
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cationic molybdenum oxo alkylidenes stabilized by N-heterocyclic carbenes: from molecular systems to efficient supported metathesis catalysts†
Janis V.
Musso‡
a,
Jordan
De Jesus Silva‡
 b,
Mathis J.
Benedikter
a,
Jonas
Groos
a,
Wolfgang
Frey
c,
Christophe
Copéret
*b and
Michael R.
Buchmeiser
*ad
b,
Mathis J.
Benedikter
a,
Jonas
Groos
a,
Wolfgang
Frey
c,
Christophe
Copéret
*b and
Michael R.
Buchmeiser
*ad
aInstitute of Polymer Chemistry, University of Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany. E-mail: michael.buchmeiser@ipoc.uni-stuttgart.de
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog-Weg 1–5, CH-8093 Zürich, Switzerland. E-mail: ccoperet@ethz.ch
cInstitute of Organic Chemistry, Universität Stuttgart, Pfaffenwaldring 55, 70569 Stuttgart, Germany
dGerman Institutes of Textile and Fiber Research (DITF) Denkendorf, Körschtalstr. 26, 73770 Denkendorf, Germany
First published on 12th July 2022
Abstract
Cationic d0 group 6 olefin metathesis catalysts have been recently shown to display in most instances superior activity in comparison to their neutral congeners. Furthermore, their catalytic performance is greatly improved upon immobilization on silica. In this context, we have developed the new family of molecular cationic molybdenum oxo alkylidene complexes stabilized by N-heterocyclic carbenes of the general formula [Mo(O)(CHCMe3)(IMes)(OR)[X−]] (IMes = 1,3-dimesitylimidazol-2-ylidene; R = 1,3-dimesityl-C6H3, C6F5; X− = B(3,5-(CF3)2C6H3)4−, B(ArF)4, tetrakis(perfluoro-t-butoxy)aluminate (PFTA)). Immobilization of [Mo(O)(CHCMe3)(IMes)(O-1,3-dimesityl-C6H3)+B(ArF)4−] on silica via surface organometallic chemistry yields an active alkene metathesis catalyst that shows the highest productivity towards terminal olefins amongst all existing molybdenum oxo alkylidene catalysts.
Introduction
Since the discovery of olefin metathesis more than half a century ago,1 the development of novel catalyst families has been at the core of extensive research efforts, with the goal to increase catalytic performance, from functional group tolerance to increased activities, selectivities and stabilities.2 Apart from Ru-based “Grubbs-type” catalysts,3 d0 early-transition-metal metathesis catalysts mostly based on group 6 metals (Mo/W), i.e. Schrock-type catalysts of the general formula M(E)(CHR)(X)(Y) with E = oxo or imido and X, Y = anionic ligands of various types (alkyls, alkoxys, amidos), have emerged as a central class of olefin metathesis catalysts.4 Besides, the use of surface organometallic chemistry (SOMC)5 has enabled to greatly improve their activity and stability through generation of the corresponding well-defined active sites dispersed at the surface of oxide supports like silica.6 In parallel, DFT calculations have revealed that the right balance of the σ-donation ability of the X, Y and E ligands is key to these improved catalytic performances.7 The concurrent presence of both strong and weaker σ-donating anionic X and Y ligands for instance avoids overstabilization of the metallacyclobutane intermediates while favouring [2 + 2] cycloaddition and cycloreversion processes as well as suppressing deactivation pathways. N-Heterocyclic carbenes (NHCs) have gained increasing importance as ancillary ligands in transition metal complexes since the 1990s.8 In this regard, the introduction of NHCs in overall tetracoordinate, metathesis-active, cationic Mo and W species has illustrated how catalytic performance can be further improved.9 Charge delocalization between the NHC and the cationic metal centre renders cationic complexes rather “soft” according to the HSAB principle, which explains the high activity and functional group tolerance in cationic group 6 alkylidene complexes.10 The combination of both concepts – introduction of the strong σ-donating NHC ligands and weak siloxy ligands provided by the silica surface – has thus enabled the generation of some of the most active and stable olefin metathesis catalysts to date.9b,d,9g For the cationic tungsten-based catalysts, the oxo-bearing complexes outperformed their imido analogues by 1–2 orders of magnitude in activity;9g this can be attributed to the combination of the smaller E ligand size, allowing for easier access of the incoming olefin to the metal centre, and its strong σ-donating ability, which simultaneously enhances catalyst stability. Previously published immobilized neutral Mo oxo alkylidene complexes proved to possess high activity for internal olefins (Fig. 1a).2j,11 Consequently, we wanted to investigate whether the activity of molybdenum oxo complexes can be boosted for terminal olefins similar to what was shown for analogous tungsten oxo complexes.9g | ||
| Fig. 1 (a) Previously reported supported neutral Mo oxo alkylidene NHC species; (b) supported cationic Mo oxo NHC complex in this work. B(ArF)4 = B(3,5-(CF3)2C6H3)4. | ||
Results and discussion
Synthesis of molecular catalysts
In this context, we developed and herein report the synthesis and characterization of the long-awaited family of molecular and well-defined silica-supported cationic Mo oxo species stabilized by NHC ligands (Fig. 1b) and investigated their activity towards internal and terminal olefins. The synthesis of cationic molybdenum oxo alkylidene NHC complexes was first attempted starting from the 4-methoxybenzylidene precursor Mo-1 (Scheme 1a).12 Addition of 1 equiv. of 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene (IMes) to Mo-1 resulted in deprotonation of the alkylidene ligand. Through careful choice of a less basic chlorinated NHC,13 this side-reaction could be vastly circumvented and the desired product was isolated in 34% yield. Single crystals of Mo-2 for X-ray diffraction were grown from a mixture of dichloroethane/n-heptane at −40 °C (Fig. 2). Mo-2 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with a = 1184.11(6) pm, b = 1247.18(7) pm, c = 1537.13(8) pm, α = 80.252(2)°, β = 81.208(2)°, γ = 66.112(3)°, Z = 2. In the solid state, the complex adopts a distorted square pyramidal geometry (τ5 = 0.34)14 with a syn alkylidene (1JCH = 134 Hz) in the apical position. The Mo–oxo bond (166.4 pm) is slightly shorter than in five-coordinate W oxo alkylidene NHC complexes (169.0–176.0 pm) whereas the Mo–NHC bond distance of 226.3 pm is well in the expected range of comparable tungsten complexes.
