
Recent advances in the synthesis of axially chiral biaryls via transition metal-catalysed asymmetric C–H functionalization
Gang
Liao†
,
Tao
Zhou†
,
Qi-Jun
Yao
and
Bing-Feng
Shi
 *
*
Department of Chemistry, Zhejiang University, Hangzhou, Zhejiang 310027, China. E-mail: bfshi@zju.edu.cn
First published on 27th June 2019
Abstract
Axially chiral biaryl motifs are widely present in natural products and pharmaceuticals. Moreover, they have been broadly used as privileged ligands in asymmetric catalysis. Over the past few decades, the efficient synthesis of axially chiral biaryls has been a research topic of great interest. This feature article will provide an overview of recent advances in the synthesis of these chiral skeletons via transition metal-catalysed asymmetric C–H functionalization.
 Gang Liao | Gang Liao received a bachelor degree (2013) in pharmaceutical engineering from the University of South China and a PhD degree (2018) from Zhejiang University under the supervision of Prof. Bing-Feng Shi. And now he is working as a postdoctoral fellow in the group of Prof. Bing-Feng Shi. His current research interests are focused on the development of new methodologies for the synthesis of bioactive molecules and novel chiral aldehyde catalysts in the area of asymmetric C–H activation. |
 Tao Zhou | Tao Zhou received his BSc degree (2012) in Shandong University and PhD (2017) from Nankai University under the supervision of Prof. Bai-Quan Wang. Now he is working as a postdoctoral fellow in the group of Prof. Bing-Feng Shi in Zhejiang University. His current research interests are focused on transition-metal catalyzed asymmetric C–H activation. |
 Qi-Jun Yao | Qi-Jun Yao received his bachelor degree (2015) in organic chemistry from Zhejiang University under the supervision of Prof. Yan-Guang Wang. And now he is pursuing a doctoral degree in the group of Prof. Bing-Feng Shi. His research interests focus on the construction of new chiral skeletons via asymmetric C–H activation. |
 Bing-Feng Shi | Bing-Feng Shi was born in Shandong, China. He received his BS degree from Nankai University in 2001 and a PhD degree from the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences under the guidance of Professor Biao Yu in 2006. Following time as a postdoctoral fellow at the University of California, San Diego (2006–2007), he moved to The Scripps Research Institute working with Professor Jin-Quan Yu as a research associate. In 2010, he joined the Department of Chemistry at Zhejiang University as a professor. His research focus is transition metal-catalyzed C–H functionalization and the synthetic applications. |
1. Introduction
The axial chirality of biaryls is caused by the restricted rotation of the aryl–aryl bond (the so-called atropisomerism) resulting from the nonplanar arrangement of four ortho-substituents about the biaryl axis.1 During the past few decades, axially chiral biaryls have received considerable attention due to their widespread presence in natural products and pharmaceuticals such as the glycopeptide antibiotic vancomycin, (+)-kinpholone, and (−)-gossyopol (Scheme 1a).2 In addition, axially chiral biaryls could be used as privileged ligands in asymmetric catalysis (Scheme 1b),3,4 as highlighted by the well-known BINAP, BINOL and its derived phosphoric acids.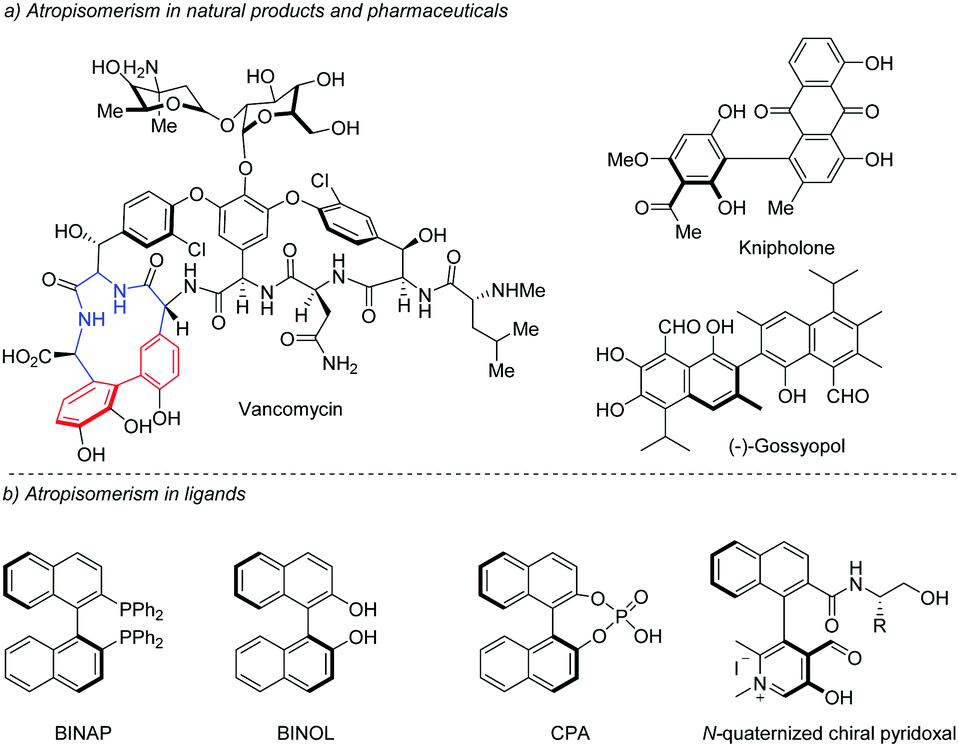 | ||
| Scheme 1 Atropisomerism in natural products and ligands. | ||
Because of the importance of these privileged scaffolds, substantial efforts have been devoted to the asymmetric synthesis of axially chiral biaryl skeletons.5 Accordingly, various catalytic systems involving metal-catalyzed approaches and organocatalyzed approaches have been developed for these asymmetric transformations. However, these methods usually suffer from several drawbacks such as the use of stoichiometric chiral auxiliaries, the requirement of prefunctionalization of starting materials and the poor efficiency of chiral induction. The growing demand for enantiopure axially chiral biaryl compounds in asymmetric catalysis and pharmaceutical industries has accelerated the development of efficient strategies for these useful structural motifs.
Recently, asymmetric C–H bond functionalization reactions have attracted much attention for their great promise as an efficient and straightforward approach to access chiral molecules.6 With this regard, transition-metal-catalysed asymmetric C–H activation would provide synthetic chemists a novel disconnection of retrosynthetic analysis logic to introduce new stereocenters to organic molecules, enabling the rapid generation of structural complexity in a single step. However, it is a formidable challenge to chemists due to the difficulty in finding suitable ligands or catalysts to control enantioselectivity during the asymmetric C–H activation process.
Although the creation of central and/or planar chirality via asymmetric C–H activation has been well investigated, methods for the asymmetric synthesis of axially chiral compounds via C–H functionalization have remained a barely explored field until recently, likely due to the possible racemization of atropoisomers under typically elevated reaction temperatures of C–H activation reactions.6 With significant advances of C–H activation reactions under mild conditions, some elegant strategies have been realized to address the bottleneck of axial chirality control. This feature article thus aims to provide an up-to-date overview of recent advances in the synthesis of axially chiral biaryls through C–H activation. We will focus exclusively on the generation of axially chiral biaryl skeletons by means of asymmetric C–H activation reactions with transition metal catalysis. That said, atroposelective C–H functionalization reactions using other organocatalysts such as peptides, and quinidine derivatives will not be discussed here.7
In general, two main strategies utilizing transition-metal-catalysed atroposelective C–H functionalization have been developed in the synthesis of axially chiral biaryls. In the first case, axially chiral biaryls were synthesized by locking a preformed axis via (dynamic) kinetic resolution or desymmetrization processes (Scheme 2a). Accordingly, this section can be further classified into four subsections: catalytic asymmetric C–H functionalization by chiral ligands, atroposelective C–H functionalization catalysed by a preformed chiral catalyst, diastereoselective C–H functionalization directed by a chiral auxiliary and enantioselective C–H functionalization enabled by a transient directing group. The second category is the creation of a biaryl axis via C–H activation/asymmetric coupling (Scheme 2b).
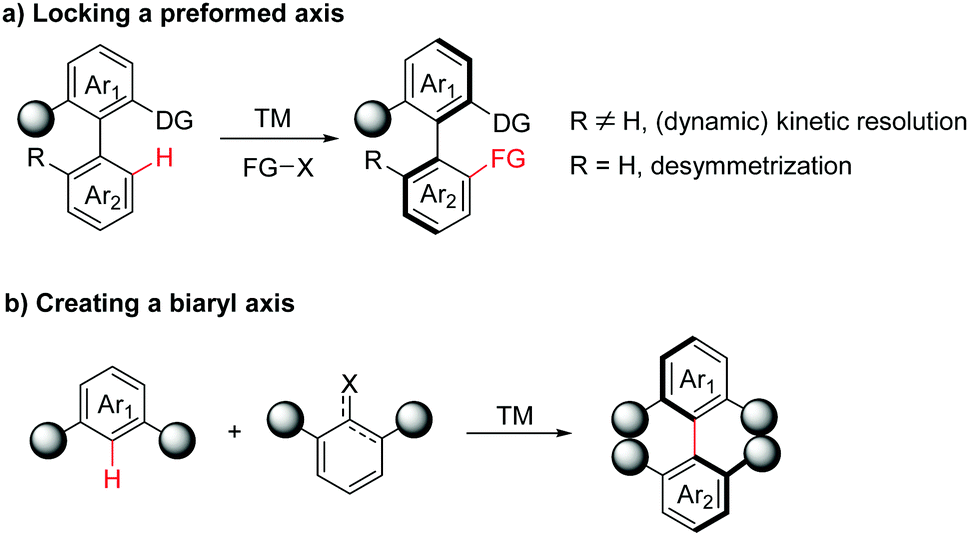 | ||
| Scheme 2 Transition-metal-catalyzed atroposelective C–H functionalization strategies towards axially chiral biaryls. | ||
2. Locking a pre-existing biaryl axis via (dynamic) kinetic resolution or desymmetrization
2.1 Catalytic asymmetric C–H functionalization cooperatively catalysed by transition metals and chiral ligands
The designation and discovery of efficient ligands for achieving high enantioselectivity has been a central topic in catalytic asymmetric synthesis. In the field of atroposelective C–H functionalization, several types of chiral ligands have already been applied for asymmetric induction of axial chirality.8–13 In 2000, the atroposelective alkylation of 2-(1-naphthyl)pyridine or 1-(1-naphthyl)isoquinoline by Rh(I)-catalysed C–H alkylation using chiral ferrocenyl phosphine as the ligand was reported by Murai and co-workers (Scheme 3).8 The low inversion barriers of biaryl substrates enabled the interconversion easily during the dynamic kinetic process. The alkylation products, however, are produced with the rise of inversion barriers to restrict the rotation. The biaryl compound rac-1 reacted with ethylene using the [{RhCl(coe)2}2]/(R),(S)-PPFOMe (L1, coe = cis-cyclooctene) catalytic system obtaining the coupling product 2 in 37% yield with 49% ee. In the case of quinoline derivative 3, the efficiency and enantioselectivity were slightly lower, giving the product 4 in 33% yield and 22% ee.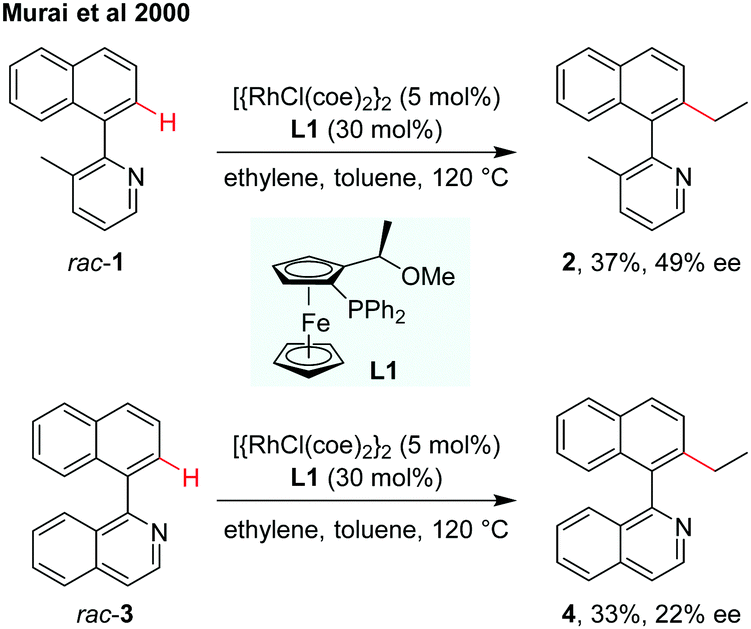 | ||
| Scheme 3 Rh(I)-Catalysed asymmetric C–H alkylation using a chiral ferrocenyl phosphine ligand (Murai et al., 2000).8 | ||
Since the pioneering work of Yu and co-workers, mono-N-protected amino acids (MPAAs) have been identified as privileged ligands for a wide variety of enantioselective C–H functionalization reactions.9 This MPAA catalytic system has also been adopted for atroposelective C–H activation. In 2014, You and co-workers achieved the first Pd-catalyzed C–H iodination for the synthesis of axially chiral biaryls via kinetic resolution (Scheme 4a).10 In their report, N-iodosuccinimide (NIS) was used as an iodinated reagent and N-monoprotected phenylalanine (L2) was used as the most effective ligand to control the stereoselectivity. A selectivity factor could be reached up to 27. The iodination products could be easily converted to aryl-substituted pyridine N-oxides by Suzuki–Miyaura coupling, which was used as a catalyst in asymmetric allylation of benzaldehyde with allyltrichlorosilane. They proposed that the asymmetric C–H bond cleavage of rac-5 may occur via a concerted metathesis deprotonation mechanism (CMD) pathway with the aid of an MPAA ligand. The Pd(II) intermediate 6a was oxidized by NIS to form a reactive Pd(IV) intermediate 6b, which then underwent reductive elimination to release the desired product (S)-7 with the regeneration of Pd(II) species.
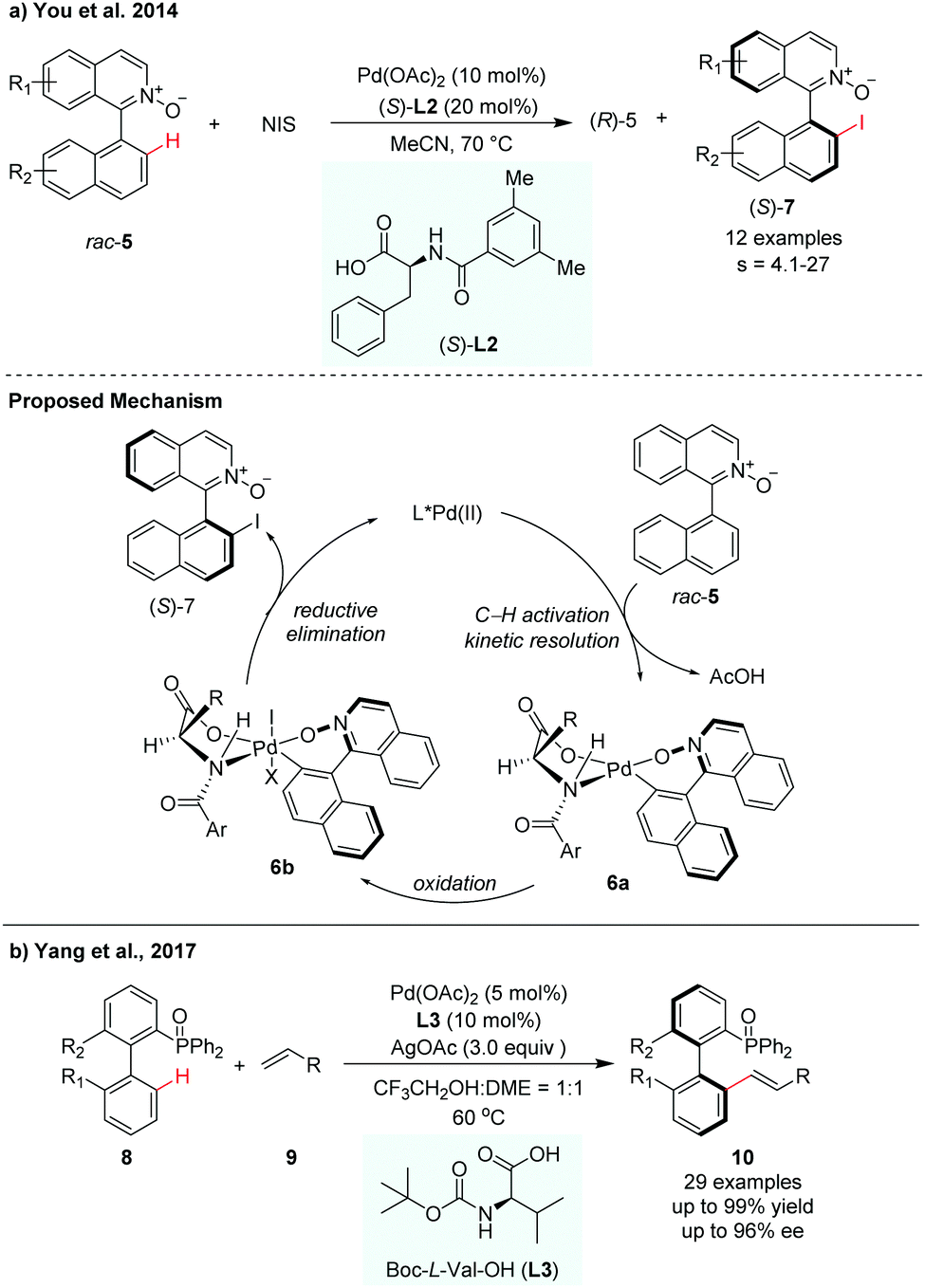 | ||
| Scheme 4 Pd-Catalysed atroposelective C–H functionalization using MPAA ligands (You et al., 2014;10 Yang, et al., 201711). | ||
In 2017, Yang and co-workers developed a P(O)R2-directed enantioselective C–H olefination for the synthesis of axially chiral phosphine–olefin biaryls (Scheme 4b).11 The reaction used Boc-L-Val-OH (L3) as the chiral ligand and Pd(OAc)2 as the catalyst. Electron-withdrawing and electron-donating substituents on the substrates were both well tolerated in this transformation. The racemic biaryl phosphine oxides underwent a dynamic kinetic resolution process in the presence of olefin, giving the corresponding chiral biaryl phosphine–olefin compounds with up to 99% yield and up to 96% ee (Scheme 4).
Indoles are a kind of important structural motif wildly occurring in natural products and bioactive molecules. Very recently, Gu et al. reported enantioselective synthesis of indole-based biaryl atropisomers via palladium-catalysed intramolecular C–H arylation (Scheme 5).12 A modified TADDOL-phosphoramidite was used as an efficient ligand for the intramolecular C–H cyclization reaction. The thermal stability of the indole-based atropisomers was also investigated.
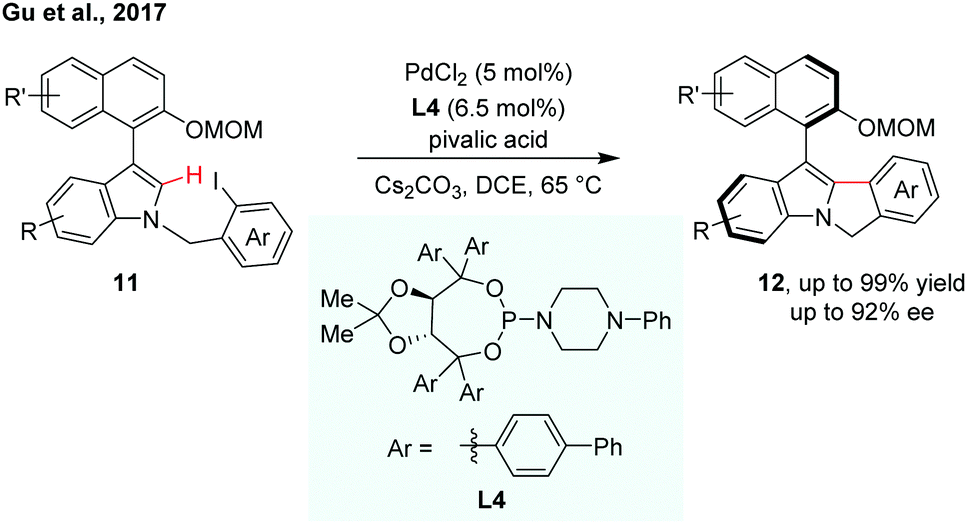 | ||
| Scheme 5 Palladium-catalyzed intramolecular dynamic kinetic C–H cyclization (Gu et al., 2017).12 | ||
Very recently, our group reported a Pd(II)-catalysed C–H olefination of quinoline-derived biaryls using chiral spiro phosphoric acids (SPAs) as chiral ligands (Scheme 6).13 (R)-STRIP (L5) was utilized as a superior ligand for the first time in atroposelective C–H functionalization and exhibited better stereocontrol than BINOL-derived phosphoric acids, probably due to the better steric interaction with the substrates. A wide range of quinoline-derived biaryls and olefins could be employed in this enantioselective transformation. In general, electron-deficient biaryls showed better reactivity than electron-rich ones. The computational results suggested that the enantioselectivity was dominated by the CMD type C–H bond cleavage step, which showed good agreement with the experimental result.
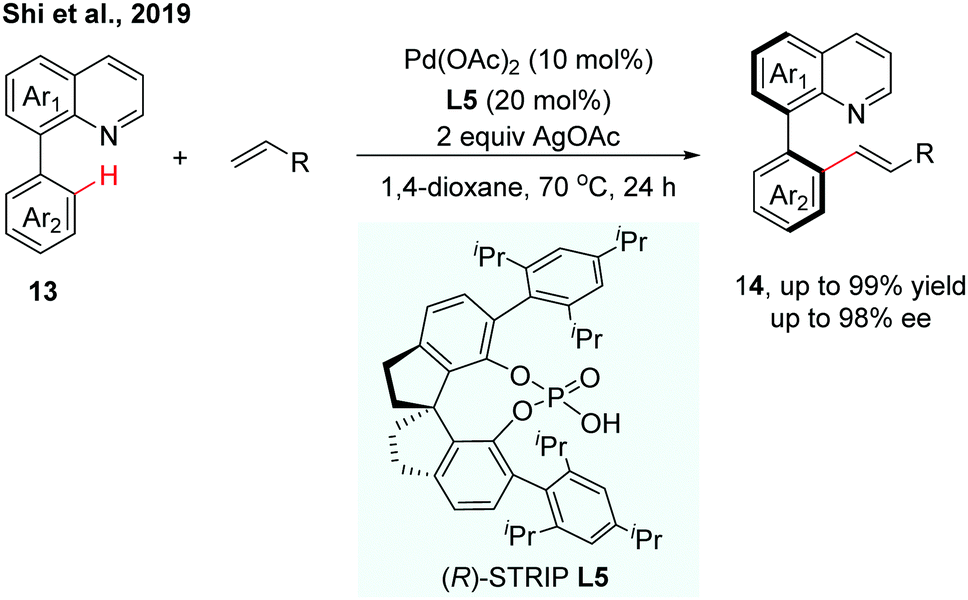 | ||
| Scheme 6 Pd-Catalyzed C–H olefination using chiral (R)-STRIP as a chiral ligand (Shi et al., 2019).13 | ||
2.2 Atroposelective C–H functionalization catalysed by a preformed chiral catalyst
In contrast to atroposelective C–H functionalization catalysed by chiral metal catalysts that were in situ generated via replacement of achiral ligands in the precatalysts with chiral ligands, asymmetric synthesis catalysed by a preformed chiral catalyst has also been realized, in the case that the achiral ligands are less easily displaced from the precatalysts. Although a preformed chiral catalyst needs extra step(s) to prepare, it could also obviate the competitive background reactions caused by achiral precatalysts.In 2012, Cramer and co-workers first introduced a series of C2-symmetric chiral [CpxRhIII] catalysts for asymmetric C–H functionalization, which were found to be highly promising catalysts for various enantioselective C–H functionalizations to create central chirality.14 In 2014, the You group elegantly adopted this type of chiral catalyst for the creation of axial chirality. They reported that the preformed chiral [CpxRhIII] catalyst (Cat1) was capable of enabling the enantioselective C–H alkenylation of isoquinoline-derived biaryls (Scheme 7).15 The formation of a five-membered cyclometalated intermediate enabled by C–H activation via dynamic kinetic resolution is challenging due to the need of two sterically hindered arenes to be coplanar. You and co-workers realized that the preformed chiral [CpxRhIII] catalyst could overcome this challenge and enhance both good reactivity and high selectivity. In this reaction, a variety of biaryl substrates and alkenes were tolerated in the presence of 5 mol% of catalyst, 5 mol% of (BzO)2 and a mixture of Cu(OAc)2 and Ag2CO3 as oxidants leading to the corresponding axially chiral biaryls in moderate to excellent yields and enantioselectivities. The potential utility of the resulting axially chiral olefinated products was further demonstrated by their application as N/olefin ligands in Rh-catalysed conjugate addition of phenylboronic acid with cyclohexanone.
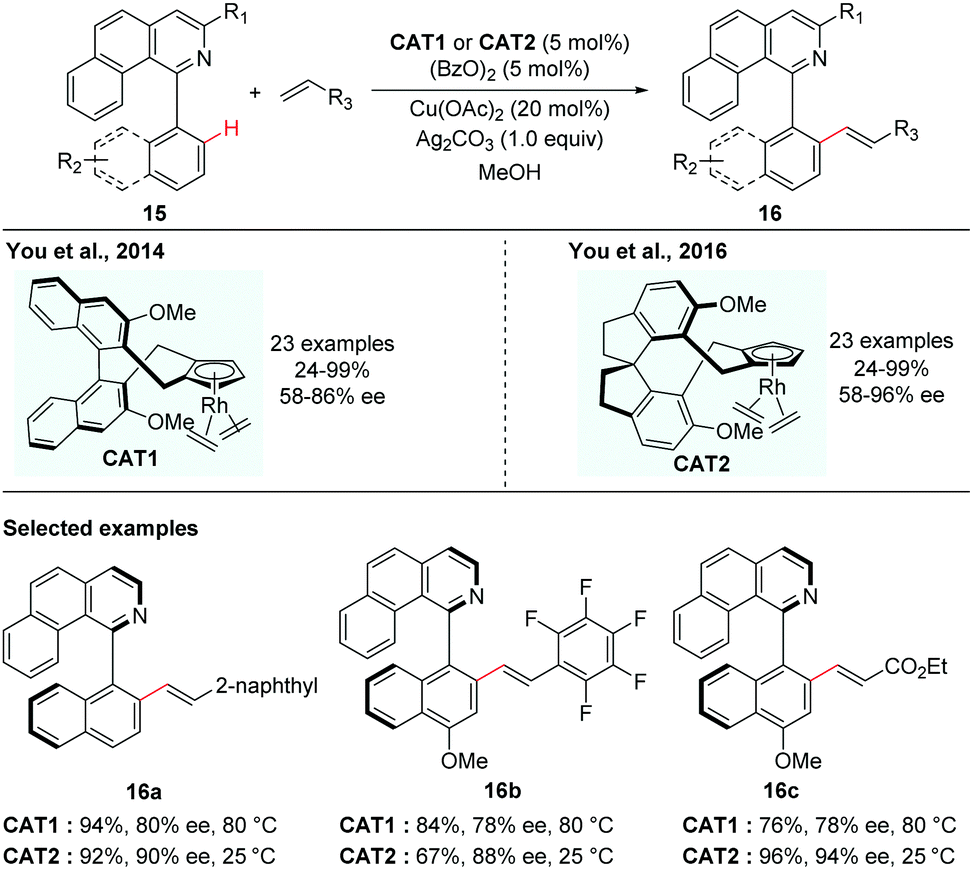 | ||
| Scheme 7 Rhodium-catalyzed enantioselective C–H alkenylation of biaryls (You et al.).15,16 | ||
You and co-workers took this innovative study one step further. In 2016, they disclosed a novel class of spirobiindane-derived Cp ligands (SCps) and found that the corresponding Rh-complexes were excellent chiral catalysts in asymmetric oxidative coupling of biaryls with alkenes (Scheme 7).16 The axially chiral biaryls were obtained in 19–97% yields with up to 96% ee. Compared to their previous studies using Cramer's C2-symmetric Cpx complexed Rh-catalyst, the newly developed [SCpRhIII] catalyst behaved as a superior catalyst and gave higher enantioselectivity.
2.3 Diastereoselective C–H functionalization directed by a chiral auxiliary
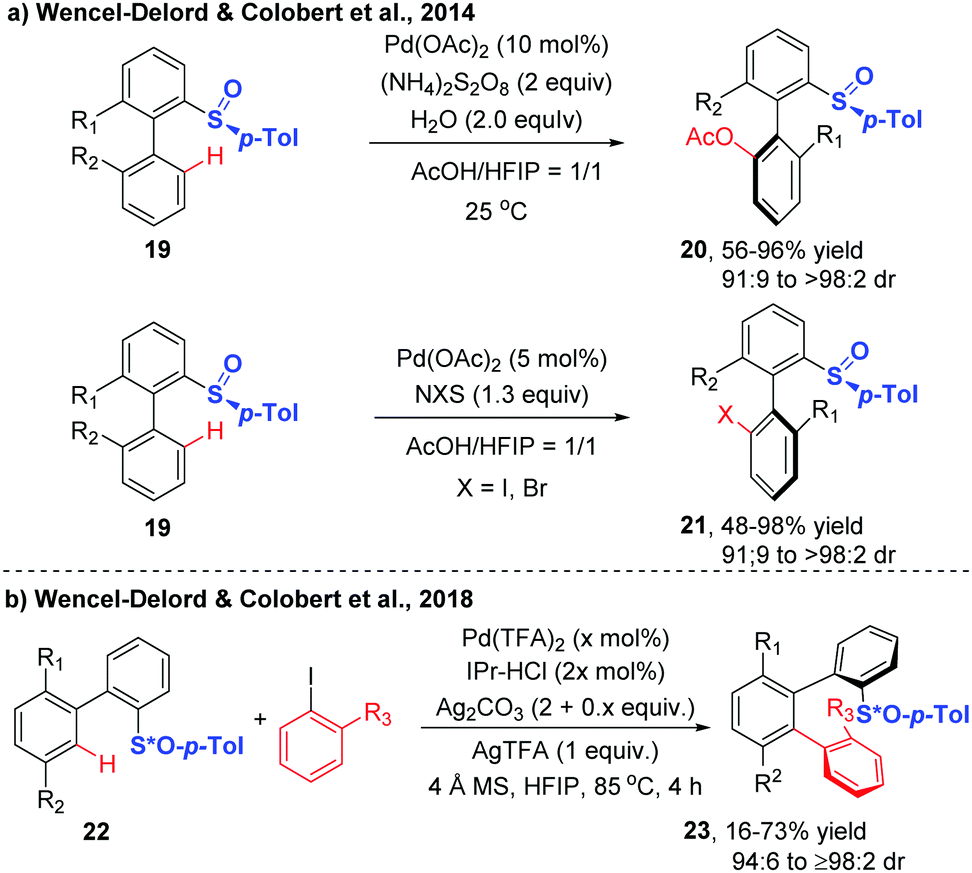
Diastereoselective C–H functionalization has also been realized as an efficient approach to access axially chiral biaryls. This strategy generally relys on the use of substrates containing chiral auxiliaries, such as sulfoxide, and menthyl phenylphosphate.17–21In 2013, the first atropodiastereoselective palladium(II)-catalyzed oxidative Heck olefination of biaryls using an enantiopure sulfoxide as both the directing group and chiral auxiliary was reported by the group of Colobert (Scheme 8).17 This reaction showed a perfect control of regioselectivity for the activation of one specific C–H bond. The observation of a total regioselectivity suggests that the soft Pd-catalyst was prone to coordinate with the softer sulfur atom (compared to oxygen), in which a privileged 6-membered palladacycle intermediate was formed. Later, they disclosed that the 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) solvent could drastically improve the efficiency and stereoselectivity of this C–H coupling.18 Mechanistic studies indicated that a hydrogen-bond was formed between the solvent and the substrate, thus accelerating the cleavage of the C–H bond and improving the stereochemical outcome of the reaction. The traceless nature of the chiral sulfoxide directing group was evidenced by a sulfoxide/lithium exchange and subsequent electrophilic trapping reaction sequence.
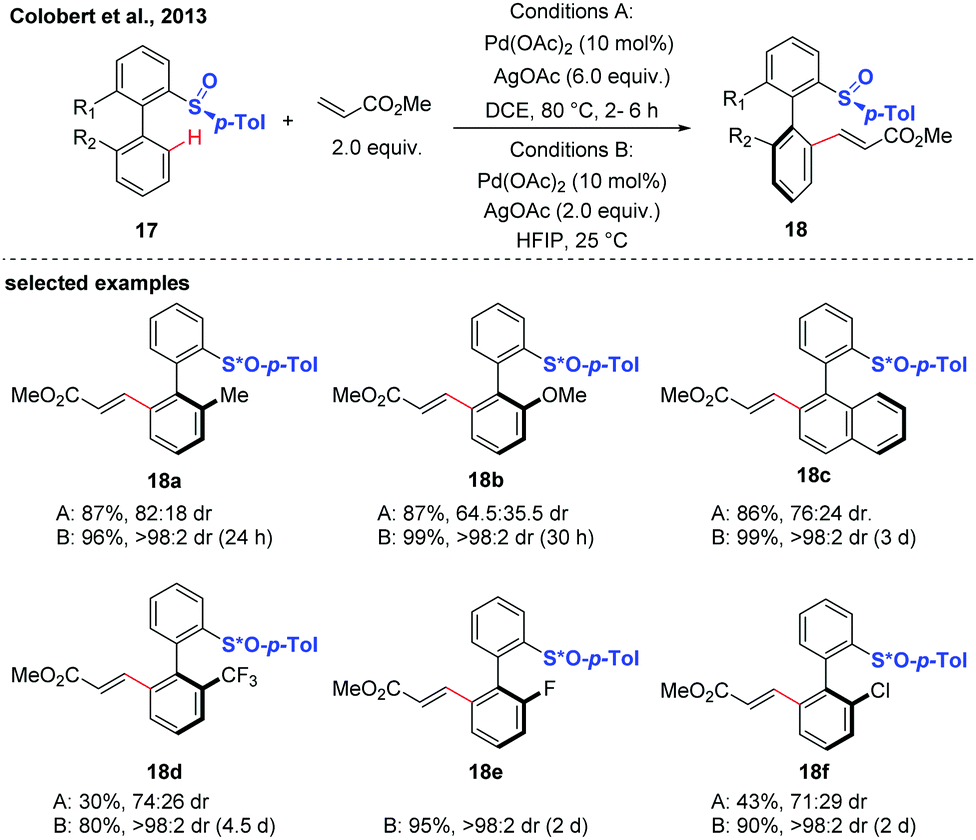 | ||
| Scheme 8 Pd-Catalyzed atroposelective sulfoxide-directed C–H olefination (Colobert et al., 2013).17 | ||
Wencel-Delord and Colobert further explored the generality of the atroposelective C–H functionalization through dynamic kinetic resolution directed by a chiral p-tolyl sulfoxide auxiliary (Scheme 9).19,20 A range of biaryls underwent Pd-catalyzed chiral sulfoxide-directed C–H acetoxylation smoothly at ambient temperature with AcOH using (NH4)2S2O8 as the stoichiometric oxidant, affording the acetoxylated biaryls in excellent diastereoselectivities. Replacing the above oxidant with N-iodosuccinimide (NIS), the catalytic system was shown to be capable of promoting the mild and highly diastereoselective C–H iodination. The iodinated biaryls were provided in high yields with diasteromeric ratios ranging from 91![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9 to > 98:2. Notably, the brominated product could also be obtained in the presence of NBS under otherwise identical conditions, albeit with a slight decrease in stereocontrol.
:9 to > 98:2. Notably, the brominated product could also be obtained in the presence of NBS under otherwise identical conditions, albeit with a slight decrease in stereocontrol.
 | ||
| Scheme 9 Chiral sulfoxide-directed diastereoselective C–H acetoxylation, halogenation and arylation (Wencel-Delord and Colobert et al.).19,20 | ||
Based on these innovative studies, Wencel-Delord and Colobert achieved the synthesis of chiral terphenyl scaffolds containing one or two chiral axes via Pd-catalysed atroposelective C–H arylation in 2018.20 Although significant advances have been achieved in chiral sulfoxide-directed C–H functionalization of biaryls, the atroposelective C–H arylation with ortho-substituted iodoarenes is extremely challenging, due to the high congestion of two sterically coupled partners as well as the difficulty in the realization of controlling two chiral inductions in one step. Using biaryls bearing a stereogenic sulfoxide moiety as substrates and ortho-substituted aryl iodides as coupling partners, the arylation reaction occurred in the presence of Pd(TFA)2, 1,3-bis(2,6-diisopropylphenyl)imidazolium chloride (IPr-HCl), Ag2CO3, AgTFA and 4 Å MS, at 85 °C in HFIP, delivering the desired products with high diastereoselectivity (Scheme 9b).
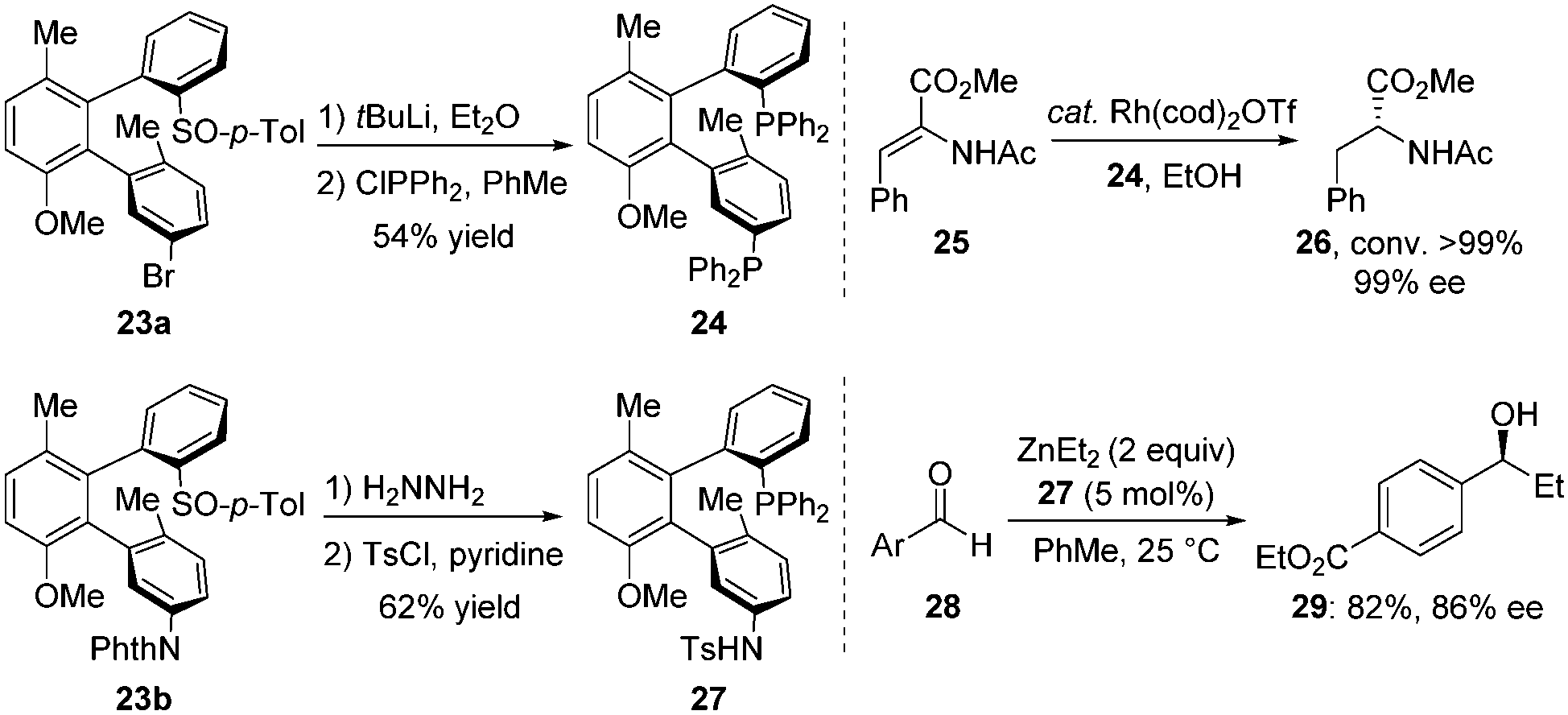
These terphenyls with two chiral axes exhibited a new tridimensional structure and thus had potential as unique chiral bidentate ligands. For example, treatment of 23a with tert-BuLi followed by PPh2Cl led to the corresponding diphosphine ligand BiaxPhos 24 in 54% yield. BiaxPhos 24 was applied in the enantioselective rhodium-catalysed hydrogenation of methyl (Z)-α-acetamidocinnamate (25), giving 26 with excellent yield and enantioselectivity. In another case, the S/N-Biax ligand 27 was synthesized in two steps from 23b, which showed good efficiency in asymmetric 1,2-addition of Et2Zn to aldehyde (Scheme 10).
 | ||
| Scheme 10 Synthesis and application of BiaxPhos and S/N-Biax ligands. | ||
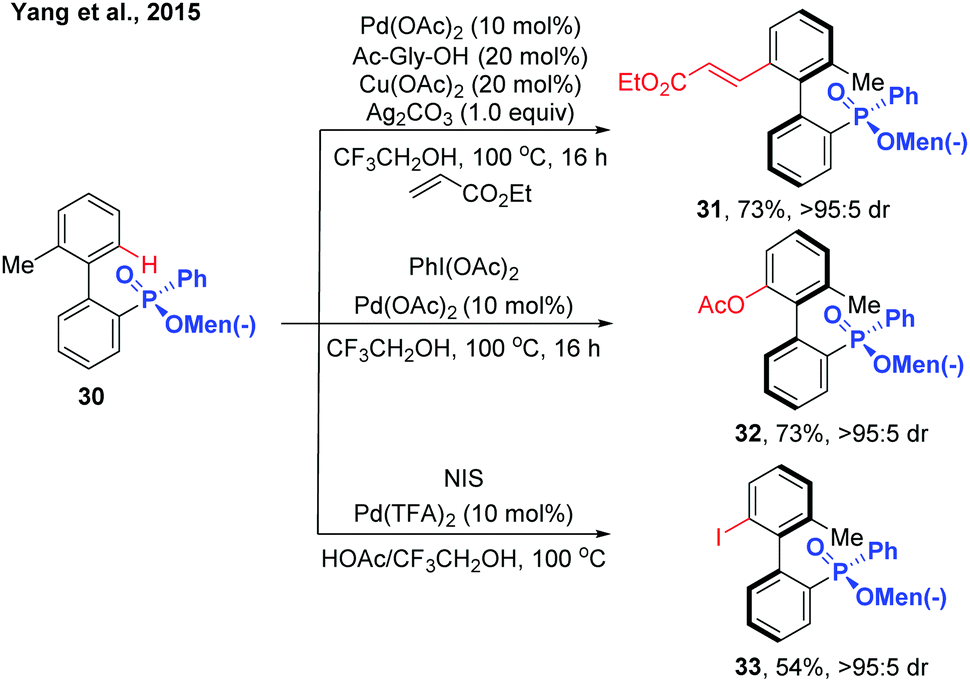
In 2015, the Yang group reported the synthesis of chiral biaryl phosphates via Pd(II)-catalysed asymmetric C–H alkenylation using an enantiopure menthylphenylphate group as the directing group (Scheme 11).21 Exploring Pd(OAc)2 and N-acyl glycine as the catalyst system, various axially chiral phosphine oxide compounds were obtained in excellent diastereomeric ratios with moderate to good yields. The asymmetric C–H functionalization can also be extended to acetoxylation and iodization using the same substrates.
 | ||
| Scheme 11 Pd(II)-Catalysed menthylphenylphate-directed C–H activation/dynamic kinetic resolution for the synthesis of chiral biaryl phosphates (Yang et al., 2015).21 | ||
2.4 Atroposelective C–H functionalization enabled by a transient chiral auxiliary
Over the past few decades, a variety of functionalities have been employed as directing groups for site-selective C–H activations.22 Recently, a transient directing group strategy has emerged as an efficient and powerful tool to functionalize the inert C–H bond for the obviation of the steps for installation/removal of the directing group.23 A breakthrough in this field was made by Yu and co-workers, who developed a transient chiral auxiliary approach for the creation of point chirality.24 Inspired by the elegance of this strategy, our group achieved several examples of Pd(II)-catalysed atroposelective C–H functionalization for the synthesis of axially chiral biaryls. Compared to the chiral ligand or chiral auxiliary enabled catalytic asymmetric C–H functionalization, as illustrated in Sections 2.1 and 2.3, this transient directing group could reversibly link with the substrate to form a better σ-donor motif, thus playing a dual role as a catalytic directing group and chiral ligand. With commercially available tert-Leucine (Tle) as a transient chiral auxiliary, we first achieved the atroposelective C–H olefination of biaryl aldehydes via a Pd(II)/Pd(0) catalytic cycle, affording the desired olefinated biaryls in good yields (up to 98%) with excellent enantioselectivities (95 to >99% ee) (Scheme 12a).25 The reaction was proposed to form the imines 35 by the dehydration of biaryl aldehydes with the chiral amino acids. Owning to steric factors, one of the imine diastereomers preferentially underwent C–H palladation to afford an axially stereo-enriched biaryl palladacycle intermediate 35c. The resulting seven-membered palladacycle underwent a Heck-type reaction with alkene to form intermediate 35d followed by hydrolysis to give the desired product 36 with high levels of enantioselectivities. The released Pd(0) species could be reoxidized back to catalytically active Pd(II) by BQ and O2.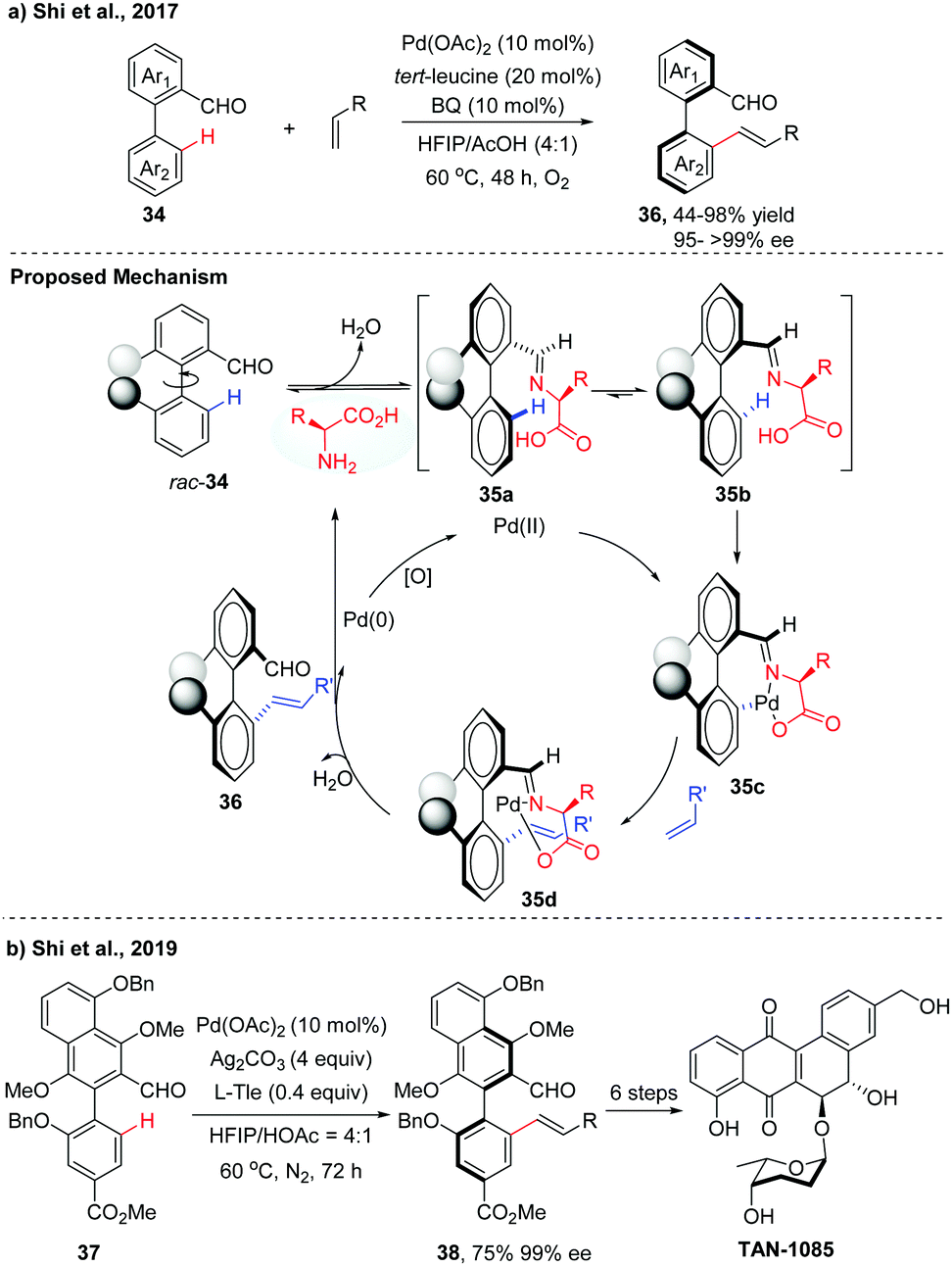 | ||
| Scheme 12 Pd-Catalyzed atroposelective C–H olefination of biaryl aldehydes using Tle as a transient chiral auxiliary (Shi et al., 2017, 2019).25,26 | ||
The potential utility of this method was demonstrated by the asymmetric total synthesis of TAN-1085. The key to the success of this total synthesis relies on the construction of the axially chiral biaryl 38 with high levels of enantiopurity via Pd-catalyzed asymmetric C–H olefination (Scheme 12b).26 Poor yield and unsatisfactory ee value were obtained using our previously reported conditions, due to the electron-rich properties of the biaryl aldehyde 37. After fine tuning of the reaction parameters, olefinated product 38 was finally obtained in 75% isolated yield with 99% ee in a 1.2 gram scale.
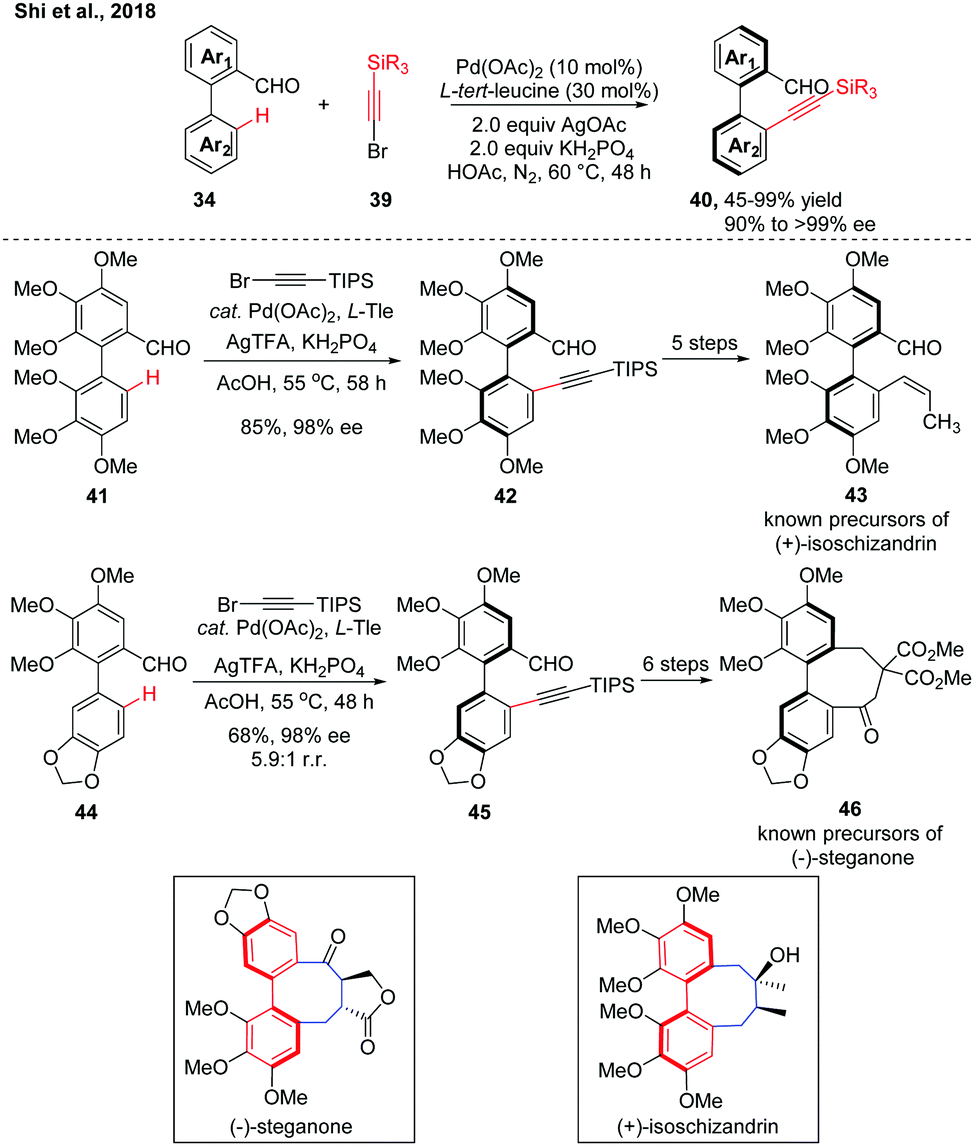
In 2018, we further adopted this strategy to Pd(II)-catalysed atroposelective C–H alkynylation. A variety of alkynylated biaryls were obtained in good to excellent yields with excellent enantioselectivities (Scheme 13).27 The potassium dihydric phosphate is a crucial additive for reaction activity, which may serve as a buffer to balance the pH value of the reaction system. Compared to the C–H olefination, this transformation may proceed using a PdII/PdIV catalytic cycle. This Pd(II)-catalyzed atroposelective C–H alkylation reaction has also found applications in the gram-scale formal syntheses of (+)-isoschizandrin and (+)-steganone. After optimization of the reaction conditions, the racemic precursors 41 and 44 were subjected to the slightly modified C–H alkynylation conditions, giving the desired products 42 and 45, respectively, in good yields and excellent enantioselecivities in gram-scale. Both intermediates were converted to known precursors of the corresponding natural products in 5–6 steps, thus completing their formal syntheses.
 | ||
| Scheme 13 Pd-Catalyzed atroposelective C–H alkynylation and its synthetic applications (Shi et al., 2018).26 | ||
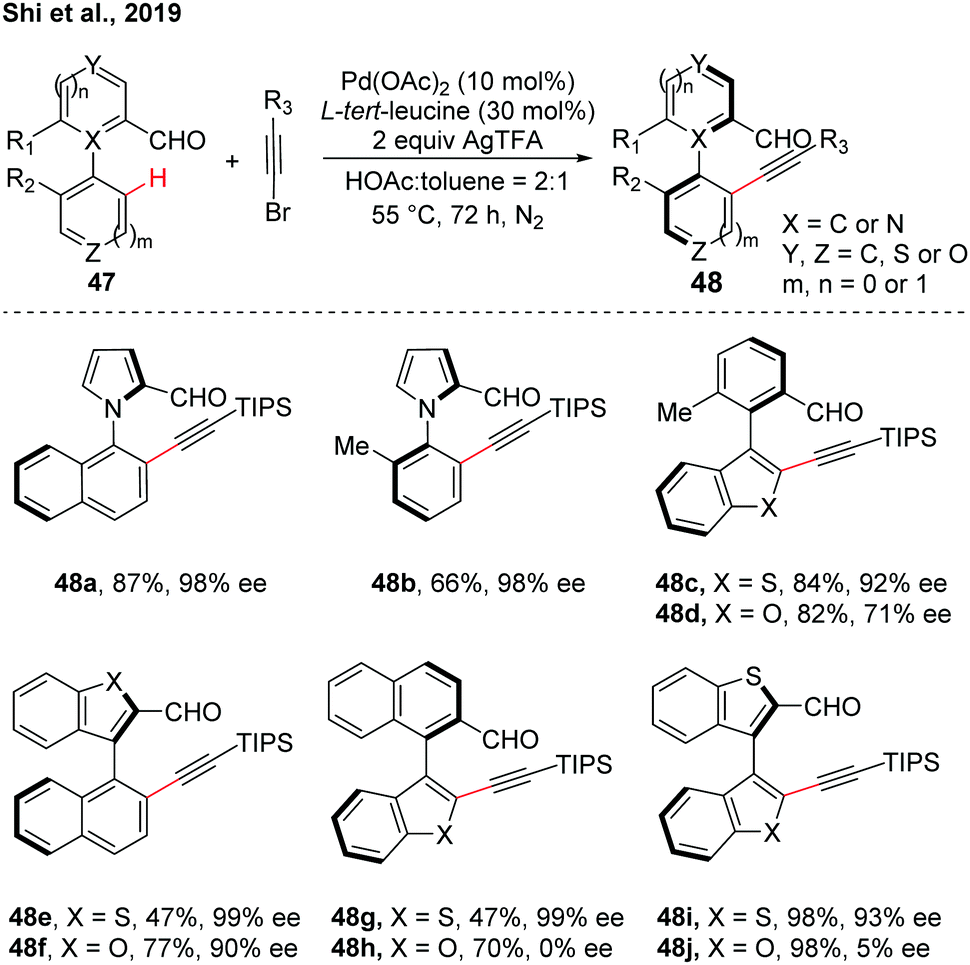
Despite the good investigations of the synthesis of the hexatomic atropisomer, the asymmetric construction of the five-membered atropisomers remains a considerable challenge due to the nature of their lower rotational barriers resulting from the relatively long distances between adjacent substituents ortho to the rotation axis.28 By using the transient chiral auxiliary strategy, a rare example of Pd-catalyzed asymmetric C–H alkynylation for the synthesis of five-membered heteroatropisomers was reported by our group (Scheme 14).29 Various five-membered heteroarenes such as pyrroles, thiophenes, benzothiophenes, and benzofurans were compatible with the reaction conditions. These atropisomers featuring one or even two five-membered rings connected through C–N or C–C bonds were obtained (up to 98% yield and up to >99% ee). Lower ee values were obtained in biaryls containing benzofuran moieties than benzofuran moieties, which was demonstrated by the calculation of the difference of the rotational barrier and half-life.
 | ||
| Scheme 14 Pd-Catalyzed enantioselective C–H alkynylation for the synthesis of atropisomers featuring pentatomic heteroaryls (Shi et al., 2019).29 | ||
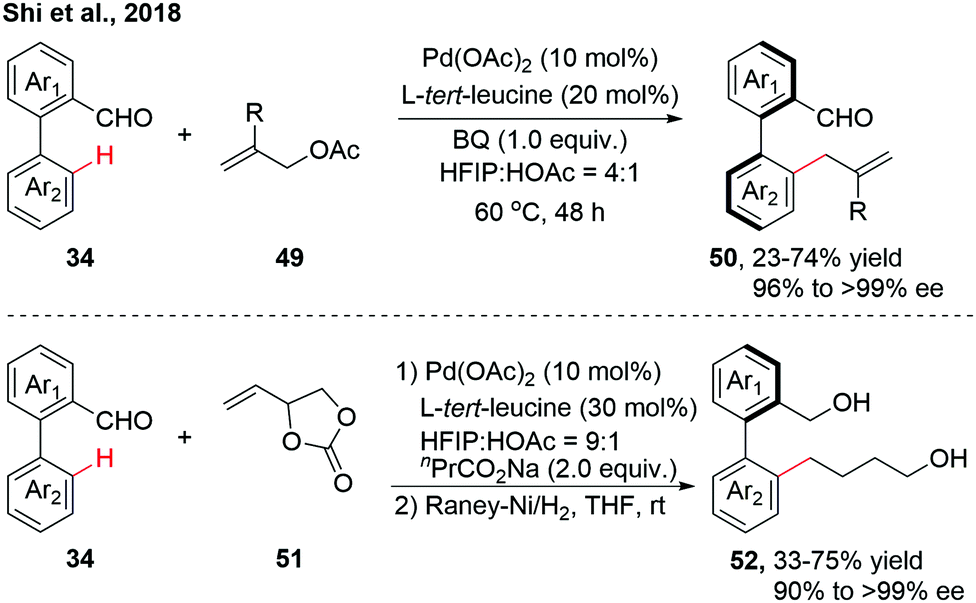
As an extension to Pd(II)-catalyzed asymmetric C–H activation reactions using a catalytic transient chiral auxiliary, our group achieved a further advance in accessing more structurally diverse axially chiral biaryls via atroposelective C–H allylation (Scheme 15).30 A variety of racemic biaryl aldehydes could be allylated with Morita–Baylis–Hillman (MBH) acetates, affording the enantioenriched biaryls through β-O elimination. 4-Vinyl-1,3-dioxolan-2-one, as a versatile allylic surrogate, was also compatible in this allylation reaction, giving a mixture of Z/E isomers instead. The mixture of allylated products was then treated with RANEY®-Ni to afford the reduced biaryls with excellent enantioselectivity.
 | ||
| Scheme 15 Pd-Catalyzed atroposelective C–H allylation (Shi et al., 2018).30 | ||
3. The creation of biaryl axis via C–H activation/asymmetric coupling
Over the past few years, transition metal-catalyzed direct C–H arylation of (hetero)arenes has been among the most attractive strategies for the construction of biaryl motifs.31 Despite the significant advances, the synthesis of axially chiral biaryls via enantioselective C–H activation remains a daunting challenge. Such an atroposelective biaryl coupling was first reported in 2012 by Yamaguchi and Itami (Scheme 16a).32 The coupling reaction proceeded between substituted thiophenes and sterically hindered naphthylboronic acids in the presence of Pd(OAc)2/biox (L6), giving the desired biaryl in 27% yield with 72% ee (Scheme 16a). A reactivity–selectivity dilemma was observed, in which the ee value increased at the expense of the reaction yield. In the following year, the same group described a second-generation catalytic system using a chiral Pd(II)-sox (sulfoxide-oxazoline) complex and iron-phthalocyanine (FePc) as a co-catalyst (Scheme 16b).33 Although this dual catalyst system could furnish the desired product in increased yield, the enantioselectivity remains to be improved. These results described above highlight the difficulty in controlling the axial chirality via a direct C–H activation/asymmetric coupling strategy. A major hurdle might be the fact that sterically demanding substituents on both coupling partners are needed to maintain the chirality of the resulting biaryls; however, the reactivity significantly decreased with those sterically hindered coupling partners.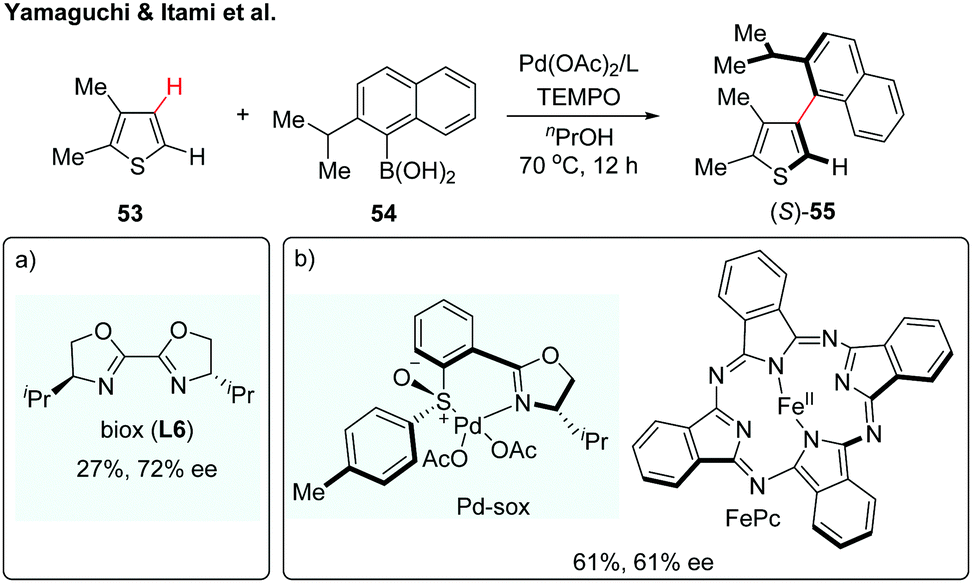 | ||
| Scheme 16 Enantioselective C–H coupling of thiophene with arylboronic acid (Yamaguchi and Itami et al.).32,33 | ||
Recently, the Waldmann and Antonchick group developed a general method for gram scale synthesis of a class of novel chiral JasCp ligands, which could be used for Rh(III)-catalyzed enantioselective C–H activation reactions.34 The chiral [JasCpRhIII] complexes (CAT3) have proven to be a good catalyst in the enantioselective C–H arylation of benzamides with diazonaphthoquinones (Scheme 17). The reaction proceeded under mild conditions and a broad range of axially chiral biaryls were formed in 37–93% yield and 79–90% ee.
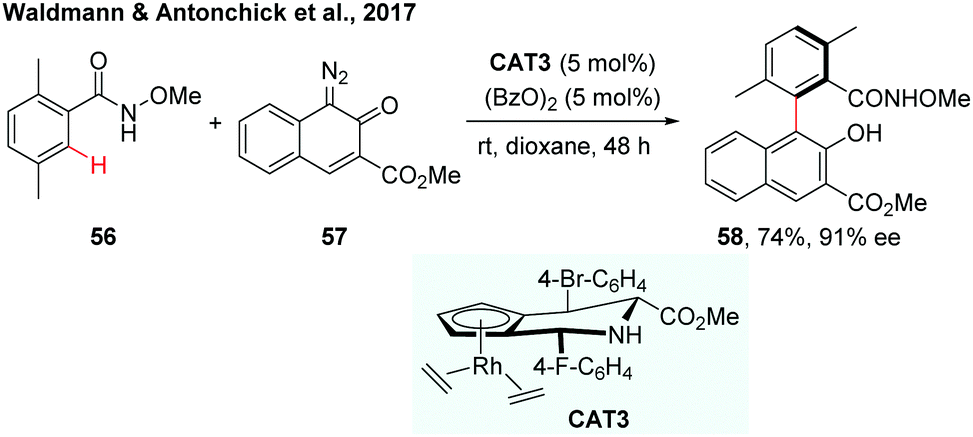 | ||
| Scheme 17 Synthesis of axially chiral biaryls by Rh-catalysed enantioselective C–H arylation (Waldmann and Antonchick et al., 2017).34 | ||
Very recently, Cramer and co-workers realized an enantioselective C–H arylation of phosphine oxides with o-quinone diazides employing a chiral iridium(III) complex (CAT4) and phthaloyl tert-leucine as a co-catalyst (Scheme 18).35 The configuration of the chiral amino acid was proved to be essential for achieving satisfactory enantioselectivity. The reaction of diazo reactants bearing sterically demanding substituents in the ortho position, afforded the stable axial and P-chiral biaryls in moderate to excellent yields (59–96%) and high enantioselectivities (up to 99% ee) (Scheme 18a). The synthesis of purely axially chiral phosphine oxides has also been investigated using tris-3,5-dimethoxyphenylphosphine oxide as a substrate (Scheme 18b). Enantiospecific reduction of the chiral phosphine oxides gave the synthetically valuable monodentate biaryl phosphorus compounds that could be of great potential as ligands in other types of asymmetric reactions.
 | ||
| Scheme 18 CpxIrIII-Catalyzed enantioselective C–H arylations of phosphine oxides (Cramer et al., 2018).35 | ||
In 2018, the asymmetric synthesis of axially chiral dibenzazepinones via Pd(0)-catalyzed atropo-enantioselective intramolecular C–H arylation of achiral amides using TADDOL-based phosphoramidite (L7) as a ligand was achieved by Cramer and co-workers (Scheme 19).36 The acid additive is essential for achieving both excellent reactivity and enantioselectivity. A board range of axially chiral dibenzazepinones were obtained in high yields and impressive enantioselectivities. Computational investigation provides a reliable prediction of the racemization barriers of the chiral axis, which also suggests that configurationally stable eight-membered palladacycles were formed through an enantio-determining CMD step during the C–H functionalization process.
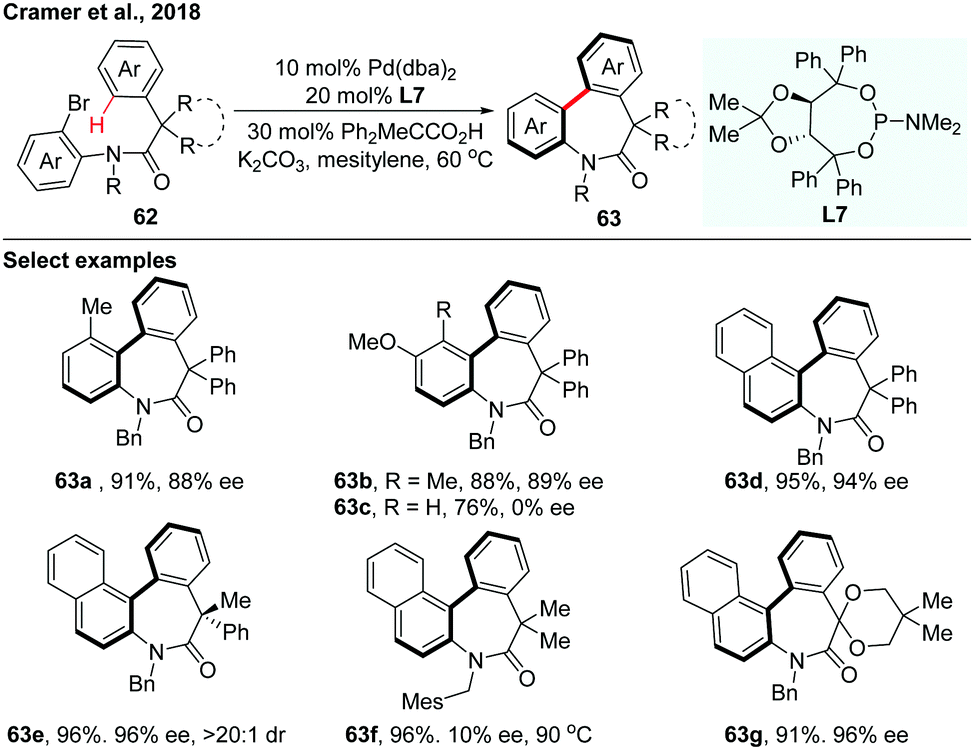 | ||
| Scheme 19 Pd(0)-Catalyzed atropo-enantioselective intramolecular C–H arylation of achiral amides (Cramer et al., 2018).36 | ||
4. Conclusions and outlook
Over the past few years, transition metal-catalysed asymmetric C–H functionalization has gradually matured into a powerful and straightforward tool for the construction of various optically active building blocks. In this review, we have summarized recent advances in the synthesis of axially chiral biaryl compounds via transition metal-catalysed asymmetric C–H functionalization.38 Although a range of biaryl backbones bearing axial chirality have become accessible via several strategies, this area is still in its infancy. The discovery of new types of chiral ligands or catalysts is still highly demanded. In addition, current studies mainly focused on the use of noble transition metals, such as Pd, Rh, and Ir. The exploration of using earth-abundant, inexpensive 3d transition metals in atroposelective C–H activation would be another promising research area.37 We hope that this review will provide some insights in this emerging field and inspire the discovery of more innovative strategies for the synthesis of axially chiral biaryls.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the NSFC (No. 21772170, 21572201, and 21422206), China Postdoctoral Science Foundation (2019M650135 and 2017M621906), the National Basic Research Program of China (No. 2015CB856600), and the Fundamental Research Funds for the Central Universities (No. 2018XZZX001-02) and Zhejiang Provincial NSFC (No. LR17B020001) is gratefully acknowledged.Notes and references
- (a) R. Adams and H. C. Yuan, Chem. Rev., 1933, 12, 261 CrossRef CAS; (b) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562 RSC.
- (a) J. Chang, J. Reiner and J. Xie, Chem. Rev., 2005, 105, 4581 CrossRef CAS PubMed; (b) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS PubMed; (c) J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398 CrossRef CAS PubMed; (d) S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505 CrossRef CAS PubMed.
- (a) W. Fu and W. Tang, ACS Catal., 2016, 6, 4814 CrossRef CAS; (b) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed; (c) Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, Wiley-VCH, Weinheim, 2011 Search PubMed; (d) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS; (e) R. Noyori and H. Takaya, Acc. Chem. Res., 1990, 23, 345 CrossRef CAS.
- (a) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155 CrossRef CAS PubMed; (b) M. Berthod, G. Mignani, G. Woodward and M. Lemaire, Chem. Rev., 2005, 105, 1801 CrossRef CAS PubMed; (c) J. M. Brunel, Chem. Rev., 2007, 107, PR1 CrossRef CAS; (d) J. Yu, F. Shi and L.-Z. Gong, Acc. Chem. Res., 2011, 44, 1156 CrossRef CAS PubMed; (e) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047 CrossRef CAS PubMed.
- (a) O. Baudoin, Eur. J. Org. Chem., 2005, 4223 CrossRef CAS; (b) G. Bringmann, A. J. Price Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384 CrossRef CAS PubMed; (c) K. Tanaka, Chem. – Asian J., 2009, 4, 508 CrossRef CAS PubMed; (d) G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615 CrossRef CAS PubMed; (e) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418 RSC; (f) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239 CrossRef CAS PubMed; (g) Y.-B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534 CrossRef CAS PubMed; (h) A. Link and C. Sparr, Chem. Soc. Rev., 2018, 47, 3804 RSC; (i) T. W. Wallace, Org. Biomol. Chem., 2006, 4, 3197 RSC; (j) P. Loxq, E. Manoury, R. Poli, E. Deydier and A. Labande, Coord. Chem. Rev., 2016, 308, 131 CrossRef CAS; (k) B. Zilate, A. Castrogiovanni and C. Sparr, ACS Catal., 2018, 8, 2981 CrossRef CAS.
- For reviews on asymmetric C–H functionalization, see: (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242 RSC; (b) L. Yang and H. Huang, Catal. Sci. Technol., 2012, 2, 1099 RSC; (c) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed; (d) J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2013, 19, 14010 CrossRef CAS PubMed; (e) C. Zheng and S.-L. You, RSC Adv., 2014, 4, 6173 RSC; (f) J. Pedroni and N. Cramer, Chem. Commun., 2015, 51, 17647 RSC; (g) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908 CrossRef CAS PubMed; (h) D.-W. Gao, Q. Gu, C. Zheng and S.-L. You, Acc. Chem. Res., 2017, 50, 351 CrossRef CAS PubMed; (i) Y. Qin, L. Zhu and S. Luo, Chem. Rev., 2017, 117, 9433 CrossRef CAS PubMed; (j) Y.-N. Ma, S.-X. Li and S.-D. Yang, Acc. Chem. Res., 2017, 50, 1480 CrossRef CAS PubMed; (k) T. G. Saint-Denis, R.-Y. Zhu, G. Chen, Q.-F. Wu and J.-Q. Yu, Science, 2018, 359, eaao4798 CrossRef PubMed; (l) Q. Zhang and B.-F. Shi, Chin. J. Chem., 2019, 37, 647 CAS.
- (a) J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251 CrossRef CAS PubMed; (b) R. Miyaji, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 6766 CrossRef CAS PubMed; (c) R. Miyaji, K. Asano and S. Matsubara, Chem. – Eur. J., 2017, 23, 9996 CrossRef CAS PubMed.
- F. Kakiuchi, P. L. Gendre, A. Yamada, H. Ohtaki and S. Murai, Tetrahedron: Asymmetry, 2000, 11, 2647 CrossRef CAS.
- (a) B.-F. Shi, N. Maugel, Y.-H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882 CrossRef CAS PubMed; (b) B.-F. Shi, Y.-H. Zhang, J. K. Lam, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 460 CrossRef CAS PubMed; (c) M. Wasa, K. M. Engle, D. W. Lin, E. J. Yoo and J.-Q. Yu, J. Am. Chem. Soc., 2011, 133, 19598 CrossRef CAS PubMed; (d) D. G. Musaev, A. Kaledin, B.-F. Shi and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 1690 CrossRef CAS PubMed.
- D.-W. Gao, Q. Gu and S.-L. You, ACS Catal., 2014, 4, 2741 CrossRef CAS.
- S.-X. Li, Y. N. Ma and S.-D. Yang, Org. Lett., 2017, 19, 1842 CrossRef CAS PubMed.
- C. He, M. Hou, Z. Zhu and Z. Gu, ACS Catal., 2017, 7, 5316 CrossRef CAS.
- J. Luo, T. Zhang, L. Wang, G. Liao, Q.-J. Yao, Y.-J. Wu, B.-B. Zhan, Y. Lan, X.-F. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2019, 58, 6708 CrossRef CAS PubMed.
- (a) B. Ye and N. Cramer, Science, 2012, 338, 504 CrossRef CAS PubMed; (b) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308 CrossRef CAS PubMed.
- J. Zheng and S.-L. You, Angew. Chem., Int. Ed., 2014, 53, 13244 CrossRef CAS PubMed.
- J. Zheng, W.-J. Cui, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2016, 138, 5242 CrossRef CAS PubMed.
- T. Wesch, F. R. Leroux and F. Colobert, Adv. Synth. Catal., 2013, 355, 2139 CrossRef CAS.
- Q. Dherbassy, G. Schwertz, M. Chess, C. K. Hazra, J. Wencel-Delord and F. Colobert, Chem. – Eur. J., 2016, 22, 1735 CrossRef CAS PubMed.
- C. K. Hazra, Q. Dherbassy, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2014, 53, 13871 CrossRef CAS PubMed.
- (a) Q. Dherbass, J.-P. Diukic, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2018, 57, 4668 CrossRef PubMed; (b) Q. Dherbass, J. Wencel-Delord and F. Colobert, Tetrahedron, 2018, 74, 6205 CrossRef.
- Y.-N. Ma, H.-Y. Zhang and S.-D. Yang, Org. Lett., 2015, 17, 2034 CrossRef CAS PubMed.
- (a) Z. Chen, B. Wang, J. Zhang, W. Yu, Z. Liu and Y. Zhang, Org. Chem. Front., 2015, 2, 1107 RSC; (b) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maesa and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603 RSC.
- (a) D.-S. Kim, W.-J. Park and C.-H. Jun, Chem. Rev., 2017, 117, 8977 CrossRef CAS PubMed; (b) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199 CrossRef CAS; (c) S. S. John-Campbell and J. A. Bull, Org. Biomol. Chem., 2018, 16, 4582 RSC; (d) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456 RSC; (e) H. Sun, N. Guimond and Y. Huang, Org. Biomol. Chem., 2016, 14, 8389 RSC.
- (a) F.-L. Zhang, K. Hong, T.-J. Li, H. Park and J.-Q. Yu, Science, 2016, 351, 252 CrossRef CAS PubMed; (b) H. Park, P. Verma, K. Hong and J.-Q. Yu, Nat. Chem., 2018, 10, 755 CrossRef CAS PubMed.
- Q.-J. Yao, S. Zhang, B.-B. Zhan and B.-F. Shi, Angew. Chem., Int. Ed., 2017, 56, 6617 CrossRef CAS PubMed.
- J. Fan, Q.-J. Yao, Y.-H. Liu, G. Liao, S. Zhang and B.-F. Shi, Org. Lett., 2019, 21, 3352 CrossRef CAS PubMed.
- G. Liao, Q.-J. Yao, Z.-Z. Zhang, Y.-J. Wu, D.-Y. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 3661 CrossRef CAS PubMed.
- (a) S. Zhang, G. Liao and B.-F. Shi, Chin. J. Org. Chem., 2019, 39, 1522 CrossRef; (b) D. Bonne and J. Rodriguez, Eur. J. Org. Chem., 2018, 2417 CrossRef CAS; (c) D. Bonne and J. Rodriguez, Chem. Commun., 2017, 53, 12385 RSC.
- S. Zhang, Q.-J. Yao, G. Liao, X. Li, H. Li, H.-M. Chen, X. Hong and B.-F. Shi, ACS Catal., 2019, 9, 1956 CrossRef CAS.
- G. Liao, B. Li, H.-M. Chen, Q.-J. Yao, Y.-N. Xia, J. Luo and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 17151 CrossRef CAS PubMed.
- For selected reviews, see: (a) D. Alberico, M. E. Scott and M. Lautens, Chem. Rev., 2007, 107, 174 CrossRef CAS PubMed; (b) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792 CrossRef CAS PubMed; (c) J. A. Ashenhurst, Chem. Soc. Rev., 2010, 39, 540 RSC; (d) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (e) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (f) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (g) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Chem. Rev., 2015, 115, 12138 CrossRef CAS PubMed; (h) Y. Yang, J. Lan and J. You, Chem. Rev., 2017, 117, 8787 CrossRef CAS PubMed.
- K. Yamaguchi, J. Yamaguchi, A. Studer and K. Itami, Chem. Sci., 2012, 3, 2165 RSC.
- K. Yamaguchi, H. Kondo, J. Yamaguchi and K. Itami, Chem. Sci., 2013, 4, 3753 RSC.
- Z.-J. Jia, C. Merten, R. Gontla, C. G. Daniliuc, A. P. Antonchick and H. Waldmann, Angew. Chem., Int. Ed., 2017, 56, 2429 CrossRef CAS PubMed.
- Y.-S. Jang, Ł. Woźniak, J. Pedroni and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 12901 CrossRef CAS PubMed.
- C. G. Newton, E. Braconi, J. Kuziola, M. D. Wodrich and N. Cramer, Angew. Chem., Int. Ed., 2018, 57, 11040 CrossRef CAS PubMed.
- (a) J. Loup, U. Dhawa, F. Pesciaioli, J. Wencel-Delord and L. Ackermann, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201904214; (b) L. Woźniak and N. Cramer, Trends Chem. DOI:10.1016/j.trechm.2019.03.013; (c) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS PubMed.
- During the revision of this review, three new reports on synthesis of axially chiral biaryls via atroposelective C–H functionalization were disclosed, highlighting the increasing importance of this exciting field: (a) M. Tian, D. Bai, G. Zheng, J. Chang and X. Li, J. Am. Chem. Soc., 2019, 141, 9527 CrossRef CAS PubMed; (b) Q. Wang, Z.-J. Cai, C.-X. Liu, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2019, 141, 9504 CrossRef CAS PubMed; (c) G. Liao, H.-M. Chen, Y.-N. Xia, B. Li, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201906700.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |