
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpiropyran in nanoassemblies as a photosensitizer for photoswitchable ROS generation in living cells†
Jinkai
Ji
,
Xiao
Li
,
Tiantian
Wu
and
Fude
Feng
 *
*
Department of Polymer Science & Engineering, School of Chemistry & Chemical Engineering, Nanjing University, Nanjing, 210023, People’s Republic of China. E-mail: fengfd@nju.edu.cn
First published on 11th June 2018
Abstract
Reversibly controlled generation of singlet oxygen from photosensitizing nanosystems has the benefits of selective cell killing and controllable effect time, but is a challenging option for photodynamic therapies. We report a strategy for integrating photochromic spiropyrans into biocompatible cationic polymers, which involved assembling nucleic acids into functional nanoparticles without introducing additional photosensitizers and imaging agents. We found that spiropyran-containing nanoparticles have photoswitching properties for both fluorescence (with a quantum yield of up to 0.27) and singlet oxygen generation (with a quantum yield of up to 0.22) in aqueous solutions and cells, and demonstrated that spiropyrans in nanoassemblies featuring aggregation-induced enhanced photosensitization and emission could be potentially applied in photodynamic therapy studies on tumor cells.
Introduction
Photodynamic therapy (PDT) is a kind of minimally invasive clinical treatment that employs photosensitizers (PSs), light, and oxygen to produce cytotoxic reactive oxygen species (ROS), especially 1O2, and induce cell damage and tissue necrosis.1–3 PDT has promising prospects in cancer therapy because of its high efficiency, selectivity, microtrauma, and negligible drug resistance.4,5 In the past few decades, a large number of PSs, in the form of small molecules or nanostructures, have been designed as key components for PDT for singlet oxygen photosensitization.6However, one of the major limitations of PDT is the undesired detrimental photosensitivity of PSs if they are distributed everywhere out of the target region or if the PSs remain photoactive after the PDT treatment. For example, approved PDT photosensitizers such as Photofrin and Temoporfin 1 have crucial defects due to their long clearance times (1–2 months).7 It is also common that PS-modified nanomaterials maintain photoactivity over weeks.8 Protection against exposure to natural light needs to be considered when PSs such as widely used porphyrins and phthalocyanines are applied.6,9 To overcome the non-specificity and undesired long-lasting of photosensitization systems, improvement in the controllability of PS activation has become an attractive goal. An ideal solution will afford a remote “press button” that can repeatedly switch on and off the ROS generation by reversibly activating the PSs at any time, without drastically disrupting the physiological environment. Nano-delivery systems have been reported to control ROS generation via heat, ultrasound, and biochemical reactions, but by involving complicated components.10–13 Recently, diarylethenes have been developed as useful photoswitches to tune the activities of target PSs in close proximity to diarylethenes,14–17 but these face concerns with water solubility and stability, particularly when used in living biological systems.
In comparison to nonemissive diarylethenes, spiropyrans are photoswitchable between two isoforms that notably differ in fluorescence properties.18 The fluorescent isoform of spiropyrans in solution has a short life span when excited, which impedes the investigation of the role of the triplet excited state that may interact with triplet oxygen. The emergence of fatigue in spiropyrans over the course of the repeated photoswitching process has been presumed to be attributable to the generation of harmful ROS.19 This implies that an understanding of the activity of ROS is needed for the treatment of cells with spiropyran-containing photoactive materials. In the past few years, spiropyrans have been well studied as a crucial component in smart nanosystems (Table S1†) for drug delivery and cell imaging purposes, but there has been a lack of interest in their possible ROS generation and ROS-mediated consequences. To date, how to remotely control the level of ROS, intracellularly in particular, remains poorly understood.
Nanoassemblies with reversibly tunable optical properties have gained increasing attention.20–22 Herein, we couple spiropyrans with cationic polymers to assemble plasmid DNA into functional nanoparticles (Fig. 1). Spiropyran nanoparticles are designed to satisfy the following requirements: (1) to tune ROS generation in solution or inside the cells in a reversible light-controlled manner without the need for extra PS species, (2) to switch within a short time into a deactivated state that does not respond to natural light, and (3) to allow for cell imaging and photo-induced cell killing.
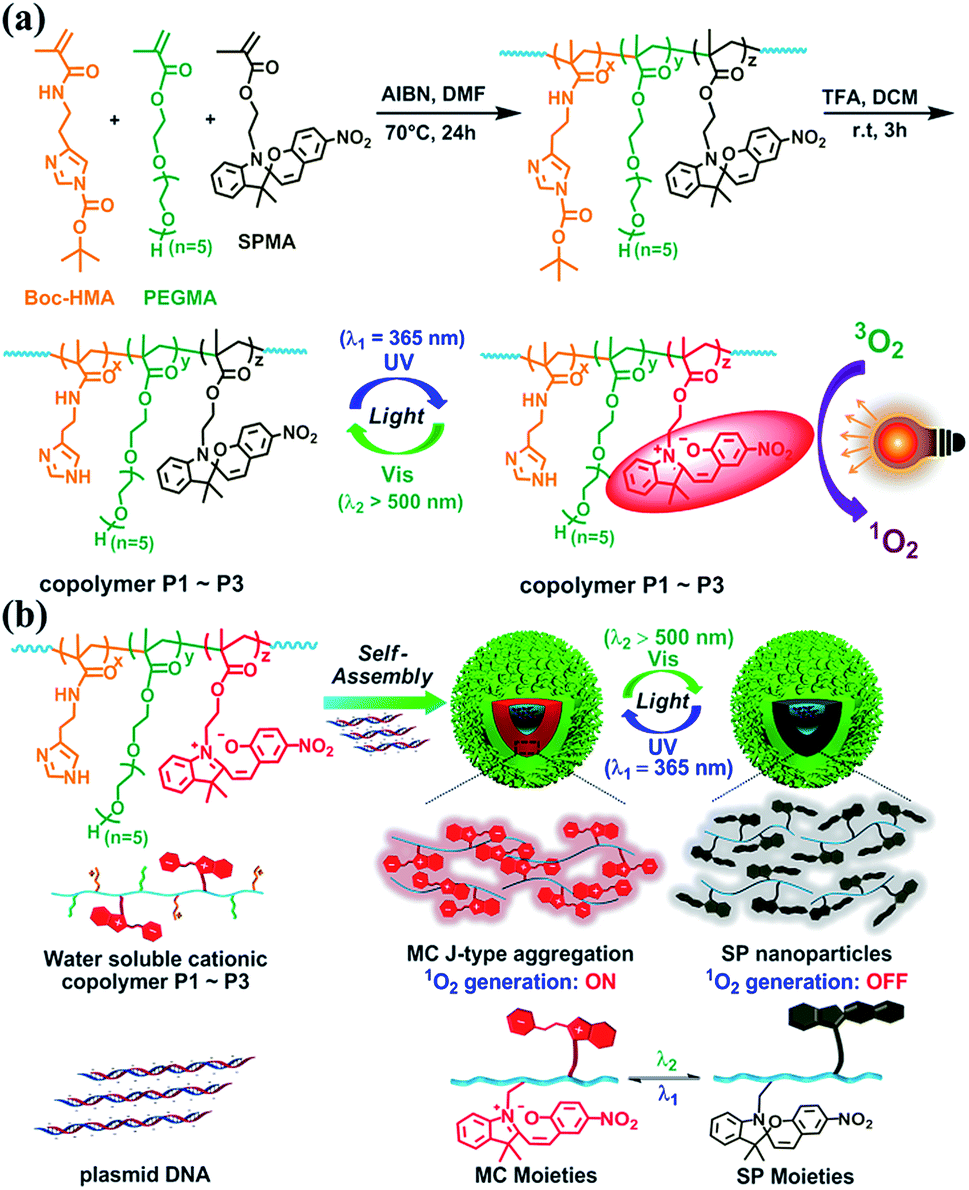 | ||
| Fig. 1 Schematic presentations of (a) the synthetic route of P1–P3, the photoswitching of SP and MC, and light-induced ROS generation and (b) the self-assembly of spiropyran-containing polymers with DNA and reversibly light-controlled ROS generation. Boc-HMA, PEGMA and SPMA represent 2-(4-tert-butyloxycarbonyl imidazolyl)ethyl methacrylamide, poly(ethylene glycol) methacrylate, and 1′-(2-methacryloxyethyl)-3′,3′-dimethyl-6-nitro-spiro-(2H-1-benzopyran-2′,2′-indoline), respectively. | ||
Results and discussion
Synthesis of cationic polymers
As shown in Fig. 1a and S1–S3,†via AIBN-initiated free radical polymerization, linear copolymers were synthesized with pendent Boc-HMA, PEGMA, and SPMA moieties (detailed information for Boc-HMA, PEGMA, and SPMA is shown in Fig. 1). The 1H NMR spectra of the monomers and the three purified polymer products indicated the successful integration of spiropyran, which accounts for 4.2, 11.7, and 21.6 mol% (Table S2†) of the products, separately. As estimated using GPC analysis (Fig. S4 and Table S2†), neutral polymers were obtained with unimodal distribution and similar molecular weights (Mn 11.7–13.1 kDa). Triflate acid treatment and purification via dialysis afforded cationic polymers P1–P3 (Fig. 1a) accordingly, as evidenced by the disappearance of the characteristic proton resonance of the Boc groups in the 1H NMR spectrum (Fig. S3†). We also prepared the control polymer P4 by starting from the radical polymerization of Boc-HMA and PEGMA and using the same procedure (Table S2, Fig. S3 and S4†).Each component of P1–P3 has a special role. Hydrophilic oligo PEG and imidazole side chains ensure an adequate water solubility of the polymers. As the dominant component (48.8–58.3 mol%), oligo PEG helps shield the positive charges of the polyplexes to minimize nonspecific cytotoxicity and immunogenicity.23 The high content of imidazole (29.6–37.5 mol%) in the polymers allows for DNA binding and condensing, and potentially boosts intracellular endolysosomal escape efficiency via the well-known proton sponge effect.24 As for the spiropyran unit, it has been well described as a photochromic material that isomerizes between a hydrophobic ring closed form (SP isoform) and a zwitterionic ring opened form (merocyanine, MC isoform).18 Generally, SP switches to MC after absorbing UV light, and this process is reversed via thermal relaxation in the dark or via excitation with visible light (>450 nm).