with a = 1184.11(6) pm, b = 1247.18(7) pm, c = 1537.13(8) pm, α = 80.252(2)°, β = 81.208(2)°, γ = 66.112(3)°, Z = 2. In the solid state, the complex adopts a distorted square pyramidal geometry (τ5 = 0.34)14 with a syn alkylidene (1JCH = 134 Hz) in the apical position. The Mo–oxo bond (166.4 pm) is slightly shorter than in five-coordinate W oxo alkylidene NHC complexes (169.0–176.0 pm) whereas the Mo–NHC bond distance of 226.3 pm is well in the expected range of comparable tungsten complexes.
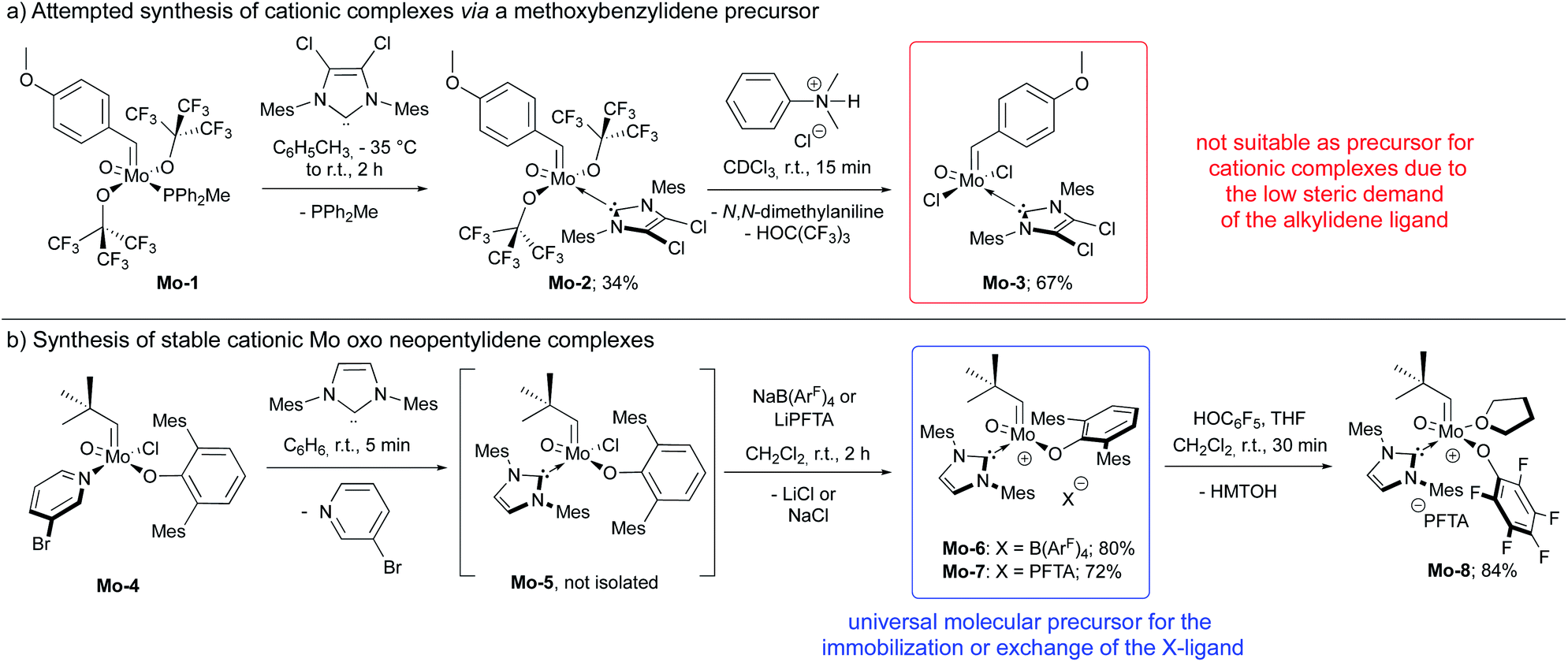 | ||
| Scheme 1 (a) Synthesis of 4-methoxybenzylidene complexes that could not be further transformed into cationic species; (b) synthesis of cationic molybdenum oxo neopentylidene complexes. | ||
 | ||
| Fig. 2 Single crystal X-ray structure of Mo-2. Selected bond lengths [pm] and angles [°]: Mo1–O(1) 166.4(2), Mo1–C(30) 192.1(3), Mo1–O(2) 203.3(2), Mo1–O(3) 206.2(2), Mo1–C(1) 226.3(3), O(1)–Mo1–C(30) 102.28(13), O(1)–Mo1–O(2) 146.29(11), C(30)–Mo1–O(2) 110.88(12), O(1)–Mo1–O(3) 95.40(10), C(30)–Mo1–O(3) 99.27(11), O(2)–Mo1–O(3) 85.36(9), O(1)–Mo1–C(1) 87.88(11), C(30)–Mo1–C(1) 92.56(12), O(2)–Mo1–C(1) 84.81(10), O(3)–Mo1–C(1) 166.73(10). | ||
Exchange of the fluorinated alkoxides was achieved by protonation using N,N-dimethylanilinium chloride to furnish complex Mo-3. Due to their electron count of 16 (counting the free electron pair at the oxo ligand, too) and their binding site occupied by an NHC, both, Mo-2 and Mo-3 were not expected to exhibit high activity in olefin metathesis. Therefore, we strived for the synthesis of highly active cationic species.9k
Abstracting one of the X-type ligands using either N,N-dimethylanilinium B(ArF)4 in the reaction with Mo-2 in the presence of pivalonitrile (pivCN) or Ag(pivCN)3B(ArF)4 in the reaction with Mo-3 resulting in complexes of the formula [MoO(CH-4-(OMe)C6H4)(X)(IMesCl2)(pivCN)][B(ArF)4] (X = OC(CF3)3, Cl) proved to be successful when conducted in CDCl3 and monitored by 1H NMR. However, the cationic species readily decomposed upon workup, which we attribute to the low steric demand of both, the 4-methoxybenzylidene moiety and the oxo ligand. And indeed, so far, all isolated cationic group 6 alkylidene complexes either bear a neophylidene or neopentylidene ligand9c in line with the “almost magic properties” of the neopentylidene ligand contributing to the stability of group 6 complexes.15
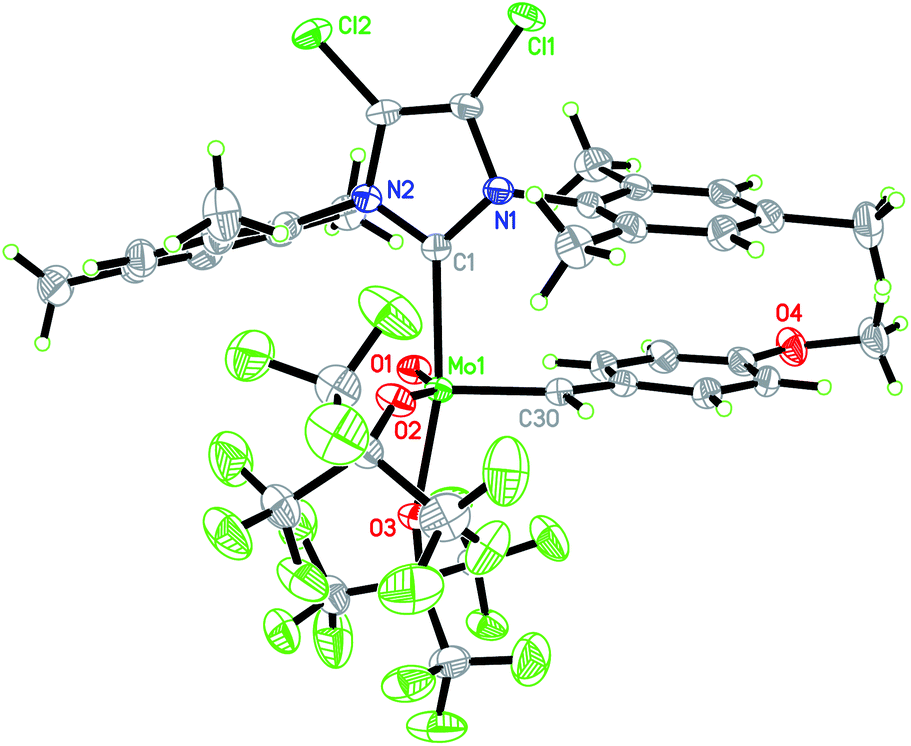
We consequently shifted our attention to a lately published procedure leading to the Mo oxo neopentylidene complex MoO(CHCMe3)Cl(OHMT)(3-Brpy) (OHMT = 2,6-dimesitylphenoxide; 3-Brpy = 3-bromopyridine).16 3-Bromopyridine as a relatively weak donor could then be readily replaced by strongly σ-donating IMes (Scheme 1b). In this case, no deprotonation of the alkylidene ligand occurred. The resulting five-coordinate NHC complex was not isolated on account of its poor crystallisation propensity. Nevertheless, the addition of NaB(ArF)4 and lithium tetrakis(perfluoro-t-butoxy)aluminate (LiPFTA), respectively, to the crude reaction mixture led to formation of the desired cationic complexes that could be purified by recrystallization and which were isolated in good yields of 72 and 80%, respectively, over two steps. The stable cationic species served as efficient and general precursors for immobilization but can also be further transformed by exchange of the X-ligand. Thus, upon addition of an excess of HOC6F5 to Mo-7, quantitative protonation of the HMTO ligand was facilitated to form Mo-8 in a high isolated yield of 84%. The coordination of THF in complex Mo-8 that lacks the sterically demanding alkoxide is imperative for its purification by recrystallization. Single crystals of Mo-8 suitable for X-ray diffraction were grown from 1,2-dichloroethane at −40 °C (Fig. 3). As for Mo-2, the triclinic space group P was found in Mo-8 with the unit cell dimensions being a = 1296.54(8) pm, b = 1388.76(9) pm, c = 1930.09(12) pm, α = 79.898(2)°, β = 78.994(2)°, γ = 86.057(3)° and two molecules in the unit cell. The distorted square pyramidal structure (τ5 = 0.29) bears a syn-alkylidene ligand in the apical position and all other ligands in the equatorial plane. All bond lengths and angles are similar to those observed for five-coordinate cationic W oxo alkylidene NHC complexes.17
