
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and electronic properties of polycyclic aromatic hydrocarbons doped with phosphorus and sulfur†
W.
Delaunay
a,
R.
Szűcs
ab,
S.
Pascal
a,
A.
Mocanu
a,
P.-A.
Bouit
a,
L.
Nyulászi
*b and
M.
Hissler
*a
aInstitut des Sciences Chimiques de Rennes, UMR 6226 CNRS-Université de Rennes 1, Campus de Beaulieu, 35042 Rennes Cedex, France. E-mail: muriel.hissler@univ-rennes1.fr
bDepartment of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, H-1111 Budapest, Hungary
First published on 20th November 2015
Abstract
In this work, we report on the synthesis of polyaromatic hydrocarbons containing phosphole and thiophene rings at the edge. The ring-closure reactions have been investigated by theoretical calculations. The optical and electrochemical properties and density functional theory calculations showed that the properties depend on the relative position of these five membered rings in the PAH structure.
Introduction
Planar π-extended organic molecules also called polycyclic aromatic hydrocarbons (PAHs) have emerged in the last decade as efficient molecular materials for opto-electronic applications.1 Their extended π-backbone affords them a reduced HOMO–LUMO gap, as well as the possibility to self-assemble via π–π interactions. These properties led to the preparation of efficient opto-electronic devices such as organic field-effect transistors (OFETs) or organic solar cells (OSCs).2 Among this class of compounds, PAHs containing hetero-elements are particularly interesting since the presence of the heteroatoms (N, B, O, S …) allows for diversifying the structures, reactivity and electronic properties.3 In particular S-containing PAHs are of interest, given the large amount of literature about thiophene derivatives for opto-electronic applications.4 Following the initial work by Müllen et al.,5 such as the preparation of A (Fig. 1), new thiophene-containing derivatives were also synthesized, displaying unique properties. For example, due to the presence of a thienyl moiety, compound B (Fig. 1) displays particular photophysical properties,6 and derivative C (Fig. 1) presents enhanced hole transport properties, which leads to the preparation of an efficient OSC.7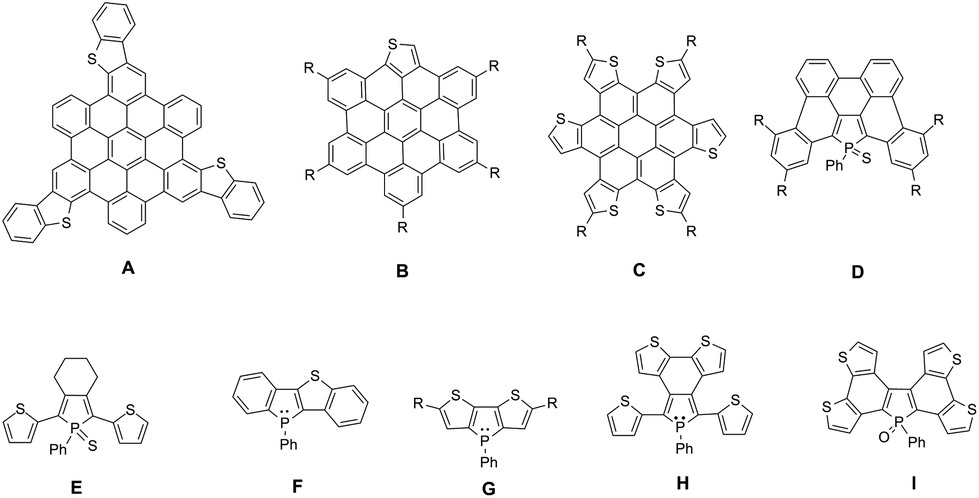 | ||
| Fig. 1 Examples of heteroatom containing PAH (S: A–C; P: D) and of mixed phosphole–thiophene π-conjugated systems E–I. | ||
We recently showed that this strategy can also be extended to organophosphorus chemistry by extending the strategies developed on P-containing π-conjugated oligomers.8 The reactivity at the P-atom of PAH D (Fig. 1) proved to be an efficient tool for the molecular engineering of this appealing family of PAHs.9 Chemical modifications via organic chemistry or coordination to Au(I) of the σ3,λ3-P center of D allowed us to prepare new compounds which exhibit different structural and electronic properties depending on the P-environment. These compounds also presented sufficient stability and adequate electronic properties to be successfully introduced into white-OLEDs.10
In order to further modify the electronic properties of these P-containing PAHs, we decided to design PAH containing both P and S atoms within their π-framework. This strategy has already been successfully used to tune the properties of mixed phosphole/thiophene linear oligomers.11 The presence of thiophene rings allowed the electropolymerization of oligomer E (Fig. 1) to prepare low-band gap polymers or by exploiting the different reactivities of the P and S heteroatoms in compound F to prepare dually switchable molecular materials (Fig. 1). Furthermore, dithieno[b,d]phospholes G,12 bithiophene-fused benzo[c]phospholes H13 and di(bithiophene)-fused benzo[b,d]phosphole I14 (Fig. 1) have also been reported to exhibit very appealing functions.
Considering the interesting properties of mixed phosphole–thiophene derivatives, there is great interest regarding the development of original structures in order to gain more insight into the structure–property relationship. In this paper, we report on the synthesis of new phosphole–thiophene based PAHs. We have investigated the impact of fusing aromatic thiophene to the antiaromatic thiooxophosphole ring at different positions on the physical properties. The electronic properties of these novel fused phosphole–thiophene derivatives are discussed on the basis of experimental and theoretical results.
Results and discussion
Synthesis
Following our previously reported strategy, substituted 3,4-biarylphospholes 3–4 (Scheme 1) featuring a C–C bond between the π-substituents (phenyls or thiophenes) at the 3,4-positions of the P-ring were targeted as key intermediates.15 To this end, dialkynes 1–2 were prepared using classical Pd-catalysed Sonogashira conditions. The usual Fagan–Nugent conditions were then applied to the precursors 1–2 (Scheme 1) resulting in the respective P-heteroles.16 The σ3,λ3-phospholes were oxidized in situ with S8 affording thiooxophospholes 3–4, respectively. These air stable compounds were isolated in moderate yields (3: 35%; 4: 48%) after column chromatography. They display 31P NMR chemical shifts in the usual range of thiooxophospholes (+56 to +58 ppm, see Experimental) and reasonable solubility in most organic solvents. Their multinuclear NMR spectroscopic and mass spectrometry data support the proposed structures. Furthermore, 3 was also characterized by X-ray diffraction (see Fig. S2†).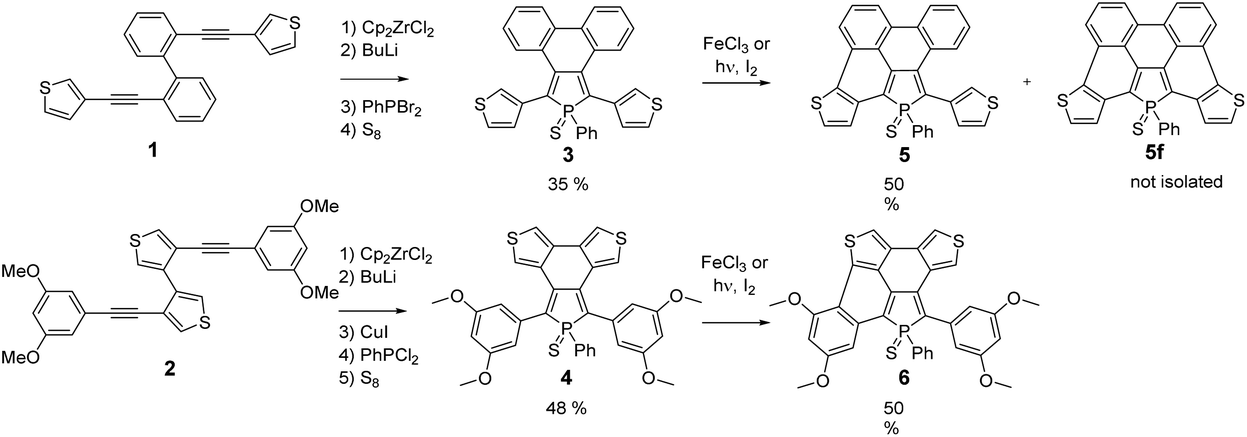 | ||
| Scheme 1 Synthesis of phosphole–thiophene PAHs 5–6. | ||
The chemical reactivity of the phospholes 3–4 under Scholl conditions (with FeCl3 or MoCl5 in CH2Cl2) was also tested. As observed for all phenyl analogues, the asymmetric phospholes 5–6 (Scheme 1) formed rapidly, but the fully cyclized product was never observed and degradation of the intermediate occurs within a few hours.17 The Pd-catalysed C–H bond activation, described as an easy method to functionalize thiophene, did not result in the formation of the desired products.18 The reaction at 100 °C between 3–4 and Pd(OAc)2 only led to a complex mixture of starting materials and uncharacterized degradation compounds.
We thus turned to the photocyclization method that successfully allowed us to prepare the P-containing PAH D.9 In the case of the thiophene containing phospholes 3–4, the photochemical method only led to the formation of the asymmetric PAHs 5–6 (Scheme 1) exhibiting 31P NMR chemical shifts which are slightly shifted to a higher field compared to those of their precursors 3 and 4. The asymmetric 1H and 13C NMR spectra of both half-fused phosphole–thiophene compounds are fully consistent with the proposed structures. For example, the transformation of compound 4 into the corresponding half-fused derivatives 6 perturbs the phosphole ring as shown by the presence of 2 signals for P-Cα and P-C-Cβ carbons respectively.
During the course of the reaction, several other products, displaying 31P NMR signals in the same range (+45 to +55 ppm) were also formed during the reaction. The analysis of the crude mixture of 5 by mass spectrometry clearly evidences the presence of the fully cyclized derivative 5f (m/z = 524.9967) (Scheme 1), however this compound could not be isolated and fully characterized (see Fig. S1†). 5 and 6 were characterized by multinuclear NMR spectroscopy and mass spectrometry. Furthermore, their structures were unambiguously confirmed by X-ray diffraction (see Fig. 2).
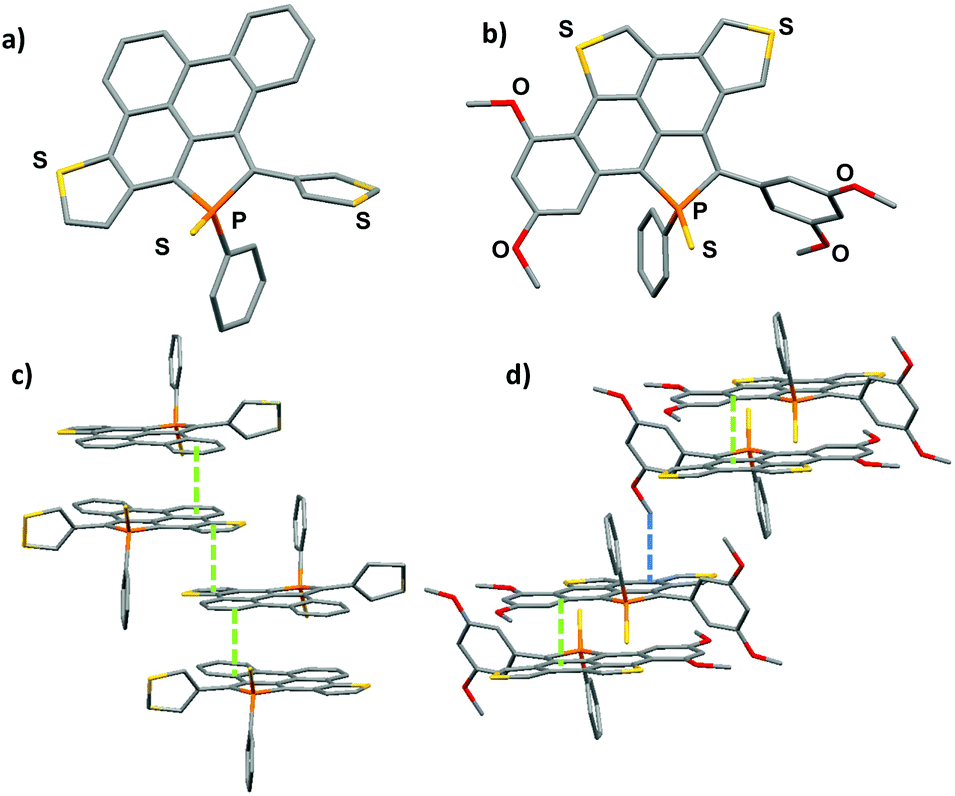 | ||
| Fig. 2 X-ray crystallographic structures of 5 (a) and 6 (b) and their molecular packing (c, d). | ||
For a better understanding of the ring-closure reactions, theoretical calculations have been carried out. First we considered a ring closure step by forming a saturated bond with the hydrogens in the trans position (reaction energy ΔE1). For investigating the overall process we used an isodesmic reaction, with the ring-closure reaction of (Z)-1,2-diphenylethene forming a phenanthrene as a reference (ΔE2). The results which are summarized in Table 1, show that ΔE2 is exothermic both in the case of 3 and 4. Furthermore, the investigation of ΔE1 values shows that the formation of the intermediate with the saturated bond is a somewhat (by ca. 20 kcal mol−1) more endothermic procedure (the corresponding transition structures are apparently at even higher energies). A further important conclusion comes from the comparison of both ΔE1 and ΔE2 values of the opened and the partly closed rings, showing that in the case of the partly closed and apparently more rigid systems the ring closure is somewhat disfavoured. The results also suggest a significantly reduced reactivity of the thiophene rings in the β positions (see 3αvs.3β and 5αvs.5β in Table 1).
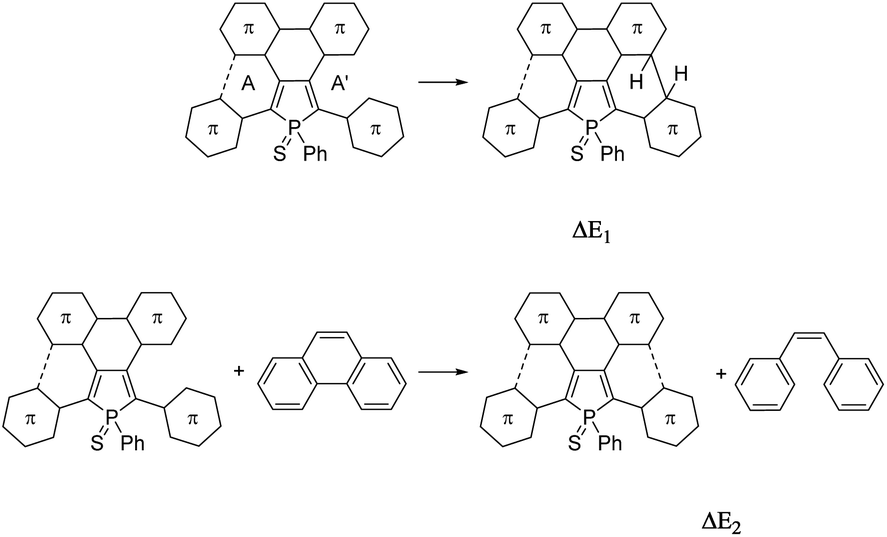
From both experimental and theoretical data, it appears that the formation of one aromatic ring is easier than the formation of two aromatic rings. Our previous experiments on star-shaped phospholes showed that only the presence of an electron-rich six-membered aryl group allowed the successful formation of PAH D.9 In the present study, when this group is replaced by a five membered electron-rich thiophene in 3 or 4, the reaction led to the formation of asymmetric PAHs 5–6 mainly. The presence of the thiophene rings, even if they are electron-rich, doesn't promote the reaction, probably due the instability of the partially or fully cyclized compound under irradiation conditions. However, this strategy allowed us to prepare unprecedented asymmetric P,S-containing PAHs 5–6. It is worth noting that due to the asymmetry of the molecule, the P-atom is stereogenic. 5 and 6 are thus constituted of a mixture of two enantiomers (vide infra). The structural and electronic properties of these derivatives are discussed in the following section.
Structural description
Compounds 5 and 6 were characterized by an X-ray diffraction study performed on a single crystal (Fig. 2). For both compounds, the unit cell is constituted of a racemic mixture of the two enantiomers (see Fig. S2 and 3†). Their P-heterocycles display a σ4,λ5-P-atom with a pyramidal shape and classical bond lengths and valence angles for phosphole rings (see Table 2).8 Remarkably, the 5-membered and 6-membered fused rings constituting the PAH scaffold are almost planar, as shown by the very low torsion angle P–C1–C2–C7 (see Table 2). These structural data confirm that these new compounds can be considered as small planar PAHs.|
|
||||
|---|---|---|---|---|
| 5 | 5 DFT | 6 | 6 DFT | |
| P–C1 | 1.790(5) | 1.82 | 1.806(2) | 1.82 |
| P–C24 | 1.820(6) | 1.85 | 1.832(2) | 1.86 |
| C1–C26 | 1.374(7) | 1.38 | 1.370(3) | 1.38 |
| C26–C25 | 1.469(7) | 1.48 | 1.478(2) | 1.48 |
| C25–C24 | 1.378(7) | 1.37 | 1.359(3) | 1.36 |
| C1–C2 | 1.406(7) | 1.41 | 1.431(3) | 1.43 |
| C26–C27 | 1.414(7) | 1.42 | 1.399(3) | 1.40 |
| C25–C28 | 1.474(8) | 1.47 | 1.476(3) | 1.47 |
| C24–C23 | 1.463(8) | 1.47 | 1.478(3) | 1.48 |
| C7–C8 | 1.429(7) | 1.43 | 1.428(3) | 1.42 |
| P–C1–C2–C7 | 2.3(9) | 2.8 | 1.0(3) | 0.6 |
| P–C24–C23–C18 | 51.1(8) | 61.8 | 68.6(2) | 69.4 |
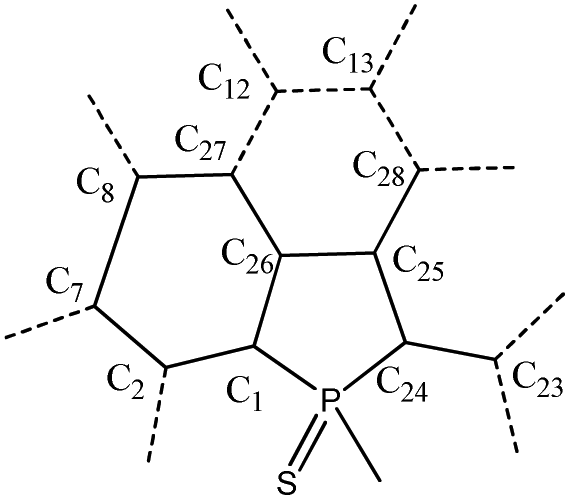
In contrast, the non-fused ring at the 2-position of the phosphole lies perpendicular to the polycyclic plane due to intramolecular H–H repulsion (see P–C24–C23–C18 torsion angle in Table 2). As observed for their phenyl analogue, C1–C2, C7–C8 and C26–C27 bonds are rather short (d ≃ 1.41–1.43 Å) with bond lengths typical of aromatic rings,10 indicating an aromatic character for the newly formed rings. The structure obtained by geometry optimization for 5 and 6 at the B3LYP/6-31+G* level is in good agreement with the X-ray data (see Fig. S5, 6† and Table 2). Rotational barriers calculated for the half-fused rings of 5 and 6 are 12.3/12.6 and 14.8/15.4 kcal mol−1 respectively, the two values correspond to the two possible transition states. These values suggest a somewhat hindered rotation.
The molecular packing of compounds 5 and 6 shows a supramolecular organization due to π-stacking. Compound 5 packs as π-dimers (d = 3.40 Å), constituted of the two different enantiomers of 5. Furthermore, these dimers are engaged in intermolecular π–π interactions (d = 3.45 Å) resulting in the formation of infinite π-stacked columns (see Fig. 2c). This long-range organization is not observed in the packing of its precursor 3. Compound 6 also packs as π-dimers (d = 3.40 Å), formed by its two enantiomers (Fig. 2d). Dimers then interact through C–H⋯ π interactions (d = 2.80 Å). These two supramolecular organizations due to π-stacking are rather unexpected for phosphole derivatives which usually aggregate poorly in the solid state (like phosphole 3 for example).8 This is clearly the effect of the large π-system fused to the phosphole ring.
Optical and electrochemical properties
The optical properties of derivatives 3–6 were studied by means of UV-Vis absorption in dichloromethane (see Fig. 3 and Table 3). Phosphole 3 presents a broad π⋯π* transition with a small extinction coefficient in the visible part of the spectrum at 486 nm (vide infra). In the case of its half-fused analogue 5, hyperchromic and hypsochromic shifts are observed together with the appearance of fine structure characteristic of polyaromatic molecules (Fig. 3 and Table 3). In the case of 4, the situation is different. The UV-Vis absorption spectrum exhibits a main band (λ1 = 340 nm) with a shoulder at lower energy (λ2 = 432 nm) with a very low extinction coefficient. The rigidification of the structure led to the appearance of a broad structured band in the visible region possessing a blue shifted absorption threshold (Table 3). The blue shift upon cyclization was not observed for PAHs D10 but these spectroscopic data are in accordance with a similar molecule reported by Matano et al.19 This behaviour was attributed to a lower conjugation as all heteroles in 4 and 6 are linked through their respective 3-positions, as a consequence the interaction between the heterocycles is weaker. The presence of the bithiophene grafted on the 3,4 position of the phosphole ring is responsible for the lack of conjugation, even in its PAH form. This observation also explains the wider optical gap of 4/6 compared to 3/5. None of these four compounds present emission properties in dichloromethane solutions and in the solid state. It is likely that non-radiative deexcitations due to intramolecular rotations are responsible for this phenomenon.20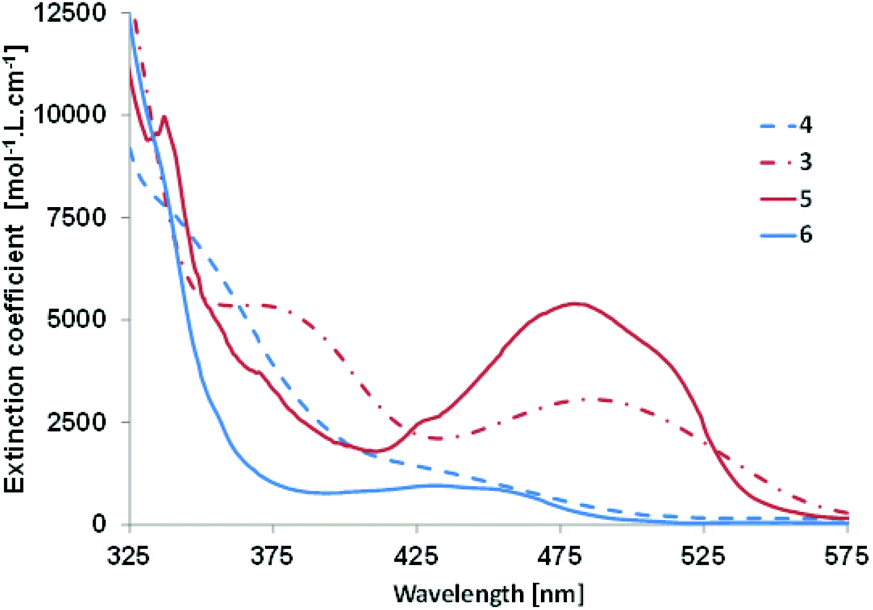 | ||
| Fig. 3 UV-Vis absorption of 3–6 in diluted DCM solutions. | ||
| Cpnd | λ abs (nm) | λ th , (nm) |
λ
DFT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (nm) c (nm) |
ε (L mol−1 cm−1) | E ox (V) | E red (V) |
|---|---|---|---|---|---|---|
| a In CH2Cl2 (10−5 M). b Absorption threshold. c TD-DFT-calculated vertical absorption wavelength. d In CH2Cl2 with Bu4N+PF6− (0.2 M) at a scan rate of 100 mV s−1. Potentials vs. ferrocene/ferrocenium. e Reversible process. | ||||||
| 3 | 486 | 572 | 553 | 3000 | +0.78 | −1.61e |
| 4 | 432(sh) | 506 | 471 | 1300 | +1.01 | −1.80e |
| 5 | 482 | 543 | 549 | 6000 | +0.73 | −1.67e |
| 6 | 434 | 494 | 452 | 1000 | +0.88 | −1.82e |
The electrochemical properties of these derivatives were also studied by cyclic voltammetry in dichloromethane. Compounds 3, 4, 5 and 6 display an irreversible mono-electronic oxidation wave at low potential and a reversible mono-electronic reduction wave. As observed by UV-Vis, the nature of the substituents (bithiophene versus biphenyl) grafted on the 3,4 position of the phosphole ring influences the redox properties. Compounds 3 and 5 exhibit lower oxidation and reduction potential compared to the corresponding compounds 4 and 6 showing that the π electrons are less efficiently spread over the fused rings for 4 and 6. The formation of one intramolecular bond (3 → 5, 4 → 6) induces a decrease of the oxidation potential and an increase of the reduction potential (Table 3). These effects lead to a similar gap or an increase of the gap, as observed by UV/Vis absorption and is also supported by TD-DFT calculations.
Theoretical calculations
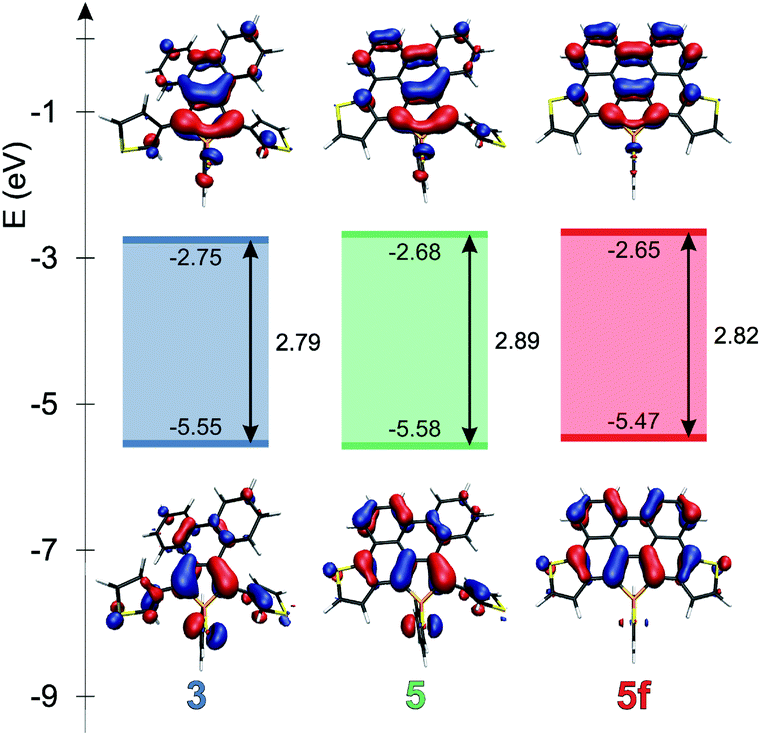
Theoretical studies carried out on the systems are in reasonable agreement with the measured optical properties. In all cases, the calculations suggest that the absorption maximum belongs to the HOMO–LUMO transition. Therefore the energetics and the shape of these orbitals were studied in depth. Fig. 4 and 5 display the change of the frontier orbitals in the two series of molecules after the first, and the second ring closure. The FMOs are typical of thiooxophosphole, as we noted before for D.9 It is noteworthy that with the extension of the conjugation the participation of the S lone pair decreases in the HOMO. This observation might contribute to the small stabilization of the HOMO between 3 and 5. Since the LUMO is also somewhat destabilized the observed unusual blue shift (see above) is understandable. Also the electrochemical observations are in accordance with the changes of the FMO energies.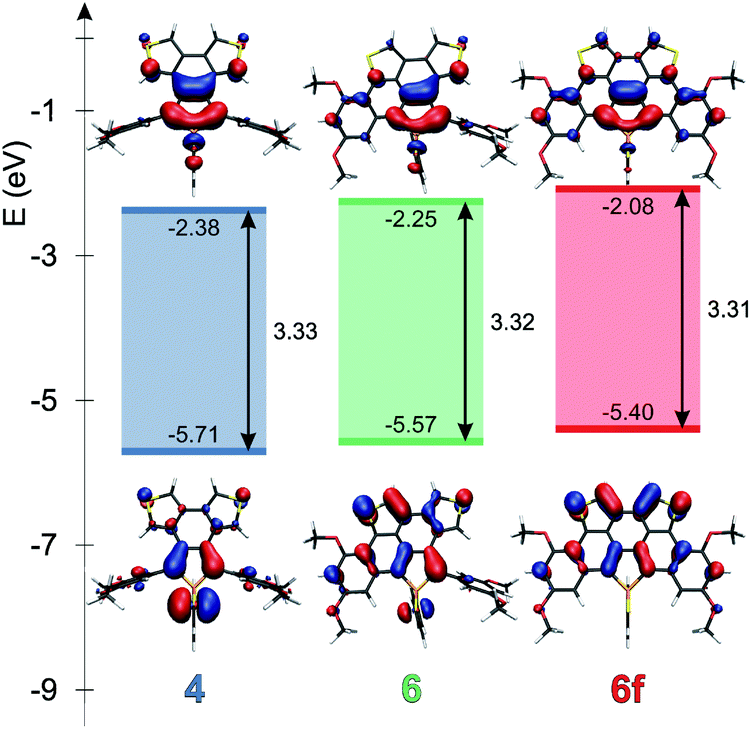 | ||
| Fig. 4 Frontier molecular orbitals of 3, 5 and 5f at the B3LYP/6-31+G* level of theory. | ||
 | ||
| Fig. 5 Frontier molecular orbitals of 4, 6 and 6f at the B3LYP/6-31+G* level of theory. | ||
The main difference between the two series lies in the shifted LUMO levels, while the HOMOs are nearly the same. The conjugation between the phosphole and the thiophene units is always weak, concluded from the small contribution of the thiophene units to the FMOs, and it is also evidenced by the small MO coefficients of the β carbon in the thiophene FMOs (see Fig. 5). However, the conjugation between the phenyl groups and the phosphole unit strongly depends on the phenyls’ positions. In the case where there is a biphenyl unit grafted on the 3 and 4 positions of the phosphole ring (3, 5, 5f), there is a strong interaction: the LUMO orbital shows significant contribution from the phenyl units. However, this interaction is significantly weakened when the phenyl rings are connecting to the 2 or 5 positions of the phosphole unit (4, 6, 6f) (for numeric results of the orbital composition analyses see Table S2†).
The electronic properties of a system are usually linked to its aromaticity. In order to investigate this property, NICS aromaticities were calculated for 3–6 (for NICS(1) representations see Fig. 6, for numeric results see Table S3†).
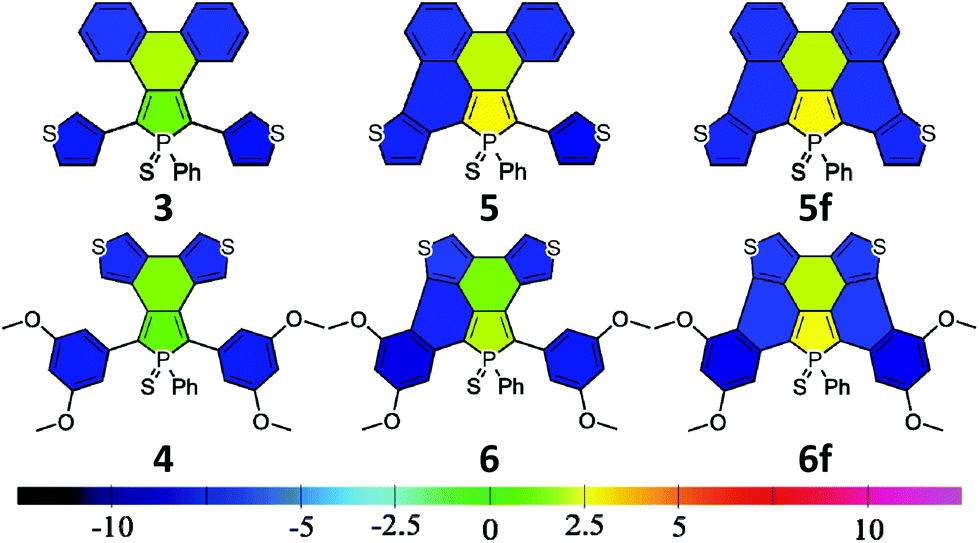 | ||
| Fig. 6 Schematic representation of the NICS(1) results for 3–6. | ||
The non-aromatic (or slightly antiaromatic) character of the thiooxophosphole unit spreads toward the ring fused to its 3,4 position.21 By increasing the number of the fused rings (3 → 5f; 4 → 6f) the antiaromaticity of these rings strengthens in both series, but regardless of this phenomenon, in good agreement with the conclusions obtained from X-ray crystallography, the formed new rings are highly aromatic. This study confirms that insertion of an antiaromatic ring inside a PAH backbone impacts its properties.
Conclusion
In conclusion, we detailed the synthesis of two mixed thiophene–phosphole PAHs featuring seven fused rings. Electronic properties of these derivatives have been studied by means of UV-Vis absorption, cyclic voltammetry and the results were rationalized by DFT calculations. These results allow a deeper understanding of the electronic properties of heteroatom containing PAHs.Experimental
General information
All experiments were performed under an atmosphere of dry argon using standard Schlenk techniques. Commercially available reagents were used as received without further purification. Solvents were freshly purified using MBRAUN SPS-800 drying columns. Irradiation reactions were conducted using a Heraeus TQ 150 mercury vapor lamp. Separations were performed by gravity column chromatography on basic alumina (Aldrich, Type 5016A, 150 mesh, 58 Å) or silica gel (Merck Geduran 60, 0.063–0.200 mm). 1H, 13C, and 31P NMR spectra were recorded on a Bruker AM300, AM400, AM500. 1H and 13C NMR chemical shifts were reported in parts per million (ppm) relative to Me4Si as the external standard. Assignment of proton and carbon atoms is based on COSY, HMBC, HMQC and DEPT-135 experiments. High-resolution mass spectra were obtained on a Varian MAT 311 or ZabSpec TOF Micromass instrument at CRMPO, University of Rennes 1. Compounds 1 and 4,4′-diethynyl-3,3′-bithiophene were synthesized according to a published procedure.19 UV-Visible spectra were recorded at room temperature on a VARIAN Cary 5000 spectrophotometer. The electrochemical studies were carried out under argon using an Eco Chemie Autolab PGSTAT 30 potentiostat for cyclic voltammetry with the three-electrode configuration: the working electrode was a platinum disk, the reference electrode was a saturated calomel electrode and the counter-electrode was a platinum wire. All potentials were internally referenced to the ferrocene/ferrocenium couple. For the measurements, concentrations of 10−3 M of the electroactive species were used in freshly distilled and degassed dichloromethane and 0.2 M tetrabutylammonium hexafluorophosphate.X-ray diffraction
Single crystals of 3, 5 and 6 suitable for X-ray crystal analyses were obtained by slow diffusion of vapors of pentane into dichloromethane solutions. Single crystal data collection was performed at 150 K with an APEX II Bruker-AXS (Centre de Diffractométrie, Université de Rennes 1, France) with Mo-Kα radiation (λ = 0.71073 Å). Reflections were indexed, Lorentz-polarization corrected and integrated by the DENZO program of the KappaCCD software package. The data merging process was performed using the SCALEPACK program.22 Structure determinations were performed by direct methods with the solving program SIR97,23 that revealed all the non-hydrogen atoms. SHELXL program24 was used to refine the structures by full-matrix least-squares based on F2. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were included in idealised positions and refined with isotropic displacement parameters. Single crystals of all these derivatives were always coated in Paratone oil once removed from the mother solution, mounted at low temperature on the diffractometer goniometer and X-ray data collection was performed at low temperature. Atomic scattering factors for all atoms were taken from International Tables for X-ray Crystallography.25 CCDC reference numbers 1430204, 1430205 and 1430206 contain the supplementary crystallographic data for 3, 5 and 6.Synthesis
:5, Rf = 0.20) gave the title compound as a brown powder in a 40% yield (407 mg). 1H NMR (CDCl3; 400 MHz): δ = 7.82 (d, J(H,H) = 3.4 Hz, 2Hthienyl), 7.61 (d, J(H,H) = 3.4 Hz, 2Hthienyl), 6.61 (d, J(H,H) = 2.2 Hz, 4Hphenyl), 6.45 (t, J(H,H) = 2.2 Hz, 2Hphenyl), 3.78 (s, 12H, OCH3) ppm. 13C NMR (CD2Cl2; 100 MHz): δ = 160.7 (s, Cphenyl), 136.2 (s, Cthienyl), 129.5 (s, CHthienyl), 124.4 (s, Cthienyl), 123.7 (s, CHthienyl), 122.2 (s, Cphenyl), 109.1 (s, CHphenyl), 101.5 (s, CHphenyl), 91.1 (s, C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 84.4 (s, CC), 55.4 (s, OCH3) ppm. HR-MS (ESI, CH3OH, m/z): [M + Na]+ calcd for C28H22O4S2Na, 509.08572; found 509.0856. Anal. Calcd for C28H22O4S2: C, 69.11, H, 4.56, S, 13.18; Found: C, 69.24, H, 4.26, S, 12.99.
C), 84.4 (s, CC), 55.4 (s, OCH3) ppm. HR-MS (ESI, CH3OH, m/z): [M + Na]+ calcd for C28H22O4S2Na, 509.08572; found 509.0856. Anal. Calcd for C28H22O4S2: C, 69.11, H, 4.56, S, 13.18; Found: C, 69.24, H, 4.26, S, 12.99.
Computational details
All calculations were carried out with the Gaussian 09 program package.26 Full geometry optimization was performed for all molecules at the B3LYP/6-31+G* level27 and subsequently harmonic vibrational frequencies were calculated at the same level to establish the nature of the stationary point obtained, for minima no negative eigenvalue of the Hessian was present. All possible rotational structures have been investigated for 3, 4, 5, 6, 5f and 6f. In cases where single crystal X-ray structure is available, the most stable optimized geometry gives a good match. NICS and TD-DFT calculations have been carried out only on the most stable rotamer. Molecular orbitals have been visualized with the VMD package.28 Molecular orbital composition analysis has been carried out with the Multiwfn 3.3.8 program.29Acknowledgements
This work is supported by the Ministère de la Recherche et de l'Enseignement Supérieur, the CNRS, the Région Bretagne, China–French associated international laboratory in “Functional Organophosphorus Materials”, Balaton PHC (830386 K) – TÉT_12_FR-1-2013-0017, the French National Research Agency (ANR)/Research Grants Council (RGC) Joint Research Scheme (ANR MOLMAT) and COST CM10302 (SIPS). The authors are grateful to Dr T. Roisnel (Centre de Diffractométrie, ISCR Université de Rennes 1, France) and Dr C. Lescop (ISCR) for X-ray diffraction studies.Notes and references
- M. D. Watson, A. Fechtenkotter and K. Müllen, Chem. Rev., 2001, 101, 1267 CrossRef CAS PubMed
; J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef PubMed
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS PubMed
-
(a) D. Wu, W. Pisula, M. C. Haberecht, X. Feng and K. Müllen, Org. Lett., 2009, 11, 5686 CrossRef CAS PubMed
-
(a) J. Roncali, Chem. Rev., 1997, 97, 173 CrossRef CAS PubMed
-
(a) R. Benshafrut, M. Rabinovitz, R. E. Hoffman, N. Ben-Mergui, K. Müllen and V. S. Iyer, Eur. J. Org. Chem., 1999, 37 CrossRef CAS
- C. J. Martin, B. Gil, S. D. Perera and S. M. Draper, Chem. Commun., 2011, 47, 3616 RSC
- A. A. Gorodetsky, C.-Y. Chiu, T. Schiros, M. Palma, M. Cox, Z. Jia, W. Sattler, I. Kymissis, M. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2010, 49, 7909 CrossRef CAS PubMed
-
(a) T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681 CrossRef CAS PubMed
- P.-A. Bouit, A. Escande, R. Szűcs, D. Szieberth, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, J. Am. Chem. Soc., 2012, 134, 6524 CrossRef CAS PubMed
- F. Riobé, R. Szűcs, P.-A. Bouit, D. Tondelier, B. Geffroy, F. Aparicio, J. Buendía, L. Sánchez, R. Réau, L. Nyulászi and M. Hissler, Chem. – Eur. J., 2015, 21, 6547 CrossRef PubMed
-
(a) C. Hay, M. Hissler, C. Fischmeister, J. Rault-Berthelot, L. Toupet, L. Nyulaszi and R. Réau, Chem. – Eur. J., 2001, 7, 4222 CrossRef CAS PubMed
- Y. Dienes, S. Durben, T. Karpati, T. Neumann, U. Englert, L. Nyulaszi and T. Baumgartner, Chem. – Eur. J., 2007, 13, 7487 CrossRef CAS PubMed
-
(a) Y. Matano, T. Miyajima, T. Fukushima, H. Kaji, Y. Kimura and H. Imahori, Chem. – Eur. J., 2008, 14, 8102 CrossRef CAS PubMed
- O. Fadhel, D. Szieberth, V. Deborde, C. Lescop, L. Nyulaszi, M. Hissler and R. Réau, Chem. – Eur. J., 2009, 15, 4914 CrossRef CAS PubMed
- Y. Matano, T. Miyajima, T. Fukushima, H. Kaji, Y. Kimura and H. Imahori, Chem. – Eur. J., 2008, 14, 8102 CrossRef CAS PubMed
- P. J. Fagan and W. A. Nugent, J. Am. Chem. Soc., 1988, 110, 2310 CrossRef CAS
- R. Szűcs, F. Riobé, A. Escande, W. Delaunay, D. Joly, P.-A. Bouit, L. Nyulászi and M. Hissler, 2015, submitted.
- D. J. Schipper and K. Fagnou, Chem. Mater., 2011, 23, 1594 CrossRef CAS
- T. Miyajima, Y. Matano and H. Imahori, Eur. J. Org. Chem., 2008, 255 CrossRef CAS
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC
- R. Szűcs, P.-A. Bouit, M. Hissler and L. Nyulászi, Struct. Chem., 2015, 26, 1351 CrossRef
-
Z. Otwinowski and W. Minor, in Methods in Enzymology, ed. C. W. Carter Jr. and R. M. Sweet, Academic Press, New York, 1997, vol. 276, p. 307 Search PubMed
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS
-
G. M. Sheldrick, SHELX97, Program for the Refinement of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed
- International Tables for X-ray Crystallography, vol. C, Kluwer, Dordrech, 1992
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS
- W. Humphrey, A. Dalke and K. Schulten, VMD – Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Computational data. CCDC 1430204–1430206. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt04154f |
| This journal is © The Royal Society of Chemistry 2016 |