
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct analysis of dried blood spots by femtosecond-laser ablation-inductively coupled plasma-mass spectrometry. Feasibility of split-flow laser ablation for simultaneous trace element and isotopic analysis
M.
Aramendía
ab,
L.
Rello
c,
S.
Bérail
d,
A.
Donnard
d,
C.
Pécheyran
d and
M.
Resano
*b
aCentro Universitario de la Defensa-Academia General Militar de Zaragoza, Carretera de Huesca s/n, 50090, Zaragoza, Spain
bDepartment of Analytical Chemistry, Aragón Institute of Engineering Research (I3A), University of Zaragoza, Pedro Cerbuna 12, 50009, Zaragoza, Spain. E-mail: mresano@unizar.es
cDepartment of Clinical Biochemistry, “Miguel Servet” Universitary Hospital, Paseo Isabel La Católica 1-3, 50009, Zaragoza, Spain
dLCABIE, IPREM UMR 5254, CNRS – Université de Pau et des Pays de l'Adour, 64053, Pau cedex 9, France
First published on 26th November 2014
Abstract
This work describes a novel procedure based on the use of a 1030 nm femtosecond (fs) laser ablation (LA) device operating at a high repetition rate (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Hz) coupled to a sector field-inductively coupled plasma-mass spectrometer (ICP-MS), enabling the complete ablation of dried blood spot (DBS) specimens in a reasonable time (200 s for samples of 5 μL). The integration of the complete signal obtained, in combination with the use of Pt as an internal standard (which can be added to the clinical filter paper prior to the blood deposition, ensuring compatibility with unsupervised sample collection schemes), permits obtaining an analytical response that is independent of the particular characteristics of every sample. On the basis of this methodology, an analytical method was developed for the direct determination of several elements (Cd, Co, Cu and Pb) in four blood reference materials as well as in three real samples, providing accurate results in all cases evaluated, at concentration levels ranging from 0.1 to hundreds of μg L−1. Detection limits of 0.043 (Cd), 0.42 (Co), 0.54 (Cu), and 0.040 (Pb) μg L−1 are achieved, and precision values most often range between 3 and 9% RSD. Finally, the potential to couple the LA device simultaneously to a multicollector-ICP-MS and a sector field-ICP-MS unit by split-flow is also demonstrated, thus allowing us to obtain both elemental (Co, Cu, Cd and Pb) and isotopic (Cu isotopic composition) information from every particular DBS, and therefore maximizing the amount of information that can be drawn from a single DBS specimen. Still, the precision of the approach is limited at this point, as RSD values of approx. 1500 ppm and delta variations of almost 4‰ were observed for five DBS specimens created from the same blood sample.
000 Hz) coupled to a sector field-inductively coupled plasma-mass spectrometer (ICP-MS), enabling the complete ablation of dried blood spot (DBS) specimens in a reasonable time (200 s for samples of 5 μL). The integration of the complete signal obtained, in combination with the use of Pt as an internal standard (which can be added to the clinical filter paper prior to the blood deposition, ensuring compatibility with unsupervised sample collection schemes), permits obtaining an analytical response that is independent of the particular characteristics of every sample. On the basis of this methodology, an analytical method was developed for the direct determination of several elements (Cd, Co, Cu and Pb) in four blood reference materials as well as in three real samples, providing accurate results in all cases evaluated, at concentration levels ranging from 0.1 to hundreds of μg L−1. Detection limits of 0.043 (Cd), 0.42 (Co), 0.54 (Cu), and 0.040 (Pb) μg L−1 are achieved, and precision values most often range between 3 and 9% RSD. Finally, the potential to couple the LA device simultaneously to a multicollector-ICP-MS and a sector field-ICP-MS unit by split-flow is also demonstrated, thus allowing us to obtain both elemental (Co, Cu, Cd and Pb) and isotopic (Cu isotopic composition) information from every particular DBS, and therefore maximizing the amount of information that can be drawn from a single DBS specimen. Still, the precision of the approach is limited at this point, as RSD values of approx. 1500 ppm and delta variations of almost 4‰ were observed for five DBS specimens created from the same blood sample.
1. Introduction
In 1963, capillary blood dried on a filter paper (FP) was introduced as a clinical specimen in the U.K. to screen for phenylketonuria in newborns.1 Since that moment, large scale neonatal screening programs based on the use of dried blood spots (DBS) have been implemented throughout the world, testing for more than thirty disorders and contributing to saving and improving many lives.2In the last few years, the importance of DBS related research has grown exponentially with more than 1200 articles published since 2010.2 At present, a large number of biomarkers are detected in DBS, not only for screening for metabolic diseases in newborns,3 but also for other purposes such as therapeutic drug monitoring,4 pharmacokinetics,5 or toxicological6,7 and forensic studies.6
The reason for this wide acceptance of DBS sampling is certainly related to its inherent advantages in different aspects, which have recently awaken the interest of the pharmaceutical sector in this methodology, thus contributing to its growing success.8,9 In this regard, minimally invasive collection of capillary blood via a finger or heel stick is probably the most important advantage, especially when applied to children under 2 years of age, for whom collection of venous blood is much more traumatic. Additionally, the simplicity of the process for preparing DBS specimens allows for non-specialized personal to carry out this procedure, even in unsupervised contexts such as at the patients' homes. The low amount of sample required to prepare a DBS is also advantageous in other situations such as discovery stage pharmacokinetics, where the use of DBS enables the pharmaceutical companies to better comply with the 3R's (Reduce, Refine, Replace) doctrine with respect to animal experiments (particularly when small rodents are deployed), allowing for serial sampling from a single individual.10
As a second main advantage, the ease of transport and conservation of DBS is also remarkable. Due to the diversity of analytes that can be targeted in blood samples, no general conclusions can be drawn about the stability of the DBS specimens with time and/or storage conditions, as those might depend on the particular analyte considered. However, several studies indicate that the filter paper has a stabilizing effect on the sample,11 such that DBS specimens do not normally require very stringent measures for conservation and/or transport such as liquid samples do (i.e. deep freezing at −20 °C to −80 °C). In fact, conservation at room temperature and for large periods of time is possible for many analytes including trace elements,12,13 so that DBS delivery can be carried out even by regular mail.
These features have obvious economical and practical implications. For instance, the ease of sample collection and the low cost of preservation and delivery can be particularly important for bringing mass screening studies to developing countries or remote locations, or for improving results of epidemiological studies for which the number of volunteers is low, as participants could donate samples without the need to go to the clinic.12 Moreover, the stability of DBS and the low storage requirements make the use of these specimens much better suited for biobanking14 than liquid samples. In conclusion, it seems clear from all of the above that extending the use of DBS sampling and analysis to as many analytes as possible would be of great interest from an economical, ethical, logistical and scientific point of view.
However, and in spite of these advantages, detractors of DBS sampling often cite the relative complexity added to the analytical process, as compared to collection and analysis of venous blood, as a major disadvantage for extending the use of this methodology.15 In this regard two issues potentially problematic are normally highlighted: (i) difficulties for sample deposition and DBS formation, and (ii) the fact that a liquid sample is converted into a solid one, a priori, more difficult to analyze.
Although potentially problematic, sample deposition for DBS preparation is well established in the clinical practice as described in detail in specialized documents from the UK National Health System16 or the US Clinical and Laboratory Standards Institute.17 When quantitative results are needed, the use of US Food and Drug Administration (FDA) approved papers,18 with a constant sample retention per surface unit, is normally relied upon, although deposition of controlled volumes by using calibrated capillary pipettes is also accepted.9 More problematic is the influence that variable hematocrit contents, which directly influence sample viscosity, have on blood diffusion and formation of the DBS. Hematocrit is the portion of blood volume occupied by red cells and varies considerably for different individuals.19 The situation becomes especially problematic when commercial blood reference materials are compared with real samples. In this regard, Cizdziel showed, by using Scanning Electron Microscopy, that red cells were absent in DBS prepared from such a reference sample.13 As a result, diffusion properties of this material were totally different from those of real samples, which hampered its use for validation or calibration purposes when Pb determination in DBS by means of laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS) was considered. The development of analytical methods overcoming this difference is therefore highly desirable in the context of DBS quantitative analysis.
As for the second problematic issue, i.e. the fact that a DBS is actually a solid sample, this might indeed introduce additional problems if conventional analysis in solution is to be carried out. In these cases, an additional step for extracting the analyte out of the DBS is normally needed, which reduces sample throughput and increases contamination risks.15 However, and particularly in some areas of application such as trace elemental analysis, direct solid sampling techniques exist that allow for accurate and precise results to be obtained with none or minimal additional sample preparation. This is the case of LA-ICP-MS, a technique providing multielemental trace and ultra-trace as well as isotopic information, the use of which in all kinds of applications,20–22 including bioanalysis,23–28 has increased dramatically in the last few years. However, and in spite of its potential interest, there are only a few papers in the literature attempting the use of LA-ICP-MS for direct (multi)elemental analysis of DBS,13,29,30 and none of them has been fully successful due to different reasons, as will be discussed in detail in Section 3.1.1.