 | ||
| Fig. 3 Single crystal X-ray structure of Mo-8, PFTA anion omitted for clarity. Selected bond lengths [pm] and angles [°]: Mo(1)–O(1) 166.4(3), Mo(1)–C(28) 189.1(3), Mo(1)–O(2) 195.8(3), Mo(1)–C(1) 220.3(3), Mo(1)–O(3) 220.5(2), O(1)–Mo(1)–C(28) 104.19(15), O(1)–Mo(1)–O(2) 146.73(13), C(28)–Mo(1)–O(2) 107.41(14), O(1)–Mo(1)–C(1) 94.78(13), C(28)–Mo(1)–C(1) 98.43(13), O(2)–Mo(1)–C(1) 90.70(12), O(1)–Mo(1)–O(3) 85.50(11), C(28)–Mo(1)–O(3) 96.81(12), O(2)–Mo(1)–O(3) 80.74(11), C(1)–Mo(1)–O(3) 164.20(11). | ||
On a final note regarding the synthesis of cationic Mo oxo alkylidene complexes, the synthesis of further species bearing fluorinated alkoxides starting from aryloxide complexes Mo-6 and Mo-7 have been attempted using HOC(CF3)3. Unfortunately, the reactions proved unsuccessful, and no conversion was observed. However, the aryloxide ligand in Mo-6 and Mo-7 was successfully protonated by the action of HCl in Et2O in the presence of pivCN to yield a mixture of complexes of the type [MoO(CHCMe3)Cl(IMes)(pivCN)][X] (X = B(ArF)4 or LiPFTA) and the protonated OHMT ligand as an inseparable oil. All attempts to isolate the complexes by crystallization or to further convert them in situ using MOC(CF3)3 (M = Li, Ag) failed.
Immobilization of complexes
The reaction of Mo-7 with HOC6F5 illustrates that protic functional groups can lead to the very selective exchange of the X-ligand without notable side-reactions. Following the development of the molecular compounds, we also sought to exploit this reactivity for grafting the complex on silica partially dehydroxylated at 700 °C (SiO2–700). For comparability with previously published complexes,9b,d,9g the B(ArF)4-containing complex Mo-6 was selected for this purpose and yielded the corresponding supported species Mo-6@SiO2 (Scheme 2). | ||
| Scheme 2 Grafting Mo-6 onto SiO2–700. | ||
Quantification by 19F solution NMR spectroscopy of the remaining B(ArF)4 of the complex indicates that ca. 29% of the surface silanols (0.32 mmol g−1) reacted with Mo-6, in line with a molybdenum loading of 0.79 wt% as determined by elemental analysis. IR spectroscopy confirms that a significant amount of isolated silanols (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3747 cm−1) are consumed upon grafting by protonolysis, although the large size of Mo-6 likely prevents full coverage.
= 3747 cm−1) are consumed upon grafting by protonolysis, although the large size of Mo-6 likely prevents full coverage.
Besides of the appearance of CH stretching bands between = 3200 and 2800 cm−1, a broad absorption band appeared at around = 3700 cm−1, typical of the non-covalent interactions of unreacted surface silanols with the aromatic ligands of the grafted complex.2h,9b,9d,11 Further characterization of Mo-6@SiO2 by 1H magic angle spinning (MAS) NMR showed the alkylidene proton resonance at 13.6 ppm, slightly upfield with respect to the molecular precursor signals in solution (13.9 ppm). The 13C cross-polarization (CP) MAS NMR spectrum shows the presence of the different methyl moieties and aromatics, whereas the alkylidene carbon, unfortunately, was not observed (for further details see ESI Fig. S1 and S2†).
Catalytic testing of molecular and immobilized complexes
Evaluation of the catalytic activity for Mo-6 was carried out for both molecular and immobilized complexes in the homo metathesis of the two benchmark substrates cis-4-nonene and 1-nonene at 30 °C (Table 1). While the homogeneous complex Mo-6 had to be tested in o-dichlorobenzene (DCB) due to solubility requirements, the immobilized analogue Mo-6@SiO2 was tested in both DCB and toluene, prototypical metathesis solvents used in many studies. In all cases, the metathesis activity towards α-olefins was higher than for internal ones. This behaviour was most pronounced for the molecular complex Mo-6, where no activity was observed for internal olefins and only 86% conversion after 8 hours was reached with 1-nonene. The lack of reactivity is likely associated with the presence of the large phenoxy (OHMT) ligand, whose steric encumbrance probably hinders the required distortion of the metal complex to a trigonal prism necessary to bind the incoming olefin.2j| Catalyst | Mol% | TOF3 mina | Time to equilibrium/maximum conversionb |
|---|---|---|---|
| a TOF at 3 min, given in min−1. b In cases where full conversion was not reached, the maximum conversion measured after a given time is provided. c Equilibrium conversion is 50%, conversion in brackets. d Maximum conversion is 100%, conversions/selectivity for the formation of hexadec-8-ene in brackets. e Experiment performed in o-dichlorobenzene. | |||
| Metathesis of cis-4-nonene | |||
| Mo-6 | 0.1 | — (<1%) | 5% after 24 h |
| Mo-6@SiO2e | 0.1 | 18 (6%) | 50% after 16 h |
| Mo-6@SiO2 | 0.1 | 26 (8%) | 50% after 4 h |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Metathesis of 1-nonene | |||
| Mo-6 | 0.1 | 47 (14%/98%) | 86% after 8 h |
| Mo-6@SiO2e | 0.1 | 124 (37%/97%) | 98% after 4 h |
| Mo-6@SiO2 | 0.1 | 118 (36%/98%) | 98% after 4 h |
| Mo-6@SiO2 | 0.02 | 254 (15%/98%) | 97% after 8 h |
| Mo-6@SiO2 | 0.005 | 225 (7%/98%) | 94% after 8 h |
In fact, switching OHMT for the smaller surface siloxy ligand leads to an increase in activity towards both olefins.2g,h Notably, the initial turnover frequency recorded for the self-metathesis of cis-4-nonene was modest in comparison to other molybdenum oxo alkylidenes (TOF3 min = 26 min−1vs. >500 min−1).2j,11 This observation is in line with the reported activity difference for the cationic tungsten analogues9b and established trends that show increased activity for internal olefins in Schrock-type metathesis catalysts with decreasing σ-donor strength of the X ligands (X = NHC, pyrrolide, alkoxide).9b,11,18 Considering also the activity for terminal olefins, the same correlation can only be observed in few cases.11 The decisive factor for a high catalytic activity might be rather the right balance of strong and weak σ-donor ligands as outlined above.7 However, the activity of Mo-6@SiO2 towards terminal olefins exceeds those previously reported for this family of supported complexes with TOF3 min = 254 min−1 at 0.02 mol% catalyst loading compared to only 75 min−1 and 170 min−1 of their neutral congeners at their optimal loadings (Fig. 4).2j,11 Indeed, this supports our working hypothesis that the catalytic activity of cationic Mo oxo alkylidenes is enhanced for terminal olefins similarly as shown previously for the corresponding tungsten oxo complexes.9g Furthermore, the catalyst remained active (94% after 8 h), when increasing the substrate to catalyst ratio to 10000:1.
 | ||
| Fig. 4 Overview of TOF3 min for the homo metathesis of cis-4-nonene (internal) and 1-nonene (terminal) of previously reported neutral Mo oxo alkylidene species2j,11 compared to the novel cationic complex Mo-6@SiO2; all reactions in toluene, at 30 °C and at optimal catalyst loadings at which the highest TOF values have been reported (Mo-6@SiO2: 0.1 mol% for cis-4-nonene and 0.02 mol% for 1-nonene; MoO(CH-4-(OMe)C6H4)(OHMT)@SiO2: 0.005 mol% for cis-4-nonene and 0.1 mol% for 1-nonene; MoO(CH-4-(OMe)C6H4)(tBuF9)(THF)@SiO2: 0.02 mol% for cis-4-nonene and 0.02 mol% for 1-nonene). | ||
The excellent performance of Mo-6@SiO2 was additionally demonstrated by recycling tests, where maximum conversion was obtained within 8 h three times, albeit accompanied by a constant loss of activity (see ESI†).
In previous studies it was shown that the homometathesis of 1-nonene using neutral immobilized Mo alkylidene catalysts can lead to low selectivities through isomerization of the position of the double bond. This can result in a selectivity of the desired hexadec-8-ene of only 80–85%, especially in closed vessels and at prolonged reaction times.2m,5b This type of isomerization can be attributed to Mo(IV) species resulting from methylidene complex decomposition that form π-complexes with olefins in the reaction mixture. Subsequently, the π-complexes undergo oxidative addition and reductive elimination processes and, thereby, lead to isomerization.19 Monitoring the selectivity for the formation of the homocoupling product in the metathesis of 1-nonene using cationic Mo oxo alkylidene complexes, in contrast, demonstrated that neither the homogeneous nor the immobilized catalyst are prone to form structural isomers. Even after long reaction times of 8 h, high selectivities of ca. 98% were obtained.
Conclusions
In conclusion, catalysts belonging to the new family of cationic Mo oxo alkylidenes NHC complexes have been developed. The immobilization on silica yielded a catalyst with preferential activity towards α-olefins over internal olefins. This particular reactivity is attributed to the nature of the NHC ligand, whose steric demand and strong σ-donor ability have an important influence on the formation and reversion of the TBP metallacyclobutane. These results further contribute to the overall understanding on how structural and electronic properties of metal alkylidenes affect the catalytic performance and further highlight the role of the NHC ligand in providing highly active cationic d0-based olefin metathesis catalysts.Experimental
General considerations
All experiments were carried out under an inert nitrogen or argon atmosphere using Schlenk techniques or an MBraun or GS glovebox equipped with a purifier unit. Water and oxygen levels were kept below 0.1 ppm. Diethyl ether, methylene chloride, pentane, toluene and tetrahydrofuran were purified using double MBraun SPS alumina columns. Benzene and benzene-d6 were distilled from Na/benzophenone. 1,2-Dichlorobenzene was distilled from CaH2. All solvents were degassed by three consecutive freeze–pump–thaw cycles.Elemental analyses were performed at Mikroanalytisches Labor Pascher, Germany, and at the Institute of Inorganic Chemistry, University of Stuttgart, Germany. All infrared (IR) spectra were recorded using a Bruker FT-IR Alpha spectrometer placed inside a glovebox, equipped with the OPUS software. The IR spectrometer had a total spectral range of 275–7500 cm−1 with a resolution <2 cm−1 and consisted of a RockSolid interferometer, a DTGS (triglycine sulfate) detector and a SiC globar source. Solid samples were investigated in a magnetic pellet holder. A typical experiment consisted of 32 consecutive transmission measurements in the region from 4000 to 400 cm−1. Solution NMR spectra were recorded on a Bruker Avance III 400. Chemical shifts are reported in ppm relative to the solvent signal (CDCl3: 7.26 ppm, C6D6 7.16 ppm, CD2Cl2 5.13 ppm).20 Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sept = septet, br = broad, m = multiplet), coupling constants (Hz) and integration. The 1H and 13C solid-state NMR spectra were acquired on a Bruker AVANCE III spectrometer operating at 400 MHz 1H frequency (9.4 T) and equipped with a 3.2 mm proton-heteronucleus magic angle spinning (1H,X MAS) probe (Bruker). Samples were packed into a 3.2 mm MAS NMR zirconia rotor closed with a VESPEL drive cap in an argon-filled glovebox, transferred to the NMR spectrometer in a tightly sealed vial under argon, rapidly inserted into the NMR spectrometer and spun under dry nitrogen. The MAS frequency was set to 16 kHz for all experiments. For the 13C CP MAS measurements, the Hartman–Hahn cross-polarization was performed by making use of a linear ramp from 70% to 100% with the contact time set to 3 ms. For decoupling, SPINAL64 was applied with 100 kHz irradiation on 1H. The acquired solid-state NMR spectra are referenced externally with respect to the downfield signal of adamantane (38.5 ppm). All MAS NMR spectra were acquired at ambient temperature.
Liquid catalytic test aliquots were analysed using a GC/FID (Agilent Technologies 7890 A) equipped with a split–splitless injector heated to 250 °C, injection volume 0.5 μL using hydrogen carrier gas. Chromatographic separations for 1-nonene and cis-4-nonene catalytic tests were performed using an HP-5 (Agilent Technologies) column (30 m, 0.32 mm, 0.25 μm stationary phase). Crystal data have been deposited with the Cambridge Crystallographic Data Centre (CCDC): Mo-2 CCDC 2151036, Mo-8 CCDC 2151037.
Synthesis of molecular precursors
Synthesis of silica-supported complex Mo-6@SiO2
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) SiO)Mo(O)(CHCMe3)(IMes)][B(ArF)4], Mo-6@SiO2.
A yellow solution of Mo-6 (78.8 mg, 0.047 mmol, 1 equiv.) was added to a suspension of SiO2–700 (160.1 mg, 0.051 mmol SiOH, 1.1 equiv.) in o-dichlorobenzene (5 mL) at 25 °C in a nitrogen filled glovebox. The suspension was slowly stirred (120 rpm) over 3 h. The supernatant was removed by decantation and the solid was washed by suspension/decantation cycles in o-dichlorobenzene (3 × 1 mL), toluene (3 × 1 mL) and pentane (3 × 2 mL). The resulting light-yellow solid was dried thoroughly under high vacuum (10−5 mbar) at room temperature for 2 hours to afford 180 mg of the title compound. All filtrate solutions were collected and analysed by 19F NMR spectroscopy in C6D6 using 4,4′-difluorobiphenyl as internal standard, indicating that 0.015 mmol of the molecular complex remained in solution, corresponding to 29% grafting. Mo 0.79%, C 5.85%, H 0.53%, N 0.47%, corresponding to 59 C/Mo (82 expected), 64 H/Mo (71 expected), 4 N/Mo (2 expected).
SiO)Mo(O)(CHCMe3)(IMes)][B(ArF)4], Mo-6@SiO2.
A yellow solution of Mo-6 (78.8 mg, 0.047 mmol, 1 equiv.) was added to a suspension of SiO2–700 (160.1 mg, 0.051 mmol SiOH, 1.1 equiv.) in o-dichlorobenzene (5 mL) at 25 °C in a nitrogen filled glovebox. The suspension was slowly stirred (120 rpm) over 3 h. The supernatant was removed by decantation and the solid was washed by suspension/decantation cycles in o-dichlorobenzene (3 × 1 mL), toluene (3 × 1 mL) and pentane (3 × 2 mL). The resulting light-yellow solid was dried thoroughly under high vacuum (10−5 mbar) at room temperature for 2 hours to afford 180 mg of the title compound. All filtrate solutions were collected and analysed by 19F NMR spectroscopy in C6D6 using 4,4′-difluorobiphenyl as internal standard, indicating that 0.015 mmol of the molecular complex remained in solution, corresponding to 29% grafting. Mo 0.79%, C 5.85%, H 0.53%, N 0.47%, corresponding to 59 C/Mo (82 expected), 64 H/Mo (71 expected), 4 N/Mo (2 expected).
Data availability
Primary and complementary data are available from https://doi.org/10.18419/darus-3013.Author contributions
JVM and JDJS contributed equally.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – Project-IDs 358283783 – SFB 1333/2 2022 and BU2174/22-1. J. D. J. S. was supported by the National Research Fund, Luxembourg (AFR Individual Ph.D. Grant 12516655). The authors thank Darryl F. Nater and Lukas Lätsch for help with IR and ssNMR measurements, respectively.References
- (a) R. L. Banks and G. C. Bailey, Ind. Eng. Chem. Prod. Res. Dev., 1964, 3, 170–173 CAS; (b) H. S. Eleuterio, J. Mol. Catal., 1991, 65, 55–61 CrossRef CAS.
- (a) F. Blanc, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage, L. Emsley, A. Sinha and R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 1216–1220 CrossRef CAS PubMed; (b) R. R. Schrock, Chem. Rev., 2009, 109, 3211–3226 CrossRef CAS PubMed; (c) S. J. Meek, S. J. Malcolmson, B. Li, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 16407 CrossRef CAS PubMed; (d) R. R. Schrock, Acc. Chem. Res., 2014, 47, 2457–2466 CrossRef CAS PubMed; (e) M. J. Koh, T. T. Nguyen, H. Zhang, R. R. Schrock and A. H. Hoveyda, Nature, 2016, 531, 459–465 CrossRef CAS PubMed; (f) M. A. B. Ferreira, J. De Jesus Silva, S. Grosslight, A. Fedorov, M. S. Sigman and C. Copéret, J. Am. Chem. Soc., 2019, 141, 10788–10800 CrossRef CAS PubMed; (g) M. P. Conley, V. Mougel, D. V. Peryshkov, W. P. Forrest, D. Gajan, A. Lesage, L. Emsley, C. Copéret and R. R. Schrock, J. Am. Chem. Soc., 2013, 135, 19068–19070 CrossRef CAS PubMed; (h) M. P. Conley, W. P. Forrest, V. Mougel, C. Copéret and R. R. Schrock, Angew. Chem., Int. Ed., 2014, 53, 14221–14224 CrossRef CAS PubMed; (i) V. Mougel, M. Pucino and C. Copéret, Organometallics, 2015, 34, 551–554 CrossRef CAS; (j) M. Pucino, F. Zhai, C. P. Gordon, D. Mance, A. H. Hoveyda, R. R. Schrock and C. Copéret, Angew. Chem., Int. Ed., 2019, 58, 11816–11819 CrossRef CAS PubMed; (k) M. Pucino, W.-C. Liao, K. W. Chan, E. Lam, R. Schowner, P. A. Zhizhko, M. R. Buchmeiser and C. Copéret, Helv. Chim. Acta, 2020, 103, e2000072 CrossRef CAS; (l) J. De Jesus Silva, M. A. B. Ferreira, A. Fedorov, M. S. Sigman and C. Copéret, Chem. Sci., 2020, 11, 6717–6723 RSC; (m) P. A. Zhizhko, V. Mougel, J. De Jesus Silva and C. Copéret, Helv. Chim. Acta, 2018, 101, e1700302 CrossRef; (n) D. F. Nater, B. Paul, L. Lätsch, R. R. Schrock and C. Copéret, Helv. Chim. Acta, 2021, 104, e2100151 CrossRef CAS.
- (a) R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed; (b) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746–1787 CrossRef CAS PubMed; (c) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS PubMed; (d) O. M. Ogba, N. C. Warner, D. J. O'Leary and R. H. Grubbs, Chem. Soc. Rev., 2018, 47, 4510–4544 RSC; (e) S. P. Nolan and H. Clavier, Chem. Soc. Rev., 2010, 39, 3305–3316 RSC; (f) C. Samojłowicz, M. Bieniek and K. Grela, Chem. Rev., 2009, 109, 3708–3742 CrossRef PubMed; (g) J. Morvan, M. Mauduit, G. Bertrand and R. Jazzar, ACS Catal., 2021, 11, 1714–1748 CrossRef CAS.
- (a) A. S. Hock, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2006, 128, 16373–16375 CrossRef CAS PubMed; (b) A. Sinha, L. P. H. Lopez, R. R. Schrock, A. S. Hock and P. Müller, Organometallics, 2006, 25, 1412–1423 CrossRef CAS; (c) R. Singh, C. Czekelius, R. R. Schrock, P. Müller and A. H. Hoveyda, Organometallics, 2007, 26, 2528–2539 CrossRef CAS PubMed; (d) M. M. Flook, A. J. Jiang, R. R. Schrock, P. Müller and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7962–7963 CrossRef CAS PubMed; (e) I. Ibrahem, M. Yu, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3844–3845 CrossRef CAS PubMed; (f) A. J. Jiang, Y. Zhao, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 16630–16631 CrossRef CAS PubMed; (g) E. S. Sattely, S. J. Meek, S. J. Malcolmson, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 943–953 CrossRef CAS PubMed; (h) M. R. Reithofer, G. E. Dobereiner, R. R. Schrock and P. Muller, Organometallics, 2013, 32, 2489–2492 CrossRef CAS PubMed.
- (a) C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef PubMed; (b) C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef PubMed; (c) J. D. A. Pelletier and J.-M. Basset, Acc. Chem. Res., 2016, 49, 664–677 CrossRef CAS PubMed.
- (a) C. Copéret, Z. J. Berkson, K. W. Chan, J. de Jesus Silva, C. P. Gordon, M. Pucino and P. A. Zhizhko, Chem. Sci., 2021, 12, 3092–3115 RSC; (b) M. M. Stalzer, M. Delferro and T. J. Marks, Catal. Lett., 2015, 145, 3–14 CrossRef CAS; (c) A. Chapovetsky, R. R. Langeslay, G. Celik, F. A. Perras, M. Pruski, M. S. Ferrandon, E. C. Wegener, H. Kim, F. Dogan, J. Wen, N. Khetrapal, P. Sharma, J. White, A. J. Kropf, A. P. Sattelberger, D. M. Kaphan and M. Delferro, Organometallics, 2020, 39, 1035–1045 CrossRef CAS.
- A. Poater, X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, J. Am. Chem. Soc., 2007, 129, 8207–8216 CrossRef CAS PubMed.
- (a) C. Romain, S. Bellemin-Laponnaz and S. Dagorne, Coord. Chem. Rev., 2020, 422, 213411 CrossRef CAS; (b) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619–636 RSC; (c) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS; (d) Q. Zhao, G. Meng, S. P. Nolan and M. Szostak, Chem. Rev., 2020, 120, 1981–2048 CrossRef CAS PubMed; (e) Q. Liang and D. Song, Chem. Soc. Rev., 2020, 49, 1209–1232 RSC; (f) S. C. Sau, P. K. Hota, S. K. Mandal, M. Soleilhavoup and G. Bertrand, Chem. Soc. Rev., 2020, 49, 1233–1252 RSC; (g) C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C.-H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed; (h) A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed; (i) Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed; (j) E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed; (k) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed; (l) N. Sinha and F. E. Hahn, Acc. Chem. Res., 2017, 50, 2167–2184 CrossRef CAS PubMed; (m) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (n) D. Zhang and G. Zi, Chem. Soc. Rev., 2015, 44, 1898–1921 RSC; (o) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- (a) R. Schowner, I. Elser, M. Benedikter, M. Momin, W. Frey, T. Schneck, L. Stöhr and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2020, 59, 951–958 CrossRef CAS PubMed; (b) J. De Jesus Silva, D. Mance, M. Pucino, M. J. Benedikter, I. Elser, M. R. Buchmeiser and C. Copéret, Helv. Chim. Acta, 2020, 103, e2000161 CrossRef CAS; (c) M. J. Benedikter, F. Ziegler, J. Groos, P. M. Hauser, R. Schowner and M. R. Buchmeiser, Coord. Chem. Rev., 2020, 415, 213315 CrossRef CAS; (d) M. Pucino, M. Inoue, C. P. Gordon, R. Schowner, L. Stöhr, S. Sen, C. Hegedüs, E. Robé, F. Tóth, M. R. Buchmeiser and C. Copéret, Angew. Chem., Int. Ed., 2018, 57, 14566–14569 CrossRef CAS PubMed; (e) M. R. Buchmeiser, Chem.–Eur. J., 2018, 24, 14295–14301 CrossRef CAS PubMed; (f) D. A. Imbrich, I. Elser, W. Frey and M. R. Buchmeiser, ChemCatChem, 2017, 9, 2996–3002 CrossRef CAS; (g) M. Pucino, V. Mougel, R. Schowner, A. Fedorov, M. R. Buchmeiser and C. Copéret, Angew. Chem., Int. Ed., 2016, 55, 4300–4302 CrossRef CAS PubMed; (h) I. Elser, W. Frey, K. Wurst and M. R. Buchmeiser, Organometallics, 2016, 35, 4106–4111 CrossRef CAS; (i) M. R. Buchmeiser, S. Sen, C. Lienert, L. Widmann, R. Schowner, K. Herz, P. Hauser, W. Frey and D. Wang, ChemCatChem, 2016, 8, 2710–2723 CrossRef CAS; (j) S. Sen, R. Schowner, D. A. Imbrich, W. Frey, M. Hunger and M. R. Buchmeiser, Chem.–Eur. J., 2015, 21, 13778–13787 CrossRef CAS PubMed; (k) R. Schowner, W. Frey and M. R. Buchmeiser, J. Am. Chem. Soc., 2015, 137, 6188–6191 CrossRef CAS PubMed; (l) M. R. Buchmeiser, S. Sen, J. Unold and W. Frey, Angew. Chem., Int. Ed., 2014, 53, 9384–9388 CrossRef CAS PubMed.
- M. Benedikter, J. Musso, M. K. Kesharwani, K. L. Sterz, I. Elser, F. Ziegler, F. Fischer, B. Plietker, W. Frey, J. Kästner, M. Winkler, J. van Slageren, M. Nowakowski, M. Bauer and M. R. Buchmeiser, ACS Catal., 2020, 10, 14810–14823 CrossRef CAS.
- J. De Jesus Silva, M. Pucino, F. Zhai, D. Mance, Z. J. Berkson, D. F. Nater, A. H. Hoveyda, C. Copéret and R. R. Schrock, Inorg. Chem., 2021, 60, 6875–6880 CrossRef CAS PubMed.
- F. Zhai, K. V. Bukhryakov, R. R. Schrock, A. H. Hoveyda, C. Tsay and P. Müller, J. Am. Chem. Soc., 2018, 140, 13609–13613 CrossRef CAS PubMed.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef CAS PubMed.
- F. Zhai, R. R. Schrock, A. H. Hoveyda and P. Müller, Organometallics, 2020, 39, 2486–2492 CrossRef CAS.
- J. V. Musso, R. Schowner, L. Falivene, W. Frey, L. Cavallo and M. R. Buchmeiser, ChemCatChem, 2022, 14, e202101510 CrossRef CAS.
- D. F. Nater, C. J. Kaul, L. Lätsch, H. Tsurugi, K. Mashima and C. Copéret, Chem.–Eur. J., 2022, 28, e202200559 CrossRef CAS PubMed.
- S. C. Marinescu, A. J. King, R. R. Schrock, R. Singh, P. Müller and M. K. Takase, Organometallics, 2010, 29, 6816–6828 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530–5534 CrossRef CAS.
- A. J. Arduengo, F. Davidson, H. V. R. Dias, J. R. Goerlich, D. Khasnis, W. J. Marshall and T. K. Prakasha, J. Am. Chem. Soc., 1997, 119, 12742–12749 CrossRef CAS.
- D. L. Reger, T. D. Wright, C. A. Little, J. J. S. Lamba and M. D. Smith, Inorg. Chem., 2001, 40, 3810–3814 CrossRef CAS PubMed.
- I. Krossing, Chem.–Eur. J., 2001, 7, 490–502 CrossRef CAS PubMed.
- F. Zhai, R. R. Schrock, A. H. Hoveyda and P. Müller, Organometallics, 2020, 39, 2486–2492 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2151036 (Mo-2) and 2151037 (Mo-8). For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc03321f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |