
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalysis by hybrid sp2/sp3 nanodiamonds and their role in the design of advanced nanocarbon materials
Yangming
Lin
*ab,
Xiaoyan
Sun
b,
Dang Sheng
Su
 *bc,
Gabriele
Centi
d and
Siglinda
Perathoner
*e
*bc,
Gabriele
Centi
d and
Siglinda
Perathoner
*e
aMax-Planck-Institut für Chemische Energiekonversion, Stiftstraße 34-36, 45470, Mülheim an der Ruhr, Germany. E-mail: yang-ming.lin@cec.mpg.de
bShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, 72 Wenhua Road, Shenyang 110016, China
cDalian Institute of Chemical Physics, Chinese Academy of Sciences, 457 Zhongshan Road, Dalian, China. E-mail: dssu@dicp.ac.cn
dUniversity of Messina, ERIC aisbl and CASPE/INSTM, Dept.s MIFT – Industrial Chemistry, V.le F. Stagno D’Alcontres 31, 98166 Messina, Italy. E-mail: centi@unime.it
eUniversity of Messina, Dept.s ChiBioFarAm – Industrial Chemistry, V.le F. Stagno D’Alcontres 31, 98166 Messina, Italy. E-mail: perathon@unime.it
First published on 29th October 2018
Abstract
Hybrid sp2/sp3 nanocarbons, in particular sp3-hybridized ultra-dispersed nanodiamonds and derivative materials, such as the sp3/sp2-hybridized bucky nanodiamonds and sp2-hybridized onion-like carbons, represent a rather interesting class of catalysts still under consideration. Their characteristics, properties and catalytic reactivity are presented, with an analysis of the state-of-the-art of their use in gas- and liquid-phase reactions, including photo- and electro-catalysis. It is remarked that intrinsic differences exist between these and other nanostructured carbon catalysts. The analysis shows how different features make nanocarbons unique with respect to other types of catalysts and are the bases for an advanced design of nanocarbon-type catalysts. The aspects discussed regard the presence of hybrid sp2/sp3 configurations, nano-engineering related to the role of defects and vacancies in their catalytic behaviour, the creation of active sites by modification in the charge density at carbon atoms or C–C bonds, the generation of strained C–C bonds by curvature and other mechanisms, and the formation of semiconducting areas and defect sites at the interface with supported nanoparticles. The advanced strategies for identifying and quantifying active sites of carbon catalysts are highlighted.
 Yangming Lin | Dr Yangming Lin received his PhD degree from the Institute of Metal Research, Chinese Academy of Sciences, in 2016, under the supervision of Prof. Dang Sheng Su and became a scientist at the Max-Planck-Institut für Chemische Energiekonversion (Germany). His main research interest is in the fabrication of carbon materials and semiconductor materials for catalysis and energy conversion. |
 Xiaoyan Sun | Dr Xiaoyan Sun obtained her PhD degree from the Institute of Metal Research, Chinese Academy of Sciences, in 2015 and became a Postdoc at Technische Universität Berlin (Germany). She has broad research interests in heterogeneous catalysis, materials chemistry and surface chemistry. |
 Dang Sheng Su | Prof. Dang Sheng Su completed his PhD at the Technical University of Vienna (Austria) in 1991, and moved to the Fritz-Haber-Institut der Max-Planck-Gesellschaft in the Department of Electron Microscopy. After a short stay at Hahn-Meitner Institut GmbH and Humboldt Universität zu Berlin (Germany) he joined the Fritz-Haber-Institut in 1999, where he worked on nanomaterials in heterogeneous catalysis and energy storage. He was then Professor and Head of the Catalysis and Materials Division of the Shenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, and now is Professor in Physical Chemistry at the Dalian Institute of Chemical Physics, Chinese Academy of Sciences. |
 Gabriele Centi | Gabriele Centi is full professor of Industrial Chemistry at the University of Messina (Italy), President of IACS (International Association of the Catalysis Societies) and of ERIC aisbl (European Research Institute of Catalysis). He was the coordinator of the Network of Excellence on catalysis IDECAT and of several EU projects. He is Chair of the editorial board of ChemSusChem, and co-Editor in chief of the Journal of Energy Chemistry (Elsevier) and of various book series. He was Chairperson of Europacat 2017 in Florence, Italy. He has published several reviews on catalysis and green energy, and he is author of over 450 scientific publications. The current h-index (Google Scholar) is 78 with about 22 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 citations.
500 citations. Siglinda Perathoner | Siglinda Perathoner obtained her PhD in Chemical Science in 1988 working on the photophysics and photochemistry of supramolecular systems with V. Balzani and Nobel Laureate J. M. Lehn. In 2001 she joined the University of Messina and is associate professor of Industrial Chemistry presently. She has coordinated many EU projects. Among the awards and recognitions, the participation in 2011 to the “Nanolife” movie produced for the European Commission to show to the public the results of nanotechnology. Her research interests include nanostructured oxides and nanocarbon materials for heterogeneous and photo- and electro-catalytic applications. The h-index (Google Scholar) is 63 with about 14 |
1. Introduction
Nanocarbon-based catalysts have represented an area of increasing interest in recent years.1–30 The term ‘nanocarbons’ defines materials where nano-dimensionality largely influences their characteristics and catalytic performances. Between the motivations of this interest15,31–35 may be cited the facts that these materials (i) represent catalysts with characteristics different from classical types of materials, such as oxides, supported metals, zeolites and other types of micro/mesoporous materials, (ii) can be synthesized in a large variety of nanostructures, today often in a cost-competitive way, (iii) can be tuned by different modalities, particularly by doping and functionalization, and (iv) show often good conductivity characteristics, an essential property for the development of electrocatalysts and materials for energy, an area of growing relevance due to the on-going transition to a new energy and chemistry production.36,37To address some of the demanding challenges arising from this transition, it is necessary to develop also disruptive catalysts,38i.e. catalysts that radically change the current concepts of catalysis and related reaction mechanisms, thus creating new market opportunities and disrupting the existing ones. Nanocarbon-based materials, particularly those metal-free,24,35,39–41 have many of the characteristics to be considered disruptive-type catalysts, because their reaction mechanism is often conceptually different from that present in current catalysts, for example in ODH (oxidative dehydrogenation) reactions.24
They can be synthesized in a variety of nanostructures (for example, carbon nano-fibres, -tubes, -coils, -horns, -diamonds, -onions, graphene, etc.) and dimensionalities, from nearly zero dimension (0D) (carbon nanodots), to 1D (nanotubes and nanofibers, and related classes of materials), 2D (graphene and graphene-like materials) and 3D (mesoporous carbons, and a large variety of 3D structures, such as exfoliated carbon structures, graphene scaffolds, hierarchical porous carbon foams, carbon nanofiber arrays).2,27,42–47 In addition, their properties can be largely modified by doping and functionalization, and the creation of hybrid materials between different carbon nanostructures or with different catalytic elements, such as metals or metal-oxide nanoparticles.48–59 Furthermore, they can host even isolated metal atoms, an area of increasing interest recently.60–67
Nanocarbons thus offer a wide range of possibilities for an advanced design of catalytic sites.68 However, notwithstanding the relevant advances in their understanding as remarked in several reviews1–30 in this area, there are still several aspects, which are underestimated.
A graphene sheet is the base structural element to understand the catalytic reactivity of nanocarbon materials. The carbon atoms in an ideal graphene sheet are essentially catalytically inactive due to their electronic structure. Only at the edges of graphene patches, where the symmetric structure is broken, are reactive sites present. This is somewhat similar to the catalytic chemistry of solid crystals, where the reactive sites are located at the edges, corners and similar coordinatively unsaturated sites. However, in nanocarbons, a larger number of possibilities are present with respect to “classical” heterogeneous catalysts. In fact, in nanocarbons, the ideal symmetric electronic configuration can be broken by bending the ideal graphene sheet, as occurs, for example at the ends of carbon nanotubes (CNTs), where several local distortions, such as pentagonal or seven-membered rings, are necessary to minimize the energy necessary for the bending.
These defects are a key crucial aspect to understand the chemistry and catalytic reactivity of nanocarbons and are related to the possibility of easy change in the hybridization of carbon atoms, leading also to a large variety of structures possible in nanocarbons. In addition to defects where distortion leads to a rehybridization (for example, the formation of coupled C5 and C7 rings instead of a C6 ring, with consequent rehybridization between sp2 and sp3), more classical defects related to missing C-atom defects (for example, vacancies or dislocations) are possible. Although defect chemistry is present and relevant in other classes of catalysts, such as oxides, it is a more relevant aspect in nanocarbons due to the larger possibilities of the change in the hybridization of C-atoms.
Another important characteristic in nanocarbons, which again differentiates them from “classical” catalysts, is the possibility of doping with heteroatoms. O and N are the most common doping heteroatoms and can be introduced into nanocarbons to substitute C (inducing changes in the local coordination and hybridization) or to saturate dangling bonds. These heteroatoms are electronegative with respect to carbon, inducing consequently a rupture of the charge neutrality. Other types of heteroatoms, such as B, can instead accept electrons from carbon because of their valence of three, thus inducing a shift in the Fermi level of the conduction band.
These heteroatoms form functional reactive groups (aldehydic, acid, phenolic, chetonic, epoxy or quinone groups for oxygen doping, and quaternary N, pyridine, amine, or pyrrole for nitrogen doping, just as examples) which induce acid or redox catalytic functionalities, although often the different stabilities of these functional groups (by annealing or during the catalytic reaction) are not taken into proper account. In addition, the introduction of these heteroatoms induces local distortion in the structure of sp2-hybridized carbons, thus inducing changes in the electron density at carbon atoms. Heteroatoms thus not only introduce reactive sites but also induce changes in the electronic local structure of the nanocarbons. Both aspects are relevant for the catalytic reactivity, although the second aspect is often underestimated.
There is thus rich catalytic chemistry deriving from doping with heteroatoms, which can be differentiated based on (a) reactive sites directly introduced (for example, Brønsted acid or redox sites) and (b) active sites deriving from the introduction of changes in the electronic structure of nearlying carbon atoms. The latter include aspects such as Lewis acid–base sites, like pyridinic N and carbon atoms next to them (the electro-negative nitrogen induces high positive charge density on adjacent carbon atoms).69 While the first type of catalytic chemistry in nanocarbons has been extensively discussed, the second type of catalytic chemistry related to how the introduction of heteroatoms creates reactive carbon atoms has been discussed in much less detail. In general, there is catalytic chemistry in nanocarbons related to the presence of reactive C sites related not only to dangling bonds, but also to the creation of reactive C atoms of C–C bonds associated with heteroatoms, defects, strains and curvature effects. In nanocarbons with hybrid sp2/sp3 configurations, such as the nanodiamond family, it may be expected that this type of chemistry could be enhanced with respect to sp2-type nanocarbons (like graphene, fullerenes, CNTs and derivatives). However, the catalytic chemistry of NDs and derivatives of this carbon family has been less studied with respect to other nanocarbon materials, and never reviewed in detail.
The scope of this review is thus to discuss the characteristics and catalytic behaviour of nanodiamonds and related materials to highlight how the results indicate the general role of reactive carbon sites in determining their catalytic behaviour, an aspect that we consider underestimated in understanding the possibilities opened by nanocarbons as novel catalytic materials.
2. Hybrid sp2/sp3 nanocarbons
Metal-free carbocatalysis1,24,35,39,40,49,70–76 has inspired many studies in recent years, but most centred on the use of functional sp2-type nanocarbons, such as graphene and CNT families. Less attention has been instead paid to other types of nanocarbon materials, particularly to sp3-hybridized ultra-dispersed nanodiamonds (NDs) and their derivatives (UNDDs), including sp3/sp2-hybridized bucky nanodiamonds (BNDs) and sp2-hybridized onion-like carbons (OLCs). They are emerging as a new category of the nanocarbon family, with interesting physicochemical properties, such as superior thermal and chemical stability, high surface energy, and unique π and σ bonding configurations. The latter aspects allow rich surface chemistry with characteristics not present in other nanocarbon materials.15 These properties have made UNDDs competitive candidates for catalytic reactions besides conventional metal-based catalysts. In addition, they provide excellent materials to understand how the catalytic performance or the molecular structure and surface chemistry depend on the presence of hybrid sp2/sp3 configurations rather than a predominantly sp2 configuration as in most of the nanocarbons reported in the cited studies. Current applications of UNDDs are mainly limited to their use as support, biocarrier, lubricant and fluorescent materials.77–81Ultra-dispersed NDs are the raw materials for UNDDs. NDs have been found in interstellar dusts, meteorites and protoplanetary nebulae82 and were first produced by a detonation method.83,84 Different methods, such as chemical detonation, high-energy ball-milling of diamond microcrystals grown under high-pressure high-temperature conditions, laser ablation and plasma-assisted chemical vapor deposition (CVD), are now known to produce high sp3-hybridized NDs. So far, chemical detonation has been the most cost-effective route to achieve large-scale production of NDs with high quality and uniform particle size (4–8 nm, the average value is about 5 nm, as determined by HRTEM). The obtained NDs can be purified effectively using liquid oxidants (such as HNO3, or H2SO4–HNO3 mixtures) and HCl to remove non-diamond carbon and trace metal impurities (Ag, Cu, Fe and other metals), respectively.81 NDs produced in this way result in a high-purity metal-free carbon material.
2.1 Characteristics of UNDDs
High temperature annealing (T ≤ 1500 °C, vacuum or inert atmosphere) of NDs produces BNDs (Fig. 1), which are sp2/sp3 hybrid materials with a sp3 carbon core covered with few sp2 graphite-like shells. Depending on the particle size and surface nature of original NDs, the number of graphitic shells of BNDs ranges from three to seven layers. BNDs combine the remarkable surface properties of graphitic materials with the characteristics of a diamond core. These promising hybrid materials have highly defective surfaces, high stability and a large presence of functional surface groups. BNDs can be further transformed into onion-like carbons (OLCs) at higher annealing temperature, generally >1500 °C (Fig. 1). OLC is an interesting non-planar self-enclosed graphitic material with multiple layers of curved sp2 concentric shells (Fig. 1).88,89 Detonation of NDs is the preferred method for the synthesis of large amounts of BNDs and OLCs at low cost.
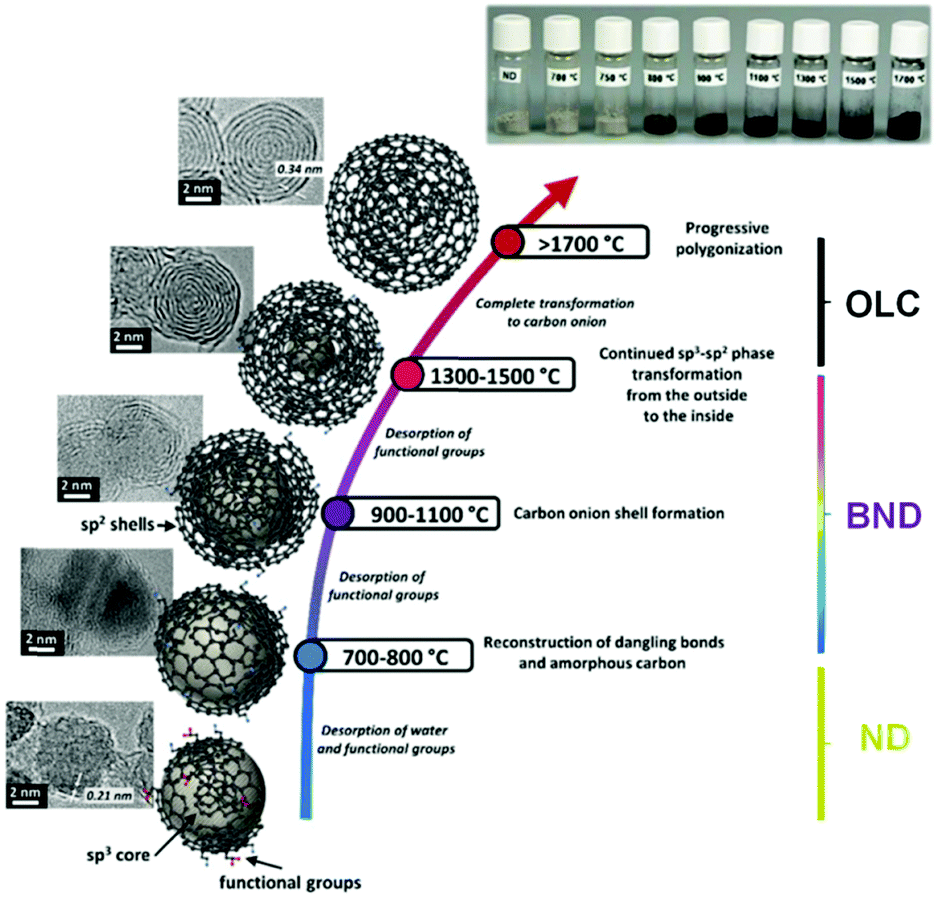 | ||
| Fig. 1 Phase transformation from ND to OLC by annealing treatment together with HRTEM images and optical images of UNDDs, schematic illustrations of intermediate steps, and the assignment of possible physical effects depending on the annealing temperature. Reproduced from ref. 89 with permission from Royal Society of Chemistry, copyright 2016. | ||
X-ray diffraction (XRD) shows a significant (002) graphite peak for temperatures higher than 1100 °C, whose intensity increases with increasing annealing temperature, while the intensity of the (111) diamond peak decreases (Fig. 2a). The rate of phase transformation increases with temperature and the transformation is nearly complete for temperatures higher than 1500 °C, as illustrated by a small (111) diamond peak in Fig. 2a.
 | ||
| Fig. 2 (a) XRD spectra and (b) vis Raman spectra of UNDDs annealed at different temperatures. (c) ADF-STEM image of an OLC and (d) the corresponding intensity profile. The difference in the number of graphitic shells is clearly shown. (e) Schematic model of a single ∼5 nm ND. The diamond core is covered with various oxygen groups and a thin layer of amorphous and disordered carbon, where sp2 carbon atoms are shown in black, oxygen atoms in red, and nitrogen in blue. The particles are stabilized by terminating the dangling bonds. Reproduced from ref. 88 with permission from Nature Publishing Group. (f) Schematic model of a single ∼5 nm OLC. Reproduced from ref. 89 and 90 with permission from Royal Society of Chemistry, copyright 2018. | ||
Raman spectroscopy is a powerful tool to characterize the structural transformations of NDs and their derivatives (UNDDs). Raman spectra of BNDs and OLCs annealed above 800 °C exhibit a G-mode (1585 cm−1) deriving from the vibration of carbon atoms in sp2-hybridized carbon networks and a broad D-mode (1325 cm−1) correlated with the breathing of hexagonal carbon rings with defects (Fig. 2b). At lower synthesis temperatures, such as 800 °C and 1100 °C, the Raman spectra of the material resemble an amorphous carbon A-mode (∼1490 cm−1) covering the surfaces of the particles, similar to the ND precursor. Due to the limited susceptibility of visible light excitation to insulating NDs, UV-Raman spectroscopy is often preferred. The diamond sp3-mode and G-mode of NDs upon UV laser excitation are located at 1336 cm−1 and 1647 cm−1, respectively.94 When the synthesis temperature is 800 °C, the occurrences of the graphitic G-mode and the disordered D-mode derived from the ND precursor imply that the sp3-hybridized carbon starts to transform into sp2-hybridized OLC. The G-mode shows a relatively broad shape due to the large bond length variation in the material.
Higher annealing temperature or a longer annealing time will make the D- and G-modes sharper and lower the intensity of the secondary A-mode signal. For example, the ID/IG value of 1300 BND (calculated by the intensity ratio) decreases from 1.31 (4 h annealing) to 1.23 (8 h). The ID/IG value of the OLC decreases from 1.12 (4 h) to 1.02 (12 h).
The ADF-STEM image clearly shows the onion structure of the OLC (Fig. 2c and d). The corresponding intensity profile provides a direct and striking representation of the shell numbers (7–10 layers) of the OLC structure.90 Mochalin and co-workers built the representative models of NDs and OLCs on the basis of the presence of surface functional groups and their variability, sp2 carbon and the shape of the particles (Fig. 2e and f).81 After annealing at high temperature, the average particle size of OLCs does not change obviously and surface functional groups will be removed together with phase transformation.
In general, the thermal phase transformation of sp3-hybridized NDs into sp2-hybridized OLC involves a multistep process. It begins with the desorption of water and the detachment of surface oxygen functional groups from the NDs when heating up to around 200 °C. Upon increasing the temperature up to 1300–1500 °C, some functional groups like carboxyl, anhydride, and lactone groups are progressively removed with the formation of CO and CO2. The dangling bonds on carbon atoms deriving from the progressive detachment of functional oxygen groups above 800 °C may further change with the formation of new π-bonds. The onset temperature of graphitization (namely, phase transformation) is typically in the 800–900 °C range. The HRTEM of BNDs further supports this indication. The phase transformation generates sp2-hybridized carbon shells on the outside of the NDs, followed by continuous graphitization inside the particles.94 Both the existing structural defects in ND surfaces and newly produced deriving from the detachment of surface functional groups increase the reactivity of surface carbon atoms and thus facilitate the phase transformation process.91 For temperatures above 1100 °C, the initially highly disordered carbon shells become increasingly more graphitic with a lower defect density. A highly ordered OLC can be obtained when the temperature is above 1500 °C.
Therefore, by controlling the annealing temperature and time, the structural defect degree and surface oxygen groups of BNDs or OLCs can be easily tuned. The color of the UNDDs gradually changes from the gray of the NDs to black of the OLCs, as shown in the optical images of Fig. 1.
Two faces (111) and (110) of NDs have recently been considered to be the most likely starting points for phase transformation at elevated temperature, as described by Kuznetsov et al.92 As shown in Fig. 3a, the graphitization process occurring at these two faces involves two steps: B/C or F/G. The binding energy per bond is found to be 2.75 eV for the (111) face and 3.92 eV for the (110) face, suggesting that graphitization of the (111) face is thermodynamically preferred over that of the (110) surface. The latter has to overcome a substantially higher barrier. It was proposed that the difference between interlayer binding energies over (111) and (110) originates from the different “packing” patterns of interlayer bonds in a volume between “parallel” planes. For the (111) planes, all such bonds are parallel in contrast to those of the (110) planes (B/C and F/G of Fig. 3a). More recently, as shown in Fig. 3b, the transformation of a ND particle into a large fullerene (like OLC) has been studied with the C–C bond rotation as the primary transformation step. The result shows a process of surface sp2 hybridization → sp2 C cage encapsulated diamond particle → enlargement of the fullerene.93
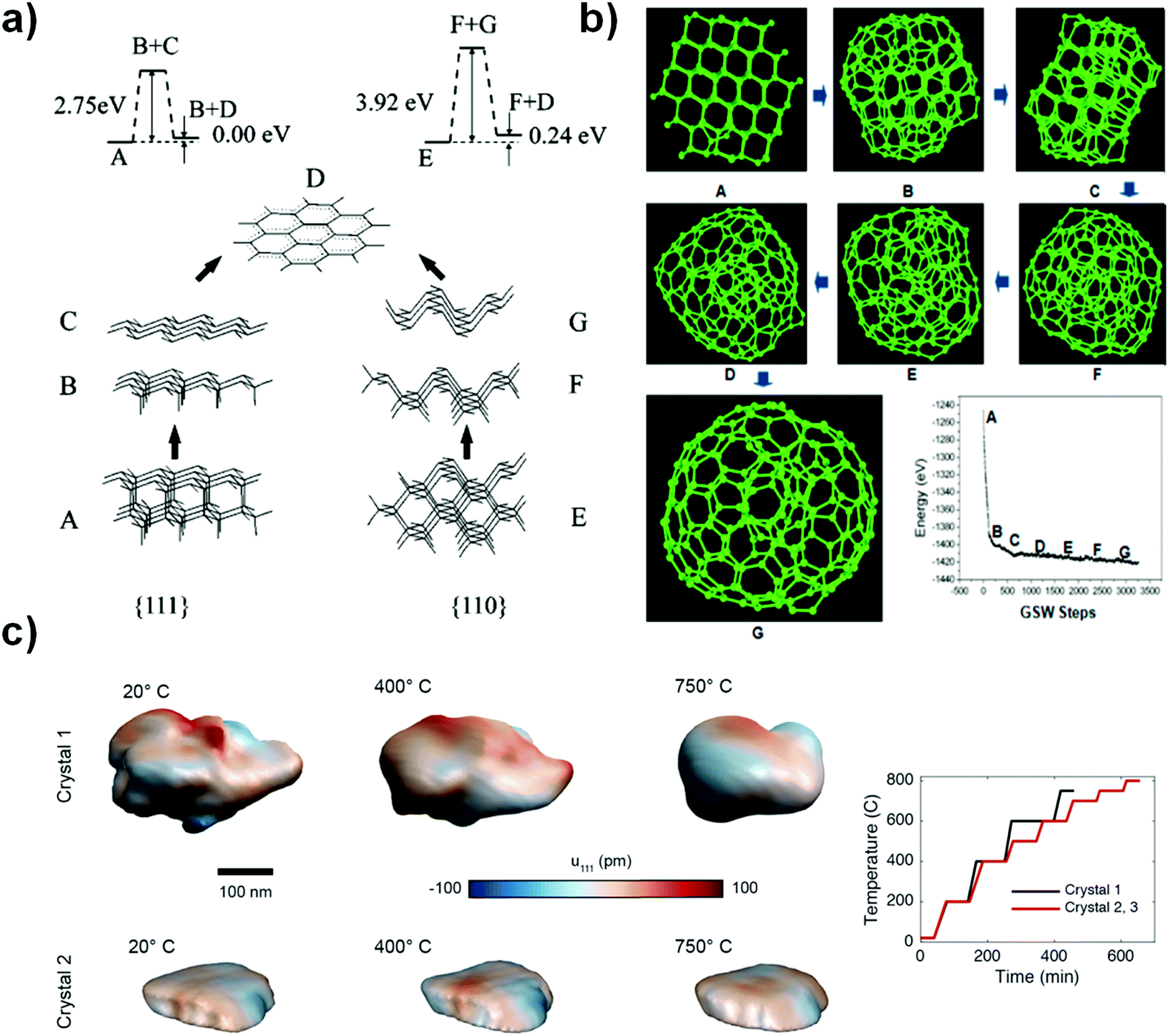 | ||
| Fig. 3 (a) Phase transformation study of UNDDs using two-layer cluster modeling of the graphitization of diamond (111) and (110) faces, reprinted from ref. 92 with permission from the AIP Publishing, copyright 1999. (b) Transformation of a ND into a large fullerene (namely, OLC) simulated by the EDKMC method, reprinted from ref. 93 with permission from American Chemical Society, copyright 2014. (c) In situ Bragg coherent X-ray diffraction imaging (BCDI) reconstructions of two kinds of NDs (Crystals 1 and 2) at different stages during annealing, reprinted from ref. 94 with the AIP Publishing, copyright 2017. | ||
Bragg coherent diffraction imaging (BCDI) was used for in situ study of the strain relaxation of NDs induced by annealing temperature (Fig. 3c). Before that, the selected NDs were drop-cast onto a (100)-surface-oriented silicon substrate. BCDI measurements were performed at a (111) Bragg peak of ND crystals at various temperatures in a helium environment. Upon increasing the temperature, the morphology and internal strain state of the NDs change, indicating the presence of lattice distortions and surface graphitization. The improvements in the homogeneity of the crystal lattice depend on the initial strain state. This method provides a direct 3D image to obtain intuitive understanding of the phase transformation of NDs into OLCs.94 However, the detailed mechanism of transformation from an almost pure sp3 ND to a mixed sp2/sp3 BND and then to a neat sp2 OLC is still not fully clear, despite also computational efforts.95–97
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups rather than aromatic-type carbon.98 The content of the latter is only 1.1 ± 0.4%. sp3-Hybridized C–H and C–OH carbons, which cover most of the ND particle's surface, accounting for ∼5% each. The concentrations of CO and –COOH groups were measured to be about 1.5%. Therefore, the total content of carbon (C–H, C–OH, CO, and CC groups) is up to 12–14% of all carbon, which matches the surface fraction expected for bulk terminated ∼5 nm diameter ND particles.
O groups rather than aromatic-type carbon.98 The content of the latter is only 1.1 ± 0.4%. sp3-Hybridized C–H and C–OH carbons, which cover most of the ND particle's surface, accounting for ∼5% each. The concentrations of CO and –COOH groups were measured to be about 1.5%. Therefore, the total content of carbon (C–H, C–OH, CO, and CC groups) is up to 12–14% of all carbon, which matches the surface fraction expected for bulk terminated ∼5 nm diameter ND particles.
The amount of surface oxygen groups of NDs approximates 9.8% as determined by XPS (Fig. 4a).99 It can be fitted into carbonyl groups (CO, 531.8 eV), carbon–oxygen ether-like single bonds (C–O, 532.8 eV) and phenolic groups (–OH, 533.7 eV). After annealing, the concentration of oxygen species of all BND samples decreases, however, higher than those of sp2-hybridized reduced graphite oxide (GR, sample from Tanmei Ltd, China) and multi-walled carbon nanotubes (MWCNTs, sample from Shandong Dazhan Technology Ltd, China). Besides, it should be remarked that the specific characteristics of NDs largely depend on many aspects, such as the size, shape and synthesis as well as purification methods. The surface oxygen groups of NDs, including carboxylic acids, ketones, phenols and lactones, endorse the surface negative charge of the NDs over a pH range of 2–12.81 Such abundant surface oxygen-terminated structures and potential disordered carbon shells provide the potential sites for catalysis.100–102
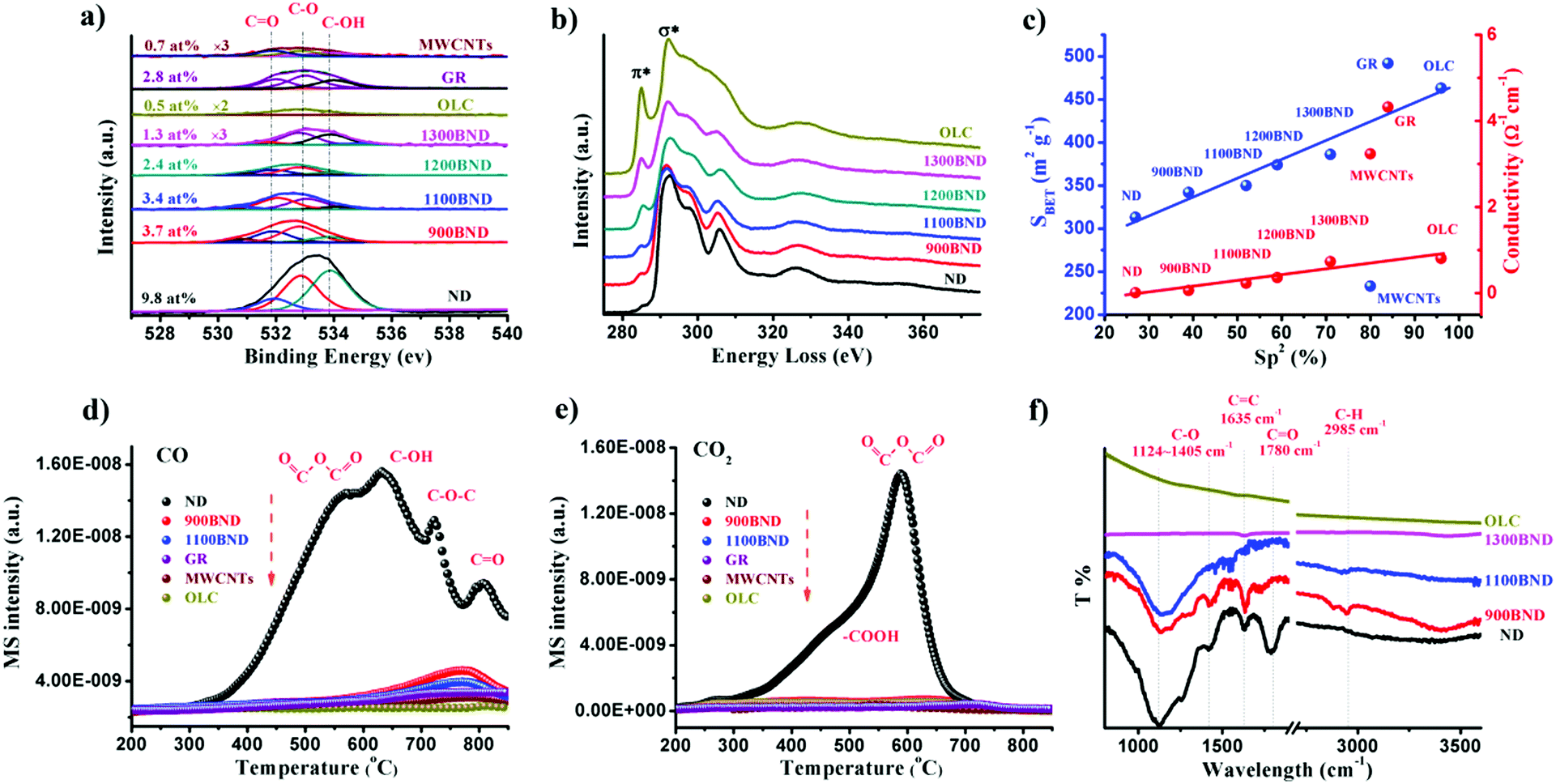 | ||
| Fig. 4 (a) XPS O 1s spectra of representative carbon materials. (b) EELS spectra of UNDDs. (c) Specific surface area and conductivity in NDs and derived materials as functions of the relative fraction of sp2 carbon (sp2, %). (d and e) TPD-CO (d, m/z = 28) and TPD-CO2 (e, m/z = 44) profiles of various carbon materials in a He atmosphere using a heating rate of 10 K min−1. (f) FTIR spectra of UNDDs. Reproduced from ref. 99 with permission from Royal Society of Chemistry, copyright 2017. | ||
Using electron energy-loss spectroscopy (EELS), the sp2 content of the OLC is found to be about 96% (Fig. 4b and c). In addition, the sp2 graphitization degree of the OLC is much higher than those of GR (84%) and MWCNTs (80%). There is a good linear trend in the ND, BND and OLC series with respect to the surface area, which increases from 313 m2 g−1 (NDs) to about 460 m2 g−1 (OLC). The change in surface groups during the transformation of the NDs into OLC can be analysed by TPD. Generally, the thermal transformation of the NDs begins with the desorption of water and detachment of oxygen-containing surface functional groups from sp3-hybridized carbon at around 200 °C. Further increase in the temperature to 700 °C will effectively remove functional groups such as carboxyl (–COOH, ∼450 °C), anhydride (OC–O–CO, ∼550 °C), ether (C–O–C, ∼650 °C) and phenol (C–OH, ∼700 °C) groups, with the formation of CO and CO2 gases (Fig. 4d and e). When the annealing temperature is above 800 °C, only surface CO remain. The intense signals of CO and CO2 in the NDs indicate a large amount of oxygen functionalities with respect to other types of carbon materials.
The surface functional groups are also evidenced by FTIR. The spectrum of the NDs shows strong peaks due to the oxygen functional groups (Fig. 4f), in particular C–O (1124–1405 cm−1) and CO (1780 cm−1) bands.103 The red-shift and intensity weakening of the signal in the BNDs are attributed to its specific sp3/sp2 structure and the minor amount of surface groups, respectively. The lack of bands associated with the oxygen functional groups in the OLC is related to the ordered sp2 carbon structure (96% graphitization degree) and the negligible amount of surface heteroatoms.
The thermal stability of UNDDs is extraordinary. Barnard and coworkers, by applying a model based on the calculated heat of formation, proposed that the thermostability of BNDs should range between the upper limit of fullerenes and the lower limit of NDs.104 Costa et al. studied the standard enthalpies of formation at 25 °C in various nanocarbons obtaining the following ranking in thermostability: graphite > BNDs > NDs > SWCNTs > OLCs > C60. The authors emphasized that the high stability of BNDs may be attributed to the oxygen-containing functional groups bonded to the sp2 structure.105 It is worthy of note that the results of TGA indicate that the OLC exhibits the best thermostability and oxidation resistance among other nanocarbons under an Ar atmosphere and in air, as shown by the shift to higher temperatures of the start of weight losses. The oxidation onset temperature of the OLC in air reached 600 °C. Upon increasing the annealing temperature, the particle size of the UNDDs does not show changes related to structural rearrangement. A typical particle size of 4–8 nm is maintained.
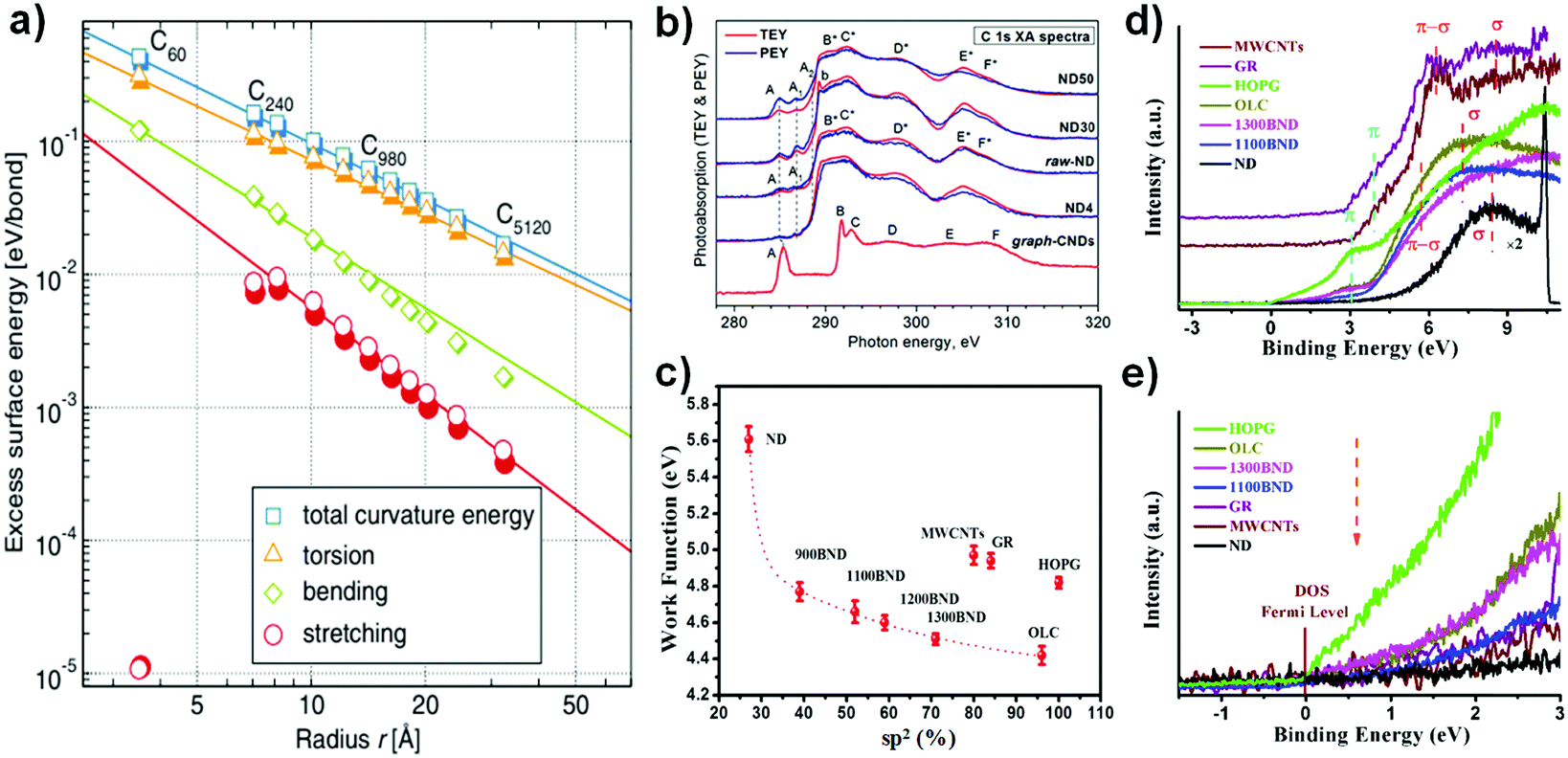 | ||
| Fig. 5 (a) Dependence of excess surface energy on carbon radius, with the graphene plane as a reference. Reproduced from ref. 108 with permission from The American Physical Society, copyright 2010. (b) Near extended X-ray absorption fine structure (NEXAFS) C 1s spectra of NDs with different particle sizes (30, 50 nm) recorded in the TEY (red lines) and PEY (blue lines) modes. Here, ‘graph-CNDs’ represents graphitized carbon. Reproduced from ref. 109 with permission from The American Physical Society, copyright 2015. (c) Relationship between sp2 and the work function in NDs and derived materials. The work function was calculated from the cut-off of secondary electrons in the UPS spectra, using Ni as a reference. (d) Valence band spectra of various carbon materials. (e) Magnified region near the Fermi level of various carbon materials. Reproduced from ref. 99 with permission from Royal Society of Chemistry, copyright 2017. | ||
The electronic structures of NDs with different particle sizes were studied with near extended X-ray absorption fine structure (NEXAFS, Fig. 5b).109 The absorption structure at A2 at a photon energy of ∼288.6 eV was regarded as a result of the C 1s → π(CO) transitions in carbonyl or carboxyl groups, CO or COOH. Moreover, the absorption structures A1 and A2 in the spectra of NDs are attributed to chemical bonding between the carbon and oxygen atoms on the surfaces of the NDs, such as C–OH, CO, and OC–OH functional groups. The intense absorption bands B*–F* can be associated with C 1s transitions in the bulk and the near-surface region of the ND particles. The lower contrast of B*–F* in the PEY (partial photoelectron yield) spectra compared with TEY (total photoelectron yield) is due to the presence of defects in the near surface region of the NDs. The authors proposed that the surfaces of all NDs with different particle sizes are covered with graphite-like carbon clusters, which are highly defective and partially oxidized. The weak intensity of the π* peak of the NDs at 285 eV in EELS spectra also points out this fact (Fig. 4b).
UPS (ultraviolet photoelectron spectroscopy) is a powerful tool to measure relevant properties such as the work function, the electron distribution in the valence band and the density of states (DOS) near the Fermi level. Fig. 5d and e shows that both BND (1100, 1300) and OLC samples have lower binding energies of the π (∼3.0 eV) and σ (∼7.2 eV) electronic transition and a steeper increase of DOS near the Fermi level as compared to MWCNTs and GR (∼3.8 eV and ∼8.6 eV).110,111 Moreover, the binding energy of the σ electronic transition of the NDs (∼8.4 eV) is also slightly lower than those of MWCNTs and GR. These results suggest that UNDDs may be activated at a lower energy than that necessary for MWCNTs and GR. The work function (ϕ) represents the minimum energy required for electrons to escape from the valence shell. A lower work function of a surface means that the material has a lower excitation energy barrier.112 The surface graphitized BNDs and OLC exhibit lower work function values than MWCNTs, GR and HOPG (Fig. 5f). This can be explained by the destabilization of the surface π-electrons (lower π electronic binding energy) due to the significant curvature of the graphitic shell, rather than to surface functional groups or the sp2 content of the nanocarbons.113–115 The effects of curvature on the work function in the case of CNTs have been previously reported.115
2.2 Catalytic reactivity of UNDDs
Typically, the catalysts used in DH and ODH reactions are transition metals and metal oxides, such as Fe2O3, Pt-, Cr2O3-, V2O5-, MoOx-, Ga2O3-based catalysts, and so on.119,120 To date, various hydrocarbons have been tested by DH or ODH using UNDDs or doped UNDDs, including ethylbenzene (EB),121–125n-butane,126 propane127–130 and methane.131 Due to their unique structure and surface properties, UNDDs and doped UNDDs not only exhibit a superior catalytic activity to other carbon materials, but in some cases they also provide even better performances than conventional metal-based catalysts. However, it is beyond the scope here to show that NDs and related materials are commercially better catalysts for these reactions. We will remark here on only their interesting catalytic chemistry related to the peculiar structure, surface and electronic properties of UNDDs discussed in the previous section.
2.2.1.1 DH reactions catalyzed by UNDDs. Zhang et al. reported excellent catalytic performance of NDs in EB dehydrogenation to styrene under steam-free conditions,125 superior to that of a commercial iron oxide catalyst used as reference, whose activity decreased rapidly from 20.2 to 9.2% within 2 h due to surface fouling by carbonaceous deposits. NDs show an activity nearly three times higher with respect to iron oxide and stable even after 60 h stream time (Fig. 6a). Note, however, that this K-promoted iron oxide used as reference was originally developed for DH of ethylbenzene strongly diluted with steam. It was also found that the ND catalyst is several times more active and selective than other nanocarbon materials such as nanographites, MWCNTs, and mesoporous carbons (Fig. 6b). In situ XPS and DRIFT tests show that the fraction of OC
O (at 530.9 eV) decreases with parallel increase of OC–O (at 532.9 eV), suggesting the reduction of CO to C–OH by the hydrocarbon and a possible relationship between ketonic CO species and the dehydrogenation reaction (Fig. 6c).
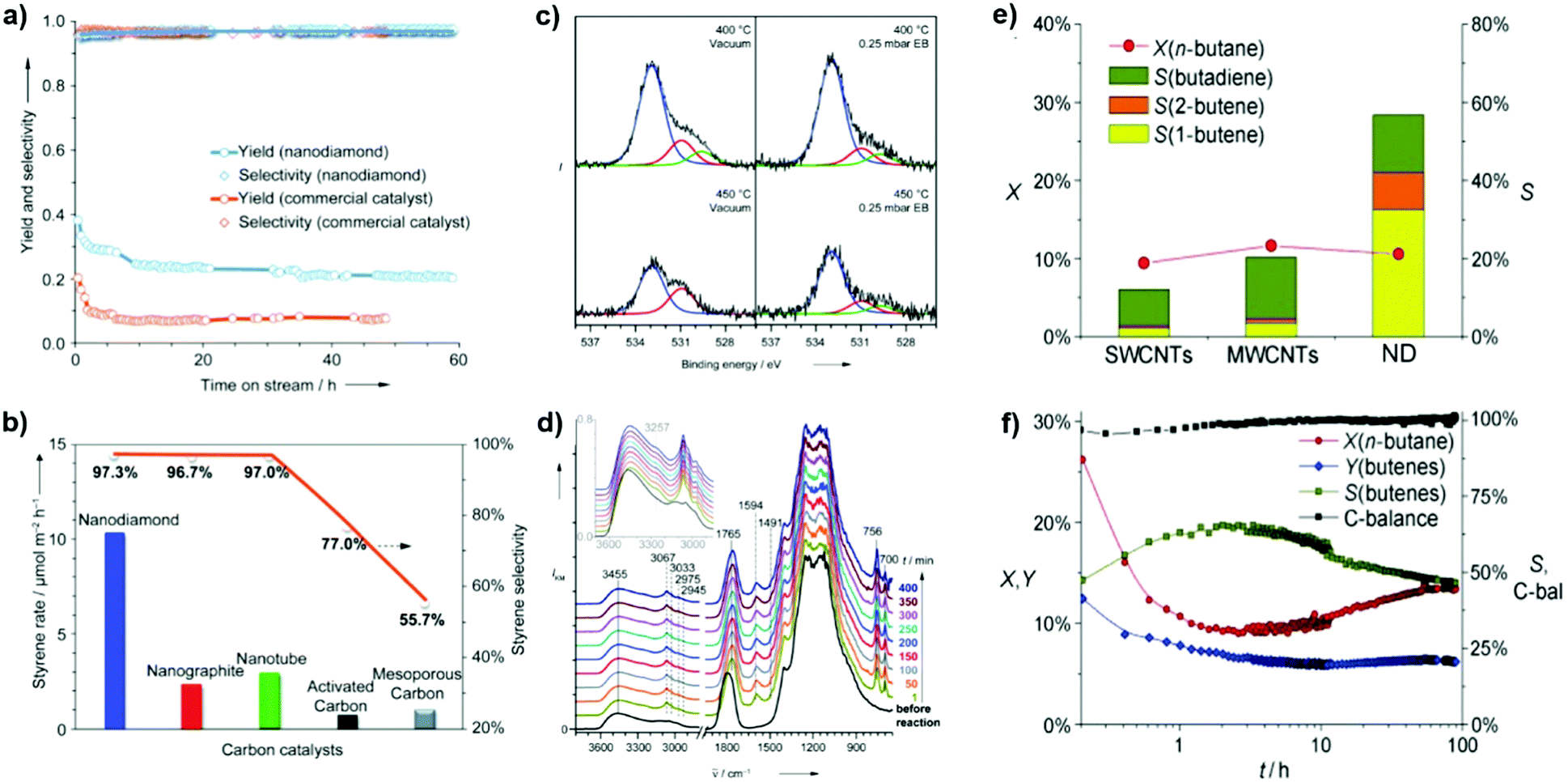 | ||
| Fig. 6 Activity of NDs in ethylbenzene (EB) dehydrogenation. (a) Long-term performance with the activity of a commercial Fe2O3 catalyst for comparison. (b) Steady-state activities of various carbons. Reaction conditions: 0.05 g, 550 °C, 2.8% EB in helium, 10 mL min−1. (c) In situ O (1s) XPS spectra in a vacuum and 0.25 mbar EB vapour. (d) In situ diffuse-reflectance infrared Fourier transform (DRIFT) spectra in EB at 450 °C. Reproduced from ref. 125 with permission from Wiley, copyright 2010. (e) n-Butane oxidative dehydrogenation (ODH) activities of various nanocarbons after 10 h time-on-stream, where X is conversion, Y is yield and S is selectivity. (f) Catalytic performances of NDs. Reproduced from ref. 126 with permission from Wiley, copyright 2011. | ||
In situ DRIFT shows that the α- or β-H of ethylbenzene interacts with ketonic O atoms, as evidenced by a significant red-shift from 1790 to 1765 cm−1, while the benzene nucleus is mostly skewed to the surface. Besides, vibrations of the benzene nucleus of EB do not change, similar to those in the gas phase (Fig. 6d). It is thus suggested that unsaturated ketone-/diketone-type carbonyl groups (CO) have substantial electron density at the oxygen atom, and thus can serve as Lewis bases to activate saturated hydrocarbons. Later, it was reported that BNDs with different graphitized degrees (sp2/sp3 carbon ratios) have distinct reactivity in the DH of propane.
The unique structural defects of the sp2/sp3 hybridized BND structure are likely responsible for the enhanced activation rate in propane dehydrogenation.128
The DH of methane, which is usually referred to as catalytic methane decomposition, has been often indicated as a valuable route to produce COx-free hydrogen.132 In this case, carbon atoms from methane are completely transformed into a carbonaceous solid which may be eventually used. Although transition metal catalysts produce gaseous products with high initial hydrogen concentration,133–135 their activity rapidly decreases due to the surface deposition of carbon.
Zhong et al. made an in-depth study on the metal-free catalysed CMD process using UNDDs as novel catalysts.131 NDs show superior activity and stability with respect to other carbon materials, including activated carbon (AC), SWCNTs and MWCNTs. The catalytic activity was largely dependent on the defective sites in the graphene sheets of UNDDs.
The possible mechanism was proposed by theoretical simulation as a series of surface stepwise dissociation reactions of CHx (x = 1–4) to form layered graphene and molecular H2.
2.2.1.2 ODH reactions catalyzed by UNDDs. The poor alkene selectivity is the major issue in the ODH reaction, even though much effort has been made towards understanding the alkane activation process. Keller and co-workers reported for the first time in 2002 that OLC is an active and selective catalyst in EB oxidative dehydrogenation.121 A 62% styrene yield was achieved with this catalyst. The authors attributed the superior activity to the absence of inner particle porosity, favoring styrene desorption and limiting secondary conversion. The activity was shown to be better than those of traditional K-promoted iron oxide catalysts for this reaction.
Recently, NDs (specifically ultradispersed nanodiamonds) were found to show superior catalytic performance for the ODH of n-butane, while in situ transforming into OLC.126 The transformation, which is activated from the catalytic reaction, leads to an enhanced selectivity. This is perhaps a unique example in the catalysis of the in situ generation of the catalytically active phase induced by the catalytic reaction itself. A high alkene selectivity (56%) in a stable n-butane conversion can be achieved over this catalyst, significantly larger than that shown by SWCNTs (12%) and MWCNTs (20%) (Fig. 6e). The transformation from the sp3 hybridization state of NDs to the sp2 hybridization state of OLC is accompanied by the generation of quinoidic carbonyl groups. The authors suggested that the regeneration of the active site (CO) is achieved by oxidation of C–OH instead of thermal dehydrogenation in the presence of O2.126
Using in situ methodologies, Sun and co-workers found that the initial oxygen groups present on BND surfaces are not active in the propane to propylene conversion, while new active oxygen species are generated in situ by the chemisorption of O2 on the surfaces of the BNDs.127 The latter oxygen functionalities are those involved directly in the catalytic redox mechanism of dehydrogenation. NDs show in EB dehydrogenation, under oxygen-lean conditions, 40% EB conversion, 92% styrene selectivity and 240 h stability. Also for NDs, it is suggested that the active sites are generated in situ.136
In both DH and ODH reactions, the surface nucleophilic ketonic carbonyl groups (CO) present in UNDDs are indicated as the active sites for the conversion of EB, propane and n-butane. The active oxygen species of the UNDDs in the reaction can be regenerated in situ by O2 chemisorption for the ODH reaction. Defects at the boundary may be play a positive role in forming CO active sites by stabilizing the O2. We will continue to discuss these reactions in the following sections.
2.2.1.3 Peculiarities of UNDDs with respect to other nanostructured carbon catalysts. The kinetic parameters for the ethylbenzene ODH reaction over NDs are similar to those of three different types of MWCNT materials, despite the different structures and hybridization states of the catalysts (Table 1). The investigation was extended to the reaction mechanism on the proposed active sites present in these nanocarbons. The reaction was demonstrated to involve a dual-site Langmuir–Hinshelwood mechanism, based on the dissociative adsorption of oxygen molecules and the non-competitive adsorption between EB and oxygen molecules. Isotopic tracer studies further indicated that the reaction pathway and the nature of the active sites are identical over both sp2-MWCNTs and sp3-NDs. This can be explained by the fact that the initial carbon surface would restructure into a state where sp2-graphitic carbon is stabilized against combustion by strongly attached ketonic oxygen functional groups. As far we know, there is no equivalent in situ mechanism of transformation in DH or ODH “conventional” catalysts (based on metals or metal-oxides). This further demonstrates the rather unique characteristics of nanocarbon catalysts with respect to other types of catalysts.
| Kinetic parametera | MWCNTs-1 | MWCNTs-2 | MWCNTs-3 | NDs |
|---|---|---|---|---|
| a Experimental conditions: 2.8–6.4 and 1.4–8.4 kPa of EB and O2, respectively (He as balance); 5 mg catalyst. b The values are normalized for the surface area. The ODH rate is measured at 723 K. | ||||
| Activation energy [kJ mol−1] | 75 | 71 | 68 | 75 |
| Reaction order, EB | 0.51 ± 0.05 | 0.52 ± 0.06 | 0.55 ± 0.04 | 0.61 ± 0.10 |
| Reaction order, oxygen | 0.30 ± 0.03 | 0.34 ± 0.04 | 0.32 ± 0.03 | 0.33 ± 0.07 |
| Reaction rateb [mmol m−2 h−1] | 4.6 × 10−2 | — | — | 4.9 × 10−2 |
| Pre-exponential factorb [mmol m−2 h−1 kPa−1] | (5.3 ± 0.5) × 103 | — | — | (5.5 ± 0.6) × 103 |
In fact, the local structure of the sp2-bonded nanocarbons is of paramount importance to ODH reaction performance and stability.122 Among various nanocarbon catalysts, the attention was initially focused on AC due to its low cost, high surface area and rich surface chemistry.137 However, the short-range disordered sp2 carbon structure, being vulnerable to oxidation under reaction conditions, could result in the combustion of the AC catalyst itself. Hence, AC is not suitable for use in the long-time ODH reaction of EB.
For the DH reaction of EB, as commented on above, NDs show superior activity with respect to other nanocarbon materials (Fig. 6b). The improved performances are related to the long-range disorder with a mixture of sp2- and sp3-hybridized carbon, which gives rise to a variety of surface groups and defects that allow α-or β-scission of the ethyl group of EB into benzene and toluene. The spatial arrangement of spherical NDs, with much smaller size and higher surface area, provides good access for the reactants, and hence is favourable for interaction with the surrounding substrates. The curvature of the surface graphene layer provides superior activity for the following reasons:
– the partial delocalization of the π-electrons induces an optimized distribution of active sites
– the abundant CO groups, acting as electron donor sites (nucleophilic sites), are able to activate EB's lateral chain.
The high activity of NDs in the endothermic DH reaction may be attributed also to the very high thermal conductance of the sp3 phase at the core. It improves the heat-transfer efficiency close to the active sites on the surface.
For the ODH reaction of n-butane, the great difference in the selectivity between CNTs (MWCNTs, SWCNTs; see Fig. 6e) and NDs suggests that the well-graphitized surface strongly enhances the selective alkane activation owing to the controllable activation of oxygen. It was found that the surfaces of the NDs tend to transform into three to ten layers of graphitic shells during the n-butane ODH reaction. This transformation process from the sp3 to sp2 configuration can selectively generate the quinoidic carbonyl groups and effectively suppress the formation of electrophilic oxygen species. The electrophilic oxygen species are believed to be responsible for the combustion of alkenes on the carbon surface,117 thus resulting in a higher product selectivity to the target butenes.
2.2.1.4 Structural effects of UNDDs on the catalytic activity. NDDs with different surface hybridized configurations (sp3, sp3/sp2 and sp2) show different electronic states (Fig. 5c–e) and surface properties (Fig. 4). These differences reflect in the different natures of defects and anchored oxygen groups. For example, NDs and OLCs (as representative sp3- and sp2-hybridized structures of UNDDs) follow different ODH pathways for EB, even though they have similar sizes and shapes.124 In the case of NDs, the sp3 carbon surface initially induces C–C bond cleavage and benzene formation, while the main reaction pathway would turn into styrene formation in parallel with the formation of sp2 carbons on the ND surface, which resulted in similar styrene selectivity to OLCs.
The significant role of structural transformation on the UNDD surface for catalytic activity and selectivity can be demonstrated also in the ODH of propane. By systematically controlling the formation of graphitic shells on NDs via the annealing treatment, which resulted in UNDDs with different sp2/sp3 ratios, a good linear relationship between the sp2-hybridized carbon fraction and the reactivity of the in situ formed surface oxygen species can be found in the ODH of propane.127 For different hybridized nanostructures of UNDDs, the catalytic capabilities of the same types of active oxygen groups generated during the reaction depend on the sp2-hybridized carbon fraction. The active oxygen groups formed on a graphitic OLC surface could provide 20% higher propene selectivity than that of an initial ND catalyst (Fig. 7). In addition, 30% higher selectivity than traditional sp2-hybridized MWCNTs gives evidence that the unique curved and strained graphitic surfaces of the UNDDs were essential for the selective alkane activation.
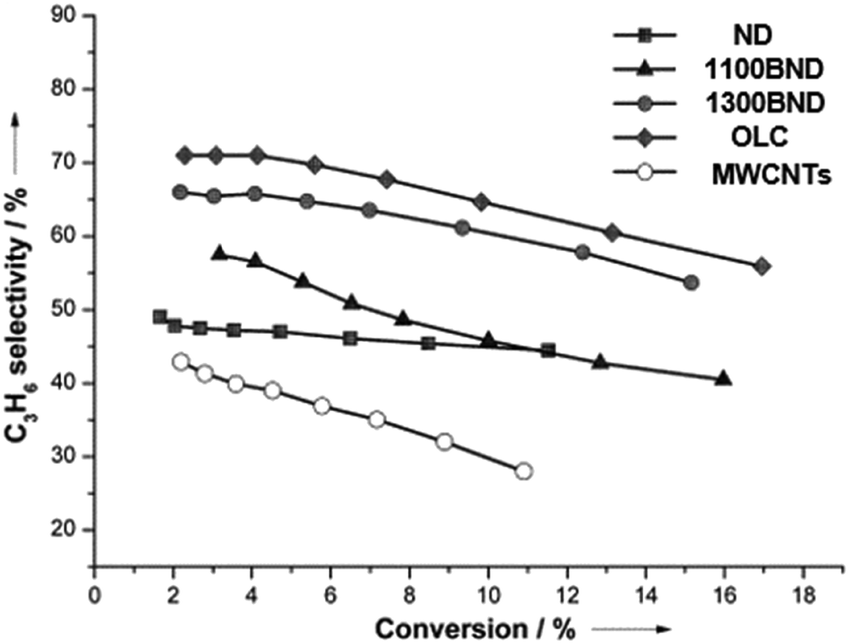 | ||
| Fig. 7 Propylene selectivity versus propane conversion curves of UNDD catalysts with different annealing temperatures and MWCNTs in the ODH process. Reproduced from ref. 127 with permission Wiley, copyright 2014. | ||
For the DH reaction of propane, Wang et al. reported that the original structure of ND particles is characterized by a diamond core covered with an amorphous carbon layer, the latter predominantly terminating with electrophilic oxygen species, such as carboxylic acids and their anhydrides. For this reason, it shows a low catalytic activity. Upon increasing the annealing temperature, the sp2-carbon on the ND surface reconstructed from the amorphous form into a well-ordered OLC via an intermediate composite BND structure. The latter is characterized by a diamond core covered with a highly defective, curved graphene outer shell. Such a hybrid may act as an appropriate matrix for the formation of ketone-type functional groups and thus exhibits superior catalytic activity.128
2.2.1.5 Creation of heteroatom-based active sites in UNDDs. Since nanocarbon catalysts often may have poor stability at high temperature and low selectivity for olefin products, engineering of the carbon surface to achieve a good overall performance is highly desirable. Heteroatom modification of nanocarbon surfaces is an effective method for such purposes. Recently, doping with P, B and N elements has been demonstrated to be an excellent methodology to enhance product selectivity in the ODH reaction of ethane,138 propane139,140 and n-butane.117,141 Recently, BNDs modified with phosphate were applied as an active and selective catalyst for the ODH of propane.129 Phosphate groups efficiently inhibit the dissociative adsorption of O2 and improve the catalyst stability, while simultaneously promoting the amount of selective oxygen species. The phosphate addition thus results in an enhanced selectivity. This promotion effect originates from the formation of a covalent C–O–P bond on the modified BND surface. The observed excellent activity could compete with state-of-the-art metal oxide catalysts, with the advantage of no carbon deposition or catalyst deactivation during the reaction.142
Borate modification on BND surfaces was used to demonstrate the roles of heteroatoms in catalytic propane ODH.130 Differently from phosphate doping, borate modification resulted in a volcano-type trend of propene selectivity upon increasing the loading of borate. At a low loading (<15 wt%), the B species mainly interacted with the defects of BNDs by forming an O–H bond. This interaction suppresses the formation of electrophilic oxygen species, thus leading to an increased propene selectivity. However, for a higher boron amount, the formation of tetrahedral coordinated boron leads to a significant blockage of the active carbonyl groups. It is worth noting that these results indicate new mechanisms of interaction between the heteroatoms and the carbon surface, and the related influence on the catalytic behaviour, different from those usually considered in analysing the effect of doping nanocarbons with heteroatoms. These new mechanisms of interaction are specifically enhanced in NDs and related materials, due to their structural and surface characteristics.
2.2.1.6 Catalytic activity of UNDD hybrid materials. The powder form of UNDDs cannot be used directly in fixed-bed industrial-size reactors due to the high-pressure drop. Besides, they are difficult to handle and may create safety issues. It is thus necessary to develop catalysts with suitable shape and characteristics, while at the same time maintaining the properties of NDs and creating the suitable pore structure to avoid diffusional limitations. Recently, immobilization of NDs onto the surfaces of porous materials or blending NDs with other carbon materials into a monolith or hierarchical composites has been shown to be a valuable strategy. For example, Liu and co-workers found that commercial ND aggregates anchored into 3D CNT/SiC networks by the sonication-assisted impregnation method could effectively improve the selectivity in the DH of EB to styrene. The styrene selectivity reached 97.1%. The authors suggested that the better performance over the 3.4%ND/CNT–SiC monolith is associated with high effective surfaces and short diffusion lengths, as well as excellent heat transfer over the SiC support during the reaction.143
Later, a new hybrid structure consisting of a ND-decorated porous β-SiC foam matrix was shown to have an EB dehydrogenation activity ten times higher than that of a commercial K–Fe catalyst and 3.8 times higher than that of pure ND powder. High stability was also observed for at least 100 h, due to the open porosity of the catalyst, which efficiently prevents coke deposition during the reaction.144
In addition, few-layer graphene as a promising scaffold has attracted considerable attention over the past few years. Such material contains a large number of vacancies and defects, which can promote the dispersion of ND particles. Some attempts have been made towards improving catalytic activity by using few-layer graphene as a support. Diao et al. reported that the superior activity of a ND/graphene hybrid with respect to pristine NDs is due to the highly dispersed NDs on the graphene surface, which offers a relatively higher density of active sites for DH reaction of EB.145 Similar results for the DH reaction of EB were reported by Pham-Huu's group.146 Zhao and co-workers used instead a N-doped mesoporous graphene as a scaffold to obtain a ND/nitrogen-doped mesoporous graphene hybrid which displayed an improved activity. A synergy between graphene and NDs in the unique mesoporous nanoarchitecture was responsible for the enhanced reaction performance.147 Recently, carbon nanofibers/graphene composites, graphene/graphene oxide composites and carbon nitrides as new composites to support ND have been studied in the DH of EB.148–151 These hybrid catalysts exhibit higher catalytic activity than those of the pristine components and reference transition metal-based catalysts. Roldán et al. found that an hybrid material formed by a reduced graphene aerogel and NDs (2 wt%) shows a high performance catalyst in the ODH of propane.152 The authors supposed that the higher number of defects in the graphitic lattice, higher oxygen content and more accessible carbonyl/quinone groups were beneficial to attaining high yields of the desired product propylene.152
Due to the milder reaction conditions (in contrast to gas-phase reactions) and the presence of solvents, carbocatalysts should have different characteristics for the two classes of reactions. On one hand, catalysts can be chosen from a broader range of materials, including those with average thermal stability. On the other hand, the active sites should be stable against reactions with solvents under the chosen reaction conditions, including leaching, solvolysis, strong chemisorption, etc. The surface hydrophilicity and hydrophobicity of catalysts are also important parameters in the liquid phase reactions that directly affect the catalytic performance. The abundant surface oxygen groups of NDs, including carboxylic acid, ketone, phenol and lactone groups, make the ND surface hydrophilic. However, the particle–particle interaction of NDs causes strong aggregation of NDs, leading to a poor dispersion of the nanoparticles in aqueous solutions. Being quite hydrophilic, NDs also cannot be well dispersed in most organic solvents. Therefore, the controlled high temperature treatment of NDs can lead to their partial or complete surface graphitization and can tune the population of certain surface oxygen functional groups.
The surface hydrophilicity/phobicity of BNDs and OLCs is slightly different from that of NDs and BNDs and OLCs show better dispersibility in organic media. It is certain that a variety of the surface groups of UNDDs or dopant-modified UNDDs (e.g., N and B) are capable of providing the underlying access to achieve effective interfacial contact between the substrate and catalyst itself in the liquid-phase reactions. Up to now, UNDDs and dopant-modified UNDDs have been proved as effective heterogeneous catalysts in many important liquid-phase reactions. Some typical liquid-phase reactions of industrial relevance are reviewed in this section.
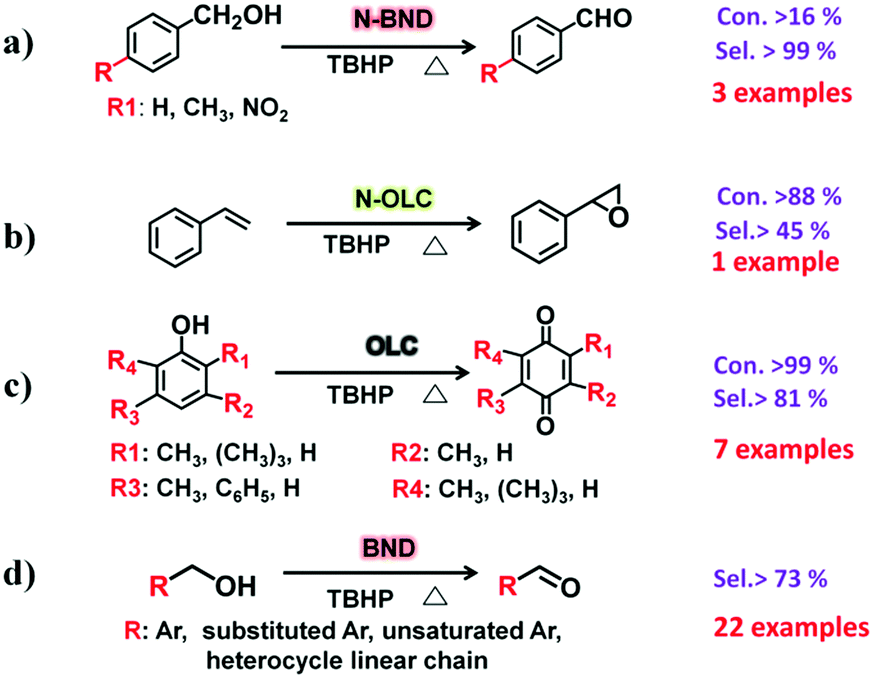
2.2.2.1 Oxidation reactions. Lin et al. demonstrated that nitrogen-modified BNDs (N-BNDs) with tuneable functional groups can be prepared via three different preparation methods in a calcination process (Fig. 8a, pathway I).156 The authors proposed a detailed mechanism of formation and dynamic behaviours of the nitrogen species in the N-BNDs. A series of complicated chemical reactions involving condensation, cyclization and dehydrogenation were proposed in view of the substitution reactions between oxygen groups and nitrogen sources (Fig. 8b). For example, the reaction of NH3 with carbonyl groups would generate intermediate lactam species and form simultaneously CO and H2. On the other hand, the lactam species may self-evolve directly into pyridinic and pyrrolic N species combined with the formation of H2O and CO. All these results support the fact that surface oxygen groups play a crucial role in the nitrogen-modified process. Meanwhile, lactam and cyano species are important intermediates in the formation of various N species. Therefore, a high content of nitrogen can be achieved by varying the surface oxygen concentration of the starting BND material. The selective oxidation of benzylic alcohols serves as a probe reaction to evaluate the catalytic performance of the N-BND catalysts (3 examples of reactions with different substituents, see Fig. 9a). The highest yield of benzaldehyde on the N-BND catalysts could reach 18.6%, which is twice higher than that of the pristine BND catalyst. This is attributed to the nitrogen-doping modification. It appears that the bonding behaviour between the nitrogen species with lone-pair electron properties and the free radicals derived from the oxidant may be responsible for the higher reactivity.
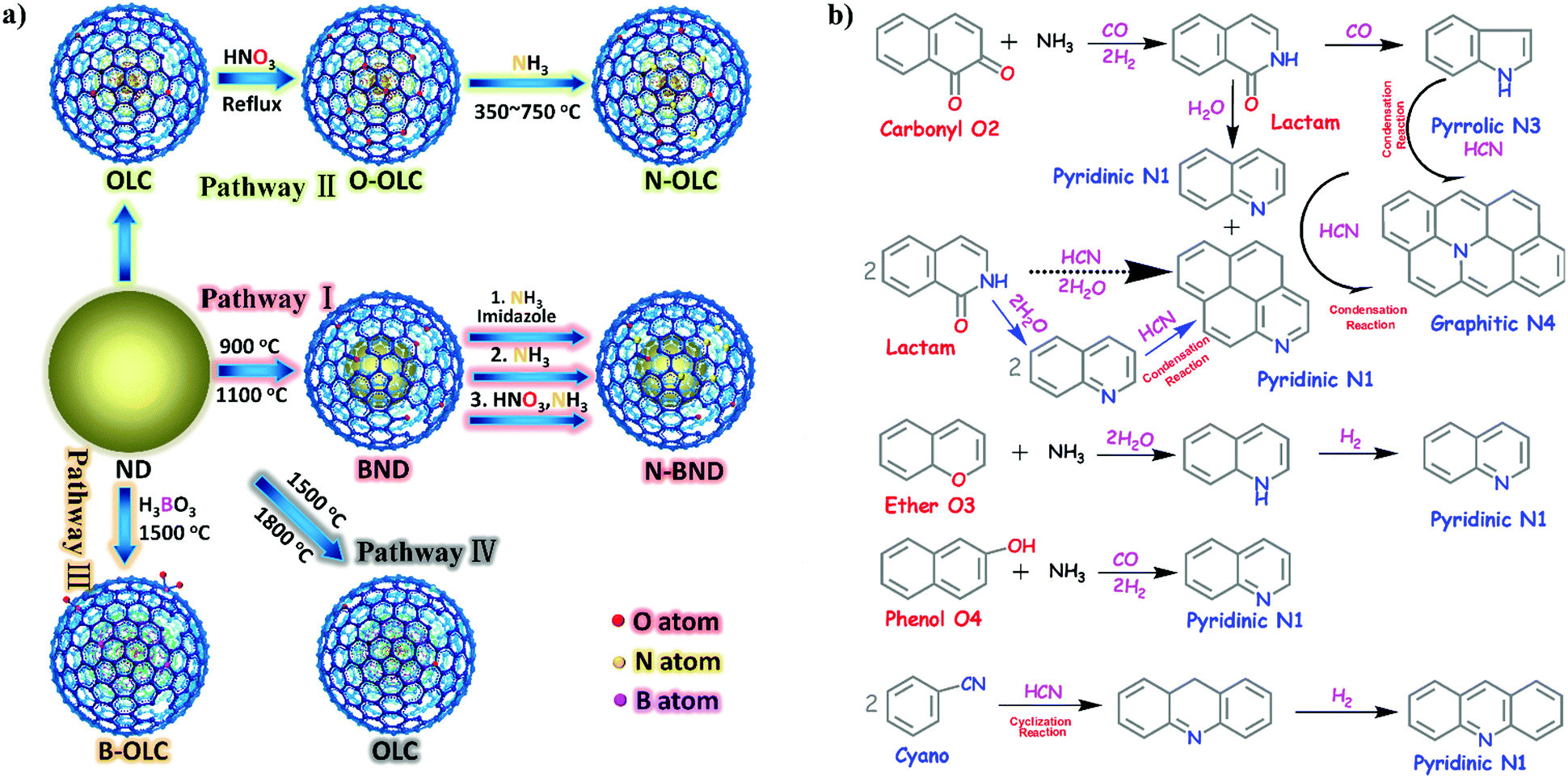 | ||
| Fig. 8 (a) Sketch for the production of UNDDs and related materials. (b) The possible evolution pathways of different nitrogen species during the preparation process of nitrogen-modified UNDDs (two hexatomic rings as ideal starting model structures in different pathways). Reproduced from ref. 156 with permission from American Chemical Society, copyright 2014. | ||
 | ||
| Fig. 9 Schematic illustration for typical liquid-phase oxidation reactions over UNDDs and modified UNDDs. | ||
Epoxides are important intermediates for fine chemicals like perfumes, drugs, epoxy resins, sweeteners, etc. Current research on olefin epoxidation generally relies on the use of metal-based catalysts, such as Au/TS-1.157,158 Depending on the calcination temperature, nitrogen-doped OLC (N-OLC) catalysts can be synthesized under an ammonia atmosphere (Fig. 8a, pathway II) and the highest nitrogen content could reach 3.95 at%.159 The N-OLC exhibits superior activity compared to pristine OLC, N-graphene, N-CNTs, Au/TS-1 and Co-OMS-2 in styrene epoxidation (Fig. 9b).160,161 It was suggested that the graphitic nitrogen species play an important role rather than defects and oxygen species in the epoxidation reaction. In fact, graphitic nitrogen does not directly participate in the activation of styrene, but instead it changes the electronic structure of the adjacent carbon atoms, making them active for the generation of reactive oxygen species such as peroxides.162 When TBHP (tBuOOH) is used as the oxidant, the adjacent carbon atom to the graphitic nitrogen in N-OLC (Cat) could undergo a one-electron oxidation to form Cat-OH species. Subsequently, the Cat-OH species is converted into intermediate species A (Cat-OOtBu), which is favourable for triggering the breakup of the double olefinic bond of styrene and the formation of a transition state, B. The latter species finally complete the whole ring-closure process of the epoxide product (Fig. 10). Although the roles of nitrogen species are confirmed, a synergistic effect between the oxygen and nitrogen species over the N-BNDs and N-OLCs in the selected catalytic reactions has not been studied.
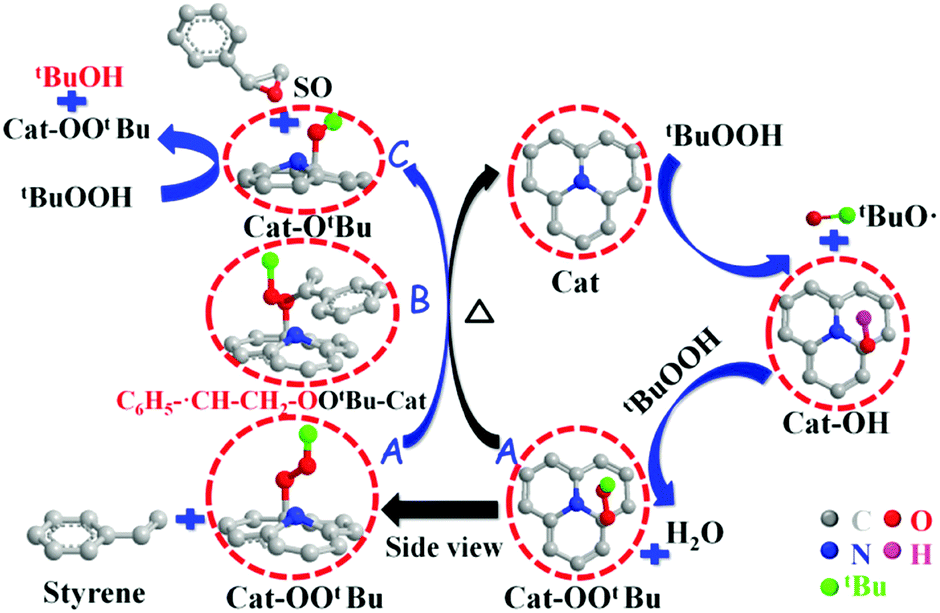 | ||
| Fig. 10 The proposed mechanistic pathways for epoxidation on N-OLC. Reproduced from ref. 159 with permission from Royal Society of Chemistry, copyright 2014. | ||
2.2.2.2 Aromatic organic molecules as models. Nanocarbon materials can be considered as carbon atoms grouped into layers of fused aromatic rings, and containing different types of oxygenated groups (Fig. 11a). Therefore, aromatic organic molecules with specific oxygen functional groups (Fig. 11b) represent model systems having a similar carbon network structure, surface properties and a tuneable electronic conjugated π system (by extending the benzene unit). They thus mimic the intrinsic structures of oxidized carbon catalysts and allow identifying specific oxygen active components, such as C
O functional groups.
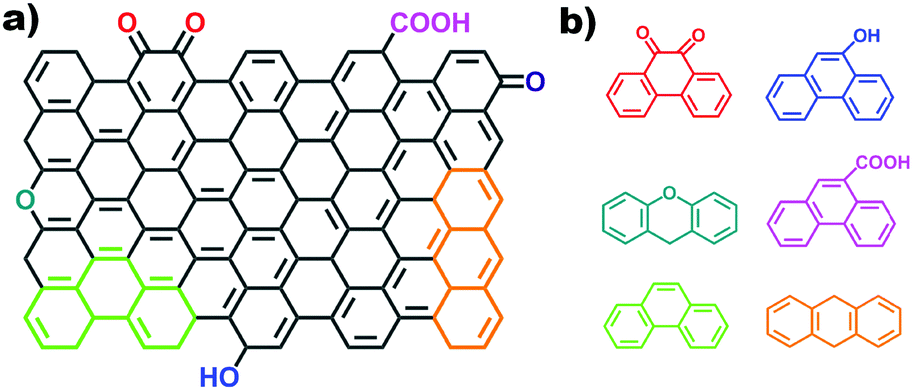 | ||
| Fig. 11 (a) Surface structure of carbon materials with typical oxygen species and edge defects. (b) Structures of different aromatic organic molecules with single oxygen groups (e.g., CO, C–OH, C–O–C, –COOH) and designed defect configurations (e.g., armchair and zigzag). | ||
Organic molecules with designated edge configurations (Fig. 11b) can also be used to simulate the armchair and zigzag edge structures of carbocatalysts and to probe the roles of defects in catalysis. This approach can thus provide valuable information for studying the nature of active centers in carbocatalysts and their behaviour in between organocatalysis and solid catalysis.