It is the purpose of this paper to develop a methodology for the direct multielemental analysis of DBS at trace and ultra-trace levels by means of LA-ICP-MS, trying to overcome the problem of differential responses for real samples and reference materials, and following as much as possible the recommendations for DBS collection common in the clinical practice as indicated, e.g., by the European Bioanalysis Forum.9 Four analytes covering a wide range of masses, Cd, Co, Cu and Pb, were selected for proving the concept. Determination of these analytes is interesting in the clinical setting for different reasons, such as monitoring prosthesis degradation in implanted patients (Co),31 revealing exposure to toxic metals (Cd and Pb)6 or because the elements targeted play a major role in a given medical condition (Cu in the case of Wilson's Disease).32 On the other hand, and considering recent advances in the field of isotopic analysis of biological samples that demonstrate the potential interest of such isotopic information for diagnosis of metal-related diseases, such as Cu isotopes and Wilson's disease,33,34 simultaneous determination of Cu isotope ratios with high precision by split-flow coupling of the LA system to a multicollector (MC)-ICP-MS was also attempted.
2. Experimental
2.1. Chemicals and reagents
All multielement and single element solutions used for preparation of matrix-matched calibration standards and preparation of the Pt internal standard solution were prepared by serial dilution of 1 g L−1 mono elemental standards for ICP-MS (Inorganic Ventures, Christiansburg, USA) with diluted nitric acid until a final concentration of 0.14 M HNO3. Pro-analysis nitric acid was purchased from Merck (Darmstadt, Germany). Ultrapure water with a resistivity of ≥18.2 mΩ cm was obtained from a Milli-Q system (Millipore, Île-de-France, France).Whatman no. 903 paper cards (lot W-092) were obtained from Whatman International Ltd (Maidstone, U.K.). This is one of the two commercial paper sources registered by the FDA as Class II medical devices.18 4.5 cm diameter discs fitting in the LA cell were manually cut out of totally empty cards (non-inked) using ceramic scissors.
2.2. Samples and standards
Four blood reference samples with certified values for the elements targeted in this study were analyzed for validation purposes. Clincheck Whole Blood Control level II was obtained from Recipe Chemicals + Instruments GmbH (Munich, Germany). Seronorm Trace Element Human Whole Blood, level II was obtained from Sero (Billingstad, Norway). Lyphochek Whole Blood Control, level 1 and level 2 were obtained from BioRad (Hercules, USA). All of these samples are provided as a lyophilized material and were reconstituted in purified water.Real venous-blood samples from four healthy volunteers were obtained from the Hospital Universitario Miguel Servet (Zaragoza, Spain). Three blood samples were characterized for their trace elemental composition either via pneumatic nebulization (PN)-ICP-MS, after 20-fold sample dilution with 0.14 M HNO3 (Cd and Co), or by means of high-resolution continuum source graphite furnace atomic absorption spectrometry (HR CS GFAAS), after 10-fold sample dilution with 0.14 M HNO3 (Pb and Cu). Although precipitation of blood proteins was not evident to the bare eye after acid dilution of the samples, proper sample homogenization was ensured before analysis in order to avoid biased results caused by potential precipitates. For analysis by means of PN-ICP-MS, on the other hand, larger rinsing times between samples were allowed to prevent nebulizer clogging. In all cases external calibration with aqueous standards was used for analysis, with In added as an internal reference in the case of PN-ICP-MS.
Another real sample, which was also analyzed as described above, was used for the preparation of matrix-matched calibration standards. For this purpose, 1 mL aliquots were spiked with small amounts of multielement solutions prepared in 0.14 M HNO3 so as to obtain a concentration range of spiked amounts ranging from 1 to 2000 μg L−1, depending on the element. The same dilution factor (lower than 2%) was introduced in all cases. The standards prepared in this way were kept for 24 h in an automated orbital shaker before deposition on the clinical filter papers was carried out.
Filter paper discs for LA-ICP-MS analysis were prepared following two different procedures, depending on the way the internal standard used as a reference (Pt) was added to the DBS specimens. In a first protocol, Pt was directly spiked into the liquid samples, standards and controls until a final Pt concentration of 100 μg L−1. For this purpose, Pt was added to the matrix-matched standards in a multielement solution together with the rest of the spiked elements as described above. The same protocol was used for spiking the samples and controls but with a monoelemental Pt solution. Afterwards, 5 or 15 μL aliquots of the spiked standards, samples and controls were carefully deposited onto the surface of the filter paper discs, trying to fit as many blood spots as possible on each disc while ensuring enough blank space among them (see Fig. 1 for details). Samples were left to dry at room temperature for at least 4 h and, once dried, were kept in sealed plastic bags at room temperature until analysis.
 | ||
| Fig. 1 Filter paper discs with 5 μL dried blood spots (DBS) from different CRMs (highlighted in the figure) and real samples (spiked and unspiked). (A) Before ablation. The different dispersion properties of the CRMs and the real samples can be clearly appreciated; (B) after ablation. Each ablated area completely covers the DBS plus a variable part of blank paper depending on the DBS size. | ||
In a second protocol,35 the filter paper discs used for sample deposition were impregnated with a 100 μg L−1 Pt solution and were left to dry at room temperature for at least 4 h before deposition of blood was carried out. Once dried, 5 or 15 μL aliquots of matrix-matched standards, samples and controls (non-spiked with Pt) were deposited onto their surface following the same procedure described above. Samples were left to dry at room temperature for at least 4 h and, once dried, were kept in sealed plastic bags at room temperature until analysis.
2.3. Instrumentation and experimental setup for split-flow LA measurements
All measurements were carried out using a Lambda 3 femtosecond laser ablation system (Nexeya SA, Canejan, France). This laser is fitted with a diode-pumped Yb:KGW crystal laser source (HP2, Ampitudes Systèmes, Pessac, France) delivering 400 fs pulses. Three wavelengths can be selected: 1030 nm (fundamental), 515 nm (2nd harmonic) and 257 nm (4th harmonic). The 1030 nm wavelength was used in this study. The laser source operates within a wide range of repetition rates (up to 100 kHz) and energy (<2 mJ per pulse below 1 kHz and <80 μJ per pulse at 100 kHz at this wavelength), which represents a different approach in analytical applications where high energy and low repetition rate are commonly used. The laser beam is focused with a 100 mm objective, and it can be rapidly moved (up to 2 m s−1) with high repositioning precision thanks to a 2D galvanometric scanning module fitted to the optical line. Further details of a previous and similar model (operating in the IR region only) of this laser ablation system are described elsewhere.36,37Splitting of the carrier gas flow through the LA system has been previously used for simultaneous isotopic and elemental analysis using two ICP-MS systems,38 and this possibility was also tested in the present work. Cu isotopic analysis was carried out on a Nu Plasma HR-MC-ICP-MS instrument (Nu instruments, Wrexham, UK), while multielemental analysis was carried out on an Element XR sector field (SF) ICP-MS instrument (Thermo Scientific, Germany) fitted with a Jet Interface for improved sensitivity. This Jet Interface consists of a high capacity dry interface pump and a specially designed set of cones. Additionally, N2 has to be added to the dry LA aerosol before entering the ICP-MS. A N2 flow of 10 mL min−1 was added in all the experiments carried out in this work. The LA coupling to the two ICP-MS systems was implemented using two 10 m long antistatic tubes (PTFE electroconductive, Fisher Bioblock, Illkirch Cedex, France) of 6 mm external diameter and 4 mm internal diameter into the ICP torches of both instruments, using He as the carrier gas. The He carrier gas flow was split after the LA sample cell with a Y-piece. To adjust sensitivity, 80% of the He flow was directed to the MC-ICP-MS while the remaining 20% was sent to the SF-ICP-MS. Considering the high amount of sample ablated and transported to the instruments per second, especially to the MC-ICP-MS, a glass fiber filter (37 mm diameter Millipore cassette partially filled with chromatographic grade glass wool) was placed before the entrance to the ICP torch of this instrument to prevent clogging of the injector tube. A schematic drawing of this experimental setup is shown in Fig. 2.
