
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormal Giese addition of C(sp3)–H nucleophiles enabled by visible light mediated Ni catalysis of triplet enone diradicals†
Geun Seok
Lee
and
Soon Hyeok
Hong
 *
*
Department of Chemistry, College of Natural Science, Seoul National University, 1 Gwanak-ro, Seoul 08826, South Korea. E-mail: soonhong@snu.ac.kr
First published on 11th June 2018
Abstract
An unprecedented utilization of triplet excited enones in Ni-catalysis enabled a formal Giese addition of C(sp3)–H nucleophiles. This mechanism-based approach has greatly widened the reaction scope, allowing the synthesis of previously inaccessible structures. In this process, the enone diradical acted as two distinct reaction centers, participating in both metalation and hydrogen atom transfer, ultimately furnishing a range of formal Giese addition products in a highly general context. This reaction provides complementary access to traditional 1,4-addition reactions of enones, with a future perspective to develop triplet diradical-based transition metal catalysis.
Introduction
For a number of decades, enones have been considered one of the most widely used and versatile building blocks in organic chemistry.1 In addition to their well-recognized classical reactivities, enones have been independently applied in transition metal catalysis and photochemistry to achieve salient chemical transformations. However, the application of the triplet diradical species in transition metal catalysis remains elusive due to the extremely short lifetime (25–35 ns for 2-cyclohexen-1-one)2 of the triplet diradical (Scheme 1a).3 We therefore postulated that such a new reactivity could potentially be realized by adopting recent advances in visible light-mediated nickel catalysis.4 | ||
| Scheme 1 Reactivities of enones and the Giese reaction of C(sp3)–H nucleophiles. | ||
The Giese addition of carbon-centered radicals directly generated from C–H bond oxidation has been actively investigated in the context of atom-economic C–C bond formation (Scheme 1b). Classically, the stoichiometric use of oxidants such as hydroperoxides and/or Lewis acids5 has restricted the scalability of such processes, in addition to presenting environmental concerns. More recently,6 the visible light-mediated photooxidative generation of α-amino radicals was reported by Pandey–Reiser7 and Yoon8 to overcome such shortcomings. However, the scope of this reaction was restricted to N-arylisoquinolines, since polymerizable or β-substituted acceptors proved less effective or unsuitable. Furthermore, Albini9 and Fagnoni10 reported a series of analogous reactions of α-oxy C–H bonds, but these reactions required the use of UV light, and only limited donor–acceptor combinations were applicable. Moreover, Fagnoni and Melchiorre reported the addition of dioxolanyl or α-amino radicals to iminium intermediates; however, this process was restricted to cyclic enones.11 Indeed, the utilization of enones as Giese acceptors often yielded poor results due to their high tendency to undergo telomerization or polymerization.12
To overcome such limitations, we envisioned an unprecedented utilization of triplet excited enones, generated by Ir-mediated energy transfer,13 based on the fact that the two resulting radicals have different electronic characters; the α-radical is electrophilic and the β-radical is nucleophilic.14 In such a system, the nucleophilic β-radical could be selectively trapped by a Ni catalyst, while the electrophilic α-radical could participate in hydrogen atom transfer (HAT) involving the most hydridic C–H bond of the Giese donor to generate a donor radical. Rapid trapping of the donor radical by the Ni catalyst would block any unproductive chain propagation reactions, at the same time giving rise to a high-valent Ni species, which could undergo facile reductive elimination to deliver formal Giese addition products (Scheme 1c).
Results and discussion
To realize our hypothesis, the readily available β-substituted enone cyclohexen-1-one (1a) was chosen as the model acceptor and tetrahydrofuran (THF) was selected as the donor, since this reaction had yet to be achieved through visible light-mediated catalysis (Table 1). Among the various ligands screened, 4,4′-di-tert-butyl-bipyridyl (dtbbpy) was examined initially, and furnished the desired product in a low yield (entry 2). Further screenings with similarly structured ligands enabled us to identify 2,2′-bis(2-oxazoline) (BiOx) as the optimal ligand (entry 1). Other Ni(II) sources such as NiCl2·glyme exhibited significantly diminished activities (entry 3). It is also notable that Ni(0) complexes failed to furnish the desired product (entry 4), and control experiments showed that all components were essential for the reaction (entries 5–8). In addition, only a moderate diastereoselectivity was achieved in the optimization process, with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of stereoisomers being obtained.
