
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solar H2 generation in water with a CuCrO2 photocathode modified with an organic dye and molecular Ni catalyst†
Charles E.
Creissen
,
Julien
Warnan
and
Erwin
Reisner
 *
*
Christian Doppler Laboratory for Sustainable SynGas Chemistry, Department of Chemistry, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: reisner@ch.cam.ac.uk
First published on 27th November 2017
Abstract
Dye-sensitised photoelectrochemical (DSPEC) cells have emerged in recent years as a route to solar fuel production. However, fuel-forming photocathodes are presently limited by photo-corrodible narrow band gap semiconductors or the small range of available wide bandgap p-type semiconductors such as NiO that display low performance with dyes. Here, we introduce CuCrO2 as a suitable p-type semiconductor for visible light-driven H2 generation upon co-immobilisation of a phosphonated diketopyrrolopyrrole dye with a Ni-bis(diphosphine) catalyst. The hybrid CuCrO2 photocathode displays an early photocurrent onset potential of +0.75 V vs. RHE and delivers a photocurrent of 15 μA cm−2 at 0.0 V vs. RHE in pH 3 aqueous electrolyte solution under UV-filtered simulated solar irradiation. Controlled potential photoelectrolysis at 0.0 V vs. RHE shows good stability and yields a Ni catalyst-based turnover number of 126 ± 13 towards H2 after 2 h. This precious metal-free system outperforms an analogous NiO|dye/catalyst assembly and therefore highlights the benefits of using CuCrO2 as a novel material for DSPEC applications.
Introduction
Artificial photosynthesis offers a platform to produce a storable energy supply from fossil fuel-free resources.1–4 This sustainable, carbon–neutral approach can produce a ‘solar fuel’ such as H2 or carbon-based molecules from water or CO2 using solar light. This process can be realised using semiconductor electrodes modified with suitable electrocatalysts in a photoelectrochemical (PEC) cell.5–9 Electrodes featuring a molecular catalyst have advantages over ‘conventional’ heterogeneous systems as their ‘single site catalysis’ is atom-efficient,10,11 they offer tunability and selectivity for challenging chemical transformations,12–15 and can be rationally designed to enhance activity.16–19 Their molecular nature also enables kinetic and mechanistic studies to reveal how they operate under various conditions, outlining routes to improvement.20–23 Despite these advantages, the development of molecular-based photocathodes is held back by severe material limitations as state-of-the-art electrodes currently lack the requirements of visible light absorption, mesoporosity, p-type conductivity, and/or stability in aqueous solution.5,24–27To bypass these limitations, a modular approach can be adopted where a visible light-absorbing dye and a molecular catalyst are co-anchored to a stable wide bandgap semiconductor platform.7,28–30 In this dye-sensitised photoelectrochemical (DSPEC) system, the p-type semiconductor serves as the anchor site for the dye, which typically permits ultra-fast hole extraction following visible light excitation of the dye and minimises energy loss. The photoreduced dye is subsequently responsible for electron transfer to the co-immobilised electrocatalyst, where the reduction half-reaction takes place. The separation of light harvesting, charge transport, and catalysis allows the components to be individually tuned for optimal performance, where the rate of each transfer step influences the overall device efficiency.30 A suitable pair of photoelectrodes in a tandem DSPEC cell could provide an efficient and inexpensive means of solar fuel production, exploiting simple and adaptable preparation techniques.31–35
The requirements for a robust DSPEC photocathode material are high p-type conductivity, propensity to anchor molecular moieties, high surface area, and a valence band position capable of readily accepting a hole from the photoexcited dye.29,30,36 Several DSPEC photocathodes have already been reported with the majority relying on NiO,18,37–43 and the only other examples being modified ITO44 and CuGaO2.34 NiO is stable and easily synthesised in mesoporous form,45–47 but suffers from the drawbacks of low charge carrier mobility and fast charge recombination between valence band holes and the reduced dye.28,48–50 Despite many efforts and different approaches to enhance the PEC properties of dye-sensitised NiO photoelectrodes,51–53 improvements in performance are hindered by these limitations and there is a crucial need for better alternatives.
Wide bandgap Cu(I)-based mixed metal oxides such as CuIMIIIO2 delafossites (M = Co, B, In, Sc, Cr, Al, Ga) have been employed in p-type dye-sensitised solar cells (p-DSSCs),54,55 but their incorporation in solar fuel devices is limited.56–58 The sole example of their use with a co-immobilised dye and molecular catalyst in solar fuel generation was reported for CO2 reduction to CO with an anchored precious metal-based Ru–Re dyad on a CuGaO2 delafossite electrode.34 Delafossite CuCrO2 has shown promise in p-DSSCs but application has yet to be extended to DSPEC cells despite it showing clear benefits such as a low-lying valence band, high hole mobility, and simple and scalable synthesis.59–63
In this study, we report solar H2 generation with dye-sensitised CuCrO2 and demonstrate the feasibility of solar fuel synthesis with a CuMO2 delafossite using precious metal-free dye/catalyst molecules. This was achieved by first modifying CuCrO2 with a phosphonic acid-bearing diketopyrrolopyrrole-based organic dye (DPP-P) and characterising the PEC reduction of a soluble electron acceptor in aqueous conditions. Then, a tetraphosphonated Ni-bis(diphosphine), [Ni(P2N2)2]2+, molecular catalyst (NiP) was co-immobilised to determine the PEC activity for the reduction of aqueous protons (Fig. 1a). The resulting hybrid DSPEC photocathode produces H2 at moderate applied voltages with good photocurrents. Direct comparison with a corresponding NiO photocathode highlights the benefits of CuCrO2 and encourages the search for new DSPEC cathode materials.
