
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-performance biphasic NaxMnO2 electrodes for cost-effective and high-power aqueous sodium batteries and capacitors†
Andrii
Boichuk
 *ab,
Tetiana
Boichuk
a,
Mahesh
Eledath-Changarath
a,
Marie
Krečmarová
a,
Rafael
Abargues
a,
Pablo P.
Boix
c,
María C.
Asensio
de,
Saïd
Agouram
f and
Juan F.
Sánchez-Royo
*ae
*ab,
Tetiana
Boichuk
a,
Mahesh
Eledath-Changarath
a,
Marie
Krečmarová
a,
Rafael
Abargues
a,
Pablo P.
Boix
c,
María C.
Asensio
de,
Saïd
Agouram
f and
Juan F.
Sánchez-Royo
*ae
aICMUV, Instituto de Ciencia de Materiales, Universidad de Valencia, 46071 Valencia, Spain. E-mail: Andrii.Boichuk@uv.es; Juan.F.Sanchez@uv.es
bKing Danylo University, 76000, Ivano-Frankivsk, Ukraine
cInstituto de Tecnología Química (Universitat Politècnica de València-Agencia Estatal Consejo Superior de Investigaciones Científicas), Valencia, 46022, Spain
dInstituto de Ciencia de Materiales de Madrid ICMM, CSIC, 28049 Madrid, Spain
eMATINÉE: CSIC Associated Unit (ICMM-ICMUV), Universidad de Valencia, Valencia, Spain
fDepartment of Applied Physics and Electromagnetism, University of Valencia, 46100 Valencia, Spain
First published on 27th December 2024
Abstract
Aqueous sodium batteries and capacitors offer a low-cost and sustainable alternative to lithium-based energy storage systems, with their performance crucially dependant on the choice of electrode materials. Among the candidates commonly used in sodium-ion devices, various phase modifications of presodiated manganese oxide are considered promising. In this work, we synthesized biphasic (orthorhombic/monoclinic) NaMnO2 using a cost-effective sol–gel technique and investigated its performance as an electrode material for aqueous sodium electrochemical systems. The performance was evaluated through cyclic voltammetry and galvanostatic charge–discharge measurements. The results demonstrated that NaMnO2 electrodes were highly suitable for high-power energy devices, exhibiting a specific capacity of 103 mA h g−1 and high capacity retention, even under high current conditions (82% capacity retention as the current increased from 1C to 20C). The superior electrochemical performance, especially under high discharge current conditions, was attributed to the optimal combination of different pseudocapacitive mechanisms associated with the biphasic monoclinic-orthorhombic phase structure, which ensured both high capacity and stability during cycling, as well as the morphology of the samples. These results paved the way for the development of high-power, stable, and cost-effective aqueous sodium-ion storage devices.
Introduction
Despite the dominance of lithium-based energy storage devices, cheaper, more abundant, safer, and efficient sodium-ion (Na) batteries1–4 and supercapacitors5–7 have garnered increasing attention. One of the most significant challenges in the intense research on these devices is to enhance their specific capacity, power, and energy, aiming to achieve competitive operational performance. This can be accomplished by selecting Faraday-active electrode materials that favor reversible Na ion intercalation. Layered structures based on manganese oxides have emerged as promising candidates for such applications. For instance, high-voltage supercapacitors based on tetragonal NaMnO2 have already been developed, exhibiting a calculated capacity of 89.6 F g−1 at a scan rate of 5 mV s−1 and demonstrating good cycle stability.8 In the presence of Na+ cations, the electrochemical potential of manganese oxide in electrolytes shifts, allowing for the achievement of higher specific energy storage. Furthermore, NaxMnO2 has been shown to exhibit beneficial electrochemical properties, especially in terms of cycle stability, when sodium doping is applied during synthesis.9–13Two phases of sodiated manganese oxide materials have been reported to date (Fig. 1a): monoclinic (α phase, space group C2/m) and orthorhombic (β phase, space group Pmnm). Additionally, β-NaMnO2 systems have demonstrated capacity values of approximately 90 mA h g−1 at high currents, with a capacity retention of around 89% over the first 100 cycles.14 These results suggest that the α-phase is particularly effective in providing good rate capability, while the β-phase is more suited for maintaining long cycle life.14 Consequently, both phases exhibit distinct but complementary electrochemical properties, which can be leveraged through their combination in a biphasic composite. Such NaMnO2 polymorphs were observed by Artem M. Abakumov et al.15 as quasi-periodic sequences with varying concentrations of α-NaMnO2 and β-NaMnO2 phases within each NaMnO2 layer of the composite. The electrochemical behavior of these biphasic materials has also been investigated.16,17 In particular, Debasis Nayak et al.16 reported discharge capacity values of 110.5 mA h g−1 with a capacity retention of about 68% and Coulombic efficiency of 98.9% even after 120 cycles at 0.05C. The biphasic NaMnO2 compounds described above have been successfully prepared using high-temperature solid-state reactions.15–17 However, owing to the lack of control over particle size, coarse-grained powders exhibit a small specific surface area, which hampers electrochemical performance at high currents (as is required for supercapacitors) and can be solved using a bottom-up approach, such as the sol–gel technique.18–20 Moreover, electrochemical testing of these microsized NaMnO2 compounds was performed in non-aqueous electrolytes, which may increase the technological complexity of construction and, consequently, the final device cost.
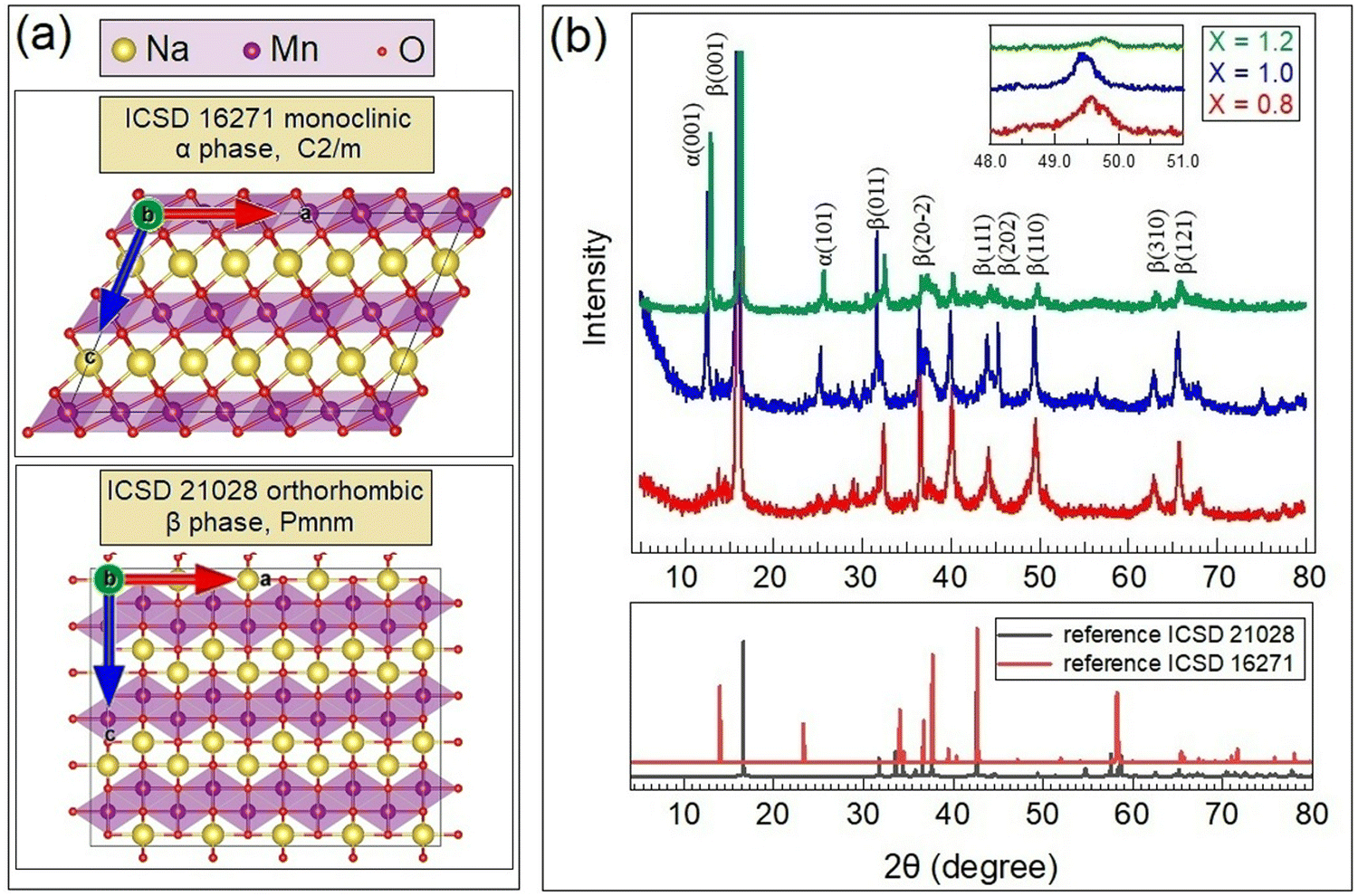 | ||
| Fig. 1 (a) Orthorhombic and monoclinic phases of NaMnO2. (b) XRD patterns of as-synthesized Na0.8MnO2 (x = 0.8), NaMnO2 (x = 1.0) and Na1.2MnO2 (x = 1.2). | ||
In this work, we overcome these limitations by demonstrating the ability of simple and cheap sol–gel techniques to synthesize biphasic NaxMnO2 compounds, which appear to exhibit capacity values as high as 103 mA h g−1 at high scan rates with their preservation for 100 cycles in aqueous electrolytes.
Experimental
Synthesis
Several samples of sodium manganese oxide with different amounts of sodium were prepared using the sol–gel method. 0.2 M water solutions of manganese acetate tetrahydrate C4H6O4Mn·4H2O (Thermo Scientific, 99%) and sodium nitrate NaNO3 (Panreac, 99%) were mixed by stirring for 5 hours. The pH was then adjusted to 8 by adding a 25% ammonia solution. The resulting solution was heated to 80 °C and kept at low pressure (200 torr) for 24 h. The obtained product was manually ground, annealed at 750 °C for 15 h at a heating rate of 3 °C min−1 and finally quenched to room temperature. The choice of the final annealing temperature is determined by DTA (Fig. S1, ESI†) and is described under the literature conditions of α and β phase formation,21.22 We obtained three samples with different Na amounts and nominal stoichiometric compositions: Na0.8MnO2 (x = 0.8), NaMnO2 (x = 1.0) and Na1.2MnO2 (x = 1.2). After cooling to room temperature, the samples were directly transferred to an Ar-filled glove-box and manually ground in an agate mortar.Characterization methods
X-ray diffraction (XRD) characterization of the samples was carried out using a Bruker D8 ADVANCE A25 diffractometer (Cu-Kα radiation source with a wavelength λ = 1.54 Å) in the 2θ range 10–80° with a step size of 0.00205°. Rietveld refinement procedures were carried out using FullProf software. The surface morphology of the powders was analyzed by SEM (Hitachi S4800 microscope at 10 kV of acceleration voltage). High-resolution X-ray photoelectron spectroscopy (XPS) measurements were performed using a SPECS GmbH system (base pressure 1.0 × 10−10 mbar) equipped with an ASTRAIOS 190 2D-CMOS hemispherical analyzer. Photoelectrons were excited with the Al-Kα line (1486.7 eV) of a monochromatic X-ray source μ-FOCUS 500 (SPECS GmbH). Measurements were taken at room temperature with a pass energy of 50 eV. Morphological, compositional and structural analyses of NaxMnO2 samples were also performed by high-resolution transmission electron microscopy (HRTEM) using a field emission gun TECNAI G2 F20 microscope operating at 200 kV, with the capabilities of selected area electron diffraction (SAED) and energy dispersive X-ray spectroscopy (EDX) in the facilities of the Servei Central de Suport a la Investigació Experimental (SCSIE) at the University of Valencia. To prepare the TEM samples, the NaxMnO2 samples were scratched, removed from the substrate and deposited onto a carbon film supported on a copper grid.Electrochemical measurement technology
The active mass of NaMnO2 (80 wt%) was mixed with 15 wt% of carbon black and 5 wt% of PTFE binder and ground in a mortar. An electrode was prepared by repeated brush coating onto nickel foam current collectors. The electrode area was 0.3 cm2, and the loading of the active mass was 8 mg independent of the active material and electrolyte. The cyclic voltammetry (CV) tests were performed (Gamry 5000P equipment) using a three-electrode cell with Pt wire and Ag/AgCl electrode as counter and reference electrodes in an aqueous 1 M solution of Na2SO4(NS), Na2CO3(NC) and NaNO3(NN) (scan rate region of 0.5–50 mV s−1). The specific capacitance of electrode (C) estimated from the cyclic voltammogram was calculated as follows:| C = S/(2ΔUmV), |
Results and discussion
Structure, morphology and stoichiometry of NaxMnO2
The XRD patterns of the as-synthesized and annealed at 750 °C NaxMnO2 samples (x = 0.8, 1.0, and 1.2) are shown in Fig. 1b. The spectra obtained were compared with the data reported for the monoclinic and orthorhombic NaMnO2 phases, revealing that the sol–gel samples studied here comprised a combination of β (ICSD 21028) and α (ICSD 16271) polymorphous phases of sodiated manganese dioxide in a proportion depending on the sodium content. Based on Rietveld refinement processes, low Na-content samples, such as the Na0.8MnO2 ones, show a single β phase structure. Samples x = 1.0 and x = 1.2 can be described as two-phase systems although stoichiometric NaMnO2 samples tend to present a higher ratio of the α phase (rough estimation of the ratio of α-phase to β-phase is 69![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :31 for x = 1.0 and 76:24 for x = 1.2). The cell parameters of all prepared samples are summarized in Table 1. The orthorhombic phases present parameters compatible with those previously reported.23 In the case of monoclinic α phase samples, the c lattice parameter results are larger than those reported in,24,25 which is attributed to the presence of a higher amount of sodium between MnO2 layers (Fig. 1a). The diffraction peak shift has similar values (Δ2θ ≈ 0.2°), and it leads to proportional changing of lattice parameters for both phases. As shown in,15 such a correlation of lattice constant can be explained by quasi-periodic modulated sequences using the known α- and β-NaMnO2 polymorphs as the two limiting cases.
:31 for x = 1.0 and 76:24 for x = 1.2). The cell parameters of all prepared samples are summarized in Table 1. The orthorhombic phases present parameters compatible with those previously reported.23 In the case of monoclinic α phase samples, the c lattice parameter results are larger than those reported in,24,25 which is attributed to the presence of a higher amount of sodium between MnO2 layers (Fig. 1a). The diffraction peak shift has similar values (Δ2θ ≈ 0.2°), and it leads to proportional changing of lattice parameters for both phases. As shown in,15 such a correlation of lattice constant can be explained by quasi-periodic modulated sequences using the known α- and β-NaMnO2 polymorphs as the two limiting cases.
| Sample | Phases | a, Å | b, Å | c, Å |
|---|---|---|---|---|
| Na0.8MnO2 | β | 4.934 | 2.852 | 6.091 |
| NaMnO2 | α | 4.462 | 2.654 | 7.023 |
| β | 5.004 | 2.893 | 6.207 | |
| Na1.2MnO2 | α | 4.457 | 2.647 | 7.012 |
| β | 4.926 | 2.880 | 6.193 |
Fig. 2(a)–(c) shows low magnification TEM micrographs of NaxMnO2 samples with different Na/Mn ratios together with selected area electron diffraction (SAED) patterns acquired in these samples. As revealed by TEM micrographs, the morphology of the synthesized product is made of relatively large crystals. Diffraction spots observed in the SAED patterns shown at the bottom of Fig. 2(a)–(c) are produced by the small monocrystals randomly orientated, giving an angular distribution of (hkl) spots at a distance proportional to 1/dhkl from the (000) spot for each allowed reflection. The SAED pattern produced by Na0.8MnO2 can be indexed only by invoking an orthorhombic phase and is in good agreement with 00-025-0844 in JCPPS files (database). In contrast, the high-resolution TEM images acquired in NaMnO2 and Na1.2MnO2 samples evidence the co-existence of both monoclinic and orthorhombic phases, in agreement with that observed by XRD (Fig. 1). In fact, interplanar distances and lattice parameters, as extracted from SAED patterns and HRTEM images of NaxMnO2 (see Fig. S2 and Table S1 of the ESI†), are also in good agreement with those obtained by XRD.
 | ||
| Fig. 2 TEM micrographs with corresponding selected area electron diffraction (SAED) patterns of Na0.8MnO2 (a), NaMnO2 (b), Na1.2MnO2 (c). SEM images of Na0.8MnO2 (d), NaMnO2 (e) and N1.2MnO2 (f). | ||
SEM micrographs (Fig. 2d, e and f) suggest a high degree of crystallinity, with the presence of layered particles of sodium manganese oxide clearly distinguishable in all cases, while samples with deviations from sodium stoichiometry present more irregular particles with more defect morphology. In fact, samples with x = 1.0 tend to be composed of regular hexagons with a surface size of about 1.5 × 1.5 microns. Energy-dispersive spectroscopy (EDS) measurements were performed to map the elemental distribution on the NaxMnO2 surface (Fig. S3, ESI†). Because we observed deviation from the homogeneous distribution of Na and Mn elements for non-stoichiometric samples (x = 0.8, x = 1.2), XPS measurements were performed in all NaxMnO2 samples to obtain additional information about their chemical composition and atomic bonding nature.
The XPS spectra of Na 1s, Mn 2p, and O 1s core levels acquired in these samples are depicted in Fig. 3a–c, respectively, which are deconvoluted by assuming Gaussian lineshape components. From this analysis, it appears that the Na 1s XPS spectra (Fig. 3a) are composed of different components with an intensity ratio that depends on the sample stoichiometry. A first component, centered at a binding energy of 1071.7 eV, is observed in all analyzed powders, which is attributed to Na+ cations intercalated between MnO2 layers in orthorhombic or/and monoclinic NaxMnO2 configurations.1,26 On the low energy side of the Na 1s spectra, an additional component centered at 1070.1 eV in binding energies can be resolved in non-stoichiometric compounds NaxMnO2 (i.e., x = 0.8 and 1.2), attributable to the presence of metallic Na0.2 In the case of the Na0.8MnO2 sample, the formation of metallic sodium can be explained by complications of sodium migration inside the grains at the initial step of synthesis owing to stable orthorhombic phase formation, which causes the appearance of oxygen defects. For the sample Na1.2MnO2, the obvious reason is an excess of sodium and its accumulation in the intergranular space. These results show that NaxMnO2 powders with x = 1 sodium concentration exhibit a higher crystal quality, as observed by SEM measurements (Fig. 2i) and an efficient Na incorporation into the lattice.
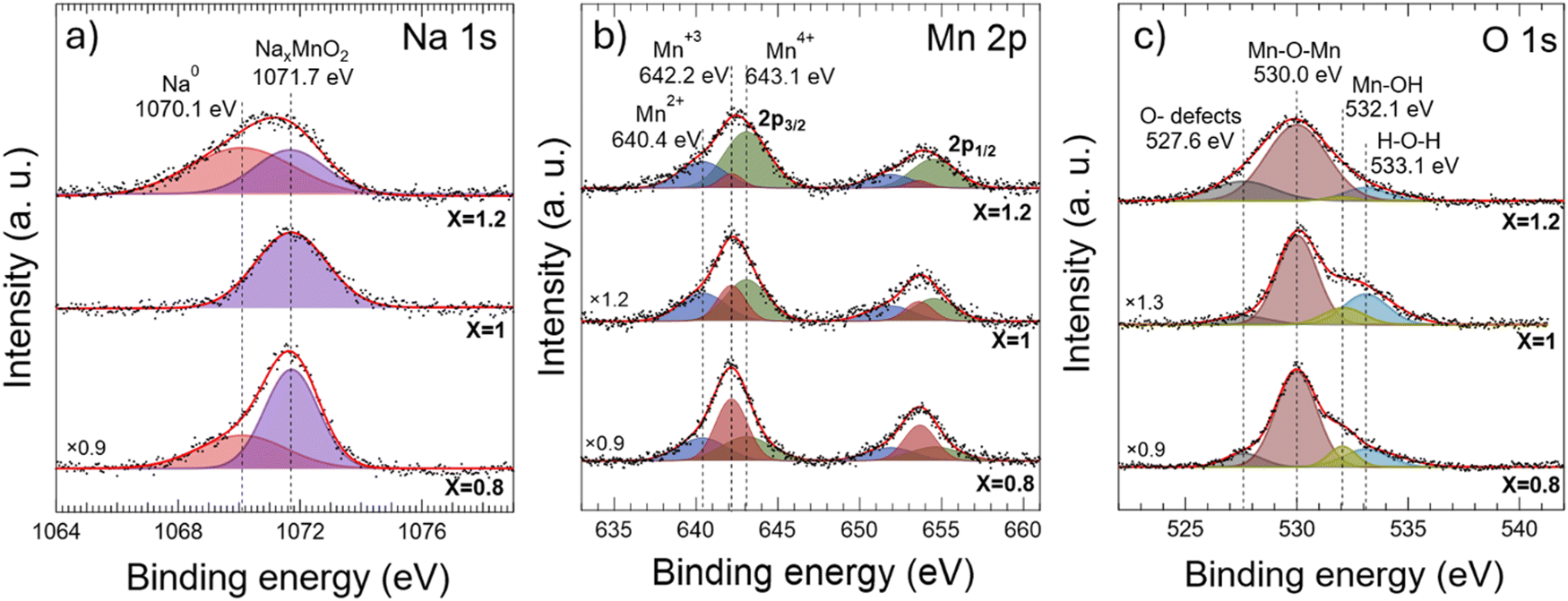 | ||
| Fig. 3 Na 1s (a), Mn 2p (b) and O 1s (c) core-level spectra acquired by XPS for Na0.8MnO2, NaMnO2 and Na1.2MnO2 powders. The fitting curves of the experimental data (red solid lines) were obtained through deconvolution, assuming Gaussian lineshape components. Shadowed Gaussian peaks correspond to the components identified through the deconvolution processes. | ||
Because in-site Mn cations are only surrounded by oxygen anions (in both orthorhombic and monoclinic structures), the observation of any XPS signal attributable to oxidation states different from the Mn3+ one expected in stoichiometric NaMnO2 provides valuable information about the effects of Na incorporation into the host lattice. Deconvolution processes performed on the Mn 2p spectra (Fig. 3b) allow for the resolution of up to three different Mn 2p3/2 and Mn 2p1/2 spin–orbit doublets whose spin–orbit splitting is ≈11.5 eV.27–29 These three doublets, whose Mn 2p3/2 components are centered at 640.4, 642.2, and 643.1 eV, appear to come from Mn2+, Mn3+, and Mn4+ related species, respectively,28,30,31 which indicates the presence of MnO and MnO2 in our samples, coexisting with the NaxMnO2 host lattice.32 Moreover, the evolution of the relative weight of each Mn 2p doublet in the Mn 2p XPS signal with the Na content reveals a continuous enhancement of the presence of Mn4+-related species to the detriment of the Mn3+-related ones, as sodium content increases in the NaxMnO2 powder, while the presence of Mn2+-related species remain stable.
To obtain more insight into the chemical state of the elements of the compounds studied here, we show in Fig. 3c the O 1s XPS signal acquired in the NaxMnO2 powders. Deconvolution of the O 1s spectra (Fig. 3c and Table S2, ESI†) allows the resolution of at least four singlet peaks, which appear at different binding energy (BE) positions: A first singlet related to the presence of oxygen defects (O-defects) and impurities (BE = 527.6 eV);33,34 a second one attributable to the host Mn–O–Mn lattice (BE = 530.0 eV);27,30,35–37 a third one attributable to manganese hydroxide Mn–OH (BE = 532.1 eV);29,35,38 and a fourth one related to adventitious adsorbed H–O–H water (BE = 533.1 eV).29,35,36,38 From the XPS results shown in Fig. 3a and c, a clear correlation can be established between the presence of O-defects density, the linewidth of the O 1s peak coming from the Mn–O–Mn lattice, and non-ionic metallic sodium atoms (1070.1 eV) (see also Table S2, ESI†), indicating that NaMnO2 powders prepared with x = 1 Na content exhibit a higher crystal quality (lower defect density and narrower O 1s linewidth of the Mn–O–Mn related peak) than the non-stoichiometric ones, which is in agreement with morphological SEM results reported here (Fig. 2). Moreover, the stoichiometric NaMnO2 powders present the highest Na+/Mn3+ content ratio among the powders prepared (see Fig. S5d of the ESI†), which suggests that optimal ionic sodium incorporation processes occur in the stoichiometric NaMnO2 powders.
We calculated Na, Mn and O atomic ratios of the powder's surface composition by considering core-level cross-sections at the used photon energy (Fig. S5d, ESI†). The atomic percentage of total chemical composition (Fig. S5d upper panel ESI†) agrees with those measured by EDS (Fig. S3, ESI†). By considering just the chemical composition of ionic elements bounded in the NaxMnO2 powder crystal (Na+, Mn3+ and O−2), i.e., not including non-ionic elements, amorphous components and defects (see Fig. S5d bottom panel ESI†) show a decreasing amount of Mn3+ oxidation state with increasing sodium amount. The calculated atomic Na+/Mn3+ ratios depicted in Fig. S5f of the ESI† linearly increase with increasing sodium amounts. It is worth mentioning that the maximal amount of incorporated ionic sodium is found in powders prepared with x = 1 sodium concentration (see Fig. S5d bottom panel ESI†).
Electrochemical testing of aqueous electrolytes
As described in the experimental section, NaxMnO2 powders of different Na contents are introduced in aqueous electrolytes to analyze their electrochemical properties using the Ag/AgCl reference electrode. Cyclic voltammograms are measured in a voltage range from −0.25 V to 0.8 V. For all samples and electrolytes (Fig. 4), we obtain similar cyclic voltammograms with two parts of pseudocapacitance and corresponding pairs of peaks: intercalation/deintercalation and reversible redox reactions at or near the surface of the electrode material, as described below. | ||
| Fig. 4 Cyclic voltammetry (CV) curves of Na0.8MnO2 (a), (d) and (g), NaMnO2 (b), (e) and (h) and Na1.2MnO2 (c), (f) and (i) in different electrolytes. | ||
At low scanning rates, a cathodic peak can be noticed at voltage values of 0.3–0.35 V irrespective of the powder and electrolyte employed, reflecting an incorporation of Na ions into the NaxMnO2 lattice, which is particularly more pronounced at slow voltage changes (Fig. S4, ESI†). Additionally, two peaks can be observed, one cathodic at 0.5 V and the other anodic at 0.6 V, whose intensities increase as the scan rates increase. The small voltage difference between the cathodic and anodic peaks indicates fast redox/oxidation reactions, mostly on the electrode surface. Particularly, there is a marked intensity in these peaks for all samples in Na2CO3 electrolyte as well as for samples with non-stoichiometric compositions in other electrolytes probably due to the presence of surface defects (as suggested in XPS and shown in Fig. 3). Additionally, in the particular case of sodium carbonate electrolytes in the three-electrode configuration, CV curves versus the current zero line are asymmetric, which implies the low reversibility of intercalation/absorption reactions.
