
Ni3S2 nanosheet-anchored carbon submicron tube arrays as high-performance binder-free anodes for Na-ion batteries†
Xueke
Xia
a,
Jian
Xie
*ab,
Shichao
Zhang
c,
Bin
Pan
d,
Gaoshao
Cao
b and
Xinbing
Zhao
 ab
ab
aState Key Laboratory of Silicon Materials, School of Materials Science and Engineering, Zhejiang University, Hangzhou 310027, P. R. China. E-mail: xiejian1977@zju.edu.cn; Fax: +86-571-87951451; Tel: +86-571-87951451
bKey Laboratory of Advanced Materials and Applications for Batteries of Zhejiang Province, Hangzhou 310027, P. R. China
cSchool of Materials Science and Engineering, Beijing University of Aeronautics and Astronautics, Beijing 100191, P. R. China
dIndustrial Technology Research Institute of Zhejiang University, Hangzhou 310058, P. R. China
First published on 23rd November 2016
Abstract
Owing to the increasing concerns regarding limited lithium reserves, sodium-ion batteries (SIBs) have attained worldwide attention in recent years. However, there is still a challenge to find suitable anodes for SIBs with high capacity and a long cycle life. In this work, we propose a unique feather-like array-type anode composed of thin Ni3S2 nanosheets anchored on cracked carbon submicron tubes (CSTs) on a porous Ni foam substrate. The porous structure of the Ni foam facilitates electrode wetting by the electrolyte. The voids in between the Ni3S2/CST arrays provide free space for buffering volume changes upon sodiation/desodiation. The CSTs not only act as the support for Ni3S2 growth but also uniformly disperse the Ni3S2 nanosheets, leading to high capacity and good capacity retention. The Ni-supported Ni3S2/CSTs can deliver a high initial reversible capacity of 887 mA h g−1 at 50 mA g−1. The reversible capacity can be kept at 212 mA h g−1 after 260 cycles. This work will shed light on the design of high-performance SIB anodes.
Introduction
Li-ion batteries (LIBs) are now the prime power sources for electric vehicles (EVs) due to them possessing the largest energy density among the available rechargeable batteries.1,2 However, the large-scale use of LIBs in EVs will cause rapid consumption of lithium resources, with low reserves, uneven distribution and a high cost to obtain.3–5 In contrast, sodium is one of the most abundant elements in the Earth's crust. Considering the resource abundance and low cost, sodium-ion batteries (SIBs) have been considered as promising alternatives to LIBs for sustainable energy storage.3–9 One of the large challenges for SIBs is to develop suitable anode materials, since the graphite anodes used in commercial LIBs show low activity towards Na-ion intercalation due to the larger ionic radius and higher atomic mass of sodium compared to lithium.10Hard carbon materials have been recognized as potential anodes for SIBs because of their moderate capacity and suitable electrode potential.11,12 However, the available capacity of hard carbon materials is not sufficient yet, usually below or around 300 mA h g−1.12 Much attention has been shifted to some high-capacity anodes with a conversion mechanism, typically transition metal oxides or sulfides. Various oxides such as Fe2O3,13–15 Fe3O4,16,17 MnFe2O4,18 FeOOH,19 VO2,20,21 NiO,22 CuO,23 Co3O4,24,25 and NiCo2O4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26 have been investigated as promising anodes for SIBs. Research on some oxides showed that their Na-storage activity is somewhat lower compared with the corresponding Li-storage activity although both obey similar conversion reactions.14,16–18,20,22,24,26 Interestingly, some sulfides exhibit higher Na-storage capacity than the oxide counterparts.27,28 A report by Choi et al. showed that a WS2-based hybrid can yield an initial capacity of 356 mA h g−1, considerably higher than its oxide counterpart (130 mA h g−1).27 Given their high Na-storage activity, low cost and environmental friendliness, great interest has been paid to some sulfides, MoS2,29–31 CoS2,32 Co3S4,33 MnS,34 Ni3S2,35,36 WS2,27,37 FeS2,38 VS4,39etc.
26 have been investigated as promising anodes for SIBs. Research on some oxides showed that their Na-storage activity is somewhat lower compared with the corresponding Li-storage activity although both obey similar conversion reactions.14,16–18,20,22,24,26 Interestingly, some sulfides exhibit higher Na-storage capacity than the oxide counterparts.27,28 A report by Choi et al. showed that a WS2-based hybrid can yield an initial capacity of 356 mA h g−1, considerably higher than its oxide counterpart (130 mA h g−1).27 Given their high Na-storage activity, low cost and environmental friendliness, great interest has been paid to some sulfides, MoS2,29–31 CoS2,32 Co3S4,33 MnS,34 Ni3S2,35,36 WS2,27,37 FeS2,38 VS4,39etc.
Despite their high capacity, the cycling stability of the sulfides is not satisfactory due to the large volume changes during the conversion reactions. Various strategies have been adopted to improve the cycling stability of the sulfides. One of the simplest methods is to use a carbon-based matrix to buffer the volume changes.40 Graphene is an ideal matrix for conversion materials due to its large surface area, high electronic conductivity and super mechanical strength.41 Choi et al. designed a high-performance anode composed of three-dimensional graphene and few-layered MoS2, which could maintain a capacity of 322 mA h g−1 after 600 cycles at 1.3 A g−1.30 Building pores or voids purposely into active materials provides another effective method to buffer the volume changes and thereby improve the cycle life.42–44 Liu et al. reported a yolk–shell Sn4P3@C hybrid which could maintain a high discharge capacity of 360 mA h g−1 after 400 cycles at 1.5 C.44 The excellent cycling stability can be attributed to the voids in the yolk–shell structure. Using array-type electrodes has proved to be a practical/facile approach for improving the cycle life since the voids in between the arrays supply buffering room for the volume changes.20,23,26,45,46
In this work, we design a binder-free array-type electrode by directly growing Ni3S2 nanosheet-loaded cracked carbon submicron tube (Ni3S2/CST) arrays on a Ni foam substrate. The growth of the cracked CST arrays on the Ni foam is through a controllable templating route, and the loading of the Ni3S2 nanosheets on the CSTs is through a facile hydrothermal route with the Ni sourced directly from the Ni substrate. The Ni3S2 nanosheets anchor both on the surface of CSTs and inside the cracked CSTs, exhibiting a unique feather-like architecture. The CSTs act as the support to grow Ni3S2 and uniformly disperse the Ni3S2. The volume changes of Ni3S2 upon sodiation/desodiation can be buffered by the voids inside the CSTs and the free space between the CST arrays. As a result, the Ni-supported Ni3S2/CST arrays exhibit a high initial reversible capacity of 887 mA h g−1 at 50 mA g−1. The array electrode also displays a long cycle life with a reversible capacity of 212 mA h g−1 retained after 260 cycles. This work provides a new design concept for high-performance SIB anodes.
Experimental section
Electrode preparation and characterization
For the preparation of the Ni-supported Ni3S2/CST array electrodes, Ni-supported CST arrays were first prepared by a templating route using ZnO arrays as the templates. The preparation of the Ni-supported CSTs was described in detail in the ESI.† The precursor solution for Ni3S2 was prepared by adding 30 mg of thiourea (CN2H4S) and 142 mg of Na2SO4 to 35 mL of deionized (DI) water under vigorous stirring for 30 min. The solution was then sealed in a Teflon-lined stainless steel autoclave (50 mL) with the Ni-supported CST piece immersed. The hydrothermal reaction for Ni3S2 growth was performed at 120 °C for 3 h, where the Ni source is the Ni substrate directly. The Ni-supported Ni3S2/CSTs were finally obtained after washing the hydrothermal product repeatedly with DI water and drying at 60 °C in air overnight. The loadings of Ni3S2 and CSTs on the Ni substrate are around 1 and 0.1 mg cm−2, respectively. The weight of Ni3S2 was calculated using the equation: m(Ni3S2) = δm × 240.2/64.13,47 where δm is the weight gain of the Ni foam after the hydrothermal reaction, which was measured by a precise balance. For comparison, Ni-supported Ni3S2 was also prepared by a similar route using Ni foam as the substrate directly instead of Ni-supported CSTs. X-ray diffraction (XRD) patterns were measured on a Rigaku D/Max-2550pc powder diffractometer equipped with Cu Kα radiation (λ = 1.541 Å). The morphology of the electrodes or the electrode components was observed by field-emission scanning electron microscopy (SEM) on a Hitachi S-4800 microscope and transmission electron microscopy (TEM) on a JEM 2100F microscope. The chemical composition of the NaOH etched sample was measured by inductively coupled plasma-atomic emission spectrometry (ICP-AES) on an IRIS Intrepid II XSP system. Raman spectra were recorded on a JOBIN-Yvon Labor Raman HR-800 Raman system using a 514.5 nm Ar-ion laser at 10 mW.Electrochemical measurements
CR2025 coin cells were fabricated in an Ar-filled glovebox with Na foils as counter electrodes, Ni-supported Ni3S2/CSTs (or Ni-supported CSTs, Ni3S2) as working electrodes, and Whatman GF/D glass fiber filters as separators. Before cell fabrication, the working electrodes were dried at 110 °C in a vacuum overnight. The electrolyte used was 1 M NaPF6 in ethylene carbonate (EC)/diethyl carbonate (DEC) (1:1 in volume). Fluoroethylene carbonate (FEC) was used as an electrolyte additive with a FEC/EC + DEC volume ratio of 5%. Charge and discharge cycling was conducted on a Neware battery cycler (Shenzhen, China) between 0.005 and 3.0 V (vs. Na/Na+). For Ni3S2/CSTs, the current density and specific capacity were normalized to the total mass of Ni3S2 and CSTs. Cyclic voltammetry (CV) tests were carried out on a VersaSTAT3 electrochemistry workstation (Princeton Applied Research) in a voltage window of 0.005–3 V (vs. Na/Na+) at 0.1 mV s−1. All of the electrochemical tests were carried out at 25 °C.
