
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCaged bulky organic dyes in a polyaromatic framework and their spectroscopic peculiarities†
Mayuko
Ueda
,
Natsuki
Kishida
,
Lorenzo
Catti
 and
Michito
Yoshizawa
*
and
Michito
Yoshizawa
*
Laboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: yoshizawa.m.ac@m.titech.ac.jp
First published on 5th July 2022
Abstract
Host–guest structures and properties have been widely studied using relatively small dyes (<1 nm) without bulky groups, due to their smooth incorporation, efficient host–guest interactions, and high analytical accessibility. In this report, on the other hand, three types of sterically demanding organic dyes trapped by a polyaromatic cage were investigated by spectroscopic analyses on the basis of supramolecular interactions. Coumarins with two bulky substituents are bound by the cage in aqueous solution. The resultant caged dyes show unusual emission enhancement, depending on the difference of a single heteroatom in their substituents. The color of perylene bisimides with two bulky substituents is remarkably changed from yellow to red upon caging. This peculiarity stems from the twist of the substituents in the cage, revealed by the combination of absorption and theoretical studies. Furthermore, tetrasubstituted, bulky porphyrins are caught by the cage in aqueous solution. The caged bulky dyes also display altered color and absorption properties, which remain intact even under acidic conditions. In contrast to typical covalent functionalization and previous host–guest studies toward small and non-bulky dyes, the unusual, non-covalent spectroscopic modulation of the large and bulky dyes can be accomplished for the first time by the present cage, featuring a prolate polyaromatic framework with four openings.
Introduction
Organic dyes displaying characteristic colors are ubiquitous in our daily lives.1 The majority of these dyes possess relatively bulky substituents and rigid core frameworks bearing extended π-conjugation systems.2 Modulation and alteration of their colors and spectroscopic properties have been widely demonstrated by covalent functionalization of the frameworks as a common synthetic approach. Non-covalent functionalization of dyes upon encapsulation by molecular cages has recently been regarded as an alternative approach,3 without multistep and time-consuming synthetic protocols. Coordination-driven self-assembly is one of the most promising methods for the preparation of the desired cages, from the viewpoints of high designability and synthetic accessibility.4 However, coordination capsules and cages capable of binding such large and bulky dyes have been rarely reported so far (Fig. 1a, left), owing to size/shape mismatch as well as poor host–guest interactions in the cavity,5 whereas the embedding of various dyes (e.g., perylene bisimide and porphyrin) into the coordinative ligands is a well-established approach. We considered that a coordination-driven molecular cage featuring both a large cavity and several medium-sized windows would be a useful molecular tool to bind a variety of organic dyes with two and more bulky substituents (Fig. 1a, right). Here we report that, as a proof-of-concept, the open large cavity of M2L4 polyaromatic cage 1 (Fig. 1b and c) can bind sterically demanding organic dyes (≥1.5 nm in maximum length) with (i) coumarin, (ii) perylene bisimide, and (iii) porphyrin cores in aqueous solution. Notably, the spectroscopic properties of the caged dyes are facilely modulated through effective supramolecular interactions in the cavity, without the aid of commonly employed host–guest π-stacking interactions.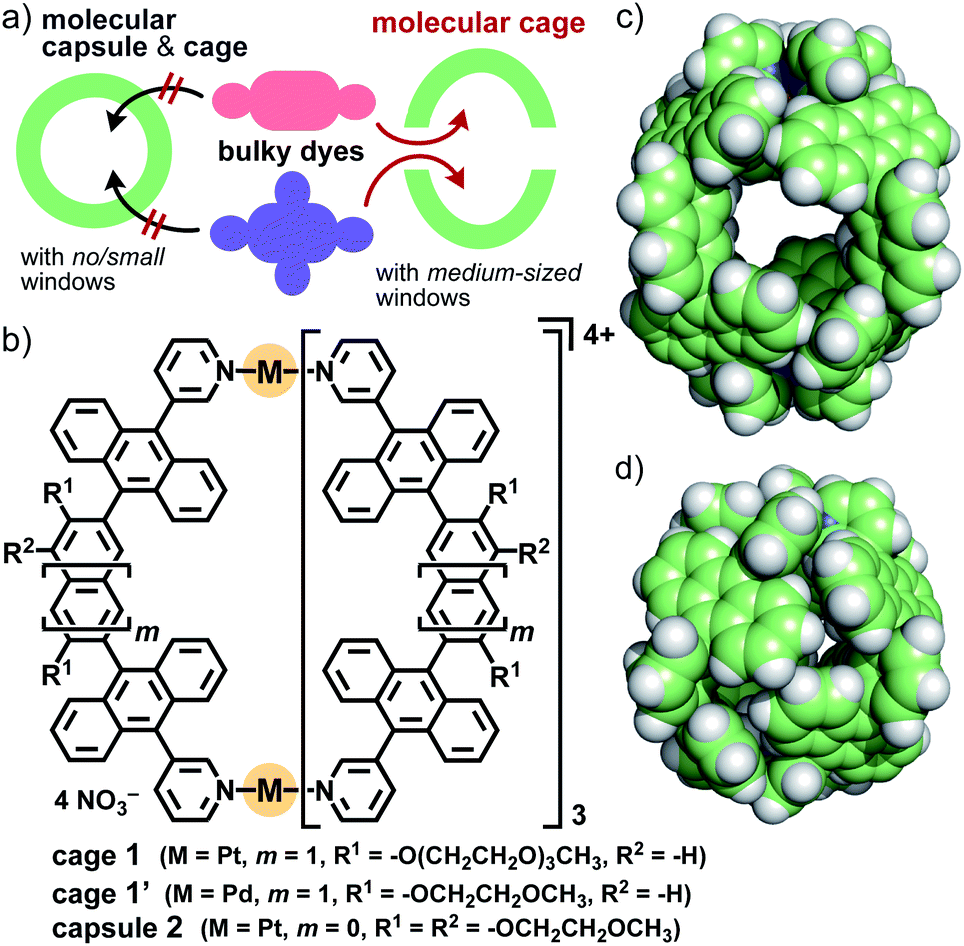 | ||
| Fig. 1 (a) Strategy for the binding of bulky organic dyes by a molecular cage with medium-sized windows for their spectroscopic modulation. (b) Present aqueous cage 1, and previous cage 1′ and capsule 2. The crystal structures of (c) 1′ and (d) 2 (R1 = R2 = H for clarity).8,10 | ||
Host functions of the polyaromatic cavities of coordination capsules have been highlighted recently.6,7 For example, the closed polyaromatic cavity of M2L4 capsule 2 (Fig. 1b) shows efficient/selective binding abilities toward various synthetic compounds8 as well as biomolecules in aqueous solution, through efficient hydrophobic effect and multipoint π–π/CH–π/hydrogen-bonding interactions.6,9 However, like other coordination cages reported previously,4 capsule 2 exhibits no binding ability toward synthetic dyes with multiple bulky substituents, whose sizes are larger than that of the cavity (d = 1.2 nm), owing to the closed spherical framework (Fig. 1d). We thus herein employed the analogous, prolate M2L4 cage 1′, with a cavity size of dmax. = 1.6 nm and a window size of dmax. = 0.7 nm (Fig. 1c).10 As the key molecular design in this study, the replacement of the short methoxyethoxy (MOE) side-chains with long hydrophilic ones (i.e., methoxytriethylene glycol (TEG)) allows cage 1 to be used in aqueous solution as well as to bind bulky, hydrophobic organic dyes efficiently for the first time. Crystallographic insights into the host–guest structures are, on the other hand, inaccessible in this system, due to the flexibility of the side-chains.
It should be noted that, in contrast to small organic dyes, relatively large organic dyes with bulky substituents and rigid core-frameworks, studied in the present work, have several disadvantages regarding their host–guest investigations. Their low or no solubility in common solvents reduces analytical accessibility, e.g., for typical Job's plot and binding constant analyses. The product yield and stability are often compromised by inefficient host–guest interactions. Accordingly, the investigations of new host–guest structures and properties using large and bulky dyes have been severely limited so far, especially in aqueous media.
Results and discussion
Synthesis of aqueous cage 1
Bispyridine ligand 3 bearing two TEG groups was synthesized from brominated, bent anthracene dimers with two MOE groups in four steps (Fig. S1–S10†).11 TEG-substituted cage 1 was facilely formed from Pt(II) ions and ligands 3 in DMSO-d6 (88% isolated yield), as confirmed by 1H NMR and ESI-TOF MS analyses (Fig. 2a and S11–S15†).11 The combination of DOSY, DLS, and molecular modeling studies suggested the core and outer diameters of 1 being ∼2 and ∼4 nm, respectively (Fig. 2c).12 The multiple, long and flexible side-chains suppress its self-aggregation in solution and ordered packing in the solid state. Whereas MOE-substituted cage 1′ is insoluble in water, present cage 1 is slightly soluble in water (∼0.04 mM; Fig. S17†) and well-soluble in 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/acetonitrile solution at room temperature. A Pd(II) analogue, obtained in the same way, showed lower water-solubility relative to 1. The fully-assignable, sharp proton signals of 1 in CD3CN (Fig. 2a) were characteristically broadened in D2O/CD3CN (Fig. 2b) in the 1H NMR spectra. The significant broadening of the NMR signals of 1 in the aqueous solution is caused by the restricted motion of the multiple polyaromatic panels through efficient intramolecular, panel–panel interactions. Such NMR properties prompted us to investigate the host–guest structures mainly through UV-visible and fluorescence analyses.
:1 water/acetonitrile solution at room temperature. A Pd(II) analogue, obtained in the same way, showed lower water-solubility relative to 1. The fully-assignable, sharp proton signals of 1 in CD3CN (Fig. 2a) were characteristically broadened in D2O/CD3CN (Fig. 2b) in the 1H NMR spectra. The significant broadening of the NMR signals of 1 in the aqueous solution is caused by the restricted motion of the multiple polyaromatic panels through efficient intramolecular, panel–panel interactions. Such NMR properties prompted us to investigate the host–guest structures mainly through UV-visible and fluorescence analyses.
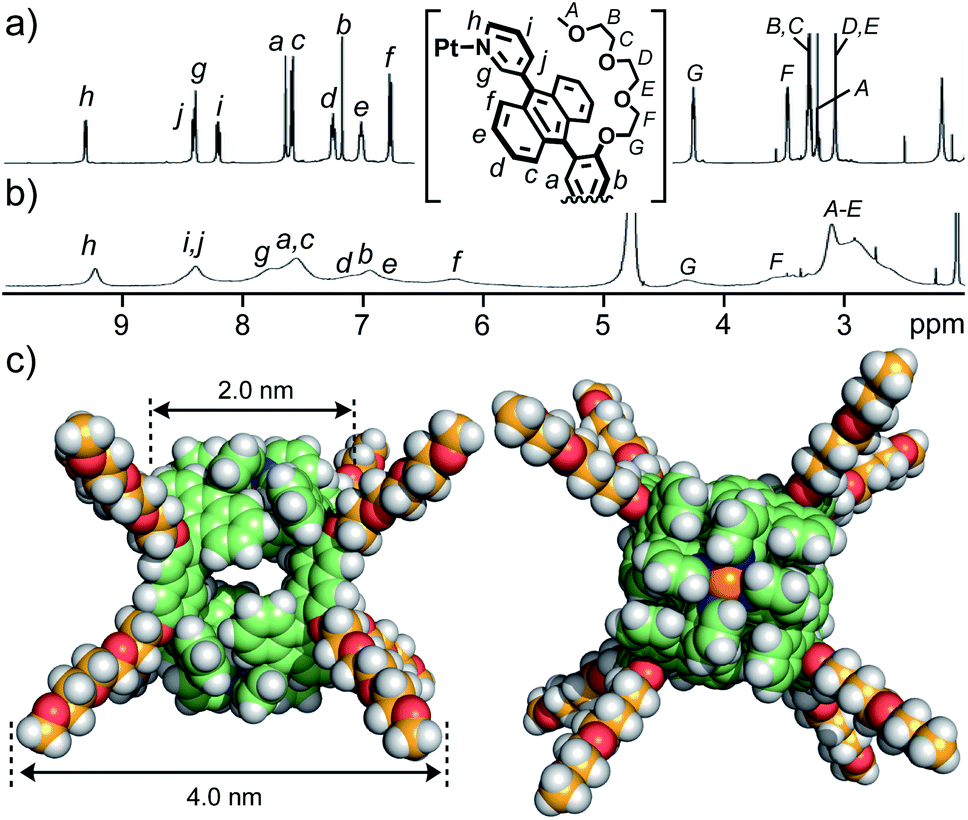 | ||
| Fig. 2
1H NMR spectra (400 MHz, r.t.) of cage 1 in (a) CD3CN and (b) 15:1 D2O/CD3CN. (c) Optimized structure of 1 (side and top views; DFT calculation).12 | ||
Caged bulky coumarin dyes
As the first result, the open polyaromatic cavity of cage 1 could bind one molecule of disubstituted coumarin dyes 4 (X = S, O, and NH; Fig. 3a, left), which are hardly encapsulated by previous capsule 2 even under various conditions. The resultant caged dyes displayed unusual substituent-dependent emission enhancement, whereas these dyes virtually provide an isostructure and low to moderate emission abilities in aqueous solutions without 1. When cage 1 (0.14 μmol) and benzothiazolyl-based coumarin dye 4a (X = S; 0.41 μmol) were stirred in a 15:1 water/acetonitrile (AN) solution (0.45 mL) at 80 °C for 2 h, a clear pale yellow solution of 1:1 host–guest complex 1·4a was obtained in 50% yield after filtration (Fig. 3a).11 Rigid and hydrophobic dye 4a itself, with a maximum length of 1.5 nm, is insoluble in the aqueous solution (15:1 water/AN) yet soluble in a 2:1 water/AN solution (up to 8 μM). Whereas the 1H NMR spectrum of 1·4a was broadened like that of empty 1 (Fig. S26† and 2b), the ESI-TOF MS and UV-visible analyses clearly indicated the 1:1 host–guest complex formation (Fig. S26c and S19†) and the binding of 4a in the hydrophobic cavity of 1, respectively. New absorption bands derived from caged 4a were found in the range of 430 to 530 nm, which are slightly red-shifted (Δλ = +9 nm) relative to the bands of uncaged 4a in 2:1 water/AN (Fig. 3b). The emission bands of 4a were also red-shifted upon binding (λmax = 519 nm, Δλ = +6 nm; Fig. 3c). These rather small shifts indicated the absence of host–guest π-stacking and charge-transfer (CT) interactions in the polyaromatic cavity. The host–guest structure of 1·4a is stable under ambient dilution conditions (∼35 μM; Fig. S19c†), implying a moderate binding constant (∼2.5 × 104 M−1).11
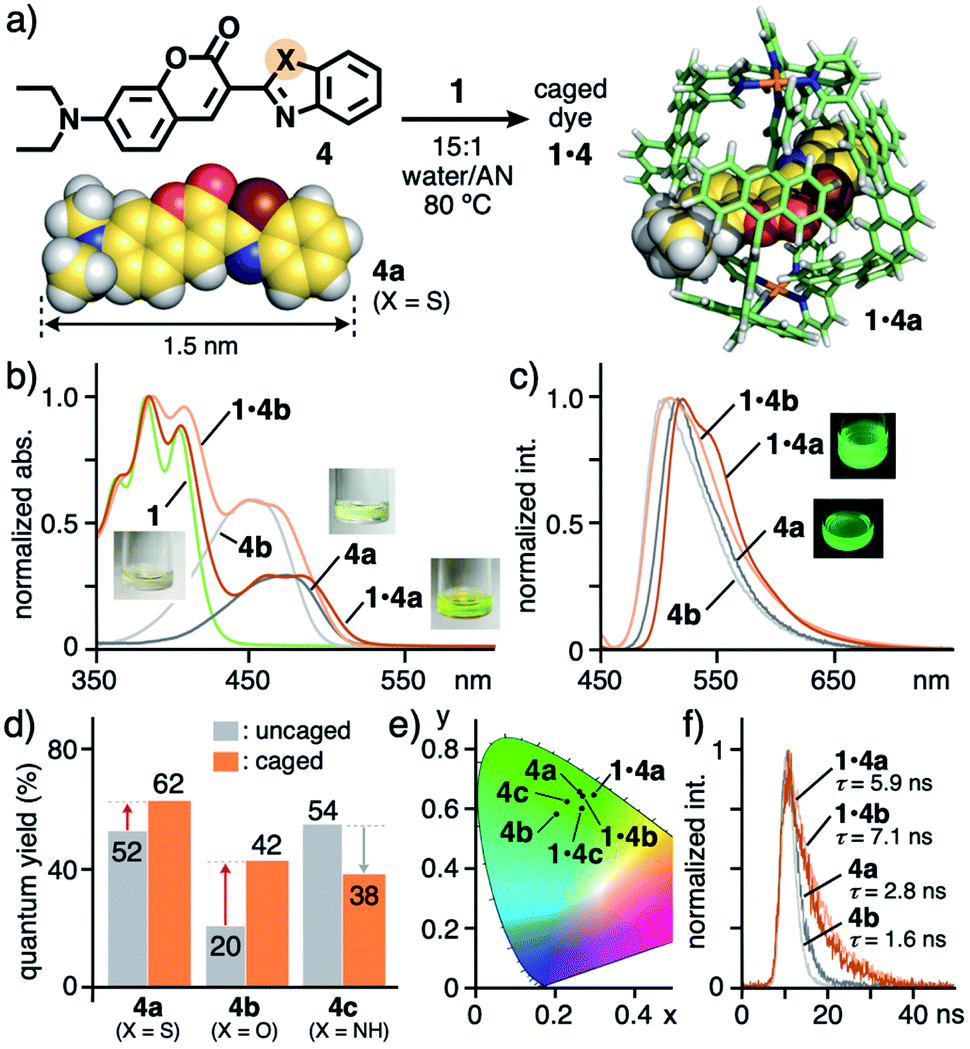 | ||
| Fig. 3 (a) Formation of caged dyes 1·4 (X = S, O, and NH) and the optimized structures of 4a and 1·4a (R1 = H for clarity).11 (b) UV-visible spectra and photographs (r.t., 15:1 water/acetonitrile (AN)) of 1·4a, 1·4b, 1, and 4a, 4b (in 2:1 water/AN), (c) their fluorescence spectra (λex = 450 nm) and (d) quantum yields, and (e) their CIE diagram (before and after binding by 1). (f) Fluorescence lifetimes (r.t., λex = 355 nm, 15:1 or 2:1 water/AN) of 1·4a, 1·4b, and 4a, 4b. | ||
Isostructural coumarin dyes such as benzoxazolyl derivative 4b (X = O) and benzimidazolyl derivative 4c (X = NH) were also bound by 1 under the same conditions (Fig. 3a). New absorption and emission bands of the caged coumarins were observed in their spectra (Fig. 3b, c and S19–S21†),11 in a manner similar to the spectroscopic properties of 4a. Again, slight band shifts (Δλ = +3–17 nm) for 1·4b and 1·4c suggested the absence of host–guest π–π and CT interactions.
The solution of caged dye 1·4a emitted strong green fluorescence with high quantum yield (ΦF = 62%) in water/AN solution, upon light irradiation (λex = 450 nm; Fig. 3d), whereas cage 1 itself shows no fluorescence despite possessing multiple anthracene fluorophores. Notably, the emission quantum yield of 4a (ΦF = 52%) was enhanced by 1.2-fold within 1, in contrast to the majority of previous coordination hosts, which quench guest fluorescence to a large degree, owing to the heavy metal effect and host–guest π-stacking/CT interactions.4,6 The emission properties of 1·4a were maintained for at least 7 d at ambient temperature in the dark (Fig. S19a†).
The green emission of coumarin dyes 4b and 4c was further enhanced by 2.1-fold (ΦF = 20 → 42%) yet slightly suppressed (ΦF = 54 → 38%), respectively, within the cage (Fig. 3d).11 The CIE diagram displayed a minor change of the emission colors of the dyes within the cage, due to weak host–guest steric and electronic interactions (Fig. 3e). The fluorescence lifetime (τ) of 1·4a was estimated to be 5.9 ns, which is longer than that of free 4a (τ = 2.8 ns; Fig. 3f), indicating the stabilization of the excited state upon caging. Further prolongation of the guest emission lifetime was observed for 1·4b (τ = 7.1 ns) under the same conditions. The opposite emission behavior of 4c is likely derived from a weakening of the intramolecular hydrogen-bonding interaction between the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H (Fig. S27b†).
O and N–H (Fig. S27b†).
The optimized structure of caged dye 1·4a indicated that the rod-shaped dye is threaded through the opposite two windows of the host framework (Fig. 3a, right). Importantly, no aromatic–aromatic stack between the host and guest frameworks is observed, consistent with small band shifts in the UV-visible and fluorescence spectra and the high quantum yield. The coumarin core can be effectively isolated from aqueous solvent, acting as an emission-quenching medium, by the polyaromatic shell. The bulky aromatic substituent of 4a is situated at the window with close host–guest contact, which most probably alters the guest emission in the cavity, depending on the difference in the heteroatom (i.e., S, O, and NH).
Caged bulky perylene bisimide dyes
Next, perylene bisimide (PBI) dyes 5 with two substituted aromatic rings were employed as longer and more rigid organic dyes (2.2 nm in maximum length) than 4. Although the binding of these bulky dyes by synthetic hosts has been quite uncommon,13,14 one molecule of 5 was selectively trapped by the open framework of 1 in a threading fashion like 4, accompanying a unique change in color. The sonication (40 kHz, 20 min) of a mixture of cage 1 and 2,6-diisopropylphenyl-based PBI dye 5a (0.14 μmol each) in 5:1 water/AN (1.0 mL) gave rise to a red clear solution of 1:1 host–guest complex 1·5a in 48% yield, through removal of suspended free 5avia simple filtration (Fig. 4a).11 The broad 1H NMR spectrum of the red solution at room temperature partially became sharp at elevated temperature (e.g., 75 °C). The host aromatic signals were split complicatedly and the guest signal HA was observed at −0.96 ppm with a large upfield shift (Δδ = −2.2 ppm; Fig. 4b–d), due to the desymmetrization and aromatic shielding effect of the host framework, respectively. The 1:1 host–guest complexation was confirmed by the ESI-TOF MS analysis, which showed molecular ion peaks corresponding to the [1·5a – n·NO3−]n+ (n = 4 and 3) species (Fig. 4e). Owing to the very broad DOSY spectrum of 1·5a at room temperature, the product size was determined by the DLS analysis (d = 4.3 nm; Fig. S32c†).11
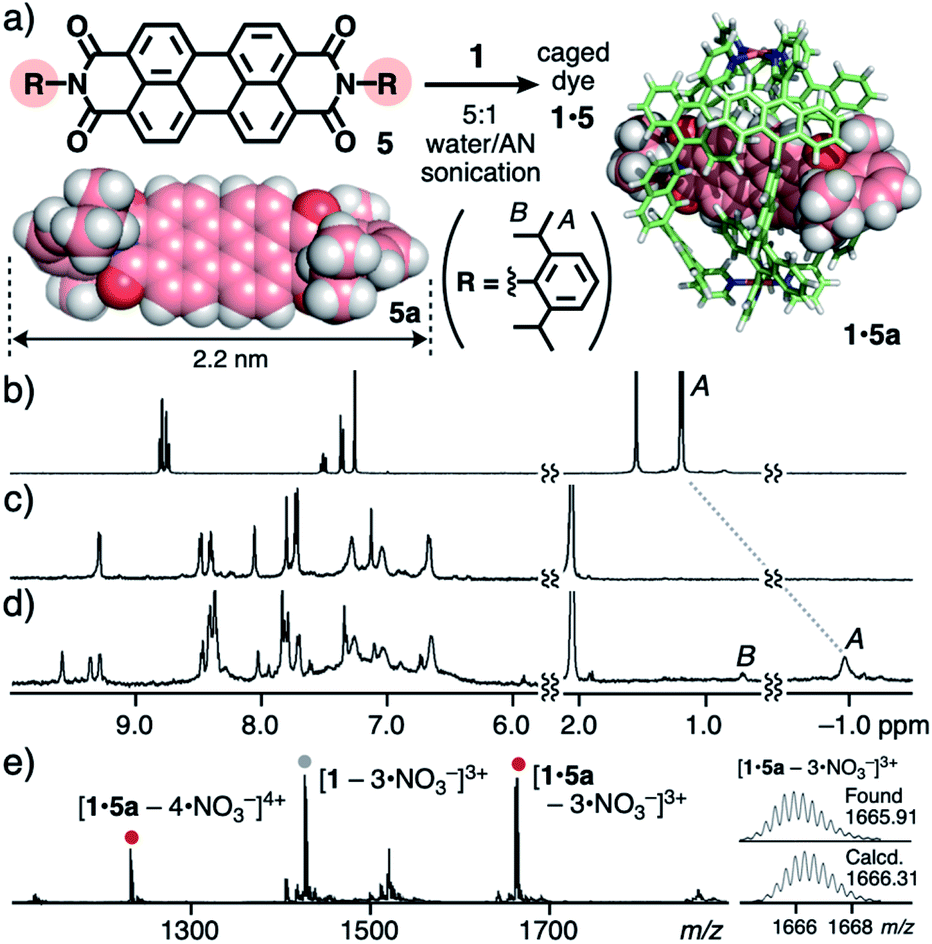 | ||
| Fig. 4 (a) Formation of caged dye 1·5 and the optimized structures of 5a and 1·5a (R1 = H for clarity).111H NMR spectra (500 MHz, 5:1 D2O/CD3CN, 75 °C) of (b) 5a (in CDCl3, r.t.), (c) 1, and (d) 1·5a. (e) ESI-TOF MS spectrum (5:1 water/AN) of 1·5a. | ||
In the same way, caged dyes 1·5b and 1·5c were obtained by the treatment of cage 1 with 2,5-di(tert-butyl)phenyl-based PBI 5b and 3,5-dimethylphenyl-based PBI 5c, respectively (Fig. S28a†).11 Although no host–guest titration studies were performed due to the insolubility of 5a–c in 5:1 water/AN without 1, the concentration-dependent UV-visible analysis of 1·5a revealed the stability of the host-complex at ∼3.5 μM (Fig. S28c†), suggesting a binding constant of >108 M−1.11 Again, no encapsulation of bulky PBIs 5a–c took place with capsule 2 even under various conditions, owing to its closed cavity.
Drastic color change of diisopropylphenyl derivative 5a from yellow to red through binding by 1 was observed and the present unusual phenomenon was elucidated by theoretical spectroscopic studies. Monomeric free 5a is yellow in less polar solvent (e.g., CH2Cl2; Fig. 5a) and orange in polar solvent (e.g., DMSO; Fig. S28b†) with similarly shaped absorption bands.11 In sharp contrast, caged dye 1·5a displayed red color in aqueous solution. The visible difference was qualified by UV-visible and fluorescence analyses. The relatively sharp absorption bands of uncaged 5a were recorded in CH2Cl2 in the range of 420 to 550 nm, which were significantly broadened and red-shifted (up to ∼610 nm) within 1 (Fig. 5a). The emission band of caged 5a was further broadened and shifted (λmax = 625 nm, Δλ = +78 nm) relative to free 5a (Fig. 5b). A similar yellow-to-red color change was observed for bulky di(tert-butyl)phenyl derivative 5b. The sharp absorption bands (415–550 nm) of 5b in CH2Cl2 were largely broadened and red-shifted (up to ∼600 nm) through caging in aqueous solution (Fig. 5c).
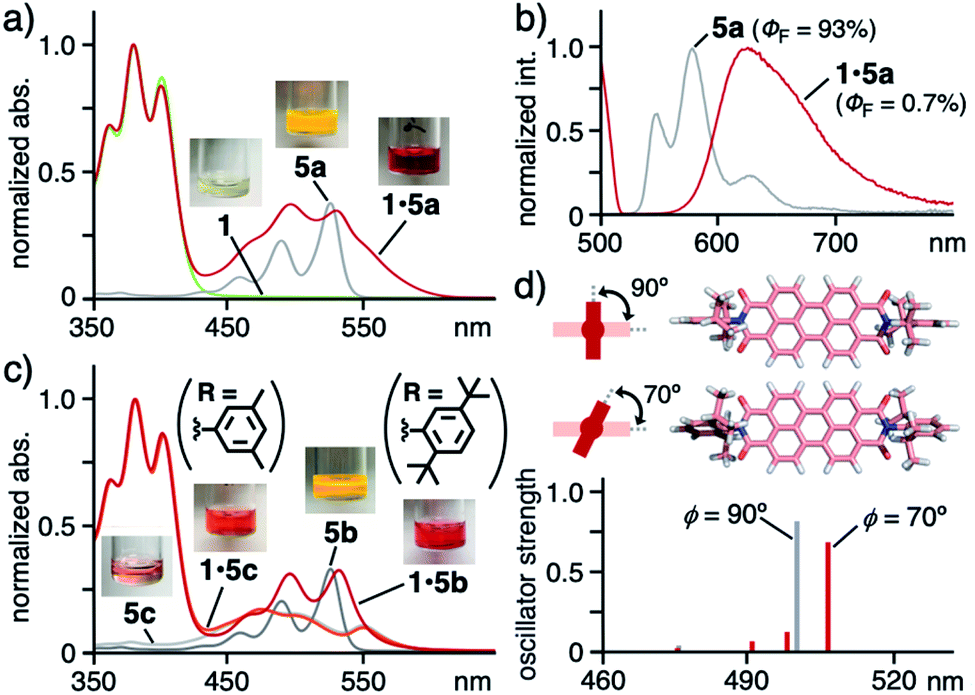 | ||
| Fig. 5 (a) UV-visible spectra and photographs (5:1 water/AN, r.t.) of 1·5a, 1, and 5a (in CH2Cl2) and (b) their fluorescence spectra (λex = 500 nm) except for 1. (c) UV-visible spectra (5:1 water/AN, r.t.) of 1·5b, 1·5c, 5b (in CH2Cl2) and 5c (in DMSO). (d) Optimized structures of 5a with ϕ = 90° and 70° and their predicted, electronic absorption bands.15 | ||
In contrast, neither color change nor spectral change occurred for less bulky and planar dimethylphenyl derivative 5c, featuring broad absorption bands at 430–600 nm (Fig. 5c), through binding by 1 under the same conditions. The absorption bands derived from 1 at 330–430 nm remained also unchanged upon caging. These results suggest that no donor–acceptor π-stacking interactions exist between the cage cavity and the PBI framework of 5a–c.
The theoretical studies indicated that the observed band shifts and broadening stem from the twisted conformation between the bulky substituents (R) and the PBI core of dye 5 within cage 1.15 The dihedral angle (ϕ) about the phenyl and imide rings of 5 corresponds to its electronic absorption features. For instance, the most stable conformer 5a with ϕ = 90° theoretically provides an intense, single electronic absorption band at 500 nm (Fig. 5d). The band is red-shifted by +6 nm through its transformation into one of the metastable conformers with ϕ = 70°. The estimated energy is +75 kJ mol−1 higher than that of the most stable conformer. In the optimized host–guest structure of 1·5a, dye 5a displayed ϕ = 70° within 1 (Fig. 4a, right). Accordingly, the observed, unusual color changes of caged 5a and 5b were induced through non-covalent twisting of the bulky substituents in the open cavity.
Caged bulky porphyrin dyes
Finally, to our surprise, the open polyaromatic cavity enabled cage 1 to bind one molecule of porphyrin dyes 6 bearing four or eight substituents. The encapsulation of porphyrins and metalloporphyrins by large proteins has been intensively studied so far16 yet a non-biological approach using coordination cages and capsules is complicated by the large and bulky dye frameworks.5 In a manner similar to 5, tetraphenylporphyrin 6a (R = Ph; 1.8 nm in maximum length) in purple was bound by 1via sonication (40 kHz) of the mixture in 8:1 water/AN for 20 min (Fig. 6a).11 The resultant brown solution including 1:1 host–guest complex 1·6a (70% yield) exhibited new absorption bands, assignable to caged 6a in the UV-visible spectrum (Fig. 6b). The characteristic Soret and the first Q bands of 6a were red-shifted by +3 and +4 nm, respectively, as compared with those of free 6a in CH2Cl2, through the binding. These bands remained unchanged even after 7 d under ambient conditions, suggesting adequate stability of the host–guest structure (Fig. S37a†). The insolubility of dye 6a in aqueous solution prevented titration, but concentration-dependent UV-visible analysis of 1·6a in 8:1 water/AN indicated high stability (Ka > 108 M−1) against high dilution (up to ∼3.5 μM; Fig. S39c†).11 In the 1H NMR and ESI-TOF MS spectra of the resultant solution, the guest and host–guest signals were scarcely observed owing to significant signal broadening and instability under MS conditions (e.g., extremely low concentration and high vacuum), respectively (Fig. S39†).11 The DLS analysis supported the product size with a single peak at d = 4.7 nm, slightly larger than that of empty 1 (d = 3.6 nm, Fig. 6c). Unfortunately, no crystals of the host–guest complex were obtained even under various conditions.
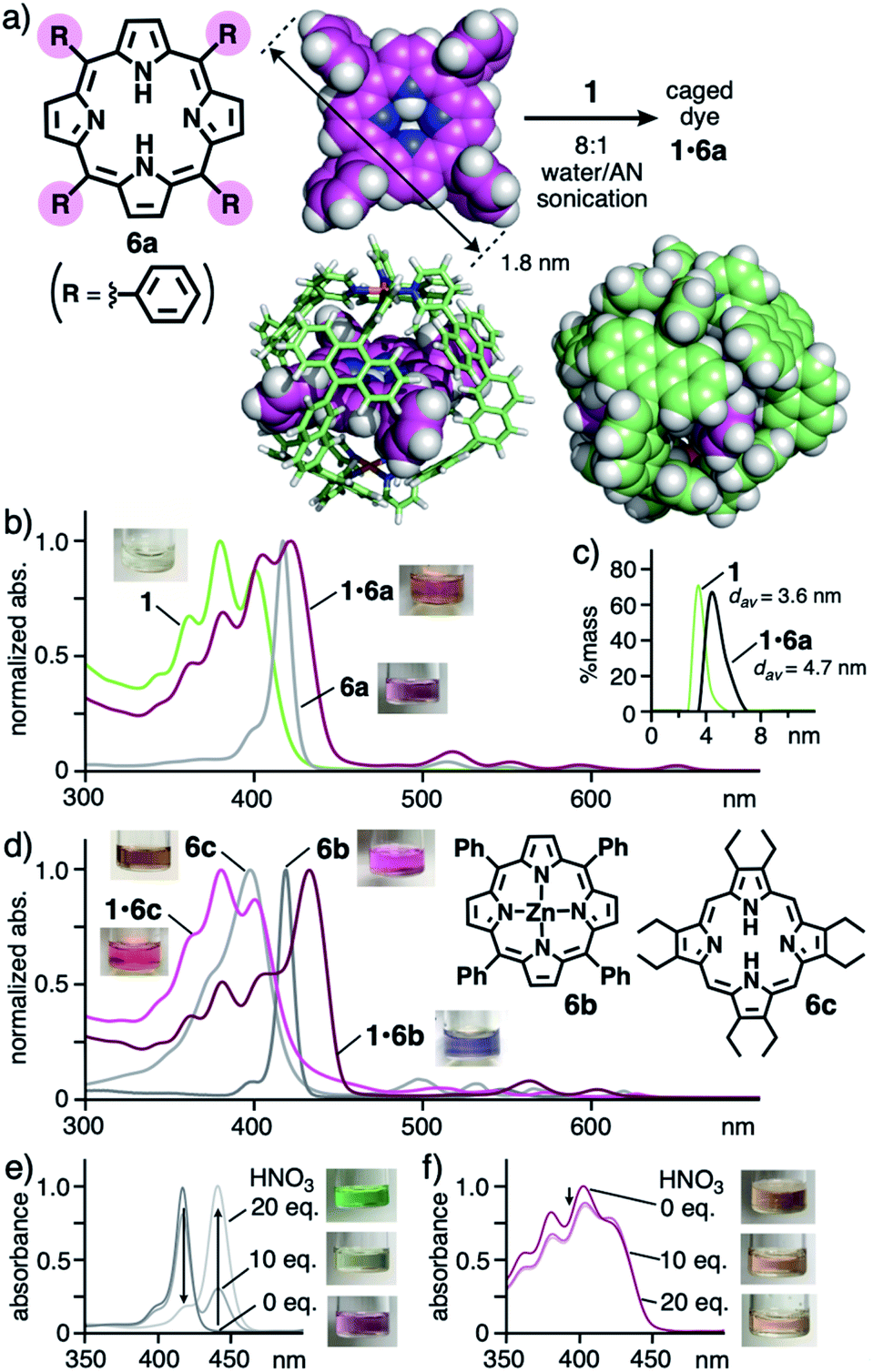 | ||
| Fig. 6 (a) Formation of caged dye 1·6a and the optimized structures of 6a and 1·6a (R1 = H for clarity).11 (b) UV-visible spectra and photographs (8:1 water/AN, r.t.) of 1·6a, 1, and 6a (in CH2Cl2) and (c) their DLS charts (r.t.) except for 6a. (d) UV-visible spectra and photographs (8:1 water/AN, r.t.) of 1·6b, 1·6c, and 6b, 6c (in CH2Cl2). UV-visible spectra and photographs (r.t.) of (e) 6a in CH2Cl2 and (f) 1·6a in 8:1 water/AN, after addition of HNO3 (0–20 equivalents). | ||
The structure of obtained, caged dye 1·6a was further supported by the theoretical calculation, where the four bulky substituents of 6a are located at the four openings of 1, whereas the large π-conjugated core of 6a is effectively surrounded by the polyaromatic cage-like framework of 1 without direct contacts (Fig. 6a, right). The complimentary symmetry facilitates the formation of unusual host–guest complex 1·6a. Because of the absence of host–guest π-stacking interactions in the optimized structure, the observed red-shifts of caged 6a are most probably derived from the stabilization of its planar conformation over its butterfly-shaped one16 within 1.
Other bulky porphyrin dyes were also trapped by cage 1 in aqueous solution with selectivity17 and the caged dyes were protected by the polyaromatic frameworks against acid. Further color and absorption changes were found through caging of Zn(II)-tetraphenylporphyrin 6b by 1 through the same sonication protocol. Although 6b itself shows no solvatochromic properties, the resultant solution of 1·6b is bluish-purple in contrast to a pink solution of free 6b in CH2Cl2 (Fig. 6d). In addition, distinct shifts of the Soret and first Q bands of 6b by approximately +15 nm in each case were observed in the UV-visible spectrum upon trapping. These unusual shifts are most likely derived from the cage-directed, non-covalent planarization of the metalloporphyrin framework. Caged octaethylporphyrin dye 1·6c and tetrakis(pentafluorophenyl)porphyrin dye 1·6d also displayed modulated color and altered absorption bands, relative to the corresponding free dyes (Fig. 6d and S37b†). Furthermore, the polyaromatic cage-like framework of 1 could prevent the π-conjugated core of 6a from protonation under acidic conditions. Uncaged 6a was fully protonated upon addition of 20 equivalent of HNO3, accompanying its color and spectral changes (Fig. 6e).18 On the other hand, the color and spectrum of caged 6a remained intact, except for a slight intensity change, even after the addition of excess HNO3 (up to 100 equivalent; Fig. 6f and S41a†).11 The present protection effect for the porphyrin dye, which was also found for 1·6c (Fig. S41b†), has not been reported with the previous synthetic hosts, except for a covalent cage from the Stoddart group.5,19 The theoretical studies indicated that the host–guest complexation of 1 and diprotonated dye 6a′ is unfavorable (ΔE > 800 kJ mol−1 based on PM6 calculation; Table S2†), owing to its sterically demanding, largely distorted framework, rather than long-range, host–guest cation–cation repulsion (d = 0.7 nm; Fig. S43†).11
Conclusions
We have demonstrated the binding and spectroscopic modulation of three types of bulky organic dyes (≥1.5 nm in maximum length) by a polyaromatic cage in aqueous solution. Binding dyes with several bulky substitutes has faced many difficulties so far, because neither too large nor too small cavities/windows are suitable for the desired host compounds. We herein employed an M2L4 cage with an open polyaromatic cavity, after simple modification of its hydrophilic side-chains, to successfully bind sterically demanding dyes with coumarin, perylene bisimide, and porphyrin cores. Thanks to size and shape host–guest matching as well as effective host–guest interactions (i.e., hydrophobic effect and multiple CH–π interactions), these dyes were straightforwardly caught by the cage in a threading fashion. Notably, the caged dyes displayed unusual fluorescence/color and spectroscopic features due to the polyaromatic cage effect, without π-stacking interactions, which was revealed by detailed theoretical and spectroscopic studies. As compared with typical covalent functionalization and previous host–guest systems targeting small and less bulky dyes, the present non-covalent modification of large and bulky organic dyes provides various advantages, since the coordination framework can for example be easily modulated via doping with heteroatoms and attachment of interactive side chains. Biological and materials applications of the present and other caged bulky dyes will be our next research targets.Data availability
The experimental procedures and analytical data are available in the ESI.†Author contributions
M. U., N. K., L. C., and M. Y. designed the work, carried out research, analyzed data, and wrote the paper. M. Y. is the principal investigator. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (Grant No. JP20K22552/JP21K18951/JP22H00348/JP22H0556), AMED (Grant No. H421TS), and The Mitsubishi Foundation (Research Grants in the Natural Sciences).Notes and references
- (a) M. Daniel, S. D. Bhattacharya, A. Arya and V. M. Raole, Natural Dyes: Scope and Challenges, Scientific Publishers, Jodhpur, 2006 Search PubMed; (b) Industrial Dyes: Chemistry, Properties, Applications, ed. K. Hunger, Wiley-VCH, 2002 Search PubMed.
- (a) F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed; (b) D. Cao, Z. Liu, P. Verwilst, S. Koo, P. Jangjili, J. S. Kim and W. Lin, Chem. Rev., 2019, 119, 10403–10519 CrossRef CAS PubMed; (c) C. J. Kingsbury and M. O. Senge, Coord. Chem. Rev., 2021, 431, 213760 CrossRef CAS.
- R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941–7980 CrossRef CAS PubMed.
- Recent reviews on coordination cages: (a) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001–7045 CrossRef CAS PubMed; (b) C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed; (c) L.-J. Chen, H.-B. Yang and M. Shionoya, Chem. Soc. Rev., 2017, 46, 2555–2576 RSC; (d) I. Sinha and P. S. Mukherjee, Inorg. Chem., 2018, 57, 4205–4221 CrossRef CAS PubMed; (e) F. J. Rizzuto, L. K. S. von Krbek and J. R. Nitschke, Nat. Rev. Chem., 2019, 3, 204–222 CrossRef; (f) A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS PubMed; (g) E. G. Percástegui, T. K. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480–13544 CrossRef PubMed; (h) H. Takezawa and M. Fujita, Bull. Chem. Soc. Jpn., 2021, 94, 2351–2369 CrossRef CAS; (i) S. Pullen, J. Tessarolo and G. H. Clever, Chem. Sci., 2021, 12, 7269–7293 RSC.