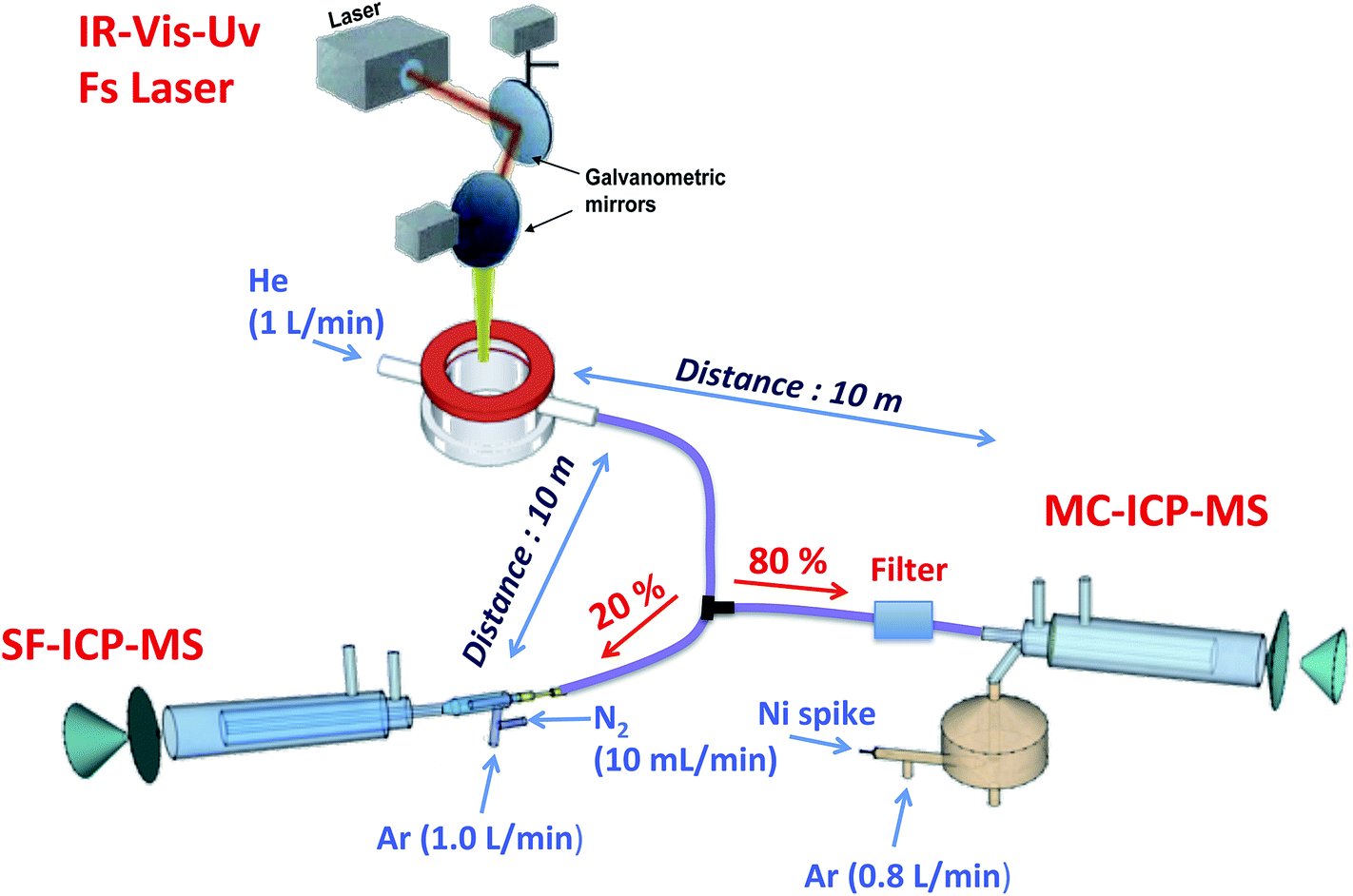 | ||
| Fig. 2 Experimental setup for split-flow simultaneous elemental and isotopic analysis of the DBS samples by means of LA-ICP-MS, using two different types of ICP-MS devices (SF-ICP-MS and MC-ICP-MS). | ||
For isotope ratio measurements with the Nu instrument, a two-inlet torch was used to mix the laser-generated aerosol together with a liquid aerosol (nebulized by means of a pneumatic 200 μL min−1 micro-concentric nebulizer combined with a mini-cyclonic Cinnabar spray chamber) before introduction into the plasma.39,40 This dual-flow introduction system enables easy optimization of the MC-ICP-MS by nebulizing a suitable solution for tuning. Furthermore, during laser ablation analyses, the plasma was kept under wet conditions by the continuous nebulization of a Ni standard solution (1 mg L−1 in 0.14 M HNO3), which was used to correct for mass bias on Cu.
Measuring conditions of the three instruments, summarized in Table 1, were adjusted for maximum sensitivity, stability and plasma robustness while minimizing the influence of interference on the analyte signals.
| Laser ablation conditions | |
|---|---|
| Laser ablation system | Lambda 3 fitted with a HP2 fs Laser source |
| Wavelength | 1030 nm |
| Pulse duration | 400 fs |
| Repetition rate | 30000 Hz |
| Spot diameter (Airy 1/e2) | 21 μm |
| Energy/fluence | 135 μJ/39 J cm−2 |
| Scan speed (y-axis) | 10 mm s−1 |
| Stage speed (x-axis) | 30 μm s−1 |
| Ablation strategy | Circles (5 or 6 mm Ø), made with 250–285 concentric circumferences |
| Transport gas | He, 1 L min−1 |
| Element XR SF-ICP-MS | |
|---|---|
| RF power | 1400 W |
| Ar plasma/auxiliary flow rates | 15.0/1.0 L min−1 |
| Sampling cone and skimmer | Ni |
| N2 flow for jet interface | 10 mL min−1 |
| % of total He flow split | 20% |
| Lens voltages | Optimized daily for maximum sensitivity |
| Scanning mode | E-scan |
| Settling time | Variable depending on the mass jump 1–100 ms |
| Dwell time per acquisition point | 10 ms |
| Acquisition points per peak | 100 |
| Mass window scanned | 5% |
| Acquisition time per replicate | 260 s |
| Resolution mode | Low res (m/Δm ∼ 400) |
| Nuclides monitored | 59Co+, 65Cu+, 111Cd+, 195Pt+, 208Pb+ |
| Nu MC-ICP-MS | |
|---|---|
| RF power | 1300 W |
| Instrument resolution | Pseudo-high resolution ∼5700 (edge resolved power) |
| Source slit width | 0.05 mm |
| Alpha 1 slit | 80 mA |
| Alpha 2 slit | 90 mA |
| Integration time | 5 s |
| Plasma gas flow rate | 13.0 L min−1 |
| Auxiliary gas flow rate | 0.80 L min−1 |
| Nebulizer pressure | 28.8 psi |
| % of total He flow split | 80% |
| Faraday cup configuration | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Collector | H7 | H6 | H5 | H4 | H3 | H2 | H1 | Ax | L1 | L2 | IC0 | L3 | IC1 | IC2 | L4 | ||
| m/z | 65 | 63 | 62 | 61 | |||||||||||||
A high-resolution continuum source graphite furnace atomic absorption spectrometer ContrAA 700 from Analytik Jena (Jena, Germany) and a quadrupole-ICP-MS device (Nexion 300x, Perkin-Elmer, Waltham, USA) were used for validation purposes when required. A Sartorius (Goettingen, Germany) model BP211D analytical balance with a precision of 10−5 g was used for weighing.
2.4. Procedure for simultaneous multielemental and Cu isotope ratio analysis of the samples of DBS by means of split flow LA-ICP-MS
The instrumental parameters used for the analysis of the samples are summarized in Table 1. The filter paper discs containing the DBS specimens were mounted onto plastic holders with the form of an empty cylinder for introduction into the LA cell. For avoiding contamination issues, only the rim of the disc was in contact with the holder, so that complete ablation of the DBS specimens could be carried out without co-ablating the plastic. Each 5 μL DBS was completely ablated by a series of 250–285 concentric circumferences (depending on its size), ablated from the inside to the outside of the blood spot (see Fig. 3). For every replicate measurement a transient signal of 200–260 seconds duration was obtained. For quantitative analysis with the Element XR SF-ICP-MS instrument, integration of this transient signal was carried out for quantification purposes as described in detail in Section 3.1.2. For Cu isotopic analysis with the Nu MC-ICP-MS, on the other hand, a linear regression slope (LRS) method was used, where the signal intensities for 65Cu were plotted against the signal intensities for 63Cu. The slope of the regression curve provides the raw 65Cu/63Cu ratio, which is later corrected for mass bias applying Russell's exponential law41 and the signal obtained for a Ni solution that was simultaneously nebulized and admixed with the ablated aerosol for this purpose. More details of this procedure are provided elsewhere.42,43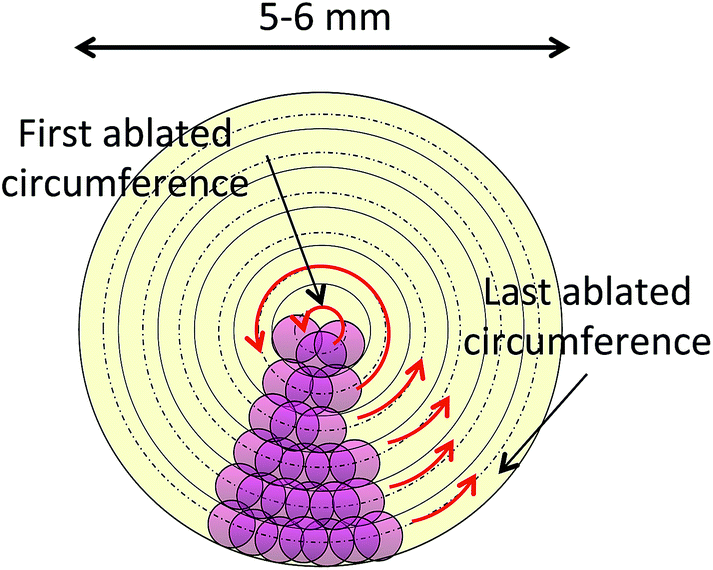 | ||
| Fig. 3 Schematic representation of the ablation protocol used for analysis of the 5 μL dried blood spots. 5–6 mm diameter craters (depending on the DBS size) were constructed by ablating 250–285 concentric circumferences from the inside to the outside of the DBS. All circles were ablated at the same speed. | ||
3. Results and discussion
3.1. Quantitative multielemental analysis
To the best of the authors' knowledge, only three papers have been published attempting this challenge. The first of them explored the technique's possibilities and limitations, studying different DBS specimens prepared with real and reference samples. However, no attempt whatsoever to obtain quantitative information was carried out.13
On another paper by Hsieh et al.,29 Pb could be quantified (in a reference sample) by means of conventional LA-ICP-MS in very small (0.5 μL) dried blood spots deposited onto the surface of a hydrophobic (PTFE) filter membrane, and using aqueous standards for calibration. Using this sample deposition, complete desorption of the dried blood/standard droplets could be accomplished by programming a grid of 4 × 5 spots covering the surface of the droplet and using special ablation conditions: 2 mm laser defocus distance, maximum laser energy available, 20 Hz repetition rate and 8 s dwell time on each ablation spot. On a second paper by the same authors30 the same methodology was used to successfully quantify 8 additional elements in reference samples. For Cd determination, however, significant matrix effects were observed and matrix-matched calibration (obtained by serial dilution of a reference material) had to be deployed.