Characterization of the P1–P4 nanoassemblies
Atomic Force Microscopy (AFM) was employed to monitor the morphology changes of the polyplex assemblies prepared by complexing plasmid DNA with MC-dominating P3 by changing the mass ratio (r) of P3 to pDNA ranging from 1 to 30. As compared to the random coiled naked pDNA (Fig. 2a), plasmid forms a mesh-like structure (Fig. 2b) at r 1 due to the bridging of the DNA segments by the abundant imidazole groups in the polymer side chains.25 As r increases from 1 to 5, node-like nuclei show up and grow in height from 3–8 nm (Fig. 2b) to 15–23 nm (Fig. S5†). The DNA coils disappear at r 15, and the increase of particle heights and sizes suggests the existence of aggregates. The increase in r from 15 to 30 results in a reduction in height from 40–70 nm to 30–45 nm and the formation of globular particles with an average diameter of approx. 126 nm (Fig. 2c–e). At r 30, the morphology of the P3 NPs was consistent with the observations from the transmission electron microscopy (TEM), which clearly showed similar results to the P1–P4 NPs in which the P1–P4 polymers had similar molecular weights (Fig. 2f). The DNA condensation behaviors are comparable to the extensively studied polylysine/DNA system due to the polycation character.26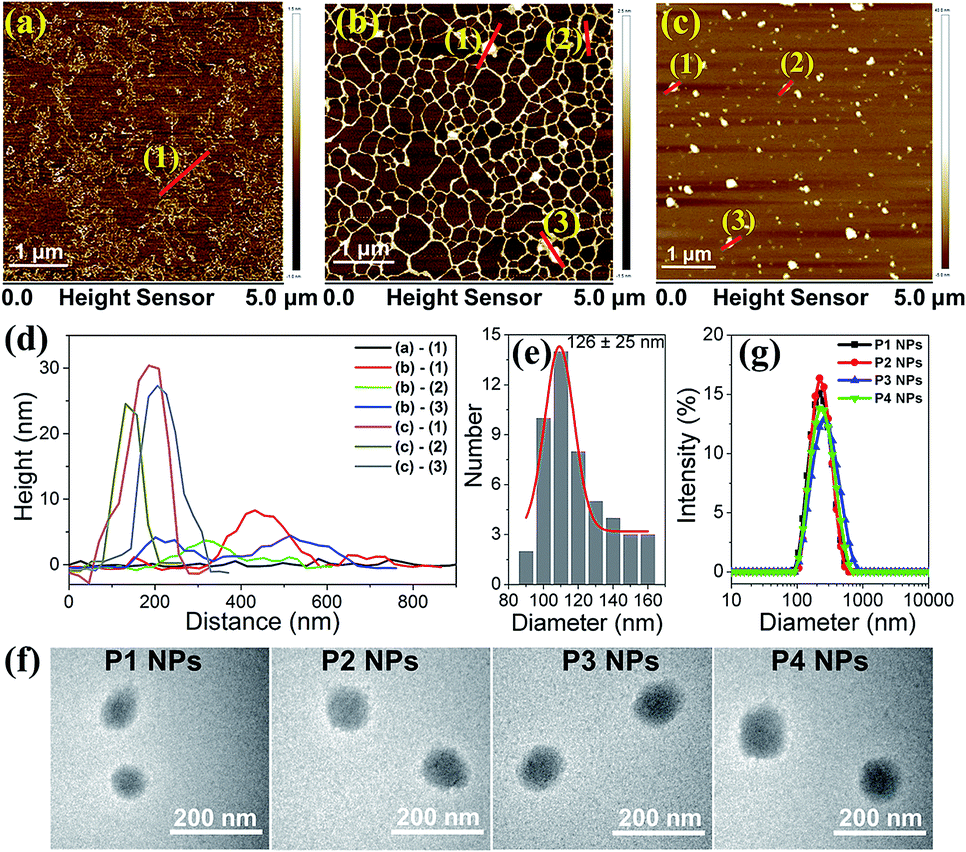 | ||
| Fig. 2 AFM images of P3 NPs formed at r 0 (a), 1 (b), and 30 (c). (d) The height cross-section along the lines shown with numbers in panels (a–c). (e) Size distribution of P3 NPs (r = 30) shown in (c), determined using AFM. (f) TEM images of the P1–P4 NPs (r = 30). (g) DLS analysis of the P1–P4 NPs (r = 30). | ||
At r 30, dynamic light scattering (DLS) analysis reveals the unimodal distribution of the P1–P4 NPs and an average hydrodynamic diameter of approx. 200 nm (Fig. 2g), which is reasonably larger than the AFM and TEM results due to the difference in the solvation and dry states. We selected r 30 for all of our investigations on the nanoparticles unless stated otherwise. Due to the surface charge shielding effect of the oligo PEG chains,27 the P1–P4 NPs exhibit zeta potentials at 6–8 mV (Table S3†), which are less positive than for typical polyplexes and so favor the weakening of the nonspecific biomacromolecular interactions and the cytotoxicity.28 It is worth noting that UV and visible light irradiation did not induce changes in the morphologies and hydrodynamic diameters of the P1–P4 NPs, as evidenced from the TEM, AFM and DLS analyses (Fig. S6–8†). Alternative manipulation of the nanoassemblies (r 30) by assembling pure P1–P4 polymers and P1–P4 NPs with high amounts of DNA (at r 1 and r 5) leads to similar morphologies and sizes (Fig. S9 and S10†), which implies that efficient polymer binding to the intermediate assembly forms results in similar final nanostructures.
Photochromic properties of spiropyrans in P3 and the P3 NPs
We investigated the photochromic properties of the spiropyran moieties in the polymers, with P3 as the example, by monitoring the changes in the UV-vis absorption spectra in HEPES buffer (10 mM, pH 7.0). The thermostable SP state of P3 has minimal absorption at 549 nm, while the MC isoform of P3 absorbs maximally with a 37 nm redshift relative to the small water soluble spiropyran molecule, SPQ (Fig. S1c and S11a†). The notable redshifts in the MC absorptions originate from the MC stacks in the polymers and nanoparticles. Brief LED UV light irradiation (λ1 = 365 nm, 2 mW cm−2, 2 min) led to rapid SP to MC isomerization, which could be identified via the appearance of a new absorption peak at 549 nm without visible shoulders or wavelength shifts (Fig. S11b†). In the reverse direction, MC to SP isomerization in P3 was achieved via slow thermal relaxation in the dark or rapid photoisomerization under LED visible light exposure (λ2 = 520 nm, 20 mW cm−2) (Fig. S11c and d†).The absorption spectroscopic and photochromic properties of spiropyrans were well preserved after P3 was complexed with pDNA (Fig. S11a and 3a–b). The difference lies in the relaxation rates that were estimated using pseudo first order reaction kinetics. In the dark, the half life (t1/2) of the MC state in the P3 NPs was prolonged to 2.8 h as compared to 1.5 h in P3 (Fig. 3c), presumably due to the slowed relaxation caused by steric factors.29 In contrast, under visible light (λ2 = 520 nm, 20 mW cm−2), the t1/2 of the MC state was sharply reduced to 3 min and 1.5 min for the P3 NPs and P3 (Fig. S12†), respectively.