- Binding of bulky porphyrin dyes via coordination bonds: (a) M. L. Merlau, M. del P. Mejia, S. T. Nguyen and J. T. Hupp, Angew. Chem., Int. Ed., 2001, 40, 4239–4242 CrossRef CAS; (b) M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanović-Burmazović, J. N. H. Reek and B. de Bruin, Chem.–Eur. J., 2013, 19, 10170–10178 CrossRef CAS PubMed; (c) F. J. Rizzuto, W. J. Ramsay and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 11502–11509 CrossRef CAS PubMed.
- (a) M. Yoshizawa and J. K. Klosterman, Chem. Soc. Rev., 2014, 43, 1885–1898 RSC; (b) M. Yoshizawa and M. Yamashina, Chem. Lett., 2017, 46, 163–171 CrossRef CAS; (c) M. Yoshizawa and L. Catti, Acc. Chem. Res., 2019, 52, 2392–2404 CrossRef CAS PubMed; (d) L. Catti, R. Sumida and M. Yoshizawa, Coord. Chem. Rev., 2022, 460, 214460 CrossRef CAS.
- Other polyaromatic capsules and tubes: (a) K. Kondo, J. K. Klosterman and M. Yoshizawa, Chem.–Eur. J., 2017, 23, 16710–16721 CrossRef CAS PubMed; (b) K. Yazaki, L. Catti and M. Yoshizawa, Chem. Commun., 2018, 54, 3195–3206 RSC.
- (a) N. Kishi, Z. Li, K. Yoza, M. Akita and M. Yoshizawa, J. Am. Chem. Soc., 2011, 133, 11438–11441 CrossRef CAS PubMed; (b) M. Yamashina, Y. Sei, M. Akita and M. Yoshizawa, Nat. Commun., 2014, 5, 4662 CrossRef CAS PubMed.
- Multiple host–guest interactions supported by X-ray crystallographic analysis: (a) K. Yazaki, M. Akita, S. Prusty, D. K. Chand, T. Kikuchi, H. Sato and M. Yoshizawa, Nat. Commun., 2017, 8, 15914 CrossRef CAS PubMed; (b) S. Kusaba, M. Yamashina, M. Akita, T. Kikuchi and M. Yoshizawa, Angew. Chem., Int. Ed., 2018, 57, 3706–3710 CrossRef CAS PubMed; (c) M. Yamashina, S. Kusaba, M. Akita, T. Kikuchi and M. Yoshizawa, Nat. Commun., 2018, 9, 4227 CrossRef PubMed; (d) M. Yamashina, T. Tsutsui, Y. Sei, M. Akita and M. Yoshizawa, Sci. Adv., 2019, 5, eaav3179 CrossRef CAS PubMed; (e) N. Kishida, K. Matsumoto, Y. Tanaka, M. Akita, H. Sakurai and M. Yoshizawa, J. Am. Chem. Soc., 2020, 142, 9599–9603 CrossRef CAS PubMed; (f) R. Sumida, Y. Tanaka, K. Niki, Y. Sei, S. Toyota and M. Yoshizawa, Chem. Sci., 2021, 12, 9946–9951 RSC.
- M. Yamashina, T. Yuki, Y. Sei, M. Akita and M. Yoshizawa, Chem. Eur. J., 2015, 21, 4200–4204 CrossRef CAS PubMed.
- See the ESI.† The yields of the resultant host–guest complexes (uptake ratios) were estimated by UV-visible analysis (with a calibration curve method) in organic solvent (i.e., DMSO), after the removal of suspended unbound dyes by filtration and the lyophilization of the aqueous solutions of cage 1 and its host–guest complexes. The geometry optimizations of host–guest complexes were performed with molecular mechanics calculations (forcite module, BIOVIA Materials Studio 2020, version 20.1.0.5).
- 1H DOSY NMR spectrum showed the presence of a single product with a diffusion coefficient (D) of 4.68 × 10−10 m2 s−1 (Fig. S14†). The optimized structure of 1 was obtained by DFT calculation (CAM-B3LYP/LanL2DZ (for Pt), 6-31G(d,p) (for organic atoms) level of theory).
- Binding of PBI dyes with hydrophilic side chains by covalent hosts: (a) F. Biedermann, E. Elmalem, I. Ghosh, W. M. Nau and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 7739–7743 CrossRef CAS PubMed; (b) W. Liu, S. Bobbala, C. L. Stern, J. E. Hornick, Y. Liu, A. E. Enciso, E. A. Scott and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 3165–3173 CrossRef CAS PubMed.
- Encapsulation of multiple PBI dyes in a stacked fashion: K. Ito, T. Nishioka, M. Akita, A. Kuzume, K. Yamamoto and M. Yoshizawa, Chem. Sci., 2020, 11, 6752–6757 RSC.
- The optimized structure of 5a (ϕ = 90°) was obtained by DFT calculation (CAM-B3LYP/6-31G(d,p) level of theory). The predicted absorption bands were obtained by TD-DFT calculation (B3LYP/6-31G(d) level of theory; Fig. 5d and S35a†). Additional calculations of 5a including dispersion effects were examined using DFT (CAM-B3LYP+D3BJ/6-31G(d,p) level of theory) and TD-DFT (B3LYP+D3BJ/6-31G(d) level of theory; Fig. S35b†).11.
- T. L. Poulos, Chem. Rev., 2014, 114, 3919–3962 CrossRef CAS PubMed.
- The incorporation efficiency of cage 1 depends on the substituents on the porphyrin core.11 Competitive binding experiments revealed that 1 selectively binds 6a (>80% selectivity) from a mixture of 6a and octaethylporphyrin (6c), due to steric effects (Fig. S45†). More rigid 6b (∼30% selectivity) was inferior to 6a regarding the incorporation by 1 (Fig. S45†).
- O. Finikova, A. Galkin, V. Rozhkov, M. Cordero, C. Hägerhäll and S. Vinogradov, J. Am. Chem. Soc., 2003, 125, 4882–4893 CrossRef CAS PubMed.
- W. Liu, C. Lin, J. A. Weber, C. L. Stern, R. M. Young, M. R. Wasielewski and J. F. Stoddart, J. Am. Chem. Soc., 2020, 142, 8938–8945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02308c |
| This journal is © The Royal Society of Chemistry 2022 |