
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron(II) β-ketiminate complexes as mediators of controlled radical polymerisation†
Benjamin R. M.
Lake
 and
Michael P.
Shaver
*
and
Michael P.
Shaver
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: michael.shaver@ed.ac.uk
First published on 9th May 2016
Abstract
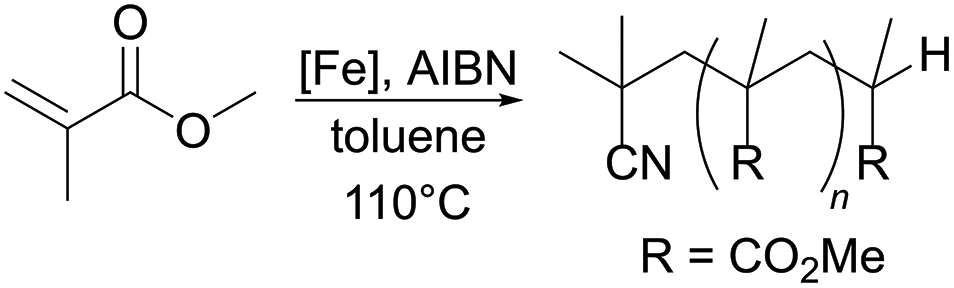
A series of tridentate, ONO- and ONN-chelating β-ketiminate ligands were synthesised via condensation reactions, and complexed with iron(II) using [Fe(N(SiMe3)2)2THF]. The complexation reactions proceeded in high yields to generate novel, monomeric, tetracoordinate iron(II) complexes, each bearing a bis(trimethylsilyl)amide ligand, as confirmed by X-ray crystallography. These complexes were amenable to further reaction (protonolysis) with alcohols and phenols, generating alkoxide/phenolate-containing complexes that were dimeric in the solid state. All complexes synthesised were screened as potential mediators of the controlled radical polymerisation (CRP) of styrene and methyl methacrylate under both atom transfer radical polymerisation (ATRP) and organometallic mediated radical polymerisation (OMRP) conditions. Whilst all of the complexes were relatively poor ATRP mediators under the conditions used here, regardless of monomer choice, dispersities (Đ) as low as 1.58 for styrene and 1.23 for methyl methacylate polymerisation under OMRP conditions could be achieved. The better performance in methacrylate polymerisation suggests the formation of a stronger metal–carbon bond in these systems. In particular, the use of a β-ketiminate ligand functionalised with an N,N-dimethylethylene pendant arm and a 2,6-diphenylphenolate ligand affords a potential Fe-based mediator of methyl methacrylate OMRP.
Introduction
The application of iron compounds as catalysts in organic chemistry is a vibrant area of research.1 This is due, in part, to the low cost of iron, its high crustal abundance, and its biocompatibility. Furthermore, the ability of iron to adopt a range of oxidation states (−2 to +5) and spin-states allows it to display remarkably variable reactivity, thus allowing it to participate in a wide range of chemical reactions. Indeed, soon after the pioneering initial reports by the groups of Matyjaszewski2 and Sawamoto3 on the controlled radical polymerisation (CRP) of alkene monomers, the first reports of the use of iron complexes as mediators of this important reaction began to appear.4 The use of iron complexes as mediators of CRP has now grown so considerably, that a number of reviews covering this thriving field have been recently published.5As part of our continuing studies on utilising new iron-based mediators for CRP6 and understanding the underlying mechanisms by which these complexes impart control over polymerisation reactions,7 we sought to develop a new series of iron complexes based on the β-ketiminate ligand scaffold. Iron-β-ketiminate complexes have been scarcely used as mediators of CRP,5a,c with only one report to the best of our knowledge published to date.8 We specifically chose β-ketiminates to support our iron complexes, due to the ease with which these ligands are synthesised and the inexpensive starting materials required to make them, both of which would be especially attractive features for use at production scale.9 Furthermore, the electronic and steric characteristics can be controlled by tuning the β-ketiminate scaffold, including through the introduction of additional donors. This ability to shape the coordination sphere could be especially important as we pursue systems that can reversibly trap radicals via the formation of a new metal–carbon bond.
In this report, we detail the synthesis of α,β-unsaturated-β-ketoamines bearing pendant amine, ether and pyridyl donors. The reaction of these compounds with the iron-containing precursor [Fe(N(SiMe3)2)2THF] produced a series of four-coordinate complexes, with bound bis(trimethylsilyl)amide donors. A second family of catalysts was generated through protonolysis of these compounds with benzyl alcohol or 2,6-diphenylphenol, yielding dimeric (in the solid state) iron(II) complexes bearing alkoxide or phenolate ligands. All of the iron complexes synthesised were examined as mediators of CRP under both atom transfer radical polymerisation (ATRP, alkyl halide initiator) and organometallic mediated radical polymerisation (OMRP, azo initiator) conditions.
By using iron(II) rather than the more stable iron(III) complexes, we are able to separate out the halogen and organometallic mechanisms and draw conclusions about the role of both ATRP and OMRP equilibria (vide infra) in imparting control over the polymerisation reactions described herein. The information gained during the course of these studies is additionally of potential relevance to all metal mediated CRP reactions, and is helping to guide our development of effective iron-based mediators.
Results and discussion
Synthesis and characterisation
The synthesis of α,β-unsaturated-β-ketoamines, precursors to β-ketiminates, bearing pendant donor functionalities was achieved via the condensation of acetylacetone and the appropriate primary amine at reflux (Scheme 1). Contrary to previously published reports,10 we found that the reactions proceeded efficiently in methanol without the need for an acid catalyst. The ligand precursors L1H and L2H were obtained as pale yellow oils following purification by vacuum distillation, while L3H and L4H could be obtained as colourless crystalline solids by recrystallisation. All four α,β-unsaturated-β-ketoamines display broad singlet resonances between 10.77–11.26 ppm (CDCl3), characteristic of the hydrogen-bonded amine proton.11 Ligand precursors L3H and L4H also each show a doublet resonance (J ≈ 6.5 Hz) at 4.57 and 4.52 ppm (CDCl3), respectively, corresponding to the picolyl-CH2 protons coupling to the amine proton. | ||
| Scheme 1 Synthesis of L1H–L4H. | ||
Previous publications have reported the synthesis of transition metal complexes bearing donor-tethered β-ketiminates using various strategies, including by reaction of the ligand precursor and metal salt in the presence of a base10a and without a base,12 by reaction of the ligand precursor with a highly basic metal starting reagent13 and by transmetalation from an alkali metal-β-ketiminate complex.10c,14 We decided to use a simple strategy and form our desired FeII(L)(N(SiMe3)2)-type complexes in a one-pot reaction using [Fe(N(SiMe3)2)2THF] as the basic metalating reagent. Indeed, reaction of the α,β-unsaturated-β-ketoamines, L1H–L4H, with an equimolar amount of [Fe(N(SiMe3)2)2THF] in hexane or toluene led to the formation of green-yellow solutions/suspensions, from which, highly oxygen and moisture-sensitive solids could be obtained upon work-up (Scheme 2). The products (1a–4a) were obtained in good yields (>72%) and isolated as green crystalline solids. Characterisation of 1a–4a by 1H NMR spectroscopy revealed paramagnetically-shifted spectra, with a series of broad resonances between approximately −60 and 180 ppm present in each spectrum (see ESI†). The solution magnetic moments of these complexes were suggestive of d6 high-spin electron configurations at ambient temperature, with calculated values (4.9–5.4μB) congruent with the spin-only magnetic moment of a high-spin iron(II) centre (4.90μB). Single crystals of all four complexes (1a–4a) suitable for X-ray diffraction analysis could be obtained, either directly from the bulk material, or by cooling a saturated n-hexane solution of the product to −35 °C. The molecular structures of complexes 1a–4a are provided in Fig. 1 and 2 along with selected bond lengths and angles. All four complexes have crystallised as monomeric, four-coordinate species, with the coordination sphere of each comprising the N,O-donors of the β-ketiminate backbone, the heteroatom of the tethered donor and the N atom of a bis(trimethylsilyl)amide. The coordination geometries about all four iron(II) centres (1a–4a) can be described as distorted seesaw according to the four-coordinate geometry index proposed by Houser in the pages of this journal,15 with τ4 values of 0.60, 0.56, 0.57 and 0.54 respectively. The coordination geometries can alternatively be described using Alvarez's system,16 which suggests a tendency towards an intermediate (spread) geometry. This geometry is extremely rare indeed for d6 metal ions,16a and it is likely that the unusual coordination geometry is imposed by a combination of the conformational requirements of the relatively rigid tridentate β-ketiminate ligand, and the extreme steric bulk imparted by the bis(trimethylsilyl)amide donor. The bond metrics of 1a–4a are comparable to those of the iron(II)-β-ketiminates (though most of these are iron(II)-bis-β-ketiminate complexes) reported in the literature so far,8,11,17 with Oketiminate–Fe bond lengths of 1.96 Å and Nketiminate–Fe bond lengths of between 2.04–2.06 Å. The Nhmds–Fe bond lengths of 1.95–1.96 Å are slightly longer than those of the three-coordinate starting material (1.92 Å), [Fe(N(SiMe3)2)2THF],18 but compare reasonably well with the Nhmds–FeII bond lengths in other reported four-coordinate complexes containing a coordinated bis(trimethylsilyl)amide.19
 | ||
| Scheme 2 Synthesis of 1a–4a. | ||
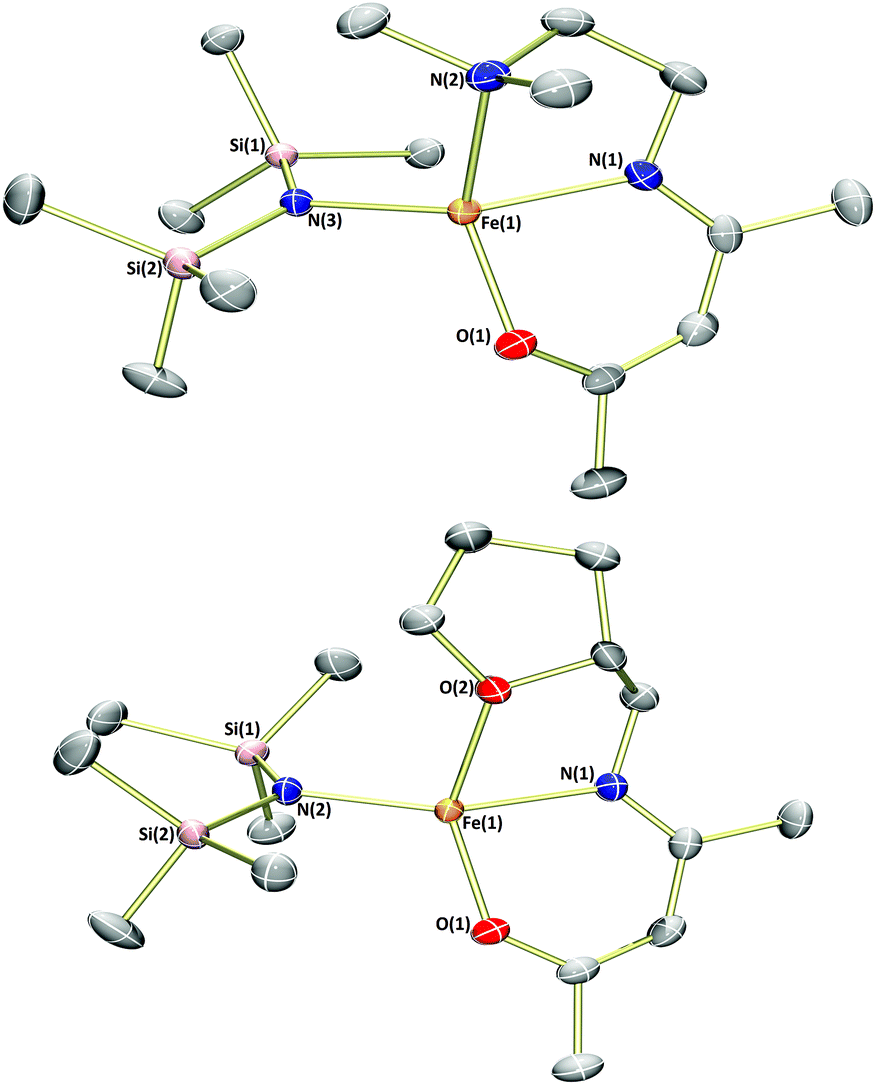 | ||
| Fig. 1 Molecular structures of 1a (top) and 2a (bottom) with ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): 1a – Fe1–O1 1.9601(11), Fe1–N1 2.0409(13), Fe1–N2 2.2342(13), Fe1–N3 1.9596(13), O1–Fe1–N2 134.54(5), O1–Fe1–N1 90.24(5), N3–Fe1–O1 110.48(5), N3–Fe1–N2 105.08(5), N3–Fe1–N1 140.40(5), N1–Fe1–N2 79.40(5). 2a – Fe1–O1 1.9614(9), Fe1–N1 2.0559(10), Fe1–O2 2.1418(9), Fe1–N2 1.9472(9), O1–Fe1–O2 133.40(4), O1–Fe1–N1 89.56(4), N1–Fe1–O2 76.66(4), N2–Fe1–O1 113.98(4), N2–Fe1–O2 98.99(4), N2–Fe1–N1 148.22(4). | ||
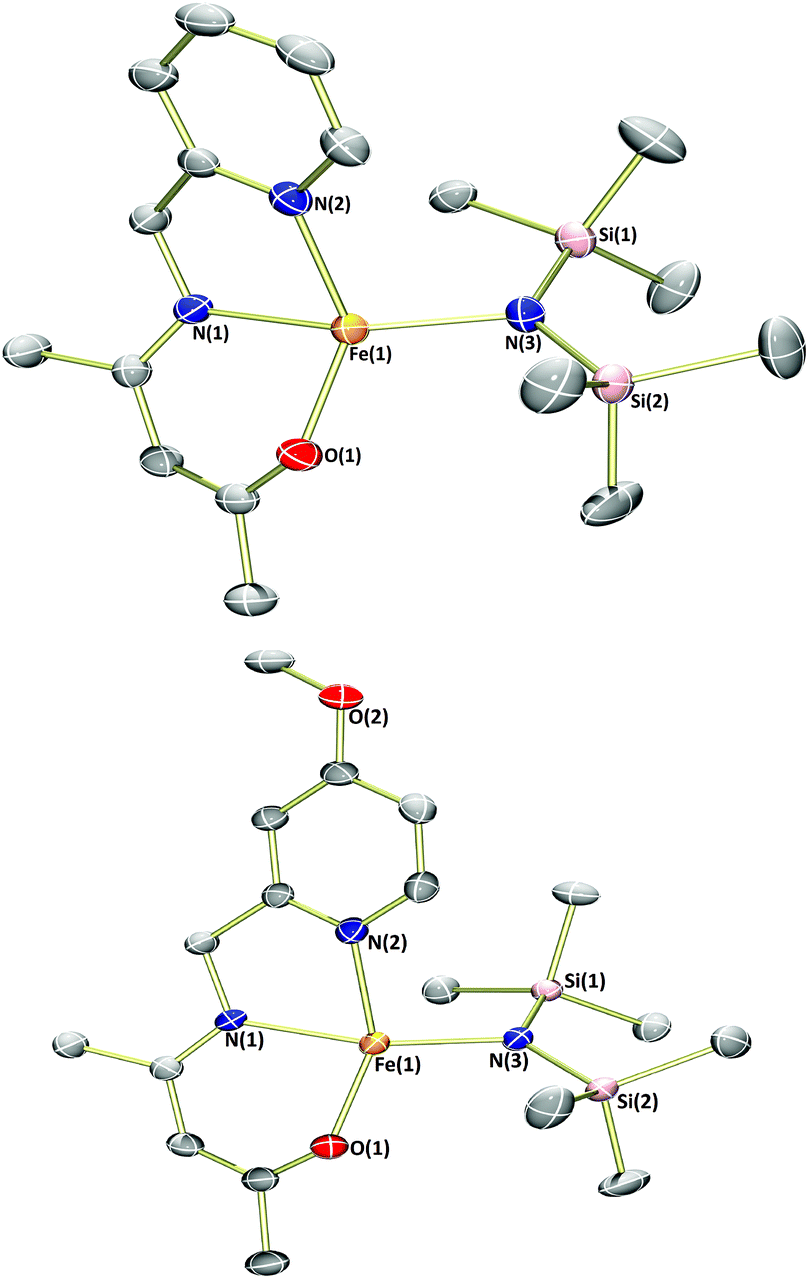 | ||
| Fig. 2 Molecular structures of 3a (top) and 4a (bottom) with ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (°): 3a – Fe1–O1 1.9599(14), Fe1–N1 2.0598(16), Fe1–N2 2.1443(17), Fe1–N3 1.9574(17), O1–Fe1–N1 88.95(6), O1–Fe1–N2 132.28(6), N1–Fe1–N2 77.67(7), N3–Fe1–O1 112.75(7), N3–Fe1–N1 147.36(7), N3–Fe1–N2 101.77(7). 4a – Fe1–O1 1.9638(13), Fe1–N1 2.0601(14), Fe1–N2 2.1430(16), Fe1–N3 1.9547(14), O1–Fe1–N1 89.02(6), O1–Fe1–N2 140.71(6), N1–Fe1–N2 77.97(6), N3–Fe1–O1 111.83(6), N3–Fe1–N1 143.53(6), N3–Fe1–N2 99.82(6). | ||
The bis(trimethylsilyl)amide-containing complexes 1a and 3a were amenable to protonolysis reactions (complexes 2a and 4a were not examined). Reaction of either of these two complexes with a hexane solution of benzyl alcohol led to an immediate and distinct colour change from green-yellow to red or orange. After a simple work-up, the products 1b and 3b were isolated as orange and red solids respectively (Scheme 3).
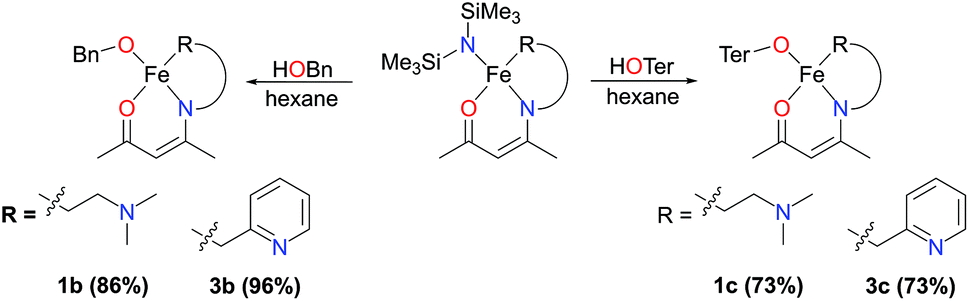 | ||
| Scheme 3 Synthesis of complexes 1b, 3b, 1c and 3c. | ||
Analysis of the products by 1H NMR spectroscopy again revealed a series of paramagnetically-shifted resonances, with solution magnetic moment data (μeff = 5.4 and 5.2μB, respectively) indicative of d6 high-spin electron configurations. Single crystals of complex 1b suitable for X-ray diffraction analysis were obtained on cooling of a saturated n-hexane solution from reflux (Fig. 3). The solid-state structure of 1b reveals a dimer, with (μ2-OBn)2 bridging the two iron(II) centres. The two halves of the dimer are related to each other through a crystallographic inversion centre, located in the centre of the Fe2O2 rhombus, with each five-coordinate iron(II) possessing a coordination geometry best described as square pyramidal. Single crystals of complex 3b, obtained from a dilute toluene/n-hexane solution stored at −35 °C, show that this complex is isostructural with 1b.
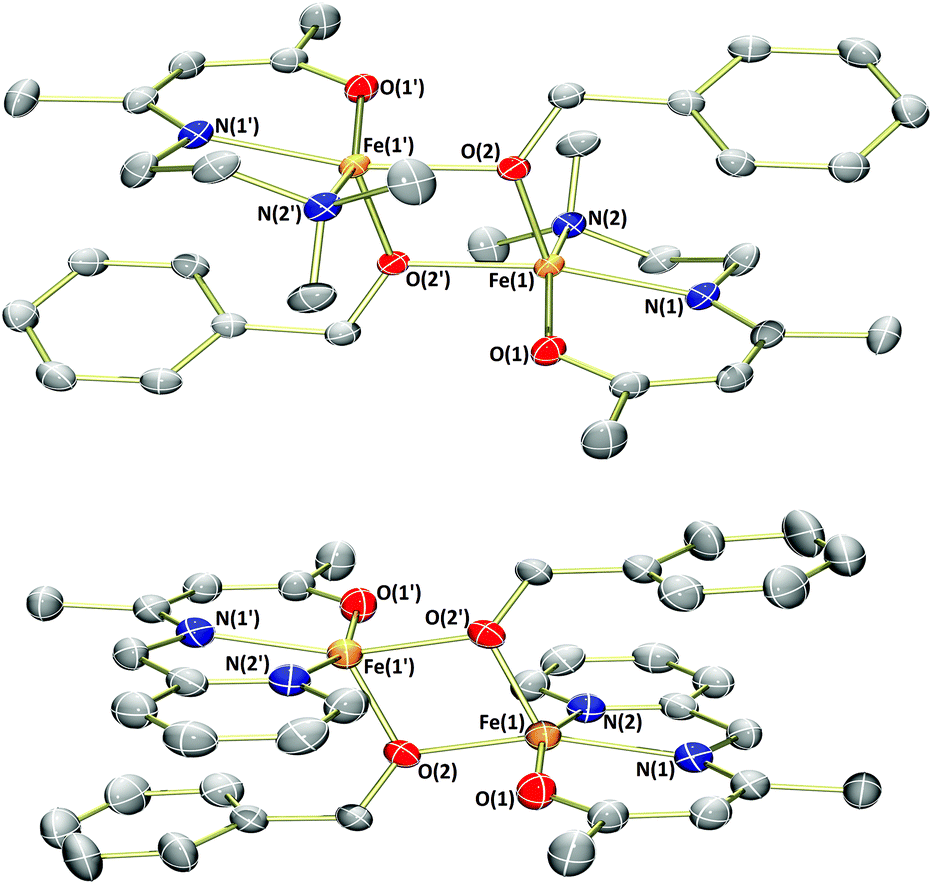 | ||
| Fig. 3 Molecular structures of 1b (top) and 3b (bottom) with ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): 1b – Fe1–O1 2.0223(10), Fe1–O2 2.0354(9), Fe1–O2′ 2.0436(9), Fe1–N1 2.0908(12), Fe1–N2 2.2864(12). 3b – Fe1–O1 1.994(4), Fe1–O2 2.047(4), Fe1–O2′ 2.061(4), Fe1–N1 2.102(4), Fe1–N2 2.171(5). | ||
As an electronic and steric contrast between the bulky amide-containing complexes 1a–4a, and the relatively non-bulky alkoxide-containing complexes 1b and 3b, we decided to synthesise bulky phenoxide-containing complexes of iron(II) bearing our tridentate β-ketiminate ligands. Terphenolate was chosen in this case, as it has been shown to support iron in the +2 and +3 oxidation states and in a number of different coordination geometries.20 Furthermore, it has been shown that the steric bulk provided by terphenolate ligands can enhance reactivity by protecting a coordination site at the metal centre.21 Similarly to the synthesis of benzyl alkoxide-substituted complexes 1b and 3b, reaction of parent complexes 1a and 3a with 2,6-diphenylphenol (HOTer) led to immediate colour changes, and isolation of yellow/orange solids following work-up (Scheme 3). The terphenolate-substituted complexes 1c and 3c were obtained in good yields (73%), and could be fully characterised, including by single crystal X-ray diffraction analysis (Fig. 4). The solid-state structure of 1c again reveals a dimer, with the crystallographic asymmetric unit containing two structurally similar molecules of dimer and two molecules of solvent (see ESI†), though both the solvent and one dimer have been omitted for clarity's sake in Fig. 4. Complex 1c contains iron(II) centres bridged by the two oxygen atoms of the two substituted β-ketiminates, with the terphenolate ligands bound in a terminal fashion. The central Fe2O2 rhombus of both dimers is noticeably puckered, while those of complexes 1b and 3b are planar, which presumably helps alleviate steric clashing between adjacent terphenolate aromatic rings. The four iron(II) centres contained within the two dimers of the asymmetric unit possess a range of coordination geometries, from slightly distorted square pyramidal to intermediate. Given the bridging nature of the β-ketiminate oxygen atoms, it is not surprising that the Oketiminate–Fe bond distances are typically somewhat longer than those found in either 1a or 1b, with lengths of between 2.05–2.18 Å. In contrast to 1c, the two iron(II) centres of complex 3c are bridged by an oxygen atom of one of the β-ketiminates and an oxygen atom of one of the terphenolates. The reason for this bridging mode is somewhat unclear, though maximising intramolecular π–π stacking interactions between the terphenolate and pyridyl rings could be a contributory factor.
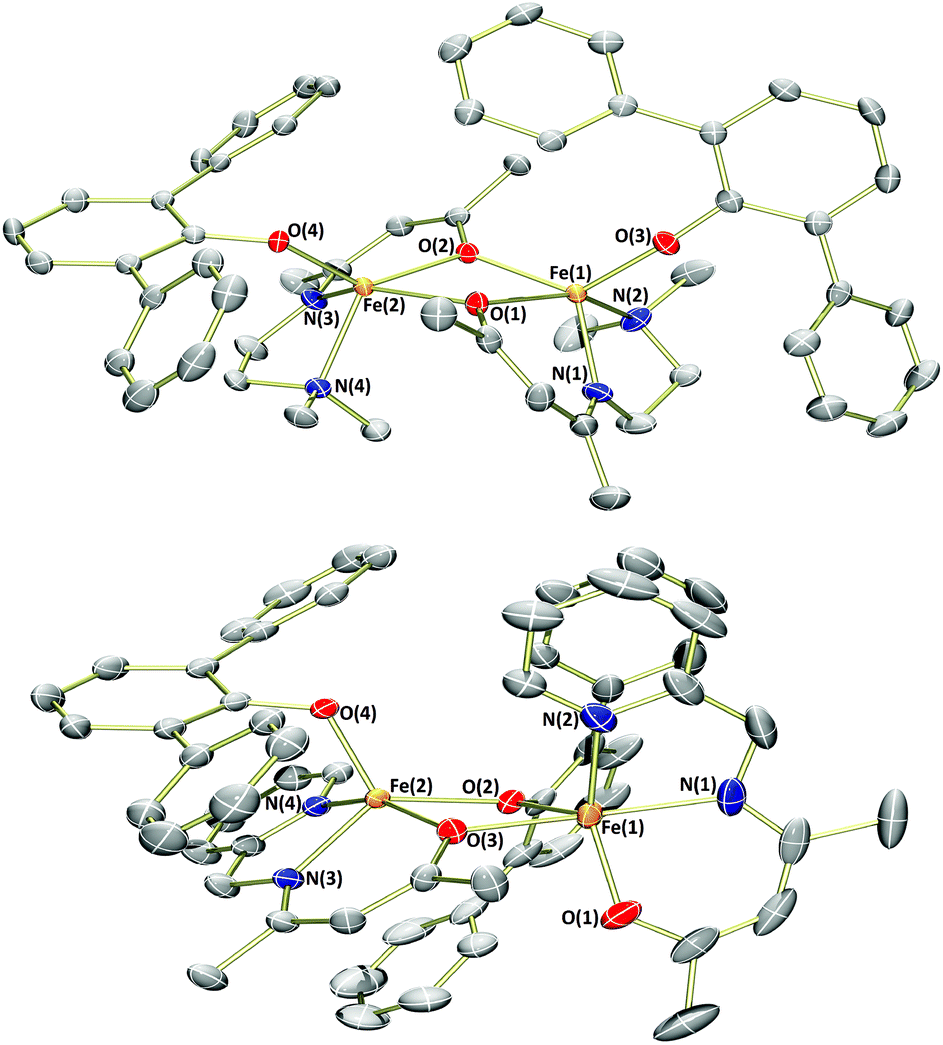 | ||
| Fig. 4 Molecular structures of 1c (top) and 3c (bottom) with ellipsoids set at the 50% probability level. Hydrogen atoms and co-crystallised solvent have been omitted for clarity. Selected bond lengths (Å): 1c – Fe1–O1 2.097(2), Fe1–O2 2.100(2), Fe1–O3 1.894(2), Fe1–N1 2.109(3), Fe1–N2 2.248(3), Fe2–O1 2.176(2), Fe2–O2 2.049(2), Fe2–O4 1.942(2), Fe2–N3 2.126(3), Fe2–N4 2.228(3). 3c – Fe1–O1 1.985(2), Fe1–O2 2.045(2), Fe1–O3 2.120(2), Fe1–N1 2.084(3), Fe1–N2 2.133(3), Fe2–O2 2.107(2), Fe2–O3 2.077(2), Fe2–O4 1.970(2), Fe2–N3 2.080(3), Fe2–N4 2.154(3). | ||
Controlled radical polymerisation
The two main equilibria by which metal-mediated CRP proceeds are ATRP and OMRP (Scheme 4).22 ATRP involves the reversible transfer of a halogen between a metal centre and propagating radical, while metal–carbon bonds are reversibly formed in OMRP. ATRP and OMRP equilibria are able to operate simultaneously via the same iron(II) species when reactions are performed under ATRP conditions (alkyl halide initiator), though an OMRP-only regime can be accessed in the absence of alkyl halide (using an azo initiator). For reactions set-up under ATRP conditions, the degree of involvement of an OMRP mechanism is highly dependent on the metal centre, ligand environment and monomer involved.5b,7a,b,d,9,23 | ||
| Scheme 4 Equilibria implicated in CRP.6b | ||
Given this paucity of literature on iron-mediated OMRP, it is not surprising that only a single report has described the polymerisation of either styrene or MMA under purely OMRP conditions (i.e. in the complete absence of halide).7d Table S3† presents the data obtained for the polymerisation of styrene under OMRP conditions using our range of iron(II) complexes and 1 equivalent of AIBN as the initiator. Most of the complexes tested exhibit little or no control over the OMRP of styrene, with the Đ of these reactions being greater than 1.8. However, picolyl-containing complexes 3a and 4a display a moderate degree of control over the reactions, with Đs of 1.58 and 1.61 being achieved. This represents a much higher degree of control under these conditions than we were able to achieve using our best performing iron(II) amine-bis(phenolate) complex,7d and may suggest improved carbon radical trapping by these systems. The presence of a picolyl donor and HMDS ligand appear important, since switching either the tethered donor (to a tertiary amine or ether) or the ancillary donor (to an alkoxide/phenoxide) reduces control significantly. However, in all cases the theoretical molecular weights are somewhat lower than the values obtained via GPC, indicating loss of a significant number of radicals before an OMRP equilibrium is established.
Based on our previous findings,7d we anticipated that control over the OMRP of MMA would be much easier to achieve (than styrene) given the apparent greater affinity of iron complexes for MMA-type radicals. Table 1 illustrates the screening data obtained for the OMRP of MMA under the same conditions as those used for styrene OMRP. It is evident that many of the complexes are reasonably efficient mediators of MMA OMRP, with Đs of <1.53 for all complexes tested. The picolyl-substituted complexes (3a, 4a, 3b and 3c) lead to significantly lower conversions, however, than the amine and ether-tethered complexes. Complex 3c specifically gave a very low conversion (9%) to polymer, with minimal solid visible after attempted precipitation in acidified methanol. The top performing complex was 1c (Đ = 1.33), where the electron-withdrawing terphenolate ligand can both sterically protect the iron centre and promote increased Lewis acidity and a stronger metal–carbon bond. Attempts were made to improve the performance of complex 1c (entry 7, Tables 1 and 2). It was observed that halving the number of equivalents of AIBN (entry 9, Table 2) led to a decrease in Đ (to 1.23) and, as expected, a decrease in conversion. Since each molecule of AIBN generates two radicals upon thermal decomposition, at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (AIBN:Fe) there are two radicals per iron(II) centre. This excess of radicals (with respect to iron(II)) should result in more termination at the early stages of the reaction. However, by halving the amount of AIBN (entry 9, Table 2), there is no longer an excess of radicals per iron(II) centre, which may help reduce termination reactions and thus improve dispersity. The use of the alternative radical initiators, V-601 (10 hour t1/2 = 66 °C), a non-nitrile initiator with a similar decomposition profile to AIBN or V-65 (10 hour t1/2 = 51 °C), a lower temperature radical initiator, led to very similar results to those obtained with AIBN (entries 10 and 11, Table 2). Note that the reaction using V-65 was performed for 2 hours at a lower temperature (90 °C), since we anticipated a lower rate of propagation and thus slower conversion to polymer. At this lower temperature, it appears that trapping of the propagating radicals is as efficient and reversible as at the higher reaction temperature. Given the rapid rate of initiator decomposition at these temperatures, this should leave the decomposition of the so-formed organometallic complex as the only source of radicals. Thus it is likely that the reaction proceeds via an RT-OMRP (reversible termination-OMRP) mechanism rather than a DT-OMRP mechanism (degenerative transfer-OMRP). The use of THF (entry 12) as solvent, or performing the reaction in the bulk (entry 13) both had deleterious on reaction control, raising Đ to 1.57 and 5.13 respectively. These results together imply that minimal stabilisation of the metal centre occurs in the presence of coordinating solvent, and that in the absence of solvent (or in the presence of a relatively volatile solvent (THF)), propagation and bimolecular coupling of radical chains are rapid giving high conversions and broad Đ. However, doubling the volume of additional solvent (entry 15) had no further positive effect on control over the reaction.
:1 (AIBN:Fe) there are two radicals per iron(II) centre. This excess of radicals (with respect to iron(II)) should result in more termination at the early stages of the reaction. However, by halving the amount of AIBN (entry 9, Table 2), there is no longer an excess of radicals per iron(II) centre, which may help reduce termination reactions and thus improve dispersity. The use of the alternative radical initiators, V-601 (10 hour t1/2 = 66 °C), a non-nitrile initiator with a similar decomposition profile to AIBN or V-65 (10 hour t1/2 = 51 °C), a lower temperature radical initiator, led to very similar results to those obtained with AIBN (entries 10 and 11, Table 2). Note that the reaction using V-65 was performed for 2 hours at a lower temperature (90 °C), since we anticipated a lower rate of propagation and thus slower conversion to polymer. At this lower temperature, it appears that trapping of the propagating radicals is as efficient and reversible as at the higher reaction temperature. Given the rapid rate of initiator decomposition at these temperatures, this should leave the decomposition of the so-formed organometallic complex as the only source of radicals. Thus it is likely that the reaction proceeds via an RT-OMRP (reversible termination-OMRP) mechanism rather than a DT-OMRP mechanism (degenerative transfer-OMRP). The use of THF (entry 12) as solvent, or performing the reaction in the bulk (entry 13) both had deleterious on reaction control, raising Đ to 1.57 and 5.13 respectively. These results together imply that minimal stabilisation of the metal centre occurs in the presence of coordinating solvent, and that in the absence of solvent (or in the presence of a relatively volatile solvent (THF)), propagation and bimolecular coupling of radical chains are rapid giving high conversions and broad Đ. However, doubling the volume of additional solvent (entry 15) had no further positive effect on control over the reaction.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Complex | Conv. (%) | M n,th [AIBN] (Da) | M n,th [Fe] (Da) | M n (Da) | Đ |
|
a Conditions: [MMA]:[FeII]:[AIBN] = 100:1.00:1.00, MMA:toluene = 1:1 (v/v), 110 °C, 1 hour. Conversion determined by 1H NMR spectroscopy. Mn,th [AIBN] = [MMA]0/(2 × [AIBN]0) × M(MMA) × conversion. Mn,th [Fe] = [MMA]0/[Fe] × M(MMA) × conversion.
b Too little polymer obtained for GPC analysis.
|
||||||
| 1 | 1a | 73 | 3654 | 7308 | 11764 |
1.47 |
| 2 | 2a | 69 | 3454 | 6908 | 11704 |
1.37 |
| 3 | 3a | 13 | 651 | 1302 | 13862 |
1.49 |
| 4 | 4a | 22 | 1101 | 2202 | 11128 |
1.53 |
| 5 | 1b | 56 | 2803 | 5606 | 10229 |
1.45 |
| 6 | 3b | 36 | 1802 | 3604 | 10123 |
1.41 |
| 7 | 1c | 54 | 2703 | 5406 | 10734 |
1.33 |
| 8 | 3c | 9 | 451 | 902 | ||
| Entry | Initiator/equiv. | Solvent | Conv. (%) | M n,th [AIBN] (Da) | M n,th [Fe] (Da) | M n (Da) | Đ |
|---|---|---|---|---|---|---|---|
|
a Conditions: [MMA]:[FeII]:[initiator] = 100:1.00:X, MMA:solvent = 1:1 (v/v), 110 °C, 1 hour. Conversion determined by 1H NMR spectroscopy. Mn,th [AIBN] = [MMA]0/(2 × [AIBN]0) × M(MMA) × conversion. Mn,th [Fe] = [MMA]0/[Fe] × M(MMA) × conversion.
b Reaction performed for 2 hours at 90 °C.
c Bimodal distribution.
d 0.5 equivalents of 1c.
e MMA:toluene = 1:2 (v/v).