Recently, Lin and co-workers demonstrated that pristine OLC (Fig. 8a, pathway IV) exhibits outstanding catalytic performance in the selective oxidation of mono-, di- and tri-substituted phenols (7 examples for substrates with different substituents, see Fig. 9c) to the corresponding p-benzoquinones. The pristine OLC shows better efficiency than reference systems such as metal-based catalysts (e.g., Ti-HMS, Ti-Si) and industrial catalysts (e.g., 40% H2SO4, CuCl2/MgCl2).163 The 99.0% conversion for phenols and 81.5–92.5% selectivity for the corresponding benzoquinones can be achieved with OLC under milder reaction conditions.
The authors used different molecules as model catalysts to exclude the roles of oxygen species in the reaction and found that the edge zigzag configuration assists the reaction by stabilizing the intermediate phenoxy radicals. The catalytic behaviour of OLC was better than those of other carbon materials, such as AC, graphite, graphene, etc. More recently, sp3/sp2-hybridized BNDs as a potential green catalyst have been shown to exhibit excellent chemoselectivity and cycling stability for the selective oxidation of complex primary alcohols to their corresponding aldehydes (22 examples, Fig. 9d). The observed activity results are comparable to those of conventional Pt/C and Ru/C catalysts for certain substrates under solvent-free conditions. Using the same method of using aromatic model compounds as references, the zigzag edges of surface sp2 carbon planes on the BNDs were shown to play a catalytic role in the reaction.164
Therefore, aromatic organic molecules with a larger conjugated system and tuneable edge configuration represent a valuable model to understand the nature of carbocatalysts and to develop new applications.
2.2.2.3 Advanced oxidation processes (AOPs). A variety of organic compounds was recently detected in industrial and municipal wastewater. Some of these compounds cause serious problems in biological treatment systems due to their resistance to biodegradation. Consequently, finding alternative treatment technologies to mineralize or totally transform refractory molecules into degradable products is a matter of great concern. Among them, advanced oxidation processes (AOPs) have been demonstrated as powerful techniques to produce highly reactive oxygen species which could effectively degrade toxic organic pollutants into harmless products.165 Transition metal-based materials have been employed as the most effective catalysts for activation of some common oxidants, such as peroxymonosulfate (PMS), peroxydisulfate (PDS), hydrogen peroxide (H2O2) and ozone (O3) to form sulfate radicals (SO4˙−), hydroxyl radicals (˙OH), and/or superoxide radicals (O2˙−) with higher redox potentials to achieve rapid decomposition of a wide range of contaminants.166 Metal-free carbon materials represent alternative valuable and less toxic alternatives to catalyze these reactions. They avoid secondary contamination (e.g. metal leaching) relative to the use of transition metal-containing catalysts.
Phenol is a model molecule and organic pollutant itself, widely present in industrial wastewater, typically used to test the behaviour in AOP reactions. In 2016, Duan and co-workers showed that different carbon materials including GO, GR, MWCNTs, BNDs and mesoporous carbon as catalysts exhibit excellent catalytic activity for phenol removal in the presence of PMS.167 Among these nanocarbons, BNDs were found to show better performances. Later, BNDs were shown to promote free radical pathways, with BNDs being able to activate PMS to produce both SO4˙− and ˙OH, the latter species being highly active in phenol degradation.168 The activity is higher than those of typical metal oxide particles, such as Fe2O3, Fe3O4, Co3O4, Ga2O3 and CuO. The unique sp2/sp3-hybridized interfaces of BNDs are capable of inducing electron transport from the ND core to the shell surface, enhancing the O–O breakup efficiency in PMS.168
Lee et al. proposed instead a non-radical oxidation pathway, based on PDS activation and phenol removal via electron transfer from phenol to PDS.169 BNDs only facilitate the electron transfer and reactant adsorption. Lee et al. indicated that radical intermediates such as SO4˙− and ˙OH are not involved in this persulfate-driven oxidation of phenol.169
More recently, a series of BNDs with different graphitized degrees (annealed at different temperatures) were investigated by Duan et al. (Fig. 12a).170 The number of graphitic shells of BNDs, which depends on the annealing treatment, determines the activation pathway of PMS. The radical quenching tests (using MeOH as a radical scavenger, Fig. 12b) suggest that the higher graphitic degree on 1100 BND gives rise to a greater proportion of the non-radical oxidation pathway. This indication is supported by the weaker intensity of the ESR of 1100 BND (Fig. 12c). On increasing the number of graphitic shells, the phenol removal mechanism transforms from a radical-dominated oxidation (for the 900 BND catalyst) to a non-radical-dominated pathway (for the 1100 BND catalyst, see Fig. 12d).
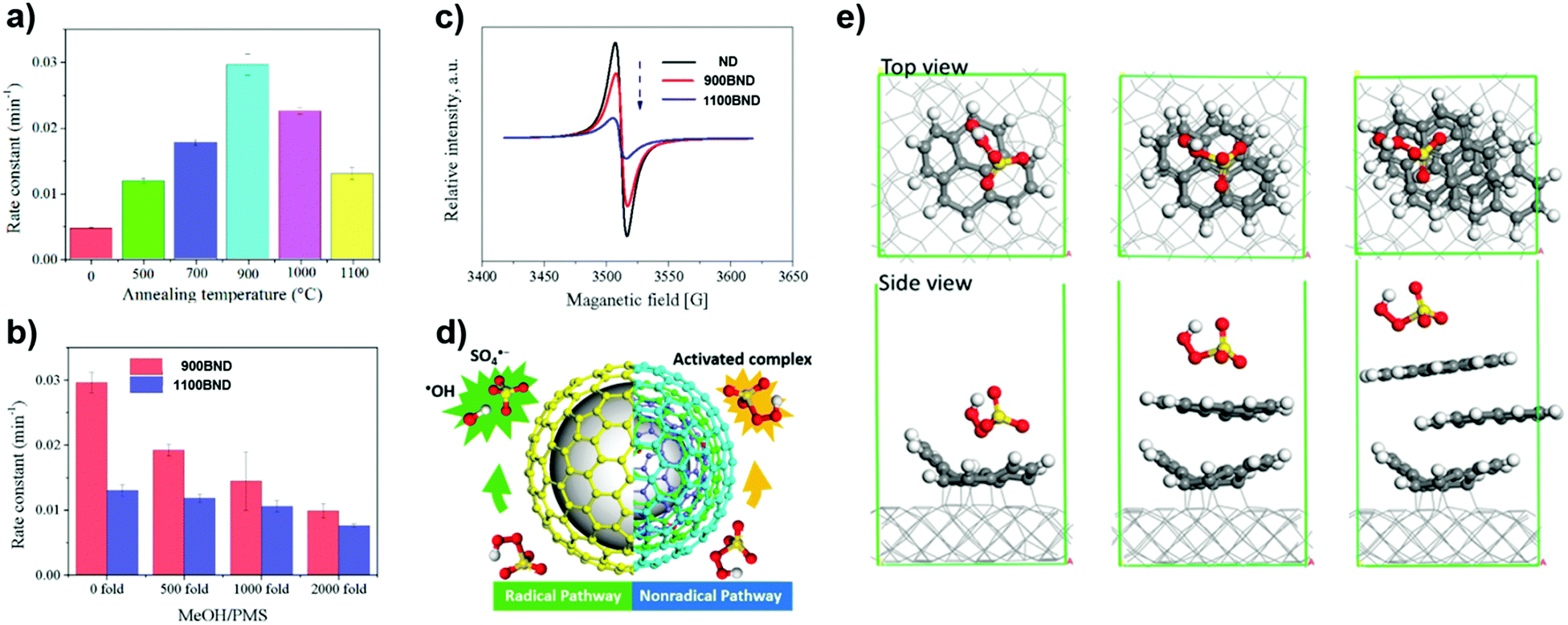 | ||
| Fig. 12 (a) Influence of different annealing temperatures on phenol removal. (b) Quenching effect of phenol removal on BNDs with different graphitic degrees. (c) ESR spectra of UNDDs. (d) Illustrations of radical and non-radical oxidations on UNDDs. (e) Theoretical modulation of BNDs with different graphitic layers, reprinted from ref. 170 with permission from Elsevier, copyright 2018. | ||
Theoretical calculations indicate that the diamond core would excite electrons to the graphitic shell of one-layer graphene/diamond via strong covalent bonds, giving rise to a denser electron population of the carbon sphere, which would promote the charge migration to PMS to generate SO4˙−. However, it is difficult for electrons to go through multiple shells in a three-layer graphene/diamond model (e.g. 1100 BND), leading to a greater adsorption energy of PMS on the outermost layer of the 1100 BND to form a surface-activated complex for non-radical oxidation (Fig. 12e). Besides, edge defects imply a non-radical pathway for the PMS activation, with quinone groups located at the boundaries having a crucial role in cleaving PMS (by weakening the O–O bond) to produce SO4˙−.
A N-BND catalyst with melamine as a nitrogen precursor was proposed with the role of nitrogen in enhancing the activation of PMS to produce SO4˙− and ˙OH associated with the creation of positively charged carbon domains.171
2.2.2.4 Reduction reactions. Traditionally, the chemoselective hydrogenation of nitro groups to amines is carried out using various transition metal or precious metal catalysts.172,173 Lin and co-workers reported for the first time that boron-doped OLC (B-OLC, Fig. 8, pathway III) shows excellent catalytic activity and stability in the nitroarene reduction reaction with N2H4 (20 examples of substrates with different substituents).174 The highest yield rate for aniline was reported to be 593 μmol m−2 h−1. The lattice BC3 species was suggested to play a positive role in improving the catalytic performance. Compared with the B atom, the C atom has a larger electronegativity. It can be expected that the B atom having an electron-deficient nature in the lattice is the positively charged site. N2H4 as a strong electron-donating reagent may be adsorbed on the site of lattice B, forming a weak B–H bond together with the activation of H atoms, and thus stabilizing the hydrogen intermediate products and avoiding the fast loss of hydrogen as H2. Once nitroarene substrates are added into the reaction system, the O atoms of the nitro group would abstract the activated H atoms from N2H4, leading finally to the high selectivity for desired products and achieving the efficient utilization of N2H4.
Importantly, an inverse relationship between the work function and the catalytic performance of UNDDs in the nitroarene reduction reaction was recently proposed.89 The lower work functions of UNDDs probably induced by their high curvatures inevitably endow them with a higher surface energy that may be beneficial to improve the catalytic activity.
Additionally, Zhang et al. developed a novel method to efficiently produce aniline by electrochemically assisted reduction of nitrobenzene using a NDD material as the cathode electrocatalyst.175 The dominant reduction pathway on the NDD electrode involves the conversion of nitrobenzene to aniline via phenylhydroxylamine.
UNDDs also show an interesting behaviour in the Suzuki coupling of organic molecules. Yeap and co-workers indicated that the spontaneous diazonium coupling of bromophenyl or boronic ester phenyl occurs on hydrogenated NDs.176 The reaction products change the surface properties of the original NDs, leading to a material showing excellent fluorescence and improved dispersion in ethanol and hexane. These results provide also useful information on how to realize new procedures for the surface functionalization of nanocarbon materials.
At the present stage, the use of UNDDs or dopant-modified UNDDs as catalysts for liquid-phase catalysis has been still in a sprouting stage. The unique surface properties and thermal reconstruction of UNDDs during the preparation process are beneficial for the modification with a higher dopant concentration (e.g., B and N) than other nanocarbons (e.g., CNTs, graphite). The superior catalytic activity of UNDDs or dopant-modified UNDDs in comparison to conventional carbon materials and other metal-based catalysts has been proved, but due to the still large range of possible tuning of these carbocatalyst materials, the possibility of further development can be indicated.
2.3 Electrochemical reactions
The on-going energy and chemistry transition characterized by the progressive electrification and substitution of raw materials with alternative sources to decrease fossil fuel use, as briefly outlined in the Introduction, has spurred the development of nanomaterials to enhance the reaction performance in electrocatalysis. Metal-free nanocarbons offer valuable alternatives as electrode materials with low cost, abundant structural varieties, tailorable surface chemistry and stability. Heteroatom-modified nanostructured carbons with tuneable surfaces further provide promising opportunities for realizing sustainable applications.31Choi et al. proposed a novel nitrogen-doped onion-like carbon (N-OLC) synthesized using a mixture of urea and oxidized OLC.178 The incorporation of nitrogen species did not change the surface morphology of the OLC raw material. The ORR activity of such a catalyst could be improved by increasing the number of active sites (e.g. pyridinic N and graphitic N groups).178 Although the onset and half-wave potentials of the prepared catalysts were still not comparable to commercial Pt/C, the long-term stability and higher resistance to methanol crossover of these N-OLC materials make them promising electrocatalysts for the ORR in alkaline media.178
N-OLCs were also prepared starting from renewable bio-based resources and collagen as precursors.179 The final material contains a high percentage of nitrogen (7.5%). The porous graphitic carbon with the higher nitrogen content shows good catalytic ORR activity, with the pyridinic nitrogen acting as an effective promoter for the ORR, as deduced from XPS comparative experiments.179
Quan and co-workers reported that B and N co-doped NDs (BNNDs) grown on a Si rod array substrate could be prepared combining thermal oxidation and dry etching processes with plasma (Fig. 13). The obtained material is a highly efficient metal-free catalyst for the ORR under alkaline conditions,180 showing a higher kinetic current density (Fig. 14a and b), stability, and methanol tolerance better than those of a commercial Pt/C catalyst. The optimized B and N contents, estimated by XPS, were 1.73% and 1.94%, respectively. The synergistic effect of the boron and nitrogen species in the catalyst was proposed to be responsible for improving the ORR activity, although the authors did not perform a key experiment to distinguish the roles of boron atoms or possible boron–oxygen compounds in the ORR.
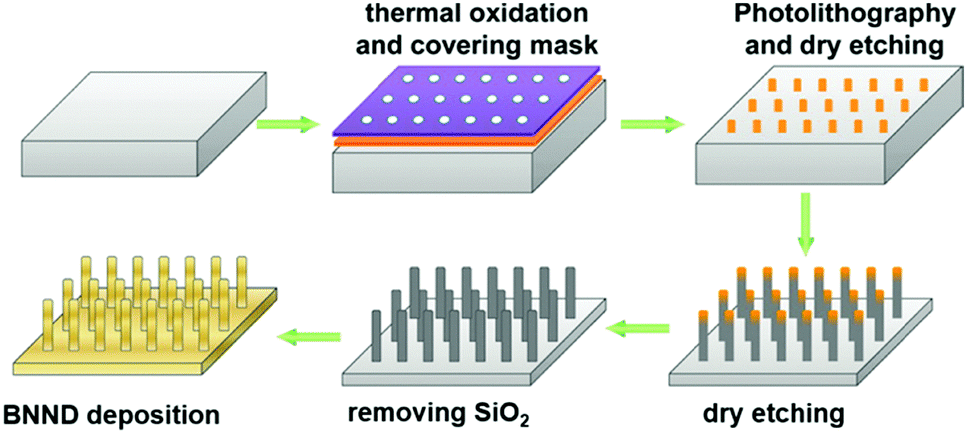 | ||
| Fig. 13 Preparation of BNND/Si rod arrays, reproduced from ref. 180 with permission from American Chemical Society, copyright 2013. | ||
 | ||
| Fig. 14 (a) ORR polarization curves and (b) the calculated Jk (kinetic current density) values of various catalysts recorded at −0.30 and −0.35 V, in O2-saturated 0.1 M KOH, reproduced from ref. 180 with permission from American Chemical Society, copyright 2014. (c) ORR polarization curves and (d) mass activities of N-OLC and Pt/C catalysts recorded at 0.8 V, in O2-saturated 0.1 M HClO4, reprinted from ref. 192 with permission from Royal Society of Chemistry, copyright 2016. | ||
Zhu et al. used pristine BNDs with different sp2/sp3 ratios to study the relationship between the ORR performance and the specific characteristics of the pristine BND (Fig. 14b).181 With the increase of the sp2 content of nanocarbon on the pristine BND (obtained by annealing), a higher catalytic activity for the ORR could be attained. Moreover, the role of nitrogen species in N-BNDs in the catalytic performance was studied in detail (the synthetic route is shown in Fig. 8, pathway I). The results show that the N-BNDs exhibit a four-electron ORR behaviour with high stability. Using EELS, the authors suggest that the relevant stability of the N-BNDs derives from the transformation of a part of sp3 carbon into sp2 carbon during the durability test.
Kannari et al. reported a similar study on the role of the sp2/sp3 content in BND and OLC materials in their catalytic performances.182 The authors proposed that the onset potential was proportional to the degree of defects. OLCs obtained by annealing at 1673 K show the best activity. Dong and co-workers used melamine as a nitrogen source to obtain N-BNDs and found that the onset potential of the catalyst reached 42 mV vs. Hg/HgO (as the reference electrode). A further study reported that N-BNDs have a similar oxygen adsorption mechanism to Pt electrocatalysts.183 The authors concluded that the enhanced catalytic performance may be attributed to the synergistic effect among graphitic N, pyrrolic N and pyridinic N.
Using laser irradiation of ND colloidal dispersion in various solvents, OLCs were prepared. These materials were then treated with NH3 and H2S to synthesize N- or S-doped OLC (N-OLC or S-OLC), respectively.184 The presence of the heteroatoms improves the catalytic performance. The electron transfer values for the N-OLC and S-OLC were calculated to be constant (3.8 and 3.3, respectively) over a potential range from −0.3 V to −0.8 V. The authors proposed that S might enrich electron or spin density in adjacent carbon atoms and promote ORR activity. The pyrrolic N structure can increase the electron delocalization because of their high electron-withdrawing ability and ensure the four-electron pathway of the ORR. Interestingly, this viewpoint was different from most of the previously reported indications on the active sites of nitrogen-doped carbon materials (generally associated with pyridine or graphitic N species).31,185,186 More recently, N-OLC was also confirmed to be an efficient and robust electrocatalyst in the ORR.187
Due to the difference in electronegativity and the number of valence electrons between carbon and boron atoms, the incorporation of boron atoms into the lattice carbon network theoretically should play a positive role in enhancing the ORR activity with respect to boron species located at the edge of the carbon network. A class of B-OLCs (Fig. 8, pathway III) with high substitutional boron shows high ORR stability and tolerance for methanol.188 The correlation between the catalytic performance and the electronic structures of doped catalysts was analysed in detail by UPS measurements. The results indicate that the presence of boron atoms is capable of lowering the work function, shifting the valence band edge and improving the density of states of the OLCs. All of these aspects are expected to play a crucial role in enhancing the catalytic performance. Recently, a hot filament assisted CVD technology has been utilized to obtain porous boron doped diamonds (BDDs) deposited on a nickel foam substrate. A certain amount of boron atoms can be successfully incorporated into the crystal lattice of diamonds and thus influence the catalytic activity.189
Additionally, the incorporation of P into OLC was studied in an effort to promote the catalytic performance in the ORR and to adjust the electronic structure of the carbon network.190 Different phosphorus chemical states were investigated, and C–O–P bonds were proven to be responsible for the enhanced activity.
The physicochemical properties of UNDDs, and behaviour in the ORR, can also be tuned by forming nanocomposites with other nanocarbons. Liu et al. applied N,B co-doped graphitic carbons as shell materials to fabricate ND/N,B co-doped graphitic carbon with a core/shell structure.191 This method utilizes effectively the chemical stability of the sp3 carbon in NDs for electrocatalysis, allowing the use of NDs with low conductivity.
Interestingly, the most recent works on the ORR involve alkaline media and the possible application in acidic media has rarely been described. A nitrogen doped diamond@carbon-onion hybrid nanostructure (actually, it is also NOLC) was found to show a remarkably enhanced ORR performance, although the mass activity calculated from the Tafel plot for the NOLC catalyst was only about 40% of that of reference Pt/C (Fig. 14c and d). The authors proposed that the hybrid structure of the graphitized OLC, with N-doped edges at the shell and residual sp3-hybridized ND structure at the core, conceived a synergistic effect with a considerably enhanced ORR.192 This work sheds light on the possibility of metal-free carbons as promising catalysts in the ORR with acidic media.
Up to now, UNDDs or doped UNDDs have been proven potential competitors to Pt-based catalysts for the ORR in alkaline media or acidic media, even if performances have to be further improved. Although few studies on the understanding of the O2 activation mechanism and the formation of intermediates were carried out on nanodiamond-type electrocatalysts, the roles of different species or edge structures in UNDDs or dopant-modified UNDDs have been in general still limitedly studied. In order to understand better the intrinsic active sites and relevant reaction processes, it is useful to review briefly below some fundamental attempts to unravel ORR mechanisms on other nanocarbon catalysts. These insights could provide indications on how to translate the findings to UNDDs and derived ORR electrocatalysts, even if it is out of the scope here to discuss in depth the ORR mechanism on carbon materials.
In 2009, Dai and co-workers reported that N-doped carbon nanotubes (NCNTs) with a vertically aligned structure can act as a metal-free electrocatalyst.193 Such NCNT materials exhibited much better electrocatalytic activity, long-term operational stability, and tolerance to the crossover effect than the Pt–C/GC catalyst. The enhanced catalytic performance was attributed to nitrogen dopant atoms neighboring to carbon atoms, resulting in a substantially higher positive charge density (left-hand side of Fig. 15). Meanwhile, the O2 activation process during the ORR was also proposed. The presence of nitrogen induces charge delocalization and thus changes the chemisorption mode of O2 from the common end-on adsorption (Pauling model) on the CNT surface (top right of Fig. 15) to a side-on adsorption (Yeager model) onto the NCNT catalysts (top right of Fig. 15). This modality of adsorption allows effectively weakening the O–O bonding and facilitates the ORR. Later, Hu et al. found that pristine CNTs are not favorable for realizing O2 adsorption.194 In contrast, the adsorption of O2 on the NCNTs is exothermic in nature and involves electron transfer to O2 related to the promotion of the energy level of the highest-occupied molecular orbital (HOMO) and the simultaneous reduction of the gap between the HOMO and the lowest-unoccupied molecular orbital (LUMO) after nitrogen doping. The authors suggested that the electron configuration of O2 on the NCNTs was similar to that of O2-species.194
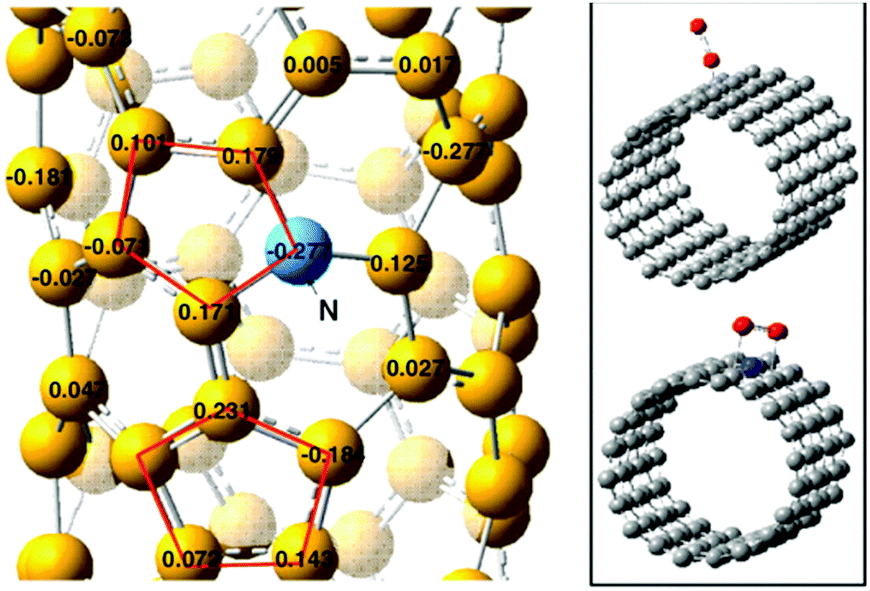 | ||
| Fig. 15 Calculated charge density distribution for the NCNTs and schematic representations of possible adsorption modes of an oxygen molecule at the CNTs (top) and NCNTs (bottom), reprinted from ref. 193 with permission from Science, copyright 2009. | ||
Actually, due to the lack of specific doping of selected N species, different types of N species coexist in nitrogen-doped nanocarbons. At least three nitrogen forms were considered (pyrrolic, pyridinic and graphitic N) to be possible active sites. The O2 dissociation behavior on different N species or different N active sites is still unclear. Additionally, the local structure of N in the carbon network is still an open question.
The energy barriers of the oxygen molecule dissociation on graphitic N, nitrogen-substituted Stone–Wales defect and pyridinic N were studied by Yang's group.195 When one nitrogen atom was incorporated into the SWCNT lattice, the oxygen adsorption energy did not obviously change compared with pure SWCNTs at the initial stage. The highest energy barrier of the O2 dissociation decreased to 0.86 eV from 1.61 eV of SWCNTs (Fig. 16a and b). Upon increasing the concentration of nitrogen and tuning the arrangement modes of nitrogen (meta-, para-positions) in the carbon lattice, the corresponding energy barriers of the O2 dissociation were reduced to 0.42 and 0.69 eV (Fig. 16c and d). The carbon atom adjacent to both nitrogen atoms had a more positive charge. The O–O bond length became 1.37 Å, longer than the previous two cases (a and b). After introducing one nitrogen atom to substitute in the Stone–Wales defect (a common defect), in the final dissociated state, one oxygen atom would adsorb on a carbon top site and the other oxygen atom would break the C–C bond, forming a C–O–C ether linkage (Fig. 16e). The energy barrier of the O2 dissociation was 0.49 eV. In the case of O2 dissociation on pyridinic N (Fig. 16f), the corresponding energy barrier was 1.28 eV, also lower than that for pristine SWCNTs.
 | ||
| Fig. 16 Optimization energy pathways of oxygen molecule dissociation on (a) pure (8,0) SWNCTs, (b) one nitrogen atom substituted (8,0) SWCNTs, (c) two meta nitrogen atom substituted (8,0) SWCNTs, (d) two para nitrogen atom substituted (8,0) SWCNTs, (e) one nitrogen atom substituted (8,0) SWCNTs with a Stone–Wales defect, and (f) (8,0) SWCNTs with pyridine-like nitrogen atoms. Gray dots, blue dots and red dots represent carbon, nitrogen and oxygen atoms, respectively, reprinted from ref. 195 with permission from Royal Society of Chemistry, copyright 2011. | ||
Therefore, the energy barriers can be reduced efficiently by common types of nitrogen doping. Graphitic N and Stone–Wales defect nitrogen were found to be more efficient in decreasing the energy barrier with respect to pyridinic N, due to their lower dissociation barrier. We will further discuss the activation of a single Stone–Wales defect nitrogen for O2 later.
The preparation of doped nanocarbons with single nitrogen species is the best way to determine directly the actual roles of each nitrogen species in the ORR, but it is still challenging. Recently, Guo et al. utilized HOPG model catalysts with well-defined π conjugation and well-controlled doping of N species to identify the real active sites.69 The measured concentration ratios of graphitic (grap) N and pyridinic (pyri) N in grap-HOPG and pyri-HOPG are up to 82% and 95%, respectively (Fig. 17a). ORR results show that the pyri-HOPG model catalyst exhibits high activity at high voltages. The pyri-HOPG sample with lower N concentration (N: 0.60 at%) is much more active than the grap-HOPG sample with higher N concentration (N: 0.73 at%). The results indicate that pyridinic N rather than graphitic N reduces the ORR overpotential and creates the active site (Fig. 17b).
 | ||
| Fig. 17 (a) N 1s XPS spectra of various catalysts. (b) ORR results for various catalysts with high concentrations of pyridinic N or graphitic N species. The inset shows the nitrogen contents of the catalysts. (c) Schematic pathway for the oxygen reduction reaction on nitrogen-doped carbon materials, reprinted from ref. 69 with permission from Science, copyright 2016. | ||
Using the TPD-CO2 methodology, it is found that the Lewis base site (i.e. the carbon atom neighboring the pyridinic N dopant atom) is the active site for the ORR. Oxygen molecules can be adsorbed on these Lewis base sites to achieve the initial step of the ORR followed by protonation of the adsorbed O2 (A–B steps of Fig. 17c). Two pathways are then possible: (i) the first is the common 4e− mechanism occurring at a single site; and (ii) the second is a 2e− mechanism. In the former mechanism, two H+ attack the two oxygen atoms, leading to the breakage of the O–OH bond (C step) and the formation of OH species (D step). In the last E step, the additional H+ would react with the adsorbed OH to form H2O. In the case of the 2e− pathway, intermediate H2O2 is generated by reaction of the adsorbed OOH species with another H+ (F step). Subsequently, H2O2 is readsorbed and reduced by two H+ to form H2O.
Several experimental results in combination with theoretical calculations have pointed out that the enhanced activity of nitrogen-doped carbon materials is related to modification in the charge density at C-atoms lying near to N dopants. This effect induces Lewis acidity on these carbon atoms due to both the electronic effect and local deformation of aromaticity.
For B-doping, the vacant 2pz orbitals of B conjugates with the carbon π system to extract the electrons were reported. These electrons would become quite active due to the low electronegativity of B, and thus O2 molecules would be adsorbed on the positively charged B sites to achieve the ORR process.196
B,N-Codoping is a possible route to optimize metal-free nanocarbon ORR electrocatalysts. However, theoretical calculations indicate that when N with extra electrons interacts with B atoms having a vacant orbital, mutual neutralization may occur. In contrast, separated B and N doping can turn CNTs into excellent ORR electrocatalysts.197
Not only is the role of heteroatoms still controversial, but the role of edges in nanocarbons for ORR behavior is also still a matter of debate. Shen and co-workers suggested that edge carbon atoms are more active than the basal-plane ones for the ORR. In their ORR experiment, an air-saturated electrolyte solution droplet (diameter ca.15 mm) was deposited at a specified position either on the edge or on the basal plane of HOPG. It was found that the ORR on the edge occurs at a more positive onset potential and with a higher current density than that on the basal plane of HOPG.198 In addition, this group applied an Ar plasma etching method to prepare a series of edge-rich nanocarbon materials. After plasma treatment, the ID/IG value is obviously improved from 1.16 to 1.51. The onset potential and the reduction peak potential of edge-rich nanocarbons significantly shift to positive values with respect to the pristine nanocarbon electrode, demonstrating the positive role of edges in the ORR.199
To date, carbon-based or doped carbon-based materials have been extensively investigated as efficient metal-free electrocatalysts for the ORR. Various single-, dual- or multi-doped systems including N, B, P and S catalysts have been made. A lot of theoretical calculations have been used to identify active sites and to explain the ORR mechanism. However, the exact interpretation of the mechanism and active site of the ORR on nanocarbons is still rife with controversy. Consequently, it is even more difficult to derive unique conclusions in the case of nanocarbons with intrinsically higher degrees of complexity, as the nanodiamond and related materials discussed here. More importantly, the possible rate-determining step (RDS) has been rarely studied using experimental methods so far. The roles of electron transfer (ET) and proton coupled electron transfer (PCET) processes in the ORR should be highlighted. One of them unavoidably involves the RDS depending on catalysts used. Furthermore, the possible application of carbon-based materials in acidic media should be further demonstrated.
In most of the studies on heterogeneous catalysis made using metal electrodes (e.g. Cu), the main products obtained through the CRR have been CO or formic acid.204 Some other high value-added products, including formaldehyde, methanol, and methane, have also been achieved using semiconductor or other metal electrodes under atmospheric or high-pressure conditions. These products generally involve direct transfer of electrons to a CO2 molecule, which requires a high reduction overpotential. Therefore, it means that the hydrogen evolution reaction is a competitive process. In fact, the protons and electrons for CO2 reduction should derive from the electrolysis of H2O on the anodic part of the electrochemical cell, but the produced H+/e− in the water oxidation (anodic) step may recombine to form H2 (on the cathodic side) rather than being used for CO2 reduction and hydrogenation. This side reaction to H2 reduced the faradaic selectivity in CO2 conversion.
It is of note that often high faradaic selectivities have been reported, but at very low current densities, i.e. very low productivities. Often the faradaic efficiencies were not achieved under the same conditions where reaction rates in the target product of CO2 reduction are reported. Using a microwave plasma assisted CVD method, a BDD electrode producing a high yield of formaldehyde (C1 product) in the CRR was obtained.205 The highest faradaic efficiency for formaldehyde reached 74% at −1.7 V vs. Ag/Ag+ at room temperature and atmospheric pressure even if formic acid was also observed with the best faradaic efficiency of 15% at −1.5 V vs. Ag/Ag+ in a MeOH electrolyte. When seawater is applied as electrolyte, formaldehyde could be still formed with a faradaic efficiency of 36%. In comparison with the MeOH electrolyte, the lower faradaic efficiency is attributed to the narrow potential window in water and the influence of the inorganic and organic impurities in seawater. The authors proposed that the high faradaic efficiency was attributed to the sp3-carbons in the BDDs.
Liu et al. synthesized a NDD/Si rod array (NDD/Si RA), i.e. analogous to that shown in Fig. 13, observing high efficiency in the CRR to formate (C1) and acetate (C2) products.206 It was found that CO2 would be preferentially and rapidly converted to acetate over formate with an onset potential of −0.36 V (vs RHE) in 0.5 M NaHCO3 solution. The behavior overcomes the common limitation of low selectivity for C2 products. Upon increasing the concentration of nitrogen from 0.93% to 2.12%, the production rates for acetate and formate increase significantly. Faradic efficiencies of about 91–92% for formate and acetate can be achieved at −0.8 to −1.0 V. The superior performance was attributed to the high overpotential for hydrogen evolution and to the presence of N-sp3C active sites (e.g. sp3 type C atoms near to a N doping atom). The main pathway for the CRR is indicated as follows:206
| CO2 → CO2˙− → (COO)2˙→ CH3COO− |
| *COOH → *CO |
| *COCHOH → *COCH2OH → *CHOCH2OH → *CH2OCH2OH |
| CO2 → *COOH → *CO → *COCO → *COCH2OH → *CH2OCH2OH → CH3CH2OH |
 | ||
| Fig. 18 (a) Faradaic efficiencies for CH3CH2OH in 16 consecutive CRR runs on BNND3 at 1.0 V. (b) Faradaic efficiencies for the CRR on different catalysts at 1.0 V. (c) Free energy diagrams for the CRR on BBNDs. (d) Energetically favorable structures for elementary steps of the CRR on BBNDs. Gray dots, pink dots, blue dots, red dots and white dots represent C, B, N, O and H atoms, respectively, reprinted from ref. 207 with permission from Wiley, copyright 2017. | ||
N- or B-(co)-doped UNDDs have shown promising behaviors in obtaining high-value added chemicals (C1 and C2) in the CRR. Similar to the ORR, the recent development with doped UNDDs as electrocatalysts in the CRR is still in its infancy. The roles of various N species or boron species have not been investigated. Ma et al. summarized the present status and future challenge of metal-free carbon materials in the CRR.208
Some examples of the catalytic behavior of nanocarbons other than UNDDs will be introduced here for a better understanding of the reaction mechanism in the CRR, although a detailed discussion of carbon materials different from UNNDs is out of scope here.
A range of 3D nitrogen-doped graphene nanoribbon networks (N-GRW) with tunable N species were designed to reveal the site-dependent CRR activity/selectivity reported by Liu et al.209 The N-GRW catalyst exhibits a high CO faradaic efficiency of 87.6% at a mild overpotential of 0.49 V. A linear regression can only be produced between current density and the concentration of pyridinic N. After using post-hydrogen treatment to reduce the content of oxidized N and graphitic N species, the change in the CRR activity along with pyridinic N content can also be fitted into a good linear relation (Fig. 19a), suggesting that pyridinic N could be the active site. Phosphoric acid treatment was further used to reduce selectively the pyridinic N of the N-GRW by adsorption. The activity of the soaked N-GRW is lower than that of the original N-GRW (Fig. 19c), further indicating that the CRR activity should be determined by the pyridinic N sites. The plausible reaction mechanism mainly involves two intermediates (*COOH and *CO). Compared with pyrrolic N, graphitic N and oxidized N have higher ΔG values. Pyridinic N has a lower ΔG value of 0.33 eV, implying that it is beneficial for the transformation of CO2 to *COOH and further to *CO (Fig. 19d).
 | ||
| Fig. 19 (a) The relationship between the CO current density and the pyridinic N content in various N-GRW catalysts at 0.5 V (vs. RHE). (b) N 1s XPS spectra of N-GRW before and after being soaked in 1 M H3PO4 aqueous solution. (c) The multipotential curves of the N-GRW, N-GRW soaked in H3PO4 aqueous solution with different concentrations for half an hour. ORR results for model catalysts. (d) Illustration of the CRR processes on N-GRW catalysts. (e) Free energy diagram of the CRR on various N species, reprinted from ref. 209 with permission from Wiley, copyright 2018. | ||
The origin of activity in N-doped nanocarbons for the CRR has also been investigated by Xu et al.210 These electrocatalysts show excellent performances, with about 90% current efficiency for CO formation and stability over 60 hours. The N-doping was realized using a specific surface methodology, based first on the creation of O-functionalities and then their reaction with a positively charged polyelectrolyte (poly(diallyldimethyl-ammonium chloride), PDDA) as the N source. Depending on PDDA loading and the temperature of annealing, different N species are formed. It is possible to quantify their amount (for example, by XPS) and then correlate the amount of the different species with the onset potential for the CRR. A good correlation with the amount of quaternary N species was observed, while no relationship was found with the other N species. The role of these quaternary N doping atoms, as indicated by DFT calculations, is to stabilize the carbon dioxide radical anion (˙CO2−), formed by one-electron transfer, thereby lowering the initial reduction barrier and improving the intrinsic activity.
For the CRR, the presence of many pathways of transformation with analogous energy and the presence of the side reaction of hydrogen evolution significantly influence the yield of the target products. However, at the same time, literature data show that selectivity could be achieved beyond what can be expected from a thermodynamic analysis of the possible reaction pathways. This indicates that it is thus the electrocatalysis, rather than just electrochemistry, which determines the possibility of achieving high performances.203 On the other hand, literature data are often not presented in the right way to understand the potential applicability, which is the faradaic efficiency at high current densities per geometrical area of the electrode.
Note also that it is often not clearly excluded that the products observed in the CRR do not derive from the nanocarbon itself, either from organic contaminations or from the direct reduction of the more reactive carbon species. At the same time, reconstruction of the carbon species may occur during the electrocatalytic reaction, as noted before for dehydrogenation reactions. A more extended use of in situ/operando methods is necessary, but using specific model electrocatalysts, which can unravel the complex electrocatalytic chemistry present in CRR systems. Compared with the ORR, the CRR system is more complicated (more H+ and e− transfers, depending on products) and thus more intense investigation on the nature of the active sites and the reaction mechanism is necessary. Studies on interfacial effects including CO2 adsorption, diffusion, activation and product formation, and desorption processes on the catalyst remain challenging.
At the same time, however, it is necessary to parallel these fundamental studies with others with practical character, where relevant data for exploitability of the results (faradaic efficiency at high current densities, productivity to target products per geometrical electrode surface, stability over extended periods) are presented. In addition, often the results in the literature are obtained in electrocatalytic reactors, which are not relevant for industrial use (the use of large electrolyte volumes, batch cells, for example) or not taking into account important aspects such as product recovery, cell fluidodynamic and current homogeneous distribution in larger cells, formation of gas caps, etc.203
2.4 Photocatalytic reactions
In general, photocatalysis is the catalysis of a photochemical reaction on a solid surface.215 Photocatalysis includes two main branches: one involves energy-related photocatalysis (e.g. water splitting, CO2 reduction, CH4 activation), and the other is environmentally-related photocatalysis (e.g. organic pollutant degradation). To date, photocatalysis has been considered as a promising sustainable technology, because it allows the direct use of solar light, although with the main limitation of low reaction rates, making commercial photocatalytic processes to be used to a limited extent. Current studies on photocatalysis are largely using oxide or derived materials (oxynitrides, for example). Their development is mainly focused on three crucial aspects: (i) improving the full visible-light harvesting and use, (ii) enhancing the charge separation of the photogenerated species, and (iii) increasing the efficiency in the use of the photogenerated charges for the chemical reactions, particularly by adding co-catalytic functionalities. Carbon-based materials have been limitedly considered as possible photocatalysts.Due to the specific electron structure and fast charge carrier combination, metal-free carbon materials (except C3N4) are not suitable options for photocatalysts in principle. However, the great flexibility of modification of nanocarbons, with the possibility of creating semiconducting zones as commented on later, nanodots and other photoactive functionalities, combined with the presence of a chemically active surface with unique properties induced by doping/defect engineering as well as by the presence of edges provides new possibilities for photocatalysis still largely unexplored.
Fig. 20a compares the energy band diagram of H-terminated diamonds with those of some other typical semiconductors, such as GaN, TiO2, CdS and SiC. It is evident that the H-terminated NDs present rather interesting possibilities for overcoming the theoretical energy barrier to initiate the one-electron reduction of CO2, the reduction of H+ to form the solvated H˙, or to form the solvated e− (aq) even if the band gap of the ND is up to 5.47 eV, which lies in the insulator range. It was shown that H-terminated NDs dispersed in 18.2 MΩ cm nanopure water can selectively reduce aqueous CO2 to CO in the presence of ultraviolet light and Na2SO3 as a sacrificial oxidation and electron donor.216 The presence of a hole scavenger (SO32−) in solution facilitates the continuous electron emission to reduce CO2 to ˙CO2−. Subsequently, CO is produced along with the formation of O˙− under ultraviolet light. Alternatively, O˙− could react with H+ in solution to produce the ˙OH that is able to react with H from the H-terminated surface to form H2O to finish the diamond oxidation process (Fig. 20b).
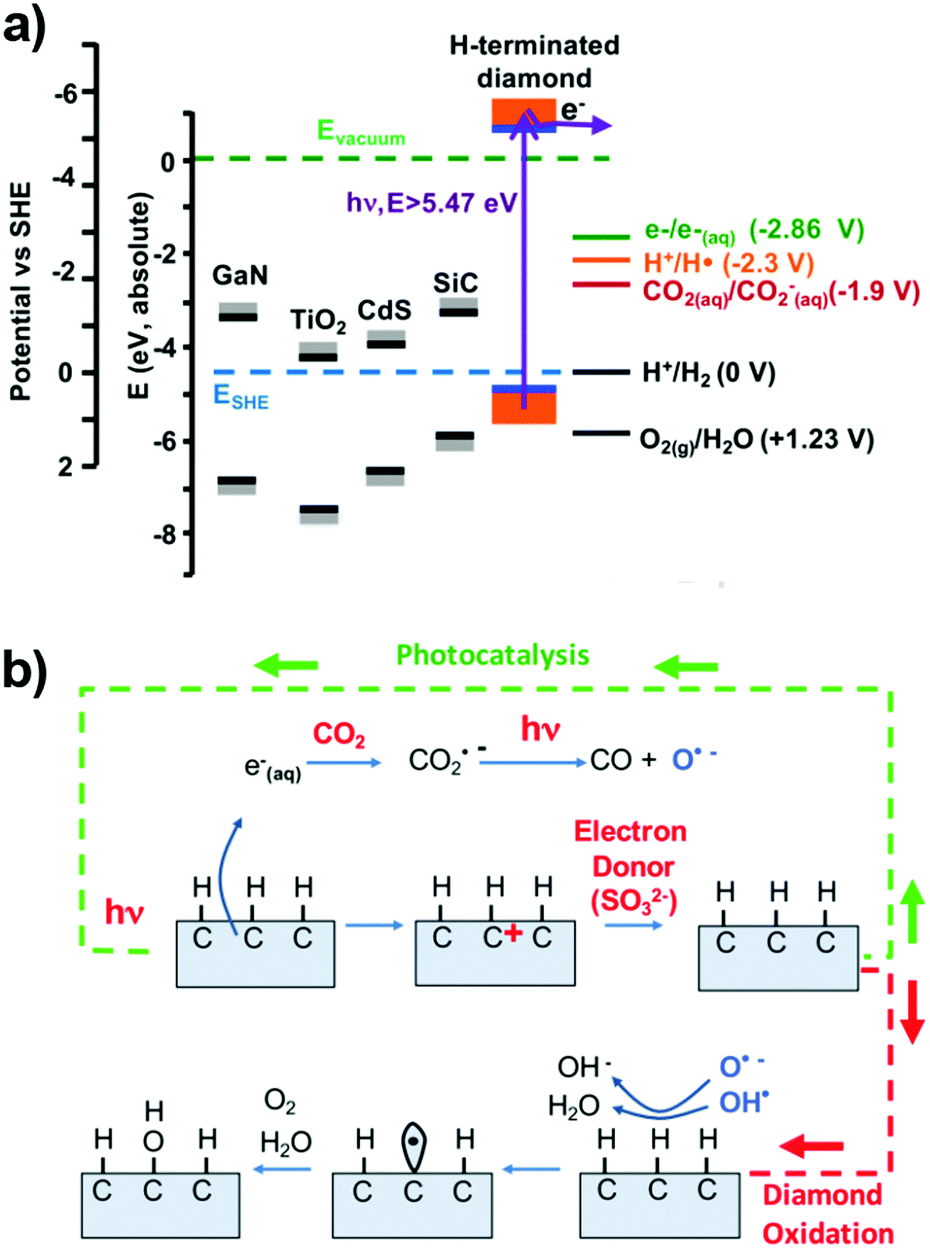 | ||
| Fig. 20 (a) Energy band diagram of H-terminated diamonds and some other semiconductors, together with some related redox reactions. (b) Schematic illustration of relevant chemical and photochemical processes, reprinted from ref. 216 with permission from Elsevier, copyright 2017. | ||
NDs were also demonstrated to be a good photocatalyst in H2 generation from H2O splitting upon 532 nm laser irradiation.217 The research showed that H-terminated NDs show better activity for H2 generation than O-terminated NDs, because the H-terminated sites operate as electron reservoirs. They can lead to the surface electric bilayer that could absorb H3O+. Once the H-terminated NDs generate the e−/h+ pairs upon the multiphoton absorption, the e− reduces the H3O+ to produce H2. Meanwhile, the H-terminated NDs were used as an excellent photocatalyst to reduce the graphene oxide to form ND-reduced graphene oxide composites which are linked by non-covalent bonds.
More recently, Tripathi and co-workers proposed that a water soluble OLC exhibits better catalytic activity for the degradation of methylene blue (MB, a typical organic pollutant dye) than that of pristine OLC under visible light irradiation.218 The higher performance of the water soluble OLC may be attributed to the surface abundant carboxylic groups (charged negatively under acid conditions) that are beneficial to provide higher binding affinity for hydrophilic MB molecules as compared to the pristine OLC. Band gap tuning and photocatalytic application of other nanostructured carbon materials will be further highlighted later.
There are thus limited studies on the use of NDs and derived materials as photocatalysts, but the preliminary indications suggest that this is an area of potentially larger interest. There is, in general, increasing interest in the use of nanocarbon materials as photocatalysts or components in photoanodes.
2.5 Active sites in UNDDs
Although some excellent achievements have been realized using UNDD or doped UNDD catalysts, there is in general still a lack of in-depth mechanistic studies on the reaction and on UNDDs to identify the specific peculiarity of this novel class of carbocatalysts. From the previous discussion, it was evident that the charge density on specific C atoms or C–C bonds induced by the curvature of the surface graphene layer, associated with the presence of a hybrid sp2/sp3 configuration and/or doping with heteroatoms, plays an important role, eventually in synergy with other functional groups (e.g., CO). The amount of functional groups in general is higher than those in other nanocarbon materials, as well as the presence of defects. The highly defective surface could:
– facilitate absorption of the reactants, stabilize intermediates or enhance the desorption rate of the products;
– generate the active sites, which may be associated, for example, with the zigzag edge configuration that was proof to play a key role in some liquid phase reactions;
– lead to the in situ formation of active sites (e.g., CO) by dissociating oxidants/reductants during the reaction.
All these characteristics are associated with the peculiar electronic structure of these highly defective materials and the additional effect of the local curvature in the graphene-like plane. These structural characteristics make possible also an in situ reconstruction during the catalytic reaction, with generation of new active sites induced by these transformations.
The introduction of heteroatoms (e.g. N) in UNDD catalysts improves largely the catalytic activity. Taking the ORR and CRR as examples, several common N species including pyridinic N, graphitic N, pyrrolic N or N-sp3C species were speculated to be active sites, but their exact roles as well as defect species, the evolution of the related species and surface microstructure changes (e.g., arrangement) during the electrochemical reaction are largely not understood.
For instance, the CO group can alter the electronic distribution of the surrounding carbon atoms on the UNDD surfaces and facilitate the adsorption/desorption of catalytic reaction intermediates, and thus may affect the electrocatalytic activity. Moreover, the roles of carbon corrosion and carbon restructuring, particularly in electrochemical applications, should be taken into account. The reaction pathways and nature of the active sites involved in the formation of different products (C1, C2) during the CRR should be explored in detail. There is, in general, a lack of mechanistic studies on these carbocatalysts for their knowledge-based development.
2.6 Nano-structuring the catalytic sites
This term defines the design of the optimal nano-scale spatial arrangement of multiple functional centers, which are necessary in a catalytic site for complex multistep catalytic reactions. One of the research directions for a future sustainable chemical production is the process intensification realized through a reduction in the number of steps.37,154 Realizing multifunctional catalysts able to make one-pot multistep (cascade) reactions, requiring different catalytic functionalities, is thus one of the areas of development for catalysis in general. In addition, it is also necessary to optimize the diffusion of the reactants (and the retrodiffusion of the products) as well as eventually define an optimal path of diffusion in the solid in order to have the target sequence of active centers and reactions.This is one of the actual frontiers of research in catalysis and nanocarbons, particularly NDs and related materials, have several of the requirements to develop these types of materials. However, it will be necessary to develop novel procedures of synthesis and post-modifications to develop the aimed characteristics and catalytic functionalities, together with the methodologies to determine the optimal 3D-configurations for multifunctional active centers. This requires combining advanced modelling and specific methodologies for determination of the nature of active sites in carbons at the nano-scale level. It will also be necessary to assemble specific carbon nano-units to create 3D-like structures with specific porosity characteristics, allowing both mass and heat transfer limitations to be avoided. This controlled nano-morphology, from the nano- to macro-scale, is necessary to limit the rates of the parallel or consecutive side reactions, which is sometimes even more critical than in “classical” catalysts, due to the presence of a charged surface and charged species deriving from electron transfer.
This concept of nano-engineering is present in the literature, but a solid and more systematic use in the development of nanocarbon catalysts is still lacking. The starting point is to design an efficient and robust mechanism to synthetize and assemble nano-components, based on an improved understanding in the nature of the active sites in nanocarbons. This is a concept going beyond that of hierarchically organized materials, which consider essentially aspects related to mass-transfer of reactants and products. It will also include the possibility of the development of nano-hybrid architectures, based on the combination of nanocarbons and other (such as oxide nanoparticles) elements.
Notwithstanding research on nanocarbons has largely focused on the synthesis procedures, as emerged also from the previous sections, further efforts in this direction are necessary.
3. From NDs to catalysis by nanocarbons
The previous section showed how novel types of nanocarbons with respect to the common classes of materials (such as graphene, CNTs and fullerene families, i.e. the most used and discussed in recent reviews) provide valuable opportunities to improve the performances and to extend the use of metal-free nanocarbons for new applications. Both these aspects are largely associated with the possibility of going beyond the concept of doping with heteroatoms, the most common aspect analysed in the literature. Although clearly this is an important aspect, the results show that the doping with heteroatoms creates a localized charge density on C atoms or C–C bonds, and these centers represent the active sites by themselves or acting in cooperation with the functionalities introduced by the heteroatoms.This is an area largely at the beginning of understanding, requiring also the development of specific methodologies of analysis. From this perspective, however, it is evident that there is a wider range of possibilities for changing the surface properties of nanocarbons than just by doping/co-doping:
– curvature and creation of strains in C–C bonds;
– creation of hybrid sp2/sp3 systems (eventually with stabilized sp1 centers as well);
– design of tailored defects and vacancies.
These elements integrate with the functionalities generated by doping to form a powerful platform of possibilities for tailoring the surface properties, and for this reason making nanocarbons a unique, potentially disruptive class of catalysts. The combination of all these tailoring methods may be called defect engineering in nanocarbons.20,29
Many aspects have been already discussed in the previous section. Here we analyse how some of the above elements have been considered in the analysis of the catalytic behaviour of other nanocarbons, although in our opinion there is a lack of a more holistic and integrated analysis of their role in understanding the catalytic behaviour of nanocarbons.
The previous section has remarked on how understanding the catalytic chemistry of NDs and related materials requires considering the synergy between catalytic functionalities created by (i) doping, (ii) nanostructure effects (curvature associated with nanostructures or other elements), (iii) edge effects and (iv) creation of reactive carbon atoms or bonds due to electron density localization. The same concept is valid to understand catalysis by nanocarbons in general.
We will present some aspects in the following sections to support this generalized concept to understand catalysis by nanocarbons, in particular in association with the last aspect often underestimated. However, it is out of the scope of a systematic analysis, being limited to evidence that this synergy is not just a peculiar property of NDs and related materials, but a general and crucial aspect to understand nanocarbon reactivity.
3.1 Defect-related catalytic reactivity
Nanocarbons contain the highest number of defects with respect to other types of catalysts such as oxides.219 This is related to the higher structural flexibility in nanocarbons, particularly NDs and related materials, to minimize energy due to defects through local instead of extended restructuration. For example, an isolated pentagon transforms a SWCNT into a sharp cone, and a heptagon converts it to a horn.220 This also explains why hybrid nanocarbons such as the nanodiamond family described in the previous section contain typically a higher number of defects. A common defect in nanocarbons is a pair of pentagon–heptagon (5/7). Their presence causes changes in the chirality of SWCNTs.220 The formation energy of a 5/7 defect site in SWCNTs ranges between 2 and 4 eV and thus is quite low. The chirality in the growth of SWCNTs is determined from defect healing.221 A generalized model for the growth of defective MWCNTs was proposed by Hembram and Rao.222 Defects such as pentagons, heptagons, vacancies, or dopants are found to modify drastically the electronic properties of these nanosystems.223The presence of defects and vacancies in nanocarbon materials is thus well established, but their role in the catalytic properties is often underestimated, in part due to the difficult characterization. Most of the studies on carbocatalysis do not analyze the presence and role of these sites.
Nevertheless, there are indications that defects and vacancies play both a direct role, inducing the formation of active sites (dangling bonds, which have characteristics different from those present on edge sites, and may easily have a cooperative effect), and an indirect role, by (i) inducing change in the local charge density at nearlying C atoms or C–C bonds, (ii) creating semiconducting states, as discussed later, and (iii) hosting metal ions or atoms. DFT calculations indicate that defects in graphene sheets can reach a concentration of up to about 1%. This effect creates also localized energy levels near to the Fermi level. Consequently, an effect on the local charge and catalytic reactivity is observed.224 Defects in graphene sheets thus generate three types of modifications:
– create reactive sites;
– change the electronic state and adsorption characteristics;
– change the conduction characteristics, which are clearly relevant not only in electrocatalysis, but also for catalysis itself, due to the possibility of delocalizing electrons (in redox reactions), generating radical species and even transporting the heat of reaction (thus reducing, for example, local increases in temperature – electron and heat transfer in carbon materials depend on similar parameters).
It was reported that defects in CNTs determine their degree of functionalization and types of functional groups generated, with a consequent change in the behavior in biomass conversion.225 The production of reactive oxygen species during irradiation and thus the photocatalytic behavior in CNTs depend on the type of defect.226 Highly defective CNTs show high photocatalytic activity.227 A base and acid washing of graphene creates a defective material showing high activity in the oxidative coupling of amines to imines.228 Hole defects in the conjugated domain were indicated as the active sites in this reaction, or alternatively the carboxylic acid terminating edges in the hole defect. Characterization of these materials by ESR indicates a relationship between the catalytic behavior and the localized spins in the edges related to the p-electron system. These sites with unpaired electrons activate molecular oxygen by a sequence of electron transport and reduction to superoxide radicals.
Mechanically induced defects in CNTs greatly modify their characteristics and the reactivity in derived Pt/CNT electrocatalysts used as anodes in PEM-type fuel cells.229 It is worth noting that it was demonstrated how these defects induce many modifications in these electrocatalysts:
– change the dispersion of Pt particles;
– improve the tolerance of the catalyst to CO poisoning, due to the carbon functional groups in close proximity to Pt particles and which catalyze the reactivation of Pt sites poisoned by CO;
– affect the electronic conductivity of CNTs;
– modify the hydrophilic character, with a consequent influence on the three-phase boundary and the protonic transport to the active metal particles.
Therefore, the effect of creation of defects cannot be correlated with specific parameters typically used to analyze the electrocatalytic behavior, such as the geometrical surface area of Pt particles or the electrochemical active surface area determined by cyclic voltammetry (CV) tests.
Zhong et al. showed the direct role of edge sites and defects in determining the catalytic behavior of NDs in the decomposition of methane.131 By quantifying the amount of defect sites through SIMS, i.e. by analyzing the signal of C2H2− (acetylene-like fragment – it is a fingerprint of hydrogenated defects) with respect to C2H− and C2− signals (fingerprint for aromatic C–H groups and aromatic/graphitic carbon, respectively), it was possible to estimate the concentration of defect sites in ND samples annealed at different temperatures. The initial rate of methane decomposition was observed to depend linearly on the number of –CHCH– defective sites (Fig. 21).
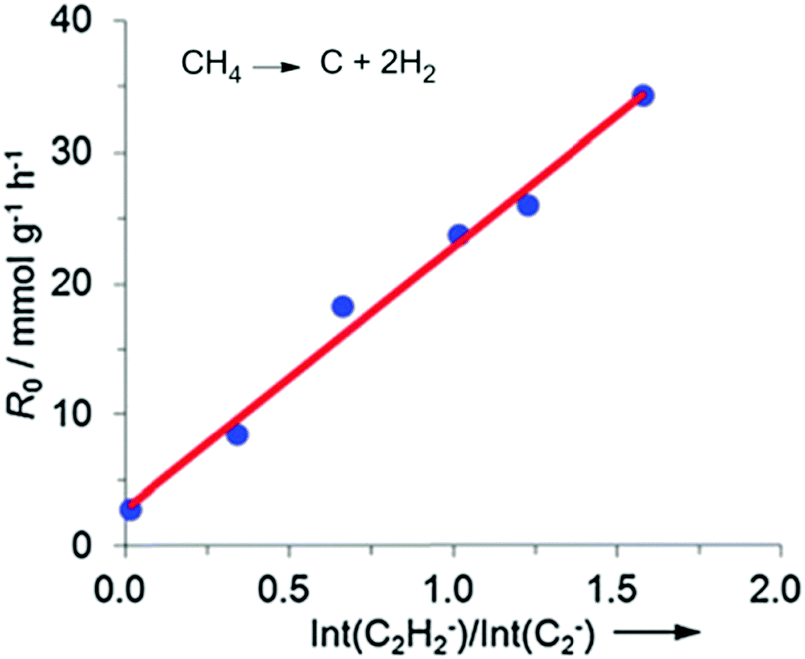 | ||
| Fig. 21 Initial rate of reaction in CH4 decomposition at 850 °C versus the C2H2−/C2− ratio in SIMS spectra of ND samples, which were annealed in the 900–2000 °C temperature range. Adapted from ref. 131 with permission from Royal Society of Chemistry, copyright 2014. | ||
DFT studies give indications on the reaction pathways and mechanisms. The rate-limiting step is the initial C–H bond cleavage in methane having an energy of about 2.47 eV. The transition state interacts with one of the carbon atoms in the vacancy, producing a –CH3 group which further loses the hydrogen at another vacancy site. The process then further continues at the carbon vacancy site, as shown in Fig. 22.
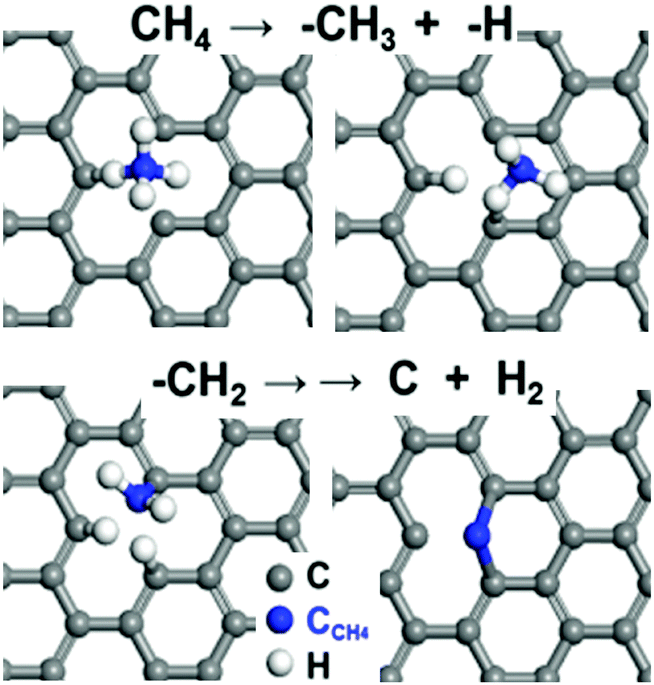 | ||
| Fig. 22 Mechanism of reaction in methane decomposition at defect sites in nanocarbons. Based on indications of ref. 131 with permission from Royal Society of Chemistry, copyright 2015. | ||
There are thus some examples in the literature on defect-related catalytic reactivity in nanocarbons, but in general attention on these aspects is limited, due to the difficulty in analyzing and characterizing the defects and vacancies in nanocarbons, their high dependence on the specific preparation conditions and the strong correlation with other characteristics of nanocarbons and thus the intrinsic difficulty in enucleating the specific aspects.
The introduction of defects in nanocarbons influences, among others, the (i) surface reactivity, (ii) mobility of adspecies, (iii) hydrophilicity, and (iv) electron conductivity. Thus, a proper determination of the relation to the catalytic behavior requires investigating all these aspects and their relative contribution to the catalytic performances.139
3.2 Modification in the charge density at carbon atoms
As briefly commented on above, the presence of defects, curvature and heteroatoms induce changes in the charge density at localized C atoms or C–C bonds. This results in the creation of potential active sites, whose role has been scarcely investigated up to now. Sidik et al. indicated that the reduction of O2 (ORR) is facilitated on the in-plane carbon atom adjacent to the substitutional N atom.230 Kim et al. also indicated that oxygen activation occurs on a C atom adjacent to a pyridinic-N at the edge of a graphene nanoribbon.231 The proposed catalytic cycle involves a ring-opening of the cyclic C–N bond which can be restored after the 4e-reduction of the chemisorbed oxygen (Fig. 23). Although also different ORR mechanisms (as discussed in Section 2.3.1) have been proposed,31,232 the one given in Fig. 23 shows both the role of defects sites/edges and the direct participation in the reaction mechanism of C atoms having nearlying heteroatoms.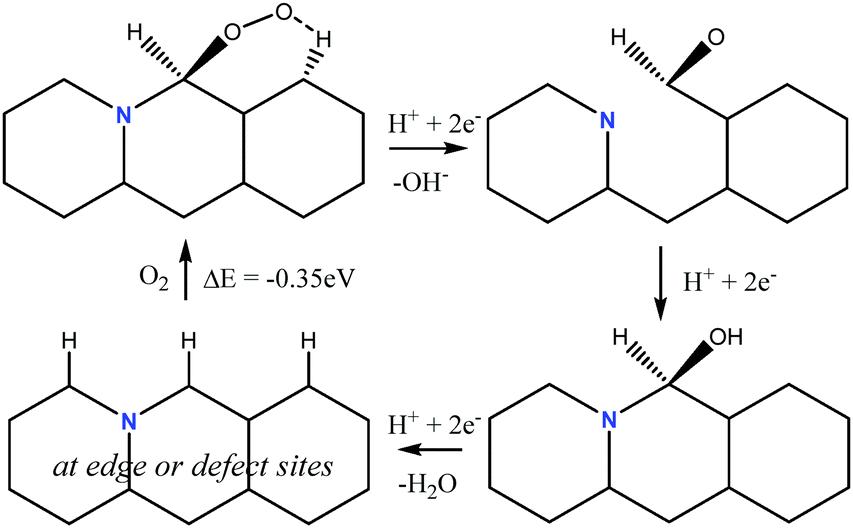 | ||
| Fig. 23 The ORR mechanism suggested for N-doped graphene edges, evidencing how O2 adsorbs at carbon adjacent to a N-atom. Based on indications of ref. 231 with permission from Royal Society of Chemistry, copyright 2016. | ||
Stone–Wales defects are a common type of nanocarbon defect. They are characterized by the presence of two five-membered rings and two seven-membered rings, substituting four adjacent six-membered rings of a pyrene-like region. The C–C bond bridging two of the adjacent rings rotates, creating a reactive C–C bond bridging the two five-membered rings. When nearlying heteroatoms are present, this may create a different charge density at the carbon atoms in this reactive C–C bond, as illustrated in Fig. 24a, where the numbers at the C atoms indicate the effective charge density. Chai et al. indicated that nitrogen pair doped Stone–Wales defect provides a good active site for the ORR, with the activity being tuned by the curvature around the active site.233 The maximum limiting potential (0.80 V) in the volcano plot for the ORR activity can be estimated, in agreement with experimental indications. Fig. 24b illustrates the proposed reaction mechanism of O2 activation, which is followed by breaking of the O–O bonds and stabilization of the surface species by the concerted 2H+/2e− transfer and formation of a C(OH)–C(OH) species, which then desorbs water.
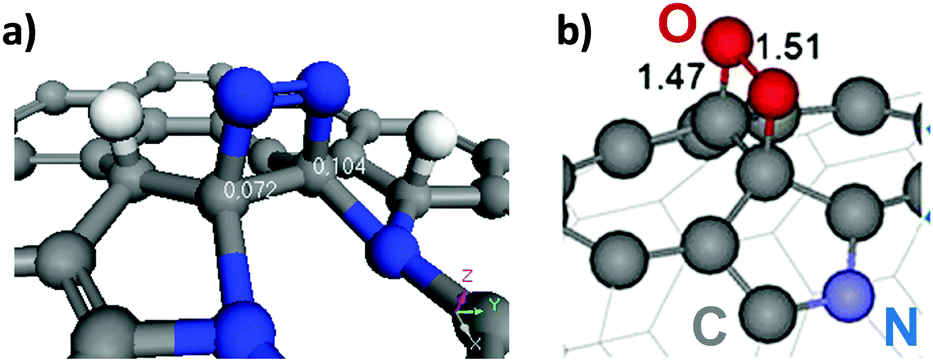 | ||
| Fig. 24 (a) Model of N2 activation at Stone–Wales defects with nearlying doping N sites (in blue). (b) Model of O2 activation at reactive C–C sites in Stone–Wales defects on the surfaces of graphene-like materials with specific curvature. Based on the model given in ref. 233 with permission from American Chemical Society, copyright 2014. | ||
The curvature plays a potential important role in determining the reactivity of C–C bonds, although very limitedly investigated in terms of catalytic reactivity. Sabirov et al. showed by DFT that in the fullerene series C20–C76 there is a correlation between the heat of 1,3-dipolar addition reaction and the carbon surface curvatures at reaction sites.234 Yang et al. using the radial deformation of SWNTs caused by the strong interactions with quartz lattice tubes reported that samples with larger degrees of radial deformation show more delocalized partial electrons at certain sidewall sites with high local curvature, suggesting a relationship with their reactivity.235 By studying using DFT the reactivity of the C20–C80 fullerene series, Anafcheh et al. indicated a correlation in various reactions between the H-abstraction from the primary and secondary C–H bonds of hydrocarbons with the curvature degree.236,237
There are thus limited data, which indicate the role of the curvature (in graphene or equivalent structures) in creating reactive sites, which is schematically illustrated in Fig. 25, although clearly should be differentiated by irreversible reaction (as often occurs on fullerene materials) and catalytic reaction, in which a mechanism close to the catalytic site should be present.
 | ||
| Fig. 25 Visualization of the effect of curvature in creating reactive C–C sites. | ||
An example of the role of curvature in influencing the behavior of carbocatalysts is given by the use of OLC-like carbon nanospheres. These materials, showing a high surface area and porosity interesting for catalytic applications, are characterized by a spherical macro shape with relevant structural disorder in the stacking of the graphitic layers together with the presence of small ordered domains.238 They are active in reactions such as glycerol etherification,239 and carbon monoxide and methane chlorination.240 The role of strained C–C bonds related to curvature was well demonstrated for the last case, with differentiation between irreversible and reversible (catalytic) chemisorption.239 The characterization of these catalysts evidences the presence of strained and highly-strained C–C bonds. The latter were associated with the disordered nano-onion type structure, and react irreversibly with Cl2 with the dissociation of the Cl–Cl bond. These sites are analogous to those in fullerenes, where high bending is present and which also react irreversibly with Cl2. The other strained C–C bonds form from the first during thermal annealing nano-ordering. These sites are reactive towards Cl2, but form a reversibly coordinated Cl2 molecule. The coordination of CO forms COCl2 and that of CH4 leads to chlorinated methane products. It was observed that C–C bonds with moderate to weak straining character instead do not or only very weakly coordinate Cl2.
The proposed catalytic mechanism of CO chlorination on these catalysts, after thermal annealing to control the strain character of the C–C bonds, is schematically illustrated in Fig. 26 assuming a fullerene hemisphere as a model of the effective nanostructure.
 | ||
| Fig. 26 Catalytic mechanism of CO chlorination on a fullerene hemisphere with controlled curvature, as a model for OLC-like carbon nanospheres. Reproduced from ref. 240 with permission from Wiley, copyright 2015. | ||
3.3 Creation of semiconducting areas
As discussed above, UNDDs were demonstrated to be a category of promising photocatalysts in water splitting, CO2 reduction and organic pollutant degradation. A perhaps unique characteristic of nanocarbon materials is the possibility of creating interesting photocatalytic properties through generation of semiconducting areas with the band gap tunable even within the visible-light region. These aspects, not having counterparts in oxide-based conventional semiconductors, derive from the possibility of local band engineering through a combination of controlled introduction of defects, doping with heteroatoms and modification of the curvature.70To understand these possibilities, it is useful to introduce first the electronic characteristics of an ideal graphene structure. The in-plane ordering of hexagonal carbon units, with all C-atoms with sp2 hybridization, leads to the formation of π-orbitals below and above the C-plane. The π-electrons can move freely and thus this material has a semi-metal electrical character. When the graphene sheet is rolled up to form a tube (SWCNT), the conduction properties will markedly depend on the diameter of the tube and chirality. While a part (about one third) of the possible conformations of the SWCNT are metallic, the remaining conformations have a semiconductor character, with the band gap being inversely proportional to the diameter. It decreases from about 0.8 to 0.4 eV in passing from 1 to 5 nm tube diameter.
This band gap is too small for photocatalytic applications, but may be modified to have wider band-gap semiconductors by inducing controlled defects.241 Small rotations of a bond, periodically repeated through the lattice, collapse the Dirac cones at the graphene K point. The electronic structure of graphene is characterized by bands, which are cones intersecting at the Fermi surface, e.g. at the K points in the Brillouin zone. The band gap is related to the distance between the apex of these cones. Patterned defects can break the graphene sublattice symmetry, creating a band gap. Small area ordering of defects thus generates a local semiconducting area.
Yeh et al. showed that N-doped graphene oxide quantum dots could act as visible-light photocatalysts for overall water splitting.242 They suggested the idea that p- and n-type semiconducting domains form in these nanocarbons. The different domains are associated with different types of doping and defects, as exemplified in Fig. 27. Although this mechanism was not jet proof, Yeh et al. indicated that the nanostructure reported in Fig. 27 shows a bandgap of around 2.2 eV, suitable for photocatalysis with visible light. In addition, the p- and n-type domains act as Ohmic spaces, which are p–n junctions allowing an efficient electron–hole separation. Water splitting may occur at the edges of the n- and p-type domains.
 | ||
| Fig. 27 Mechanism242 suggested for the photocatalytic water splitting on nitrogen-doped graphene. Reproduced from ref. 70 with permission from Elsevier, copyright 2017. | ||
Yeh et al. reported for these carbocatalysts (loaded with small amounts of Pt) a quantum efficiency of 12.8% at 420 nm in the photocatalytic H2 evolution in aqueous triethanolamine.243 Even in the presence of a sacrificial agent, the results are promising and superior to those of g-C3N4 and other Pt-doped conventional photocatalysts.
Although more solid proof is necessary about the presence of these n- and p-type domains and their structures, these indications outline how nanocarbons can have unique properties, characterized by local semiconducting domains in a conductive matrix. There is no equivalent in conventional semiconductors to design advanced photocatalysts based on the possibility of creating nanosized and tunable n- and p-type domains. In addition, semiconducting nanocarbons differentiate from conventional semiconductors in particular in terms of the following aspects: band structure and pinning at the interface, formation of heterostructures, charge separation, quantum confinements, and charge carrier mobility.
3.4 Strains and defects induced by nanocarbon interaction with supported metal nanoparticles
Carbon materials are well-known supports for metal nanoparticles and the development of carbon–oxide nanoparticle hybrids or composites.58,71,244–249 However, attention is always paid to the reactivity of the supported nanoparticles, although it is well established that the properties largely depend on the specific nature of the nanocarbons, and that the presence of functional groups, doping heteroatoms and defects (although the latter aspect is often not considered) influence largely the catalytic reactivity. Depending on these characteristics, the shape and the reactivity of the supported nanoparticles can be largely influenced.250–252The interaction between the metal nanoparticles depends on both the functional groups in CNTs and the degree of graphitization.253,254 What is interesting is that a closer inspection by HRTEM of the supported nanoparticles indicates that (i) not only are the nanoparticles themselves sitting at defect sites, but they also introduce new deformations and defects (indicated by smaller white arrows in Fig. 28), and (ii) also highly defective carbon shells (indicated by large white arrows in Fig. 28) may wrap the nanoparticles during the thermal treatment or catalytic reaction. The effect depends on the type of functionalization and nanostructure of the nanocarbon.
 | ||
| Fig. 28 HRTEM image of a Pd nanoparticle supported over a functionalized CNT. Small arrows indicate new defective areas created at the carbon–nanoparticle interfaces, while large arrows indicate carbon shells wrapping the nanoparticle. | ||
This indicates that the carbon–nanoparticle interaction not only influences the characteristics of the supported nanoparticles, but also changes the feature of the interface itself, with generation of a highly defective carbon region around the nanoparticles, and the possible generation of carbon shells wrapping the nanoparticles. This could stabilize the nanoparticles against the sintering and change their available area, but eventually creating access to a smaller area or crystal faces of the nanoparticles, influencing the activity and selectivity. There are very few data in the literature on these aspects, especially regarding how the catalytic reactivity depends on these aspects. However, it can explain the very large influence of the modification of carbon characteristics and functionalization over the catalytic behavior of the supported nanoparticles.
Another aspect related to the previous observation is that the creation of a highly defective area, or of very thin carbon shells (likely highly defective) wrapping the nanoparticles, creates new carbon-related active sites, as discussed previously. This is an aspect largely not considered in the literature, but some recent evidence indicates that this aspect could be the critical one.
The direct electrocatalytic synthesis of NH3 from N2 and H2O could be realized at room temperature and pressure using iron nanoparticles supported over oxygen-functionalized CNTs.255,256Operando electrocatalytic studies indicate that iron particles transform into FeOOH nanoparticles during the reaction,257 which is important because they may allow realizing a multi-electron transfer of electrons/protons to chemisorbed N2, with a reaction mechanism similar to that present in nitrogenase enzyme.203 N2 activation, however, occurs at carbon sites at the interface with the iron nanoparticles, possibly C atoms with specific charge density nearlying defects or doping heteroatoms. When a potential of −2.0 eV is applied, a carbon shell wraps the iron nanoparticles, as shown by HRTEM images, leading to an increase of 4–5 times the reaction rate in ammonia formation.
Therefore, although this area is just at the beginning and more in depth studies are required, there are some initial indications that the carbon interface around the nanoparticles could play a direct role in catalysis and electrocatalysis, even in highly challenging reactions such as ammonia direct synthesis.
4. Identification and quantification of active sites
A variety of characterization methods are employed to study various active sites (e.g. CO, defects, N species) as well as reaction mechanisms. Among them, some advanced in situ XPS and DRIFT technologies are used to reveal the real role of CO in gas phase reactions. The role of defects is preliminarily investigated by SIMS. Identification and quantification of active sites, however, is still rife with controversy because of the surface complexity of carbon materials. To date, although DFT is the most common way to simulate the intermediates and analyze reaction pathways at a molecular level, but there are various limitations including the use of real reaction conditions (temperature, solvent, etc.) and simulation of complex structures with different types of defects, etc. It is thus highly desired to develop new technologies to identify and quantify the active sites to understand their role in reaction mechanisms. In this section, we will highlight some promising methods.
4.1 Model catalysts
In Section 2.2.2.2, the potential application of aromatic organic molecules as model catalysts in determining active sites was briefly discussed. Although the feasibility of this strategy has been demonstrated in some catalytic reactions involving defects or CO active sites, more in-depth investigation on other heteroatom species should be emphasized, such as pyridinic N species. It should be noted that aromatic organic molecules with a shorter π-conjugated system (upper part of Fig. 29) might be only appropriate for liquid phase reactions, because of their limited solubility in solvents. With regard to high temperature-driven gas phase reaction and electrically-driven electrochemical reaction, better thermostability and conductivity for aromatic organic molecules are essential.
 | ||
| Fig. 29 Polycyclic aromatic hydrocarbon (PAH) organic molecules with single (a) armchair, (b) CO and (c) pyridinic N configurations as model catalysts. | ||
By chemical synthesis, a series of polycyclic aromatic hydrocarbon (PAH) organic molecules with single armchair, CO and pyridinic N configurations and enhanced π-conjugated systems could be prepared (lower part of Fig. 29). We believe that these model catalysts can be used directly in most reactions to experimentally identify real active sites of carbon catalysts or doped carbon catalysts. It can also be expected that designing other PAHs with different B, P or S species is an effective procedure to unravel the specific reactivity of the active sites formed in nanocarbons by doping.
4.2 Chemical titration measurements
Although active sites can be identified using model catalysts, the quantification of active sites on carbon catalysts is still challenging because of the detection limit of current common measurements, such as the locality of XPS for catalyst surfaces. Herein a chemical titration method to determine the surface concentration of three kinds of typical oxygen functional groups (CO, C–OH and COOH) on the surfaces of carbon nanotubes was proposed by Qi et al.258 During the titration process, some chemicals (Fig. 30, PH, BA and BrPE) with the ability to selectively react with different oxygen groups are introduced. Through selective deactivation of these specific oxygen functional groups and the assessment of the catalytic activity of titrated CNTs for EB ODH reactions, the intrinsic active sites (CO) can be identified and quantified simultaneously. Importantly, the chemical titration measurement method has the potential to be in situ operated in some catalytic reactions.
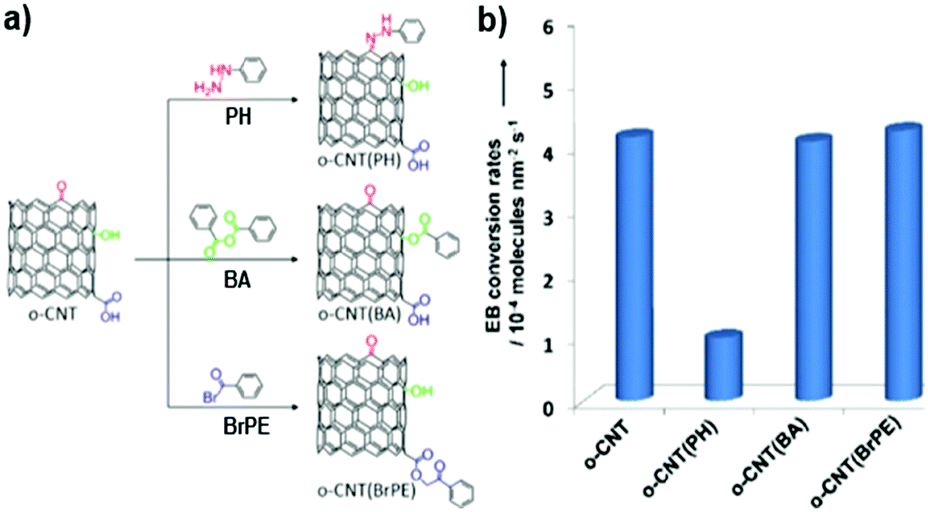 | ||
| Fig. 30 (a) Schematic drawings of the chemical titration process for oxygen functionalities on oxidized CNTs (o-CNT). (b) EB ODH conversion rates of o-CNT and titrated o-CNT. Adapted from ref. 258 with permission from the Wiley, copyright 2013. | ||
4.3 Isotopic labeling measurements
A better understanding of the reaction mechanism requires determining the reaction kinetics of the catalytic process. The common research scope contains the reaction order, reaction rate and activation energy. The use of the kinetic isotope effect (KIE) is an established experimental technique to study chemical reactions involving H, C, O, N and so on.For instance, the substitution of hydrogen with deuterium has been implemented extensively due to the large differences in reaction rates originating from the reduced mass differences between the isotopes. The observed reaction kinetics will be changed in the catalytic process. For instance, the substitution of hydrogen with deuterium has been implemented extensively due to the large difference in reaction rates when C–H bond dissociation is the RDS. Taking EB ODH as an example, when EB (C8H10) is displaced by deuterated EB (C8D10), the corresponding reaction rates decrease from 3.3 to 0.9 (theoretical KIE = 3.3/0.9 = 3.6), because the reaction is kinetically controlled by the C–H activation process – namely, the breaking of the C–H bond is the RDS.259 This strategy can be employed in liquid phase, electrochemical (ORR, OER, CRR) and photocatalytic reactions (CO2/C18O2 reduction, CH4/CD4 activation) to determine the possible RDS.
In short, more and more measurements will be developed and promoted as research progresses, such as in situ/operando NEXAFS/ESR spectroscopy. Here, we just highlight three kinds of facile methods to gain insight into carbon catalytic essence.
5. Perspectives
UNDDs offer, in our opinion, distinct characteristics from other types of nanocarbon materials and the scope of this review was to highlight how they offer not only the possibility of developing improved thermal, electro- and photo-catalysts, but also of showing how they open new conceptual perspectives for the design of advanced nanocarbon catalysts.The key aspects discussed in this review regarded the presence of hybrid sp2/sp3 configurations, defect and nano-engineering in the various aspects related to the role of defects and vacancies in the catalytic behaviour, the creation of active sites by modification in the charge density at carbon atoms or C–C bonds, the generation of strained C–C bonds by curvature and other mechanisms, and the formation of semiconducting areas and defect sites at the interface with supported nanoparticles.
We believe that UNDDs represent unique catalytic materials and a unique class of nanocarbons, where these aspects and their synergistic interaction could be demonstrated to have a relevant role in understanding their behaviour and the development of catalysts with improved performances. This is related to their peculiar structural, surface and electronic characteristics. However, they are features relevant in catalysis by all types of nanocarbons. The better understanding of these aspects that we consider often underestimated will lead to new or improved applications. Even if the roles of the single aspects have been reported in the literature, we feel that a more comprehensive picture is missing, so the necessary further step is to exploit the potential disruptive character of these materials.
For UNDDs, we also have to make the following observations:
(1) The abundant surface groups and different basic and acid sites (oxygen species) can serve as a promising platform for rational design of efficient and stable catalytic systems relevant to these different groups.
(2) A unique surface energy, electronic structure and high curvature on the molecular level is present. There are relevant catalytic implications, as discussed in this review.
(3) The exploitation of UNDD-based catalysts requires that they be shaped into industrially usable pellets. It is necessary to develop hierarchically structured materials, which can be used as catalysts or 3D-type electrodes.
(4) The highly defective surface curve shell and surface area of UNDDs are favourable for using them also as supports. The deposition of nanoparticles creates also new reactive carbon areas at the interface or at carbon shells on these nanoparticles. This is an area still largely unexplored. Nanoparticles themselves may be thus not reactive, but could create reactive sites in nanocarbons, or a valuable synergy could be achieved between the nanoparticles and the carbon itself, thus acting as a co-catalyst, not just as a support.
(5) It is possible to prepare a class of pristine or heteroatom modified-carbon quantum dots with tuneable properties via a facile chemistry dispersion approach, but their use to develop advanced catalysts is still quite limited. Hybrid sp2/sp3 nanocarbon materials offer new possibilities in this direction, although in UNDDs, for example, achieving a tuneable band gap and controlling the electron characteristics is still a challenge. The development of new practical applications, such as photocatalysts, semiconductor devices, diodes based on nanocarbons and novel photoelectrodes to produce solar fuels,260 requires understanding the role of defect engineering and the presence of hybrid sp2/sp3 nanostructures in the rather unique characteristic of creation of mixed semiconductor domains with n/p character. In general, the role of nano-scale design in the photo-activated properties of these materials is a crucial, but still requires large effort and better guidelines on the methodologies and assessment of the results.
(6) Heteroatom co-doping offers interesting possibilities for developing UNDD materials with Lewis acid (e.g., B) and Lewis base sites (e.g., N, P, F), which will provide important opportunities for reactions such as borylation, hydrosilylation, hydrogenation, cycloaddition of CO2, and C–H activation. These materials also offer new opportunities to design catalysts with frustrated Lewis pair character. In general, there are unique possibilities for creating multifunctional catalysts able to realize in one-pot multistage reactions.
In conclusion, UNDD materials have been demonstrated to exhibit richer surface chemistry properties and peculiar electronic structures in comparison to other carbon materials. Some recent achievements have shown their outstanding and stable performance in many catalytic reactions, making them alternative options to metal or metal-based catalysts.
6. Conclusions
The interest in nanocarbon materials as novel catalysts has been largely increasing recently (as an example, the number of entries in SciFinder for the term “metal-free carbon catalysis” was 59 in the 2000–2005 period and over thousand in the 2012–2017 period). Various reviews have been published on nanocarbons, as commented on in the Introduction. However, most of them were focused on describing the synthesis and application of families of materials such as graphene and CNTs, and their modification by doping, with the discussion of the catalytic behaviour related mainly to the functional groups deriving from the introduction of heteroatoms.UNDDs have been studied in less detail with respect to other types of nanocarbons, such as graphene, CNTs and derived materials. They were also never specifically reviewed. We thus hope that this review has opened new possibilities for a better understanding of these catalysts. At the same time, these results are creating new clues for a better design of the nanocarbon catalyst family.
It should be remarked that systematic studies on the relationship between the nature of these active sites in UNDDs and their catalytic reactivity are still limited. Intensified effort is necessary in advanced in situ/operando techniques to understand the catalytic mechanism, but integrated with direct advanced catalytic methodologies, such as combination of surface functional group titration, isotopic labelling methods and high-energy X-ray imaging technology. Attention should be paid also to the effect induced in surface charge or electron density localization on reactive C atoms or C–C bonds. A proper identification and quantification of the active sites is necessary. There are novel methodologies available, as summarized in Section 4, although with still limited utilization.
Compared with conventional carbon materials (e.g., CNTs, graphene, AC), the superior catalytic activities of UNDDs derive from their unique microstructure, electronic structure and surface properties, such as tuneable sp2/sp3 ratios upon temperature changes, local curvature effects, stabilization of defects and vacancy sites, low π and σ electronic binding energies, enhanced DOS, low work functions, controllable surface modifications and bulk doping. However, the respective role of sp2 and sp3-bonded carbon and the in-depth mechanism of heteroatoms in the catalytic process need to be further explored. Specific methodologies to single-out the specific contributions also have to be developed. The interaction between active domains and substrates on the surfaces of catalysts, the solvent effect and the kinetics in liquid-phase reactions should be investigated for a better understanding. To date, preparing model carbon catalysts with applicable π conjugated structures (e.g., seven benzene units or above) and designated single species (e.g., only CO, pyridinic N, zigzag or armchair configurations) has been considered as the best and direct way to comprehend the nature of metal-free catalysis.
This review thus forecasts a bright future in catalysis by UNDD family materials and the use of unconventional concepts of active sites such as C atoms and C–C bonds with specific charge density due to the presence of nearlying heteroatoms, defects or vacancies, and curvature in the graphene-like plane. It is an emerging and conceptually new area of catalytic materials. However, as shown above, there is still a lot of work to be done before this fascinating class of nanocarbons can be brought to light for real practical applications.
Acronyms
| AC | Activated carbon |
| AOP | Advanced oxidation process |
| BCDI | Bragg coherent X-ray diffraction imaging |
| BDD | Boron-doped diamond |
| BNND | Boron and nitrogen co-doped diamond |
| BND | Bucky nanodiamond |
| 900 BND | Annealing nanodiamond at 900 °C |
| 1100 BND | Annealing nanodiamond at 1100 °C |
| 1200 BND | Annealing nanodiamond at 1200 °C |
| 1300 BND | Annealing nanodiamond at 1300 °C |
| CMD | Catalytic methane decomposition |
| CNT | Carbon nanotube |
| CRR | CO2 reduction reaction |
| CV | Cyclic voltammetry |
| CVD | Chemical vapour deposition |
| DFT | Density functional theory |
| DH | Direct dehydrogenation |
| DRIFT | Diffuse-reflectance infrared Fourier transform |
| EB | Ethylbenzene |
| EELS | Electron energy loss spectroscopy |
| ESR | Electron spin resonance |
| ET | Electron transfer |
| NEXAFS | Near extended X-ray absorption fine structure |
| FTIR | Fourier transform infrared spectrum |
| GR | Reduced graphite oxide |
| GO | Graphite oxide |
| HOPG | Highly ordered pyrolytic graphite |
| HRTEM | High-resolution transmission electron microscopy |
| MOF | Metal–organic framework |
| MWCNT | Multi-walled carbon nanotube |
| ND | Nanodiamond |
| NDD | Nitrogen-doped diamond |
| N-BND | Nitrogen-modified BND |
| NMR | Nuclear magnetic resonance |
| N-OLC | Nitrogen-doped OLC |
| ODH | Oxidative dehydrogenation |
| OLC | Onion-like carbon |
| ORR | Oxygen reduction reaction |
| PAH | Polycyclic aromatic hydrocarbon |
| PCET | Proton coupled electron transfer |
| PDDA | Poly(diallyldimethylammoniumchloride) |
| PEM | Proton-exchange membrane |
| PMS | Peroxymonosulfate |
| PDS | Peroxydisulfate |
| RDE | Rotating disk electrode |
| RDS | Rate-determining step |
| SIMS | Secondary-ion mass spectrometry |
| SWCNT | Single-walled carbon nanotube |
| TBHP | tert-Butyl hydroperoxide |
| TGA | Thermogravimetric analysis |
| TPD | Temperature programmed desorption |
| UNDDs | Ultradispersed nanodiamonds and their derivatives |
| UPS | Ultraviolet photoelectron spectroscopy |
| XRD | X-ray diffraction |
| XPS | X-ray photoelectron spectroscopy |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Jian Zhang, Xi Liu, Robert Schlögl and Linhui Yu for their great contributions to this study. The authors thank Kuang-Hsu Wu (University of New South Wales) for his comments on this review. The authors thank Dr Vadym N. Mochalin (Missouri University of Science & Technology) and Prof. Y. Gogotsi (Department of Materials Science and Engineering and A. J. Drexel Nanotechnology Institute, Drexel University) for providing the original schematic model of nanodiamond. YL, XS and DS acknowledge the “Strategic Priority Research Program” of the Chinese Academy of Sciences, Grant No. XDA09030103. GC and SP acknowledge the Italian MIUR through the PRIN Project 2015K7FZLH SMARTNESS “Solar driven chemistry: new materials for photo- and electro-catalysis”. Open Access funding provided by the Max Planck Society.References
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro, M. Antonietti and H. Garcıa, Chem. Soc. Rev., 2017, 46, 4501 RSC.
- S. Navalòn, J. R. Herance, M. Alvaroa and H. Garcıa, Mater. Horiz., 2018, 5, 363 RSC.
- X.-H. Li and M. Antonietti, Chem. Soc. Rev., 2013, 42, 6593 RSC.
- Y. Yan, J. Miao, Z. Yang, F.-X. Xiao, H. B. Yang, B. Liu and Y. Yang, Chem. Soc. Rev., 2015, 44, 3295 RSC.
- S. A. Miners, G. A. Rance and A. N. Khlobystov, Chem. Soc. Rev., 2016, 45, 4727 RSC.
- X.-K. Kong, C.-L. Chen and Q.-W. Chen, Chem. Soc. Rev., 2014, 43, 2841 RSC.
- Y. Xu, M. Kraft and R. Xu, Chem. Soc. Rev., 2016, 45, 3039 RSC.
- X. Fan, G. Zhang and F. Zhang, Chem. Soc. Rev., 2015, 44, 3023 RSC.
- W. Qi, P. Yan and D. S. Su, Acc. Chem. Res., 2018, 51, 640 CrossRef CAS PubMed.
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2010, 150, 151 CrossRef CAS.
- B. Qiu, M. Xing and J. Zhang, Chem. Soc. Rev., 2018, 47, 2165 RSC.
- M.-M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250 RSC.
- D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782 CrossRef CAS PubMed.
- J. Li, Z. Zhao, Y. Ma and Y. Qu, ChemCatChem, 2017, 9, 1554 CrossRef CAS.
- C. Genovese, C. Ampelli, S. Perathoner and G. Centi, J. Energy Chem., 2013, 22, 202 CrossRef CAS.
- A. Primo, V. Parvulescu and H. Garcia, J. Phys. Chem. Lett., 2017, 8, 264 CrossRef CAS PubMed.
- E. Perez-Mayoral, V. Calvino-Casilda and E. Soriano, Catal. Sci. Technol., 2016, 6, 1265 RSC.
- G. Centi, S. Perathoner and D. S. Su, Catal. Surv. Asia, 2014, 18, 149 CrossRef CAS.
- M. Li, F. Xu, H. Li and Y. Wang, Catal. Sci. Technol., 2016, 6, 3670 RSC.
- J. Zhang and L. Dai, ACS Catal., 2015, 5, 7244 CrossRef CAS.
- E. Lam and J. H. T. Luong, ACS Catal., 2014, 4, 3393 CrossRef CAS.
- W. Qi and D. S. Su, ACS Catal., 2014, 4, 3212 CrossRef CAS.
- A. Sarapuu, E. Kibena-Poldsepp, M. Borghei and K. Tammeveski, J. Mater. Chem. A, 2018, 6, 776 RSC.
- C. Tang and Q. Zhang, Adv. Mater., 2017, 29, 1604103 CrossRef PubMed.
- G. Centi and S. Perathoner, ChemSusChem, 2011, 4, 913 CrossRef CAS PubMed.
- D. S. Su and G. Centi, J. Energy Chem., 2013, 22, 151 CrossRef CAS.
- C. Ampelli, S. Perathoner and G. Centi, Chin. J. Catal., 2014, 35, 783 CrossRef CAS.
- D. S. Su, S. Perathoner and G. Centi, Catal. Today, 2012, 186, 1 CrossRef CAS.
- D.-W. Wang and D. S. Su, Energy Environ. Sci., 2014, 7, 576 RSC.
- B. Frank, in Nanocarbon-Inorganic Hybrids, ed. D. Eder and R. Schlögl, De Gruyter, 2014, p. 393 Search PubMed.
- R. Schlögl, Adv. Catal., 2013, 56, 103 Search PubMed.
- D. S. Su and R. Schlögl, ChemSusChem, 2010, 3, 136 CrossRef CAS PubMed.
- D. S. Su, J. Zhang, B. Frank, A. Thomas, X. Wang, J. Paraknowitsch and R. Schlögl, ChemSusChem, 2010, 3, 169 CrossRef CAS PubMed.
- P. Lanzafame, S. Abate, C. Ampelli, C. Genovese, R. Passalacqua, G. Centi and S. Perathoner, ChemSusChem, 2017, 10, 4409 CrossRef CAS PubMed.
- S. Perathoner, S. Gross, E. J. M. Hensen, H. Wessel, H. Chraye and G. Centi, ChemCatChem, 2017, 9, 904 CrossRef CAS.
- S. Abate, K. Barbera, G. Centi, P. Lanzafame and S. Perathoner, Catal. Sci. Technol., 2016, 6, 2485 RSC.
- T. Zhang, Y. Zhu and J. Y. Lee, J. Mater. Chem. A, 2018, 6, 8147 RSC.
- L. Liu, Y.-P. Zhu, M. Su and Z.-Y. Yuan, ChemCatChem, 2015, 7, 2765 CrossRef CAS.
- D. Yu, E. Nagelli, F. Du and L. Dai, J. Phys. Chem. Lett., 2010, 1, 2165 CrossRef CAS.
- Q. Wu, L. Yang, X. Wang and Z. Hu, Acc. Chem. Res., 2017, 50, 435 CrossRef CAS PubMed.
- V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Chem. Rev., 2015, 115, 4744 CrossRef CAS PubMed.
- H. Tang, C. M. Hessel, J. Wang, N. Yang, R. Yu, H. Zhao and D. Wang, Chem. Soc. Rev., 2014, 43, 4281 RSC.
- K. Dirian, M. A. Herranz, G. Katsukis, J. Malig, L. Rodriguez-Perez, C. Romero-Nieto, V. Strauss, N. Martin and D. M. Guldi, Chem. Sci., 2013, 4, 4335 RSC.
- L.-F. Chen, Y. Feng, H.-W. Liang, Z.-Y. Wu and S.-H. Yu, Adv. Energy Mater., 2017, 7, 1700826 CrossRef.
- H. Jiang, P. S. Lee and C. Li, Energy Environ. Sci., 2013, 6, 41 RSC.
- T. F. Liu, S. Ali, Z. Lian, B. Li and D. S. Su, J. Mater. Chem. A, 2017, 5, 21596 RSC.
- C. Hu and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736 CrossRef CAS PubMed.
- Y. Zhang, J. Zhang and D. S. Su, ChemSusChem, 2014, 7, 1240 CrossRef CAS PubMed.
- K. N. Wood, R. O'Hayre and S. Pylypenko, Energy Environ. Sci., 2014, 7, 1212 RSC.
- J. Zhu, A. Holmen and D. Chen, ChemCatChem, 2013, 5, 378 CrossRef CAS.
- G. Tuci, A. Rossin, L. Luconi, C. Pham-Huu, S. Cicchi, H. Ba and G. Giambastiani, Catal. Sci. Technol., 2017, 7, 5833 RSC.
- G. Tuci, C. Zafferoni, A. Rossin, L. Luconi, A. Milella, M. Ceppatelli, M. Innocenti, Y. Liu, C. Pham-Huu and G. Giambastiani, Catal. Sci. Technol., 2016, 6, 6226 RSC.
- G. Tuci, C. Zafferoni, P. D'Ambrosio, S. Caporali, M. Ceppatelli, A. Rossin, T. Tsoufis, M. Innocenti and G. Giambastiani, ACS Catal., 2013, 3, 2108 CrossRef CAS.
- R. Lv, E. Cruz-Silva and M. Terrones, ACS Nano, 2014, 8, 4061 CrossRef CAS PubMed.
- H. Wang and H. Dai, Chem. Soc. Rev., 2013, 42, 3088 RSC.
- G. Centi and S. Perathoner, in Nanocarbon-Inorganic Hybrids, ed. D. Eder and R. Schlögl, De Gruyter, 2014, p. 429 Search PubMed.
- Y. Cheng, Y. Fan, Y. Pei and M. Qiao, Catal. Sci. Technol., 2015, 5, 3903 RSC.
- D. Zhang, W. Chen, Z. Li, Y. Chen, L. Zheng, Y. Gong, Q. Li, R. Shen, Y. Han, W.-C. Cheong, L. Gu and Y. Li, Chem. Commun., 2018, 54, 4274 RSC.
- Y. Chen, T. Kasama, Z. Huang, P. Hu, J. Chen, X. Liu and X. Tang, Chem. – Eur. J., 2015, 21, 17397 CrossRef CAS PubMed.
- G. Vilé, D. Albani, M. Nachtegaal, Z. Chen, D. Dontsova, M. Antonietti, N. López and J. Pérez-Ramírez, Angew. Chem., Int. Ed., 2015, 54, 11265 CrossRef PubMed.
- H. Fei, J. Dong, M. J. Arellano-Jiménez, G. Ye, N. D. Kim, E. L. Samuel, Z. Peng, Z. Zhu, F. Qin and J. Bao, Nat. Commun., 2015, 6, 8668 CrossRef CAS PubMed.
- H.-W. Liang, S. Brüller, R. Dong, J. Zhang, X. Feng and K. Müllen, Nat. Commun., 2015, 6, 7992 CrossRef CAS PubMed.
- G. Zhao, H. Liu and J. Ye, Nano Today, 2018, 19, 108 CrossRef CAS.
- D. Liu, C. Wu, S. Chen, S. Ding, Y. Xie, C. Wang, T. Wang, Y. A. Haleem, Z. ur Rehman, Y. Sang, Q. Liu, X. Zheng, Y. Wang, B. Ge, H. Xu and L. Song, Nano Res., 2018, 11, 2217 CrossRef CAS.
- L.-L. Liu, C.-P. Chen, L.-S. Zhao, Y. Wang and X.-C. Wang, Carbon, 2017, 115, 773 CrossRef CAS.
- S. Navalon, A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Rev., 2014, 114, 6179 CrossRef CAS PubMed.
- D. Guo, R. Shibuya, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2016, 351, 361 CrossRef CAS PubMed.
- S. Perathoner, C. Ampelli, S. Chen, R. Passalacqua, D. Su and G. Centi, J. Energy Chem., 2017, 26, 207 CrossRef.
- C. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275 CrossRef CAS PubMed.
- P. Tang, G. Hu, M. Li and D. Ma, ACS Catal., 2016, 6, 6948 CrossRef CAS.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594 CrossRef CAS PubMed.
- C. K. Chua and M. Pumera, Chem. – Eur. J., 2015, 21, 12550 CrossRef CAS PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823 CrossRef CAS PubMed.
- H. Hu, J. H. Xin, H. Hu, X. Wang and Y. Kong, Appl. Catal., A, 2015, 492, 1 CrossRef CAS.
- N. Matsumoto, L. Joly-Pottuz, H. Kinoshita and N. Ohmae, Diamond Relat. Mater., 2007, 16, 1227 CrossRef CAS.
- J. Zang, Y. Wang, L. Bian, J. Zhang, F. Meng, Y. Zhao, X. Qu and S. Ren, Int. J. Hydrogen Energy, 2012, 37, 6349 CrossRef CAS.
- M. Chen, X.-Q. Zhang, H. B. Man, R. Lam, E. K. Chow and D. Ho, J. Phys. Chem. Lett., 2010, 1, 3167 CrossRef CAS.
- Y.-R. Chang, H.-Y. Lee, K. Chen, C.-C. Chang, D.-S. Tsai, C.-C. Fu, T.-S. Lim, Y.-K. Tzeng, C.-Y. Fang, C.-C. Han, H.-C. Chang and W. Fann, Nat. Nanotechnol., 2008, 3, 284 CrossRef CAS PubMed.
- V. N. Mochalin, O. Shenderova, D. Ho and Y. Gogotsi, Nat. Nanotechnol., 2012, 7, 11 CrossRef CAS PubMed.
- Z. R. Dai, J. P. Bradley, D. J. Joswiak, D. E. Brownlee, H. G. M. Hill and M. J. Genge, Nature, 2002, 418, 157 CrossRef CAS PubMed.
- V. V. Danilenko, Phys. Solid State, 2004, 46, 595 CrossRef CAS.
- N. R. Johnson, N. R. Greiner, D. S. Phillips, J. D. Johnson and F. Volk, Nature, 1988, 333, 440 CrossRef.
- V. L. Kuznetsov, A. L. Chuvilin, E. M. Moroz, V. N. Kolomiichuk, S. K. Shaikhutdinov, Y. V. Butenko and I. Y. Mal'kov, Carbon, 1994, 32, 873 CrossRef.
- S. Osswald, G. Yushin, V. Mochalin, S. O. Kucheyev and Y. Gogotsi, J. Am. Chem. Soc., 2006, 128, 11635 CrossRef CAS PubMed.
- O. O. Mykhaylyk and Y. M. Solonin, J. Appl. Phys., 2005, 97, 074302 CrossRef.
- D. Pech, M. Brunet, H. Durou, P. Huang, V. Mochalin, Y. Gogotsi, P.-L. Taberna and P. Simon, Nat. Nanotechnol., 2010, 5, 651 CrossRef CAS PubMed.
- M. Zeiger, N. Jackel, V. N. Mochalin and V. Presse, J. Mater. Chem. A, 2016, 4, 3172 RSC.
- M. Li, W. Liu, H. Zhang, Z. Liang, P. Duan, X. Yan, P. Guan, B. Xu and J. Guo, Phys. Chem. Chem. Phys., 2018, 20, 2022 RSC.
- T. Petit, J.-C. Arnault, H. A. Girard, M. Sennour and P. Bergonzo, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 233407 CrossRef.
- V. L. Kuznetsov, I. L. Zilberberg, Yu. V. Butenko and A. L. Chuvilin, J. Appl. Phys., 1999, 86, 863 CrossRef CAS.
- F. Ding and B. I. Yakobson, J. Phys. Chem. Lett., 2014, 5, 2922 CrossRef CAS PubMed.
- S. O. Hruszkewycz, W. Cha, P. Andrich, C. P. Anderson, A. Ulvestad, R. Harder, P. H. Fuoss, D. D. Awschalom and F. J. Heremans, APL Mater., 2017, 5, 026105 CrossRef.
- V. L. Kuznetsov, A. L. Chuvilin, Y. V. Butenko, I. Y. Mal'kov and V. M. Titov, Chem. Phys. Lett., 1994, 222, 343 CrossRef CAS.
- L. Hawelek, A. Brodka, S. Tomita, J. C. Dore, V. Honkimäki and A. Burian, Diamond Relat. Mater., 2011, 20, 1333 CrossRef CAS.
- M. Todt, R. D. Bitsche, M. A. Hartmann, F. D. Fischer and F. G. Rammerstorfer, Int. J. Solids Struct., 2014, 51, 706 CrossRef CAS.
- J.-F. Cui, X.-W. Fang and K. Schmidt-Rohr, J. Phys. Chem. C, 2014, 118, 9621 CrossRef CAS.
- Y. Lin, Z. Feng, L. Yu, Q. Gu, S. Wu and D. S. Su, Chem. Commun., 2017, 53, 4834 RSC.
- Y. V. Butenko, V. L. Kuznetsov, E. A. Paukshtis, A. I. Stadnichenko, I. N. Mazov, S. I. Moseenkov, A. I. Boronin and S. V. Kosheev, Fullerenes, Nanotubes, Carbon Nanostruct., 2006, 14, 557 CrossRef CAS.
- G. P. Bogatyreva, M. A. Marinich, E. V. Ishchenko, V. L. Gvyazdovskaya, G. A. Bazalii and N. A. Oleinik, Phys. Solid State, 2004, 46, 738 CrossRef CAS.
- B. Moosa, K. Fhayli, S. Li, K. Julfakyan, A. Ezzeddine and N. M. Khashab, J. Nanosci. Nanotechnol., 2014, 14, 332 CrossRef CAS PubMed.
- V. L. Kuznetsov, M. N. Aleksandrov, I. V. Zagoruiko, A. L. Chuvilin, E. M. Moroz, V. N. Kolomiichuk, V. A. Likholobov, P. M. Brylyakov and G. V. Sakovitchet, Carbon, 1991, 29, 665 CrossRef CAS.
- A. S. Barnard, S. P. Russo and I. K. Snook, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 073406 CrossRef.
- G. C. C. Costa, J. K. McDonough, Y. Gogotsi and A. Navrotsky, Carbon, 2014, 69, 490 CrossRef CAS.
- S. Tomita, T. Sakurai, H. Ohta, M. Fujii and S. Hayashi, J. Chem. Phys., 2001, 114, 7477 CrossRef CAS.
- L. G. Bulusheva, A. V. Okotrub, V. L. Kuznetsov, A. L. Chuvilin, Y. V. Butenko and M. I. Heggie, MRS Proc., 2002, 703, V9.22 Search PubMed.
- D. Holec, M. A. Hartmann, F. D. Fischer, F. G. Rammerstorfer, P. H. Mayrhofer and O. Paris, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 235403 CrossRef.
- E. M. Zagrebina, A. V. Generalov, A. Yu. Klyushin, K. A. Simonov, N. A. Vinogradov, M. Dubois, L. Frezet, N. Martensson, A. B. Preobrajenski and A. S. Vinogradov, J. Phys. Chem. C, 2015, 119, 835 CrossRef CAS.
- D. S. Sutar, G. Singh and V. D. Botcha, Appl. Phys. Lett., 2012, 101, 103103 CrossRef.
- S. H. Lim, H. I. Elim, X. Y. Gao, A. T. S. Wee, W. Ji, J. Y. Lee and J. Lin, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045402 CrossRef.
- M. Shiraishi and M. Ata, Carbon, 2001, 39, 1913 CrossRef CAS.
- H. Ago, T. Kugler, F. Cacialli, W. R. Salaneck, M. S. P. Shaffer, A. H. Windle and R. H. Friend, J. Phys. Chem. B, 1999, 103, 8116 CrossRef CAS.
- A. Siokou, F. Ravani, S. Karakalos, O. Frank, M. Kalbac and C. Galiotis, Appl. Surf. Sci., 2011, 257, 9785 CrossRef CAS.
- M. Shiraishi and M. Ata, Mater. Res. Soc. Symp. Proc., 2001, 72, 633 Search PubMed.
- X. Li, X. Pan, L. Yu, P. Ren, X. Wu, L. Sun, F. Jiao and X. Bao, Nat. Commun., 2014, 5, 3688 CrossRef CAS PubMed.
- J. Zhang, X. Liu, R. Blume, A. Zhang, R. Schlogl and D. S. Su, Science, 2008, 322, 73 CrossRef CAS PubMed.
- B. Dai, K. Chen, Y. Wang, L. Kang and M. Zhu, ACS Catal., 2015, 5, 2541 CrossRef CAS.
- A. Schüle, U. Nieken, O. Shekhah, W. Ranke, R. Schlögl and G. Kolios, Phys. Chem. Chem. Phys., 2007, 9, 3619 RSC.
- J. H. B. S. Jesper, R.-M. Javier, S.-J. Eduardo and M. W. Bert, Chem. Rev., 2014, 114, 10613 CrossRef PubMed.
- N. Keller, N. I. Maksimova, V. V. Roddatis, M. Schur, G. Mestl, Y. V. Butenko, V. L. Kuznetsov and R. Schlögl, Angew. Chem., Int. Ed., 2002, 41, 1885 CrossRef CAS PubMed.
- D. S. Su, N. Maksimova, J. J. Delgado, N. Keller, G. Mestl, M. J. Ledoux and R. Schlögl, Catal. Today, 2005, 102–103, 110 CrossRef CAS.
- J. Zhang, D. S. Su, A. Zhang, D. Wang, R. Schlögl and C. Hebert, Angew. Chem., Int. Ed., 2007, 46, 7319 CrossRef CAS PubMed.
- D. S. Su, N. I. Maksimova, G. Mestl, V. L. Kuznetsov, V. Keller, R. Schlögl and N. Keller, Carbon, 2007, 45, 2145 CrossRef CAS.
- J. Zhang, D. S. Su, R. Blume, R. Schlögl, R. Wang, X. Yang and A. Gajovic, Angew. Chem., Int. Ed., 2010, 49, 8640 CrossRef CAS PubMed.
- X. Liu, B. Frank, W. Zhang, T. P. Cotter, R. Schlögl and D. S. Su, Angew. Chem., Int. Ed., 2011, 50, 3318 CrossRef CAS PubMed.
- X. Sun, R. Wang, B. Zhang, R. Huang, X. Huang, D. S. Su, T. Chen, C. Miao and W. Yang, ChemCatChem, 2014, 6, 2270 CrossRef CAS.
- R. Wang, X. Y. Sun, B. S. Zhang, X. Y. Sun and D. S. Su, Chem. – Eur. J., 2014, 20, 6324 CrossRef CAS PubMed.
- X. Sun, Y. Ding, B. Zhang, R. Huang, D. Chen and D. S. Su, ACS Catal., 2015, 5, 2436 CrossRef CAS.
- X. Sun, Y. Ding, B. Zhang, R. Huang and D. S. Su, Chem. Commun., 2015, 51, 9145 RSC.
- B. Zhong, J. Zhang, B. Li, B. Zhang, C. Dai, X. Sun, R. Wang and D. S. Su, Phys. Chem. Chem. Phys., 2014, 16, 4488 RSC.
- U. P. M. Ashik, W. M. A. Wan Daud and H. F. Abbas, Renewable Sustainable Energy Rev., 2015, 44, 221 CrossRef CAS.
- R. Guil-Lopez, J. A. Botas, J. L. G. Fierro and D. P. Serrano, Appl. Catal., A, 2011, 396, 40 CrossRef CAS.
- N. Q. Zhao, C. N. He, J. Ding, T. C. Zou, Z. J. Qiao, C. S. Shi, X. W. Du, J. J. Li and Y. D. Li, J. Alloys Compd., 2007, 428, 79 CrossRef CAS.
- R. Aiello, J. E. Fiscus, H.-C. Zur Loye and M. D. Amiridis, Appl. Catal., A, 2000, 192, 227 CrossRef CAS.
- J. Diao, Z. Feng, R. Huang, H. Liu, S. B. A. Hamid and D. S. Su, ChemSusChem, 2016, 9, 662 CrossRef CAS PubMed.
- G. C. Grunewald and R. S. Drago, J. Mol. Catal., 1990, 58, 227 CrossRef CAS.
- B. Frank, M. Morassutto, R. Schomäcker, R. Schlögl and D. S. Su, ChemCatChem, 2010, 2, 644 CrossRef CAS.
- B. Frank, J. Zhang, R. Blume, R. Schlögl and D. S. Su, Angew. Chem., Int. Ed., 2009, 48, 6913 CrossRef CAS PubMed.
- C. L. Chen, J. Zhang, B. S. Zhang, C. L. Yu, F. Peng and D. S. Su, Chem. Commun., 2013, 49, 8151 RSC.
- V. Schwartz, H. Xie, H. M. Meyer, S. H. Overbury and C. D. Liang, Carbon, 2011, 49, 659 CrossRef CAS.
- F. Cavani, N. Ballarini and A. Cericola, Catal. Today, 2007, 127, 113 CrossRef CAS.
- H. Y. Liu, J. Y. Diao, Q. Wang, S. Y. Gu, T. Chen, C. X. Miao, W. M. Yang and D. S. Su, Chem. Commun., 2014, 50, 7810 RSC.
- H. Ba, Y. Liu, X. Mu, W. H. Doh, J. M. Nhuta, P. Granger and C. Pham-Huu, Appl. Catal., A, 2015, 499, 217 CrossRef CAS.
- J. Diao, H. Liu, Z. Feng, Y. Zhang, T. Chen, C. Miao, W. Yang and D. S. Su, Catal. Sci. Technol., 2015, 5, 4950 RSC.
- H. Ba, S. Podila, Y. Liu, X. Mu, J. M. Nhuta, V. Papaefthimiou, S. Zafeiratos, P. Granger and C. Pham-Huu, Catal. Today, 2015, 249, 167 CrossRef CAS.
- Z. Zhao, Y. Dai, G. Ge, Q. Mao, Z. Rong and G. Wang, ChemCatChem, 2015, 7, 1070 CrossRef CAS.
- T. T. Thanh, H. Ba, T. P. Lai, J. M. Nhut, O. Ersen, D. Begin, I. Janowska, D. L. Nguyen, P. Granger and C. Pham-Huu, J. Mater. Chem. A, 2014, 2, 11349 RSC.
- H. Ba, L. Truong-Phuoc, Y. Liu, C. Duong-Viet, J.-M. Nhut, L. Nguyen-Dinh, P. Granger and C. Pham-Huu, Carbon, 2016, 96, 1060 CrossRef CAS.
- Z. K. Zhao and Y. T. Dai, J. Mater. Chem. A, 2014, 2, 13442 RSC.
- Z. Zhao, W. Li, Y. Dai, G. Ge, X. Guo and G. Wang, ACS Sustainable Chem. Eng., 2015, 3, 3355 CrossRef CAS.
- L. Roldán, A. M. Benito and E. García-Bordejé, J. Mater. Chem. A, 2015, 3, 24379 RSC.
- D. Su, G. Wen, S. Wu, F. Peng and R. Schlögl, Angew. Chem., Int. Ed., 2017, 56, 936 CrossRef CAS PubMed.
- F. Cavani, G. Centi, S. Perathoner and F. Trifirò, Sustainable Industrial Chemistry: Principles, Tools and Industrial Examples, Wiley-VCH, 2009 Search PubMed.
- G. Centi and R. A. van Santen, Catalysis for Renewables: From Feedstock to Energy Production, Wiley-VCH, 2007 Search PubMed.
- Y. Lin and D. Su, ACS Nano, 2014, 8, 7823 CrossRef CAS PubMed.
- S. C. Laha and R. Kumar, J. Catal., 2001, 204, 64 CrossRef CAS.
- X. Deng and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 17178 CrossRef CAS PubMed.
- Y. Lin, X. Pan, W. Qi, B. Zhang and D. S. Su, J. Mater. Chem. A, 2014, 2, 12475 RSC.
- J. Huang, C. Liu, D. Sun, Y. Hong, M. Du, T. Odoom-Wubah, W. Fang and Q. Li, Chem. Eng. J., 2014, 235, 215 CrossRef CAS.
- R. Ghosh, X. Shen, J. C. Villegas, Y. Ding, K. Malinger and S. L. Suib, J. Phys. Chem. B, 2006, 110, 7592 CrossRef CAS PubMed.
- Y. Gao, G. Hu, J. Zhong, Z. Shi, Y. Zhu, D. S. Su, J. Wang, X. Bao and D. Ma, Angew. Chem., Int. Ed., 2013, 52, 2109 CrossRef CAS PubMed.
- Y. Lin, B. Li, Z. Feng, Y. A. Kim, M. Endo and D. S. Su, ACS Catal., 2015, 5, 5921 CrossRef CAS.
- Y. Lin, K.-H. Tim Wu, L. Yu, S. Heumann and D. S. Su, ChemSusChem, 2017, 10, 3497 CrossRef CAS PubMed.
- A. S. Stasinakis, Global NEST J., 2008, 10, 376 Search PubMed.
- W.-D. Oh, Z. l. Dong and T.-T. Lim, Appl. Catal., B, 2016, 194, 169 CrossRef CAS.
- X. Duan, Z. Ao, L. Zhou, H. Sun, G. Wang and S. Wang, Appl. Catal., B, 2016, 188, 98 CrossRef CAS.
- X. Duan, C. Su, L. Zhou, H. Sun, A. Suvorova, T. Odedairo, Z. Zhu, Z. Shao and S. Wang, Appl. Catal., B, 2016, 194, 7 CrossRef CAS.
- H. Lee, H.-i Kim, S. Weon, W. Choi, Y. S. Hwang, J. Seo, C. Lee and J.-H. Kim, Environ. Sci. Technol., 2016, 50, 10134 CrossRef CAS PubMed.
- X. Duan, Z. Aob, H. Zhang, M. Saunders, H. Sund, Z. Shao and S. Wang, Appl. Catal., B, 2018, 222, 176 CrossRef CAS.
- X. Duan, Z. Ao, D. Li, H. Sun, L. Zhou, A. Suvorova, M. Saunders, G. Wang and S. Wang, Carbon, 2016, 103, 404 CrossRef CAS.
- A. Corma and P. Serna, Science, 2006, 313, 332 CrossRef CAS PubMed.
- R. V. Jagadeesh, A. E. Surkus, H. Junge, M. M. Pohl, J. Radnik, J. Rabeah, H. Huan, V. Schunemann, A. Bruckner and M. Beller, Science, 2013, 342, 1073 CrossRef CAS PubMed.
- Y. Lin, S. Wu, W. Shi, B. Zhang, J. Wang, Y. A. Kim, M. Endo and D. S. Su, Chem. Commun., 2015, 51, 13086 RSC.
- Q. Zhang, Y. Liu, S. Chen, X. Quan and H. Yu, J. Hazard. Mater., 2014, 265, 185 CrossRef CAS PubMed.
- W. S. Yeap, S. Chen and K. P. Loh, Chem. Mater., 2009, 25, 185 CAS.
- X. Sun, R. Wang and D. Su, Chin. J. Catal., 2013, 34, 508 CrossRef CAS.
- E. Y. Choi and C. K. Kim, Sci. Rep., 2017, 7, 4178 CrossRef CAS PubMed.
- K. Chatterjee, M. Ashokkumar, H. Gullapalli, Y. Gong, R. Vajtai, P. Thanikaivelan and P. M. Ajayan, Carbon, 2018, 130, 645 CrossRef CAS.
- Y. Liu, S. Chen, X. Quan, H. Yu, H. Zhao, Y. Zhang and G. Chen, J. Phys. Chem. C, 2013, 117, 14992 CrossRef CAS.
- Y. Zhu, Y. Lin, B. Zhang, J. Rong, B. Zong and D. S. Su, ChemCatChem, 2015, 7, 2840 CrossRef CAS.
- N. Kannari, T. Itakura and J. Ozaki, Carbon, 2015, 87, 415 CrossRef CAS.
- L. Dong, J. Zang, J. Su, Y. Jia, Y. Wang, J. Lu and X. Xu, Electrochim. Acta, 2015, 174, 1017 CrossRef CAS.
- D. M. Jang, H. S. Im, S. H. Back, K. Park, Y. R. Lim, C. S. Jung, J. Park and M. Lee, Phys. Chem. Chem. Phys., 2014, 16, 2411 RSC.
- R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., 2010, 122, 2619 CrossRef.
- D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760 RSC.
- Y. Zhang, A. Reed and D. Y. Kim, Curr. Appl. Phys., 2018, 18, 417 CrossRef.
- Y. Lin, Y. Zhu, B. Zhang, Y. A. Kim, M. Endo and D. S. Su, J. Mater. Chem. A, 2015, 3, 21805 RSC.
- N. Suo, H. Huang, A. Wu, G. Cao, X. Hou and G. Zhang, Appl. Surf. Sci., 2018, 439, 329 CrossRef CAS.
- X. Sun, J. Xu, Y. Ding, B. Zhang, Z. Feng and D. S. Su, ChemSusChem, 2015, 8, 2872 CrossRef CAS PubMed.
- X. Liu, Y. Wang, L. Dong, X. Chen, G. Xin, Y. Zhang and J. Zang, Electrochim. Acta, 2016, 194, 161 CrossRef CAS.
- J. Koh, S. H. Park, M. W. Chung, S. Y. Lee and S. I. Woo, RSC Adv., 2016, 6, 27528 RSC.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760 CrossRef CAS PubMed.
- X. Hu, Y. Wu, H. Li and Z. Zhang, J. Phys. Chem. C, 2010, 114, 9603 CrossRef CAS.
- S. Ni, Z. Li and J. Yang, Nanoscale, 2012, 4, 1184 RSC.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132 CrossRef CAS PubMed.
- Y. Zhao, L. Yang, S. Chen, X. Wang, Y. Ma, Q. Wu, Y. Jiang, W. Qian and Z. Hu, J. Am. Chem. Soc., 2013, 135, 1201 CrossRef CAS PubMed.
- A. Shen, Y. Zou, Q. Wang, R. A. W. Dryfe, X. Huang, S. Dou, L. Dai and S. Wang, Angew. Chem., 2014, 126, 10980 CrossRef.
- L. Tao, Q. Wang, S. Dou, Z. Ma, J. Huo, S. Wang and L. Dai, Chem. Commun., 2016, 52, 2764 RSC.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711 RSC.
- E. A. Quadrelli, G. Centi, J. L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194 CrossRef CAS PubMed.
- C. Ampelli, S. Perathoner and G. Centi, Philos. Trans. R. Soc., A, 2015, 373, 20140177 CrossRef PubMed.
- S. Perathoner and G. Centi, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.03.005.
- C. Costentin, M. Robert and J.-M. Savėant, Chem. Soc. Rev., 2013, 42, 2423 RSC.
- K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., 2014, 126, 890 CrossRef.
- Y. Liu, S. Chen, X. Quan and H. Yu, J. Am. Chem. Soc., 2015, 137, 11631 CrossRef CAS PubMed.
- Y. Liu, Y. Zhang, K. Cheng, X. Quan, X. Fan, Y. Su, S. Chen, H. Zhao, Y. Zhang, H. Yu and M. R. Hoffmann, Angew. Chem., Int. Ed., 2017, 56, 15607 CrossRef CAS PubMed.
- X. Duan, J. Xu, Z. Wei, J. Ma, S. Guo, S. Wang, H. Liu and S. Dou, Adv. Mater., 2017, 29, 1701784 CrossRef PubMed.
- S. Liu, H. Yang, X. Huang, L. Liu, W. Cai, J. Gao, X. Li, T. Zhang, Y. Huang and B. Liu, Adv. Funct. Mater., 2018, 28, 1800499 CrossRef.
- J. Xu, Y. Kan, R. Huang, B. Zhang, B. Wang, K. H. Wu, Y. Lin, X. Sun, Q. Li, G. Centi and D. S. Su, ChemSusChem, 2016, 9, 1085 CrossRef CAS PubMed.
- Y. Liu, S. Chen, X. Quan, X. Fan, H. Zhao, Q. Zhao and H. Yu, Appl. Catal., B, 2014, 154–155, 206 CrossRef CAS.
- L. H. Chen, J. B. Zang, Y. H. Wang and L. Y. Bian, Electrochim. Acta, 2008, 53, 3442 CrossRef CAS.
- C. Shu, Y. Lin and D. Su, J. Mater. Chem. A, 2016, 4, 2128 RSC.
- C. Shu, Y. Lin, B. Zhang, S. B. A. Hamid and D. Su, J. Mater. Chem. A, 2016, 4, 6610 RSC.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515 CrossRef CAS.
- L. Zhang and R. J. Hamers, Diamond Relat. Mater., 2017, 78, 24 CrossRef CAS.
- D. M. Jang, Y. Myung, H. S. Im, Y. S. Seo, Y. J. Cho, C. W. Lee, J. Park, A.-Y. Jeeb and M. Lee, Chem. Commun., 2012, 48, 696 RSC.
- K. M. Tripathi, T. S. Tran, Y. J. Kim and T. Y. Kim, ACS Sustainable Chem. Eng., 2017, 5, 3982 CrossRef CAS.
- G. Centi and S. Perathoner, Comprehensive Inorganic Chemistry II, Section 7.18 “Mixed-Metal Oxides”, 2014, p. 153 Search PubMed.
- Q. Yuan, Z. Xu, B. I. Yakobson and F. Ding, Phys. Rev. Lett., 2012, 108, 245505 CrossRef PubMed.
- J. Kim, A. J. Page, S. Irle and K. Morokuma, J. Am. Chem. Soc., 2012, 134, 9311 CrossRef CAS PubMed.
- K. P. S. S. Hembram and G. M. Rao, Mater. Lett., 2012, 72, 68 CrossRef CAS.
- J.-C. Charlier, Acc. Chem. Res., 2002, 35, 1063 CrossRef CAS PubMed.
- J. M. Carlsson and M. Scheffler, Phys. Rev. Lett., 2006, 96, 046806 CrossRef PubMed.
- J.-P. Tessonnier, A. Villa, O. Majoulet, D. S. Su and R. Schlögl, Angew. Chem., Int. Ed., 2009, 48, 6543 CrossRef CAS PubMed.
- C.-Y. Chen and C. T. Jafvert, Energy Environ. Sci., 2010, 44, 6674 CAS.
- Y. Luo, Y. Heng, X. Dai, W. Chen and J. Li, J. Solid State Chem., 2009, 182, 2521 CrossRef CAS.
- C. Su, M. Acik, K. Takai, J. Lu, S. J. Hao, Y. Zheng, P. Wu, Q. Bao, T. Enoki, Y. J. Chabal and K. P. Loh, Nat. Commun., 2012, 3, 1298 CrossRef PubMed.
- G. Centi, M. Gangeri, M. Fiorello, S. Perathoner, J. Amadou, D. Bégin, M. J. Ledoux, C. Pham-Huu, M. E. Schuster, D. S. Su, J.-P. Tessonnier and R. Schlögl, Catal. Today, 2009, 147, 287 CrossRef CAS.
- R. A. Sidik, A. B. Anderson, N. P. Subramanian, S. P. Kumaraguru and B. N. Popov, J. Phys. Chem. B, 2006, 110, 1787 CrossRef CAS PubMed.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505 RSC.
- K.-H. Wu, D.-W. Wang, D. S. Su and J. R. Gentle, ChemSusChem, 2015, 8, 2772 CrossRef CAS PubMed.
- G.-L. Chai, Z. Hou, D.-J. Shu, T. Ikeda and K. Terakura, J. Am. Chem. Soc., 2014, 136, 13629 CrossRef CAS PubMed.
- D. Sh. Sabirov, S. L. Khursan and R. G. Bulgakov, Russ. Chem. Bull., 2008, 57, 2520 CrossRef CAS.
- J. Yang, Y. Liu, D. Zhang, X. Wang, R. Li and Y. Li, Nano Res., 2015, 8, 3054 CrossRef CAS.
- M. Anafcheh, Z. Khodadadi, F. Ektefa and R. Ghafouri, J. Phys. Chem. Solids, 2016, 92, 26 CrossRef CAS.
- M. Anafcheh, Mol. Phys., 2018, 116, 179 CrossRef CAS.
- K. Barbera, L. Frusteri, G. Italiano, L. Spadaro, F. Frusteri, S. Perathoner and G. Centi, Chin. J. Catal., 2014, 35, 869 CrossRef CAS.
- L. Frusteri, C. Cannilla, G. Bonura, A. L. Chuvilin, S. Perathoner, G. Centi and F. Frusteri, Catal. Today, 2016, 277, 68 CrossRef CAS.
- G. Centi, K. Barbera, S. Perathoner, N. K. Gupta, E. E. Ember and J. A. Lercher, ChemCatChem, 2015, 7, 3036 CrossRef CAS.
- D. J. Appelhans, Z. Lin and M. T. Lusk, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 073410 CrossRef.
- T. F. Yeh, C. Y. Teng, S. J. Chen and H. S. Teng, Adv. Mater., 2014, 26, 3297 CrossRef CAS PubMed.
- T. F. Yeh, S. J. Chen and H. S. Teng, Nano Energy, 2015, 12, 476 CrossRef CAS.
- Y. Zhai, Z. Zhu and S. Dong, ChemCatChem, 2015, 7, 2806 CrossRef CAS.
- X. Zhou, J. Qiao, L. Yang and J. Zhang, Adv. Energy Mater., 2014, 4, 1301523 CrossRef.
- Y. Yang, K. Chiang and N. Burke, Catal. Today, 2011, 178, 197 CrossRef CAS.
- V. Calvino-Casilda, A. J. Lopez-Peinado, C. J. Duran-Valle and R. M. Martin-Aranda, Catal. Rev.: Sci. Eng., 2010, 52, 325 CrossRef CAS.
- J. J. Vilatela and D. Eder, ChemSusChem, 2012, 5, 456 CrossRef CAS PubMed.
- D. Eder, Chem. Rev., 2010, 110, 1348 CrossRef CAS PubMed.
- R. Arrigo, M. E. Schuster, S. Abate, S. Wrabetz, K. Amakawa, D. Teschner, M. Freni, G. Centi, S. Perathoner, M. Hävecker and R. Schlögl, ChemSusChem, 2014, 7, 179 CrossRef CAS PubMed.
- S. Abate, M. Freni, R. Arrigo, M. E. Schuster, S. Perathoner and G. Centi, ChemCatChem, 2013, 5, 1899 CrossRef CAS.
- S. Abate, R. Arrigo, M. E. Schuster, S. Perathoner, G. Centi, A. Villa, D. Su and R. Schlögl, Catal. Today, 2010, 157, 280 CrossRef CAS.
- B. Zhang and D. S. Su, ChemCatChem, 2015, 16, 3639 CrossRef.
- B. Zhang, L. Shao, W. Zhang, X. Sun, X. Pan and D. S. Su, ChemCatChem, 2014, 6, 2607 CrossRef CAS.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, ACS Sustainable Chem. Eng., 2017, 5, 7393 CrossRef CAS.
- S. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. Su and G. Centi, Angew. Chem., 2017, 129, 2743 CrossRef.
- C. Genovese, M. E. Schuster, E. K. Gibson, D. Gianolio, V. Posligua, R. Grau-Crespo, G. Cibin, P. P. Wells, D. Garai, V. Solokha, S. K. Calderon, J. J. Velasco-Velez, C. Ampelli, S. Perathoner, G. Held, G. Centi and R. Arrigo, Nat. Commun., 2018, 9, 935 CrossRef PubMed.
- W. Qi, W. Liu, B. Zhang, X. Gu, X. Guo and D. Su, Angew. Chem., Int. Ed., 2013, 52, 14224 CrossRef CAS PubMed.
- X. Guo, W. Qi, W. Liu, P. Yan, F. Li, C. Liang and D. Su, ACS Catal., 2017, 7, 1424 CrossRef CAS.
- R. Passalacqua, S. Perathoner and G. Centi, J. Energy Chem., 2017, 26, 219 CrossRef.
| This journal is © The Royal Society of Chemistry 2018 |