Following one of the main objectives of the present work, the preparation of a stable biphasic structure with optimized morphology to operate under high current conditions with high reversibility, the specific capacity values are calculated based on the results obtained from cyclic voltammograms. Table 2 shows the calculated specific values and their retention after 100 CVA cycles (scan rate of 50 mV s−1) for NaxMnO2 samples with x = 0.8, 1.0 and 1.2 in three different electrolytes selected. The maximum value (103 mA h g−1) is obtained for NaMnO2 sample cycling in sodium sulfate-based electrolytes. In addition, the highest capacity retention rate (93 percent after 100 cycles) was found for the NaMnO2 sample in a sodium sulfate solution.
| Sample | Electrolyte | |||||
|---|---|---|---|---|---|---|
| NaNO3 | Na2SO4 | Na2CO3 | ||||
| Capacity, mA h g−1 | Capacity retention, % | Capacity, mA h g−1 | Capacity retention, % | Capacity, mA h g−1 | Capacity retention, % | |
| Na0.8MnO2 | 13 | 66 | 63 | 82 | 60 | 72 |
| NaMnO2 | 23 | 89 | 103 | 93 | 61 | 83 |
| Na1.2MnO2 | 18 | 70 | 42 | 80 | 39 | 42 |
The different behavior of the electrochemical performance of the devices prepared can be found in the structural and morphological properties of the electrode materials, which bend the balance towards one of the two possible pseudocapacitance mechanisms: intercalation (diffusion-controlled processes) and surface absorption. According to the discharge curves (Fig. 5a–c) obtained for cells based on NaxMnO2 in different electrolytes (discharge current 100 mA g−1), charge accumulation occurs more commonly via adsorption, but the presence of a weakly evident horizontal plateau at 0.3 volts (versus Ag/AgCl electrode) indicates Faraday reactions. For diffusion-controlled processes, the magnitude of cathodic and anodic currents, which was proportional to the mass transport rate, resulted in an increase in the scan rate. As shown in Fig. 5(d)–(f), we observe linear dependencies between measured based on CVA cathodic and anodic current peaks and the scan rate square, indicating a diffusion-controlled behavior.
 | ||
| Fig. 5 Discharge curves of NaxMnO2 electrode in NaNO3 (a), Na2SO4 (b), Na2CO3 (c). The relationship between the cathodic (Ic) and anodic (Ia) current peaks and square root of the scan rate for NaNO3 (d), Na2SO4 (e), and Na2CO3 (f). | ||
Among all samples studied, samples in NaNO3 electrolyte showed the lowest capacity values (Table 2), which is attributable to the small difference found between anode and cathode current peaks and their low values (from −1 mA to −1.5 mA for cathodic and from 1.2 mA to 2 mA for anodic), even in a high scan rate region (Fig. 5d). Cycling in Na2CO3 electrolytes (Fig. 5f) demonstrates low cathodic peak values for all electrode materials employed here, as well as significantly high anodic peak values (up to 5 mA at high scan rates), which demonstrates low Coulombic efficiency and capacity retention. Fig. 5e illustrates that sample cycling in Na2SO4 electrolytes reflects a symmetrical behavior whose highest values of cathodic and anodic peaks occur for NaMnO2 (x = 1.0).39,40
Intercalation and absorption pseudocapacity mechanisms monitored by XPS
Typically, upon cycling, the solid–electrolyte interface (SEI) degrades upon unwanted reactions at the electrode/electrolyte interface defects and dislocations, blocking local Na+/electrons diffusion.41 To determine the stability degree of the devices prepared in this work, the XPS technique is employed to determine the effects of pseudocapacitive charge accumulation processes. Hereinafter, we focus on NaxMnO2-based devices exhibiting the highest operation values observed here (i.e., devices prepared with Na2SO4 electrolytes).Fig. 6 shows XPS spectra acquired in NaxMnO2 electrodes (x = 0.8, 1.0, and 1.2) after 100 operation cycles at 50 mV s−1 in Na2SO4 electrolytes. To unravel the chemical effects of cycling on electrodes, acquired XPS spectra are deconvoluted by following procedures similar to those carried out for the powder materials. Comparing the XPS results obtained in the powders (Fig. 3) with those obtained in the cycled electrodes (Fig. 6), it appears that, first, two new Na 1s components develop at the high binding-energy side of the XPS spectra of the cycled electrodes, which is detrimental to the Na0-related XPS signal detected in the powders. Note that the powders were not in contact with the electrolytes. However, the samples in Fig. 6 are cycled and, therefore, in contact with the electrolytes, a second source of Na. One of the new Na 1s-related peaks, the one centered at 1072.9 eV, stems from the Na2SO4 electrolyte,42 while the one centered at ∼1074 eV can be attributed to residual NaOH surface layer formed during cycling.38,41 Regarding the XPS Mn 2p core-level spectra measured in the cycled electrodes (Fig. 6b), deconvolution processes performed in these spectra reveal a notable enhancement of the presence of Mn4+ species to the detriment of Mn2+ ones in the cycled electrodes compared to the powder samples. This can be confirmed by a closer examination of the atomic ratios of manganese oxidation states before and after cycling (see Fig. S5e of the ESI,† bottom panel). A ratio between both Mn oxidized states, Mn4+/Mn3+, is 2.7 for Na0.8MnO2 (x = 0.8) and Na1.2MnO2 (x = 1.2) cycled electrodes and 12.0 for NaMnO2 (x = 1) cycled electrode (Fig. S5e in the ESI†). These facts (i.e., the absence of a metallic Na component after cycling, attributable to its oxidation by interacting with the electrolyte,37 and the relative enhancement of both the Mn4+-like species and NaOH at the surface of the samples) suggest that cycling promotes the accumulation of NaOH at the surface of the samples, partly substituting for Na+ ions in the starting NaxMnO2 powder. Deconvolution of the O 1s core-level XPS spectra acquired in the cycled NaxMnO2 electrodes (Fig. 6c) seems to support these observations. Besides the observation of the O 1s XPS signal coming from adsorbed H–O–H water and Na2SO4 electrolyte residuals at BE ∼533.9 ± 0.4 eV,29,35,36,38 a notable enhancement of the XPS signal from Mn- and Na-hydroxides29,35,38 is produced.
 | ||
| Fig. 6 High-resolution X-ray photon emission spectroscopy (XPS) analysis of Na0.8MnO2 (x = 0.8), NaMnO2 (x = 1.0) and Na1.2MnO2 (x = 1.2) electrodes in Na2SO4 electrolyte after 100 CVA cycles at 50 mV s−1, with corresponding Na 1s (a), Mn 2p (b) and O 1s (c) core-level spectra. Note the presence of the Na KLL sodium Auger peak (BE = 533.9 eV– 537.3 eV)26,28,33,38 which was absent in the O 1s spectra of the powders. The detection of this Auger peak in the cycled samples suggests the accumulation of Na atoms at the electrode surface, likely originating from the Na2SO4 electrolyte. | ||
The intercalation of NaOH into the electrode structure after cycling explains the increase in the interlayer distance between the nanosheets, as observed elsewhere.8,9 This widening of the interlayer distance explains the improvement in capacity with cycling (Table S2 of the ESI†) as a consequence of the better circulation of electrolyte ions within the crystal. The high capacity and its retention with enhanced circulation properties of NaMnO2 (x = 1) cathode in Na2SO4 electrolyte can be attributed to the transformation of α-NaMnO2 crystal phase into the stable hydrated Na-birnessite phase,10,8,9 containing interlayer structural water between separated individual nanosheets without significant change in their initial crystal structure. The interlayer water mediates the interaction between the intercalated cation and the birnessite.8,9 This structural modification allows for the enhancement of cation absorption not only on the electrode planar surface but also between the interlayers of individual electrode nanosheets, thus boosting cathode electrochemical properties.
Sample operation at different currents: results and their representation
The XPS and electrochemical results obtained here for NaMnO2-based devices indicate the coexistence of the two types of pseudocapacitance. Our samples can be used under conditions of low operating current values and devices with fast charge–discharge processes. For this purpose, we performed a more detailed study of CVA at different scanning rates and galvanostatic discharges at different currents. In CV measurements, the total current measured under a potential sweep rate can be interpreted as the sum of the current related to the slow diffusion-controlled process (id) and the current required to charge the double layer at the electrolyte interface or to initiate fast faradaic reactions on the exposed electrode surface (ic).36 An empirical description of the charge/discharge processes can be summarized as follows:| i(v) = ic + id = avb, |
| logi(v) = loga + blog(v), |
i(v) versus log(v) and is used to provide kinetic information about the electrochemical reactions. In the case of cycling of the three samples in the Na2SO4 electrolyte (Fig. 7d), the calculated b values for the cathodic peaks are within 0.63–0.74 and anodic 0.59–0.69. The smallest among cathodic values (0.63) was observed for NaMnO2. This means that for this sample, we have a charge accumulation mechanism closer to the diffusion control than for the other samples. Consequently, we achieve higher values of capacity and discharge time (Fig. 5a–c).
 | ||
| Fig. 7 Capacity rate (in percent compared with capacity at 1C) at different currents (5C, 10C, and 20C) for cells based on NaxMnO2 in NaNO3 (a), Na2SO4 (b), and Na2CO3 (c). Linear fitting of the relationships between cathodic and anodic peak currents and scan rate for NaxMnO2 in Na2SO4 electrolyte (d). Dependencies between current peak differences and peak voltage differences (f) for NaMnO2 at different scan rates (0.5, 2, 5, 10, 20 and 50 mV s−1) and explanation of each elements (e). | ||
For high power device applications, it is important to have high capacity values and save these values under higher discharge current. Based on the galvanostatic discharge results recorded here (Fig. S6 ESI†), the capacity rate (in percent compared with the capacity at 1C) on the discharge current for all electrolytes is shown in Fig. 7a–c. Even at 20C, cells based on NaMnO2 demonstrate capacity retention at the level of 84% (NaNO3) and 82% (Na2SO4). From these results, it appears that the best opportunities for application in aqueous sodium devices are biphasic NaMnO2 with the best stoichiometry, larger lattice constant and optimized morphology based on an increase in interlayer spacing, enhancing cathode performance, especially using these structures as an electrode material for high power devices.
Ragone plots are employed to quantify the energy–power relationship of the electrode material or full electrochemical systems.43,44 Usually, based on measurements of charge–discharge characteristics, these plots show the dependency existing between specific power and energy. In our case, comparing the results of CVA and discharge characteristics of samples at different currents, we noted the relationship between CVA curve shapes at different scan rates and discharge curves. This became the basis for our approach to constructing an analogue of the “power-capacitance” diagram based only on cyclic voltammetry, which can quickly provide information on specific capacities and their values under high current conditions. This plot (Fig. 7f) represents the relationship between ΔIp and ΔUp that could be taken exclusively from cyclic voltammetry at different scan rates. A detailed explanation of this approach is presented in Fig. S7 (ESI†). ΔIp is the difference between anodic Ipa and cathodic Ipc current peaks for each scan rate, and ΔUp is the difference between cathodic Upc and anodic current peak Upa voltage positions (Fig. 7e).
In the case of a cell based on NaMnO2 electrode (Fig. 7f), the maximal value of ΔIp (responsible for transferred charge amount during the cycle) is observed for Na2SO4, and it is completely consistent with the calculated specific capacity based on CVA (Table 2). The ability to work under high currents can be estimated using ΔUp: systems with better power ability show smaller values of ΔUp with a gradual increase in the scan rate (For example, NaMnO2 (Fig. 7a–c) has capacity retention of 84% (NaNO3), 82% (Na2SO4) and 64% (Na2CO3) at a current of 20C, which correlates with the change in ΔUp with increasing scan rate (Fig. 7f)). It is important to note that this approach does not provide real values of capacity or power but can be successfully used in the case of studying the electrochemical properties of a certain cathode material in different electrolytes or their concentrations, or in symmetrical or asymmetrical systems with different anodes.
Conclusions
In this work, we describe the synthesis of biphasic sodiated manganese oxide using the sol–gel technique. Based on XRD, XPS and TEM, it is shown that it is possible to obtain stoichiometric compounds with both polymorphous monoclinic and orthorhombic phases and a layered structure that is stable during electrochemical sodium insertion and extraction. Based on the CVA in aqueous electrolytes at different scan rates, it has been established that the total capacity is provided mostly by a diffusion-controlled process. Technologically, promising values of the specific capacity (103 mA h g−1) are observed for stoichiometric biphasic systems in Na2SO4 electrolytes with high retention during the first 100 cycles (93%). Moreover, these samples appear to exhibit a high ability to operate at high currents (82% capacity retention when the current increases from 1C to 20C). These results open new opportunities for using these materials in high power, highly stable and cheap aqueous sodium storages due to the possibilities of surface optimization during synthesis.Author contributions
AB: conceptualisation, investigation, methodology, validation, visualization, writing (original draft preparation), writing (review and editing). TB: conceptualisation, investigation, methodology, validation, visualization, writing (original draft preparation). MEC: investigation, validation, data curation. MK: investigation, data curation, visualization. RA: methodology, validation. PPB: methodology, review and editing. MCA: formal analysis, funding acquisition, review and editing. SA: investigation, data curation, visualization, writing. JFSR: conceptualisation, supervision, funding acquisition, writing (review and editing).Data availability
All data generated or processed during this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The present work has been supported by the projects PID2023-146181OB-100 of the Ministerio de Ciencia, Innovación y Universidades, MFA 2022/009 (SPINO2D) and Prometeo/2021/082 of the Generalitat Valenciana as well as the project TED 2021-132656B-C21 and TED 2021-132656B-C22 granted by the Call 2021 – “Ecological Transition and Digital Transition Projects” promoted by the Ministry of Science and Innovation, funded by the European Union within the “NextGeneration” EU program, the Recovery, Transformation, and Resilience Plan, and the State Investigation Agency. A. B and T. B acknowledge financial support from the University of Valencia through the Program “Universitat de València with Ukraine”, the Generalitat Valenciana through the Program “Acoge CV-UCRANIA personal investigador” of the Conselleria de Innovación, Universidades, Ciencia y Sociedad Digital, and the Ministerio of Universidades through the program “Plan de Acción Universidad-Refugio”. M. K. acknowledges the APOSTD programme (contract APOSTD/2020/103). M. E. C. acknowledges financial support from the Santiago Grisolia Program of the Generalitat Valenciana (grant CIGRIS/2021/150). Authors are grateful to the SCSIE, University of Valencia, for providing HRTEM facility.References
- L. Zhao, et al., Engineering of Sodium-Ion Batteries: Opportunities and Challenges, Engineering, 2023, 24, 172–183, DOI:10.1016/j.eng.2021.08.032.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Research Development on Sodium-Ion Batteries, Chem. Rev., 2014, 114(23), 11636–11682, DOI:10.1021/cr500192f.
- R. Usiskin, et al., Fundamentals, status and promise of sodium-based batteries, Nat. Rev. Mater., 2021, 6(11), 1020–1035, DOI:10.1038/s41578-021-00324-w.
- K. M. Abraham, How Comparable Are Sodium-Ion Batteries to Lithium-Ion Counterparts?, ACS Energy Lett., 2020, 5(11), 3544–3547, DOI:10.1021/acsenergylett.0c02181.
- H. Wang, C. Zhu, D. Chao, Q. Yan and H. J. Fan, Nonaqueous Hybrid Lithium-Ion and Sodium-Ion Capacitors, Adv. Mater., 2017, 29(46), 1702093, DOI:10.1002/adma.201702093.
- J. Ding, W. Hu, E. Paek and D. Mitlin, Review of Hybrid Ion Capacitors: From Aqueous to Lithium to Sodium, Chem. Rev., 2018, 118(14), 6457–6498, DOI:10.1021/acs.chemrev.8b00116.
- T. Boichuk, A. Boichuk, V. Vashchynskyi, M. E. Changarath, M. Krečmarová, R. Abargues, J. P. Martínez-Pastor and J. F. Sánchez-Royo, Highly porous activated carbon derived from plant raw material as high-performance anode for aqueous sodium-ion supercapacitors, J. Power Sources, 2024, 622, 235356, DOI:10.1016/j.jpowsour.2024.235356.
- K. S. Kumar, D. Pandey and J. Thomas, High Voltage Asymmetric Supercapacitors Developed by Engineering Electrode Work Functions, ACS Energy Lett., 2021, 6(10), 3590–3599, DOI:10.1021/acsenergylett.1c01484.
- M. A. Khan, D. Han, G. Lee, Y.-I. Kim and Y.-M. Kang, P2/O3 phase-integrated Na0.7MnO2 cathode materials for sodium-ion rechargeable batteries, J. Alloys Compd., 2019, 771, 987–993, DOI:10.1016/j.jallcom.2018.09.033.
- S. Wang, et al., Enhancing the capacitive deionization performance of NaMnO2 by interface engineering and redox-reaction, Environ. Sci.: Nano, 2019, 6(8), 2379–2388, 10.1039/C9EN00545E.
- M. Palluzzi, L. Silvestri, A. Celeste, M. Tuccillo, A. Latini and S. Brutti, Structural Degradation of O3-NaMnO2 Positive Electrodes in Sodium-Ion Batteries, Crystals, 2022, 12(7), 885, DOI:10.3390/cryst12070885.
- Q. Ding, W. Zheng, A. Zhao, Y. Zhao, K. Chen, X. Zhou, H. Zhang, Q. Li, X. Ai, H. Yang, Y. Fang and Y. Cao, W-Doping Induced Efficient Tunnel-to-Layered Structure Transformation of Na0.44Mn1-xWxO2: Phase Evolution, Sodium-Storage Properties, and Moisture Stability, Adv. Energy Mater., 2023, 13, 2203802, DOI:10.1002/aenm.202203802.
- Y. Lai, H. Xie, P. Li, B. Li, A. Zhao, L. Luo, Z. Jiang, Y. Fang, S. Chen, X. Ai, D. Xia and Y. Cao, Ion-Migration Mechanism: An Overall Understanding of Anionic Redox Activity in Metal Oxide Cathodes of Li/Na-Ion Batteries, Adv. Mater., 2022, 34, 2206039, DOI:10.1002/adma.202206039.
- J. Billaud, et al., β-NaMnO2: A High-Performance Cathode for Sodium-Ion Batteries, J. Am. Chem. Soc., 2014, 136(49), 17243–17248, DOI:10.1021/ja509704t.
- A. M. Abakumov, A. A. Tsirlin, I. Bakaimi, G. Van Tendeloo and A. Lappas, Multiple Twinning As a Structure Directing Mechanism in Layered Rock-Salt-Type Oxides: NaMnO2 Polymorphism, Redox Potentials, and Magnetism, Chem. Mater., 2014, 26(10), 3306–3315, DOI:10.1021/cm5011696.
- D. Nayak, J. K. Dora, S. Ghosh and V. Adyam, Superior diffusion kinetics and electrochemical properties of α/β–type NaMn0.89Co0.11O2 as cathode for sodium-ion batteries, J. Solid State Electrochem., 2022, 26(5), 1231–1239, DOI:10.1007/s10008-022-05157-3.
- D. Nayak, et al., Biphasic α/β-Type NaMn0.89Fe0.11O2 as a Cathode for Sodium-Ion Batteries: Structural Insight and High-Performance Relation, ACS Appl. Energy Mater., 2022, 5(1), 116–125, DOI:10.1021/acsami.1c02576.
- M. Borlaf and R. Moreno, Colloidal sol-gel: A powerful low-temperature aqueous synthesis route of nanosized powders and suspensions, Open Ceram., 2021, 8, 100200, DOI:10.1016/j.oceram.2021.100200.
- O. M. Uhorchuk, V. U. Uhorchuk, M. V. Karpets, L. S. Kaykan, B. J. Deputat, A. M. Boychuk and M. I. Hasyuk, Lithium ferrite as the cathode of the electrochemical power sources: the perspectives of sol-gel synthesis method, J. Nano-Electron. Phys., 2015, 7, 02012 Search PubMed [Google Scholar].
- R. Lisovsky, B. Ostafiychuk, I. Budzulyak, V. Kotsyubynsky, A. Boychuk and B. Rachiy, Nanostructured Iron-Substituted Lithium-Manganese Spinel as an Electrode Material for Hybrid Electrochemical Capacitor, Acta Phys. Pol. A, 2018, 133, 876–878, DOI:10.12693/APhysPolA.133.876.
- L. Zhang, Y. Zhang, Z. Su, H. Tian and L. Wang, Synthesis and Electrochemical Characterization of α-NaMnOi as a Cathode Material for Hybrid Na/Li-Ion Batteries, Int. J. Electrochem. Sci., 2019, 14, 2422–2429, DOI:10.20964/2019.03.82.
- Q. Zhao, A. Song, S. Ding, R. Qin, Y. Cui, S. Li and F. Pan, Preintercalation Strategy in Manganese Oxides for Electrochemical Energy Storage: Review and Prospects, Adv. Mater., 2020, 32, 2002450, DOI:10.1002/adma.202002450.
- A. A. Nechikott, H. D. Yoo and P. K. Nayak, A mixed dual-ion electrolyte for high-rate performance and remarkable cycling stability of NaMnO2 in hybrid supercapacitors, J. Power Sources, 2024, 591, 233825, DOI:10.1016/j.jpowsour.2023.233825.
- T. Sato, K. Yoshikawa, W. Zhao, T. Kobayashi, H. B. Rajendra, M. Yonemura and N. Yabuuchi, Efficient Stabilization of Na Storage Reversibility by Ti Integration into O′3-Type NaMnO2, Energy Mater. Adv., 2021, 2021/9857563, DOI:10.34133/2021/9857563.
- Z. Guo, X. Li, Y. Lyu and S. Xu, Improved electrochemical performance of B doped O’3-NaMnO2 for Na-ion battery, Electrochim. Acta, 2022, 430, 141079, DOI:10.1016/j.electacta.2022.141079.
- T. Sun, et al., Micron-sized Na0.7MnO2.05 as cathode materials for aqueous rechargeable magnesium-ion batteries, Ionics, 2019, 25(10), 4805–4815, DOI:10.1007/s11581-019-03057-7.
- J. Li, et al., Na–Mn–O@C yolk–shell nanorods as an ultrahigh electrochemical performance anode for lithium ion batteries, J. Mater. Chem. A, 2017, 5(35), 18509–18517, 10.1039/C7TA06046G.
- Z. Huang, et al., High performance of Mn-Co-Ni-O spinel nanofilms sputtered from acetate precursors, Sci. Rep., 2015, 5(1), 10899, DOI:10.1038/srep10899.
- Z. Yang, et al., Facile Synthesis of Coaxial CNTs/MnOx-Carbon Hybrid Nanofibers and Their Greatly Enhanced Lithium Storage Performance, Sci. Rep., 2015, 5(1), 17473, DOI:10.1038/srep17473.
- X. Feng and D. F. Cox, Oxidation of MnO(100) and NaMnO2 formation: Characterization of Mn2+ and Mn3+ surfaces via XPS and water TPD, Surf. Sci., 2018, 675, 47–53, DOI:10.1016/j.susc.2018.04.022.
- G. Xu, C. Xie, Y. Wen, A. Tang and H. Song, Mn(OH)2 electrodeposited on secondary porous Ni nano-architecture foam as high-performance electrode for supercapacitors, Ionics, 2019, 25(7), 3287–3298, DOI:10.1007/s11581-018-2824-8.
- D. Delgado, M. Minakshi, D.-J. Kim and C. Kyeong, Influence of the Oxide Content in the Catalytic Power of RANEY® Nickel in Hydrogen Generation, Anal. Lett., 2017, 50(15), 2386–2401, DOI:10.1080/00032719.2017.1300806.
- W. Wei, et al., Subsurface oxygen defects electronically interacting with active sites on In2O3 for enhanced photothermocatalytic CO2 reduction, Nat. Commun., 2022, 13(1), 3199, DOI:10.1038/s41467-022-30958-5.
- M. Han, et al., Oxygen Defects in β-MnO2 Enabling High-Performance Rechargeable Aqueous Zinc/Manganese Dioxide Battery, iScience, 2020, 23(1), 100797, DOI:10.1016/j.isci.2019.100797.
- J. D. A. Pereira, et al., Tuning the morphology of manganese oxide nanostructures for obtaining both high gravimetric and volumetric capacitance, Mater. Adv., 2020, 1(7), 2433–2442, 10.1039/D0MA00524J.
- G. Kucinskis, et al., Enhanced Electrochemical Properties of Na0.67MnO2 Cathode for Na-Ion Batteries Prepared with Novel Tetrabutylammonium Alginate Binder, Batteries, 2022, 8(1), 6, DOI:10.3390/batteries8010006.
- X. Feng and D. F. Cox, Synthesis of a planar, multicomponent catalytic surface of Na2CO3/MnO, Surf. Sci., 2021, 707, 121807, DOI:10.1016/j.susc.2021.121807.
- F. Xiao and Y. Xu, Pulse Electrodeposition of Manganese Oxide for High-Rate Capability Supercapacitors, Int. J. Electrochem. Sci., 2012, 7(8), 7440–7450, DOI:10.1016/S1452-3981(23)15795-6.
- V. Hiremath, M. Cho and J. G. Seo, Self-assembled Mn3O4 nano-clusters over carbon nanotube threads with enhanced supercapacitor performance, New J. Chem., 2018, 42(24), 19608–19614, 10.1039/C8NJ04850A.
- J. Liu, et al., Advanced Energy Storage Devices: Basic Principles, Analytical Methods, and Rational Materials Design, Adv. Sci., 2018, 5(1), 1700322, DOI:10.1002/advs.201700322.
- Y.-F. Liu, et al., Layered oxide cathodes for sodium-ion batteries: From air stability, interface chemistry to phase transition, InfoMat, 2023, 5(6), e12422, DOI:10.1002/inf2.12422.
- N. Zhao, C. Li and X. Guo, Long-life Na–O2 batteries with high energy efficiency enabled by electrochemically splitting NaO2 at a low overpotential, Phys. Chem. Chem. Phys., 2014, 16(29), 15646, 10.1039/c4cp01961j.
- T. Christen and C. Ohler, Optimizing energy storage devices using Ragone plots, J. Power Sources, 2002, 110(1), 107–116, DOI:10.1016/S0378-7753(02)00228-8.
- I. Beyers, A. Bensmann and R. Hanke-Rauschenbach, Ragone plots revisited: A review of methodology and application across energy storage technologies, J. Energy Storage, 2023, 73, 109097, DOI:10.1016/j.est.2023.109097 Part D.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma01150c |
| This journal is © The Royal Society of Chemistry 2025 |