Although the method proposed by Hsieh et al. permitted to overcome potential chromatographic effects on the DBS by complete desorption of the sample and yielded good analytical results for the quantification of most of the target analytes, blood collection on PTFE slides instead of on FDA approved filter paper cards can be hardly considered as an ideal method for maintaining sample integrity over time and/or through delivery by regular mail, as the sample is not embedded in the PTFE membrane but only sticks to its surface. Moreover, accurate deposition of such small (0.5 μL) sample volumes (needed for ensuring sample stability as well as minimal matrix effects and for maintaining short drying times) onto such a membrane does not seem straightforward and would probably imply collection of a small quantity of venous blood and sample deposition with a micropipette, which eliminates one of the main advantages of the DBS methodology. In any case, and even though the method by Hsieh might not be the most suited for routine analysis due to the potential difficulties discussed above, the idea of ablating the whole DBS for overcoming chromatographic effects and accurately quantifying the analytes irrespective of the sample origin (reference material or real blood samples) is worth exploring with specimens deposited on FDA-approved filter papers and following as much as possible the usual recommendations for bioanalysis in the clinical practice.9 While this possibility seems overly difficult with conventional LA systems delivering high energy laser pulses but at very low repetition rates (up to 20–50 Hz), the use of the Lambda 3 laser, delivering lower energy laser pulses at very high repetition rates (up to 100 KHz), would potentially allow for this ablation strategy.34
For carrying out this test, deposition of relatively small DBS specimens was considered advantageous for keeping the ablation time at a manageable level, taking into account that the full paper depth should be pierced for avoiding any in-depth variations to affect the final quantification. As a result, deposition of 5 μL blood spots, eventually allowing complete ablation of the DBS in a reasonable time (about 200 s as described below), was considered best for initial optimization of the analytical method.
As could be anticipated from the results published in the literature,13 transversal chromatographic effects were visually detectable for the different types of samples under study. As shown in Fig. 1, DBS specimens of similar volume from reconstituted reference blood samples expand over a considerably larger area than those from real blood samples, with some of them even presenting two clearly differentiated areas with different colors. In that situation, complete ablation of the DBS specimen seems totally justified for obtaining comparable quantitative results for both the samples and the reference materials, one of the main goals of the work. For total ablation of the DBS, a series of contiguous concentric circles ablated from the inside to the outside of the blood spot was programmed (see Fig. 3), always ablating a small part of the blank paper surrounding the DBS for ensuring complete introduction of the blood sample into the ICP. Depending on the DBS size, different for CRMs and real samples as seen from Fig. 1, 250–285 concentric circles were needed for complete sample ablation, thus providing transient signals of 200–260 seconds duration for the 5 μL DBS specimens. In all cases, the complete sample was ablated with the same sampling speed.
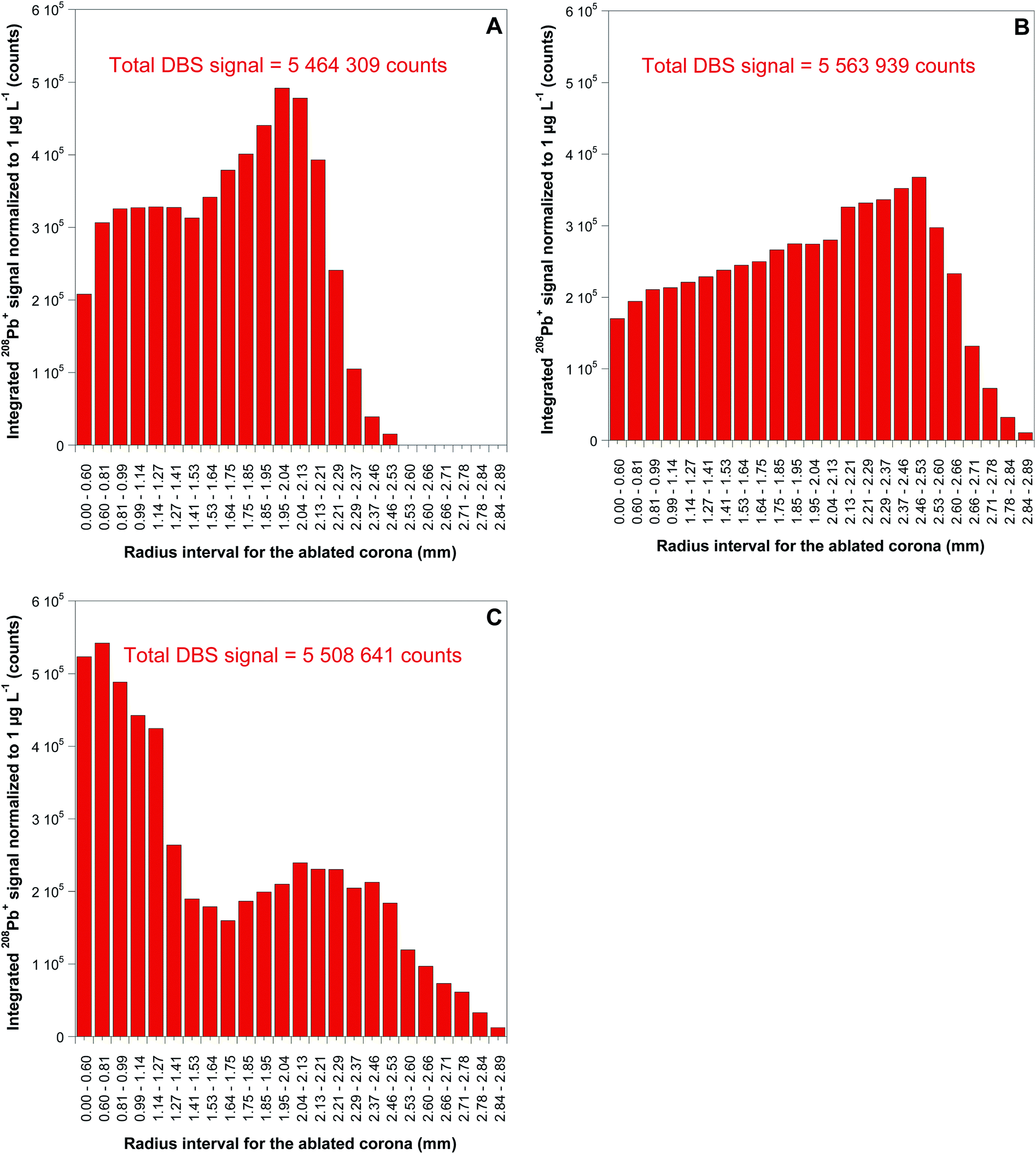 | ||
| Fig. 4 Approximated radial distribution of Pb in 5 μL DBS obtained from: (A) a real sample (B) Lyphochek level I reference sample (C) Seronorm level II reference sample. Each bar represents the integrated 208Pb+ signal, normalized for a concentration of 1 μg L−1, for the ablated corona with internal and external radius indicated in the x-axis in the figure. | ||
At this point, the possibility for using blood volumes greater than 5 μL was considered. In fact, although venous-blood samples were used in this work for optimization of the analytical method, sample volumes that could be easily picked in a finger or heel prick by using calibrated capillary pipettes should be deployed for the method to fully exploit the advantages of DBS sampling, thus having the chance of being implemented in the clinical practice. In this regard, it is interesting to highlight that there are commercially available calibrated pipettes accurately picking and delivering small sample volumes starting from 5 μL ± 0.3%,44 thus making the proposed methodology suited for practical implementation with capillary blood collection. However, and considering the general practice in the clinical setting for neonatal screening studies, where larger volumes of capillary blood (up to 50–75 μL) are normally collected, the possibility for using the same working strategy for larger DBS specimens was considered to be an interesting point for research.
For testing this possibility, 15 μL DBS specimens were prepared from one real sample, following the same protocol deployed for 5 μL DBS and detailed in Section 2.2. The total ablation of this 15 μL DBS needed 365 concentric circles, providing transient signals of 430 seconds duration. Integration of the signals thus obtained was carried out in a similar way to that described before, and the results were compared to those obtained with 5 μL DBS of the same sample for all the analytes. Comparison of these results is summarized in Table 2. As seen from this table, the average signal values obtained for 15 μL DBS specimens correspond to roughly three times those observed for 5 μL DBS, with deviations in the range of 10–15% that were improved down to less than 10% if internal standard standardization is carried out, as will be described in detail in Section 3.1.3. This proves that good results can be obtained with the working methodology proposed irrespective of the blood volume deployed for depositing the DBS.
| Internal standard | Cd | Co | Cu | Pb | |
|---|---|---|---|---|---|
| Ratio signal 5 μL/15 μL | No IS | 3.4 | 3.4 | 2.6 | 2.5 |
| % Deviation | 12.3 | 14.7 | −14.0 | −15.4 | |
| Ratio signal 5 μL/15 μL | Pt as IS | 2.9 | 3.2 | 2.8 | 2.8 |
| % Deviation | −2.1 | 6.3 | −8.3 | −7.4 |
At this point, addition and use of an adequate internal standard (IS) was evaluated. Use of an IS is highly recommended when working with LA-ICP-MS for quantitative purposes for correcting for different ablation efficiencies or sensitivity drifts.24 For this purpose, an element present in all samples and standards for which concentration is constant or known in advance needs to be used. In this regard, and considering the good results obtained in our previous work with DUS,26 addition of a constant Pt amount to all samples and calibration standards was contemplated. This element was considered ideal also in this case, as it is not typically found in blood samples, and should not be affected by spectral overlaps when monitored in a blood matrix by means of ICP-MS. The challenge at this point is to find a way for spiking samples and standards with the IS but without compromising the inherent advantages of DBS sample collection.
Three different approaches for IS application on DBS have been described in the literature for analysis of biomolecules, as detailed in the work by Abu Rabie and co-workers:45 (i) direct application of the IS onto the DBS; (ii) addition of the IS to the blood matrix before spotting it onto the paper; and (iii) use of paper discs that were previously pretreated with IS. These options were thus evaluated in the context of the current work. Option (i) was not considered feasible with the means available at our lab. In the work by Abu Rabie, a very fine IS aerosol was obtained by using a piezoelectric system (Touch Spray) especially designed for the purpose, providing satisfactory results for quantitative bioanalysis after sample extraction. Unfortunately, the Touch Spray device is not available at our lab, and the application of the IS over the filter paper card already impregnated with different blood samples and standards with a simple pipette, or by a similar method, could lead to DBS blurring and sample mixing. From the other two application methodologies, option (iii) is the only one that is fully compatible with DBS sample collection. Self evidently, option (ii) is not applicable to direct sampling from finger tip blood droplets using calibrated pipettes, as described in the introduction, but it may be argued that, a priori and from an analytical point of view, option (ii) would be preferred, as it ensures that the IS is fully incorporated into the sample matrix together with the analyte before deposition on the filter paper. Hence, the performance of both options, (ii) and (iii), was compared in this work. The addition of the IS to the samples, calibration standards and reference materials following these two deposition methodologies is fully described in Section 2.2.
The way in which IS normalization was carried out was different depending on which method had been followed for IS deposition. For DBS where the IS was added to the liquid sample before deposition on the filter paper, the 195Pt+ signal was integrated in the same way as those recorded for the rest of the analytes. The response value for samples and standards was then obtained by dividing the analytes' integrated signals by the 195Pt+ integrated signal. This methodology, however, cannot be deployed when papers pretreated with the IS are used for DBS deposition. In this case, Pt is distributed along the whole filter paper surface and hence, the integrated 195Pt+ signal varies with the size of the DBS ablated, so that direct normalization of the analyte signals with the integrated 195Pt+ signal will lead to erroneous results. In order to avoid this potential problem, the average 195Pt+ signal recorded during the integration interval considered for the analytes was used for normalization instead.
The performance of the two different IS deposition methods considered was next compared following the protocol described above for data treatment. For this purpose, analysis of the Lyphochek Whole Blood Control level 2 reference material was carried out using the two different IS deposition strategies. This material was selected as it exhibits comfortable concentration levels for all the analytes. As seen for Pb in Fig. 5, satisfactory calibration curves were obtained with both approaches for all the analytes, showing good linearities (R2 > 0.99) in the concentration ranges monitored (spiked concentrations: 1–100 μg L−1 for Cd and Co; 1–500 μg L−1 for Pb; 1–2000 μg L−1 for Cu). Please notice that the slopes of both calibration curves should not be directly compared as the amount of Pt that is incorporated into the filter paper using options (ii) and (iii) is not necessarily the same, although it should remain constant for every particular option.
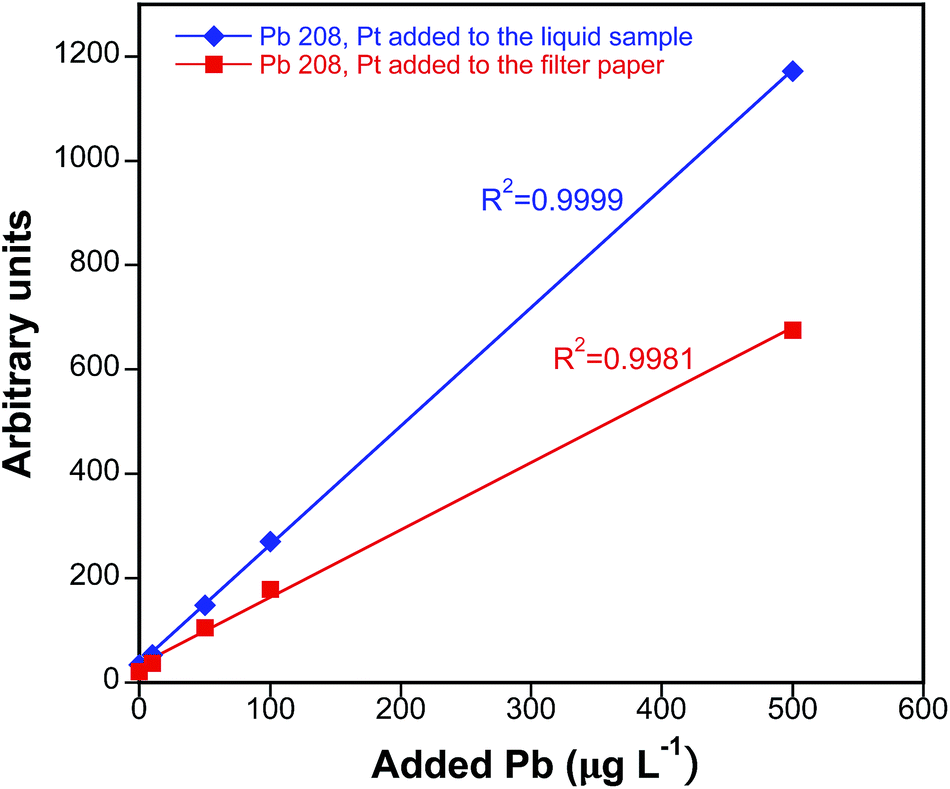 | ||
| Fig. 5 Calibration curves for Pb obtained with matrix-matched standards (5 μL DBS specimens) prepared from a real sample spiked with known Pb amounts, with either Pt added to the liquid sample (blue diamonds) or Pt previously added to the filter paper (red squares). | ||
The results obtained after analysis of the reference material, on the other hand, are gathered in Table 3. As seen from this table, similar results, within experimental error, were obtained for all the analytes using both IS deposition methodologies. Moreover, results are in good agreement with the reference values (obtained from the certificate of analysis, when available, or from analysis of the blood samples by PN-ICP-MS or HR CS GFAAS, as described in Section 2.2), which demonstrates the validity of the analytical protocol developed. RSD values are typically below 10%, except for Co (approx. 12%), probably owing to the lower content of this element. Therefore, and considering that use of paper discs that were previously pretreated with the IS is the only methodology fully compatible with unsupervised sample collection schemes, such a method of IS addition was chosen for further experiments.
