew. Chem., Int. Ed. Engl., 1988, 27, 1624 CrossRef; (b) B. Lecachey, H. Oulyadi, P. Lameiras, A. Harrison-Marchand, H. Gérard and J. Maddaluno, J. Org. Chem., 2010, 75, 5976 CrossRef CAS; (c) E. Hevia and R. E. Mulvey, Angew. Chem., Int. Ed., 2011, 50, 6448 CrossRef CAS; (d) Y. Gimbert, D. Lesage, C. Fressigné and J. Maddaluno, J. Org. Chem., 2017, 82, 8141 CrossRef CAS; (e) S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332 CrossRef CAS.
- Reviews: (a) R. E. Mulvey, F. Mongin, M. Uchiyama and Y. Kondo, Angew. Chem., Int. Ed., 2007, 46, 3802 CrossRef CAS; (b) R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743 CrossRef CAS.
- (a) L. M. Jackman and B. C. Lange, J. Am. Chem. Soc., 1981, 103, 4494 CrossRef CAS; (b) L. M. Jackman and T. S. Dunne, J. Am. Chem. Soc., 1985, 107, 2805 CrossRef CAS; (c) A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef CAS; (d) C. Unkelbach, D. F. O'Shea and C. Strohmann, Angew. Chem., Int. Ed., 2014, 53, 553 CrossRef CAS; (e) C. Feng, D. W. Cunningham, Q. T. Easter and S. A. Blum, J. Am. Chem. Soc., 2016, 138, 11156 CrossRef CAS; (f) Y.-H. Chen, M. Ellwart, G. Toupalas, Y. Ebe and P. Knochel, Angew. Chem., Int. Ed., 2017, 56, 4612 CrossRef CAS.
- (a) Y. Ma, A. C. Hoepker, L. Gupta, M. F. Faggin and D. B. Collum, J. Am. Chem. Soc., 2010, 132, 15610 CrossRef CAS; (b) A. C. Hoepker, L. Gupta, Y. Ma, M. F. Faggin and D. B. Collum, J. Am. Chem. Soc., 2011, 133, 7135 CrossRef CAS; (c) A. D. Benischke, G. Le Corre and P. Knochel, Chem. – Eur. J., 2017, 23, 778 CrossRef CAS.
- V. H. Gessner, C. Däschlein and C. Strohmann, Chem. – Eur. J., 2009, 15, 3320 CrossRef CAS.
- (a) P. García-Álvarez, R. E. Mulvey and J. A. Parkinson, Angew. Chem., Int. Ed., 2011, 50, 9668 CrossRef; (b) Review: R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 44, 11470 CrossRef; (c) C. Su, R. Hopson and P. G. Williard, J. Am. Chem. Soc., 2013, 135, 14367 CrossRef CAS; (d) R. Neufeld, M. John and D. Stalke, Angew. Chem., Int. Ed., 2015, 54, 6994 CrossRef CAS; (e) D. Li, I. Keresztes, R. Hopson and P. G. Williard, Acc. Chem. Res., 2009, 42, 270 CrossRef CAS; (f) G. Barozzino-Consiglio, G. Hamdoun, C. Fressigné, A. Harrison-Marchand, J. Maddaluno and H. Oulyadi, Chem. – Eur. J., 2017, 23, 12475 CrossRef CAS; (g) R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354 RSC; (h) D. R. Armstrong, P. García-Álvarez, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 6725 CrossRef CAS.
- (a) C. Däschlein, J. O. Bauer and C. Strohmann, Angew. Chem., Int. Ed., 2009, 48, 8074 CrossRef; (b) K. S. Lokare, N. Frank, B. Braun-Cula, I. Goikoetxea, J. Sauer and C. Limberg, Angew. Chem., Int. Ed., 2016, 55, 12325 CrossRef CAS; (c) J. O. Bauer and C. Strohmann, J. Am. Chem. Soc., 2015, 137, 4304 CrossRef CAS; (d) C. Golz, P. Steffen and C. Strohmann, Angew. Chem., Int. Ed., 2017, 56, 8295 CrossRef CAS; (e) K. S. Lokare, B. Braun-Cula, C. Limberg, M. Jorewitz, J. T. Kelly, K. R. Asmis, S. Leach, C. Baldauf, I. Goikoetxea and J. Sauer, Angew. Chem., Int. Ed., 2019, 58, 902 CrossRef CAS.
- J. Greindl, J. Hioe, N. Sorgenfrei, F. Morana and R. M. Gschwind, J. Am. Chem. Soc., 2016, 138, 15965 CrossRef CAS.
- (a) A. Kermagoret, F. Tomicki and P. Braunstein, Dalton Trans., 2008, 2945 RSC; (b) S. Liu, R. Peloso and P. Braunstein, Dalton Trans., 2010, 39, 2563 RSC; (c) G. Jin, L. Vendier, Y. Coppel, S. Sabo-Etienne and S. Bontemps, Dalton Trans., 2015, 44, 7500 RSC; (d) C. Hettstedt, M. Unglert, R. J. Mayer, A. Frank and K. Karaghiosoff, Eur. J. Inorg. Chem., 2016, 1405 CrossRef CAS.
- (a) J. O. Bauer, Main Group Met. Chem., 2020, 43, 1 CAS; (b) N. A. Espinosa-Jalapa and J. O. Bauer, Z. Anorg. Allg. Chem., 2020, 646, 828 CrossRef CAS.
- (a) M. Devillard, C. A. Lamsfus, V. Vreeken, L. Maron and J. I. van der Vlugt, Dalton Trans., 2016, 45, 10989 RSC; (b) M. Devillard, B. de Bruin, M. A. Siegler and J. I. van der Vlugt, Chem. – Eur. J., 2017, 23, 13628 CrossRef CAS; (c) M. Devillard, A. Ehlers, M. A. Siegler and J. I. van der Vlugt, Chem. – Eur. J., 2019, 25, 3875 CrossRef CAS; (d) A. Fernandes, C. Laye, S. Pramanik, D. Palmeira, Ö. Ö. Pekel, S. Massip, M. Schmidtmann, T. Müller, F. Robert and Y. Landais, J. Am. Chem. Soc., 2020, 142, 564 CrossRef CAS.
- (a) J. Manzur and W. K. Musker, Inorg. Nucl. Chem. Lett., 1973, 9, 841 CrossRef CAS; (b) S. Schwieger, R. Herzog, C. Wagner and D. Steinborn, J. Organomet. Chem., 2009, 694, 3548 CrossRef CAS.
- (a) H. Ott, U. Pieper, D. Leusser, U. Flierler, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 2978 CrossRef CAS; (b) A. R. Kennedy, R. E. Mulvey, R. I. Urquhart and S. D. Robertson, Dalton Trans., 2014, 43, 14265 RSC; (c) E. V. Brouillet, A. R. Kennedy, T. Krämer, R. E. Mulvey, S. D. Robertson, A. Stewart and S. Towie, Z. Anorg. Allg. Chem., 2020, 646, 726 CrossRef CAS.
- (a) A. Kermagoret, F. Tomicki and P. Braunstein, Dalton Trans., 2008, 2945 RSC; (b) I. Objartel, N. A. Pott, M. John and D. Stalke, Organometallics, 2010, 29, 5670 CrossRef CAS; (c) I. Objartel, D. Leusser, F. Engelhardt, R. Herbst-Irmer and D. Stalke, Z. Anorg. Allg. Chem., 2013, 639, 2005 CrossRef CAS.
- J. O. Bauer and C. Strohmann, Chem. Commun., 2012, 48, 7212 RSC.
- R. Neufeld, T. L. Teuteberg, R. Herbst-Irmer, R. A. Mata and D. Stalke, J. Am. Chem. Soc., 2016, 138, 4796 CrossRef CAS.
- U. Kroesen, L. Knauer and C. Strohmann, Angew. Chem., Int. Ed., 2017, 56, 6232 CrossRef CAS.
- (a) H. Zhang, R. Kerssebaum and R. M. Gschwind, Magn. Reson. Chem., 2009, 47, 568 CrossRef CAS; (b) K. Schober, E. Hartmann, H. Zhang and R. M. Gschwind, Angew. Chem., Int. Ed., 2010, 49, 2794 CrossRef CAS.
- D. R. Armstrong, A. H. Khandelwal, L. C. Kerr, S. Peasey, P. R. Raithby, G. P. Shields, R. Snaith and D. S. Wright, Chem. Commun., 1998, 1011 RSC.
- (a) E. C. Constable, M. J. Doyle, J. Healy and P. R. Raithby, J. Chem. Soc., Chem. Commun., 1988, 1262 RSC; (b) D. M. Seo, T. Afroz, J. L. Allen, P. D. Boyle, P. C. Trulove, H. C. De Long and W. A. Henderson, J. Phys. Chem. C, 2014, 118, 25884 CrossRef CAS; (c) T. Afroz, D. M. Seo, S.-D. Han, P. D. Boyle and W. A. Henderson, J. Phys. Chem. C, 2015, 119, 7022 CrossRef CAS; (d) S.-D. Han, S.-H. Yun, O. Borodin, D. M. Seo, R. D. Sommer, V. G. Young Jr. and W. A. Henderson, J. Phys. Chem. C, 2015, 119, 8492 CrossRef CAS.
- Early studies concerning Li–F interactions: (a) D. Stalke, U. Klingebiel and G. M. Sheldrick, J. Organomet. Chem., 1988, 344, 37 CrossRef CAS; (b) D. Stalke, U. Klingebiel and G. M. Sheldrick, Chem. Ber., 1988, 121, 1457 CrossRef CAS; (c) D. Stalke and K. H. Whitmire, J. Chem. Soc., Chem. Commun., 1990, 833 RSC.
- C. M. Burba and R. Frech, J. Phys. Chem. B, 2005, 109, 15161 CrossRef CAS.
- J. Seyden-Penne, Chiral Auxiliaries and Ligands in Asymmetric Synthesis, John Wiley & Sons, New York, 1995 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2018217–2018220. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0cc05682k |
| This journal is © The Royal Society of Chemistry 2020 |