:2 mixture of stereoisomers being obtained.
|
|
||
|---|---|---|
| Entry | Reaction conditions | Yieldb (%) |
|
a
1a (0.20 mmol), NiBr2·glyme (5 mol%), BiOx (15 mol%), Ir[dF(CF3)ppy]2(dtbbpy)PF6 (1 mol%), and THF (2.0 mL) irradiated with a 34 W blue LED.
b GC yields calculated using dodecane as the internal standard. d.r. = 3:2.
c Isolated yield.
|
||
| 1 | As shown | >99 (91c) |
| 2 | dtbbpy instead of BiOx | 31 |
| 3 | NiCl2·glyme as the Ni precatalyst | 26 |
| 4 | Ni(COD)2 as the Ni precatalyst | 0 |
| 5 | No NiBr2·glyme | 0 |
| 6 | No BiOx | 0 |
| 7 | No Ir[dF(CF3)ppy]2(dtbbpy)PF6 | 0 |
| 8 | No light source | 0 |

With the optimized conditions in hand, the scope of the reaction was evaluated (Table 2). For cases where the photocatalyst was insoluble, acetonitrile was used as a co-solvent. Various ethers including tetrahydropyran (THP), diethyl ether, and tert-butyl methyl ether were compatible with this reaction system (3a–5a). To our delight, the reaction scope was not restricted to ethers, with toluene delivering the corresponding benzylated product in a moderate yield (6a). The allylic C–H bond was also successfully functionalized in a good yield (7a).

In terms of the enone scope of this reaction, cyclic enones provided excellent yields (2a, 2b), with the reactivity being mostly preserved even when addition occurred at a sterically congested neopentyl center (2c). For acyclic enones, alkyl or aryl substituents were compatible on both sides (2d–2g). In addition, the functional group tolerance of this system was assessed using a range of chalcones, which represent a key structural motif in medicinal chemistry.15 Furthermore, electron donating groups generally promoted high reactivity, likely due to enhanced stabilization of the excited triplet state (2h–2n, 2u, 2x). Gratifyingly, the hydroxy functional group, which is key to the bioactivity,16 was also tolerated in our system (2n). Chalcones bearing fluorine and chlorine substituents also gave the desired products in good yields (2o, 2p, 2s, 2t). However, chalcones bearing strongly electron withdrawing groups reulted in reduced yields (2q, 2r). Disubstituted chalcones with a range of electronic properties gave the desired products in good yields (2v–2y). Finally, naphthyl and heteroaromatic moieties were also compatible with the developed conditions (2z–2ab). To highlight the unprecedented scope of the developed methodology, we note that the majority of these products were reported for the first time herein.
We then moved on to examine the mechanism of this reaction system. To determine whether any photoredox processes occur under the reaction conditions, we initially examined 1a by cyclic voltammetry both in the presence and the absence of stoichiometric amounts of the in situ-generated Ni(BiOx)Br2 complex. In both cases, the voltammograms indicated that neither oxidation nor reduction took place in the active range17 of the photocatalyst (Fig. S1–S3†) as expected from the reduction potential,18 thereby implying that no photoredox process involving 1a took place.
To determine the possibility of photocatalytic energy transfer, the same reaction was conducted under UV-C (254 nm) irradiation in the presence of UV triplet sensitizers (Scheme 2a).2,19 Although the desired products were obtained in lower yields, photosensitizers exhibiting higher triplet energies gave higher product yields, strongly suggesting that an energy transfer mechanism is indeed involved.20 In addition, Mn powder (Eo = −1.42 V vs. SCE (saturated calomel electrode)),21 which was used as an external reductant (comparable to E1/2 (Ir(III)/Ir(II)) = −1.37 V vs. SCE),17 also failed to promote the desired reaction, again indicating that a photoredox-mediated pathway does not prevail.
 | ||
| Scheme 2 Mechanistic studies. | ||
However, for the above experiments, it must be noted that the triplet energy of benzil was not sufficient to excite 1a. This indicated that the Ni complex assisted the triplet state excitation, analogous to the Sc-mediated process reported by Yoon.13a Thus, to assess the role of Ni in the generation of enone triplets, photodimerization experiments were conducted (Scheme 2b). The [2+2]-self-dimerization of 1a was efficient under neat conditions, furnishing the head-to-head dimer 3 and the head-to-tail dimer 4 in a combined yield of over 70% accompanied by other isomers. The result implies that the energy transfer from the photocatalyst to the enone is a facile process. To observe the Ni effect on the energy transfer process, the reactions were checked in a shortened reaction time (2 h) with a reduced loading of the Ir photocatalyst (1 mol%). Over five-fold enhancement of the reaction yields was obtained in the presence of the Ni catalyst, suggesting that the Ni complex can activate enones, facilitating the energy transfer. The possibility of Ni-catalyzed dimerization was dismissed, as the dimerization product was not observed without light or the Ir photocatalyst. In addition, first order kinetics were observed with respect to the Ni catalyst (Fig. S4†), thereby indicating the involvement of Ni prior to the rate-limiting steps (vide infra), and implying Ni involvement in the HAT process.
To check the possibility of a reaction mechanism which involves Lewis acid activation of enones followed by a direct Giese propagation process as proposed by the Yoon group,8 the reaction progress was monitored with alternating periods of illumination and darkness (Fig. S5†). The formation of the product was only observed during illumination, thereby excluding any persistent chain-propagation pathway. Moreover, the experimentally measured quantum yield (0.18) conclusively ruled out the chain propagative mechanism (see the ESI† for details).22 In addition, all examined Lewis acids or Brønsted acids with the exception of Ni(II) sources provided unmanageable mixtures, likely due to polymerization (Table S1†).
Subsequently, to further examine the HAT event, the reaction was conducted using THF-d8 (Scheme 3). Notably, the isolated adduct showed full incorporation of a single deuterium atom at the α-position. Furthermore, the addition of water to the reaction mixture had no effect on the deuterium content, thereby confirming that the α-radical is responsible for the HAT process, and excluding the involvement of enolate intermediates and a further possible mechanism involving Ni-hydride intermediates, as recently reported by the Wu group.23 Moreover, a halogen radical-mediated HAT process24 was also excluded as all deuterium was incorporated at the α-position even in the presence of water with the fact that NiCl2·glyme exhibits a lower reactivity (Table 1, entry 3).
 | ||
| Scheme 3 Deuterium incorporation study. | ||
The absolute regioselectivity of the HAT process is believed to result from the initially postulated difference in radical polarity, as in the case of cycloaddition reactions, by achieving the highest degree of HOMO–LUMO matching.25 Following generation of the diradical, a polarity-matched pseudo-[2+2] metalation-HAT process is proposed (Scheme 4). Nevertheless, it is currently unclear whether this is a stepwise or concerted process.
 | ||
| Scheme 4 Depiction of the HAT process. | ||
To further unveil the underlying photocatalytic process, Stern–Volmer quenching experiments of the Ir photocatalyst were conducted with the in situ generated Ni(BiOx)Br2 complex, and with 1a (Fig. S10 and S11†). Both showed similar degrees of quenching, indicating that both the Ni catalyst and enone could interact with the photocatalyst. These results not only support the energy transfer between the enone and the photocatalyst but also indirectly indicate the initial photoreduction of the Ni(II) precatalyst to a Ni(I) species which participates in a Ni(I)/Ni(III) cycle,26 excluding the involvement of a highly unlikely Ni(IV) species in the catalytic cycle.
Competition experiments were then conducted to verify the possibility of selective C–H functionalization (Scheme 5). Based on the reaction rates and product yields observed under the standard conditions, the reactivity order ranks as follows: THF, cyclohexene, and then toluene. Indeed, the experimental results supported this order, with the exception of the THF/cyclohexene competition experiment. Thus, to determine the origin of this result, kinetic isotope effects (KIEs) were measured (Scheme 6), and a primary KIE (kH/kD = 3.13) was observed for THF, indicating that the HAT step is the rate-limiting step (rls). In contrast, secondary KIEs (toluene, 1.40; cyclohexene, 1.45) were observed for the other donors, indicating a kinetic shift in the reaction mechanism. Presumably, the rls shifted to the reductive elimination step, owing to the relative weakness of π-conjugated C–H bonds, resulting in non-rate-limiting HATs. The comparatively late rls accounts for the second entry, since the kinetically favored cyclohexenyl-bound Ni complex would dominate.
 | ||
| Scheme 5 Competition experiments. | ||
 | ||
| Scheme 6 Kinetic isotope effect experiments. | ||
Based on the experimental results, a plausible reaction mechanism was proposed, as outlined in Scheme 7. After the photoreduction of the Ni(II) precatalyst, the Ni(I) catalyst binds to the enone substrate, followed by facile triplet excitation via Ir-mediated energy transfer. The resulting diradical then participates in a [2+2]-like metalation-HAT process, controlled in a perfectly regioselective fashion by the matching of radical characters. This gives rise to a dialkyl Ni(III) species, which undergoes reductive elimination to furnish the desired product, thereby completing the catalytic cycle. A catalytic cycle involving Ni(0) species can be ruled out since the Ni(0) source fails to promote the reaction (Table 1, entry 4).
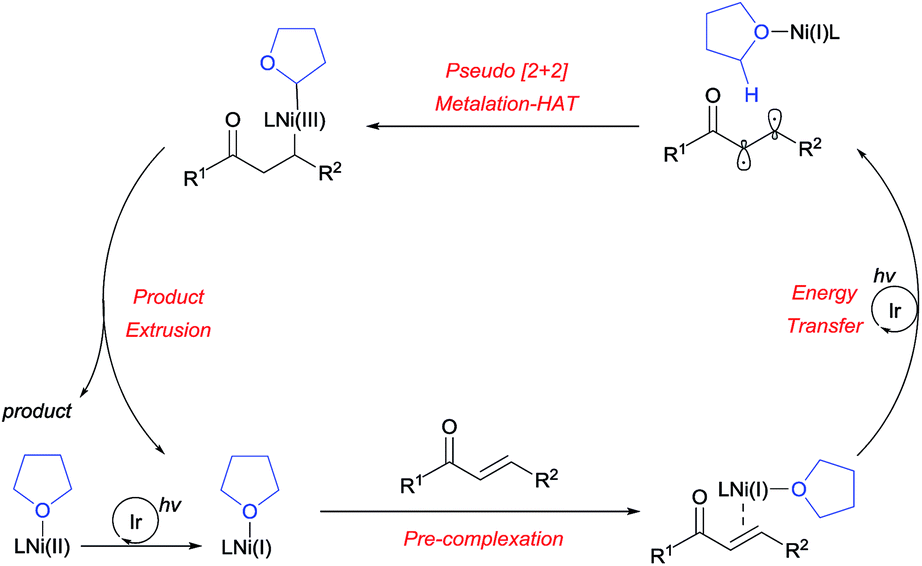 | ||
| Scheme 7 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we herein described a novel reactivity of enones, namely a direct C–H bond activation/formal Giese addition reaction, where the enone triplet diradical acts as two distinct reaction centers. More specifically, the diradical appears to participate in a pseudo-[2+2] metalation-HAT process, which ultimately enables net C–H bond addition across activated olefins for a broad range of substrates in the presence of various functional groups. The developed reaction not only serves as an efficient method for the preparation of functionalized ketones, but can also be considered an innovative platform for the elusive application of excited enone triplets in transition metal catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank S. Y. Yeon of Prof. T. D. Chung's group for assistance with the cyclic voltammetry measurements. We thank H. Kim of Prof. D. Lee's group for assistance with the quantum yield measurements and fluorescence quenching experiments. This work was supported by the Samsung Science and Technology Foundation under Project Number SSTF-BA1601-12.Notes and references
- S. Patai and Z. Rappoport, Chemistry of Enones: Pts 1 & 2, John Wiles & Sons Ltd, 1989 Search PubMed.
- D. I. Schuster, G. E. Heibel, R. A. Caldwell and W. Tang, Photochem. Photobiol., 1990, 52, 645–648 CrossRef PubMed.
- (a) B. E. Rossiter and N. M. Swingle, Chem. Rev., 1992, 92, 771–806 CrossRef; (b) C. Chen, S.-F. Zhu, X.-Y. Wu and Q.-L. Zhou, Tetrahedron: Asymmetry, 2006, 17, 2761–2767 CrossRef; (c) S. E. Shockley, J. C. Holder and B. M. Stoltz, Org. Process Res. Dev., 2015, 19, 974–981 CrossRef PubMed; (d) Y. Yamamoto, T. Nishikata and N. Miyaura, J. Synth. Org. Chem., Jpn., 2003, 64, 1112–1121 CrossRef; (e) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef PubMed; (f) H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 18323–18326 RSC; (g) T. E. Schmid, S. Drissi-Amraoui, C. Crevisy, O. Basle and M. Mauduit, Beilstein J. Org. Chem., 2015, 11, 2418–2434 CrossRef PubMed; (h) R. Shrestha, S. C. Dorn and D. J. Weix, J. Am. Chem. Soc., 2013, 135, 751–762 CrossRef PubMed; (i) K. M. Huihui, R. Shrestha and D. J. Weix, Org. Lett., 2017, 19, 340–343 CrossRef PubMed.
- For selected reviews, see: (a) J. C. Tellis, C. B. Kelly, D. N. Primer, M. Jouffroy, N. R. Patel and G. A. Molander, Acc. Chem. Res., 2016, 49, 1429–1439 CrossRef PubMed; (b) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef PubMed; (c) L. N. Cavalcanti and G. A. Molander, Top. Curr. Chem., 2016, 374, 39–61 CrossRef PubMed.
- A. Citterio, A. Arnoldi and A. Griffini, Tetrahedron, 1982, 38, 393–395 CrossRef.
- For direct β-functionalization of ketones using photoredox catalysis, see: (a) M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593–1596 CrossRef PubMed; (b) F. R. Petronijevic, M. Nappi and D. W. MacMillan, J. Am. Chem. Soc., 2013, 135, 18323–18326 CrossRef PubMed.
- P. J. Kohls, D. Pandey and G. Reiser, Org. Lett., 2012, 14, 672–675 CrossRef PubMed.
- L. Ruiz Espelt, E. M. Wiensch and T. P. Yoon, J. Org. Chem., 2013, 78, 4107–4114 CrossRef PubMed.
- D. Dondi, M. Fagnoni and A. Albini, Chem.–Eur. J., 2006, 12, 4153–4163 CrossRef PubMed.
- (a) R. Mosca, M. Fagnoni, M. Mella and A. Albini, Tetrahedron, 2001, 57, 10319–10328 CrossRef; (b) D. Ravelli, M. Zoccolillo, M. Mella and M. Fagnoni, Adv. Synth. Catal., 2014, 356, 2781–2786 CrossRef.
- J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Nature, 2016, 532, 218–222 CrossRef PubMed.
- G. C. Eastmond, Comprehensive Chemical Kinetics, Free-Radical Polymerisation, Elsevier, New York, 1976 Search PubMed.
- For visible-light mediated enone triplet excitation using photoredox catalysts, see: (a) T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391–1395 CrossRef PubMed; (b) S. K. Pagire, A. Hossain, L. Traub, S. Kerres and O. Reiser, Chem. Commun., 2017, 53, 12072–12075 RSC.
- F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2721–2724 CrossRef PubMed.
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef PubMed.
- S. Radhakrishnan, R. Shimmon, C. Conn and A. Baker, Bioorg. Chem., 2015, 62, 117–123 CrossRef PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712–5719 CrossRef.
- H. O. House, L. E. Huber and M. J. Umen, J. Am. Chem. Soc., 1972, 94, 8471–8475 CrossRef.
- For triplet energy values, see: W. G. Herkstroeter, A. A. Lamola and G. S. Hammond, J. Am. Chem. Soc., 1964, 86, 4537–4540 CrossRef.
- In the absence of the Ni catalyst, less than 3% yield of 2a was obtained in all entries in Scheme 2a, demonstrating the crucial role of the Ni complex.
- D. C. Harris, Quantitative Chemical Analysis, Freeman, 9th edn, 2015 Search PubMed.
- M. A. Cismesia and T. P. Yoon, Chem. Sci., 2015, 6, 5426–5434 RSC.
- H. P. Deng, X. Z. Fan, Z. H. Chen, Q. H. Xu and J. Wu, J. Am. Chem. Soc., 2017, 139, 13579–13584 CrossRef PubMed.
- (a) D. R. Heitz, J. C. Tellis and G. A. Molander, J. Am. Chem. Soc., 2016, 138, 12715–12718 CrossRef PubMed; (b) B. J. Shields and A. G. Doyle, J. Am. Chem. Soc., 2016, 138, 12719–12722 CrossRef PubMed; (c) M. K. Nielsen, B. J. Shields, J. Liu, M. J. Williams, M. J. Zacuto and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 7191–7194 CrossRef PubMed.
- For selected studies, see: (a) K. N. Houk, Acc. Chem. Res., 1975, 8, 361–369 CrossRef; (b) E. J. Corey, J. D. Bass, R. LeMahieu and R. B. Mitra, J. Am. Chem. Soc., 1964, 86, 5570–5583 CrossRef.
- N. Ishida, Y. Masuda, N. Ishikawa and M. Murakami, Asian J. Org. Chem., 2017, 6, 669–672 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization of new compounds. See DOI: 10.1039/c8sc01827h |
| This journal is © The Royal Society of Chemistry 2018 |