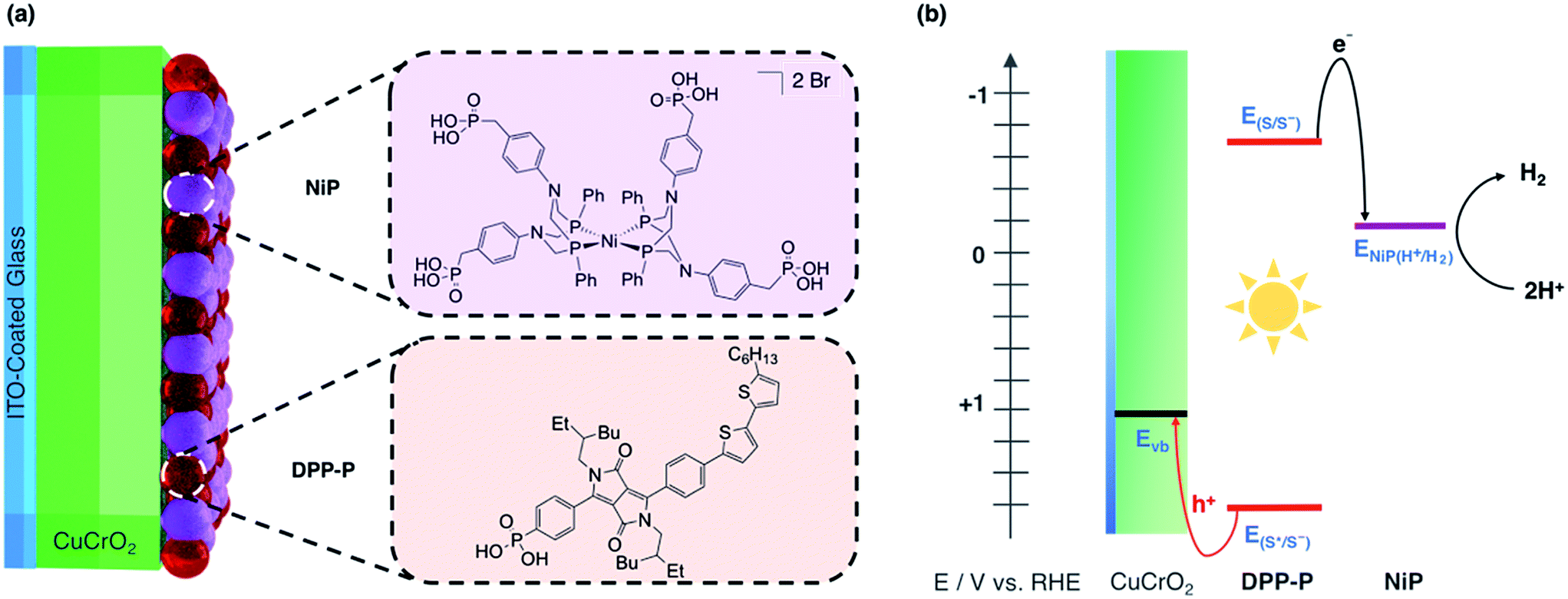 | ||
| Fig. 1 (a) Dye (DPP-P) and catalyst (NiP) co-immobilised on the CuCrO2 electrode with their molecular structures. (b) Energy diagram showing movement of electrons with black and holes with red arrows. S represents the dye sensitiser where E(S/S−) is the ground state reduction potential and E(S*/S−) is the reduction potential in the excited state. ENiP(H+/H2) is the catalytic onset potential for NiP and Evb is the CuCrO2 valence band potential. | ||
Results and discussion
Synthesis and characterisation of CuCrO2
Scalable and straightforward procedures for preparation of CuCrO2 make it a highly accessible material, and its metal oxide character ensures that molecular species can be easily attached to the surface using anchoring groups such as phosphonic acids or carboxylic acids.59–63 In this study, CuCrO2 films were grown directly on ITO-coated glass following a previously established sol–gel route.59,60 In brief, a mixture of Cu(acetate)2·H2O (0.2 M), Cr(NO3)3·9H2O (0.2 M), and triethanolamine (0.4 M) in absolute ethanol was spin-coated on an ITO-coated glass substrate. These samples were annealed in air at 400 °C for 45 min before repeating the spin-coating and annealing steps to obtain a total of 6 layers. Post-annealing was carried out under N2 at 600 °C for 45 min to form the delafossite structure. NiO films (2 μm thick) were prepared for comparison using a previously reported hydrothermal growth method.37CuCrO2 crystallises in a rhombohedral unit cell (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) and is a wide bandgap p-type semiconductor (Eg = 3.1 eV) exhibiting a low-lying valence band and high hole mobility.63,64 The structure consists of ‘infinite’ [CrO2] layers of edge-sharing [CrO6] octahedra linked by linear O–Cu–O dumbbells and the p-type conductivity stems predominantly from Cu+ vacancies in the crystal lattice.65,66 Favourable mixing of Cr 3d states with O 2p states increases the covalent nature of this interaction in the valence band, hence holes are more delocalised than in other corresponding delafossite structures, accounting for the intrinsic high hole mobility.64,66
m) and is a wide bandgap p-type semiconductor (Eg = 3.1 eV) exhibiting a low-lying valence band and high hole mobility.63,64 The structure consists of ‘infinite’ [CrO2] layers of edge-sharing [CrO6] octahedra linked by linear O–Cu–O dumbbells and the p-type conductivity stems predominantly from Cu+ vacancies in the crystal lattice.65,66 Favourable mixing of Cr 3d states with O 2p states increases the covalent nature of this interaction in the valence band, hence holes are more delocalised than in other corresponding delafossite structures, accounting for the intrinsic high hole mobility.64,66
X-ray diffraction (XRD) analysis confirmed the rhombohedral delafossite structure for CuCrO2 (Fig. S1†) and scanning electron microscopy (SEM) images showed individual rods with a length of 73.3 ± 16.5 nm and thickness of 20.7 ± 3.7 nm, leaving a pore diameter of 16.7 ± 4.8 nm (Fig. 2a). The CuCrO2 film (resulting from 6 layers) was approximately 500 nm thick. N2 gas adsorption isotherms showed type IV behaviour consistent with a mesoporous material and gave a BET surface area of 25 m2 g−1 (Fig. S2†), which is similar to that obtained with other mesoporous structures.47 The direct bandgap of CuCrO2 was estimated from a Tauc plot as 3.1 eV (Fig. S3†) and the flatband potential, Efb, of +1.0 V vs. RHE with Mott–Schottky analysis from consecutive impedance scans (Fig. S4†). This is 0.25 V more positive than the Efb of our NiO electrodes.37 See Experimental section for more details about synthesis and characterisation of the electrodes.
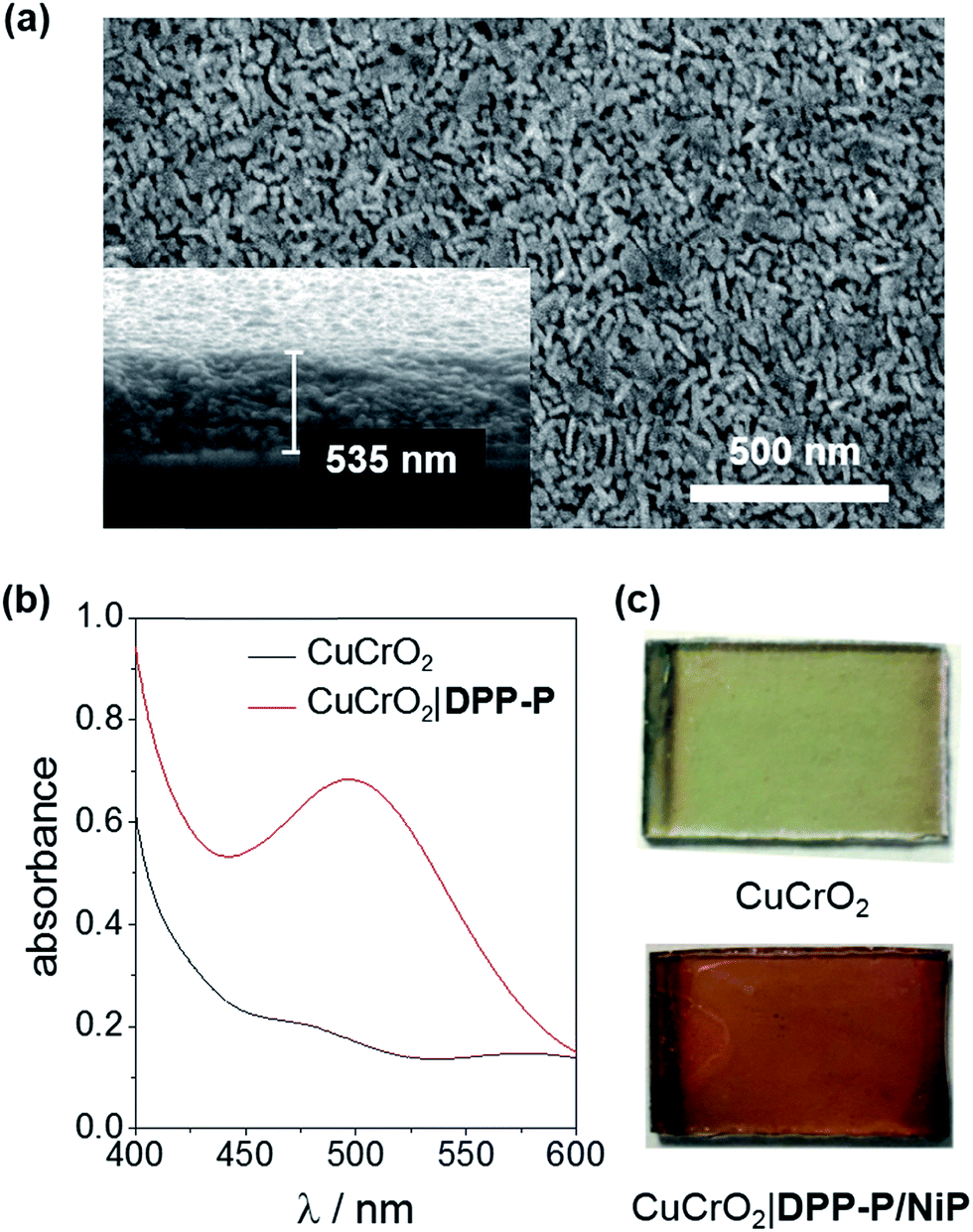 | ||
| Fig. 2 (a) Top-down and cross-sectional (inset) images of a 6-layer CuCrO2 electrode, (b) transmission UV-Vis spectrum of CuCrO2 and CuCrO2|DPP-P electrodes (ITO-glass background subtracted), (c) photographs of the as-prepared CuCrO2 and CuCrO2|DPP-P/NiP electrodes. | ||
Components of the molecule-loaded CuCrO2 photoelectrode
As dye and catalyst species, we selected DPP-P and NiP respectively, both recently synthesised in our group (Fig. 1a).67,68 For the most suitable light absorber, a dye with sufficient driving force to reduce the H2 evolution catalyst as well as a thermodynamically accessible reduction potential for the extraction of holes by CuCrO2 is required. Diketopyrrolopyrrole (DPP) chromophores have recently displayed high activity with NiO in p-DSSCs and are considered suitable candidates due to their high photostability, simple synthesis and modification, and lack of precious metal elements.36DPP-P absorbs strongly in the visible range (ε496 nm = 2.6 × 104 M−1 cm−1, DMF)67 and is expected to undergo reductive quenching when immobilised on a p-type semiconductor due to fast hole injection originating from the proximity and good electrical communication between the dye and semiconductor.69–74 In this pathway, the first step upon dye excitation is the reduction of DPP-P* by hole injection into the valence band of CuCrO2, followed by oxidation of DPP-P− by the catalyst, which ultimately performs the chemical reaction. NiP, a Dubois-type Ni-catalyst75,76 featuring four phosphonic acid anchoring groups, has previously demonstrated reduction of aqueous protons both in solution and when immobilised on a semiconductor surface whilst maintaining molecular integrity during photocatalysis.5,6,67,68DPP-P has a reduction potential in the excited state of +1.57 V vs. RHE and the reduced dye has an oxidation potential of −0.7 V vs. RHE, thus DPP-P− can provide sufficient driving force for the reduction of NiP to a catalytically active state (onset of catalytic current for NiP = −0.21 V vs. RHE).68 The respective electrochemical potential of each component and the hole and electron transfer pathways for the fully assembled DPP-P/NiP-modified CuCrO2 electrode is shown in Fig. 1b and the corresponding energy diagram with possible recombination routes in Fig. S5.†Photoelectrochemistry of CuCrO2|DPP-P
To evaluate the compatibility of DPP-P with CuCrO2 and to ensure this interface could function without the kinetic limitations imposed by immobilisation of a molecular catalyst, PEC measurements were conducted on dye-sensitised electrodes in the presence of a soluble electron acceptor. These photocathodes were prepared by soaking CuCrO2 electrodes in a DPP-P solution (1 mM, DMF) for 15 h. The UV-Vis spectrum of the electrodes with immobilised DPP-P displays an absorption maximum at approximately 500 nm, consistent with the electronic transition of the free dye (Fig. 2b and c).67 Linear sweep voltammetry (LSV) and chronoamperometry experiments were carried out in an aqueous Na2SO4 electrolyte solution (0.1 M, pH 3) at room temperature in a N2-purged one-compartment three-electrode electrochemical cell using a Pt counter electrode and a Ag/AgCl/KClsat reference electrode. UV-filtered simulated solar light was used for all PEC measurements (100 mW cm−2, AM 1.5G, λ > 420 nm). In control experiments without the acceptor, the bare CuCrO2 electrodes displayed a small cathodic dark current, which has previously been attributed to the reduction of Cu2+ impurities to Cu+ with oxygen deintercalation (Fig. 3a).77 Irradiation of the unmodified and DPP-P modified electrodes resulted in only minor photocurrents without a soluble acceptor (|j| < 3 μA cm−2, 0.0 V vs. RHE) (Fig. 3a).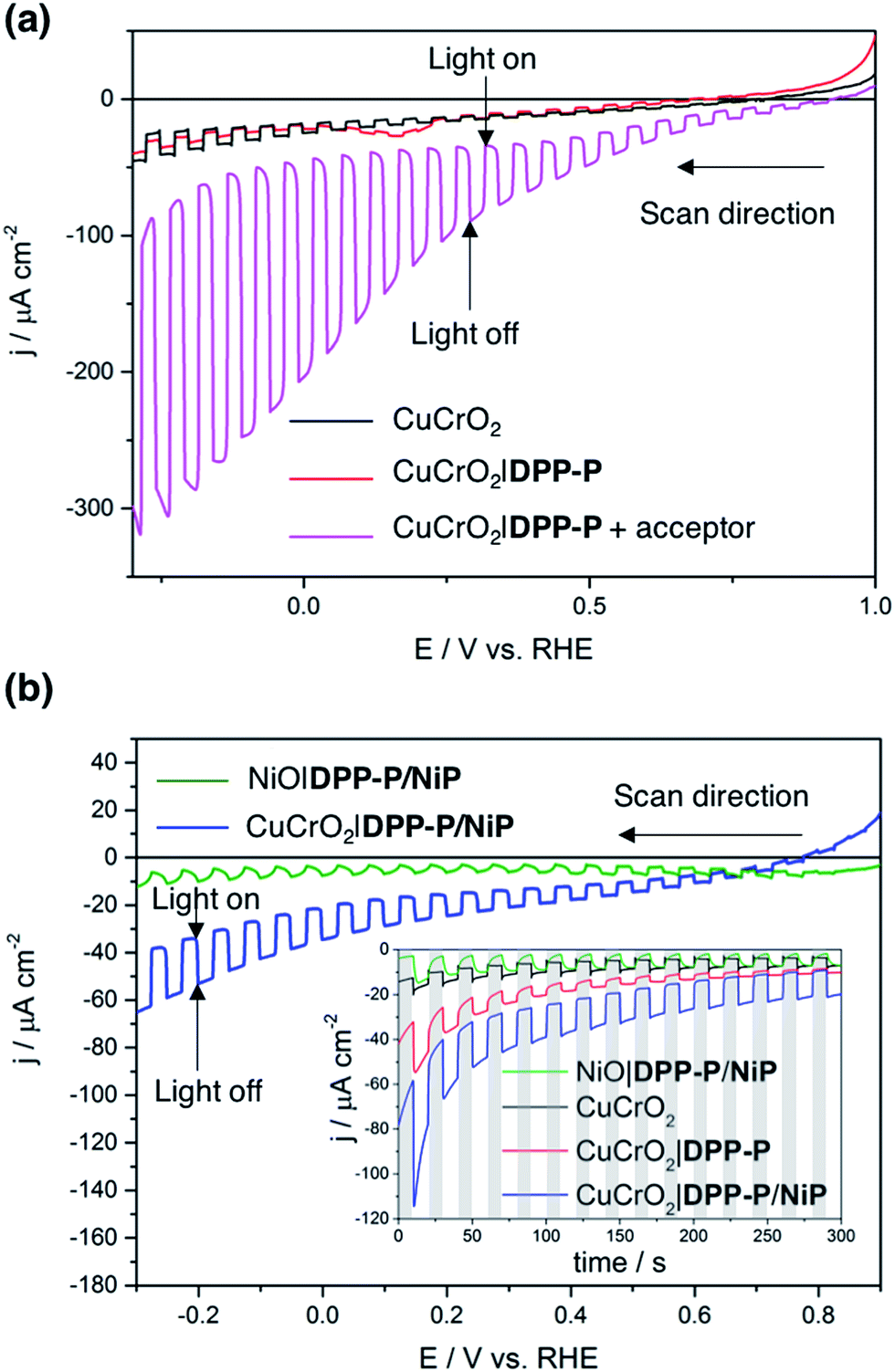 | ||
| Fig. 3 Linear sweep voltammograms under chopped light illumination of (a) CuCrO2 (black) and CuCrO2|DPP-P (red) electrodes, and a CuCrO2|DPP-P electrode with 5 mM DTDP acceptor in solution (magenta), (b) LSV scans of CuCrO2|DPP-P/NiP (blue) and NiO|DPP-P/NiP (green) electrodes along with chronoamperograms (inset) of all relevant electrode compositions. The dark chops are shown with grey lines for the chronoamperograms. All experiments were performed in aqueous Na2SO4 electrolyte solution (0.1 M) adjusted to pH 3. Illumination with 100 mW cm−2, AM 1.5G, with a 420 nm cutoff filter at room temperature. An active electrode area of 0.25 cm2 was used with a scan rate of 5 mV s−1 for voltammograms. | ||
Addition of the electron acceptor 4,4′-dithiodipyridine (DTDP, 5 mM) in the electrolyte solution allows for estimation of a maximal attainable photocurrent as DTDP is known to be easily reduced in solution (Ered,DTDP = −0.06 V vs. RHE).37 The electron acceptor allows the photoreduced dye to dispose of photo-electrons and to regenerate the ground state, thereby limiting the effects of reductive dye decomposition and charge recombination, and dramatically enhancing the photocathodic response for CuCrO2|DPP-P. An absolute photocurrent response of ≈160 μA cm−2 (0.0 V vs. RHE, Fig. 3a) was observed, which indicates efficient light-induced hole injection from the dye to the valence band of CuCrO2 with reduction of the acceptor by DPP-P−. For comparison, a NiO electrode sensitised in the same manner displayed a lower maximum photocurrent (|j| ≈ 80 μA cm−2, 0.0 V vs. RHE), suggesting lower susceptibility to recombination between the reduced dye and holes in CuCrO2 (Fig. S6†). Thus, DPP-P displays excellent electronic communication with CuCrO2, which suggests that co-anchoring of a catalyst could be a viable approach to exploit the reductive power of DPP-P− for solar H2 production.
Photoelectrochemistry with CuCrO2|DPP-P/NiP
Catalyst and dye molecules were co-immobilised on CuCrO2 electrodes through soaking in a solution of NiP (0.5 mM) and DPP-P (1 mM), in DMF for 15 h. The loading of DPP-P was quantified by UV-Vis spectroscopy following desorption in alkaline solution and the amount of immobilised NiP determined using inductively coupled plasma optical emission spectroscopy (ICP-OES) measurements. This resulted in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of dye to catalyst on the electrodes (Table S1†). Co-immobilisation of NiP and DPP-P on CuCrO2 resulted in a five-fold enhancement in photocurrent compared to the bare electrode (|j| = 15.1 μA cm−2, 0.0 V vs. RHE) (Fig. 3b). This increased response is attributed to the ability of DPP-P− to reduce NiP and ultimately protons.67 This is supported by the incident photon-to-current efficiency (IPCE) spectrum, which displays a maximum photocurrent at the same wavelength as the absorption maximum of DPP-P (λmax = 500 nm, Fig. S7†). For comparison, CuCrO2 electrodes showed low efficiency and no peak at this wavelength, demonstrating the essential role of the sensitiser.
:1 ratio of dye to catalyst on the electrodes (Table S1†). Co-immobilisation of NiP and DPP-P on CuCrO2 resulted in a five-fold enhancement in photocurrent compared to the bare electrode (|j| = 15.1 μA cm−2, 0.0 V vs. RHE) (Fig. 3b). This increased response is attributed to the ability of DPP-P− to reduce NiP and ultimately protons.67 This is supported by the incident photon-to-current efficiency (IPCE) spectrum, which displays a maximum photocurrent at the same wavelength as the absorption maximum of DPP-P (λmax = 500 nm, Fig. S7†). For comparison, CuCrO2 electrodes showed low efficiency and no peak at this wavelength, demonstrating the essential role of the sensitiser.
H2 generation was studied using controlled potential photoelectrolysis (CPPE) under constant light illumination with an applied potential of 0.0 V vs. RHE. The CPPE trace of the CuCrO2|DPP-P/NiP electrode showed high stability over a 2 hour period (Fig. S8†) with 94 ± 10 nmol of H2 generated, corresponding to a turnover number of the NiP catalyst (TONcat) of 126 ± 13 and a faradaic efficiency (FE) of 34 ± 8%. Possible explanations for the modest FE are the dark current originating from Cu2+ reduction and oxygen deintercalation,77 as well as capacitive currents due to the mesoporous structure or from electrons trapped in surface states.56,78–80 The FE is lowered by probable photobleaching/decomposition and desorption of the dye species, and is overall comparable to previously reported dye-sensitised photocathodes (Table 1). Control experiments without dye (CuCrO2|NiP) or catalyst (CuCrO2|DPP-P) produced no detectable hydrogen, confirming that the full assembly is required for catalysis. A comparable NiO|DPP-P/NiP electrode modified in the same manner only yielded 35 ± 2 nmol of H2 after 2 hours, with a FE of 31 ± 8%, demonstrating the superior performance (2–3 times) of CuCrO2 (Table 1). Accurate quantification of the Ni-catalyst loading on NiO was not possible by ICP-OES (same element in catalyst and substrate) or by UV-Vis spectroscopy following desorption (low molar absorption of NiP).
| Substrate | Dye | Catalyst | pH | Electrolyte solution | |j|/μA cm−2@Eapp/V vs. RHE | Faradaic efficiency | TONcat | Reference |
|---|---|---|---|---|---|---|---|---|
| a n. r.: not reported. b Calculated from the reference. | ||||||||
| CuCrO2 | DPP-P | NiP | 3 | Na2SO4 | 15@0.00 | 34 ± 8% | 126 ± 13.3 (2 h) | This work |
| NiO | DPP-P | NiP | 3 | Na2SO4 | 5.8@0.00 | 31 ± 8% | n. r.a | This work |
| NiO | Ru complex (RuP3) | NiP | 3 | Na2SO4 | ∼10@0.30 | 8.6 ± 2.3% | ≈1 (2 h)b | 37 |
| NiO | Dyad organic dye-cobalt diimine-dioxime | 5.5 | MES/NaCl | 15@0.14 | 9.5% | ≈4 (2 h)b | 18 | |
| NiO | Supramolecular Ru complex-cobaloxime assembly | 7 | Phosphate | 8@0.51 | 68% | n. r.a | 40 | |
| NiO | Organic dye (P1) | Cobaloxime | 7 | Phosphate | ∼5@0.21 | 68% | n. r.a | 39 |
| NiO | Ru complex (RuP) | Cobaloxime (CoHEC) | 7 | Phosphate | 13@0.21 | n. r.a | n. r.a | 41 |
| NiO | Coumarine C343 | Fe2(CO)6(bdt) | 4.5 | Acetate | ∼10@0.16 | 50% | ≤3 (18 min) | 38 |
| NiO | CdSe | Cobaloxime | 6.8 | Na2SO4 | 100@0.40 | 81% | n. r.a | 42 |
| ITO | Supramolecular Ru complex-(RuP2)-NiP assembly | 5.1 | MES | 56@0.05 | 53 ± 5% | ≈16 (3 h)b | 43 | |
| NiKO|NiCuO|ioITO | Supramolecular Ru complex (RuP2)-NiP assembly | 5.0 | MES/KCl | ∼59@0.05 | ∼90% | ≈20 (2 h) | 44 | |
Post-electrolysis characterisation of CuCrO2|DPP/NiP electrodes using ICP-OES showed that the amount of NiP retained on the surface after 2 h of CPPE was 54% of the initial loading (Table S1†). This is in part due to the relatively low surface area exhibited by the delafossite particulates (25 m2 g−1), which accounts for low loadings of catalyst and dye, and allows for their easy desorption into the media. Nanostructuring of the surface would ensure higher loadings of dye and catalyst species, enhancing both stability and activity in the future. Alternate methods such as atomic layer deposition (ALD)52,81–83 or polymeric assembly84–87 could also be employed as additional stabilisation methods.
Comparison with state-of-the-art
Limited improvements in photocathode development for DSPEC proton reduction are largely due to p-type materials with low performance. Since the first report in 1999 towards p-type DSSC, dye-sensitised NiO electrodes have generated a range of beneficial research on dye architecture and electrolyte composition.48,88,89 Despite this, their performance remains significantly lower than their n-type counterparts, highlighting the limitations of NiO and the need for a better alternative. Table 1 highlights relevant examples as a comparison for our system.The TONcat is a good measure of catalytic activity for a molecular catalyst-based system but remains unreported in most cases. A TONcat > 125 after 2 h for our CuCrO2 system in water compares favourably with the currently highest reported value of ≈20 for a NiO DSPEC photocathode.43
With NiP as the catalyst, an ITO electrode produced higher photocurrents and more H2,44 but PEC activity has only been demonstrated for an applied potential of +0.05 V vs. RHE. CuCrO2 allows for a much higher working voltage due to the onset potential being situated at +0.75 V vs. RHE and therefore shows greater suitability for energy storage and implementation in tandem DSPEC cells. This photocurrent onset is also more favourable than other commonly used narrow bandgap p-type semiconductors such as GaP,90,91 and p-Si,5,92 highlighting the benefits of moving to dye-sensitised systems for H2 generation.
CdSe-sensitised NiO produces the highest amount of H2 of these electrodes over the duration of 2 hours of CPPE,42 but a large portion of the photocurrent stems from the bare quantum dots. Despite this, sensitisation with quantum dot species is a viable approach to further enhance the H2 producing capability of a CuCrO2-based photocathode in the future. In comparing these properties, it is clear that material alteration can have a great influence on activity, and that transferring from NiO to CuCrO2 has advantages for DSPEC applications.
Conclusions
We have introduced CuCrO2 co-sensitised with an organic dye (DPP-P) and molecular catalyst (NiP) for DSPEC H2 generation under aqueous conditions. CuCrO2|DPP-P/NiP showed a photocurrent onset at +0.75 V vs. RHE and a photocurrent density of 15 μA cm−2 at 0.0 V vs. RHE with a TONcat of 126 ± 13 achieved in controlled potential photoelectrolysis under UV-filtered simulated solar light irradiation. The molecule-loaded delafossite electrode therefore surpasses the performance of benchmark NiO electrodes in side-by-side comparison. We also show that the phosphonated organic DPP dye allows for high performance in aqueous conditions on an electrode and is able to electronically cooperate with NiP, which enabled us to assemble a fully precious metal-free DSPEC photocathode. The photocathode displays a higher photovoltage than other current state-of-the-art materials such as p-Si and GaP, making it well suited for coupling with a photoanode in tandem water splitting. Co-immobilisation of a dye and a CO2 reduction catalyst on this p-type semiconductor may allow photocathodic production of carbon based fuels and chemical feedstocks.The synthesis of CuCrO2 by sol–gel techniques is straightforward and scalable. Nanostructuring would enhance the molecular loading and provide another avenue to increase photocurrents and the H2 producing capability of the photocathode. Material alteration, for example through Mg2+ doping,62 could also improve the activity by further enhancing conductivity and therefore charge extraction through the film. Other methods to improve the separation between catalyst and the delafossite surface would also enhance the efficiency by reducing charge recombination.37 This work demonstrates the benefit of adopting new delafossite structures as p-type semiconductors for solar fuel generation.
Experimental section
Materials and methods
NiP 68 and DPP-P67 were synthesised according to previously reported methods. Cu(acetate)2·H2O (ACROS Organics, ACS reagent), Cr(NO3)3·9H2O (Sigma-Aldrich, ≥99%), and triethanolamine (Sigma-Aldrich, ≥99.5%) were used to prepare CuCrO2. ITO-coated glass substrates (Vision Tek Systems Ltd., R = 12 Ω cm−2, thickness = 1.1 mm) were cut into 3 × 3 cm2 squares and scored into 1 × 1.5 cm2 divisions before cleaning. Milli-Q® H2O (R > 18.2 MΩ cm) was used for all electrochemical and analytical measurements. DTDP (Sigma-Aldrich, 98%) was used as an electron acceptor at a concentration of 5 mM. Addition of DTDP resulted in a change in pH of the electrolyte solution from 3 to 4.6.Preparation of CuCrO2 electrodes
ITO-coated glass was cleaned through successive sonication in isopropanol, ethanol, and acetone for 15 min each, followed by drying at 100 °C in air before use. A mixture of Cu(acetate)2·H2O (0.2 M), and triethanolamine (0.4 M) in absolute ethanol was stirred for 1 h before addition of Cr(NO3)3·9H2O (0.2 M). This solution was kept stirring for 15 h before being spin-coated on the ITO-glass slides (Laurell WS-650MZ spin coater, 1500 rpm, 15 s, 3000 rpm s−1 acceleration, 0.4 mL volume). The slides were annealed in air to 400 °C for 45 min with a ramp rate of 10 °C min−1 in a chamber furnace (Carbolite Gero). These steps were repeated to form 6 layers. The final annealing step involved heating in a N2 atmosphere to 600 °C for 45 min with a ramp rate of 5 °C min−1 using a tube furnace fitted with a quartz tube, end seals, and insulation plugs (Carbolite Gero). The electrodes were left to cool to room temperature and used as-prepared without any additional treatment.Material characterisation
XRD measurements were conducted using a PANalytical BV X'Pert Pro X-ray diffractometer. SEM images were taken using a FEI Phillips XL30 sFEG microscope. UV-Vis absorption spectra were obtained using a Varian Cary 50 spectrophotometer in transmission mode.N2 gas adsorption measurements
Adsorption isotherms were carried out using a Micromeritics 3 Flex (Micromiretics, Norcross, GA, USA) with N2 as the adsorbate. Samples were prepared on glass slides then scraped from the surface. Degassing for 10 h at 110 °C was required prior to measurements, which were carried out in liquid N2. The BET specific surface area was obtained by fitting N2 isotherms using the Microactive software.Mott–Schottky analysis
Electrochemical impedance spectroscopy (EIS) measurements were conducted using an IviumStat potentiostat at 25 °C using a 3-necked round-bottomed flask under dark conditions. A three-electrode setup using a Pt mesh counter, Ag/AgCl/KClsat reference, and a CuCrO2 working electrode (0.25 cm2 active area) was used with an electrolyte solution of Na2SO4 (0.1 M, pH 3). The frequency range was 10 kHz to 0.01 Hz, with an excitation voltage of 10 mV. Nyquist plots obtained in the potential range 1.1 V to 0.3 V vs. RHE (15 mV step) were fitted using ZView® (Scribner Associates Inc.) to a Randles circuit (inset Fig. S4†) to obtain interfacial capacitance (Csc) values. The Mott–Schottky equation,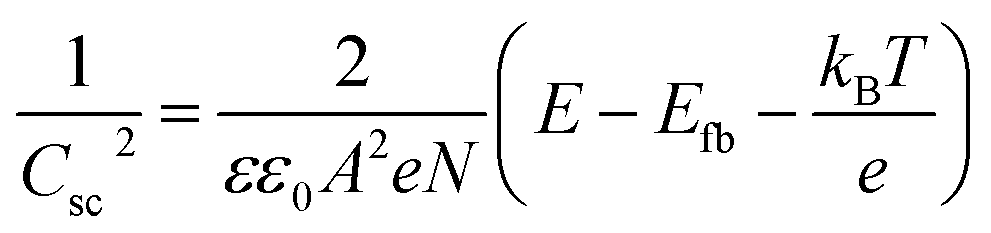 , was used to obtain an estimate of the flatband potential through a plot of 1/Csc2 against the applied potential. A negative slope indicated p-type character and the x-intercept is equal to Efb + kBT/e.37
, was used to obtain an estimate of the flatband potential through a plot of 1/Csc2 against the applied potential. A negative slope indicated p-type character and the x-intercept is equal to Efb + kBT/e.37
Electrochemical measurements
Cyclic voltammetry was used to determine the reduction potential of the DPP-P dye, E(S/S−), from the half-wave potential. This was performed in a 3-electrode setup with a glassy carbon working electrode, Pt-mesh counter electrode, and a Ag/AgCl/KClsat reference electrode with a scan rate of 50 mV s−1. The electrolyte solution consisted of tetrabutylammonium tetrafluoroborate (0.1 M) in dry DMF with the addition of DPP-P (around 0.1 M). Addition of the E00 to E(S/S−) provides an estimate for the excited state reduction potential, E(S*/S−).Modification of electrodes with dye and catalyst species
Molecular species were co-immobilised through soaking in a bath consisting of DPP-P (1 mM) and NiP (0.5 mM) in DMF for 15 h. For CuCrO2|DPP-P and CuCrO2|NiP electrodes the concentration was 1 mM but all other conditions kept the same. All electrodes were rinsed with DMF and H2O then dried in air and stored in the dark before use.Quantification of loaded DPP-P and NiP
DPP-P was desorbed from CuCrO2|DPP-P/NiP electrodes using a solution of 0.1 M tetrabutylammonium hydroxide 30-hydrate in DMF (1 mL) and the absorption at 500 nm was determined using UV-Vis spectroscopy. A calibration curve was used to fit values and determine the loading for 4 different electrodes. NiP was quantified by ICP-OES after digestion of CuCrO2|DPP-P/NiP electrodes (1 cm2 film area) in aqueous HNO3 (70%, 1 mL) overnight and dilution to 10% v/v with MilliQ® water. CuCrO2|DPP-P/NiP electrodes pre- and post-electrolysis were analysed along with blanks for nitric acid, CuCrO2, and CuCrO2|DPP-P in triplicate. Errors represent standard deviation from the mean.37PEC measurements
Photoelectrochemical measurements were carried out using an Ivium CompactStat potentiostat in a one-compartment three-necked custom made cell equipped with a flat borosilicate glass window. A three-electrode setup was used with a Pt-counter electrode, a Ag/AgCl/KClsat reference, and the working electrode consisted of the CuCrO2 platform with an illuminated area of 0.25 cm2 confined using electrical tape. All measurements were conducted using aqueous Na2SO4 electrolyte solution (0.1 M, pH 3) and the cell was purged with N2 for 15 min prior to experiments. Frontside illumination was used for all experiments using a calibrated Newport Oriel solar light simulator (150 W, 100 mW cm−2, AM 1.5G) fitted with a UQG Optics UV Filter (λ > 420 nm) and IR water filter.CPPE experiments were carried out in a custom two-compartment airtight electrochemical cell separated by a Nafion membrane and featuring a flat quartz glass window. The volume of electrolyte solution in the working compartment was 12 mL with a gas headspace of 5 mL while the counter compartment consisted of 4.5 mL solution and a 3.5 mL headspace. Prior to electrolysis, the gas headspace was purged for 30 min with 2% CH4 in N2. An Agilent 7890A series gas chromatograph with a 5 Å molecular sieve column and a thermal conductivity detector was used to quantify the amount of H2 produced. The oven temperature was kept constant at 45 °C and the flow rate was 3 mL min−1. The partial pressure of H2 was calculated to account for dissolved H2 and this was added to the overall amount of hydrogen generated to obtain the faradaic efficiency. All CPPE experiments were carried out in triplicate with an applied potential of 0.0 V vs. RHE.
IPCE measurements
IPCE spectra were recorded in a N2-purged three-necked one-compartment custom cell with a flat borosilicate glass window. A three-electrode setup with Pt counter, Ag/AgCl/KClsat reference, and working electrode was used with pH 3 Na2SO4 electrolyte solution (0.1 M). Monochromatic light was provided using a 300 W Xenon lamp solar light simulator coupled to a monochromator (MSH300, LOT Quantum design) and the intensity calibrated to 0.8 mW cm−2 for each wavelength. The potential was maintained at 0.0 V vs. RHE for all wavelengths and photocurrents were recorded in triplicate with different electrodes (0.25 cm2 active area) for both CuCrO2 and CuCrO2|DPP-P/NiP arrangements.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Christian Doppler Research Association (Austrian Federal Ministry of Science, Research, and Economy and the National Foundation for Research, Technology and Development), the OMV Group (E. R. and J. W.), and the EPSRC NanoDTC in Cambridge (EP/L015978/1; E. R. and C. E. C.). Thanks to Dr Benjamin Martindale (Department of Chemistry, University of Cambridge) for providing NiP, Mr Alan Dickerson (Department of Chemistry, University of Cambridge) for ICP-OES measurements, and Mohammad Hadi Modarres and Dr Michael de Volder (Department of Engineering, University of Cambridge) for help with BET measurements. We appreciate the help of Dr Bertrand Reuillard and Miss Jane J. Leung for their suggestions and comments on the manuscript.References
- Y. Tachibana, L. Vayssieres and J. R. Durrant, Nat. Photonics, 2012, 6, 511–518 CrossRef CAS.
- J. Willkomm, K. L. Orchard, A. Reynal, E. Pastor, J. R. Durrant and E. Reisner, Chem. Soc. Rev., 2015, 45, 9–23 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- J. J. Leung, J. Warnan, D. H. Nam, J. Z. Zhang, J. Willkomm and E. Reisner, Chem. Sci., 2017, 8, 5172–5180 RSC.
- T. E. Rosser, M. A. Gross, Y.-H. Lai and E. Reisner, Chem. Sci., 2016, 7, 4024–4035 RSC.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- N. Queyriaux, N. Kaeffer, A. Morozan, M. Chavarot-Kerlidou and V. Artero, J. Photochem. Photobiol., C, 2015, 25, 90–105 CrossRef CAS.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 15010 CrossRef CAS.
- M. P. Stewart, M. H. Ho, S. Wiese, M. Lou Lindstrom, C. E. Thogerson, S. Raugei, R. M. Bullock and M. L. Helm, J. Am. Chem. Soc., 2013, 135, 6033–6046 CrossRef CAS PubMed.
- G. Sahara and O. Ishitani, Inorg. Chem., 2015, 54, 5096–5104 CrossRef CAS PubMed.
- D.-I. Won, J. S. Lee, J. M. Ji, W. J. Jung, H. J. Son, C. Pac and S. O. Kang, J. Am. Chem. Soc., 2015, 137, 13679–13690 CrossRef CAS PubMed.
- M. F. Kuehnel, K. L. Orchard, K. E. Dalle and E. Reisner, J. Am. Chem. Soc., 2017, 139, 7217–7223 CrossRef CAS PubMed.
- S. Roy, B. Sharma, J. Pécaut, P. Simon, M. Fontecave, P. D. Tran, E. Derat and V. Artero, J. Am. Chem. Soc., 2017, 139, 3685–3696 CrossRef CAS PubMed.
- B. Reuillard, K. H. Ly, T. E. Rosser, M. F. Kuehnel, I. Zebger and E. Reisner, J. Am. Chem. Soc., 2017, 139, 14425–14435 CrossRef CAS PubMed.
- J. Willkomm, N. M. Muresan and E. Reisner, Chem. Sci., 2015, 6, 2727–2736 RSC.
- K. Sekizawa, K. Maeda, K. Domen, K. Koike and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 4596–4599 CrossRef CAS PubMed.
- N. Kaeffer, J. Massin, C. Lebrun, O. Renault, M. Chavarot-Kerlidou and V. Artero, J. Am. Chem. Soc., 2016, 138, 12308–12311 CrossRef CAS PubMed.
- Y. Tamaki and O. Ishitani, ACS Catal., 2017, 7, 3394–3409 CrossRef CAS.
- A. Reynal, F. Lakadamyali, M. A. Gross, E. Reisner and J. R. Durrant, Energy Environ. Sci., 2013, 6, 3291–3300 CAS.
- G. Neri, M. Forster, J. J. Walsh, C. M. Robertson, T. J. Whittles, P. Farràs and A. J. Cowan, Chem. Commun., 2016, 52, 14200–14203 RSC.
- E. Pastor, F. Le Formal, M. T. Mayer, S. D. Tilley, L. Francàs, C. A. Mesa, M. Grätzel and J. R. Durrant, Nat. Commun., 2017, 8, 14280 CrossRef CAS PubMed.
- T. E. Rosser and E. Reisner, ACS Catal., 2017, 7, 3131–3141 CrossRef CAS.
- M. Schreier, J. Luo, P. Gao, T. Moehl, M. T. Mayer and M. Grätzel, J. Am. Chem. Soc., 2016, 138, 1938–1946 CrossRef CAS PubMed.
- L. Ji, M. D. McDaniel, S. Wang, A. B. Posadas, X. Li, H. Huang, J. C. Lee, A. A. Demkov, A. J. Bard, J. G. Ekerdt and E. T. Yu, Nat. Nanotechnol., 2014, 10, 84–90 CrossRef PubMed.
- S. Hu, N. S. Lewis, J. W. Ager, J. Yang, J. R. McKone and N. C. Strandwitz, J. Phys. Chem. C, 2015, 119, 24201–24228 CAS.
- M. Crespo-Quesada and E. Reisner, Energy Environ. Sci., 2017, 10, 1116–1127 CAS.
- P. Xu, N. S. McCool and T. E. Mallouk, Nano Today, 2017, 14, 42–58 CrossRef CAS.
- Z. Yu, F. Li and L. Sun, Energy Environ. Sci., 2015, 8, 760–775 CAS.
- E. A. Gibson, Chem. Soc. Rev., 2017, 46, 6194–6209 RSC.
- M. S. Prévot and K. Sivula, J. Phys. Chem. C, 2013, 117, 17879–17893 Search PubMed.
- G. Sahara, H. Kumagai, K. Maeda, N. Kaeffer, V. Artero, M. Higashi, R. Abe and O. Ishitani, J. Am. Chem. Soc., 2016, 138, 14152–14158 CrossRef CAS PubMed.
- F. F. Abdi, L. Han, A. H. M. Smets, M. Zeman, B. Dam and R. van de Krol, Nat. Commun., 2013, 4, 2195 Search PubMed.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- K. Fan, F. S. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. C. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- Y. Farré, L. Zhang, Y. Pellegrin, A. Planchat, E. Blart, M. Boujtita, L. Hammarström, D. Jacquemin and F. Odobel, J. Phys. Chem. C, 2016, 120, 7923–7940 Search PubMed.
- M. A. Gross, C. E. Creissen, K. L. Orchard and E. Reisner, Chem. Sci., 2016, 7, 5537–5546 RSC.
- L. J. Antila, P. Ghamgosar, S. Maji, H. Tian, S. Ott and L. Hammarström, ACS Energy Lett., 2016, 1, 1106–1111 CrossRef CAS.
- F. Li, K. Fan, B. Xu, E. Gabrielsson, Q. Daniel, L. Li and L. Sun, J. Am. Chem. Soc., 2015, 137, 9153–9159 CrossRef CAS PubMed.
- Z. Ji, M. He, Z. Huang, U. Ozkan and Y. Wu, J. Am. Chem. Soc., 2013, 135, 11696–11699 CrossRef CAS PubMed.
- K. Fan, F. Li, L. Wang, Q. Daniel, E. Gabrielsson and L. Sun, Phys. Chem. Chem. Phys., 2014, 16, 25234–25240 RSC.
- P. Meng, M. Wang, Y. Yang, S. Zhang and L. Sun, J. Mater. Chem. A, 2015, 3, 18852–18859 CAS.
- B. Shan, B. D. Sherman, C. M. Klug, A. Nayak, S. L. Marquard, Q. Liu, R. M. Bullock and T. J. Meyer, J. Phys. Chem. Lett., 2017, 8, 4374–4379 CrossRef CAS PubMed.
- B. Shan, A. Das, S. L. Marquard, B. H. Farnum, D. Wang, R. M. M. Bullock and T. J. Meyer, Energy Environ. Sci., 2016, 9, 3693–3697 CAS.
- L. Lepleux, B. Chavillon, Y. Pellegrini, E. Blart, L. Cario, S. Jobic and F. Odobel, Inorg. Chem., 2009, 48, 8245–8250 CrossRef CAS PubMed.
- E. A. Gibson, M. Awais, D. Dini, D. P. Dowling, M. T. Pryce, J. G. Vos and A. Hagfeldt, Phys. Chem. Chem. Phys., 2013, 15, 2411–2420 RSC.
- C. J. Wood, G. H. Summers, C. A. Clark, N. Kaeffer, M. Braeutigam, L. R. Carbone, L. D'Amario, K. Fan, Y. Farré, S. Narbey, F. Oswald, L. A. Stevens, C. D. J. Parmenter, M. W. Fay, A. La Torre, C. E. Snape, B. Dietzek, D. Dini, L. Hammarström, Y. Pellegrin, F. Odobel, L. Sun, V. Artero and E. A. Gibson, Phys. Chem. Chem. Phys., 2016, 18, 10727–10738 RSC.
- F. Odobel, Y. Pellegrin, E. A. Gibson, A. Hagfeldt, A. L. Smeigh and L. Hammarström, Coord. Chem. Rev., 2012, 256, 2414–2423 CrossRef CAS.
- A. Morandeira, J. Fortage, T. Edvinsson, L. Le Pleux, E. Blart, G. Boschloo, A. Hagfeldt, L. Hammarström and F. Odobel, J. Phys. Chem. C, 2008, 112, 1721–1728 CAS.
- Z. Huang, G. Natu, Z. Ji, M. He, M. Yu and Y. Wu, J. Phys. Chem. C, 2012, 116, 26239–26246 CAS.
- K. A. Click, D. R. Beauchamp, Z. Huang, W. Chen and Y. Wu, J. Am. Chem. Soc., 2016, 138, 1174–1179 CrossRef CAS PubMed.
- R. J. Kamire, M. B. Majewski, W. L. Hoffeditz, B. T. Phelan, O. K. Farha, J. T. Hupp and M. R. Wasielewski, Chem. Sci., 2017, 8, 541–549 RSC.
- C. J. Flynn, S. M. McCullough, E. Oh, L. Li, C. C. Mercado, B. H. Farnum, W. Li, C. L. Donley, W. You, A. J. Nozik, J. R. McBride, T. J. Meyer, Y. Kanai and J. F. Cahoon, ACS Appl. Mater. Interfaces, 2016, 8, 4754–4761 CAS.
- I. Sullivan, B. Zoellner and P. A. Maggard, Chem. Mater., 2016, 28, 5999–6016 CrossRef CAS.
- M. Yu, T. I. Draskovic and Y. Wu, Phys. Chem. Chem. Phys., 2014, 16, 5026–5033 RSC.
- M. S. Prévot, X. A. Jeanbourquin, W. S. Bourée, F. Abdi, D. Friedrich, R. van de Krol, N. Guijarro, F. Le Formal and K. Sivula, Chem. Mater., 2017, 29, 4952–4962 CrossRef.
- M. S. Prévot, N. Guijarro and K. Sivula, ChemSusChem, 2015, 8, 1359–1367 CrossRef PubMed.
- J. Gu, Y. Yan, J. W. Krizan, Q. D. Gibson, Z. M. Detweiler, R. J. Cava and A. B. Bocarsly, J. Am. Chem. Soc., 2014, 136, 830–833 CrossRef CAS PubMed.
- A. K. Díaz-García, T. Lana-Villarreal and R. Gómez, J. Mater. Chem. A, 2015, 3, 19683–19687 Search PubMed.
- S. Götzendörfer, C. Polenzky, S. Ulrich and P. Löbmann, Thin Solid Films, 2009, 518, 1153–1156 CrossRef.
- S. Powar, D. Xiong, T. Daeneke, M. T. Ma, A. Gupta, G. Lee, S. Makuta, Y. Tachibana, W. Chen, L. Spiccia, Y. B. Cheng, G. Götz, P. Bäuerle and U. Bach, J. Phys. Chem. C, 2014, 118, 16375–16379 CAS.
- D. Xiong, W. Zhang, X. Zeng, Z. Xu, W. Chen, J. Cui, M. Wang, L. Sun and Y. B. Cheng, ChemSusChem, 2013, 6, 1432–1437 CrossRef CAS PubMed.
- D. Xiong, Z. Xu, X. Zeng, W. Zhang, W. Chen, X. Xu, M. Wang and Y. B. Cheng, J. Mater. Chem., 2012, 22, 24760–24768 RSC.
- D. O. Scanlon, K. G. Godinho, B. J. Morgan and G. W. Watson, J. Chem. Phys., 2010, 132, 24707 CrossRef PubMed.
- T. Jiang, X. Li, M. Bujoli-Doeuff, E. Gautron, L. Cario, S. Jobic and R. Gautier, Inorg. Chem., 2016, 55, 7729–7733 CrossRef CAS PubMed.
- D. O. Scanlon and G. W. Watson, J. Mater. Chem., 2011, 21, 3655–3663 RSC.
- J. Warnan, J. Willkomm, J. N. Ng, R. Godin, S. Prantl, J. R. Durrant and E. Reisner, Chem. Sci., 2017, 8, 3070–3079 RSC.
- M. A. Gross, A. Reynal, J. R. Durrant and E. Reisner, J. Am. Chem. Soc., 2014, 136, 356–366 CrossRef CAS PubMed.
- A. M. Brown, L. J. Antila, M. Mirmohades, S. Pullen, S. Ott and L. Hammarström, J. Am. Chem. Soc., 2016, 138, 8060–8063 CrossRef CAS PubMed.
- F. A. Black, C. A. Clark, G. H. Summers, I. P. Clark, M. Towrie, T. Penfold, M. W. George and E. A. Gibson, Phys. Chem. Chem. Phys., 2017, 19, 7877–7885 RSC.
- B. Dhital, V. G. Rao and H. P. Lu, Phys. Chem. Chem. Phys., 2017, 19, 17216–17223 RSC.
- A. L. Smeigh, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel and L. Hammarström, Chem. Commun., 2012, 48, 678–680 RSC.
- M. Borgström, E. Blart, G. Boschloo, E. Mukhtar, A. Hagfeldt, L. Hammarström and F. Odobel, J. Phys. Chem. B, 2005, 109, 22928–22934 CrossRef PubMed.
- L. Zhang, L. Favereau, Y. Farré, E. Mijangos, Y. Pellegrin, E. Blart, F. Odobel and L. Hammarström, Phys. Chem. Chem. Phys., 2016, 18, 18515–18527 RSC.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- W. J. Shaw, M. L. Helm and D. L. Du Bois, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1123–1139 CrossRef CAS PubMed.
- W. Ketir, A. Bouguelia and M. Trari, Desalination, 2009, 244, 144–152 CrossRef CAS.
- J. Bisquert, Phys. Chem. Chem. Phys., 2003, 5, 5360–5364 RSC.
- L. Bertoluzzi, P. Lopez-Varo, J. A. Jiménez Tejada and J. Bisquert, J. Mater. Chem. A, 2016, 4, 2873–2879 CAS.
- J. Bisquert, Phys. Chem. Chem. Phys., 2008, 10, 49–72 RSC.
- A. M. Lapides, B. D. Sherman, M. K. Brennaman, C. J. Dares, K. R. Skinner, J. L. Templeton and T. J. Meyer, Chem. Sci., 2015, 6, 6398–6406 RSC.
- H. J. Son, C. Prasittichai, J. E. Mondloch, L. Luo, J. Wu, D. W. Kim, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 11529–11532 CrossRef CAS PubMed.
- D. Wang, M. V. Sheridan, B. Shan, B. H. Farnum, S. L. Marquard, B. D. Sherman, M. S. Eberhart, A. Nayak, C. J. Dares, A. K. Das, R. M. Bullock and T. J. Meyer, J. Am. Chem. Soc., 2017, 139, 14518–14525 CrossRef CAS PubMed.
- B. Reuillard, J. Warnan, J. J. Leung, D. W. Wakerley and E. Reisner, Angew. Chem., Int. Ed., 2016, 55, 3952–3957 CrossRef CAS PubMed.
- S. K. Ibrahim, X. Liu, C. Tard and C. J. Pickett, Chem. Commun., 2007, 1535–1537 RSC.
- D. H. Pool and D. L. DuBois, J. Organomet. Chem., 2009, 694, 2858–2865 CrossRef CAS.
- A. Krawicz, J. Yang, E. Anzenberg, J. Yano, I. D. Sharp and G. F. Moore, J. Am. Chem. Soc., 2013, 135, 11861–11868 CrossRef CAS PubMed.
- F. Odobel and Y. Pellegrin, J. Phys. Chem. Lett., 2013, 4, 2551–2564 CrossRef CAS.
- J. He, H. Lindström, A. Hagfeldt and S.-E. Lindquist, J. Phys. Chem. B, 1999, 103, 8940–8943 CrossRef CAS.
- A. M. Beiler, D. Khusnutdinova, S. I. Jacob and G. F. Moore, ACS Appl. Mater. Interfaces, 2016, 8, 10038–10047 CAS.
- D. Khusnutdinova, A. M. Beiler, B. L. Wadsworth, S. I. Jacob and G. F. Moore, Chem. Sci., 2017, 8, 253–259 RSC.
- C. A. Downes and S. C. Marinescu, J. Am. Chem. Soc., 2015, 137, 13740–13743 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7sc04476c. Additional data related to this publication is available at the University of Cambridge data repository (https://doi.org/10.17863/CAM.16678). |
| This journal is © The Royal Society of Chemistry 2018 |