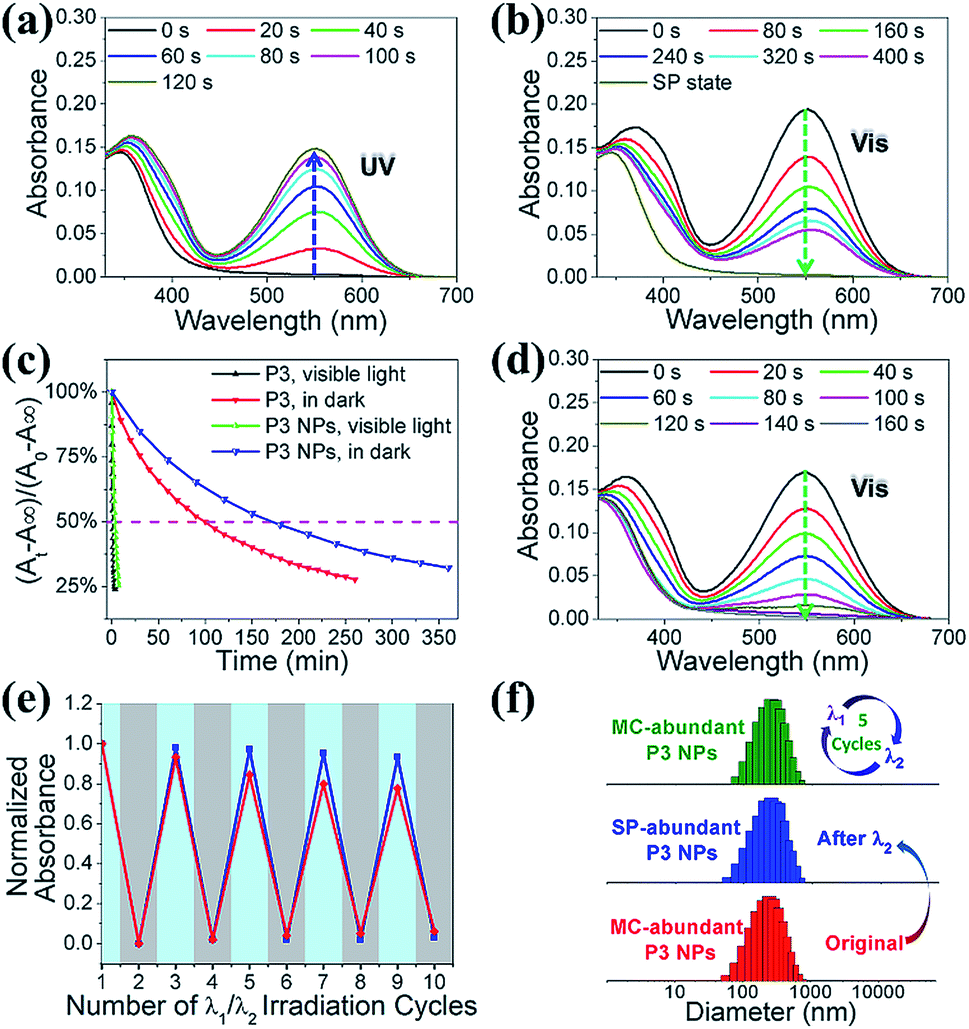 | ||
| Fig. 3 Absorption spectra of the P3 NPs under (a) UV light (λ1 = 365 nm, 2 mW cm−2, 0–120 s) irradiation and (b) visible light (λ2 = 520 nm, 20 mW cm−2, 0–400 s) irradiation. (c) MC-SP isomerization kinetics of P3 and the P3 NPs in the dark or under visible light. A0, A∞ and At are the absorption intensities at 549 nm, measured at the beginning, at the end and at the time of reaction, respectively. (d) Absorption spectra of the P3 NPs under visible light (λ2 > 500 nm, 250 mW cm−2, 0–160 s) irradiation. (e) Photoswitching cycles of the P3 NPs under irradiation by alternating 365 nm UV light and 520 nm visible light. Absorption intensities were monitored at 549 nm. The red and blue lines denote the measurements in the air-saturated and degassed solutions, respectively. (f) Comparison of hydrodynamic diameters of the P3 NPs before and after different irradiation treatments. | ||
It is of note that the MC-SP photoisomerization in the P3 NPs could be complete within a few minutes under the exposure of an elevated irradiation power (λ2 > 500 nm, 250 mW cm−2) (Fig. 3d), which alleviates concerns about the possibility of the active MC state remaining after photoswitching treatment to ensure the insensitivity of the P3 NPs toward natural light. In addition, in the dark, the full SP state showed undetectable absorption at 549 nm and the generation of the MC state over hours was nearly negligible in both P3 and the P3 NPs (Fig. S13a and b†), in contrast to the extreme sensitivity of the SP state to low power density UV light (λ1 = 365 nm, 2 mW cm−2) (Fig. S13c†). It should be mentioned that the heat generation upon alternating light irradiation was insignificant, as seen in the slight temperature fluctuation (<0.5 °C) (Fig. S14†). Thereby the concern of undesired photothermal effects that may cause alteration of nanoparticle structures, photoswitching kinetics and nonspecific cell damage is eliminated.
This efficient reversible photoisomerization allows for multiple cycles of photoswitching of the P3 NPs under irradiation by alternating 365 nm UV light and 520 nm visible light. During 5 cycles of λ1/λ2 irradiation of the P3 NPs and P3, the maximal MC absorption intensity remained unchanged in degassed HEPES buffer, and lost approx. 20% in air-saturated solution due to a slight fatigue effect (Fig. 3e). The stability of the P3 NPs in photochromic processes was confirmed using DLS analysis, which revealed unaffected hydrodynamic diameters during the irradiation cycles (Fig. 3f).
Fluorescence and light-induced ROS generation of P1–P3 and the P1–P3 NPs
Generally, spiropyrans are not strong fluorophores, and their fluorescence originates from their MC states when they are excited by visible light. While the content of MC increases, the P1–P3 polymers have a low fluorescence quantum yield (Φf) from 0.050 to 0.068, using rhodamine B as the standard (Φf = 0.89).30 The Φf of SPQ is only 0.005, which is partly due to the short life span of MC in aqueous solution. In contrast, the P1–P3 NPs display enhanced fluorescence (λem = 644 nm, Fig. S11a†) with Φf ranging from 0.18 to 0.27, which is 3.5–4.0 fold larger relative to the polymer counterparts (Fig. 4a) and means they outperform or are comparable to reported MC-containing film or particle systems (Table S1†).31–33 This phenomenon is considered to be attributed to the aggregation-induced (enhanced) emission (AIE or AIEE) effect associated with J-type rather than H-type aggregation.34,35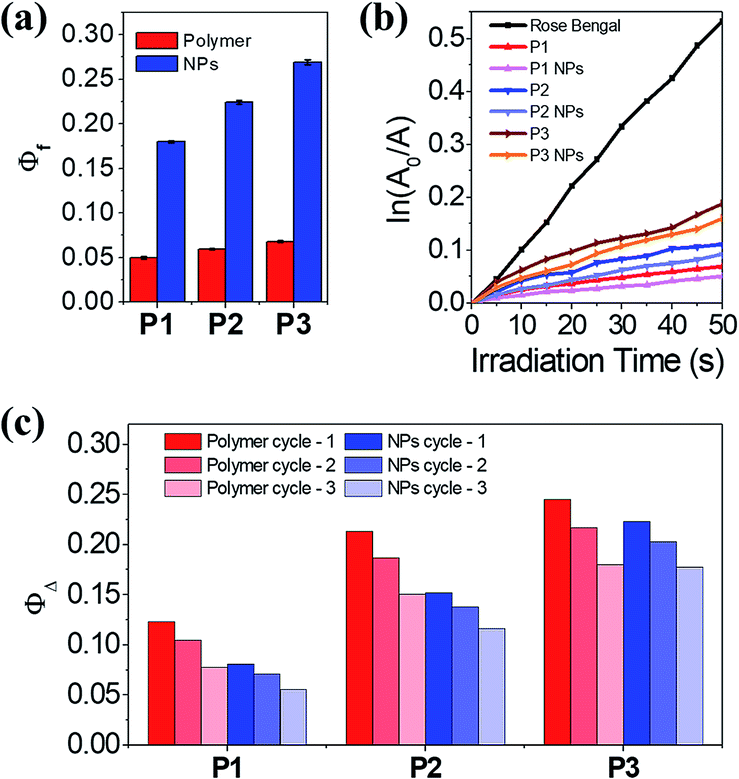 | ||
Fig. 4 (a) Φf of P1–P3 and the P1–P3 NPs in HEPES buffer (10 mM, pH 7.0) at 25 °C. (b) Plot of ln(A0/A) as a function of time of visible light irradiation on the various photosensitizers, where A0 and A are absorption intensity at 400 nm at the beginning and at irradiation time t, respectively. (c) ΦΔ of P1–P3 and the P1–P3 NPs in HEPES buffer/D2O (V/V = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9). :9). | ||
The evidence supporting the fact that the J-aggregates are dominant comes from the following multiple observations: (1) the increase of Φf with increasing MC content in the polymers, (2) the redshift of the MC absorptions relative to the well dispersed SPQ, (3) the unimodal and constant MC absorption shape during isomerization, lacking the appearance of a shoulder peak at short wavelengths, (4) the bright fluorescence at long wavelengths, and (5) the well-fitted nonexponential kinetics of the MC/SP photoisomerization in the absorption at 549 nm.19,31,36,37 It is not necessary to distinguish the dimers or multimers of the MC conformers that contribute to J-stacks in favor of irradiative relaxation.38
Interestingly, both the polymers and NPs exhibit a capability to generate 1O2 upon activating the MC units under visible light. Using tetrasodium α,α′-(anthracene-9,10-diyl)bis(methylmalonate) (termed ABMM) as the 1O2 trapping agent and Rose Bengal as the actinometer (0.75 in water),39 the 1O2 generation yields (ΦΔ) of P1–P3 in HEPES buffer (10 mM, pH 7.0)/D2O (V/V = 1:9) were determined to be 0.12, 0.21, and 0.25, respectively (Fig. 4b–c). With the same trend between ΦΔ and the MC content, the P1–P3 NPs afforded ΦΔ values of 0.08, 0.15, and 0.22, respectively, which are comparable to those of their polymer counterparts. In consideration of the big sizes of the NPs and the limited traffic distance of 1O2,40 the ΦΔ values of the NPs are prone to be underestimated as a few amounts of 1O2 are deactivated to the ground state before being accessible to the 1O2 trapping agent. Nevertheless, after three cycles of λ1/λ2 irradiation on P1–P3 and the P1–P3 NPs, the NPs indicate improved fatigue performance (Fig. 4c). Evidently, the fluorescence and light-induced ROS generation behaviors are in parallel with each other because of J-stacks. This feature would facilitate the use of NPs, particularly P3 NPs with the largest Φf and ΦΔ values, in both cell imaging and light-controlled cell killing studies.
Cell imaging with the P3 NPs
The P3 NPs have decent stability in both the HEPES buffer containing a high concentration of NaCl (0.2 M) and the complete DMEM medium containing 10% fetal bovine serum (FBS), as confirmed by the constant hydrodynamic diameters during five alternating λ1/λ2 irradiation cycles (Fig. S15a†). Similarly, as a control, the size of the P4 NPs was not affected by the same treatment (Fig. S15b†). Such stability of nanoassemblies against salt and serum arises from the high content of PEG substituents and the low zeta potentials, affording excellent compatibility with cell culturing conditions.By taking advantage of the MC fluorescence, we investigated cell uptake of the P3 NPs under a confocal laser scanning microscope (CLSM). HeLa cells were pretreated with the P3 NPs for 4 h before imaging. As shown in Fig. 5a, after brief UV light irradiation (λ1 = 365 nm, 90 mW cm−2, 1 min), the cells emitted bright red fluorescence from the MC state with excitation at 543 nm. The nuclei were not stained with red fluorescence, which is in good agreement with the frequently reported polymer-based nucleic acid delivery systems and is due to the endolysosome barrier for the internalized nanoparticles.41,42 Subsequent LED visible light irradiation (λ2 > 500 nm, 250 mW cm−2, 10 min) led to an almost entire fluorescence quenching as a result of the efficient MC-SP photoswitching in the P3 NPs. The fluorescence was found to be switched on and off again during the second and third cycles of λ1/λ2 alternating irradiation. The high level of MC fluorescence in the cells suggests that the nanoassembly structure could survive in intracellular internalization and photoswitching processes, according to the fluorescence of the MC state that has been shown to be highly dependent on the J-stacks. In contrast, the P3 polymer alone when taken up by the HeLa cells gave a drastically attenuated signal (Fig. S16†), due to the low Φf of P3 as well as the limited cell internalization.
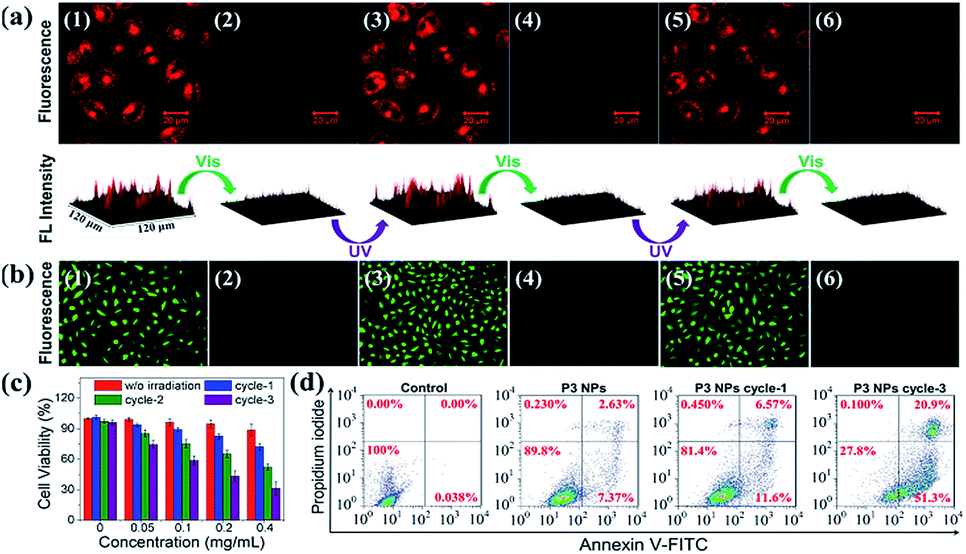 | ||
| Fig. 5 Photoswitching cycles of the P3 NPs taken up by the HeLa cells under alternating UV-light (365 nm, 90 mW cm−2, 1 min) and visible light (>500 nm, 250 mW cm−2, 10 min) treatments: (a) fluorescence images and intensity profiles (1,3,5-UV light; 2,4,6-visible light), and (b) oxidized DCF fluorescence images after visible light irradiation on the light-treated cells (1,3,5-MC state generated by UV light; 2,4,6-SP state generated by visible light). (c) MTT analysis and (d) flow cytometric analysis of the P3 NP-treated HeLa cells with various photoswitching cycles under alternating UV light (365 nm, 90 mW cm−2, 1 min) and visible light (>500 nm, 250 mW cm−2, 3 min) treatments. The final concentrations of P3 (in the form of NPs) are 0.2, 0.2 and 0.4 mg mL−1 applied in (a), (b) and (d), respectively. | ||
Reversible ROS generation in cells controlled using the P3 NPs
To investigate the light-controlled reversible 1O2 generation in cells, we detected intracellular ROS production in the P3 NP-treated HeLa cells using commercial 2,7-dichlordihydrofluorescein diacetate (termed DCFH-DA) as a ROS probe. The green fluorescence intensity of the oxidized product DCF was proportional to the level of ROS generated.43,44 After incubation with the P3 NPs over a period of 4 h, the HeLa cells were subjected to LED UV light irradiation (365 nm, 90 mW cm−2, 1 min) to accumulate the MC state. The MC-abundant cells were then subjected to photoswitching cycles before going into the ROS detection procedure, or directly went into the ROS detection procedure via incubation with DCFH-DA for 20 min in the dark, washing with PBS buffer, treatment with LED visible light (>500 nm, 250 mW cm−2, 10 min) and imaging under an inverted fluorescence microscope. As shown in Fig. 5b, the bright green emission from DCF (λex = 465–495 nm) throughout the cells was only observed with the MC-abundant cells, while the SP-abundant cells were detected with negligible fluorescence of DCF, correlating with the light-induced ROS generation capability of the MC state. As a control, DCF fluorescence was not detected in HeLa cells treated with PBS, PBS with visible light exposure, or MC-abundant P3 NPs without visible light exposure (Fig. S17†). These results demonstrate that the intracellular ROS level could be reversibly controlled using light of selective wavelengths at desired times. The generation of a high intracellular ROS level is potentially useful for PDT purposes.Accompanying the ROS generation under visible light, the P3 NPs underwent simultaneous MC-SP isomerization and gradually lost their oxygen photosensitization capability unless they were re-activated. On the one hand, the thermally stable SP state is resistant to natural light and thereby avoids cell nonspecific phototoxicity. On the other hand, the re-activatable nature of the P3 NPs allows for the inducing of cellular apoptosis via the repeated generation of ROS. To evaluate the cytotoxicity of the P3 NPs, a conventional MTT assay was performed at 24 h after the light irradiation treatment of the HeLa cells. In the dark, the P3 NPs exhibit marginal cytotoxicity in the polymer concentration range 0–0.4 mg mL−1 (Fig. 5c). However, treatment with successive 1–3 alternating irradiation cycles led to irradiation-cycle-dependent cytotoxicity, correlating with the level of accumulated ROS generated (Fig. 5c). Each cycle included sequential UV light (λ1 = 365 nm, 90 mW cm−2, 1 min) and visible light (λ2 > 500 nm, 250 mW cm−2, 3 min) treatments. To minimize the possibility of concurrent activation of the MC isoform in the SP-MC conversion process, the UV light irradiation duration time was short. Consequently, only approx. 30% of the HeLa cells survived three irradiation cycles. This result was in good agreement with the flow cytometric analysis, which detected 72.2% apoptotic cells under the same protocols (Fig. 5d). Obviously, it is convenient to alter the cytotoxic effect by controlling the number of irradiation cycles. As a control, using the same procedure except that the P3 NPs were absent or the P3 NPs were replaced by P4 NPs, cell viabilities were greater than 95% (Fig. 5d and S18†), suggesting that the tumor cells were insensitive to light treatments without spiropyrans, but vulnerable to ROS generation using oxygen photosensitization.
Conclusions
In summary, spiropyran-decorated cationic copolymers were synthesized and complexed with plasmid DNA to form nanoassemblies via electrostatic interaction. The nanoparticles were stable in buffer solutions containing salt or serum, and showed good biocompatibility. By virtue of the photochromic properties of the spiropyran units, the nanoparticles conferred the following merits of the spiropyran chromophore: (1) a suitable MC half life of up to 2.8 h in the dark; (2) a relatively high Φf of up to 0.27; (3) an applicable ΦΔ of up to 0.22; (4) reversibility in controlling the fluorescence and ROS generation using light; (5) a dual functionality in cell imaging and apoptosis. To the best of our knowledge, this is the first example that utilizes spiropyran as a photosensitizer and provides a proof-of-concept approach to reversibly controllable aggregation-induced enhanced photosensitization and emission (AIEPE). This simple concept provides a useful option for biomedical applications such as light-controlled PDT and drug delivery. The two-photon photoswitching properties of the spiropyran-containing nanosystems45 will be an additional bonus for expanding the use of AIEPE in in vivo studies using near infrared light for tissue penetration to activate the J-stacks of the MC isoform.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Basic Research Program of China (2015CB856300), the National Natural Science Foundation of China (21474046), the 1000 Young Talent Program, the Collaborative Innovation Center of Chemistry for Life Sciences, and the Program for Changjiang Scholars and Innovative Research Team in University for financial support.Notes and references
- P. R. Ogilby, Chem. Soc. Rev., 2010, 39, 3181 RSC.
- H. Chen, J. Tian, W. He and Z. Guo, J. Am. Chem. Soc., 2015, 137, 1539 CrossRef PubMed.
- M. Lismont, L. Dreesen and S. Wuttke, Adv. Funct. Mater., 2017, 27, 1606314 CrossRef.
- A. Castano, P. Mroz and M. Hamblin, Nat. Rev. Cancer, 2006, 6, 535 CrossRef PubMed.
- H. Fan, G. Yan, Z. Zhao, X. Hu, W. Zhang, H. Liu, X. Fu, T. Fu, X. Zhang and W. Tan, Angew. Chem., Int. Ed., 2016, 55, 5477 CrossRef PubMed.
- M. Ethirajan, Y. Chen, P. Joshi and R. Pandey, Chem. Soc. Rev., 2011, 40, 340 RSC.
- T. Kocki, B. Czarczynska-Goslinska, K. Kocka, M. Stolarska, D. Wachowska, S. Lijewski, T. Koczorowski and T. Goslinski, in Nurses and pharmacists in interdisciplinary team of health care providers in photodynamic therapy, ed. Y. Tanaka, Photomedicine Intech., 2017 Search PubMed.
- D. Min, D. Jeong, M. G. Choi and K. Na, Biomaterials, 2015, 52, 484 CrossRef PubMed.
- X. Ding and B. H. Han, Angew. Chem., Int. Ed., 2015, 54, 6536 CrossRef PubMed.
- Z. Zhou, J. Song, L. Nie and X. Chen, Chem. Soc. Rev., 2016, 45, 6597 RSC.
- S. Kolemen, T. Ozdemir, D. Lee, G. Kim, T. Karatas, J. Yoon and E. Akkaya, Angew. Chem., Int. Ed., 2016, 55, 3606 CrossRef PubMed.
- W. Li, C. Su, Y. Chang, Y. Lin and C. Yeh, ACS Nano, 2016, 10, 2017 CrossRef PubMed.
- J. Ge, M. Lan, B. Zhou, W. Liu, L. Guo, H. Wang, Q. Jia, G. Niu, X. Huang and H. Zhou, Nat. Commun., 2014, 5, 4596 CrossRef PubMed.
- J. Park, Q. Jiang, D. Feng and H. Zhou, Angew. Chem., Int. Ed., 2016, 128, 7304 CrossRef.
- J. Park, D. Feng, S. Yuan and H. Zhou, Angew. Chem., Int. Ed., 2015, 54, 430 CrossRef PubMed.
- L. Hou, X. Zhang, T. C. Pijper, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 910 CrossRef PubMed.
- J. Qi, C. Chen, X. Y. Zhang, X. L. Hu, S. L. Ji, R. T. K. Kwok, J. W. Y. Lam, D. Ding and B. Z. Tang, Nat. Commun., 2018, 9, 1848 CrossRef PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148 RSC.
- V. I. Minkin, Chem. Rev., 2004, 104, 2751 CrossRef PubMed.
- G. Wu, S. C. Chen, C. L. Liu and Y. Z. Wang, ACS Nano, 2015, 9, 4649 CrossRef PubMed.
- C. H. Li, Y. X. Zhang, J. M. Hu, J. J. Cheng and S. Y. Liu, Angew. Chem., Int. Ed., 2010, 49, 5120 CrossRef PubMed.
- X. R. Wang, J. M. Hu, G. H. Liu, J. Tian, H. J. Wang, M. Gong and S. Y. Liu, J. Am. Chem. Soc., 2015, 137, 15262 CrossRef PubMed.
- J. S. Suk, Q. G. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28 CrossRef PubMed.
- I. K. Park, K. Singha, R. B. Arote, Y. J. Choi, W. J. Kim and C. S. Cho, Macromol. Rapid Commun., 2010, 31, 1122 CrossRef PubMed.
- A. Muramatsu, Y. Shimizu, Y. Yoshikawa, W. Fukuda, N. Umezawa, Y. Horai, T. Higuchi, S. Fujiwara, T. Imanaka and K. Yoshikawa, J. Chem. Phys., 2016, 145, 235103 CrossRef PubMed.
- I. Nayvelt, T. Thomas and T. J. Thomas, Biomacromolecules, 2007, 8, 477 CrossRef PubMed.
- J. S. Choi, D. K. Joo, C. H. Kim, K. Kim and J. S. Park, J. Am. Chem. Soc., 2000, 122, 474 CrossRef.
- S. Venkataraman, W. L. Ong, Z. Y. Ong, S. C. J. Loo, P. L. R. Ee and Y. Yang, Biomaterials, 2011, 32, 2369 CrossRef PubMed.
- H. R. Allcock and C. Kim, Macromolecules, 1991, 24, 2846 CrossRef.
- D. D. Cheng, X. L. Liu, Y. D. Xie, H. T. Lv, Z. Q. Wang, H. Z. Yang, A. X. Han, X. M. Yang and L. Zang, Sensors, 2017, 17, 2517 CrossRef PubMed.
- L. Y. Zhu, W. W. Wu, M. Q. Zhu, J. J. Han, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2007, 129, 3524 CrossRef PubMed.
- J. L. Bahr, G. Kodis, L. de la Garza, S. Lin, A. L. Moore, T. A. Moore and D. Gust, J. Am. Chem. Soc., 2001, 123, 7124 CrossRef PubMed.
- T. Minami, N. Tamai, T. Yamazaki and I. Yamazaki, J. Phys. Chem., 1991, 95, 3988 CrossRef.
- C. H. Li and S. Y. Liu, Chem. Commun., 2012, 48, 3262 RSC.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC.
- M. Q. Zhu, L. Zhu, J. J. Han, W. W. Wu, J. K. Hurst and A. D. Q. Li, J. Am. Chem. Soc., 2006, 128, 4303 CrossRef PubMed.
- L. V. Natarajan, T. J. Bunning and S. Y. Kim, Macromolecules, 1994, 27, 7248 CrossRef.
- P. Uznanski, Synth. Met., 2000, 109, 281 CrossRef.
- M. Wang, S. Maragani, L. Y. Huang, S. Jeon, T. Canteenwala, M. R. Hamblin and L. Y. Chiang, Eur. J. Med. Chem., 2013, 63, 170 CrossRef PubMed.
- L. Xia, X. G. Kong, X. M. Liu, L. P. Tu, Y. L. Zhang, Y. L. Chang, K. Liu, D. Z. Shen, H. Y. Zhao and H. Zhang, Biomaterials, 2014, 35, 4146 CrossRef PubMed.
- N. Nishiyama, A. Iriyama, W. Jang, K. Miyata, K. Taka, Y. Inoue, H. Takahashi, Y. Yanag, Y. Tamaki, H. Koyama and K. Kataoka, Nat. Mater., 2005, 4, 934 CrossRef PubMed.
- M. Karimi, A. Ghasemi, P. S. Zangabad, R. Rahighi, S. M. M. Basri, H. Mirshekari, M. Amiri, Z. S. Pishabad, A. Aslani, M. Bozorgomid, D. Ghosh, A. Beyzavi, A. Vaseghi, A. R. Aref, L. Haghani, S. Bahramia and M. R. Hamblin, Chem. Soc. Rev., 2016, 45, 1457 RSC.
- W. O. Carter, P. K. Narayanan and J. P. Robinson, J. Leukocyte Biol., 1994, 55, 25 Search PubMed.
- Singlet Oxygen. Applications in Biosciences and Nanosciences, ed. S. Nonell and C. Flors, London, 2016 Search PubMed.
- G. Marriott, S. Mao, T. Sakata, J. Ran, D. K. Jackson, C. Petchprayoon, T. J. Gomez, E. Warp, O. Tulyathan, H. L. Aaron, E. Y. Isacoff and Y. Yan, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17789 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01148f |
| This journal is © The Royal Society of Chemistry 2018 |