Results and discussion
Ni-supported ZnO arrays were first prepared by a facile hydrothermal route, and were used as the templates to grow carbon submicron tube arrays. After ZnO etching, Ni-supported CST arrays were obtained. Then a hydrothermal reaction was performed to grow Ni3S2 on CSTs, which led to the formation of the final product, Ni-supported Ni3S2/CSTs. The formation of Ni3S2 begins with hydrolysis of thiourea, followed by sulfuration of Ni through the following reactions:48,49| SC(NH2)2 + H2O → OC(NH2)2 + H2S | (1) |
| 2H2S + 3Ni → Ni3S2 + 2H2 | (2) |
The preparation procedure is schematically shown in Fig. 1. The reaction product at each step in the above synthesis was checked by XRD, as shown in Fig. 2. The ZnO grown on the Ni substrate can be indexed to hexagonal phase ZnO with a space group of P63mc. As seen in Fig. 3a and S1a (ESI†), rod-like ZnO is uniformly distributed on the skeleton of the Ni foam and displays an array-type architecture with the porous structure of Ni foam (Fig. S1b, ESI†) kept intact. The ZnO rods have a submicron size with a diameter of 300–600 nm and a length of 2–4 μm. No carbon or ZnO diffraction peaks are detected after etching, suggesting amorphous features of the coated carbon and the sufficient removal of ZnO after the NaOH etching. The ICP-AES result shows that the Zn/C atomic ratio is below 0.02, confirming that ZnO has been sufficiently removed after the NaOH etching. The SEM image in Fig. 3b demonstrates that the CSTs have a cracked structure. The crystallization of the CSTs was checked by high-resolution TEM (HRTEM) and Raman spectroscopy. The HRTEM image (Fig. S2b, ESI†) of the tubular wall of a CST indicates that the CST is poorly crystallized. The broad G peak and the presence of a D peak in the Raman spectrum (Fig. S2c, ESI†) also suggest that the CST is poorly crystallized and is rich in defects, disorder and dopants.50 Therefore, low electronic conductivity of the CSTs is expected as the CSTs act mainly as the support for the growth of Ni3S2. The XRD results indicate that the Ni3S2 deposited on the CSTs is rhombohedral phase Ni3S2 with a space group of R32 (JCPDS no. 44-1418). Nitrogen adsorption–desorption isotherms were measured to determine the specific surface area and pore size distribution of Ni-supported Ni3S2/CSTs as shown in Fig. S3 (ESI†). As a result, Ni-supported Ni3S2/CSTs have been fabricated through a controllable route.
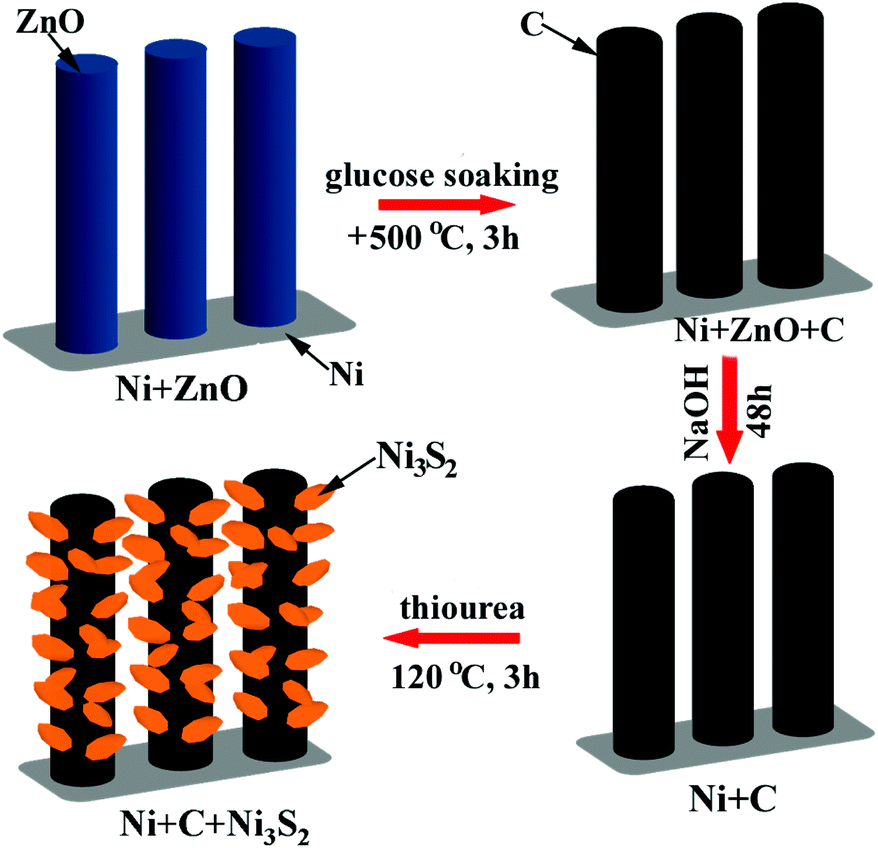 | ||
| Fig. 1 Schematic illustration of the synthetic procedure for Ni-supported Ni3S2/CST arrays. | ||
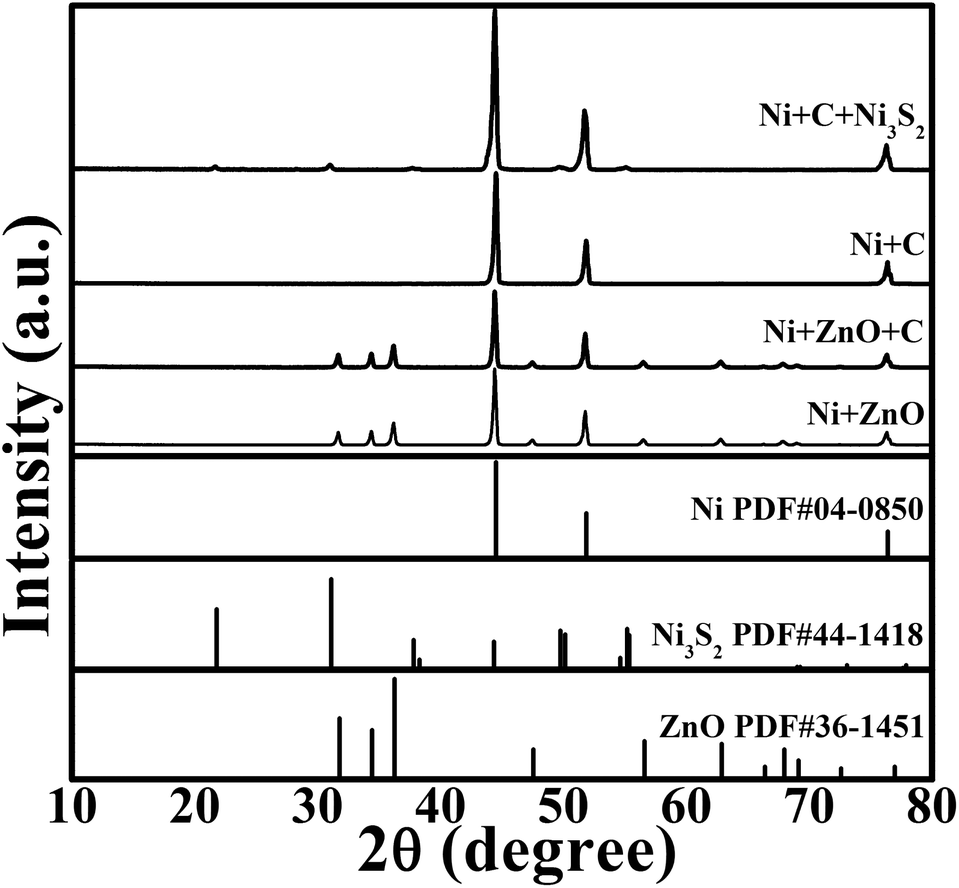 | ||
| Fig. 2 XRD patterns of the products at different synthetic stages with the final product of Ni-supported Ni3S2/CSTs. | ||
 | ||
| Fig. 3 SEM images of Ni-supported (a) ZnO and (b) CSTs, (c–e) Ni3S2/CSTs, and (f) TEM and (g–k) STEM images and the corresponding EDX mapping of Ni3S2/CSTs exfoliated from the electrode. | ||
The morphology of the Ni-supported Ni3S2/CSTs was characterized by SEM. The Ni3S2/CST arrays were grown only on the skeleton of Ni foam which facilitates wetting of the electrode by the electrolyte (Fig. 3c). The SEM image also shows that the array-type structure of the CSTs has been preserved after Ni3S2 deposition, where the free space between the arrays provides room for buffering the volume changes during sodiation/desodiation. The enlarged views in Fig. 3d and e indicate that thin Ni3S2 nanosheets are anchored on CSTs, forming a feather-like structure. It seems that the cracked structure of the CSTs is favorable for the deposition of Ni3S2 nanosheets. As shown in Fig. 3e, some Ni3S2 sheets with a smaller size are located inside the cracked CSTs due to the large diameter of the cracked CSTs. The wall thickness of the CSTs is estimated to be around 10 nm from TEM observation (Fig. S2b, ESI†). The microstructure of the Ni3S2/CSTs was further characterized by TEM, scanning transmission electron microscopy (STEM) and corresponding energy dispersive X-ray spectrometry (EDX) mapping as seen in Fig. 3f–k. The results reveal that both the surface and inside of the CSTs were anchored with Ni3S2 sheets. The above characterization confirms that an array-type Ni3S2/CST electrode has been obtained.
The electrochemical Na-storage performance of Ni-supported Ni3S2/CSTs was evaluated by galvanostatic cycling with Na cells. Fig. 4a shows the first three voltage profiles of the Ni3S2/CST–Na cell at 50 mA g−1. The first charge and discharge capacities of the Ni-supported Ni3S2/CSTs are 887 and 1452 mA h g−1, respectively. In contrast, Ni-supported CSTs deliver much lower charge and discharge capacities of 5 and 44 mA h g−1, respectively. This suggests that bare CSTs are electrochemically inactive, and that the capacity of Ni3S2/CSTs comes mainly from Ni3S2. The high capacity of the Ni-supported Ni3S2/CSTs can be attributed to the small size of Ni3S2 and the uniform dispersion by the CSTs, which enables a high utilization of active material. Fig. 4b shows the first five CV plots scanned at 0.1 mV s−1 between 0.005 and 3.0 V (vs. Na/Na+). In the first cathodic scan, there is a large peak (A peak) at around 0.3 V followed by a tail (B peak), which are attributed to the formation of a solid electrolyte interface (SEI) layer and the conversion reaction of 4Na+ + 4e− + Ni3S2 → 3Ni + 2Na2S.35,36 During the subsequent cathodic scans, a broad peak (C peak) at around 0.7 V and a tail (D peak) appear, which correspond to the two successive plateaus in Fig. 4a. For the first anodic sweep, the large peak at around 1.8 V (E peak) is ascribed to the reconstruction of Ni3S2 through the reaction 3Ni + 2Na2S → Ni3S2 + 4Na+ + 4e−.35 The XRD patterns of the electrode (Fig. S3, ESI†) confirm the reconstruction of Ni3S2 after the anodic scan to 3 V. For the subsequent cycles, the peaks become broad with reduced peak intensity (arrow F). Fig. 4c shows the cycling stability of the Ni-supported Ni3S2/CSTs at 50 mA g−1. After a rapid capacity drop in the initial cycles, the cell can keep a relatively stable cycling value during the subsequent cycles. After 260 cycles, a charge capacity of 212 mA h g−1 can be maintained for the Ni-supported Ni3S2/CSTs. For the bare CSTs, in contrast, the capacity is rather low despite the stable cycling as shown in Fig. 4d.
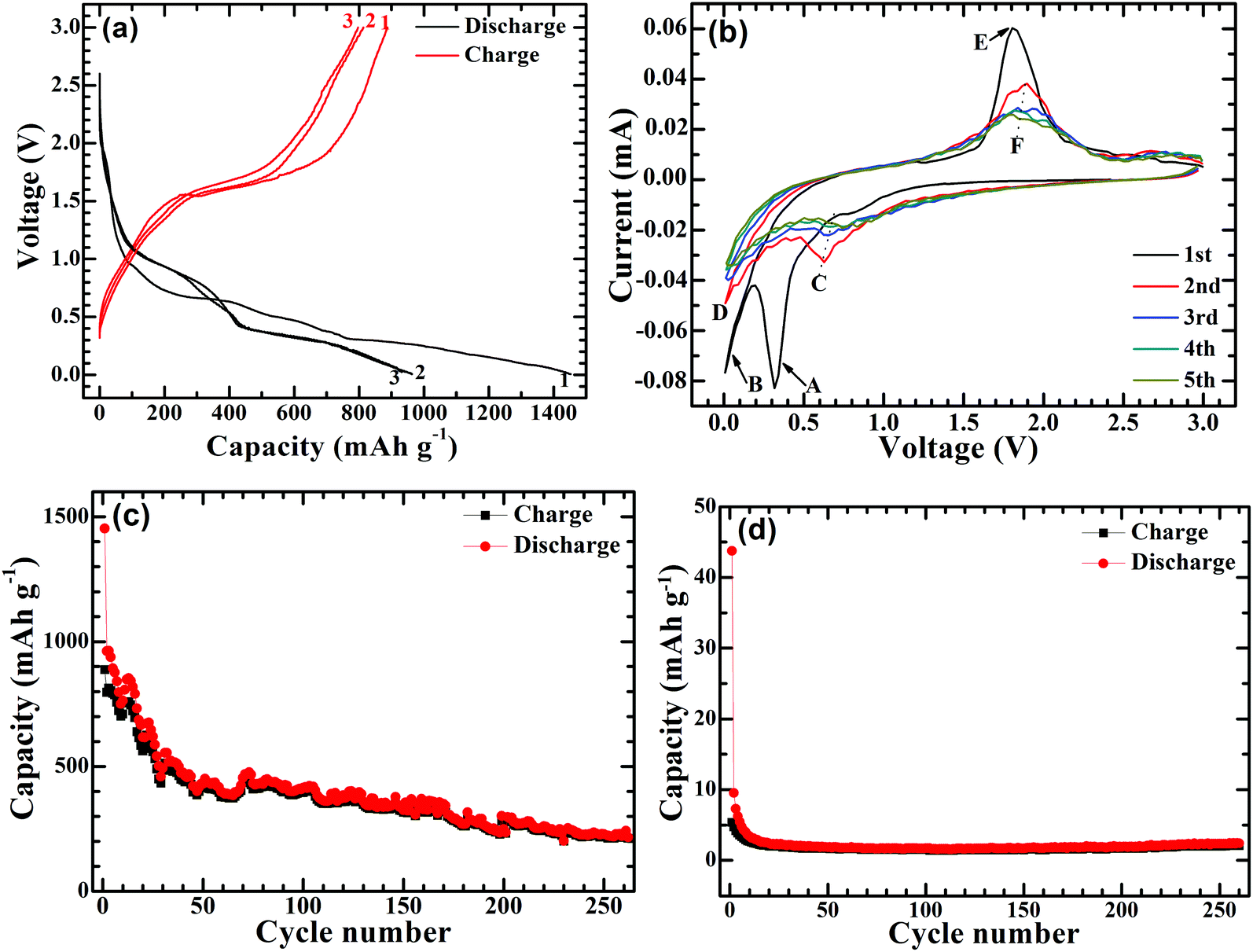 | ||
| Fig. 4 (a) Voltage profiles at 50 mA g−1 and (b) CV plots at 0.1 mV s−1 of Ni-supported Ni3S2/CSTs, and the cycling performance of (c) Ni-supported Ni3S2 and (d) Ni-supported CSTs at 50 mA g−1. | ||
It should be noted that the long-term cycling stability of the Ni-supported Ni3S2/CST arrays is not satisfactory yet and that the polarization is also increased during cycling (Fig. S4, ESI†). Nevertheless, the use of array-type electrodes may be an effective way to relieve capacity fade by supplying voids for volume expansion. For comparison, the electrochemical performance of the Ni-supported Ni3S2 was also investigated. As shown in Fig. S5 (ESI†), Ni3S2 plates can be grown directly on a Ni foam substrate. Although the Ni-supported Ni3S2 has a high capacity and exhibits a capacity increase in the initial cycles, it exhibits a rapid capacity fade for long-term cycling. After 260 cycles, the charge capacity drops to 81 mA h g−1. The more rapid capacity fade for this electrode may be attributed to the absence of large free space for volume expansion. It suggests that the large free space in the Ni-supported Ni3S2/CST arrays may underlie their better cycling stability. To justify this assumption, the proximity of the array structures was changed by loading more or less Ni3S2/CSTs onto the Ni substrate. As seen in Fig. S6a and b (ESI†), a lower loading of Ni3S2/CSTs leads to a lower initial capacity but enhanced cycling stability due to larger free space for buffering the volume changes. As expected, with a higher loading, the electrode shows quicker capacity fade (Fig. S6c and d, ESI†).
Ex situ SEM characterization was performed to understand the good electrochemical performance of the Ni-supported Ni3S2/CSTs. As seen in Fig. 5a, the array-type structure of the electrode is preserved after the first discharge. The pristine fluffy thin sheets have been converted into dense aggregates composed of thick plates after the conversion reaction. Of note is that free space still exists, although volume expansion occurs during the sodiation reaction, which proves that the array structure is desirable for the structural integrity of the electrode. After the first charge, the fluffy structure with thin sheets appears again and the electrode still exhibits an array-type structure, indicating the reversible desodiation reaction. To clarify the reason for the initial capacity fade, the morphology of the electrode after 30 cycles (in the charged state) was observed as seen in Fig. 5c. Although the array-type structure of the electrode is well retained after repeated cycling, the electrode has lost some of the Ni3S2 nanosheets. This suggests that exfoliation of the active material does occur due to the strain from the large volume changes, leading to the rapid capacity loss in the initial cycles. As expected, long-term cycling results in continuous exfoliation of the active material as shown in Fig. S7 (ESI†), which can explain the continuous capacity fade during the following cycles, although it is seen at a slower rate. The XRD results in Fig. S3† confirm the loss of active material from the electrode. It is expected that the remaining Ni3S2, which is located mainly inside the CSTs, can be fixed by the CSTs in the following cycles, leading to relatively stable cycling, where the voids inside the CSTs also offer buffering room for the volume changes. The sodiation/desodiation process of the Ni-supported Ni3S2/CST electrode is schematically shown in Fig. 6.
 | ||
| Fig. 5 SEM images of Ni-supported Ni3S2/CST arrays after (a) the first discharge, (b) the first charge, and (c) the 30th charge. | ||
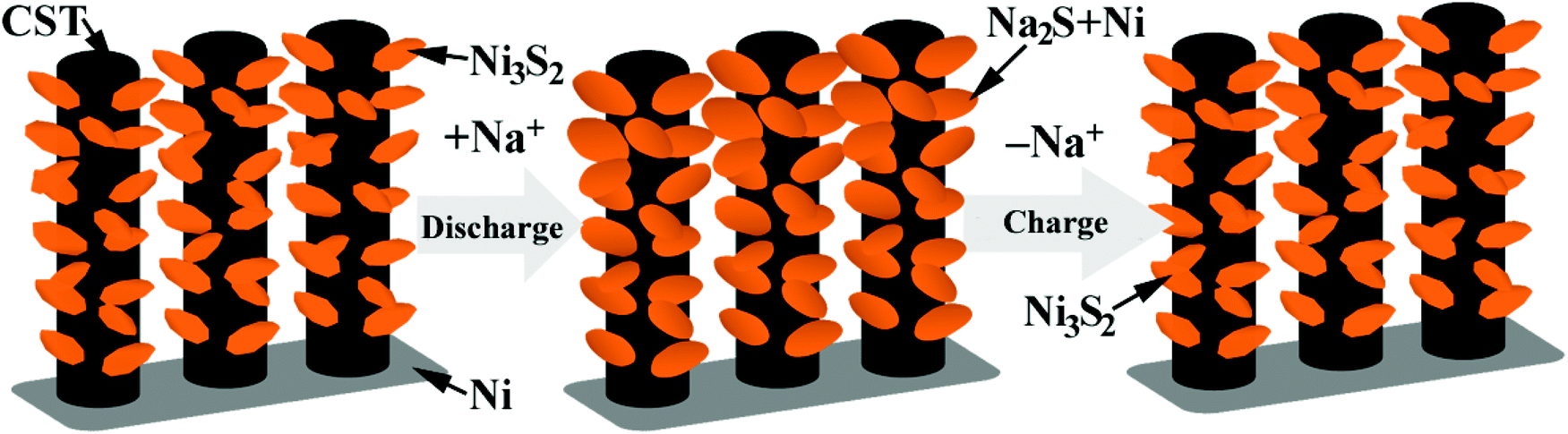 | ||
| Fig. 6 Schematic illustration of the discharge/charge process of the Ni-supported Ni3S2/CST arrays. | ||
Owing to their unique structure, the Ni-supported Ni3S2/CSTs exhibit a good cycle life which is comparable with or superior to those of recently reported sulfide materials as summarized in Table S1 (ESI†). Compared with MnS/G (497 mA h g−1)34 and FeS/C (500 mA h g−1),51 the Ni-supported Ni3S2/CSTs exhibit a higher initial capacity (887 mA h g−1). The Ni-supported Ni3S2/CSTs can maintain capacities of 401 and 368 mA h g−1 after 100 and 125 cycles, higher than those of MnS/G (308 mA h g−1 after 125 cycles) and FeS/C (365 mA h g−1 after 100 cycles). The Ni-supported Ni3S2/CSTs can deliver a higher initial capacity than other Ni3S2-based materials, such as Ni3S2/PEDOT (318.3 mA h g−1)35 and Ni3S2 + Ni7S6/G (512.7 mA h g−1).52 Compared with the above two materials, the Ni-supported Ni3S2/CSTs also exhibit a long cycle life (260 cycles vs. 30 or 50 cycles). However, Ni3S2/PEDOT can be cycled at a high current density due to the high electronic conductivity of PEDOT.35 Similarly, the Ni-supported Ni3S2/CSTs yield a higher initial capacity and longer cycle life than VS4/G (450.4 mA h g−1 in the first cycle and 237.1 mA h g−1 in the 50th cycle).39
Conclusions
In summary, we have designed a unique binder-free Na-storage anode by growing array-type Ni3S2/CSTs directly on a Ni foam substrate. The Ni3S2 exhibits a thin-sheet nanostructure and anchors both on the surface and inside of the cracked CSTs, forming a unique feather-like architecture. The Ni-supported Ni3S2/CSTs deliver a high reversible capacity of 887 mA h g−1 at 50 mA g−1 due to the small size and sheet-like structure of Ni3S2 and the dispersing effect of the CSTs. The array-type structure is preserved during repeated cycling and voids in between the arrays act as free space for volume changes upon sodiation/desodiation. It was found that the Ni3S2 on the CST surface is easy to exfoliate, resulting in a relatively rapid initial capacity fade, while the Ni3S2 inside the CSTs is firmly adhered to the CSTs, enabling good long-term cycling stability. After 260 cycles, a reversible capacity of over 200 mA h g−1 is still retained. The good electrochemical performance of the Ni-supported Ni3S2/CSTs makes them a promising anode for high-performance SIB anodes.Acknowledgements
This work was supported by the National Basic Research Program of China (2013CB934001), the National Natural Science Foundation of China (No. 51572238), Zhejiang Provincial Natural Science Foundation of China under Grant No. LY15E010004, the Fundamental Research Funds for the Central Universities (2016XZZX005-07), and the Program for Innovative Research Team in University of Ministry of Education of China (IRT13037).References
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 CAS.
- J. B. Dunn, L. Gaines, J. C. Kelly, C. James and K. G. Gallagherc, Energy Environ. Sci., 2015, 8, 158–168 CAS.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- L. P. Wang, L. H. Yu, X. Wang, M. Srinivasan and Z. J. Xu, J. Mater. Chem. A, 2015, 3, 9353–9378 CAS.
- S. W. Kim, D. H. Seo, X. H. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero González and T. Rojo, Energy Environ. Sci., 2012, 5, 5884–5901 CAS.
- V. Palomares, M. Casas Cabanas, E. Castillo Martínez, M. H. Han and T. Rojo, Energy Environ. Sci., 2013, 6, 2312–2337 CAS.
- D. Kundu, E. Talaie, V. Duffort and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3431–3448 CrossRef CAS PubMed.
- M. M. Doeff, Y. P. Ma, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 1993, 140, L169–L170 CrossRef CAS.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803–A811 CrossRef CAS.
- E. Irisarri, A. Ponrouch and M. R. Palacin, J. Electrochem. Soc., 2015, 162, A2476–A2482 CrossRef CAS.
- Z. L. Jian, B. Zhao, P. Liu, F. J. Li, M. B. Zheng, M. W. Chen, Y. Shi and H. S. Zhou, Chem. Commun., 2014, 50, 1215–1217 RSC.
- Y. Zhao, Z. X. Feng and Z. J. Xu, Nanoscale, 2015, 7, 9520–9525 RSC.
- N. Zhang, X. P. Han, Y. C. Liu, X. F. Hu, Q. Zhao and J. Chen, Adv. Energy Mater., 2015, 5, 1401123 CrossRef.
- Y. Q. Fu, Q. L. Wei, X. Y. Wang, G. X. Zhang, H. B. Shu, X. K. Yang, A. C. Tavares and S. H. Sun, RSC Adv., 2016, 6, 16624–16633 RSC.
- D. Y. Park and S. T. Myung, ACS Appl. Mater. Interfaces, 2014, 6, 11749–11757 CAS.
- P. Kollu, P. R. Kumar, C. Santosh, D. K. Kim and A. N. Grace, RSC Adv., 2015, 5, 63304–63310 RSC.
- L. H. Yu, L. P. Wang, S. B. Xi, P. Yang, Y. H. Du, M. Srinivasan and Z. J. Xu, Chem. Mater., 2015, 27, 5340–5348 CrossRef CAS.
- D. L. Chao, C. R. Zhu, X. H. Xia, J. L. Liu, X. Zhang, J. Wang, P. Liang, J. Y. Lin, H. Zhang, Z. X. Shen and H. J. Fan, Nano Lett., 2015, 15, 565–573 CrossRef CAS PubMed.
- G. He, L. J. Li and A. Manthiram, J. Mater. Chem. A, 2015, 3, 14750–14758 CAS.
- F. Zou, Y. M. Chen, K. W. Liu, Z. T. Yu, W. F. Liang, S. M. Bhaway, M. Gao and Y. Zhu, ACS Nano, 2016, 10, 377–386 CrossRef CAS PubMed.
- S. Yuan, X. L. Huang, D. L. Ma, H. G. Wang, F. Z. Meng and X. B. Zhang, Adv. Mater., 2014, 26, 2273–2279 CrossRef CAS PubMed.
- J. W. Wen, D. W. Zhang, Y. Zang, X. Sun, B. Cheng, C. X. Ding, Y. Yu and C. H. Chen, Electrochim. Acta, 2014, 132, 193–199 CrossRef CAS.
- J. P. Yang, T. F. Zhou, R. Zhu, X. Q. Chen, Z. P. Guo, J. W. Fan, H. K. Liu and W. X. Zhang, Adv. Mater. Interfaces, 2016, 3, 1500464 CrossRef.
- Y. D. Mo, Q. Ru, J. F. Chen, X. Song, L. Y. Guo, S. J. Hu and S. M. Peng, J. Mater. Chem. A, 2015, 3, 19765–19773 CAS.
- S. H. Choi and Y. C. Kang, Nanoscale, 2015, 7, 3965–3970 RSC.
- Y. C. Lu, C. Z. Ma, J. Alvarado, N. Dimov, Y. S. Meng and S. Okada, J. Mater. Chem. A, 2015, 3, 16971–16977 CAS.
- L. David, R. Bhandavat and G. Singh, ACS Nano, 2014, 8, 1759–1770 CrossRef CAS PubMed.
- S. H. Choi, Y. N. Ko, J. K. Lee and Y. C. Kang, Adv. Funct. Mater., 2015, 25, 1780–1788 CrossRef CAS.
- Y. Y. Lu, Q. Zhao, N. Zhang, K. X. Lei, F. J. Li and J. Chen, Adv. Funct. Mater., 2016, 26, 911–918 CrossRef CAS.
- Z. Shadike, M. H. Cao, F. Ding, L. Sang and Z. W. Fu, Chem. Commun., 2015, 51, 10486–10489 RSC.
- Y. C. Du, X. S. Zhu, X. S. Zhou, L. Y. Hu, Z. H. Dai and J. C. Bao, J. Mater. Chem. A, 2015, 3, 6787–6791 CAS.
- X. J. Xu, S. M. Ji, M. Z. Gu and J. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 20957–20964 CAS.
- C. Q. Shang, S. M. Dong, S. L. Zhang, P. Hu, C. J. Zhang and G. L. Cui, Electrochem. Commun., 2014, 50, 24–27 CrossRef.
- X. S. Song, X. F. Li, Z. M. Bai, B. Yan, D. J. Li and X. L. Sun, Nano Energy, 2016, 26, 533–540 CrossRef CAS.
- C. B. Zhu, P. Kopold, W. H. Li, P. A. van Aken, J. Maier and Y. Yu, J. Mater. Chem. A, 2015, 3, 20487–20493 CAS.
- A. Douglas, R. Carter, L. Oakes, K. Share, A. P. Cohn and C. L. Pint, ACS Nano, 2015, 9, 11156–11165 CrossRef CAS PubMed.
- R. M. Sun, Q. L. Wei, Q. D. Li, W. Luo, Q. Y. An, J. Z. Sheng, D. Wang, W. Chen and L. Q. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 20902–20908 CAS.
- M. S. Balogun, Y. Luo, W. T. Qiu, P. Liu and Y. X. Tong, Carbon, 2016, 98, 162–178 CrossRef CAS.
- S. P. Wu, R. Y. Ge, M. J. Lu, R. Xu and Z. Zhang, Nano Energy, 2015, 15, 379–405 CrossRef CAS.
- Y. Zhao, C. Wei, S. N. Sun, L. P. Wang and Z. J. Xu, Adv. Sci., 2015, 2, 1500097 CrossRef PubMed.
- C. B. Zhu, P. Kopold, W. H. Li, P. A. van Aken, J. Maier and Y. Yu, Adv. Sci., 2015, 2, 1500200 CrossRef PubMed.
- J. Liu, P. Kopold, C. Wu, P. A. van Aken, J. Maier and Y. Yu, Energy Environ. Sci., 2015, 8, 3531–3538 CAS.
- L. Y. Liang, Y. Xu, C. L. Wang, L. Y. Wen, Y. G. Fang, Y. Mi, M. Zhou, H. P. Zhao and Y. Lei, Energy Environ. Sci., 2015, 8, 2954–2962 CAS.
- H. K. Wang, X. P. Gao, J. K. Feng and S. L. Xiong, Electrochim. Acta, 2015, 182, 769–774 CrossRef CAS.
- W. C. Duan, W. C. Yan, X. Yan, H. Munakata, Y. C. Jin and K. Kanamura, J. Power Sources, 2015, 293, 706–711 CrossRef CAS.
- O. O. Balayeva, A. A. Azizov, M. B. Muradov, A. M. Maharramov, G. M. Eyvazova, R. M. Alosmanov, Z. Q. Mamiyevc and Z. A. Aghamaliyev, Mater. Res. Bull., 2016, 75, 155–161 CrossRef CAS.
- C. W. Su, J. M. Li, W. Yang and J. M. Guo, J. Phys. Chem. C, 2014, 118, 767–773 CAS.
- A. Morelos Gómez, P. G. Mani González, A. E. Aliev, E. Muñoz Sandoval, A. Herrera Gómez, A. A. Zakhidov, H. Terrones, M. Endo and M. Terrones, Adv. Funct. Mater., 2014, 24, 2612–2619 CrossRef.
- X. Wei, W. H. Li, J. A. Shi, L. Gu and Y. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 27804–27809 CAS.
- W. Qin, T. Q. Chen, T. Lu, D. H. C. Chua and L. K. Pan, J. Power Sources, 2016, 302, 202–209 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of ZnO on Ni and Ni foam, XRD patterns, Raman spectra and BET isotherms of Ni-supported Ni3S2/CSTs, SEM and cycling performance of the control sample, and a comparison of cycle life of some sulfides for Na storage. See DOI: 10.1039/c6qi00286b |
| This journal is © the Partner Organisations 2017 |