| Analyte | Pt added to the liquid sample | Pt added to the filter paper | Reference value |
|---|---|---|---|
| a Reference values available from the certificate of analysis. b Reference value obtained by means of PN-ICP-MS. c Reference value obtained by means of HR CS GFAAS. | |||
| Cd | 22.3 ± 1.5 | 21.6 ± 1.4 | 21.0 ± 4.2a |
| Co | 3.8 ± 0.9 | 4.2 ± 0.8 | 3.8 ± 0.4b |
| Cu | 584 ± 81 | 511 ± 88 | 526 ± 24c |
| Pb | 286 ± 22 | 301 ± 24 | 297 ± 59a |
| Seronorm | Clincheck level II | Lyphochek level I | Real sample M1 | Real sample M2 | Real sample M4 | ||
|---|---|---|---|---|---|---|---|
| a Reference values available from the certificate of analysis. b Reference value obtained by means of PN-ICP-MS. c Reference value obtained by means of HR CS GFAAS. d Defined as ten times the standard deviation of the blank filter signals divided by the sensitivity. | |||||||
| Cd | Ref. value | 6.0 ± 0.4a | 3.14 ± 0.63a | 11.6 ± 2.3a | 0.15 ± 0.02b | 0.17 ± 0.01b | 0.21 ± 0.03b |
| LA-SF-ICP-MS | 5.6 ± 0.2 | 2.75 ± 0.24 | 12.0 ± 1.4 | 0.10 ± 0.03 | 0.17 ± 0.02 | 0.18 ± 0.02 | |
| LOD | 0.043 | ||||||
| Co | Ref. value | 6.1 ± 0.4a | 4.17 ± 0.83a | 2.2 ± 0.8b | 0.48 ± 0.05b | 1.58 ± 0.19b | 0.76 ± 0.07b |
| LA-SF-ICP-MS | 6.8 ± 0.8 | 4.43 ± 0.81 | 2.0 ± 0.7 | <LOQd | 1.70 ± 0.24 | <LOQd | |
| LOD | 0.42 | ||||||
| Cu | Ref. value | 666 ± 58a | 805 ± 70c | 586 ± 66c | 818 ± 46c | 958 ± 75c | 858 ± 81c |
| LA-SF-ICP-MS | 646 ± 110 | 857 ± 136 | 594 ± 102 | 892 ± 136 | 948 ± 163 | 729 ± 126 | |
| LOD | 0.54 | ||||||
| Pb | Ref. value | 393 ± 21a | 237 ± 47a | 117 ± 23a | 22 ± 1.1c | 28 ± 2.1c | 58 ± 4.1c |
| LA-SF-ICP-MS | 381 ± 26 | 259 ± 26 | 144 ± 23 | 24 ± 2.7 | 27 ± 3.5 | 55 ± 5.3 | |
| LOD | 0.040 | ||||||
As for limits of detection (LOD), these were estimated using the signals obtained for 5 calibration standards and also monitoring 5 blank filters (only Milli-Q water was deposited onto them). LODs were calculated as 3 times the standard deviation of the blank filter signals divided by sensitivity obtained for each of the calibration standards. The final value was obtained as the average of these 5 individual values and is also displayed in Table 4 for each of the target analytes. The higher values obtained for Co and Cu are probably related to the existence of spectral interference in the ICP-MS not resolved in the low resolution mode of analysis deployed (38Ar23Na+ and 43Ca16O+ for 59Co+ and 40Ar25Mg+ for 65Cu+, respectively). Although further experiments would be needed, it seems clear that some room for improvement exists in this regard if medium resolution is deployed for determination of these two analytes. In this work, however, it was preferred to work in the low resolution mode, thus ensuring a higher sensitivity, as the LODs achieved already suffice for the vast majority of the applications intended. Furthermore, and as shown in Table 4, very low detection limits were obtained for Cd and Pb. This is not surprising considering the extremely high sensitivity that can be achieved with the experimental setup deployed, especially the LA unit that is able to ablate a very high amount of sample per second. It is worth indicating at this point that all results were obtained with the split-flow experimental setup described in Section 2.4, i.e., by only directing 20% of the LA ablated aerosol into the SF-ICP-MS instrument. Although one might consider that, by directing the full amount of ablated aerosol into the SF-ICP-MS instrument LODs could be somewhat improved, it is our belief that this would probably lead to increased matrix effects due to the high amount of blood and, especially, paper matrix introduced into the ICP-MS per second, as observed for Cu isotopic analysis in the MC-ICP-MS instrument (see Section 3.2 for more details). As a result, and even if some additional experiments would be needed to exactly determine how much sample matrix can be efficiently dealt with by the ICP-MS, we believe that the LODs provided can be considered as a realistic estimation of what can be achieved with this method of analysis. As previously indicated, only some improvements coming from the use of higher resolution modes for interfered elements could be expected.
3.2. Simultaneous Cu isotopic analysis by means of split-flow LA-MC-ICP-MS
As briefly discussed in the introduction, our recent experience in the field of isotopic analysis of biological samples has demonstrated the potential interest of such isotopic information, combined with quantitative information for the same elements, for diagnosis of metal-related diseases such as Wilson's disease and Cu isotopes.33,34 Based on these results, determination of Cu isotope ratios with high precision in the same DBS specimen used for quantitative analysis by split-flow coupling of the LA system to a MC-ICP-MS was attempted in this work, as a means to provide an analytical method capable of maximizing the information extracted from a single blood spot that might be, for instance, archived in biobanks.Unfortunately, the challenges related to this task are noteworthy. To start with, previous results obtained for Cu isotopic determination in serum samples33 showed that, for discriminating patients with a certain medical condition related to Cu metabolism from healthy individuals, the method of analysis deployed must provide uncertainty budgets below about 0.5 delta units (2s), or different populations may overlap. For achieving this goal, serum samples were acid-digested in the cited work and Cu was separated from the sample matrix by ion-exchange chromatography, which permitted to virtually eliminate Na from the measuring solutions. In fact, Na is found at high concentrations in serum and blood samples and causes significant interference problems in the ICP-MS that affect both precision and accuracy for Cu isotope ratio determination. In particular, Na is at the origin of the 40Ar23Na+ interference on the 63Cu+ signal. Considering that Na concentration in serum or blood of healthy patients is about 2000 times higher than that of Cu (even higher for Wilson's disease patients), the significance of this interference is very remarkable for this kind of analysis, where extremely good precision and accuracy values are needed.
When LA is used for sampling dried matrix spots for which minimal sample pre-treatment is aimed at, chemical analyte isolation from the sample matrix before analysis is not feasible and other ways to circumvent the problem of the 40Ar23Na+ overlap must be found. For this purpose, a method frequently used in the past when instruments fitted with reaction or collision cells were not available yet was first tested in this work. Weakly bonded polyatomic interferents such as 40Ar23Na+ are believed to be formed by condensation reactions in the interface region of the ICP-MS,46 and the use of mixed-gas plasmas (obtained by addition of molecular and inert gases to the coolant, nebulizer or auxiliary Ar gas flows) has been found to reduce the extension in which these condensation reactions occur without affecting analyte sensitivity to a large extent.47 In this particular case, N2 (g) was added in the torch injector of the ICP-MS, a solution that had proved successful in the past for reducing the 40Ar23Na+ interference on 63Cu+ for seawater analysis.48 Moreover, addition of N2 could also help to alleviate other matrix effects that might be present from the heavy sample matrix tackled in this work, through provision of more robust plasma conditions.49
This possibility was first tested with aqueous solutions, without any laser coupling. A two-inlet torch was used to introduce the Ar flow needed for nebulizing the aqueous solutions (first port), while N2 was added through the second port into the MC-ICP-MS torch, together with He to mimic the conditions used for LA analysis. In particular, three different solutions were prepared for exploring the effect of N2 addition on the measured Cu ratios (i) 200 μg L−1 Cu; (ii) 400 mg L−1 Na and (iii) 200 μg L−1 Cu + 400 mg L−1 Na, the latter solution showing similar concentration ratios for Cu and Na to those observed in real blood samples. These solutions were measured in the MC-ICP-MS at different N2 flow rates added to the main He and Ar flows, which were also finely optimized for each experiment together with the ion optics to avoid reflected power in the instrument to increase too much. The results of these experiments are summarized in Fig. 6.
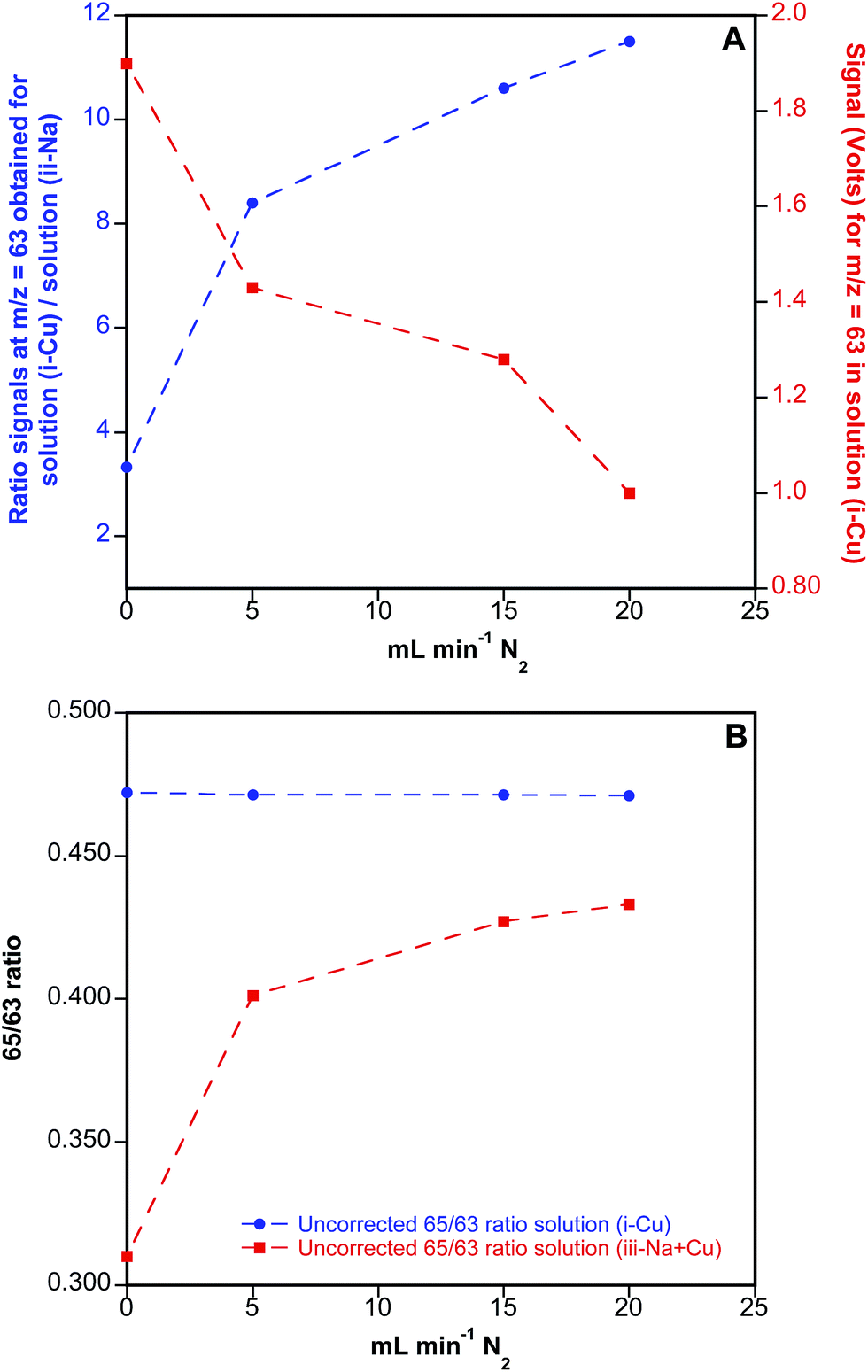 | ||
| Fig. 6 Experiments for reduction of the 40Ar23Na+ interference on the 63Cu+ signal through addition of N2 to the plasma in the Nu MC-ICP-MS instrument. (A) Left y-axis: ratio for the signals monitored at m/z = 63 obtained for solution (i), containing 200 μg L−1 Cu, vs. solution (ii), containing 400 mg L−1 Na; right y-axis: signal intensity obtained at m/z = 63 for solution (i). (B) Uncorrected 65/63 ratios obtained for solution (i) and for solution (iii), which contains 200 μg L−1 Cu + 400 mg L−1 Na. | ||
As seen from Fig. 6A, addition of N2 to the ICP-MS results in a clear reduction of 40Ar23Na+ interference on 63Cu+, as shown by the increasing ratio obtained for signals monitored at m/z = 63 in solution (i), containing only Cu, vs. solution (ii), containing only Na, when increasing amounts of N2 were added to the system. Unfortunately and as also shown in Fig. 6A, this is accompanied by a significant reduction of analyte sensitivity, reaching a factor of 2 for a flow of N2 of 20 mL min−1. On the other hand, the effect of the N2 flow added on the uncorrected 65/63 isotope ratios is displayed in Fig. 6B. As seen from this figure, 65/63 ratios obtained for solution (iii), containing both Cu and Na, are biased low when no N2 is added to the system due to the formation of 40Ar23Na+ polyatomic species. Even though addition of N2 significantly improves the 65/63 ratio obtained for this solution, the results never match those obtained for solution (i), containing only Cu, indicating that the interference is only partially resolved by using this methodological approach. As a result, and considering also the reduction in sensitivity introduced by the addition of N2 to the system, this working methodology was disregarded for further experiments with DBS specimens.
Being the previous methodology unsuccessful for completely eliminating the influence of the 40Ar23Na+ interference on the 63Cu+ signal, the same approach that had provided good results for the analysis of DUS in ref. 34, i.e. working at pseudo high resolution mode and measuring the signals at the left side of the peak, was used for the rest of the experiments. Although this working methodology permitted to circumvent the interference problem for urine samples in the cited work, this effect was achieved at the cost of reducing the analyte sensitivity by a factor of 10, which ultimately resulted in a considerable loss of precision, and this was the reason why this methodological approach was not considered from the beginning.
In addition, the first experiences with the DBS samples and the LA system showed that important matrix effects appear in the MC-ICP-MS instrument when these specimens were analyzed using the ablation conditions described in Section 2.3, selected for achieving adequate sensitivity and reasonable analysis time. In particular, the high amount of sample ablated per second and the fact that 80% of the ablated aerosol was directed to the MC-ICP-MS caused significant signal suppression in this instrument (directly observed in the 62Ni+ signal used for mass bias correction) and led to torch injector clogging after just two or three DBS ablations. To address the problem of clogging, a glass fiber filter (see Section 2.3. for details) had to be placed just before the entrance of the ablated aerosol to the instrument torch (see Fig. 2), which permitted to work without severe sensitivity fluctuations in sessions of more than 8 h. On the other hand, and as for the signal suppression observed for 62Ni+ in the MC-ICP-MS, ablation of a blank filter permitted to conclude that most suppression effects came from the filter paper. This fact is advantageous as a similar response in the ICP-MS can be expected for all the DBS samples then, despite the possible matrix differences found in the original blood samples.
A specific study for determining the best precision achievable under laboratory conditions by means of this technique was carried out next. For this purpose, a set of 5 different DBS specimens from a healthy volunteer with a Cu content of about 1 mg L−1 was analyzed. The first issue taken into consideration was the way of treating the data for correcting 65/63 isotope ratios for mass bias, and two different approaches were also tested in this occasion: linear regression slope (LRS) and point-by-point methodology.34 In the LRS method, first proposed by Fietzke,42,43 the raw 65Cu/63Cu ratio is estimated as the linear regression slope of the graphical representation of 65Cu vs.63Cu signals, which is later corrected for mass bias applying Russell's exponential law41 and the signal obtained for the Ni solution simultaneously nebulized and admixed with the ablated aerosol. Conversely, in the point-by-point strategy the 65/63 ratio for every individual measurement (1 value every 5 seconds) is calculated using the signal intensities (blank-corrected) for 65Cu and 63Cu, and those ratios are averaged at the end for the complete transient signal. More details on these correction strategies can be found elsewhere.34,43,50 The results of this experiment showed that, as was the case for the analysis of DUS,34 the linear regression slope (LRS) method provides much better precision for the transient signals obtained than the traditional way to process data based on the use of a point-by-point methodology. The results obtained with the LRS method are displayed in Table 5. As can be seen, RSD values on the corrected ratios of about 800–1500 ppm (internal precision) for 63Cu+ signal intensities of approx. 100–200 mV corresponding to Cu contents of about 1 mg L−1 are obtained, while the point-by-point methodology provided RSD values up to 4 times worse for the same samples.
| δ 65Cu (‰) | RSD (ppm) | |
|---|---|---|
| DBS 1 | 1.57 | 1160 |
| DBS 2 | 1.33 | 1330 |
| DBS 3 | −2.17 | 1560 |
| DBS 4 | 0.21 | 800 |
| DBS 5 | −0.92 | 770 |
| Overall | 0.03 | 1570 |
The delta values obtained for these specimens, using the Seronorm blood material as a reference, are also shown in Table 5. As can be noticed, the variation found for every specimen is relatively large (from 1.6 to −2.2‰), but the overall delta is very close to zero, as could be expected, because there is a priori no reason to find differences in the Cu ratios for a healthy patient and a reference material that typically exhibits the “natural” Cu composition. This delta variation of almost 4‰ is obtained in the best analytical situation, if compared to the DBS from patients suffering from different medical conditions affecting Cu metabolism, for which much lower Cu contents can be expected. Nevertheless, these values are comparable to those obtained for analysis of DUS,34 indicating that the use of the split-flow methodology does not introduce additional sources of uncertainty to the method. While these values may still not be good enough to differentiate patients suffering from different diseases affecting Cu metabolism from healthy individuals,33 it seems clear that, in spite of the challenge that coupling the three instruments used in this work might represent, the method proposed for simultaneous isotopic and quantitative analysis of DBS samples is promising and can become a reality if the new MC-ICP-MS instrumentation available in the market providing better resolution values at a lower sensitivity cost is deployed for analysis.51
4. Conclusions
In this work it has been shown that simultaneous quantitative and isotopic information can be obtained after the direct analysis of DBS specimens by split-flow LA simultaneously coupled to a MC-ICP-MS and a SF-ICP-MS instrument, without degrading the analytical performance of each of the hyphenated techniques, which could be a very interesting alternative for maximizing the information that can be drawn from a single DBS specimen that might be, e.g., archived in a biobank or difficult to obtain again.The selection of an ablation strategy implying the complete sampling of each DBS specimen has permitted to achieve for the first time similar analytical responses for DBS obtained from real and reference samples by means of LA-ICP-MS. In this way, calibration is simplified and can rely upon construction of regression curves based on in-house matrix-matched standards (prepared out of spiked pooled blood samples from healthy volunteers or from reference materials), leading to satisfactory results in terms of precision, accuracy and LOD for analysis of real blood samples in the form of DBS.
Acknowledgements
This work has been funded by the Spanish Ministry of Economy and Competitiveness (project CTQ2012-33494), the Aragón Government (Fondo Social Europeo), the Région Aquitaine and Feder.References
- R. Guthrie and A. Susi, Pediatrics, 1963, 32, 338–343 CAS.
- P. A. Demirev, Anal. Chem., 2013, 85, 779–789 CrossRef CAS.
- J. J. Pitt, Clin. Biochem. Rev., 2010, 31, 57–68 Search PubMed.
- P. M. Edelbroek, J. van der Heijden and L. M. L. Stolk, Ther. Drug Monit., 2009, 31, 327–336 CrossRef.
- P. Beaudette and K. P. Bateman, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2004, 809, 153–158 CrossRef CAS.
- C. P. Stove, A. S. M. E. Ingels, P. M. M. De Kesel and W. E. Lambert, Crit. Rev. Toxicol., 2012, 42, 230–243 CrossRef CAS.
- M. Resano, L. Rello, E. García-Ruiz and M. A. Belarra, J. Anal. At. Spectrom., 2007, 22, 1250–1259 RSC.
- N. Spooner, Bioanalysis, 2010, 2, 1343–1344 CrossRef CAS.
- P. Timmerman, S. White, S. Globig, S. Lüdtke, L. Brunet and J. Smeraglia, Bioanalysis, 2011, 3, 1567–1575 CrossRef CAS.
- S. Tanna and G. Lawson, Anal. Methods, 2011, 3, 1709–1718 RSC.
- T. W. McDade, S. Williams and J. J. Snodgrass, Demography, 2007, 44, 899–925 CrossRef.
- J. C. Rockett, G. M. Buck, C. D. Lynch and S. D. Perreault, Environ. Health Perspect., 2004, 112, 94–104 CrossRef.
- J. V. Cizdziel, Anal. Bioanal. Chem., 2007, 388, 603–611 CrossRef CAS.
- P. Elliott and T. C. Peakman, Int. J. Epidemiol., 2008, 37, 234–244 CrossRef.
- P. Abu-Rabie, Bioanalysis, 2011, 3, 1675–1678 CrossRef CAS.
- UK Newborn Screening Programme Centre, Guidelines for Newborn Blood Spot Sampling, UK National Screening Committee, February 2012, ISBN: 978-0-9562374-2-2, newbornbloodspot.screening.nhs.uk.
- W. H. Hannon, V. R. De Jesús, M. S. Chavez, B. F. Davin, J. Getchell, M. Green, P. V. Hopkins, K. B. Kelm, B. Noorgaard-Pedersen, C. Padilla, E. Plokhovy, J. V. Mei and B. L. Therrell, Blood Collection on Filter Paper for Newborn Screening Programs; Approved Standard, Clinical and Laboratory Standards Institute (CLSI), Document NBS-01–A6, West Valley Road, Suite 1400, Wayne, Pennsylvania 19087–1898 USA, 6 edn, 2013 Search PubMed.
- J. V. Mei, S. D. Zobel, E. M. Hall, V. R. de Jesús, B. W. Adam and W. H. Hannon, Bioanalysis, 2010, 2, 1397–1403 CrossRef CAS.
- A. J. Wilhelm, J. C. G. den Burger, R. M. Vos, A. Chahbouni and A. Sinjewel, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1595–1598 CrossRef CAS.
- J. Pisonero, B. Fernández and D. Günther, J. Anal. At. Spectrom., 2009, 24, 1145–1160 RSC.
- J. Koch and D. Günther, Appl. Spectrosc., 2011, 65, 155–162 CrossRef.
- R. E. Russo, X. Mao, J. J. Gonzalez, V. Zorba and J. Yoo, Anal. Chem., 2013, 85, 6162–6177 CrossRef CAS.
- I. Konz, B. Fernández, M. L. Fernández, R. Pereiro and A. Sanz-Medel, Anal. Bioanal. Chem., 2012, 403, 2113–2125 CrossRef CAS.
- D. Hare, C. Austin and P. Doble, Analyst, 2012, 137, 1527–1537 RSC.
- M. R. Flórez, M. Aramendía, M. Resano, A. C. Lapeña, L. Balcaen and F. Vanhaecke, J. Anal. At. Spectrom., 2013, 28, 1005–1015 RSC.
- M. Aramendía, L. Rello, F. Vanhaecke and M. Resano, Anal. Chem., 2012, 84, 8682–8690 CrossRef.
- W. Nischkauer, F. Vanhaecke, S. Bernacchi, C. Herwig and A. Limbeck, Spectrochim. Acta, Part B, 2014, 101, 123–129 CrossRef CAS.
- D. Pozebon, G. L. Scheffler, V. L. Dressler and M. A. G. Nunes, J. Anal. At. Spectrom., 2014, 29, 2204–2228 RSC.
- H.-F. Hsieh, W.-S. Chang, Y.-K. Hsieh and C.-F. Wang, Talanta, 2009, 79, 183–188 CrossRef CAS.
- H.-F. Hsieh, W.-S. Chang, Y.-K. Hsieh and C.-F. Wang, Anal. Chim. Acta, 2011, 699, 6–10 CrossRef CAS.
- A. Sarmiento-González, J. M. Marchante-Gayón, J. M. Tejerina-Lobo, J. Paz-Jiménez and A. Sanz-Medel, Anal. Bioanal. Chem., 2008, 391, 2583–2589 CrossRef.
- A. Ala, A. P. Walker, K. Ashkan, J. S. Dooley and M. L. Schilsky, Lancet, 2007, 369, 397–408 CrossRef CAS.
- M. Aramendía, L. Rello, M. Resano and F. Vanhaecke, J. Anal. At. Spectrom., 2013, 28, 675–681 RSC.
- M. Resano, M. Aramendía, L. Rello, M. L. Calvo, S. Bérail and C. Pécheyran, J. Anal. At. Spectrom., 2013, 28, 98–106 RSC.
- J. Mommers, Y. Mengerink, E. Ritzen, J. Weusten, J. van der Heijden and S. van der Wal, Anal. Chim. Acta, 2013, 774, 26–32 CrossRef CAS.
- E. Ricard, C. Pécheyran, G. Sanabria Ortega, A. Prinzhofer and O. F. X. Donard, Anal. Bioanal. Chem., 2011, 399, 2153–2165 CrossRef CAS.
- C. Pécheyran, S. Cany, P. Chabassier, E. Mottay and O. F. X. Donard, J. Phys.: Conf. Ser., 2007, 59, 112–117 CrossRef.
- H.-L. Yuan, S. Gao, M.-N. Dai, C.-L. Zong, D. Günther, G. H. Fontaine, X.-M. Liu and C. R. Diwu, Chem. Geol., 2008, 247, 100–118 CrossRef CAS.
- G. Ballihaut, L. Tastet, C. Pécheyran, B. Bouyssiere, O. Donard, R. Grimaud and R. Lobinski, J. Anal. At. Spectrom., 2005, 20, 493–499 RSC.
- G. Ballihaut, F. Claverie, C. Pécheyran, S. Mounicou, R. Grimaud and R. Lobinski, Anal. Chem., 2007, 79, 6874–6880 CrossRef CAS.
- F. Vanhaecke, L. Balcaen and D. Malinovsky, J. Anal. At. Spectrom., 2009, 24, 863–886 RSC.
- J. Fietzke, M. Frische, T. H. Hansteen and A. Eisenhauer, J. Anal. At. Spectrom., 2008, 23, 769–772 RSC.
- J. Fietzke, V. Liebetrau, D. Günther, K. Gürs, K. Hametner, K. Zumholz, T. H. Hansteen and A. Eisenhauer, J. Anal. At. Spectrom., 2008, 23, 955–961 RSC.
- http://www.hawksley.co.uk/blood_analysis/02i_pipettes/index.shtml, last accessed November 2014.
- P. Abu-Rabie, P. Denniff, N. Spooner, J. Brynjolffssen, P. Galluzzo and G. Sanders, Anal. Chem., 2011, 83, 8779–8786 CrossRef CAS.
- H. Niu and R. S. Houk, Spectrochim. Acta, Part B, 1996, 51, 779–815 CrossRef.
- B. S. Sheppard and J. A. Caruso, J. Anal. At. Spectrom., 1994, 9, 145–149 RSC.
- M. J. Bloxham, P. J. Worsfold and S. J. Hill, Anal. Proc., 1994, 31, 95–97 RSC.
- C. Agatemor and D. Beauchemin, Anal. Chim. Acta, 2011, 706, 66–83 CrossRef CAS.
- V. N. Epov, S. Berail, M. Jimenez-Moreno, V. Perrot, C. Pecheyran, D. Amouroux and O. F. X. Donard, Anal. Chem., 2010, 82, 5652–5662 CrossRef CAS.
- L. Yang, Mass Spectrom. Rev., 2009, 28, 990–1011 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |