DOI:
10.1039/C6QI00346J
(Research Article)
Inorg. Chem. Front., 2017,
4, 123-130
Syntheses, structures, and properties of sulfides constructed by SbS4 teeter-totter polyhedra: Ba3La4Ga2Sb2S15 and BaLa3GaSb2S10†‡
Received
30th August 2016
, Accepted 12th November 2016
First published on 15th November 2016
Abstract
Two new pentanary sulfides Ba3La4Ga2Sb2S15 (1) and BaLa3GaSb2S10 (2) have been discovered by high temperature solid state reactions. Ba3La4Ga2Sb2S15 features a new zero-dimensional structure type, which consists of unique isolated Sb2S7 dimers and GaS4 tetrahedra. In contrast, with less GaS4 tetrahedra in the formula, BaLa3GaSb2S10 forms a one-dimensional structure which is constructed by teeter-totter (SbS4)n chains and isolated GaS4 tetrahedra. On the basis of UV/Vis spectroscopy, their band gaps are determined to be 2.42 and 2.15 eV, respectively, which are much wider than that of analogue La4FeSb2S10 (1.0 eV). In addition, theoretical studies illustrate the important role of Ga in expanding the band gaps with respect to Fe atoms. The calculated crystal orbital Hamilton population and electron localization function confirm that the long Sb–S bonds in Ba3La4Ga2Sb2S15 (2.932(2) Å) and BaLa3GaSb2S10 (3.216(5) and 3.219(5) Å) have weak but nonnegligible bonding interactions.
Introduction
Over the past few decades, Sb-containing chalcogenides have attracted lots of attention because of their diverse structures and various physical properties.1–24 In terms of their crystallographic structures, the common building units of chalcogenides are MX4 tetrahedra and MX6 octahedra (M = cation of group 13 or 14 elements, d10, or d0etc.; X = chalcogen).25–29 In particular, a Sb3+ cation has a lone electron pair configuration of 5s25p0. With stereochemically active lone-pair electrons, the Sb3+ cation exhibits flexible structural chemistry with the coordination number ranging from 3 to 6 to form diverse polyhedra. SbIIIXn (n = 3–6) polyhedra can connect with second building units, which further evolve into one-dimensional (1D) chains, two-dimensional (2D) layers or three-dimensional (3D) frameworks via sharing corners, edges or faces.30 Among these SbIIIXn (n = 3–6) polyhedra, the SbIIIX4 teeter-totter polyhedron (Fig. S1‡), which can be considered as a hemioctahedron and differs from the conventional tetrahedron MX4, has aroused our interest.31 More importantly, the SbIIIS4 teeter-totter polyhedron has not yet been seen as an isolated polyhedron, but can be seen as a component of some structures, including (1) isolated finite complex anions, such as in Ba4LaGe3SbSe13,32 Ba4Sb3S8Cl,33 and Ba8Sb6S17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 34 (see Fig. S2‡); (2) infinite chains, such as in Pr4GaSbS9,5 La4InSbS9,6 La4FeSb2S10,8 Ba4SiSb2Se11,13 Na9Gd5Sb8S26,35 SrGeSb2Se8,36 and BaSb2S437 (see Fig. S3‡); and (3) layers, such as in La2Ga0.33SbS5,15 RbU2SbS8,38 and KU2SbSe838 (see Fig. S4‡). Inspired by the above phenomenon, we chose the SbIIIS4 teeter-totter polyhedron as a potential unit to build chalcogenides with novel structures and properties. In addition, with the result that La4FeSb2S108 exhibits a very narrow band gap of 1.00 eV, replacing Fe atoms with Ga atoms may be an effective way to enlarge band gaps because the latter lacks low energy d–d transitions. Meanwhile, with a relatively high positive charge, alkaline-earth metal cations such as Ba2+ can increase the ionic contributions of the lattice energy, which helps to enrich the structure and physical properties of compounds.39
34 (see Fig. S2‡); (2) infinite chains, such as in Pr4GaSbS9,5 La4InSbS9,6 La4FeSb2S10,8 Ba4SiSb2Se11,13 Na9Gd5Sb8S26,35 SrGeSb2Se8,36 and BaSb2S437 (see Fig. S3‡); and (3) layers, such as in La2Ga0.33SbS5,15 RbU2SbS8,38 and KU2SbSe838 (see Fig. S4‡). Inspired by the above phenomenon, we chose the SbIIIS4 teeter-totter polyhedron as a potential unit to build chalcogenides with novel structures and properties. In addition, with the result that La4FeSb2S108 exhibits a very narrow band gap of 1.00 eV, replacing Fe atoms with Ga atoms may be an effective way to enlarge band gaps because the latter lacks low energy d–d transitions. Meanwhile, with a relatively high positive charge, alkaline-earth metal cations such as Ba2+ can increase the ionic contributions of the lattice energy, which helps to enrich the structure and physical properties of compounds.39
We thoroughly investigated the pentanary Ba/RE/Ga/Sb/S (RE = Rare-earth metal) system where GaS4 tetrahedra and the unique teeter-totter polyhedron SbS4 are used as building units. Herein, two new pentanary chalcogenides Ba3La4Ga2Sb2S15 (1) and BaLa3GaSb2S10 (2) were discovered and characterized. The isolated teeter-totter dimer Sb2S7 is found for the first time in compound 1. Whereas, compound 2 features a branched chain of (SbS4)n which is constructed by the teeter-totter SbS4 polyhedra. In addition, structural relationships, band gaps and electronic structures are discussed.
Experimental
Synthesis
All related elements were stored in an Ar-filled glovebox (oxygen and moisture levels are all below 0.1 ppm), and all manipulations were carried out in a glovebox. La (with purities of 99.5%), S (99.999%) and CsBr (99.9%) were purchased from Aladdin China (Shanghai) Co. Ltd, Ga shot and Sb (99.999%) were purchased from Sinopharm Chemical Reagent Co. Ltd and Ba rod (99+%) and BaCl2 (99.5%) were purchased from Alfa Aesar China (Tianjin) Co. Ltd. For compound 1, a total 0.5 g of Ba, La, Ga, Sb and S in a molar ratio of 3:4:2:2:15 were mixed in a graphite crucible in a glovebox. For 2, a total 0.5 g of Ba, La, Ga, Sb and S in a molar ratio of 1:3:1:2:10 were mixed with 0.2 g of BaCl2 and 0.4 g of CsBr in a graphite crucible. The graphite crucibles were sealed in evacuated fused-silicon tubes under 10−3 Pa atmosphere, then the assemblies were heated to 1253 K for 72 h, and then held for 120 h at this temperature, before being subsequently cooled to 573 K during 120 h before the furnace was turned off. The yellowish-brown and brown crystals were collected after being washed with distilled water. Both crystals are stable in air at room temperature for at least two years.
Single-crystal X-ray crystallography
All crystal data collections were performed at 298 K on a Rigaku Saturn724 diffractometer which is equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). The data were corrected for Lorentz and polarization factors. Absorption corrections were operated by the multiscan method.40 Both of the structures were solved by direct methods and refined by the full-matrix least-squares fitting on F2 by SHELX-97.41 Both of the structures were verified using the ADDSYM algorithm with the program PLATON.42 The assignments of all atoms were determined on the basis of the interatomic distances and relative displacement parameters. These atoms were refined with anisotropic thermal parameters and a secondary extinction correction. No fractional occupancy was observed. Crystallographic data and structural refinement details are drawn in Table 1, the positional coordinates and isotropic equivalent thermal parameters are drawn in Table S1‡ and some related bond lengths (Å) are given in Table S2.‡
Table 1 Crystal data and structure refinements for Ba3La4Ga2Sb2S15 (1) and BaLa3GaSb2S10 (2)a
| Formula |
1
|
2
|
|
R
1 = ∑‖Fo| − |Fc‖/∑|Fo|, wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2.
|
|
F
w
|
1831.49 |
1187.9 |
| Crystal system |
Orthorhombic |
Monoclinic |
| Crystal color |
Yellowish-brown |
Brown |
| Space group |
lbam (no. 72) |
P21/m (no. 11) |
|
a (Å) |
23.628(2) |
7.748(4) |
|
b (Å) |
7.8863(5) |
13.507(7) |
|
c (Å) |
13.5905(4) |
15.410(8) |
|
α (°) |
90 |
90 |
|
β (°) |
90 |
90.09 |
|
γ (°) |
90 |
90 |
|
V (Å3) |
2532.5(3) |
1612.7(15) |
|
Z
|
4 |
4 |
|
D
c (g cm−3) |
4.804 |
4.893 |
|
μ (mm−1) |
16.580 |
16.373 |
| GOOF on F2 |
1.042 |
1.007 |
|
R
1, wR2 (I > 2σ(I))a |
0.0351, 0.0944 |
0.0405, 0.1105 |
|
R
1, wR2 (all data) |
0.0412, 0.1351 |
0.0557, 0.1393 |
| Largest diff peak and hole, e Å−3 |
3.558 and −4.595 |
3.174 and −3.877 |
Powder X-ray diffraction
The powder XRD patterns were collected on a Rigaku MiniFlex II diffractometer using Cu Kα radiation at 298 K. Data in the range 2θ = 10–70° were collected with scan steps of 0.02°. The measured and simulated XRD patterns which are calculated from the single crystal data are shown in Fig. S5.‡
Elemental analysis
The elemental analyses have been examined on handpicked single crystals of 1 and 2 with the aid of a field emission scanning electron microscope (FESEM, JSM6700F) equipped with an energy dispersive X-ray spectroscope (EDX, Oxford INCA). The results showed the presence of Ba, La, Ga, Sb and S, and the ratios of elements in compound 1 and 2 are close to the structure determination.
UV/Vis diffuse reflectance spectroscopy
The optical diffuse reflectance spectra of powdered samples were obtained at 298 K using a Perkin-Elmer Lambda 950 UV-Vis spectrophotometer which was equipped with an integrating sphere attachment and BaSO4 as a reference in the range of 0.25–2.5 μm. The absorption spectra were obtained from the reflection spectra according to the Kubelka–Munk function: α/S = (1 − R)2/2R, in which α is the absorption coefficient, S is the scattering coefficient, and R is the reflectance.43
Theoretical calculations
Band structures and the density of states (DOS) were calculated by the Vienna ab initio simulation package VASP.44 The generalized gradient approximation (GGA)45 was chosen as the exchange–correlation functional and a plane wave basis with the projector augmented wave (PAW) potentials was used.46 The plane-wave cutoff energy of 500 eV, and the threshold of 10−5 eV were set for the self-consistent-field convergence of the total electronic energy. Pseudo atomic calculations were performed for Ba, 5s25p66s2; La, 4p65d16s2; Ga, 4s24p1; Sb, 5s25p3 and S, 3s23p4. The k integration over the Brillouin zone was performed by the tetrahedron method47 using a 3 × 3 × 3 Monkhorst–Pack mesh for compound 1 and 5 × 5 × 16 Monkhorst–Pack mesh for 2. In addition, to analyze the bond strengths of Sb–S, the integrated crystal orbital Hamilton population (COHP) was calculated.47,48
Results and discussion
Crystal structure
Ba3La4Ga2Sb2S15 (1).
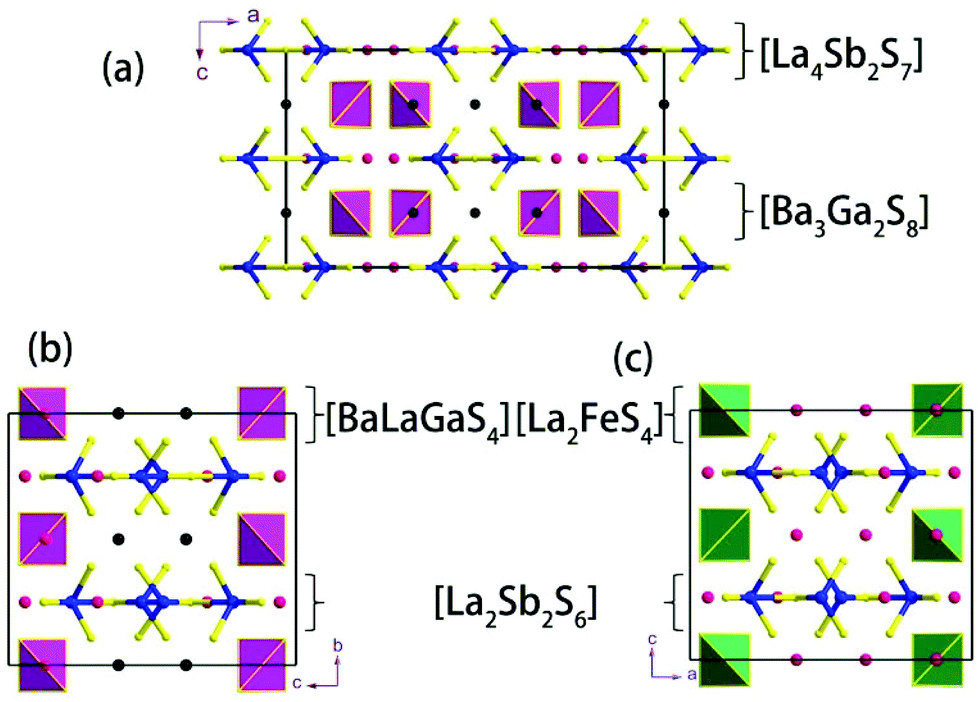
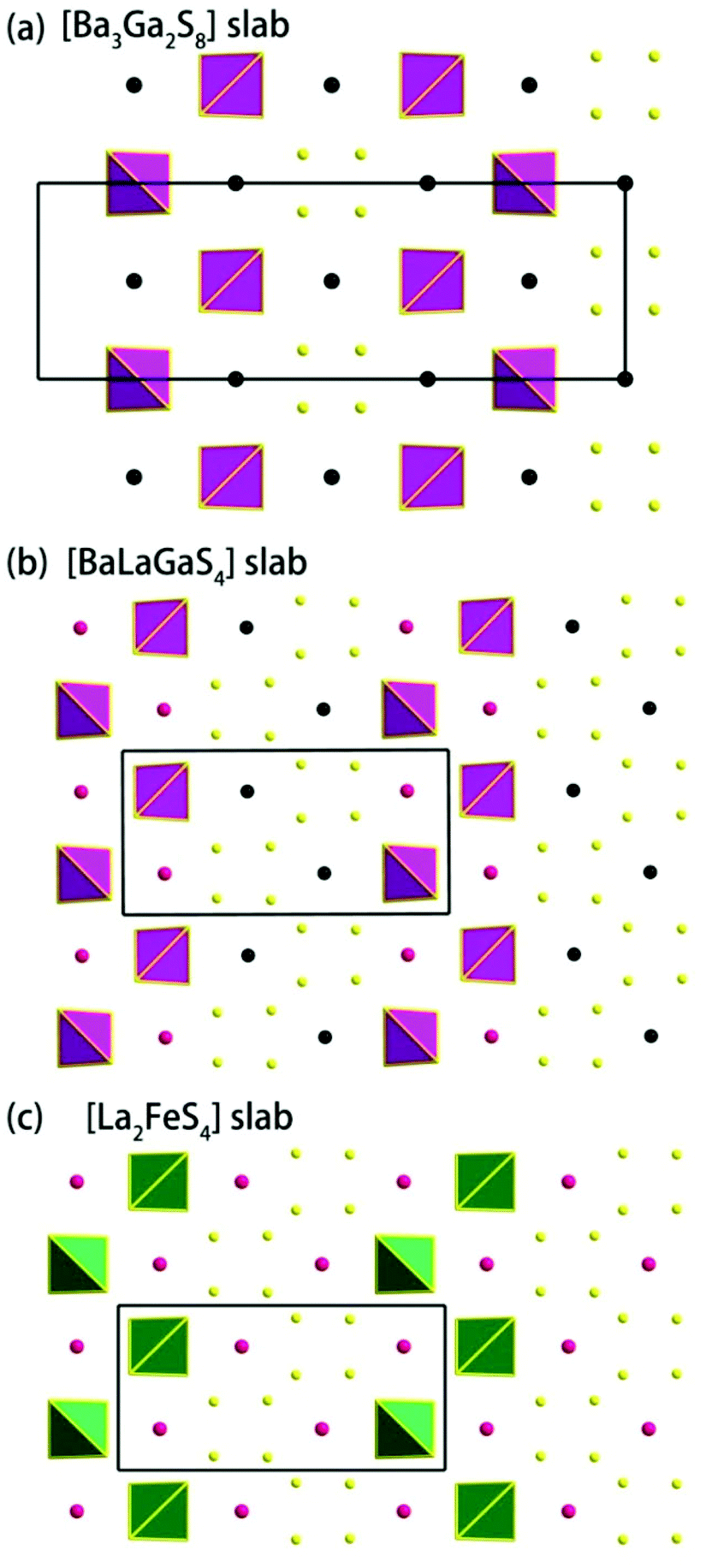
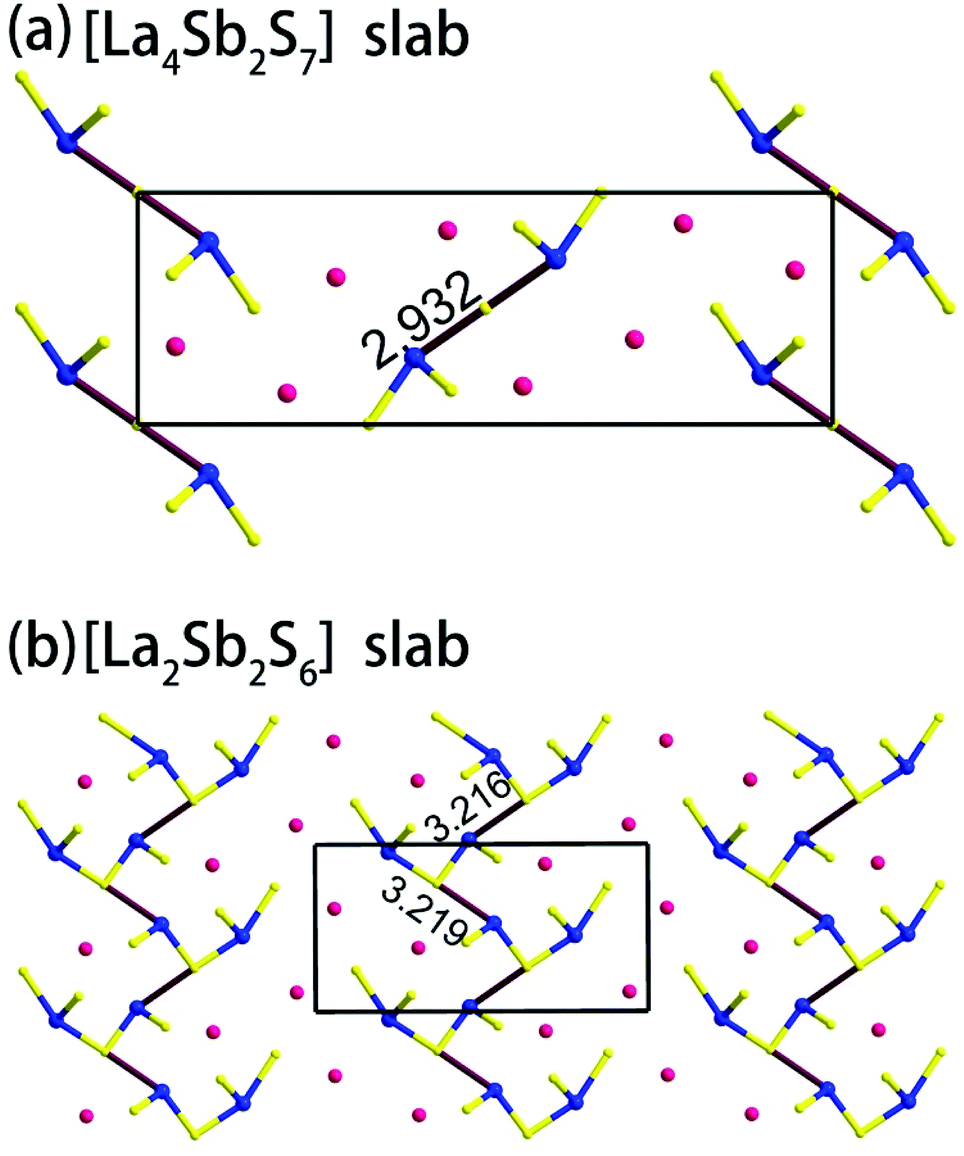
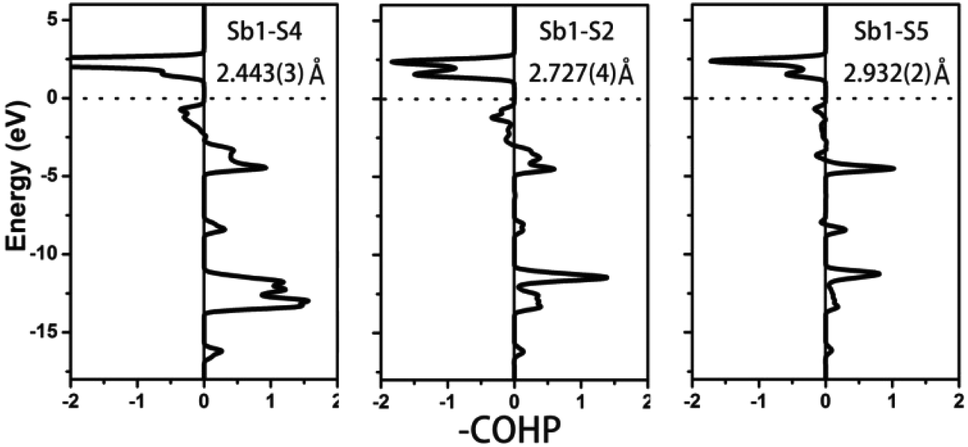
Compound 1 features a new structure type in which the isolated dimer of the teeter-totter Sb2S7 polyhedron is stabilized for the first time. The compound crystallizes in the orthorhombic space group Ibam (no. 72) with a = 23.628(2), b = 7.8863(5), c = 13.5905(4) Å and Z = 4. As shown in Fig. 1a, compound 1 contains isolated Sb2S7 dimers that are well separated by Ba2+ and La3+ cations and isolated GaS4 tetrahedra. The structure can be viewed as a stacking of slabs of Ba–Ga–S ([Ba3Ga2S8] slab, Fig. 2a) and La–Sb–S ([La4Sb2S7] slab, Fig. 3a) along the c direction. More specifically, the Ba–Ga–S slab includes two crystallographically different Ba atoms (4a, 8f), one Ga atom (8f), and two S atoms (16k), and is named as the [Ba3Ga2S8] slab. The slab consists of isolated GaS4 tetrahedra and Ba2+ cations (Fig. 2a). According to Fig. S6,‡ the two crystallographically different Ba ions in compound 1 are all surrounded by ten S atoms with Ba–S lengths of 3.182(3)–3.424(3) Å, which are common in sulfides, such as Ba23Ga8Sb2S38 (2.896(2)–3.331(2) Å),7 Ba3(BS3)1.5(SbS3)0.5 (3.097(2)–3.512(2) Å)10 and BaAgSbS3 (3.165(3)–3.369(3) Å).19 Based on Fig. 2a, two-thirds of the tetrahedral sites are occupied by GaS4 with the bonds of Ga–S ranging from 2.262(3) to 2.449(3) Å, which are comparable to that in Pr4GaSbS9 (2.272(2)–2.312(2) Å),5 Ba23Ga8Sb2S38 (2.217(2)–2.318(2) Å)7 and CsCd4Ga5S12 (2.351(4)–2.416(4) Å).25 On the other hand, Fig. 3a shows that the La–Sb–S slab includes two crystallographically different La (8j), one Sb (8j), and three S atoms (8j, 16k and 4c sites). This gives an atomic ratio of 16 La:8 Sb:28 S = 4 La4Sb2S7. The [La4Sb2S7] slab contains isolated Sb2S7 dimers and La3+ cations. The La ion is coordinated by eight S atoms to form a bicapped trigonal prism with the La–S bond lengths between 2.869(3) and 3.054(3) Å (see Fig. S6‡), which are in the normal range, for example as shown in La4InSbS9 (2.838(2)–3.040(2) Å),6 La2Ga0.33SbS5 (2.893(5)–3.3531(4) Å)15 and La7Sb9S24 (2.787 (3)–3.747(2) Å).23 In addition, as shown in Fig. 3a, the Sb atoms are coordinated by four S atoms with two short Sb–S bonds of 2.443(3) Å and two long Sb–S bonds in the range of 2.727(4)-2.932(2) Å to form the teeter-totter SbS4 polyhedra. The uncommon isolated dimer Sb2S7 is then formed by two SbS4 units via corner sharing with the long Sb–S bonds of 2.932(2) Å, which is comparable with the long Sb–S bonds of teeter-totter SbS4 polyhedra in Nd4InSbS9 (2.924(3) Å),6 La4FeSb2S10 (2.917(2) Å)8 and Ba4Sb3S8Cl (3.062(2) Å).33 Moreover, the significant bonding interaction of the long Sb–S bond (2.932(2) Å) is confirmed by the ICOHP value of −1.068 eV per bond (see Table 2, Fig. 6), which illustrates that the local geometries around Sb atoms are indeed 4-fold coordinated teeter-totter polyhedra as revealed by crystallographic analysis. Although they have similar bond lengths and angles, the Sb2S7 dimers in compound 1 are very different from the Sb2S6 dimers. The common Sb2S6 dimers are constructed by two teeter-totter SbS4 polyhedra via edge-sharing, then linked into one-dimensional (1D) chains via different second units, e.g. Ga2S6, In2S6 and SbS3 in Pr4GaSbS9,5 Nd4InSbS9,6 and Ba4Sb3S8Cl33 (see Fig. S2‡). Notably, the isolated dimer Sb2S7 in 1 is observed for the first time.
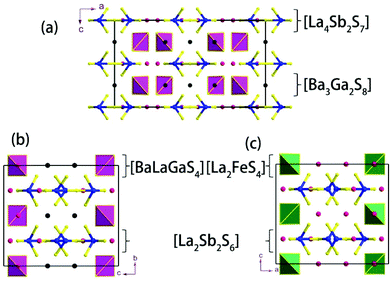 |
| | Fig. 1 View of the unit cell structures of (a) compound 1, (b) compound 2 and (c) La4FeSb2S10.8 Legend: black, Ba; red, La; blue, Sb; yellow, S; pink tetrahedron, GaS4; green tetrahedron, FeS4. | |
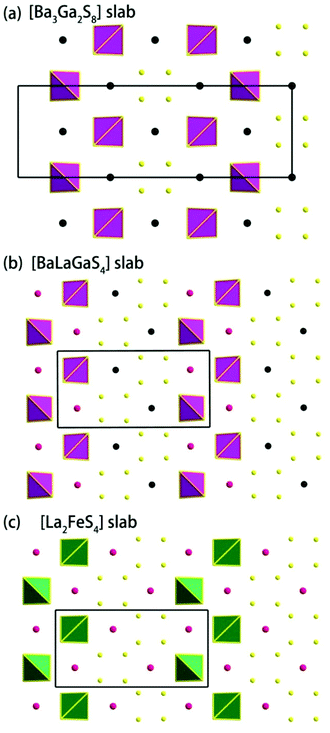 |
| | Fig. 2 (a) The [Ba3Ga2S8] slab in compound 1, (b) [BaLaGaS4] slabs in compound 2, and (c) [La2FeS4] slabs in compound La4FeSb2S10.8 | |
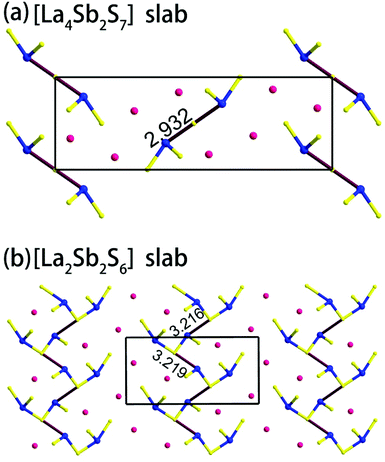 |
| | Fig. 3 (a) View of the [La4Sb2S7] slab in compound 1, and (b) the [La2Sb2S6] slab in 2. The Sb–S bond lengths are in Å. | |
Table 2 The interatomic distances (Å) and –ICOHP (eV per bond) values of Sb–S in compound 1 and 2
| Bond |
Interatomic distance |
–ICOHP |
|
1
|
| Sb1–S4 × 2 |
2.443(3) |
3.470 |
| Sb1–S2 |
2.727(4) |
1.700 |
| Sb1–S5 |
2.932(2) |
1.068 |
|
2
|
| Sb1–S3 × 2 |
2.454(3) |
3.478 |
| Sb1–S12 |
2.681(5) |
1.795 |
| Sb1–S4 |
2.747(4) |
1.357 |
| Sb2–S10 × 2 |
2.439(3) |
3.362 |
| Sb2–S11 |
2.539(4) |
2.506 |
| Sb2–S12 |
3.219(5) |
0.4035 |
| Sb3–S9 × 2 |
2.435(3) |
3.378 |
| Sb3–S12 |
2.530(4) |
2.515 |
| Sb3–S11 |
3.216(5) |
0.4064 |
| Sb4–S1 × 2 |
2.447(3) |
3.505 |
| Sb4–S11 |
2.675(5) |
1.800 |
| Sb4–S2 |
2.733(4) |
1.378 |
BaLa3GaSb2S10 (2).
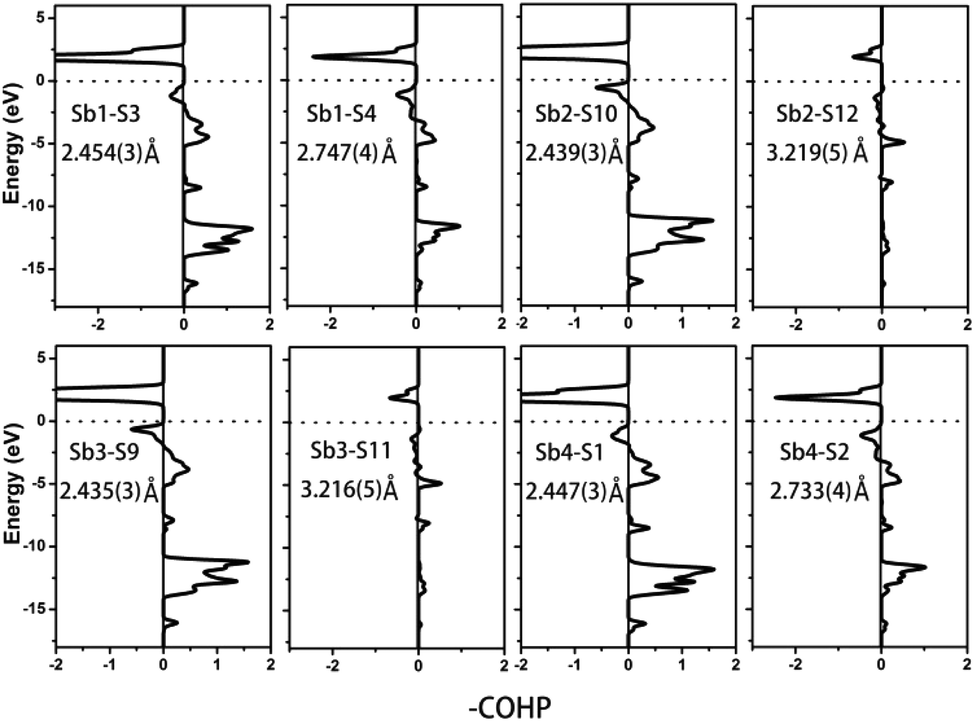
Compound 2 crystallizes in the monoclinic space group P21/m (NO. 11) with a = 7.748(4), b = 13.507(7), c = 15.410(8) Å, β = 90.05° and Z = 4. As show in Fig. 1b, this compound features a 1D structure in which the Ba–La–Ga–S and La–Sb–S slabs are propagating along the b direction. This is similar to La4FeSb2S10 which consists of La–Fe–S and La–Sb–S slabs propagating along the c direction (see Fig. 1c).8 As shown in Fig. 2b, the Ba–La–Ga–S slab in 2 features a centric 0D structure which contains isolated GaS4 tetrahedra with Ba12+ and La53+ cations located between them, named as BaLaGaS4. Compared to the slab of La–Fe–S ([La2FeS4] slab, Fig. 2c) in La4FeSb2S10, the GaS4 tetrahedron in 2 replaces the FeS4 tetrahedron with similar bond lengths and angles of 2.243(3)–2.275(3) Å and 103.8(1)–119.2(1)° vs. 2.298(1)–2.303(1) Å and 106.006(4)–112.31(4)°, respectively.8 Meanwhile, to balance the valency, the Ba atoms in 2 substitute the La atoms in La4FeSb2S10 with the similar geometry configurations of BaS10 (3.203(3)–3.422(3) Å) vs. LaS10 (3.079(1)–3.336(1) Å) (see Fig. S7‡). On the other hand, the La–Sb–S slab exhibits 1D (SbS4)n branched chains along the a axis with La3+ cations filling in them, named as La2Sb2S6, which is comparable with the La–Sb–S slab in La4FeSb2S10 (see Fig. 3b).8 In the [La2Sb2S6] slabs, there are four La atoms (2e), four Sb (2e) and eight S atoms (located at 4f, 2e, 4f, 2e, 4f, 4f, 2e and 2e sites, respectively). Sb1 and Sb4 have the same geometry configuration of SbS4 teeter-totter polyhedra (2.446(3)–2.747(4) Å). In addition, Sb2 and Sb3 all show three short Sb–S bonds (2.439(3)–2.539(4) vs. 2.435(3)–2.530(4) Å) and one long bond (3.219(5) vs. 3.216(5) Å) to generate SbS4 teeter-totter polyhedra. The 1D (SbS4)n branched chains are then connected by SbS4 teeter-totter polyhedra via corner sharing.
Remarkably, the long Sb–S bonds of the SbS4 teeter-totter polyhedra in compound 2 are longer than those in 1 and La4FeSb2S10,8 3.216(5)–3.219(5) vs. 2.932(2) vs. 2.917(2) Å. However, the long Sb–S bonds also can be seen in other Sb-containing chalcogenides, such as Pr4GaSbS9 (2.446(3)–2.851(2) Å),5 Nd4InSbS9 (2.431(3)–2.924(3) Å),6 Sr6Sb6S17 (2.428(2)–3.436(3) Å),12 La7Sb9S24 (2.433(2)–3.446(2) Å),23 Ba4Sb3S8Cl33 (2.375(2)–3.062(2) Å) and Na9Gd5Sb8S26 (2.444(2)–3.274(2) Å).35 According to Table 2, the calculated COHP values of the Sb–S bonds (3.216(5) and 3.219(5) Å) are negative (−0.40637 and −0.40350 eV per bond). This evidences the weak bonding interactions of Sb–S bonds, which demonstrate that the SbS4 polyhedra are indeed extending in a novel teeter-totter chain motif via corner sharing as shown in Fig. 3b. On the other hand, the angles of S–Sb–S in the SbS4 units in 1, 2, and La4FeSb2S108 also show obvious differences. The axial angles of S–Sb–S for 1, 2 and La4FeSb2S108 are closer and in the range of 152.63(5)–159.4(2)°. However, in compound 2, there are partial axial angles of S–Sb–S which vary from 160.9(2) to 161.1(2)°. The equatorial angles of S–Sb–S are 94.32(2) vs. 99.3(2)–103.1(2) vs. 103.38(5)–104.13(5)° in compound 1, 2 and La4FeSb2S10,8 respectively.
In addition, guided by the [Ba3Ga2S6], [BaLaGaS4] and [La2FeS4] slabs in compounds 1, 2 and La4FeSb2S10,8 the extra Ba atoms and GaS4 tetrahedra in 1 increase unit cell a from 15.066(4), to 15.410(8) and 23.628(8) Å. Meanwhile, extra La in the [La2Sb2S6] slabs of 1 also cannot be ignored in the increase of unit cell a. However, b and c of the unit cell in 1 do not show any obvious change comparable with 2 and La4FeSb2S10.8
Optical properties
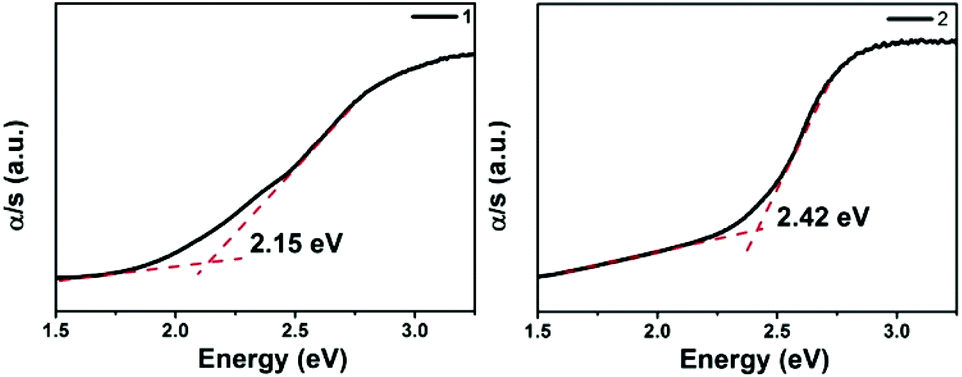
The optical absorption spectra (Fig. 4) show that the band gaps are 2.15 and 2.42 eV for 1 and 2, respectively, which is consistent with their brown colors. These values are larger than those of La2Ga0.33SbS5 (1.76 eV),15 and Na9Gd5Sb8S26 (1.63 eV),35 however similar to the values of Sm4GaSbS9 (2.23 eV),5 La4InSbS9 (2.07 eV),6 and Ba4LaGe3SbSe13 (2.0 eV).32 Compared to La4FeSb2S10 (1.0 eV),8 the band gaps of 1 and 2 are enlarged effectively by the substitution of Fe with Ga. This trend is consistent with that of the corresponding binary compounds, Ga2S3vs. FeS. The detailed reasons for the differences among the band gaps of 1, 2 and La4FeSb2S108 will be discussed below.
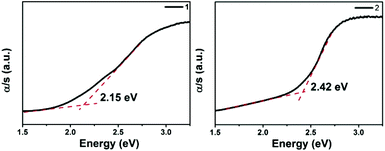 |
| | Fig. 4 The UV-vis diffuse reflectance of compound 1 and 2. | |
Theoretical results
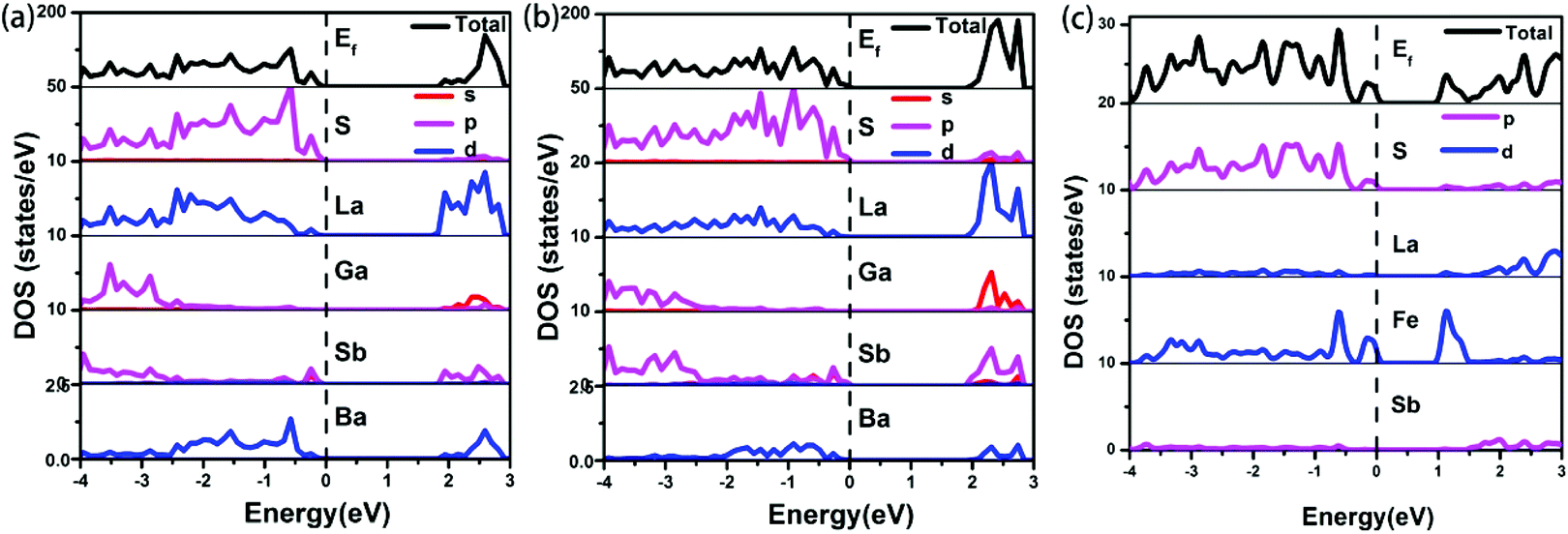
The electronic band structures (see Fig. S8a‡) reveal a direct band gap for 1, with the valence band maximum (VBM) and the conduction band minimum (CBM) located at the same X points. While, compound 2 shows the antitype (an indirect band gap) in which the VBM and CBM are located at different k points (see Fig. S8b‡). The calculated band gaps display an increasing trend with the following order: 1 (1.82 eV) < 2 (1.92 eV), which is consistent with the experimental values of 1 (2.15 eV) and 2 (2.42 eV) (see Fig. 4). A comparison of the band gaps of compounds 1, 2 and La4FeSb2S10,8 shows that there is about a 1.4 eV difference between them. The reason is shown in Fig. 5, which illustrates the total and partial densities of states (PDOS) of compound 1, 2, and La4FeSb2S10.8 In the PDOS, the contributions of the La-4f states are not shown. As shown in Fig. 5a and b, the VBM of compound 1 and 2 is dominated by the S-3p states with minor contributions from the La-5d, Sb-5p, Ba-5d and Ga-4p states. Moreover, the CBM is mainly occupied by the La-5d states with small amounts of S-3p, Ga-4s, Sb-5p and Ba-5d states. Therefore, the optical gaps of 1 and 2 are mainly determined by the electron excitation from the S-3p to La-5d states. However, the role of Ga and Sb in the overall band structures is important, e.g., Ba6La8Ga4Sb4S30 (1) has a direct band gap, whereas Ba3La9Ga3Sb6S30 (2) has an indirect band gap (see Fig. S8‡). The electron transition in an indirect band gap is usually higher in energy, which is consistent with the optical band gap measurement (Fig. 4) where 2 exhibits a wider band gap of 2.42 eV than 2.15 eV of 1. In the case of La4FeSb2S10,8 the VBM is mainly composed of the S-3p and Fe-3d states mixed with small contributions of the Sb-5p and La-5d states. Strong orbital hybridizations between the S-3p and Fe-3d orbitals are shown near VBM. In addition, the CBM is primarily derived from the Fe-3d and La-5d states with small amounts of the S-3p and Sb-5p states. The optical absorptions close to the near-IR edge are mainly from the S-3p states to the unoccupied Fe-3d states for La4FeSb2S10.8 According to the analysis above, when Fe atoms are substituted by Ga, the optical absorption responses have changed, from the S-3p to Fe-3d states in La4FeSb2S108vs. from the S-3p to La-5d states in 1 and 2. In the absence of the unoccupied Fe-3d orbitals which can trap electrons during UV-Vis excitation, the CBM is pulled away from Ef and produces a larger band gap for compound 1 and 2.49,50 Therefore, 1 and 2 which contain no transition metal showing strong electron correlation possess relatively larger band gaps than La4FeSb2S10.8
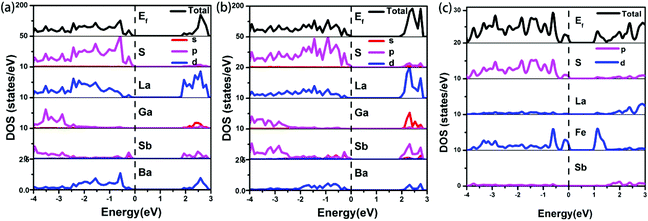 |
| | Fig. 5 Total and partial densities of state of (a) compound 1, (b) compound 2 and (c) La4FeSb2S10.8 Dashed lines show the Fermi energy (Ef). | |
To further characterize the bonding interactions of the Sb–S bonds in 1 and 2, the COHP and electron localization function (ELF)51 were calculated (see Table 2, Fig. 6, 7 and S9‡). For compound 1, the bonding states for the shortest Sb1–S4 bond (2.443(3) Å) are located at −2.7 to −17 eV. The slightly longer Sb1–S2 bond (2.727(4) Å) extends from −3 to −17 eV, and the longest Sb1–S5 bond is located at −8 to −17 eV. It is obvious that the bonding interactions become weaker and weaker as the distances of the Sb–S bonds increase. Comparable with 1, a similar situation occurs in compound 2 as is shown in Fig. 7. The shortest Sb–S bonds mainly extend from −2 to −17 eV. Nevertheless, the two longer Sb3–S11 bonds (3.216(5) Å) and Sb2–S12 bonds (3.219(5) Å) are located between −4.2 and −16.6 eV. The values of –COHP for these two longer bonds are 0.4064 and 0.4035 eV per bond, about 12% that of the strongest Sb3–S9 and Sb2–S10 bonds (3.378 and 3.362 eV per bond). Moreover, the results of the COHP calculation are in agreement with that of ELF in Fig. S9‡ (the region with an ELF value of 1.00 means fully localized electrons and the region close to 0.00 is a low electron density area). As shown in Fig. S9a,‡ the longest Sb1–S5 bonds (2.932(2) Å) in 1 show moderate bonding interactions. On the other hand, in 2 (Fig. S9b‡), the two longer Sb2–S12 (3.219(5) Å) and Sb3–S11 (3.216(5) Å) bonds show weak but nonnegligible bonding interactions, located at the region at about the value of 0.25. All of this illustrates that Sb3+ cations in 1 and 2 are indeed surrounded by four S atoms to form SbS4 teeter-totter polyhedra.
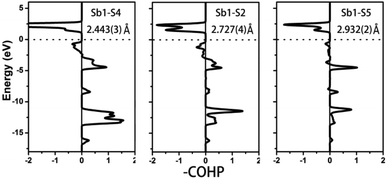 |
| | Fig. 6 COHP curves of selected bond distances for compound 1. Ef is located at 0 eV. | |
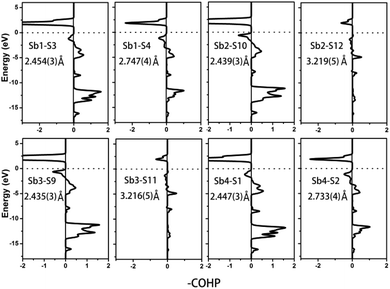 |
| | Fig. 7 COHP curves of selected bond distances for compound 2. Ef is located at 0 eV. | |
Conclusions
Two new pentanary chalcogenides with Ba/RE/Ga/Sb/S (RE = rare-earth metal), Ba3La4Ga2Sb2S15 (1) and BaLa3GaSb2S10 (2), have been synthesized and characterized. Compound 1 crystallizes with a new structure type in the space group Ibam as a 0D structure containing totally isolated GaS4 and Sb2S7 units; meanwhile, compound 2 crystallizes in the space group P21/m and has a 1D structure which is built by (SbS4)n chains and isolated GaS4 units. With a lower spacer GaS4 tetrahedron concentration, the structure changes from 0D in 1 to 1D in 2: i.e. an isolated Sb2S7 dimer vs. (SbS4)n chains. As the diffuse reflectance spectra showed, their optical band gaps were 2.12 and 2.42 eV, respectively. Moreover, theory studies demonstrate that replacing Fe atoms with Ga is an effective way to widen band gaps, compared to the analogue La4FeSb2S10. In addition, COHP and ELF calculations of the Sb–S bonds reveal that the long Sb–S bonds in 1 (2.932(2) Å) and 2 (3.216(5) and 3.219(5) Å) show weak but indispensable bonding interactions, which play significant roles in constructing the isolated Sb2S7 dimer for 1 and (SbS4)n chains for 2.
Acknowledgements
This research was supported by the NSF of China (No. 21225104, 21233009, 91422303, 21571020, 21301175, 21171168 and 21671023). We thank Prof. Yong-Fan Zhang at Fuzhou University for helping with the DFT calculations.
References
- T. Thonhauser, T. J. Scheidemantel, J. O. Sofo, J. V. Badding and G. D. Mahan, Phys. Rev. B: Condens. Matter, 2003, 68, 08521 Search PubMed.
-
(a) K. F. Hsu, S. Loo, F. Guo, W. Chen, J. S. Dyck, C. Uher, T. Hogan, E. K. Polychroniadis and M. G. Kanatzidis, Science, 2004, 303, 818 CrossRef CAS PubMed;
(b) J. H. Kim, D. Y. Chung, D. Bilc, S. Loo, J. Short, S. D. Mahanti, T. Hogan and M. G. Kanatzidis, Chem. Mater., 2005, 17, 3606 CrossRef CAS.
- E. Dashjav, A. Szczepenowska and H. Kleinke, J. Mater. Chem., 2002, 12, 345 RSC.
-
(a) J. Wang, K. Lee and K. Kovnir, Z. Anorg. Allg. Chem., 2015, 641, 1087 CrossRef CAS;
(b) J. Wang, K. Lee and K. Kovnir, J. Mater. Chem. C, 2015, 3, 9811 RSC.
- M. C. Chen, L. H. Li, Y. B. Chen and L. Chen, J. Am. Chem. Soc., 2011, 133, 4617 CrossRef CAS PubMed.
- H. J. Zhao, Y. F. Zhang and L. Chen, J. Am. Chem. Soc., 2012, 134, 1993 CrossRef CAS PubMed.
- M. C. Chen, L. M. Wu, H. Lin, L. J. Zhou and L. Chen, J. Am. Chem. Soc., 2012, 134, 6058 CrossRef CAS PubMed.
- H. J. Zhao, L. H. Li, L. M. Wu and L. Chen, Inorg. Chem., 2009, 48, 11518 CrossRef CAS PubMed.
- H. J. Zhao, L. H. Li, L. M. Wu and L. Chen, Inorg. Chem., 2010, 49, 5811 CrossRef CAS PubMed.
- Y. Y. Li, B. X. Li, G. Zhang, L. J. Zhou, H. Lin, J. N. Shen, C. Y. Zhang, L. Chen and L. M. Wu, Inorg. Chem., 2015, 54, 4761 CrossRef CAS PubMed.
- W. Y. Hao, D. J. Mei, W. L. Yin, K. Feng, J. Y. Yao and Y. C. Wu, J. Solid State Chem., 2013, 198, 81 CrossRef CAS.
- K. S. Choi and M. G. Kanatzidis, Inorg. Chem., 2000, 39, 5655 CrossRef CAS PubMed.
- K. S. Choi and M. G. Kanatzidis, Inorg. Chem., 2001, 40, 101 CrossRef CAS PubMed.
- K. S. Choi, L. Iordanidis, K. Chondroudis and M. G. Kanatzidis, Inorg. Chem., 1997, 36, 3804 CrossRef CAS.
- H. J. Zhao, J. Solid State Chem., 2016, 237, 99 CrossRef CAS.
- W. L. Yin, K. Feng, L. Kang, B. Kang, J. G. Deng, Z. S. Lin, J. Y. Yao and Y. C. Wu, J. Alloys Compd., 2014, 617, 287 CrossRef CAS.
- J. D. Breshears and M. G. Kanatzidis, J. Am. Chem. Soc., 2000, 122, 7839 CrossRef CAS.
- L. Geng, W. D. Cheng, H. Zhang, C. S. Lin, W. L. Zhang, Y. Y. Li and Z. Z. He, Inorg. Chem., 2011, 50, 2378 CrossRef CAS PubMed.
- C. Liu, Y. Y. Shen, P. P. Hou, M. J. Zhi, C. M. Zhou, W. X. Chai, J. W. Cheng and Y. Liu, Inorg. Chem., 2015, 54, 8931 CrossRef CAS PubMed.
- Z. M. Ma, F. Weng, Q. R. Wang, Q. Tang, G. H. Zhang, C. Zheng, R. P. S. Han and F. Q. Wang, RSC Adv., 2014, 4, 28937 RSC.
- H. J. Zhao and P. F. Liu, J. Solid State Chem., 2015, 232, 37 CrossRef CAS.
- E. C. Agha, C. D. Malliakas, J. Im, H. Jin, L. D. Zhao, A. J. Freeman and M. G. Kanatzidis, Inorg. Chem., 2014, 53, 673 CrossRef CAS PubMed.
- A. Assoud, K. M. Kleinke and H. Kleinke, Chem. Mater., 2006, 18, 1041 CrossRef CAS.
- A. Assoud and H. Kleinke, Solid State Sci., 2010, 12, 919 CrossRef CAS.
- H. Lin, L. J. Zhou and L. Chen, Chem. Mater., 2012, 24, 3406 CrossRef CAS.
- P. Yu, L. J. Zhou and L. Chen, J. Am. Chem. Soc., 2012, 134, 2227 CrossRef CAS PubMed.
- W. L. Yin, D. J. Mei, K. Feng, J. Y. Yao, P. Z. Fu and Y. C. Wu, Dalton Trans., 2011, 40, 9159 RSC.
- Y. F. Shi, Y. K. Chen, M. C. Chen, L. M. Wu, H. Lin, L. J. Zhou and L. Chen, Chem. Mater., 2015, 27, 1876 CrossRef CAS.
- H. Lin, J. N. Shen, Y. F. Shi, L. H. Li and L. Chen, Inorg. Chem. Front., 2015, 2, 298 RSC.
-
(a) X. Q. Wang and F. Liebau, Acta Crystallogr., Sect. B: Struct. Sci., 1996, 52, 7 CrossRef;
(b) X. Q. Wang and F. Liebau, Z. Kristallogr., 1996, 211, 437 CAS.
-
(a) X. Q. Wang and F. Liebau, J. Solid State Chem., 1994, 111, 385 CrossRef CAS;
(b) X. Q. Wang and A. J. Jacobson, J. Solid State Chem., 1998, 140, 387 CrossRef CAS;
(c) K. Vold and H. Schäfer, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1979, 34, 1637 Search PubMed;
(d) G. L. Schimek and J. W. Kolis, Inorg. Chem., 1997, 36, 1689 CrossRef CAS PubMed;
(e) R. Stähler and W. Bensch, J. Chem. Soc., Dalton Trans., 2001, 2001, 2518 RSC;
(f) L. Engelke, C. Näther and W. Bensch, Eur. J. Inorg. Chem., 2002, 2002, 2936 CrossRef;
(g) R. Stähler, C. Näther and W. Bensch, J. Solid State Chem., 2003, 174, 264 CrossRef;
(h) R. Stähler and W. Bensch, Eur. J. Inorg. Chem., 2001, 2001, 3073 CrossRef;
(i) M. L. Feng, D. N. Kong, Z. L. Xie and X. Y. Huang, Angew. Chem., Int. Ed., 2008, 47, 8623 CrossRef CAS PubMed;
(j) J. Zhou, J. Dai, G. Q. Bian and C. Y. Li, Coord. Chem. Rev., 2009, 253, 1221 CrossRef CAS;
(k) B. Seidlhofer, N. Pienack and W. Bensch, Z. Naturforsch., 2010, 65b, 937 Search PubMed;
(l) J. Zhou, L. T. An, X. Liu, L. J. Huang and X. J. Huang, Dalton Trans., 2011, 40, 11419 RSC;
(m) B. Seidlhofer, C. Näther and W. Bensch, CrystEngComm, 2012, 14, 5441 RSC;
(n) K. Z. Du, M. L. Feng, L. H. Li, B. Hu, Z. J. Ma, P. Wang, J. R. Li, Y. L. Wang, G. D. Zou and X. Y. Huang, Inorg. Chem., 2012, 51, 3926 CrossRef CAS PubMed;
(o) M. L. Feng, C. L. Hu, K. Y. Wang, C. F. Du and X. Y. Huang, CrystEngComm, 2013, 15, 5007 RSC;
(p) C. Y. Yue, X. W. Lei, H. P. Zang, X. R. Zhai, L. J. Feng, Z. F. Zhao, J. Q. Zhao and X. Y. Liu, CrystEngComm, 2014, 16, 3424 RSC;
(q) B. Zhang, M. L. Feng, H. H. Cui, C. F. Du, X. H. Qi, N. N. Shen and X. Y. Huang, Inorg. Chem., 2015, 54, 8474 CrossRef CAS PubMed;
(r) C. Anderer, C. Nather and W. Bensch, Cryst. Growth Des., 2016, 16, 3802 CrossRef CAS;
(s) A. H. Fang, F. Q. Huang, X. M. Xie and M. H. Jiang, J. Am. Chem. Soc., 2010, 132, 3260 CrossRef CAS PubMed.
- A. Assoud, N. Soheilnia and H. Kleinke, J. Solid State Chem., 2004, 177, 2249 CrossRef CAS.
- H. J. Zhao, J. Solid State Chem., 2016, 235, 18 CrossRef CAS.
- W. Dorrscheidt and H. Schafer, Z. Naturforsch., B: Chem. Sci., 2014, 36, 410 Search PubMed.
- S. Park and S. J. Kim, J. Solid State Chem., 2001, 161, 129 CrossRef CAS.
- C. Y. Yu, M. F. Wang, M. Y. Chung, S. M. Jang, J. C. Huang and C. S. Lee, Solid State Sci., 2008, 10, 1145 CrossRef CAS.
- G. Cordier, C. Schwidetzky and H. Schafer, J. Solid State Chem., 1984, 54, 84 CrossRef CAS.
- K. S. Choi and M. G. Kanatzidis, Chem. Mater., 1999, 11, 2613 CrossRef CAS.
- R. Pocha and D. Johrendt, Inorg. Chem., 2004, 43, 6830 CrossRef CAS PubMed.
-
CrystalClear, version 1.3.5, Rigaku Corp., The Woodlands, TX, 1999 Search PubMed.
-
G. M. Sheldrick, SHELXTL, version 5.1, Bruker-AXS, Madison, WI, 1998 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
-
G. Kortüm, Reflectance Spectroscopy, Springer-Verlag, New York, 1969 Search PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter, 1992, 45, 13244 CrossRef.
-
(a) G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter, 1999, 59, 1758 CrossRef CAS;
(b) P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- R. Dronskowski and P. E. Blochl, J. Phys. Chem., 1993, 97, 8617 CrossRef CAS.
- G. A. Landrum and R. Dronskowski, Angew. Chem., Int. Ed., 2000, 39, 1560 CrossRef CAS.
- S. George, S. Pohkrel, Z. X. Ji, B. L. Henderson, T. Xia, L. J. Li, J. I. Zink, A. E. Nel and L. Mädler, J. Am. Chem. Soc., 2011, 133, 11270 CrossRef CAS PubMed.
- F. Zhou, K. Kang, T. Maxisch, G. Ceder and D. Morgan, Solid State Commun., 2004, 132, 181 CrossRef CAS.
- A. Savin, R. Nesper, S. Wengert and T. F. Fassler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Mercouri G. Kanatzidis on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1500607 and 1500608 for BaLa3GaSb2S10 and Ba3La4Ga2Sb2S15. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00346j |
|
| This journal is © the Partner Organisations 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 ab,
Jin-Ni
Shen
c,
Chen-Sheng
Lin
d,
Peng-Fei
Liu
ab,
Jin-Ni
Shen
c,
Chen-Sheng
Lin
d,
Peng-Fei
Liu