|
|||||||
| 7 | AIBN/1 | Toluene | 54 | 2703 | 5406 | 10734 |
1.33 |
| 9 | AIBN/0.5 | Toluene | 20 | 2002 | 2002 | 10376 |
1.23 |
| 10 | V-601/1 | Toluene | 62 | 3104 | 6208 | 12672 |
1.35 |
| 11b | V-65/1 | Toluene | 55 | 2753 | 5506 | 11112 |
1.34 |
| 12 | AIBN/1 | THF | 67 | 3354 | 6708 | 10545 |
1.57 |
| 13 | AIBN/1 | Neat | 70 | 3504 | 7008 | 13628 |
5.13c |
| 14d | AIBN/1 | Toluene | 68 | 3404 | 13616 |
11605 |
1.56 |
| 15 | AIBN/1 | Toluenee | 43 | 2153 | 4306 | 9755 | 1.38 |
As described previously, complex 3c gave an especially poor conversion to polymer, even with the use of an excess of radical initiator. We sought to examine the fate of complex 3c and thus, presumably, the reason for this low conversion by reacting it with MMA-type radicals, which can be generated by thermal decomposition of the azo initiator, V-601. The reaction was observed to darken over time, and an amount of black solid along with a small number of black crystals could be obtained via vapour diffusion of n-hexane in to the crude reaction mixture (Fig. 5). The molecular structure of the complex obtained (3c′) illustrates a monomeric iron(III) complex, bearing two terphenolate donors. Of particular note in this structure is the substitution of a picolyl H atom for a methyl isobutyrate group, the methyl isobutyrate being derived from the decomposition of V-601. While it is difficult to speculate on the origin of this complex without further experimental corroboration, we suggest that it is likely formed via initial H atom abstraction by one equivalent of methyl isobutyrate radical, followed by radical–radical combination of the so-formed β-ketiminate-derived radical with a further methyl isobutyrate radical. The addition of a further methyl isobutyrate group to the picolyl tether is likely precluded by steric factors. The loss of radicals via reaction with the ligand scaffold in this manner will certainly suppress productive polymerisation, hence accounting for the very low conversion obtained for complex 3c. This type of reactivity may account for the lower conversions obtained using the other complexes bearing picolyl tethers (cf. entries 3, 4, 6 and 8, Table 1).
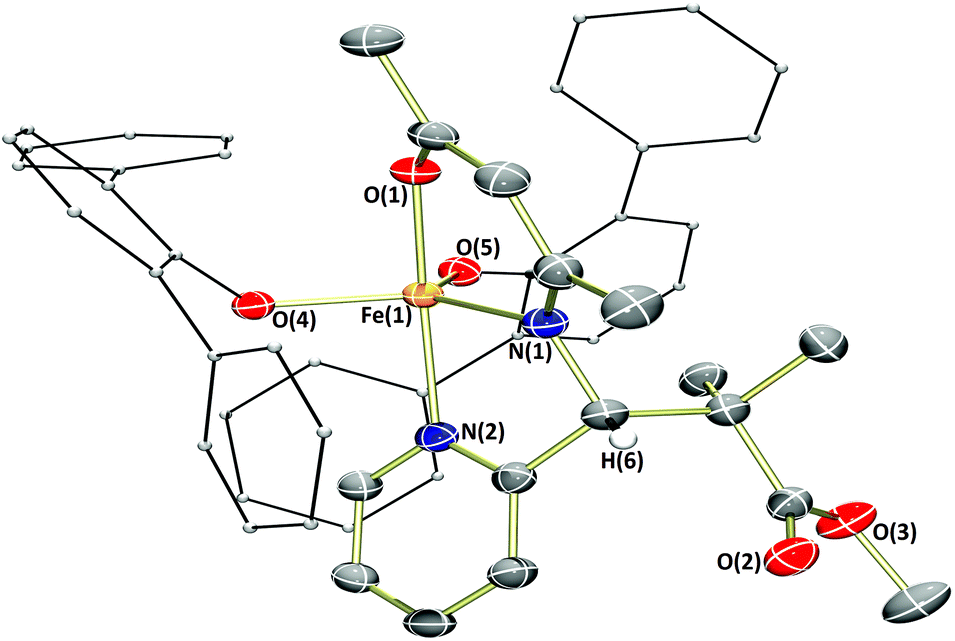 | ||
| Fig. 5 Molecular structures of 3c′ with ellipsoids set at the 50% probability level. Hydrogen atoms (except picolyl H atom) have been omitted for clarity. Terphenolate rings represented in ball and stick form for clarity. Selected bond lengths (Å): 3c′ – Fe1–O1 1.9376(12), Fe1–O4 1.8995(12), Fe1–O5 1.8554(12), Fe1–N1 2.0856(15), Fe1–N2 2.1341(14). | ||
While the lower dispersities in most OMRP reactions described here imply some level of control over the radical polymerisation, the deviation from unity also suggests that termination reactions must be occurring. This is further supported by kinetic analysis of the OMRP of MMA mediated by complex 1a (see ESI†), which shows that molecular weights top out at higher conversions. Thus, catalytic chain transfer events are kinetically competitive with propagation, as has been observed previously in α-diimine iron systems.25
Conclusions
A series of donor-tethered tridentate β-ketiminate ligands have been synthesised and coordinated to iron(II) to afford four-coordinate complexes bearing a coordinated bis(trimethylsilyl)amide group. Protonolysis of these complexes with either benzyl alcohol or 2,6-diphenylphenol led to the formation of dimeric iron(II) complexes. All of the complexes synthesised were examined as mediators of styrene and methyl methacrylate polymerisation under both ATRP, and OMRP conditions. While all complexes were generally very poor mediators of ATRP, Đs as low as 1.23 could be achieved in the OMRP of MMA. Furthermore, through crystallographic characterisation of a decomposition product, we provide evidence that the presence of reactive picolyl H atoms may have a detrimental effect on a complex's ability to act as an efficient mediator of CRP. Clear design principles to manage metal–halogen and metal–carbon bond strengths are emerging and continue to guide our design of iron-based catalysts in promoting controlled radical polymerisation and limiting chain transfer. As we understand the role of these complexes in controlling radical chemistry, we can consider applying them to other transformations. In particular, the application of these complexes (1a–3c) as catalysts for small molecule transformations invoking radical chemistry is also an area of interest to our group,26 and will underpin future studies.Experimental
Materials and methods
All experiments involving moisture- and air-sensitive compounds were performed under a nitrogen atmosphere using an MBraun LABmaster sp glovebox system equipped with a −35 °C freezer and [H2O] and [O2] analysers or using standard Schlenk techniques. Solvents used were obtained from a solvent purification system (Innovative Technologies) consisting of columns of alumina and copper catalyst and were further degassed by three freeze–pump–thaw cycles prior to use. Benzene-d6 and THF-d8 were dried by stirring over sodium/benzophenone, before being collected by distillation and degassed by three freeze–pump–thaw cycles. Chloroform-d1 was used as received. Styrene and methyl methacrylate (MMA) were dried by stirring over calcium hydride for a minimum of 24 hours, before being vacuum transferred and stored at −35 °C. 2,2′-Azobis(2-methylpropionitrile) (AIBN), V-601 (Wako) and V-65 (Wako) were recrystallised from DCM/hexane, dried under vacuum and stored at −35 °C. (1-Chloroethyl)benzene (1-PECl) and benzyl alcohol were dried by stirring over calcium hydride for a minimum of 24 hours, before being distilled. [Fe(N(SiMe3)2)2THF] was synthesised using a modified literature procedure.18 (4-Methoxypyridin-2-yl)methanamine was synthesised using a literature procedure.27 Following purification, all reagents described above were stored under an inert atmosphere. 2,6-Diphenylphenol, acetylacetone, N,N-dimethylethylenediamine, 2-picolylamine and tetrahydrofurfurylamine were all used as received. Gel permeation chromatography (GPC) was carried out in THF at a flow rate of 1 mL min−1 at 35 °C on a Malvern Instruments Viscotek 270 GPC Max triple detection system with 2× mixed bed styrene/DVB columns (300 × 7.5 mm). Absolute molar masses were obtained using dn/dc values of 0.185 for poly(styrene)28 and 0.088 for poly(methyl methacrylate).29 NMR spectra were obtained on either a 400 MHz or 500 MHz Bruker Avance III spectrometer. Solution magnetic moments were determined via NMR spectroscopy using Evans’ method.30 Mass spectra were obtained on a Bruker Daltonics micro TOF instrument operating in the positive ion electrospray mode. Elemental analyses were performed by Stephen Boyer at London Metropolitan University.Synthetic procedures
CRP procedures
:monomer, 1:1, v/v). To this was added 1-PECl (24.0 μmol) by microsyringe. The ampoule was brought out of the glovebox and heated at 120 °C for 1 hour with a stir-rate of 500 rpm. After this time, the ampoule was cooled rapidly to ambient temperature, and an aliquot removed for analysis by 1H NMR spectroscopy to determine monomer conversion. The remainder of the reaction mixture was dissolved in a small volume of THF (ca. 2 ml), and the polymer precipitated by addition of the THF solution to acidified methanol (MeOH:HCl(aq), ca. 75 ml:1 ml). The polymer was collected by filtration and dried in vacuo.
:monomer, 1:1, v/v) and AIBN (24.0 μmol). The ampoule was brought out of the glovebox and heated at 110 °C for 1 hour with a stir-rate of 500 rpm. After this time, the ampoule was cooled rapidly to ambient temperature, and an aliquot removed for analysis by 1H NMR spectroscopy to determine monomer conversion. The remainder of the reaction mixture was taken-up in a small volume of THF (ca. 2 ml), and the polymer precipitated by addition of the THF solution to acidified methanol (MeOH:HCl(aq), ca. 75 ml:1 ml). The polymer was collected by filtration and dried in vacuo.
Crystallography
X-Ray diffraction data was collected on an Agilent SuperNova diffractometer fitted with an Atlas CCD detector with Mo-Kα radiation (λ = 0.7107 Å) or Cu-Kα radiation (λ = 1.5418 Å). Crystals were mounted under paratone on MiTeGen loops. The structures were solved by direct methods using SHELXS or SHELXT interfaced through Olex2 and refined by full-matrix least-squares on F2 using SHELXL, interfaced through Olex2.34 Molecular graphics for all structures were generated using POV-RAY, POVLabel and Ortep.Acknowledgements
M. P. S. and B. R. M. L. gratefully acknowledge support from the EPSRC (EP/M000842/1), a Marie Curie Career Integration Grant (FP7-PEOPLE-2013-CIG-618372) and the University of Edinburgh for financial support. We thank Dr Gary Nichol for collection of the X-ray diffraction data. We also thank Wako Chemicals GmbH for providing us with samples of the initiators V-601 and V-65, and Synthomer for useful discussions.Notes and references
- (a) I. Bauer and H.-J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed; (b) I. Bauer and H.-J. Knölker, in Iron Catalysis in Organic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, pp. 1–27 Search PubMed; (c) C. Bolm, J. Legros, J. Le Paih and L. Zani, Chem. Rev., 2004, 104, 6217–6254 CrossRef CAS PubMed; (d) J. Gui, C.-M. Pan, Y. Jin, T. Qin, J. C. Lo, B. J. Lee, S. H. Spergel, M. E. Mertzman, W. J. Pitts, T. E. La Cruz, M. A. Schmidt, N. Darvatkar, S. R. Natarajan and P. S. Baran, Science, 2015, 348, 886–891 CrossRef CAS PubMed; (e) J. M. Hoyt, V. A. Schmidt, A. M. Tondreau and P. J. Chirik, Science, 2015, 349, 960–963 CrossRef CAS PubMed; (f) R. Pony Yu, D. Hesk, N. Rivera, I. Pelczer and P. J. Chirik, Nature, 2016, 529, 195–199 CrossRef CAS PubMed; (g) X. Jia and Z. Huang, Nat. Chem., 2016, 8, 157–161 CAS; (h) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600–604 CrossRef CAS PubMed.
- (a) J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS; (b) J.-S. Wang and K. Matyjaszewski, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- (a) M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS; (b) T. Ando, M. Kato, M. Kamigaito and M. Sawamoto, Macromolecules, 1996, 29, 1070–1072 CrossRef CAS.
- (a) T. Ando, M. Kamigaito and M. Sawamoto, Macromolecules, 1997, 30, 4507–4510 CrossRef CAS; (b) K. Matyjaszewski, M. Wei, J. Xia and N. E. McDermott, Macromolecules, 1997, 30, 8161–8164 CrossRef CAS; (c) Y. Kotani, M. Kamigaito and M. Sawamoto, Macromolecules, 1999, 32, 6877–6880 CrossRef CAS; (d) B. Göbelt and K. Matyjaszewski, Macromol. Chem. Phys., 2000, 201, 1619–1624 CrossRef; (e) J. Louie and R. H. Grubbs, Chem. Commun., 2000, 1479–1480 RSC; (f) M. Teodorescu, S. G. Gaynor and K. Matyjaszewski, Macromolecules, 2000, 33, 2335–2339 CrossRef CAS; (g) G. Moineau, P. Dubois, R. Jérôme, T. Senninger and P. Teyssié, Macromolecules, 1998, 31, 545–547 CrossRef CAS; (h) J. Xia, H.-j. Paik and K. Matyjaszewski, Macromolecules, 1999, 32, 8310–8314 CrossRef CAS.
- (a) Z. Xue, D. He and X. Xie, Polym. Chem., 2015, 6, 1660–1687 RSC; (b) B. R. M. Lake and M. P. Shaver, in Controlled Radical Polymerization: Mechanisms, American Chemical Society, 2015, vol. 1187, ch. 17, pp. 311–326 Search PubMed; (c) R. Poli, L. E. N. Allan and M. P. Shaver, Prog. Polym. Sci., 2014, 39, 1827–1845 CrossRef CAS.
- (a) L. E. N. Allan, J. P. MacDonald, A. M. Reckling, C. M. Kozak and M. P. Shaver, Macromol. Rapid Commun., 2012, 33, 414–418 CrossRef CAS PubMed; (b) L. E. N. Allan, J. P. MacDonald, G. S. Nichol and M. P. Shaver, Macromolecules, 2014, 47, 1249–1257 CrossRef CAS.
- (a) R. Poli and M. P. Shaver, Inorg. Chem., 2014, 53, 7580–7590 CrossRef CAS PubMed; (b) H. Schroeder and M. Buback, Macromolecules, 2015, 48, 6108–6113 CrossRef CAS; (c) H. Schroeder, M. Buback and M. P. Shaver, Macromolecules, 2015, 48, 6114–6120 CrossRef CAS; (d) H. Schroeder, B. R. M. Lake, S. Demeshko, M. P. Shaver and M. Buback, Macromolecules, 2015, 48, 4329–4338 CrossRef CAS.
- J.-I. Lee, T.-Y. Lee, L.-C. Chang, C.-Y. Lin, H. M. Lee, L. Hung, A. Datta and J.-H. Huang, J. Mol. Struct., 2009, 929, 207–212 CrossRef CAS.
- R. Poli, Chem. – Eur. J., 2015, 21, 6988–7001 CrossRef CAS PubMed.
- (a) S. Gulli, J.-C. Daran and R. Poli, Eur. J. Inorg. Chem., 2011, 2011, 1666–1672 CrossRef; (b) H.-C. Tseng, M. Y. Chiang, W.-Y. Lu, Y.-J. Chen, C.-J. Lian, Y.-H. Chen, H.-Y. Tsai, Y.-C. Lai and H.-Y. Chen, Dalton Trans., 2015, 44, 11763–11773 RSC; (c) Y. Yamaguchi, H. Ando, M. Nagaya, H. Hinago, T. Ito and M. Asami, Chem. Lett., 2011, 40, 983–985 CrossRef CAS.
- D. M. Granum, P. J. Riedel, J. A. Crawford, T. K. Mahle, C. M. Wyss, A. K. Begej, N. Arulsamy, B. S. Pierce and M. P. Mehn, Dalton Trans., 2011, 40, 5881–5890 RSC.
- (a) S. J. J. Titinchi, G. Von Willingh, H. S. Abbo and R. Prasad, Catal. Sci. Technol., 2015, 5, 325–338 RSC; (b) A. Ray, G. Pilet, C. J. Gómez-García and S. Mitra, Polyhedron, 2009, 28, 511–520 CrossRef CAS.
- C. Scheiper, D. Dittrich, C. Wölper, D. Bläser, J. Roll and S. Schulz, Eur. J. Inorg. Chem., 2014, 2014, 2230–2240 CrossRef CAS.
- T. Akashi, J. Nakazawa and S. Hikichi, J. Mol. Catal. A: Chem., 2013, 371, 42–47 CrossRef CAS.
- L. Yang, D. R. Powell and R. P. Houser, Dalton Trans., 2007, 955–964 RSC.
- (a) J. Cirera, P. Alemany and S. Alvarez, Chem. – Eur. J., 2004, 10, 190–207 CrossRef CAS PubMed; (b) J. Cirera, E. Ruiz and S. Alvarez, Inorg. Chem., 2008, 47, 2871–2889 CrossRef CAS PubMed.
- A. F. Lugo and A. F. Richards, Inorg. Chim. Acta, 2010, 363, 2104–2112 CrossRef CAS.
- M. M. Olmstead, P. P. Power and S. C. Shoner, Inorg. Chem., 1991, 30, 2547–2551 CrossRef CAS.
- (a) S. A. Sulway, D. Collison, J. J. W. McDouall, F. Tuna and R. A. Layfield, Inorg. Chem., 2011, 50, 2521–2526 CrossRef CAS PubMed; (b) G. Jin, L. Vendier, Y. Coppel, S. Sabo-Etienne and S. Bontemps, Dalton Trans., 2015, 44, 7500–7505 RSC; (c) B. A. Frazier, P. T. Wolczanski, E. B. Lobkovsky and T. R. Cundari, J. Am. Chem. Soc., 2009, 131, 3428–3429 CrossRef CAS PubMed; (d) B. A. Frazier, V. A. Williams, P. T. Wolczanski, S. C. Bart, K. Meyer, T. R. Cundari and E. B. Lobkovsky, Inorg. Chem., 2013, 52, 3295–3312 CrossRef CAS PubMed.
- (a) D. J. Darensbourg, C. G. Ortiz and D. R. Billodeaux, Inorg. Chim. Acta, 2004, 357, 2143–2149 CrossRef CAS; (b) P. Zhao, H. Lei, C. Ni, J.-D. Guo, S. Kamali, J. C. Fettinger, F. Grandjean, G. J. Long, S. Nagase and P. P. Power, Inorg. Chem., 2015, 54, 8914–8922 CrossRef CAS PubMed; (c) J. S. Figueroa, J. G. Melnick and G. Parkin, Inorg. Chem., 2006, 45, 7056–7058 CrossRef CAS PubMed; (d) T. J. Boyle, L. A. M. Ottley, C. A. Apblett, C. A. Stewart, S. M. Hoppe, K. L. Hawthorne and M. A. Rodriguez, Inorg. Chem., 2011, 50, 6174–6182 CrossRef CAS PubMed; (e) C. Ni and P. P. Power, Chem. Commun., 2009, 5543–5545 RSC.
- (a) E. M. Townsend, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2012, 134, 11334–11337 CrossRef CAS PubMed; (b) R. R. Schrock, A. J. Jiang, S. C. Marinescu, J. H. Simpson and P. Müller, Organometallics, 2010, 29, 5241–5251 CrossRef CAS; (c) J. McKinven, G. S. Nichol and P. L. Arnold, Dalton Trans., 2014, 43, 17416–17421 RSC.
- (a) F. di Lena and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 959–1021 CrossRef CAS; (b) R. Poli, Eur. J. Inorg. Chem., 2011, 2011, 1513–1530 CrossRef.
- (a) L. E. N. Allan, M. R. Perry and M. P. Shaver, Prog. Polym. Sci., 2012, 37, 127–156 CrossRef CAS; (b) R. Poli and M. P. Shaver, Chem. – Eur. J., 2014, 20, 17530–17540 CrossRef CAS PubMed.
- (a) J. Wang, J. Zhou, H. S. E. M. Sharif, D. He, Y. S. Ye, Z. Xue and X. Xie, RSC Adv., 2015, 5, 96345–96352 RSC; (b) Z. Xue and R. Poli, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 3494–3504 CrossRef CAS.
- (a) M. P. Shaver, L. E. N. Allan, H. S. Rzepa and V. C. Gibson, Angew. Chem., Int. Ed., 2006, 45, 1241–1244 CrossRef CAS PubMed; (b) L. E. N. Allan, M. P. Shaver, A. J. P. White and V. C. Gibson, Inorg. Chem., 2007, 46, 8963–8970 CrossRef CAS PubMed; (c) R. Poli and M. P. Shaver, Chem. – Eur. J., 2014, 20, 17530–17540 CrossRef CAS PubMed.
- (a) K. Zhu, M. P. Shaver and S. P. Thomas, Eur. J. Org. Chem., 2015, 2119–2123 CrossRef CAS; (b) K. Zhu, M. P. Shaver and S. P. Thomas, Chem. – Asian J., 2016, 11, 977–980 CrossRef CAS PubMed; (c) K. Zhu, M. P. Shaver and S. P. Thomas, Chem. Sci., 2016, 7, 3031–3035 RSC.
- C. X. Zhang, S. Kaderli, M. Costas, E.-i. Kim, Y.-M. Neuhold, K. D. Karlin and A. D. Zuberbühler, Inorg. Chem., 2003, 42, 1807–1824 CrossRef CAS PubMed.
- S. Grcev, P. Schoenmakers and P. Iedema, Polymer, 2004, 45, 39–48 CrossRef CAS.
- K. Min, H. Gao, J. A. Yoon, W. Wu, T. Kowalewski and K. Matyjaszewski, Macromolecules, 2009, 42, 1597–1603 CrossRef CAS.
- D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC.
- V. A. Williams, P. T. Wolczanski, J. Sutter, K. Meyer, E. B. Lobkovsky and T. R. Cundari, Inorg. Chem., 2014, 53, 4459–4474 CrossRef CAS PubMed.
- Due to relatively poor solubility in deuterated solvents (THF-d8, C6D6, CD3CN), we were unable to obtain a clear 1H NMR spectrum with an acceptable signal-to-noise ratio. We believe the low quality of the spectra obtained also results from solution fluxionality.
- The solution magnetic moment (4.8μB) determined for complex 3c is slightly lower than might be expected for a d6 high-spin complex. We suggest this is caused by lattice solvent, which is not accounted for in the solution magnetic moment calculation. Using the information obtained from the SQUEEZE routine in the crystallographic software, PLATON, it is calculated that there is electron density pertaining to approximately 2.5 molecules of solvent toluene (or hexane) per unit cell, which equates to approximately 0.6 molecules of toluene per dimer. If 0.6 molecules of toluene are included in the solution magnetic moment calculation, a value of 5.0μB is obtained.
- (a) O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS; (b) G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed; (c) G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: GPC data, 1H NMR spectra and single crystal X-ray diffraction data. CCDC 1470307–1470310, 1470323, 1470324 and 1470383–1470385. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01208f |
| This journal is © The Royal Society of Chemistry 2016 |