
Insights on pathways for hydrogen generation from ethanol
Sonil
Nanda
a,
Rachita
Rana
b,
Ying
Zheng
c,
Janusz A.
Kozinski
a and
Ajay K.
Dalai
 *b
*b
aDepartment of Earth and Space Science and Engineering, Lassonde School of Engineering, York University, Toronto, Canada
bDepartment of Chemical and Biological Engineering, University of Saskatchewan, Saskatoon, Canada. E-mail: ajay.dalai@usask.ca; Fax: +1-306-966-4777; Tel: +1-306-966-4771
cInstitute for Materials and Processes, The University of Edinburgh, Edinburgh, UK
First published on 31st May 2017
Abstract
Growing apprehensions on greenhouse gas emissions, global warming, and pollution problems are directly related to fossil fuels consumption. To counteract these environmental issues, global efforts are being made to diversify energy supplies towards cleaner fuels, especially for the transportation sector. Hydrogen surpasses all other biofuels such as biodiesel, bio-oil, ethanol, and butanol because of its high energy content, no greenhouse gas emissions, rapid combustion properties, non-corrosive nature, and physical state. Hydrogen production from methane and methanol, through reforming reactions, has been thoroughly studied and these are well-entrenched industrial technologies. However, ethanol is an attractive feedstock for hydrogen production because it is less toxic than methanol and can easily be produced from renewable biomass. Interest in the conversion of ethanol to hydrogen through several thermochemical, hydrothermal, photochemical, and electrochemical technologies has grown recently. In this review, different thermochemical, hydrothermal, photochemical, and electrochemical technologies for ethanol to hydrogen conversion are comprehensively discussed. Ethanol conversion technologies reviewed in this paper include steam reforming, partial oxidation, autothermal reforming, alkaline-enhanced reforming, dehydrogenation, supercritical water gasification, photocatalysis, and electrocatalysis. Current advancements, technical challenges, and future perspectives of each technology are thoroughly discussed. This technical compilation brings together various techniques that are being explored for hydrogen production from ethanol.
 Sonil Nanda | Dr Sonil Nanda is a Research Associate at the Lassonde School of Engineering in York University, Toronto, Canada. His research areas are in the production of advanced biofuels and chemicals from waste and renewable biomass through gasification, pyrolysis and fermentation. His parallel interests are in the generation of hydrothermal flames for treatment of hazardous wastes, agronomic applications of biochar, phytoremediation of heavy metal contaminated soils, as well as carbon capture and sequestration. Dr Nanda leads many transdisciplinary research projects relating to clean energy, smart materials, sustainable environment and public health. |
 Rachita Rana | Ms. Rachita Rana is a Research Assistant in the Department of Chemical and Biological Engineering at the University of Saskatchewan, Saskatoon, Canada. Her current research includes the application of supercritical fluid technology for generation of energy from renewable feedstocks and effective remediation of petrochemical waste streams. Her M.Sc. research was focused on the impact of fine particle deposition during hydrotreating of bitumen-derived gas oils. She has a keen interest in exploring the pathways for unconventional fuel generation from oil sands and biomass, as well as synthesis of smart nano-materials. |
 Ying Zheng | Prof. Ying Zheng is the Chair and Professor in Catalysis and Reaction Engineering at the School of Engineering in University of Edinburgh, UK. Her research interests lie at the development of novel catalysts and chemical processes for waste-to-energy and value-added products. Dr Zheng is a Fellow Member of the Canadian Academy of Engineering, Chemical Institute of Canada and Member of the College of New Scholars of the Royal Society of Canada. Her contribution to Chemical Engineering has been recognised by several awards, including the Syncrude Canada Innovation Award, Imperial Oil University Research Award and the University of New Brunswick Research Scholar Award. |
 Janusz A. Kozinski | Prof. Janusz A. Kozinski is the Founding Dean and Professor in the Lassonde School of Engineering at York University, Toronto, Canada. He is an internationally-renowned higher education leader, research and entrepreneur, and one of the most widely acknowledged experts in sustainable energy systems and immune building concepts applied to public safety and security. His multi-disciplinary research background relates to thermodynamics, space science, chemical and biological engineering. Some of his notable works are in supercritical water gasification for biofuel production, hydrothermal flames for toxic waste remediation, next generation nuclear energy reactors and development of immune buildings systems. |
 Ajay K. Dalai | Prof. Ajay K. Dalai is a Professor of Chemical Engineering at the University of Saskatchewan, Saskatoon, Canada. He holds the Canada Research Chair position in Bioenergy and Environmental-friendly Chemical Processing. He is a Fellow Member of the Royal Society of Chemistry, the Royal Society of Canada, Chemical Institute of Canada, American Institute of Chemical Engineers, Indian Institute of Chemical Engineers, Canadian Academy of Engineering and Engineering Institute of Canada. His wide-ranging research interests are in Fischer–Tropsch catalysis, bio-oil upgrading, biodiesel production, hydroprocessing of heavy gas oil, dry reforming of methane, gasification and pyrolysis. |
1. Introduction
There is a burgeoning concern in research and development for alternative energy sources that can be environmentally sustainable, reliable, benign, and affordable. This apprehension is related to uncertainty in fossil fuel prices, the future supply chain of crude oil, and increasing greenhouse gas emissions.1 Fossil fuels, especially crude oil, natural gas, and coal fulfill about 80% of the global energy demand.2 The recent atmospheric concentration of CO2 is 406 ppm,3 which has increased by about 32% from its 1750 levels i.e., 278 ppm.4 This high level of CO2 along with other greenhouse gas emissions is a prime cause of the greenhouse effect, depletion of the ozone layer, and global warming. Therefore, there is an immediate need for renewable fuels as alternative energy sources that could potentially counteract the greenhouse effect and replace the use of fossil fuels.Hydrogen (H2) is considered as an advanced alternative fuel, energy vector, and energy carrier. H2 has a calorific value of 141.9 kJ g−1, which is 4.8 times greater than that of ethanol (29.7 kJ g−1), 3 times greater than that of gasoline (47 kJ g−1), and 2.6 times greater than that of natural gas (54 kJ g−1). It is foreseeable that H2 can proportionate to approximately 11% of the total renewable energy share of 36% by 2025.2 H2 is also likely to contribute nearly 34% of the total renewable energy supply of 69% by 2050. This indicates that H2 has remarkable prospects in a sustainable global energy supply. H2 is considered as a clean fuel because it liberates water upon combustion. This property defines its name i.e., ‘hydro’ means ‘water’ and ‘gen’ means ‘generating’. Some of the fuel properties of H2 are compared to ethanol and gasoline in Table 1.
| Properties | Gasoline | Ethanol | Hydrogen |
|---|---|---|---|
| a References: USDE.5,6 | |||
| Auto-ignition temperature in air (°C) | 230–900 | 423 | 585 |
| Boiling point (°C) | 27–225 | 78.5 | −253 |
| Cetane number | — | 0–54 | — |
| Density at 20 °C and 1 atm (kg m−3) | 751 | 789 | 0.0838 |
| Flash point (°C) | −43 | 13 | <−253 |
| Molecular weight (kg kmol−1) | 100–105 | 46.07 | 2.02 |
| Pump octane number | 84–93 | 110 | >130 |
| Vapor specific heat at 20 °C and 1 atm | 3.66 | — | 0.0696 |
| Viscosity density at 20 °C and 1 atm (g cm−1 s−1) | 0.0037–0.0044 | 0.0119 | 8.81 × 10−5 |
| Physical state | Liquid | Liquid | Compressed gas or liquid |
Apart from ammonia production, H2 is predominantly used in petrochemical industries for hydrogenation, hydrotreating, and hydrodesulfurization. Hydrogenation employs H2 for reducing or saturating organic compounds such as converting alkenes into saturated alkanes (paraffin) and cycloalkanes (naphthene). Hydrogenation is also used to create semi-solid fats (margarine) from liquid vegetable oil.7 If used as a direct fuel, H2 can be used in combustion engines and fuel cell electric motors. It also acts as a precursor along with CO (as syngas) to generate synthetic chemicals, green diesel, and other hydrocarbon fuels via Fischer–Tropsch catalysis.8 Fuel cells convert H2 and an oxidant (usually O2) directly into electricity through a low-temperature electrochemical process. H2 also acts as an electron donner to reduce wastewater pollutants viz. arsenate, chromate, dibromochloropropane, nitrate, perchlorate, selenite, etc.9
H2 can be synthesized from diverse sources such as fossil fuels, oils, alcohols, water, and biomass. Today, 96% of H2 produced is directly from fossil fuels and the remaining 4% is from other sources.10 Commercial scale H2 synthesis is achieved through steam reforming of natural gas or methane. Steam reforming of methane is the most preferred commercial H2-producing technology, contributing 96% of the total H2 supply and having a production cost of 1.5–3.7 U.S. $ per kg.2,11 Although H2 is regarded as a green fuel, as its combustion does not produce any CO2, still, its synthesis from fossil-based resources (natural gas) imparts an irreplaceable carbon footprint to the manufacturing process.
The current annual world energy demand as cited by the U.S. Energy Information Administration is 590 quadrillion British Thermal Units (BTUs).12 The major contributors to this energy include oil, natural gas, and coal attributing nearly 82% of the total world energy supply. This gives roughly 486 quadrillion BTUs, which can be further classified on the basis of individual energy contributions: for example, by oil and other liquid fuels (41% or 193 quadrillion BTUs), coal (24% or 162 quadrillion BTUs), and natural gas (22% or 131 quadrillion BTUs). A further contribution is from renewable energy sources (hydrogen and other biofuels) but which supplement only 74 quadrillion BTUs. The remaining 28 quadrillion BTUs energy comes from nuclear sources.
In the current market, ethanol has emerged as a biofuel for blending with gasoline because it exhibits clean burning characteristics such as reduction in greenhouse gas emissions and particulate matter along with benefits of low vapor pressure.13 Ethanol has a potential to replace 32% of gasoline usage when used as E85 blend (i.e. 85% ethanol and 15% gasoline).14 Bioethanol is predominantly produced via fermentation, and the substrates for bioethanol production include directly fermentable sugars (pentose and hexose), starch-based materials, and lignocellulosic feedstocks. Bioconversion of these materials involves biomass pretreatment (acid, alkali, ionic or mechanical), delignification, enzymatic hydrolysis, and fermentation.15
Using ethanol as a fuel has several mechanical issues related to vehicle engines. Ethanol cannot be used as a drop-in fuel as most of the current vehicle engines are not capable of being powered by pure ethanol.16 Furthermore, ethanol is completely miscible in water but partially miscible in gasoline, which results in phase separation problems.17 Phase separation is unavoidable when ethanol and water mix in an ethanol-blended gasoline. Moreover, ethanol's low viscosity leads to the progressive wearing of vehicular engine parts. Besides, high concentrations of ethanol in gasoline blends are corrosive to engine components because of increased water content in the fuel and organic acids produced in oxygenates.17 Considering all these practical issues for using ethanol as a direct fuel, there is a strong possibility to use it as a precursor to produce H2. Moreover, ethanol-blended gasoline in vehicle engines results in about 20% energy efficiency, whereas ethanol reformed to H2 for fuel cells can have more than 60% efficiency.18 Ethanol-to-hydrogen conversion is not only carbon-neutral but also environmentally-friendly. Moreover, ethanol is energy dense and has relatively high H2 content, which under reaction with water during reforming can be thermodynamically achievable.
There is limited literature available on exploring new hydrocarbon feedstocks other than CH4 for reforming to H2. With this objective, the current paper is focused on exploring ethanol as a substrate for H2 generation. The H2-producing technologies reviewed in this paper include steam reforming, alkaline-enhanced reforming, partial oxidation, autothermal reforming, gasification, dehydrogenation, photocatalysis, and electrocatalysis (Fig. 1). This review gives an overview of each technology along with its benefits and drawbacks. Recent advancements and current trends for catalyst synthesis, reactor modifications, and new process developments to improve yield and energy efficiency with low-cost considerations have been highlighted through this work.
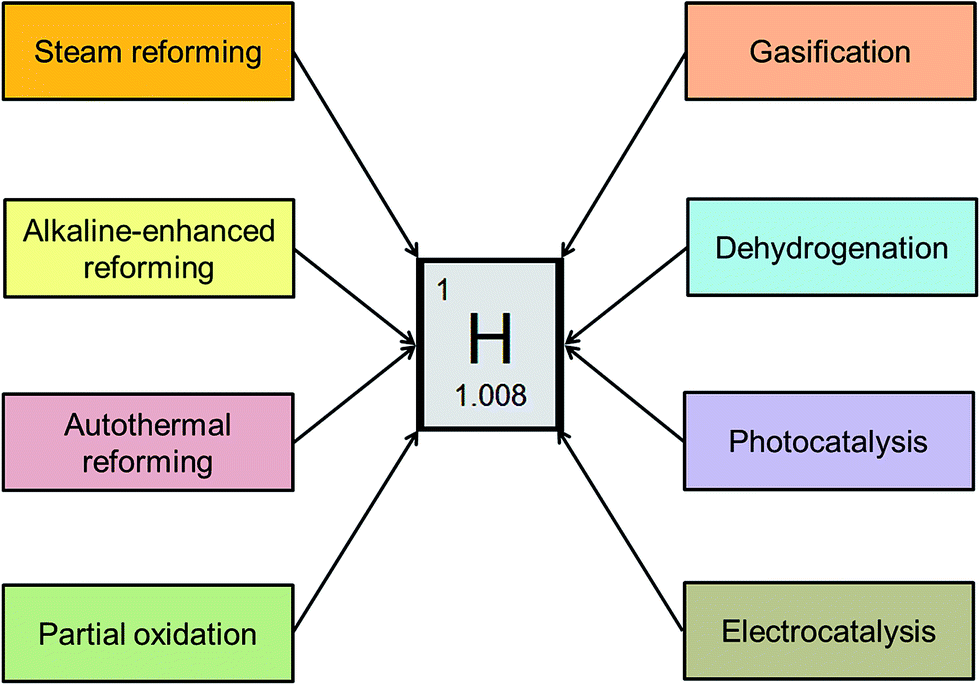 | ||
| Fig. 1 Different routes of hydrogen production from ethanol. | ||
2. Reforming of ethanol
A wide variety of feedstocks can be used to produce H2 through reforming such as carbohydrates (sugars), hydrocarbons, fossil fuels, alcohols, and bio-oils. Owing to its high energy density, low sulfur levels, and safety in handling, methanol has been one of the industrially favorite feedstocks for reforming reactions for H2 generation. Some current commercial technologies for H2 generation from ethanol include steam reforming, alkaline-enhanced reforming, partial oxidation, and autothermal reforming.Fig. 2 depicts a mechanistic representation of several reactions involved in ethanol reforming. Among all these reforming technologies, steam reforming is mostly preferred because it catalytically converts hydrocarbons or oxygenated hydrocarbons to produce a gas mixture consisting predominantly of H2 and CO.18 Steam reforming of ethanol results in H2, CO2, and CO. Dry reforming of ethanol with CO2 primarily liberates CO and H2. CO and H2 are also produced as a result of partial oxidation upon the reaction of ethanol with oxygen. CO reacts with water to form water–gas and via water–gas shift reaction at high temperatures releases H2 and CO2. Autothermal reforming also produces H2 and CO2 upon the reaction of ethanol with water and oxygen. However, in the presence of supercritical water, reforming of ethanol results in H2, CO2, and CH4. CO and CO2 may undergo methanation and hydrogenation in supercritical water to primarily produce CH4, which is undesirable when H2 is the main product of interest. CH4 and C2H4 are produced by ethanol decomposition and dehydration, respectively. Dehydrogenation of ethanol leads to the formation of H2 and acetaldehyde, which releases CH4 and CO upon decarbonylation. Polymerization and the Boudouard reaction are detrimental from an industrial perspective as they lead to coke formation, which can deactivate the catalysts via carbon deposits. These reactions are thoroughly discussed in the subsequent sections.
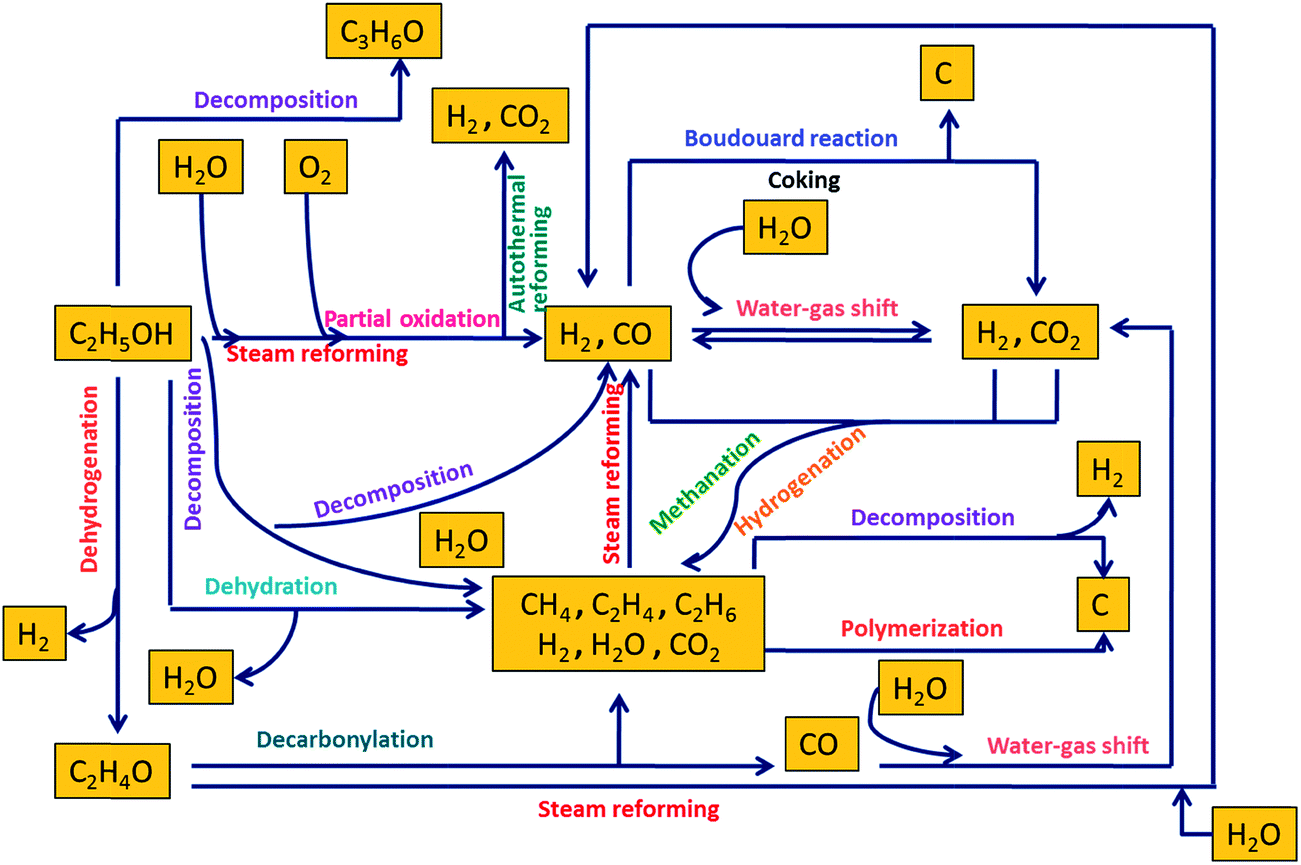 | ||
| Fig. 2 Mechanistic representation of different reactions involved in reforming of ethanol (adapted from Haryanto et al.19). | ||
2.1. Steam reforming
Steam reforming in the presence of metal catalysts leads to cleavage of chemical bonds (C–C, C–H and O–H) in hydrocarbons for enhanced thermal decomposition.20 Catalysts used for hydrocarbon reforming are classified into groups of noble metals from group VIII elements (e.g. Pt or Rh) and transition metals such as Ni.21 The steam reforming of several model biomass compounds such as glucose, xylose, sucrose, acetic acid, acetone, ethanol, methanol, phenol, cresol, dibenzyl ether, etc. has been carried out with Ni-based catalysts and noble metal catalysts (e.g. Pt, Rh, Pd, Ru, and Ir).22,23In the case of non-noble metal catalysts such as Ni-based catalysts, steam reforming reactions usually occur at high temperatures of 600–800 °C with high space velocities.22 It is further suggested that certain bimetallic catalysts composed of noble metals such as Pt–Rh/CeO2, Rh–Au/CeO2 give better H2 yields from ethanol reforming.24 Noble metals are the best catalysts for steam reforming of ethanol due to their greater ability to cleave C–C bonds, reducing the requirement of active metal loading compared to non-noble metals.24 Pt and Rh are the two noble catalysts found to exhibit high activity for breaking the C–C bond in ethanol and promoting water–gas shift reaction.23
A steam-to-carbon molar ratio of 3–5 is used in a typical steam reforming process.20 Steam reforming of hydrocarbons has been described by the following stoichiometric reactions (eqn (1)–(3)) proposed by Huber et al.22 In the first step of catalytic reforming, the hydrocarbon dissociates on the metal surface to release H2 and CO. In the second step, the methanation and water–gas shift reaction results in the formation of equilibrium concentrations of H2, CO2, CO, and CH4 along with water. The steam reforming reaction for ethanol is shown in eqn (4).
Steam reforming of hydrocarbons:
| CxHy + xH2O → xCO + (x + 0.5y)H2 | (1) |
| CO + 3H2 ↔ CH4 + H2O | (2) |
| CO + H2O ↔ CO2 + H2 | (3) |
Steam reforming of ethanol:25
| C2H5OH + 3H2O → 2CO2 + 6H2, [ΔH = 173.7 kJ mol−1] | (4) |
Steam reforming of ethanol is an endothermic process and has been reported to be thermodynamically feasible at high temperatures.26 Oxygen mobility of the catalyst support is a critical factor for consideration to favor carbon removal and avoid catalyst deactivation.27 Utmost precautions should be taken while using crude bioethanol samples for reforming because of the fact that presence of any metal impurities can weaken or deactivate the catalyst. Ideal features of a catalyst for steam reforming of ethanol are: (i) high thermal stability, (ii) high activity for ethanol reforming, (iii) greater yields and selectivity of H2, (iv) resistance to coke formation, and (v) tolerance to impurities in crude ethanol against deactivation.
Ethanol activation routes are contingent on the metal catalysts, which are generally divided into two groups: (i) less-oxophilic metals e.g., Pd and Pt that facilitate α-C–H activation, and (ii) more-oxophilic metals e.g., Co, Ni, Rh, and Ru that enhance O–H activation.23 In addition to the catalyst, it is the support that plays a significant role in determining activity and stability of the catalyst. Particularly, in the case of noble metal catalysts, where metal loading on the support is less than 1%, the support directly contributes towards the reaction mechanism.23
2.2. Dry reforming
Another approach for H2 production is through dry reforming of ethanol. The dry reforming process for producing H2 from ethanol is a series of complex reactions. There are several side-reactions that result in the formation of undesirable products along with H2. Hence, H2 yield is highly dependent on the main process parameters such as temperature, pressure, CO2, and ethanol molar ratio. Dry reforming of ethanol to produce H2 is a strongly endothermic reaction, and the products from this reaction are only H2 and CO if the reaction conditions are favorable. The reaction for dry reforming of ethanol is given in eqn (5).Dry reforming of ethanol:28
| C2H5OH + CO2 → 3CO + 3H2, [ΔH° = 296.7 kJ mol−1] | (5) |
Bej et al. studied the dry reforming of ethanol for H2 production over NiO nanoparticles supported on alumina catalyst in silica that was synthesized by the sol–gel method.28 Catalyst activity was assessed in terms of CO2 and ethanol conversion, and in terms of H2 and CO production. Jankhah et al. reported the thermodynamic equilibrium for thermal and catalytic cracking of ethanol along with dry reforming reactions at different ratios of CO2 and ethanol.29 The obtained results showed that in cracking and catalytic dry reforming of ethanol, carbon deposits were observed in the form of nanofilaments. There are several other studies that report the thermodynamics of the dry reforming process for H2 production from ethanol.30–32
2.3. Alkaline-enhanced reforming
Alkaline-enhanced reforming converts aqueous organic compounds to H2 at comparatively lower temperatures (<220 °C) and pressures than those involved in steam reforming reactions. As the name suggests, the process involves an alkali catalyst such as NaOH, which leads to faster reaction kinetics compared to steam reforming.20Eqn (6) represents alkaline-enhanced reforming of ethanol.Alkaline-enhanced reforming of ethanol:
| C2H5OH + 4NaOH + H2O ↔ 2Na2CO3 + 6H2, [ΔH = 4.2 kJ mol−1] | (6) |
Gas products generated through alkaline-enhanced reforming typically produce lower yields of CO2 because the process generates precipitates as alkali salts, especially Na2CO3. Upon precipitation, Na2CO3 particles can provide a larger surface area for catalytic reactions, which can be considered as a positive feature.33 However, there are chances of reactor corrosion due to salt formation and precipitation on the reactor walls. Nevertheless, lower yields of CO2 make alkaline-enhanced reforming beneficial as the mass fraction of H2 is found to be relatively higher as compared to steam reforming.
2.4. Partial oxidation
Partial oxidation transpires when a sub-stoichiometric fuel–air mixture reacts in a reformer to yield H2-rich syngas. In contrast to catalytic partial oxidation of hydrocarbons, non-catalytic partial oxidation in the presence of oxygen usually requires higher temperatures than stream reforming. To ensure complete conversion and reduce carbon or soot formation, flame temperatures of 1300–1500 °C are required.34 However, catalysts such as Ni and Rh can reduce temperature requirements up to 800–900 °C. Partial oxidation of a model hydrocarbon fuel and ethanol are depicted in eqn (7) and (8), respectively.Partial oxidation of hydrocarbons:
| CxHy + 0.5xO2 → xCO + 0.5H2 | (7) |
Partial oxidation of ethanol:
| C2H5OH + 0.5O2 → 2CO + 3H2, [ΔH = −551.8 kJ mol−1] | (8) |
Partial oxidation reaction is reported to have a fast start-up and response time compared to other hydrocarbon reforming technologies.35 In addition, the reaction does not require any indirect addition of heat through a heat exchanger, making it possible for more compact reactors to be used than large steam reformers. Another reason for compact reactors in partial oxidation of ethanol is self-balanced conditions rendered by ethanol and air, which are the two basic reactants.36
Unlike hydrocarbons, the partial oxidation of ethanol is slightly endothermic and mostly exothermic.36,37 Salge et al. performed partial oxidation of ethanol and a ethanol–water mixture using noble metals (Pt, Pd, and Rh) and metal plus ceria-coated alumina foam with catalyst contact times less than 10 ms.37 H2 selectivity of more than 80% and conversion of 95% were achieved from the catalytic partial oxidation of ethanol and ethanol–water using Rh–Ce catalysts because of their thermal stability and redox capabilities of Ce. H2 yields less than 50% were obtained from the partial oxidation of ethanol using Rh, Pt, Pd, and Rh–Ru catalysts. Upon addition of water to the reaction medium (i.e., ethanol–water mixture), CH4 selectivity shrunk to less than 3% and H2 selectivity exceeded 100% because both water and ethanol contribute H2. Moreover, Ni–Fe catalysts have revealed high activities for partial oxidation of ethanol to H2 with conversions reaching 87% at high temperatures (∼300 °C).36
2.5. Autothermal reforming
Selectivity of H2 from the partial oxidation of ethanol is relatively lower when compared to autothermal reforming. Autothermal reforming involves the addition of steam to catalytic partial oxidation. Autothermal reforming is a combination of exothermic partial oxidation and endothermic steam reforming, thus referred to as oxidative steam reforming and remains thermodynamically neutral.38In autothermal reforming, partial oxidation supplies the initial heat required to start the steam reforming reaction. This is advantageous in saving energy as the heat released from partial oxidation restricts the requirement for an external heat source.21 Heat released from the exothermic partial oxidation reaction balances the endothermic reforming reaction in the autothermal reforming of methanol.22 Autothermal reforming has other advantages such as compactness of reactor, low operating cost, and potential for scale-up.39 Autothermal reforming also is beneficial as coke formation is inhibited by oxidation.40 Oxygen facilitates efficient removal of carbon species formed during the reaction. Autothermal reforming of a model hydrocarbon and ethanol are shown in eqn (9) and (10), respectively.
Autothermal reforming of hydrocarbon:
| CxHy + 0.5xH2O + 0.25xO2 → xCO + (0.5x + 0.5y)H2 | (9) |
Autothermal reforming of ethanol:41
| C2H5OH + 2H2O + 0.5O2 → 2CO2 + 5H2, [ΔH = −1280 kJ mol−1] | (10) |
Oxygen-to-fuel ratio and steam-to-carbon ratio dictate the operating temperature of autothermal reforming and resulting gas composition. Catalysts used in autothermal reforming are classified as: (i) noble metal-based, (ii) non-noble metal-based, and (iii) bi-metallic catalysts.38 Ethanol conversion and H2 selectivity by autothermal reforming largely vary with the catalyst type, support material, and molar ratios of oxygen, steam, and ethanol. Several catalysts studies for the autothermal reforming of ethanol to H2 include Ni–Rh/CeO2, Pd/ZnO, Pt–Al2O3, Rh–Ce, and Cu1−xNixZnAl-mixed metal oxide.40 A molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.8:0.6 for ethanol, water, and oxygen can make autothermal reforming thermally-neutral as the exothermic oxidation of ethanol provides the heat energy necessary for endothermic steam reforming of ethanol.42
:1.8:0.6 for ethanol, water, and oxygen can make autothermal reforming thermally-neutral as the exothermic oxidation of ethanol provides the heat energy necessary for endothermic steam reforming of ethanol.42
3. Supercritical water gasification
Water exists in a supercritical state when its temperature and pressure exceeds critical points (374 °C and 22.1 MPa). Supercritical water acts as a reaction medium and reactant to convert biomass into H2-rich syngas. The non-polar characteristic of supercritical water heightens its scope to dissolve organic compounds in a homogeneous phase.43 The non-toxic nature, as well as unique solvating and transport properties of supercritical water, makes it attractive for hydrothermal gasification of waste biomass.44,45Synthesis gas or syngas is a gas mixture mainly consisting of H2 and CO alongside CO2, CH4, C2H6, and C2+ obtained through thermochemical gasification of biomass. Supercritical water gasification of biomass and other organic compounds (e.g. alcohols, carbohydrates, sugars, proteins, lipids, fats, etc.) for H2 production is a promising technology. There are several advantages of supercritical water gasification over other thermochemical technologies, a few of which include: (i) processing of feedstock containing moisture content greater than 50%, (ii) production of H2 at high pressures and greater yields, (iii) possibility to recover energy from high-temperature product gas, and (iv) reduced yield of tar and char.46
The deceleration of hydrogen bonds in supercritical water stimulates ionic mechanisms. The ionic mechanisms result in formation of H+ and OH− ions that create a favorable environment for acid–base catalyzed reactions for degradation of recalcitrant organic compounds.47 Kuhlmann et al. performed supercritical water gasification experiments with D2O instead of H2O to confirm the phenomenon that water acts as a reactant in gasification to produce H2.48 Low density, high diffusivity, and negligible surface tension of supercritical water promote reforming reactions and degradation of components in gasification.49,50
It is essential to understand the reaction mechanism, thermal events, and degradation behavior of model compounds and real biomass in supercritical conditions. Several model compounds that have been investigated in supercritical water gasification for H2 production include glucose, fructose, lactose, glycerol, glycine, guaiacol, cellulose, lignin, phenolics, ethanol, methanol, humic acid, etc.51–59 Supercritical water gasification also involves several vital sub-reactions to facilitate the thermal events towards H2 production and consumption. A few of such reactions include steam reforming, hydrolysis, water–gas shift reaction, methanation, hydrogenation, and the Boudouard reaction as summarized in the equations below.
Steam reforming reaction:
| CHxOy + (2 − y)H2O → CO2 + (2 − y + 0.5x)H2 | (11) |
| CHxOy + (1 − y)H2O → CO + (1 − y + 0.5x)H2 | (12) |
Reforming of ethanol:
| C2H5OH + H2O → CO2 + CH4 + 2H2 | (13) |
Water–gas shift reaction:
| CO + H2O → CO2 + H2 | (14) |
Methanation reaction of CO:
| CO + 3H2 → CH4 + H2O | (15) |
Methanation reaction of CO2:
| CO2 + 4H2 → CH4 + 2H2O | (16) |
Hydrogenation reaction:
| CO + 2H2 → CH4 + 0.5O2 | (17) |
Boudouard reaction:
| 2CO → C + CO2 | (18) |
| CO2 → O2 + C | (19) |
Biomass components and intermediate products (e.g., phenols) undergo steam reforming in supercritical water to produce CO, CO2, and H2 (eqn (11) and (12)). Lignin is a polymeric compound that degrades in supercritical water to produce phenolics, which further reform to produce H2, CO, and CO2. Water–gas refers to a mixture of CO and water; thus, a water–gas shift reaction indicates the reaction between CO and H2O to produce H2 and CO2 (eqn (14)). The water–gas shift reaction is weakly exothermic, which becomes dominant at high supercritical temperatures, lower feed concentrations, and moderate residence times.60 Methanation and hydrogenation reactions are mostly considered secondary reactions as they are pertinent at longer residence times and consume H2, CO2, and CO to primarily produce CH4 (eqn (15)–(17)). On the other hand, the Boudouard reaction also consumes CO to form carbon, usually in the form of char or coke (eqn (18) and (19)). Higher hydrocarbons are also formed when carbon is deposited by the Boudouard reaction.61
Reddy et al. reviewed different mechanisms and thermal reactions for biomass conversion to H2 through supercritical water gasification.62 Cellulose and hemicellulose hydrolyse to form pentose and hexose sugars, whereas lignin degrades to phenolics including guaiacols and syringols. These intermediate products further dissociate to acids (e.g., acetic acid, carboxylic acid, succinic acid, formic acid, propionic acid, etc.), alcohols (e.g. coumaryl, coniferyl, sinapyl, etc.), phenols, aromatics, ethers, esters, ketones, and aldehydes.56,57 Acids arise from hydrothermal decomposition of sugars, while alcohols, aromatics, and aldehydes result from denaturation of lignin and phenolics.
Supercritical water also acts as a favorable medium for the reforming reaction for H2 production at high pressures with short residence times. Voll et al. used Gibbs free energy minimization to calculate the equilibrium composition for supercritical water gasification of ethanol, methanol, cellulose, glucose, and glycerol.55 The results indicated that H2 yield amplified with an increase in temperature, decrease in feed concentration, and increase in residence time. Byrd et al. studied ethanol reforming in supercritical water using Ru/Al2O3 for H2 production.18 Generation of H2 was favored at a high temperature of 700 °C and high water-to-ethanol ratio. The formation of CH4 was suppressed at an optimal residence time of 4 s, high temperature of 800 °C, and low feed concentration of ethanol (5 wt%). However, decomposition and dehydration of ethanol were responsible for formation of CH4, C2H4, and C2H6 as shown in eqn (20) and (21), respectively.19 Ethylene (C2H4) formed through ethanol dehydration further polymerizes to generate coke (eqn (22)).
Decomposition of ethanol:28
| C2H5OH → CH4 + CO + H2, [ΔH = 49.6 kJ mol−1] | (20) |
Dehydration of ethanol:
| C2H5OH → C2H4 + H2O | (21) |
Polymerization reaction:
| C2H6 → C (coke) | (22) |
4. Dehydrogenation
Dehydrogenation of ethanol is another significantly potential route for H2 production, which is achieved by using catalysts. The reaction proceeds with removal of two hydrogen atoms from one ethanol molecule to form molecular hydrogen. Ethanol conversion to desired products with long carbon chains that have a high specific heat of combustion is possible through coupled condensation and dehydrogenation reactions.Freni reported that the dehydrogenation step is followed by decarbonylation, which leads to the formation of CH4 and CO.63 CH4, thus formed, undergoes steam reforming and then follows the water–gas shift reaction. They also reported that excess water in the system prevented coke formation for a prolonged working time. Rh was used for decarbonylation of acetaldehyde (C2H4O) to yield CH4 and CO. Eqn (23) and (24) show the respective sequential dehydrogenation of ethanol and decarbonylation of acetaldehyde.
Dehydrogenation of ethanol:28
| C2H5OH → C2H4O + H2, [ΔH = 68.5 kJ mol−1] | (23) |
Decarbonylation reaction:
| C2H4O → CH4 + CO | (24) |
Heterogeneous catalysts (e.g., Cu/ZrO2, Cu/Al2O3, Cu/ZnO, Cu–Zn–Zr–Al–O, and Cu–Cr–O) have a wide scope for direct synthesis of ethyl acetate and H2 from ethanol using a dehydrogenation mechanism.64–70 Owing to their optical properties, iron oxides are found to be promising materials for H2 production from ethanol dehydrogenation. The excited states formed at the surface of iron oxides, when a light of appropriate wavelength is incident, are the key to this characteristic feature. Hence, the properties of bulk-generated photoelectrons from photoreactions carried out in aqueous solutions have been reported in the literature.71 It was suggested that photoelectrons from the bulk of Fe2O3 colloids migrate to the crystallite interfaces. This phenomenon decreases the production of a comparatively large quantum yield of H2O inside the solids. Furthermore, Fe-based catalysts are preferred due to their low costs and abundant availability as a natural resource as compared to other transition metals.71
In the case of photo-dehydrogenation of ethanol, H2 is produced in the presence of catalysts and photons. Galindo-hernández et al. performed a incipient wet impregnation method to synthesize FexOy/C photocatalysts with different metal loadings and then calcined at 500 °C.72 They evaluated activity of a photocatalyst on the basis of H2 production from a ethanol photo-dehydrogenation reaction. With an increase in metal loadings from 15–30 wt% (Fe content) in the photocatalyst, the surface area of the substrate was reported to decrease from 638 to 490 m2 g−1. X-ray diffractograms indicated coexistence of wüstite and magnetite phases of Fe for samples with 15 and 20 wt% loadings. Additionally, hematite was seen to exist with two other phases in the case of 30 wt% loading. Content of Fe3+ ions was related to the reactivity of photo-dehydrogenation of ethanol for H2 production.
Lin and Chang used porous stainless steel to fabricate sequentially plated Pd–alloy membranes without employing electrodes.73 The synthesized membrane-reactors were used for dehydrogenation of ethanol to H2 and acetaldehyde. It was reported that the product yield amplified with the decrease of pressure between the membranes and increase in the flow rates of the sweep gas. The influence of sweep gas inflow was found significant on H2 fluxes at elevated temperatures. The membrane-reactor design gave higher conversions than that expected from the thermodynamic equilibrium for ethanol dehydrogenation.
Ashok et al. synthesized Co-nanoparticles using a solution combustion synthesis technique.74 Nanoparticles were used to study the decomposition mechanism of ethanol to H2. Different reducing agents such as glycine, hydrazine, citric acid, and urea were used to produce cobalt nanoparticles from cobalt nitrate along with thermodynamic studies of the process. Co catalyst revealed selectivity for acetate and aldehyde along with the production of H2, CO2, and water. Characterized nanoparticles after the dehydrogenation reaction indicated an increase in particle size due to sintering and coking on the surface of the catalyst.
Dolgykh et al. attempted to test the catalytic activity of Cu-based catalysts for dehydrogenation of ethanol to H2.75 The temperature range studied for the reaction was 150–300 °C with 12 wt% ethanol in water. It was reported that a high temperature of 300 °C was required to get an appreciable H2 yield in absence of the catalyst. However, H2 production increased from 0.3 g h−1 kg−1-cat at 400 °C in absence of the catalyst to 19–25 g h−1 kg−1-cat in the presence of Cu catalyst.
5. Photocatalysis
Heterogeneous photocatalysis holds much relevance in the sustainable production of H2. Fig. 3 illustrates a typical photocatalytic process for H2 production from an ethanol–water mixture. Activities and performances of various catalysts (e.g. Co, Cu, Ni, Pd, Pt, and Rh) have been reported for photocatalysis by several authors.41,76–78 However, photocatalysis is conceptualized and less explored due to several limitations such as the expensive catalysts and challenges in designing efficient cells.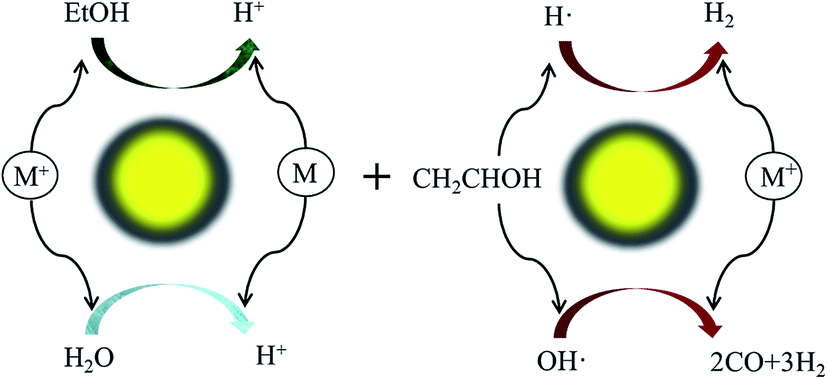 | ||
| Fig. 3 Photocatalysis of ethanol–water mixture for hydrogen production. | ||
Atoniadou et al. explained the two major categories for H2 production through a photocatalytic process.79 The first category is photocatalytic H2 production where light is absorbed by a semiconducting photocatalyst to generate electrons and holes. Holes can interact with either water or with organic/inorganic substances and split the H+ ions. The reduction of H+ ions by the electrons forms molecular H2. However, the target-substance and charge carrier interaction competes with the recombination of electron and a hole. Thus, techniques were explored to avoid many of the favored recombination mechanisms. One approach is deposition of metal nanoparticles on the surface of a photocatalyst. Metal has a tendency to take the electron which avoids recombination. Noble metals such as Au, Pd, and Pt are preferred for this purpose. A second approach is photo-electrochemical in nature where a catalyst is deposited on the anode connected to a platinum cathode. The holes generated decompose the target substance by oxidation and the electrons transfer to a counter electrode where they interact with H+ ions, producing H2 molecules. Thus, it can be inferred that a metal deposited photocatalyst functions similar to a miniature photoelectrochemical cell. In this work by Atoniadou et al., a chemically-biased two electrode reactor was used to study the usability of nanocrystalline titania films.79 The objective was to produce H2 and electricity by using ethanol as the target substance. It was found that the cell could function only in the presence of a chemical bias. Though the cell could work with water, the escalated H2 yield and electricity production indicated ethanol photodecomposition to be a more efficient process than water splitting.
Yoo et al. introduced a novel nanomaterial Au/TiO2 photocatalyst for the oxidation of ethanol to H2.80 Fabrication of a modified photocatalytic surface required a systematic overlay of self-organizing layers. In the initial step, TiO2 nanocavities were formed, while in the succeeding step Au was loaded into the cavities. Thermal dewetting of thin Au films was deposited on TiO2 by sputtering. Both steps used in the fabrication of the photocatalyst were reported to be scalable and inexpensive. It was also suggested that different metals that qualify for the self-ordering oxide and dewettability could be explored for similar applications.
Au/TiO2 was also used to manufacture microchannels of a microreactor to generate H2 from a ethanol–water mixture. In this study by Castedo et al., a silicone base was coated with Au/TiO2 photocatalyst and sealed using a thin silicone sheet.81 Different parameters such as residence time, photon irradiance, water–ethanol ratio, and photo-catalyst loading were tested for the microreactor at atmospheric pressure and room temperature. A two-day photocatalyst test indicated stable H2 yield from the photocatalytic silicone microreactor.
1,3-Dialkylimidazolium cations based ionic liquids are potential media that can be used for the synthesis and stabilization of several inorganic nanoparticles as proposed by Souza et al.82 Their pre-organized structures with polar and non-polar characteristics, specifically through structural directionality or the entropic effect displayed by H2 bonds present in these ionic liquids, enable them with such unique properties. Hence, ionic liquids have been widely used to produce Ti-based nanoparticles with modified properties to enhance photocatalytic activities. Structural directionality due to ionic liquids is suggested to be more effective in the formation of tailored nanoparticles that use a metal oxide precursor as the ionic liquid. Nanoparticles that result from this synthesis have a hybrid structure with the incorporation of ionic liquids on metal–oxide through physical or chemical means.
Souza et al. also described a route for preparing the highly active photocatalyst tantalum oxide (Ta2O5).82 Hydrolysis of ionic liquids produced hybrid Ta2O5/ionic liquid nanoparticles where the nature of ionic liquids dictated size distribution of the synthesized particles. These nanoparticles were found to be the most efficient photocatalyst for H2 generation through ethanol–water splitting. Pt was added to enhance the activity of the Ta2O5 nanoparticles. H2 amounts as high as 9.2 mmol g−1 h−1 were attained with the introduction of Pt. Therefore, this synthesized nanomaterial was considered to be a candidate photocatalyst for obtaining H2 from an ethanol–water solution.82
Antony et al. fabricated nanotubes of anatase phase TiO2 with a particle size that was comparable to the de-Broglie wavelength of TiO2 charge carriers.83 H2 generation efficiency of the photocatalyst was studied by correlating it to the photoluminescence of Pt-loaded samples. It was inferred that the particle size of Pt is critical for photocatalytic H2 generation from ethanol.
Photo-production of H2 using F-doped Co3O4 from ethanol was explored by Gasparotto et al.84 It was reported that H2 production from ethanol could be significantly enhanced by using F-doped Co3O4 films prepared by chemical vapor deposition. Particularly in the near-ultraviolet region of F-doped oxide, a five-time enhanced H2 yield was attained when compared to non-doped oxide. There was no notable improvement in H2 yield when simulated solar energy was used to test H2 production from ethanol using the F-doped Co3O4 films. However, the yield with respect to time made this material a potential photocatalyst for H2 production from ethanol. It could be concluded from the findings that doping of fluorine improved the photocatalytic activity of Co3O4 compared with the best reported H2 yields from ethanol.
Another application of photocatalysis is the synthesis of nanocrystalline PrFeO3 perovskite-type material using sol–gel, combustion, and template methods. It was reported by Tijare et al. that formation of nanocrystalline particles improved physical characteristics at lower reaction temperatures.85 The photocatalyst prepared by the sol–gel method gave a maximum H2 yield of 2847 μmol g−1 h−1 in visible light irradiation in an ethanol–water mixture.
Recently, there is an increased interest in photoelectrocatalysis, especially with applications of photoelectrochemical cells that function as H2 producers either through water splitting or oxidation of organic substrates. There is limited literature on the subject, but the area of research holds a lot of potential for an eco-friendly production of H2. Application of photoelectrochemical cells is not only realized in energy generation but also in environmental remediation of hazardous wastes. A typical photoelectrocatalytic cell operates through oxidation of an organic substrate or an inorganic sacrificial agent with the involvement of water.86 During photoelectrocatalysis, water, protons, or oxygen are reduced. The main objective behind this technology is water splitting by oxidation and/or reduction as well as oxidation of organic and inorganic wastes. Recently, Sfaelou et al. demonstrated tungsten trioxide (WO3) photoanodes as efficient photoanodes that adsorbed visible light for applications in photoelectrocatalytic cells.87 These WO3 photoanodes increased the rate of H2 production from ethanol by more than three-fold due to a higher current density. Kalamaras et al. also developed hematite photoanodes doped with titanium through electrodeposition and high-temperature annealing.88 These photoanodes were efficient in photo-reforming of ethanol and water splitting in the absence of a sacrificial agent, leading to a superior photoelectrochemical H2 production. However, the efficiency of photoelectrocatalytic fuel cells is also dependent on functionality of the counter electrode towards reduction of oxygen.86
6. Electrocatalysis
Electrocatalysis is another method to generate H2 from ethanol as shown in eqn (25) and (26). Lamy et al. investigated the feasibility of clean H2 production at higher rates by ethanol electrolysis in a Proton Exchange Membrane Electrolysis Cell (PEMEC) using several Pt-based catalysts.89 The principle of electrocatalysis of ethanol in a PEMEC is represented in Fig. 4. Ethanol mixed in water is fed to the anode to oxidize it completely. CO2 and protons are released, and the protons that reach the cathode are reduced to release H2. Both the reactions for H2 production need external energy. However, the energy needed for ethanol is less than that for water. It was reported that the thermodynamics of ethanol decomposition makes it meritorious for producing H2 of high purity in PEMEC.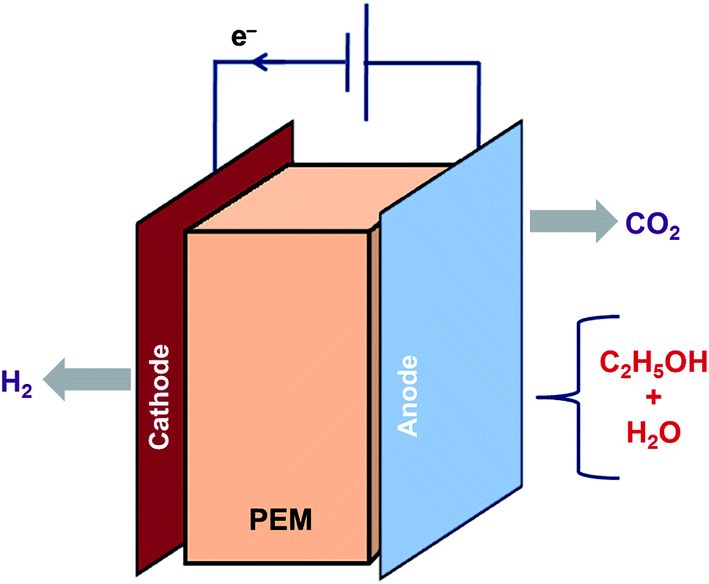 | ||
| Fig. 4 Electrocatalysis of ethanol–water mixture using a typical proton exchange membrane. | ||
Electrocatalysis of ethanol:
| C2H5OH + 3H2O → 2CO2 + 12H+ + 12e− | (25) |
| C2H5OH + 3H2O → 2H2 + 2CO2 | (26) |
The presence of poisonous species like CO that come from dissociative chemisorption of ethanol led to potential oscillations at low current. It was demonstrated through the work by Lamy et al. that the nature and structure of the electrocatalyst are essential to reduce the amount of electricity used to produce H2.89 Sn or Ru decreased the overvoltage, thereby increasing the rate of H2 generation.
Graphene as well as its N2-doped particles have great electronic properties and hence are potential supporting materials for nanoparticle synthesis of novel metals. Several methods have been reported for the synthesis of graphene and N2-doped graphene composites that are fabricated with nanoparticles of platinum applicable for fuel cell design.90–96
A one-step electrochemical method to fabricated N2-doped sheets of graphene with uniformly deposited Pt nanoparticles was introduced by Navaee et al.97 Application of the synthesized material for the oxidation of fuels such as ethanol, methanol, and formic acid was investigated. By employing a cyclic voltammetry technique it was reported that this composite had reduced current densities and onset voltage as compared to usual Pt/C for fuel oxidation. Additionally, the modified electrode using this fabricated composite showed long-term stability and great tolerance towards poisoning.
There have been several materials used for chemical or electrochemical fabrication of Pt nanostructures. However, most of the materials such as K2PtCl4 or K2PtCl6 are expensive. Navaee and Salimi proposed the use of N2-doped graphene to generate Pt nanoparticles though an economical process.98 The resulting material was used for electrocatalytic H2 generation from ethanol oxidation. Similar works were conducted by Dong et al.99 and Zhang et al.100 where new strategies opted for loading the Pt nanoparticles uniformly with high catalytic activity.
To enhance usability of Pt for electrocatalytic H2 generation from ethanol, various attempts were made by several authors as follows. The performance of nanoparticles is highly dependent on their shape, morphology, structure, and composition.101 Different nanostructures and core–shells have been designed to reduce obstacles and possible challenges. Several groups have worked to redesign the nanostructures of a Pt catalyst and attempted to modify the morphology of it for better results.102–107 A novel technique explored for H2 production is the glow discharge plasma electrolysis of ethanol solution by Yan et al.108 Apart from H2, formaldehyde is simultaneously produced with low CO2 emission.
Li et al. studied the effects of adding MgO to C-supported Pd catalyst for the electrochemical oxidation of ethanol in alkaline medium.109 The outcomes suggested that the catalyst activity and poisoning resistance during ethanol electro-oxidation improved after MgO was added to the catalyst. Best performance was attained with a weight ratio of 2:1 of Pd to MgO for the catalyst. Onset potential and peak potential on the PdMgO/C electrode were lower than that on the Pd/C electrode due to a synergistic interaction between Pd and MgO. Addition of MgO to Pd/C negatively shifted the onset potential by a value of 80 mV. Furthermore, the peak current density was enhanced by 3.4 times as compared to that with the Pd/C electrode used for ethanol electro-oxidation.
7. Challenges and prospective
The benefits and limitations of several hydrogen producing technologies have have been summarized in Table 2. Presently, the cost of H2 production from biomass though direct gasification is almost thrice that of H2 production from natural gas by steam methane reforming.110 Steam methane reforming costs 1.5–3.7 US $ per kg (natural gas price assumed to be 7 US $ per GJ) for H2 production, while the cost of H2 production from biomass is 10–14 US $ per GJ.2 However, lignocellulosic biomass is advantageous owing to its abundant availability, relatively inexpensive nature, and high H2 yields at high pressures. Furthermore, it is reported that H2 from biomass has higher energy efficiency (56–64%) and heating values.111| Technology | Advantages | Disadvantages |
|---|---|---|
| Steam reforming | • Well established and extensively used industrial technology | • Highest air emissions |
| • Process temperatures are lower | • Coking of catalysts | |
| • Best desirable ratios of H2/CO are obtained | ||
| Alkaline-enhanced reforming | • Relatively lower temperature requirement | • Reactor corrosion by salt and precipitates of alkali catalysts |
| • Faster reaction kinetics by the use of alkali | ||
| • Mass fraction of H2 is higher due to lower CO2 emissions | ||
| Autothermal reforming | • Process temperatures lower than partial oxidation | • Less commercial experience |
| • Methane slip is lower | • O2 or air required | |
| • High H2 selectivity compared to steam reforming and partial oxidation | ||
| • Compactness of reactor set-up | ||
| • Less coke formation | ||
| • Potential for scale-up | ||
| Partial oxidation | • Less desulfurization required | • Lower ratio of H2/CO obtained |
| • Catalysts reduce temperature requirement | • Processing temperatures are very high | |
| • Methane slip is lower | • Process complexity due to soot formation/handling | |
| • Fast start-up and response time | ||
| Supercritical water gasification | • Fewer impurities in product stream | • High temperature and high pressure required |
| • High water content in ethanol feed can be used | • Chances of reactor corrosion due to salt and precipitates of homogeneous catalysts | |
| • Hydrogen yield at high pressure is obtained | ||
| Dehydrogenation | • Product stream has usable byproducts along with H2 | • Challenges in scaling-up the process |
| Photocatalysis | • Use of metal-free catalyst as alternatives | • High cost of catalysts |
| • Reduced costs for H2 generation | • Challenges in designing efficient photocatalytic cells | |
| • Can be performed at ambient temperature and pressure | ||
| Electrocatalysis | • Use of metal-free catalyst as alternative | • High cost of catalysts |
| • Improved H2 yield | • Additional cost of tailoring the electrodes | |
| • Ecological process |
The capital costs and operating expenditures for supercritical fluid systems running at elevated pressures are high. Field application of technologies for large scale generation of H2 using supercritical water gasification of biomass with evaluation of net positive energy and economic analysis is limited to commercial exploitation of steam methane reforming from natural gas. Additionally, high-temperature and high-pressure requirements in supercritical water gasification necessitate the reactor materials to be resistant to extreme conditions and corrosion. Certain catalysts, especially inorganic salts, lack proper dissolution at supercritical conditions and hence precipitate on the reactor walls causing corrosion. The precipitated salts also react with tars and chars leading to reactor plugging, which is another vital problem in biomass gasification.112,113
The use of a fuel cell for H2 generation from ethanol as a source of energy is more effective due to switching to an efficient, clean, and salient alternative. Total conversion efficiency of the energy generated from H2 is twice that obtained from conventionally used combustion engines.114 There are recent developments in generating a new fuel cell that can operate at comparatively low temperatures. These constant attempts can certainly open newer opportunities for using ethanol as a source of H2 at commercial levels.115–118
The major challenge faced during ethanol reforming is the formation of CO2 and CH4 as byproducts along with H2 generation. The high levels of CO in the gaseous product results in poisoning of the Pt anode used in Proton Exchange Membrane (PEM) fuel cells.18 Additionally, for supercritical water reforming, to produce H2 selectively, high temperatures result in CO production.22 A focus is also being laid towards combined low-temperature shift/high-temperature shift (LTS/HTS) reactions to make the best use of thermodynamics and kinetics of the water–gas shift reaction.
H2 produced from bio-based materials through steam reforming is also reported to aid in synthesizing gasoline components. Yin et al. proposed a new process based on aqueous-phase dehydration and hydrogenation to directly produce liquid alkanes (C7 to C9), which are predominant constituents of gasoline.119 In the steam gas phase, a portion of cellulose was converted to H2 by steam reforming, whereas in the liquid water phase, cellulose was converted to an alkane precursor such as 5-hydroxymethylfurfural. In the subsequent reaction step, the resulting H2 reacts with 5-hydroxymethylfurfural to produce liquid alkanes through aqueous-phase dehydration or hydrogenation.
CO2 reforming of ethanol is gaining attention because it utilizes ethanol as a renewable reactant and consumes CO2 to produce syngas with a H2/CO ratio of around 1.0, which is suitable for Fischer–Tropsch synthesis. Bahari et al. investigated the effects of lanthanum promoter on the catalytic attributes and performance of Ni/Al2O3 for dry reforming of ethanol at varying partial pressures of CO2 (20–50 kPa) and a constant partial pressure of CH4 (20 kPa).120 CH4 yields declined at higher partial pressures of CO2 while a reverse trend was noticed for H2 and CO yields. This suggested the CH4 dry reforming reaction as a secondary reaction, which converted CH4 formed from ethanol decomposition to H2 and CO. Higher lanthanum loading increased ethanol conversion due to high mobile oxygen storage capacity of La2O3. Lanthanum also prevented catalyst deactivation by oxidizing carbon deposition on the catalyst surface and averting NiO particles from aggregating on the catalyst support.
There are several advancements reported on H2 production from ethanol to reduce formation of unwanted products such as CO and improve energy efficiency. Hu et al. studied H2 production from ethanol by plasma reforming using a plasma discharge reactor.121 It was reported that an increase in flow rate of ethanol feed reduced reforming efficiency. Despite rigorous attempts the developed systems fail to match the yields and conversion rates of conventionally used processes. Thus, this field needs further exploration to foster its effectiveness when compared to conventional ethanol-to-hydrogen conversion techniques.
8. Conclusions
H2 is a potential energy source and can play a significant role to decrease greenhouse gas emissions into the environment. Ethanol is considered the most efficient and sustainable alternative for H2 generation compared to methane in the field of fuel cells owing to its transportable, renewable, and non-toxic nature. Co-feeding steam with ethanol in the presence of oxygen/air, and effective production of H2 from ethanol through steam reforming and oxidation reactions can be meritoriously achieved. Different pathways for H2 production from ethanol include reforming, dehydrogenation, supercritical water gasification, photocatalysis, and electrocatalysis.Steam reforming is a mature technology for H2 production from methanol and methane. The nature of catalysts, its synthesis, reaction conditions and byproduct formation can determine the conversion of ethanol to H2via reforming. Bioethanol, containing an excess amount of water, can be directly subjected to supercritical water gasification and steam reforming, thus eliminating one unit operation (i.e., distillation) that is otherwise required to recover pure ethanol. Therefore, compared to other H2-producing technologies, supercritical water gasification and steam reforming could be economically attractive. However, the literature very rarely reports reforming and supercritical water gasification of ethanol for H2 production. Therefore, more studies can help provide new data to better understand the underlying reaction mechanisms in these pathways.
Several nanocatalysts have also been developed for application in photocatalysis and electrocatalysis of ethanol for H2 production. To enhance the usability of photocatalysts and electrocatalysts as a potential method of H2 generation from ethanol, low-cost novel catalysts (as cathode composites) and anodes are also being designed. Besides, the properties of the electrode materials are tested to avoid corrosion and carbon deposition for effective H2 production. H2 has tremendous potential to serve as the “fuel of the future” and ethanol undoubtedly is one of the next-generation precursors for its sustainable synthesis.
Acknowledgements
The authors would like to thank Natural Sciences and Engineering Research Council of Canada (NSERC) and Canada Research Chair (CRC) for funding this bioenergy research.References
- S. Nanda, J. Mohammad, S. N. Reddy, J. A. Kozinski and A. K. Dalai, Biomass Convers. Biorefin., 2014, 4, 157–191 CrossRef CAS.
- H. Balat and E. Kırtay, Int. J. Hydrogen Energy, 2010, 35, 7416–7426 CrossRef CAS.
- Earth CO2 Homepage, https://www.co2.earth/, accessed March 2017.
- European Environment Agency (EEA), Atmospheric concentration of carbon dioxide, methane and nitrous oxide. Copenhagen, Denmark, http://www.eea.europa.eu, accessed March 2016.
- Hydrogen Analysis Resource Center, U.S. Department of Energy, http://www.hydrogen.pnl.gov/, accessed March 2017.
- Alternative Fuels Data Center, U.S. Department of Energy, http://www.afdc.energy.gov/fuels/fuel_properties.php, accessed March 2017.
- M. Endisch, T. Kuchling and J. Roscher, Energy Fuels, 2013, 27, 2628–2636 CrossRef CAS.
- M. Trépanier, A. Tavasoli, A. K. Dalai and N. Abatzoglou, Fuel Process. Technol., 2009, 90, 367–374 CrossRef.
- J. Chung, B. E. Rittmann, W. F. Wright and R. H. Bowman, Biodegradation, 2007, 18, 199–209 CrossRef CAS PubMed.
- R. Kothari, D. Buddhi and R. L. Sawhney, Renewable Sustainable Energy Rev., 2008, 12, 553–563 CrossRef CAS.
- B. C. R. Ewan and R. W. K. Allen, Int. J. Hydrogen Energy, 2005, 30, 809–819 CrossRef CAS.
- U.S. Energy Information Administration (USEIA), International Energy Outlook 2016, Report Number: DOE/EIA-0484(2016), https://www.eia.gov/outlooks/ieo/world.cfm, accessed May 2017.
- S. Nanda, R. Azargohar, A. K. Dalai and J. A. Kozinski, Renewable Sustainable Energy Rev., 2015, 50, 925–941 CrossRef CAS.
- S. Kim and B. E. Dale, Biomass Bioenergy, 2004, 26, 361–375 CrossRef.
- S. Nanda, A. K. Dalai and J. A. Kozinski, Energy Sci. Eng., 2014, 2, 138–148 CrossRef CAS.
- S. Nanda, D. Golemi-Kotra, J. C. McDermott, A. K. Dalai, I. Gökalp and J. A. Kozinski, New Biotechnol., 2017, 37, 210–221 CrossRef CAS PubMed.
- V. R. Surisetty, A. K. Dalai and J. Kozinski, Appl. Catal., A, 2011, 404, 1–11 CAS.
- A. J. Byrd, K. K. Pant and R. B. Gupta, Energy Fuels, 2007, 21, 3541–3547 CrossRef CAS.
- A. Haryanto, S. Fernando, N. Murali and S. Adhikari, Energy Fuels, 2005, 19, 2098–2106 CrossRef CAS.
- D. B. Levin and R. Chahine, Int. J. Hydrogen Energy, 2010, 35, 4962–4969 CrossRef CAS.
- J. D. Holladay, J. Hu, D. L. King and Y. Wang, Catal. Today, 2009, 139, 244–260 CrossRef CAS.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- B. Cifuentes, M. F. Valero, J. A. Conesa and M. Cobo, Catalysts, 2015, 5, 1872–1896 CrossRef CAS.
- H. Idriss, Platinum Met. Rev., 2004, 48, 105–115 CrossRef CAS.
- H. Chen, H. Yu, Y. Tang, M. Pan, G. Yang, F. Peng, H. Wang and J. Yang, J. Nat. Gas Chem., 2009, 18, 191–198 CrossRef CAS.
- Y. Casas-Ledón, L. E. Arteaga-Perez, M. C. Morales-Perez and L. M. Peralta-Suárez, ISRN Thermodyn., 2012, 1–8 CrossRef.
- D. Zanchet, J. B. O. Santos, S. Damyanova, J. M. R. Gallo and J. M. C. Bueno, ACS Catal., 2015, 5, 3841–3863 CrossRef CAS.
- B. Bej, S. Bepari, N. C. Pradhan and S. Neogi, Catal. Today, 2016 DOI:10.1016/j.cattod.2016.12.010.
- S. Jankhah, N. Abatzoglou and F. Gitzhofer, Int. J. Hydrogen Energy, 2008, 33, 4769–4779 CrossRef CAS.
- A. L. da Silva, C. de Fraga Malfatti and I. L. Müller, Int. J. Hydrogen Energy, 2009, 34, 4321–4330 CrossRef.
- W. Wang and Y. Wang, Int. J. Hydrogen Energy, 2009, 34, 5382–5389 CrossRef CAS.
- S. Liu, K. Zhang, L. Fang and Y. Li, Energy Fuels, 2008, 22, 1365–1370 CrossRef CAS.
- P. Muthukumaran and R. B. Gupta, Ind. Eng. Chem. Res., 2000, 39, 4555–4563 CrossRef CAS.
- J. Rostrup-Nielsen, in Encyclopedia of Catalysis, ed. I. T. Horváth, Wiley-Interscience, 2003 Search PubMed.
- L. V. Mattos and F. B. Noronha, J. Power Sources, 2005, 145, 10–15 CrossRef CAS.
- W. Wang, Z. Wang, Y. Ding, J. Xi and G. Lu, Catal. Lett., 2002, 81, 63–68 CrossRef CAS.
- J. R. Salge, G. A. Deluga and L. D. Schmidt, J. Catal., 2005, 235, 69–78 CrossRef CAS.
- R. Baruah, M. Dixit, P. Basarkar, D. Parikh and A. Bhargav, Renewable Sustainable Energy Rev., 2015, 51, 1345–1353 CrossRef CAS.
- D. J. Wilhelm, D. R. Simbeck, A. D. Karp and R. L. Dickenson, Fuel Process. Technol., 2001, 71, 139–148 CrossRef CAS.
- M. Ni, D. Y. C. Leung and M. K. H. Leung, Int. J. Hydrogen Energy, 2007, 32, 3238–3247 CrossRef CAS.
- G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Science, 2004, 303, 993–997 CrossRef CAS PubMed.
- W. Cai, F. Wang, A. C. Van Veen, H. Provendier, C. Mirodatos and W. Shen, Catal. Today, 2008, 138, 152–156 CrossRef CAS.
- Y. Guo, S. Z. Wang, D. H. Xu, Y. M. Gong, H. H. Ma and X. Y. Tang, Renewable Sustainable Energy Rev., 2010, 14, 334–343 CrossRef CAS.
- M. Watanabe, T. Sato, H. Inomata, R. L. Smith Jr, K. Arai, A. Kruse and E. Dinjus, Chem. Rev., 2004, 104, 5803–5821 CrossRef CAS PubMed.
- A. Kruse and E. Dinjus, J. Supercrit. Fluids, 2007, 39, 362–380 CrossRef CAS.
- J. G. Van Bennekom, R. H. Venderbosch, D. Assink and H. J. Heeres, J. Supercrit. Fluids, 2011, 58, 99–113 CrossRef CAS.
- S. N. Reddy, S. Nanda and J. A. Kozinski, Chem. Eng. Res. Des., 2016, 113, 17–27 CrossRef CAS.
- B. Kuhlmann, E. M. Arnett and M. Siskin, J. Org. Chem., 1994, 59, 5377–5380 CrossRef CAS.
- A. Kruse, Biofuels, Bioprod. Biorefin., 2008, 2, 415–437 CrossRef CAS.
- Ž. Knez, E. Markočič, M. K. Hrnčič, M. Ravber and M. Škerget, J. Supercrit. Fluids, 2015, 96, 46–52 CrossRef.
- G. J. DiLeo, M. E. Neff and P. E. Savage, Energy Fuels, 2007, 21, 2340–2345 CrossRef CAS.
- Z. Fang, T. Sato, R. L. Smith Jr, H. Inomata, K. Arai and J. A. Kozinski, Bioresour. Technol., 2008, 99, 3424–3430 CrossRef CAS PubMed.
- Z. Fang, T. Minowa, R. L. Smith Jr, T. Ogi and J. A. Kozinski, Ind. Eng. Chem. Res., 2004, 43, 2454–2463 CrossRef CAS.
- D. Xu, S. Wang, X. Hu, C. Chen, Q. Zhang and Y. Gong, Int. J. Hydrogen Energy, 2009, 34, 5357–5364 CrossRef CAS.
- F. A. P. Voll, C. C. R. S. Rossi, C. Silva, R. Guirardello, R. O. M. A. Souza, V. F. Cabral and L. Cardozo-Filho, Int. J. Hydrogen Energy, 2009, 34, 9737–9744 CrossRef CAS.
- S. Nanda, S. N. Reddy, H. N. Hunter, A. K. Dalai and J. A. Kozinski, J. Supercrit. Fluids, 2015, 104, 112–121 CrossRef CAS.
- S. Nanda, S. N. Reddy, H. N. Hunter, I. S. Butler and J. A. Kozinski, Ind. Eng. Chem. Res., 2015, 54, 9296–9306 CrossRef CAS.
- M. Gong, S. Nanda, M. J. Romero, W. Zhu and J. A. Kozinski, J. Supercrit. Fluids, 2017, 119, 130–138 CrossRef CAS.
- M. Gong, S. Nanda, H. N. Hunter, W. Zhu, A. K. Dalai and J. A. Kozinski, Catal. Today, 2017 DOI:10.1016/j.cattod.2017.02.017.
- R. F. Susanti, L. W. Dianningrum, T. Yum, Y. Kim, B. G. Lee and J. Kim, Int. J. Hydrogen Energy, 2012, 37, 11677–11690 CrossRef CAS.
- J. W. Snoeck, G. F. Froment and M. Fowles, Ind. Eng. Chem. Res., 2002, 41, 4252–4265 CrossRef CAS.
- S. N. Reddy, S. Nanda, A. K. Dalai and J. A. Kozinski, Int. J. Hydrogen Energy, 2014, 39, 6912–6926 CrossRef CAS.
- S. Freni, J. Power Sources, 2001, 94, 14–19 CrossRef CAS.
- T. Tsuchida, J. Kubo, T. Yoshioka, S. Sakuma, T. Takeguchi and W. Ueda, J. Catal., 2008a, 259, 183–189 Search PubMed.
- T. Tsuchida, T. Yoshioka, S. Sakuma, T. Takeguchi and W. Ueda, Ind. Eng. Chem. Res., 2008b, 47, 1443–1452 Search PubMed.
- S. W. Colley, J. Tabatabaei, K. C. Waugh and M. A. Wood, J. Catal., 2005, 236, 21–33 CrossRef CAS.
- K. Inui, T. Kurabayashi and S. Sato, J. Catal., 2002, 215, 207–215 CrossRef.
- K. Inui, T. Kurabayashi and S. Sato, Appl. Catal., A, 2002, 237, 53–61 CrossRef CAS.
- K. Inui, T. Kurabayashi, S. Sato and N. Ichikawa, J. Mol. Catal., 2004, 216, 147–156 CrossRef CAS.
- A. G. Sato, D. P. Volanti, I. C. de Freitas, E. Longo, J. Maria and C. Bueno, Catal. Commun., 2012, 26, 122–126 CrossRef CAS.
- Y. Chen and K. Tu, Int. J. Photoenergy, 2012, 980595, DOI:10.1155/2012/980595.
- F. Galindo-hernández, J. Wang, L. Chen, X. Bokhimi, R. Gómez, A. Pérez-larios and N. Nava, J. Hazard. Mater., 2013, 263, 11–19 CrossRef PubMed.
- W. Lin and H. Chang, Catal. Today, 2004, 97, 181–188 CrossRef CAS.
- A. Ashok, A. Kumar, R. Bhosale, M. Ali and S. Saad, Int. J. Hydrogen Energy, 2017 DOI:10.1016/j.ijhydene.2017.01.175.
- L. Dolgykh, I. Stolyarchuk, I. Deynega and P. Strizhak, Int. J. Hydrogen Energy, 2006, 31, 1607–1610 CrossRef CAS.
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS PubMed.
- D. Liguras, D. Kondaridas and X. E. Verykios, Appl. Catal., B, 2003, 43, 345–354 CrossRef CAS.
- F. Mariño, E. Gerrella, S. Duhalde, M. Jobbagy and M. A. Laborde, Int. J. Hydrogen Energy, 1998, 23, 1095–1101 CrossRef.
- M. Antoniadou, P. Bouras, N. Strataki and P. Lianos, Int. J. Hydrogen Energy, 2008, 33, 5045–5051 CrossRef CAS.
- J. E. Yoo, K. Lee, M. Altomare, E. Selli and P. Schmuki, Angew. Chem., 2013, 52, 7514–7517 CrossRef CAS PubMed.
- A. Castedo, E. Mendoza, I. Angurell and J. Llorca, Catal. Today, 2016, 273, 106–111 CrossRef CAS.
- V. S. Souza, J. D. Scholten, D. E. Weibel, D. Eberhardt, D. L. Baptista and J. Dupont, J. Mater. Chem. A, 2016, 4, 7469–7475 CAS.
- R. P. Antony, T. Mathews, C. Ramesh, N. Murugesan, A. Dasgupta, S. Dhara, S. Dash and A. K. Tyagi, Int. J. Hydrogen Energy, 2012, 37, 8268–8276 CrossRef CAS.
- A. Gasparotto, D. Barreca, D. Bekermann, A. Devi, R. A. Fischer, P. Fornasiero, V. Gombac, O. I. Lebedev, C. Maccato, T. Montini, G. Van Tendeloo and E. Tondello, J. Am. Chem. Soc., 2011, 133, 19362–19365 CrossRef CAS PubMed.
- S. N. Tijare, S. Bakardjieva, J. Subrt, M. V. Joshi, S. S. Rayalu, S. Hishita and N. Labhsetwar, J. Chem. Sci., 2014, 126, 517–525 CrossRef CAS.
- P. Lianos, Appl. Catal., B, 2017, 210, 235–254 CrossRef CAS.
- S. Sfaelou, L. C. Pop, O. Monfort, V. Dracopoulos and P. Lianos, Int. J. Hydrogen Energy, 2016, 41, 5902–5907 CrossRef CAS.
- E. Kalamaras, V. Dracopoulos, L. Sygellou and P. Lianos, Chem. Eng. J., 2016, 295, 288–294 CrossRef CAS.
- C. Lamy, T. Jaubert, S. Baranton and C. Coutanceau, J. Power Sources, 2014, 245, 927–936 CrossRef CAS.
- D. Geng, Y. Hu, Y. Li, R. Li and X. Sun, Electrochem. Commun., 2012, 22, 65–68 CrossRef CAS.
- M. H. Seo, S. M. Choi, H. J. Kim and W. B. Kim, Electrochem. Commun., 2011, 13, 182–185 CrossRef CAS.
- K. Tiido, N. Alexeyeva, M. Couillard, C. Bock, B. R. Macdougall and K. Tammeveski, Electrochim. Acta, 2013, 107, 509–517 CrossRef CAS.
- B. Xiong, Y. Zhou, Y. Zhao, J. Wang, X. Chen, R. O. Hayre and Z. Shao, Carbon, 2012, 52, 181–192 CrossRef.
- X. Xu, Y. Zhou, T. Yuan and Y. Li, Electrochim. Acta, 2013, 112, 587–595 CrossRef CAS.
- D. Yu, L. Wei, W. Jiang, H. Wang, B. Sun, Q. Zhang, K. Goh, R. Si and Y. Chen, Nanoscale, 2013, 5, 3457–3464 RSC.
- Y. Zhao, Y. Zhou, R. O. Hayre and Z. Shao, J. Phys. Chem. Solids, 2013, 74, 1608–1614 CrossRef CAS.
- A. Navaee, A. Salimi, S. Soltanian and P. Servati, J. Power Sources, 2015, 277, 268–276 CrossRef CAS.
- A. Navaee and A. Salimi, Electrochim. Acta, 2016, 211, 322–330 CrossRef CAS.
- G. Dong, M. Fang, H. Wang, S. Yip, H. Cheung, F. Wang, C. Wong, S. T. Chu and J. C. Ho, J. Mater. Chem. A, 2015, 3, 13080–13086 CAS.
- C. Zhang, Y. Hong, R. Dai, X. Lin, L. Long, C. Wang and W. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 11648–11653 CAS.
- T. L. Tan, L. Wang, J. Zhang, D. D. Johnson and K. Bai, ACS Catal., 2015, 5, 2376–2383 CrossRef CAS.
- A. Arabzadeh, A. Salimi and M. Ashrafi, Catal. Sci. Technol., 2016, 6, 3485–3496 CAS.
- L. Ding, G. Li, Z. Wang, Z. Liu and H. Liu, Chem.–Eur. J., 2012, 18, 8386–8391 CrossRef CAS PubMed.
- X. Liu, Y. Zhang, M. Gong, Y. Tang and T. Lu, J. Mater. Chem. A, 2014, 2, 13840–13844 CAS.
- X. Liu, G. Xu, Y. Chen, T. Lu, Y. Tang and W. Xing, Sci. Rep., 2015, 5, 6–11 Search PubMed.
- Y. Li, Z. W. Wang, C. Y. Chiu, L. Ruan, W. Yang, Y. Yang, R. E. Palmer and Y. Huang, Nanoscale, 2012, 4, 845–851 RSC.
- Y. Yang, B. Yang, J. Peng, Z. Zhao and Y. Zhao, RSC Adv., 2015, 5, 20981–20986 RSC.
- Z. Yan, L. Chen and H. Wang, J. Phys. D: Appl. Phys., 2008, 41, 155205–155212 CrossRef.
- N. Li, Y. X. Zeng, S. Chen, C. W. Xu and P. K. Shen, Int. J. Hydrogen Energy, 2014, 39, 16015–16019 CrossRef CAS.
- P. L. Spath, M. K. Mann and W. A. Amos, Update of Hydrogen from Biomass: Determination of the Delivered Cost of Hydrogen. NREL Milestone Report, NREL/MP-510–33112, Colorado, 2003 Search PubMed.
- C. N. Hamelinck and A. P. C. Faaij, J. Power Sources, 2002, 111, 1–22 CrossRef CAS.
- L. J. Guo, Y. J. Lu, X. M. Zhang, C. M. Ji, Y. Guan and A. X. Pei, Catal. Today, 2007, 129, 275–286 CrossRef CAS.
- M. D. Bermejo and M. J. Cocero, AIChE J., 2006, 52, 3933–3951 CrossRef CAS.
- X. G. Li, Principles of Fuel Cells, Taylor & Francis, New York, 2006 Search PubMed.
- W. J. Zhou, S. Q. Song, W. Z. Li, Z. H. Zhou, G. Q. Sun, Q. Xin, S. Douvartzides and P. Tsiakaras, J. Power Sources, 2005, 140, 50–58 CrossRef CAS.
- S. Song, W. Zhou, J. Tian, R. Cai, G. Sun, Q. Xin, S. Kontouc and P. Tsiakaras, J. Power Sources, 2005, 145, 266–271 CrossRef CAS.
- S. Song and P. Tsiakaras, Appl. Catal., B, 2006, 63, 187–193 CrossRef CAS.
- F. Colmati, E. Antolini and E. R. Gonzalez, J. Power Sources, 2006, 157, 98–103 CrossRef CAS.
- S. Yin, A. K. Mehrotra and Z. Tan, Biomass Bioenergy, 2012, 47, 228–239 CrossRef CAS.
- M. B. Bahari, B. C. Goo, T. L. M. Pham, T. J. Siang, H. T. Danh, N. Ainirazali and D.-V. N. Vo, Procedia Eng., 2016, 148, 654–661 CrossRef CAS.
- Y. P. Hu, G. Li, Y. Yang, X. Gao and Z. Lu, Int. J. Hydrogen Energy, 2012, 37, 1044–1047 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |