
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanocrystalline cellulose-based mixed ionic–electronic conductor for bioelectronics†
Katharina
Matura
a,
Rosarita
D’Orsi
b,
Laura
Spagnuolo
b,
Felix
Mayr
a,
Munise
Cobet
a,
Christoph
Putz
c,
Alessandra
Operamolla
 b and
Serpil
Tekoglu
*a
b and
Serpil
Tekoglu
*a
aLinz Institute for Solar Cells (LIOS) and Institute of Physical Chemistry, Johannes Kepler University Linz, Altenberger Str. 69, A-4040, Linz, Austria. E-mail: serpil.tekoglu@jku.at
bDepartment of Chemistry and Industrial Chemistry, University of Pisa, Via Giuseppe Moruzzi 13, I-56124, Pisa, Italy
cDivision of Soft Matter Physics and Institute of Experimental Physics, Johannes Kepler University Linz, Altenberger Str. 69, A-4040, Linz, Austria
First published on 27th September 2024
Abstract
Mixed ionic–electronic conductors (MIEC) are pivotal in advancing medical diagnostics, therapeutic devices, and health monitoring solutions due to their unique properties that enable more effective interfaces between electronic devices and biological systems. Cellulose, a natural and abundant polymer, is a promising material in the development of MIECs for bioelectronics. Combining cellulose with conductive polymer components can leverage the biocompatibility, sustainability, and mechanical properties of composite materials. In this study, we highlight the sulfated cellulose nanocrystals (S-CNCs) as a template for the facile green synthesis of conductive polymer PEDOT (poly(3,4-ethylenedioxythiophene)). The PEDOT:S-CNCs nanocomposite possesses good conductivity and high dispersibility in water. The electronic conductivity is recorded up to 5 S cm−1. A comprehensive investigation for material characterization is associated with the changes in their micro- and nanostructure surface morphology. The biocomposite is deposited as a channel material in organic electrochemical transistors (OECTs) to analyze ion-to-electron transduction. The maximum transconductance values are obtained up to 13.6 mS and 44.3 mS for single-channel and interdigitated OECTs, respectively, without applying photolithography techniques. The high transconductance values reveal the great potential of PEDOT:S-CNCs composite for bioelectronics.
Introduction
Mixed ionic–electronic conductors (MIECs) are key components in the field of bioelectronics and biosensing due to their unique ability to transport both ions and electrons simultaneously.1–4 This dual conductivity enables MIECs to bridge the gap between electronic devices and biological systems, which communicate through ionic signals. Among mixed conductors, PEDOT:PSS (poly(3,4-ethylenedioxythiophene):polystyrene sulfonate) has become a benchmark conductive polymer in bioelectronics because of its excellent electrical properties, solution processing and commercial availability.5 However, its long-term biocompatibility and stability in vivo remain critical factors determining its suitability for clinical applications. PEDOT, as the electrically conductive component, has shown good initial biocompatibility in vitro and short-term in vivo studies, with minimal cytotoxicity and a low level of acute inflammatory response.6 G. Dijk et al. reported on biocompatibility of PEDOT:PSS in vitro up to 3–4 months.7 Yet, the biocompatibility of polystyrene sulfonate (PSS), a component of PEDOT:PSS, is a subject of ongoing debate in the field of bioelectronics.8 While PSS contributes to the excellent conductive properties of PEDOT, its acidity poses challenges for long-term biocompatibility and stability. Ongoing research and development efforts are focused on mitigating these drawbacks through material modifications, composite formation, and post-treatment techniques.9 To overcome mechanical, environmental stability and biocompatibility issues, PEDOT can be combined with biopolymers.10 In our previous study, we investigated deoxyribonucleic acid (DNA) as a counterion for PEDOT and polypyrrole (PPy) to create mixed conductors.11Cellulose, the most abundant polymer in nature, is emerging as a promising material in the development of biomedical applications such as tissue engineering, wound dressing, and drug delivery.12 Being a renewable resource, cellulose contributes to the development of sustainable and eco-friendly materials. The hydroxyl groups on cellulose can be chemically modified, allowing for functionalization and improved interaction with other materials. Cellulose nanocrystals (CNCs) and cellulose nanofibrils (CNFs) have been investigated as reinforcement material for conductive polymers such as PEDOT:PSS, PPy, and polyaniline (PANI) for flexible energy and electronic devices.13–17 Nanocellulose has been mostly blended with conductive polymers to enhance the mechanical stability and flexibility of conductive composites.14,15
The hydroxyl groups on cellulose can be functionalized to introduce ionic groups to template conductive polymers, improving the ionic and electronic transport properties. In particular, CNCs are promising to graft conductive polymers due to excellent hydrophilicity with polar groups, tunable surface chemistry, high mechanical strength, and biodegradability.18 Additionally, CNCs can perform as multifunctional nanoplatforms for bioelectronics and biosensing.19 In the surface grafting method, conductive materials can be tightly bonded onto nanocellulose as a biotemplate via ionic interaction. The sulfated CNCs (S-CNCs) exhibit a negative surface charge, which drastically enhances their dispersibility in aqueous media.20 The polyelectrolyte nature of S-CNCs makes it an anionically charged rigid polyelectrolyte as a counterpart for the electronically conductive and cationic polymer PEDOT, resulting in p-type mixed ionic–electronic conductors.16,21,22 S-CNCs display rod-like morphology, whereby their length and width can range from 70–500 nm and 2–20 nm, respectively.23–25 In general, these CNCs show amphiphilic characteristics, which originate from the dual presence of hydrophilic sites (polar) and hydrophobic sites (non-polar). Owing to this property, CNCs have been previously employed to increase the stability of pickering emulsions.23,26 PEDOT-CNCs samples with poor dispersibility can promote significant phase separation in their thin films. The electrical conductivity is strongly dependent on phase separation, and electronic transport between conductive sites can be a limiting factor.13–16,22 The insulating CNC-rich domains may block the pathways between the PEDOT-rich domains, resulting in low electrical conductivities up to ∼200 μS cm−1.16 Therefore, nanocellulose-based composites with stable dispersion of PEDOT and uniform thin-film formation are crucial to achieve high ionic and electronic conduction.
In this work, we incorporate biodegradable cellulose nanocrystals as rigid rod nanoparticles in a p-type MIEC. PEDOT:S-CNCs are synthesized by following two steps. The first step is the preparation of sulfated cellulose nanocrystals through acid hydrolysis. As the second step, subsequent polymerization of 3,4-ethylenedioxythiophene (EDOT) monomer is carried out by chemical oxidation. The in situ oxidative polymerization of EDOT is templated by S-CNCs and represents a facile green synthesis in aqueous solution. The resulting PEDOT:S-CNCs nanocomposite possesses good conductivity, high dispersibility in water, and beneficial phase separation morphology in the solid state. The findings reveal high electronic conductivities up to 5 S cm−1. A comprehensive study is conducted using attenuated total reflection Fourier-infrared (ATR-FTIR) spectroscopy, X-ray photoelectron spectroscopy (XPS), and Raman spectroscopy for material characterization. The morphological changes are recorded by using scanning electron microscopy (SEM), atomic force microscopy (AFM), and polarized light microscopy to examine their micro- and nanostructures. To validate their ion-to-electron transduction, the composites are successfully deposited via solution processing (spin-coating) as a channel material in organic electrochemical transistors (OECTs). The maximum transconductance (gmax) values of 13.6 mS and 44.3 mS are obtained for single-channel and interdigitated OECTs, respectively, after optimizing the device parameters, without applying photolithography techniques.
Results and discussions
Sulfated cellulose nanocrystals (S-CNCs) can be prepared from microcrystalline cellulose (MCC) via sulfuric acid hydrolysis. S-CNCs were prepared from Avicel PH-101, a form of commercial microcrystalline cellulose, by 64% sulfuric acid hydrolysis, according to previous literature reports.20,27,28 The reaction, carried out at 50 °C, allows the isolation of rod-like nanoparticles composed of cellulose I. As shown in Fig. 1a, MCC is composed of two structural motives.24 On the one side, MCC is composed of regions with a high degree of crystallinity, which is heavily influenced by the strong intra- and inter-molecular hydrogen bond interactions between the cellulose chains.24,29 On the other side, in regions with a lower level of alignment of the chains, the material is amorphous. Due to the lower structural stability, these amorphous regions exhibit a higher susceptibility to chemical attack. Consequently, during hydrolysis, acid molecules penetrate the cellulose material protonating the acetal moiety and water can break the acetal linkage between the glucopyranose rings.27 Additionally, some of the primary hydroxylic groups on the cellulose nanocrystals get converted into a negatively charged sulfate ester groups (–OSO3−) following an esterification reaction. Therefore, S-CNCs can be easily dispersed in water, thanks to their negative surface charge. The upper picture in Fig. 1b represents the aqueous dispersions of S-CNCs. Elemental analyses were carried out on the S-CNCs samples revealing C, H, N, and S content of 40.10%, 6.55%, 0.00%, and 0.60%, respectively (in the ESI,† Table S1). This allowed us to determine the sulfation degree of cellulose, which was found to be ∼0.05 with a ζ-potential of −15 ± 4 mV measured in water.30 The crystallinity index of cellulose was qualitatively determined from X-ray diffractograms (Fig. S1, ESI†) by the Segal method,31 and found to be 88%. S-CNCs were mainly composed of cellulose I allomorph. Additionally, the S-CNCs show a low density of 1.6 g cm−3.24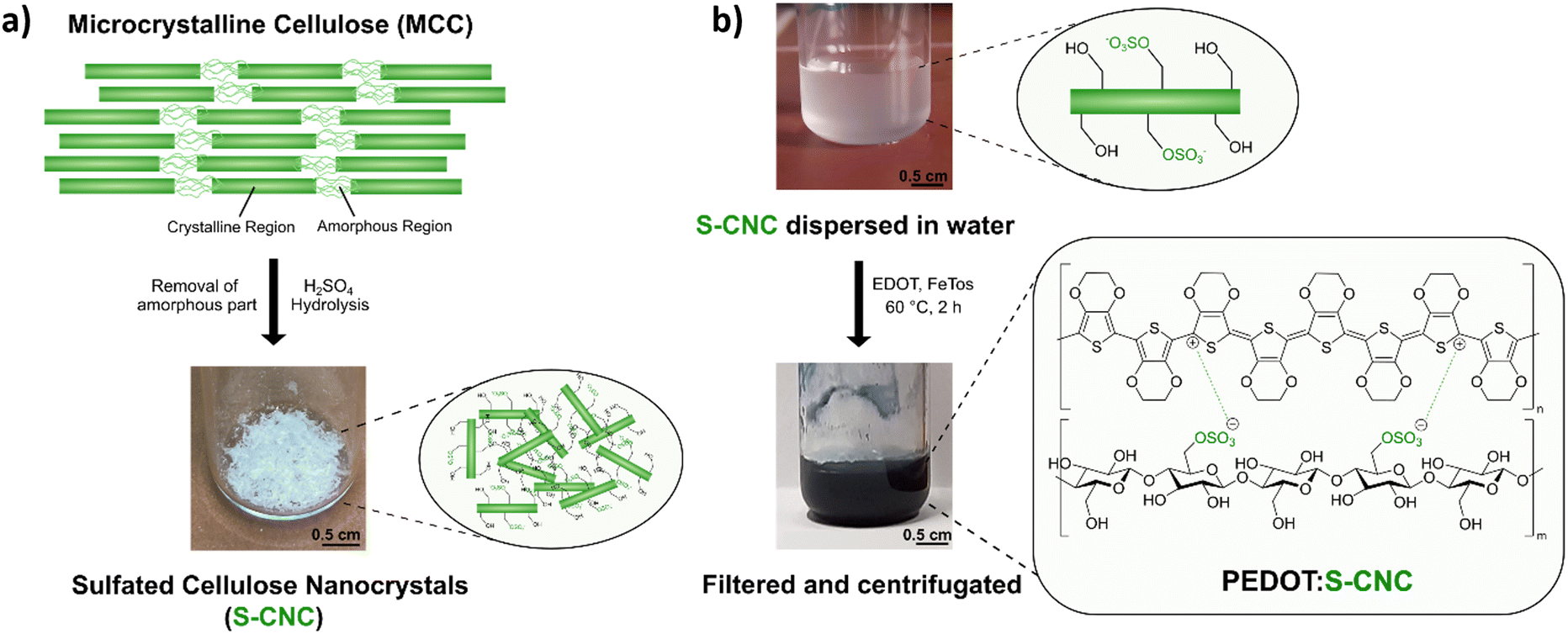 | ||
| Fig. 1 (a) Illustration of deconstruction from microcrystalline cellulose (MCC) via sulfuric acid (H2SO4) hydrolysis to the produced sulfated cellulose nanocrystals (S-CNCs). (b) The S-CNCs dispersed in water (semi-transparent) changes color to a dark blue upon oxidative chemical polymerization with EDOT and FeTos. The shown molecular structures are linked to the photos of the colloidal solutions. | ||
The novel PEDOT:S-CNCs biocomposite was synthesized by adapting the previously reported procedure for the PEDOT:DNA composites.11 Accordingly, EDOT was oxidatively polymerized in a suspension of S-CNCs after adding EDOT with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 weight-to-weight monomer ratio to S-CNCs. The EDOT/S-CNC mass ratio of 1 was chosen to obtain high electrical conductivity while maintaining the uniformity and stability of the PEDOT:S-CNC dispersion.21 During the oxidative chemical polymerization of EDOT, the as-formed PEDOT chains possess positive charges, exploited to interact with the dispersed negatively charged S-CNCs via Coulomb forces. A corresponding illustration of the composite synthesis and the final product (appears as a dark blue liquid) are shown in Fig. 1b, whereby the synthesis is described in more detail in the Experimental section.
:1 weight-to-weight monomer ratio to S-CNCs. The EDOT/S-CNC mass ratio of 1 was chosen to obtain high electrical conductivity while maintaining the uniformity and stability of the PEDOT:S-CNC dispersion.21 During the oxidative chemical polymerization of EDOT, the as-formed PEDOT chains possess positive charges, exploited to interact with the dispersed negatively charged S-CNCs via Coulomb forces. A corresponding illustration of the composite synthesis and the final product (appears as a dark blue liquid) are shown in Fig. 1b, whereby the synthesis is described in more detail in the Experimental section.
Spectroscopic characterization
Fourier transform infrared spectroscopy (FTIR) in ATR (attenuated total reflection) mode, X-ray photoelectron spectroscopy (XPS), and Raman spectroscopy were performed to examine the chemical structures of the materials.The ATR-FTIR analysis of S-CNCs, PEDOT, and PEDOT:S-CNCs is depicted in Fig. 2a. FTIR spectroscopy confirms changes in the chemical constituent and structure of composite compared to pristine S-CNCs. By observing the FTIR spectra of S-CNCs and the PEDOT:S-CNCs, the broad peak at 3310 cm−1 can be clearly attributed to the presence of O–H stretching vibrations within hydroxyl groups on the cellulose backbone.20,32,33 The bands at 2905 cm−1 are characteristic of the C–H groups of the cellulose chains, and the peak at 1516 cm−1 can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of the PEDOT backbone. Additionally, the peaks at 1311 cm−1 and 1023 cm−1 originate from C–C stretching and C–O–C vibrations, respectively, whereby both functional motives can be found within the pyranose rings as well as the PEDOT chain.32,34 The peaks at 891 cm−1 can be identified as C–S bonds of the thiophene ring of PEDOT, which can be found in both the pristine PEDOT and the PEDOT:S-CNCs composite.35,36
C stretching of the PEDOT backbone. Additionally, the peaks at 1311 cm−1 and 1023 cm−1 originate from C–C stretching and C–O–C vibrations, respectively, whereby both functional motives can be found within the pyranose rings as well as the PEDOT chain.32,34 The peaks at 891 cm−1 can be identified as C–S bonds of the thiophene ring of PEDOT, which can be found in both the pristine PEDOT and the PEDOT:S-CNCs composite.35,36
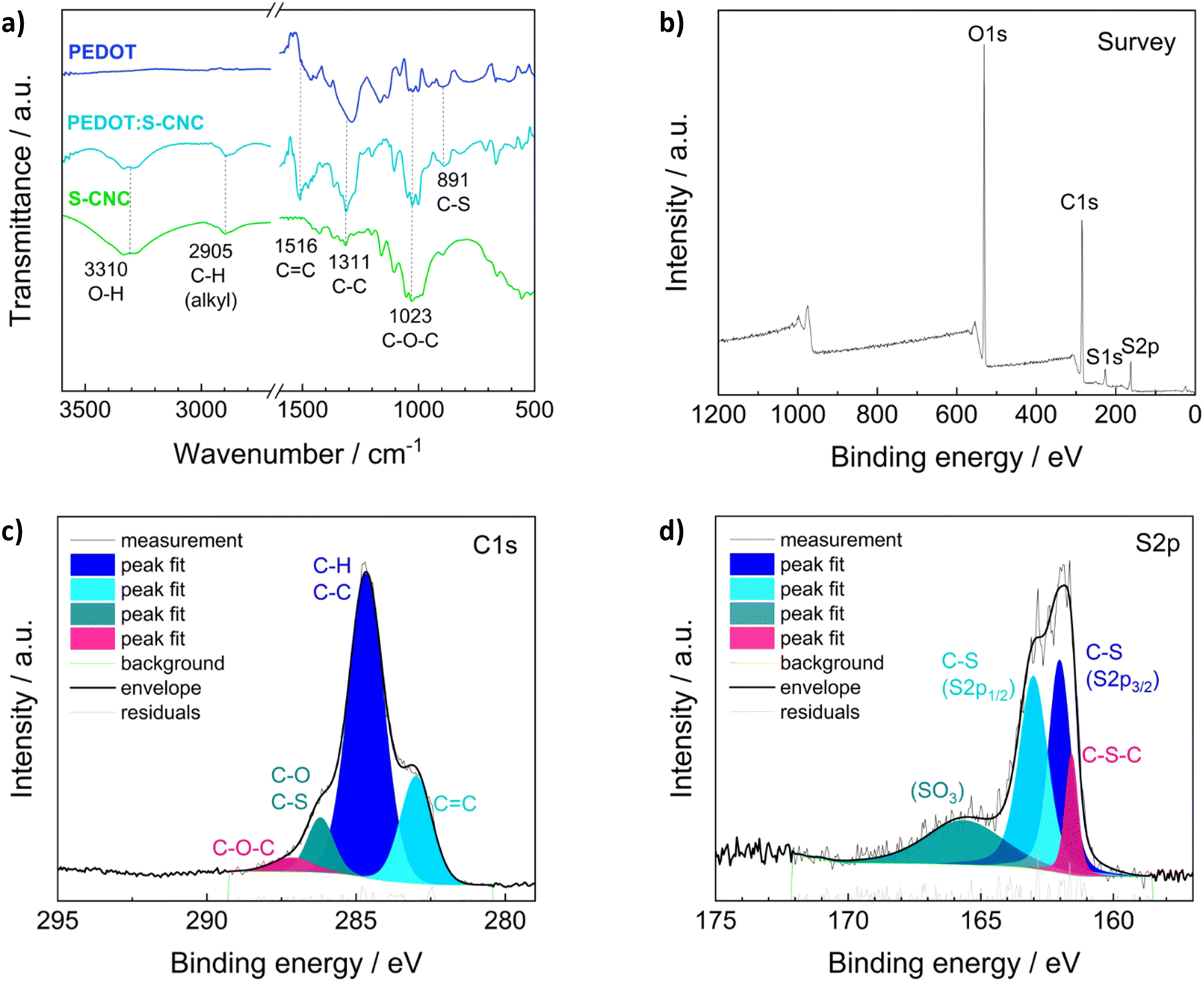 | ||
| Fig. 2 (a) ATR-FTIR spectra for the pristine S-CNCs (green line), the pristine PEDOT (dark blue line) and the composite PEDOT:S-CNCs (turquoise line). (b) XPS survey spectrum of PEDOT:S-CNCs. Peak deconvolution for the (c) C1s and (d) S2p XPS signals of PEDOT:S-CNCs. | ||
The elemental distribution of the composite was obtained from XPS spectra of the biocomposite (Fig. 2b–d). XPS confirmed the presence of the C, O, N, and S elements in PEDOT:S-CNCs samples. The characteristic C 1s core level peaks of PEDOT are C–S (284.7 eV) in the α position, CC–O (286.2 eV) in the β-position and C–O–C (287.1 eV) bonding in the ethylene bridge (Fig. 2c), in a good agreement with the values reported in the literature.37
The S 2p XPS spectra of PEDOT:S-CNCs further confirmed the content of S element (Fig. 2d). The contribution peaks S 2p correspond to sulfur atoms of the PEDOT at 161.8 and 163 eV. Peaks between 165 and 167 eV at higher binding energies are attributed to S-CNCs due to the electronegative oxygen atom attached to the electropositive sulfur atom in the S-CNCs sulfate moiety.21 A complete elemental analysis with the calculation of C, O, and S ratios can be found in the (ESI†), Table S2. We did not observe any residual Fe2+ ions from the oxidant after the polymerization of EDOT (Fig. S2, ESI†).
Raman spectroscopy was recorded to examine the conformational changes of the PEDOT chains in PEDOT:S-CNCs and pristine PEDOT films. The spectra in the 350–1600 cm−1 range are shown in ESI,† Fig. S3a. The bands at 1252 cm−1, 1364 cm−1, and 1414 cm−1 (1410 cm−1 in PEDOT:S-CNCs) correspond to the Cα–Cα, Cβ–Cβ, and symmetric CαCβ stretching vibrations, respectively for PEDOT.38 The peaks at 438 cm−1, 575 cm−1, and 988 cm−1 are associated with the deformation of the oxyethylene ring.38 The strong band at 1400–1470 cm−1 is assigned to CαCβ symmetric stretching on the five-member thiophene ring of PEDOT. In PEDOT:S-CNCs, the peak position of the symmetric stretching vibrations red-shifted from 1414 to 1410 cm−1 and became narrower (ESI,† Fig. S3b). The phenomenon is consistent with the literature and indicates the structural change in PEDOT chains from a benzoid to a quinoid structure in the PEDOT:S-CNCs biocomposite, with respect to pristine PEDOT.39,40 The systematic downshift (4 cm−1) has also been observed by Howikawa et al. for sulfated cellulose-doped PEDOT, and referred to a more extended quinoid formation.40
The presence of the characteristic bands both on FTIR and Raman for PEDOT:S-CNCs suggests that the successful oxidative polymerization in cellulose batches drastically affects the final physical–chemical properties of the composite.
Morphological characterization
The morphologies of the S-CNCs and PEDOT:S-CNCs samples were analyzed by atomic force microscopy (AFM) and scanning electron microscopy (SEM).
Fig. 3a, c and d show the image of a thin-film of S-CNCs prepared from its dilute suspensions. Field emission scanning electron microscopy (FE-SEM) was employed to investigate the size distribution of CNCs, deposited on glass from a 1 mg L−1 dispersion in DMSO (micrograph in Fig. 3a). In this image, the S-CNCs were detected with high magnification (120000×). They can be observed as individual nanocrystals with a rod-like shape with a diameter of ∼10 nm and 151 ± 15 nm length with similar dimensions that have been reported in the literature.41 The particles had a low aspect ratio (length/diameter) at around 15 ± 1.5 nm. SEM shows particle assemblies: small clusters (i) or large groups of oriented S-CNCs (ii). Random arrangement (i) is noted at low concentrations of nanorods. As the number of particles increases, crystals are densely packed in the same direction to form micro-length fibrous bundles (ii). The nanocrystals aggregate mostly laterally, however, there is also longitudinal aggregation.42 The alignment of the nanocrystals can be attributed to the intermolecular interactions due to sulfate and hydroxyl groups on the nanocrystal surface during drying process of the specimen.
 | ||
| Fig. 3 FE-SEM micrographs (120000×) (a) of the pristine S-CNCs30 and (b) the PEDOT:S-CNCs composite. Scale bar 200 nm. Atomic force microscopy (AFM) images of (c) and (d) pristine S-CNCs and (e) the PEDOT:S-CNCs composite (thickness = 250 nm), and (f) PEDOT:PSS (thickness = 260 nm). The photographs of composites thin-films on the right side of the respective AFM images. | ||
The transparent films of cellulose nanocrystals were deposited on mica substrates from aqueous suspensions as a specimen for AFM and on glass for optical microscopy. AFM results indicate the state of aggregation as separated nanocrystal domains with regions where there are no nanocrystals. As seen in Fig. 3c, the nanocrystals are smaller (i) than the features recorded by AFM. The data confirms that the images correlate with agglomerated nanocrystals, which are coarser-diameter and micronbundles (ii), as seen in SEM.
Polarized light microscopy (PLM) provides good insight into optical anisotropy or birefringence. The morphology was monitored via optical microscopy by using a polarized-light source and a representative image is shown in ESI,† Fig. S4. The optical response reveals birefringent S-CNCs film corresponding to micron-sized randomly distributed individual crystallites resulting from the self-organization of cellulose nanocrystals. The sample demonstrates a similar blue appearance of CNCs in solid film, which has been correlated to the anisotropic phase after the removal of residual water.43 PLC shows consistency with the indication of the self-assembly behavior of CNCs in dried film. The detailed structure and orientation of the aggregations were visualized by AFM at higher magnification with scan size at the nanometer level. As seen in small aggregates, particles interact with each other to create coarser-diameter fibers. The sharp edge of the micro-bundles can be recognized in Fig. 3d. Additionally, the image also proves longitudinal aggregation, i.e. between the edges of the nanorods.42
Fig. 3b depicts the FE-SEM micrograph of PEDOT:S-CNCs composite material. The film morphology is significantly different than pristine S-CNCs. Notably, the cationic PEDOT polymer was deposited on the surface of charged S-CNCs, resulting in a continuous-like phase as an indication of template synthesis. The polymer preferentially grows around the negatively charged cellulose, leading to PEDOT-coated CNC nanorods, as previously reported.44 The nanocomposite has an amorphous granular structure with nanometer size. Intensive polymerization is observed in some parts of the nanocomposite (middle), while some S-CNC crystals can be observed within the polymer matrix (bottom left). The morphology has a complex composition featuring a two-phase structure allocated to PEDOT- and S-CNC-rich regions. The two-phase morphology is also a well-known phenomenon for PEDOT:PSS in dry polymer film due to synthesis and post-treatment45,46 Additionally, the degree of microstructural heterogeneity can provide a pathway for overcoming hole-limited doping.47,48 PEDOT:PSS has shown efficient transport, indicating homogenous energy landscape for holes despite microstructural heterogeneity.48
In Fig. 3e, the AFM image of PEDOT:S-CNCs reveals a phase separation structure as islands of bright-regions as was reported for PEDOT:PSS.46 The morphology with globular domains can be correlated to the formation of phase separation with PEDOT-rich regions (bright color) and CNC-rich regions (dark color). The phase separation induces enlarged sizes of phase domains for the new composite, with relatively uniform distribution. The surface height image of PEDOT:PSS shows a smooth surface with small islands that are isolated from each other in Fig. 3f. The root-mean-square (RMS) roughness of PEDOT:S-CNCs (40 nm) is higher than PEDOT:PSS (∼4 nm) indicating that the surface of reference is smoother. The photographs of semi-transparent thin-films for each composite are depicted on the right-hand side of the corresponding AFM images.
OECT devices
In addition to morphological characterization, the electrical properties of the novel biocomposite were investigated to demonstrate its suitability for use in electrochemical transistors. Therefore, conductivity measurements were performed, and the electronic conductivity was found to be around 5 S cm−1 (±1 S cm−1) for the thin films (thickness approx. 250 nm) prepared from dispersed solutions with 94.5% (v/v) PEDOT:S-CNCs dispersion, 0.5% (v/v) 4-dodecylbenezene sulfonic acid (DBSA), 10% (v/v) glycerol and additional 1% (v/v) of (3-glycidyloxypropyl) trimethoxysilan (GOPS) with respect to the total volume of dispersion. Our result is significantly higher than the conductivity value of 1.2 S cm−1 (±0.1) S cm−1 reported by Atifi et al.20 Compared to their study on S-CNCs with 3.5 OSO3H units per 100 glucose units, we obtained the sulfation degree of S-CNCs which was elevated to 5 OSO3H units per 100 glucose units, as derived from the elemental analysis in ESI,† Table S1. This difference can be explained by the varied synthesis conditions20 as well as the additives (GOPs, DBSA, glycerol) introduced into the thin-film. The conductivity results for the respective synthesis method and used additives are shown in detail (in ESI,† Table S3).The synthesized biocomposite's applicability in electronic devices was assessed by using PEDOT:S-CNCs as the channel material in organic electrochemical transistors (OECTs). Usually, adjusting the channel length and width is challenging and requires complex fabrication methods. Traditional lithography is widely employed to fabricate planar OECTs with a channel length down to tens of microns.1–4 To avoid the time-consuming photolithography technique with multi-steps, source and drain electrodes were thermally evaporated through a hard shadow mask, which was designed to shorten the channel length. In this work, OECTs were fabricated by using the device configuration shown in Fig. 4. Additionally, two different source–drain structures were applied, namely a single pair of electrodes and an interdigitated electrode structure with 10 consecutive electrode pairs, as schematically depicted in Fig. 4a and b, respectively. In comparison to our previous work, we have successfully improved the amplitude of the device signal by optimizing the channel geometry.11
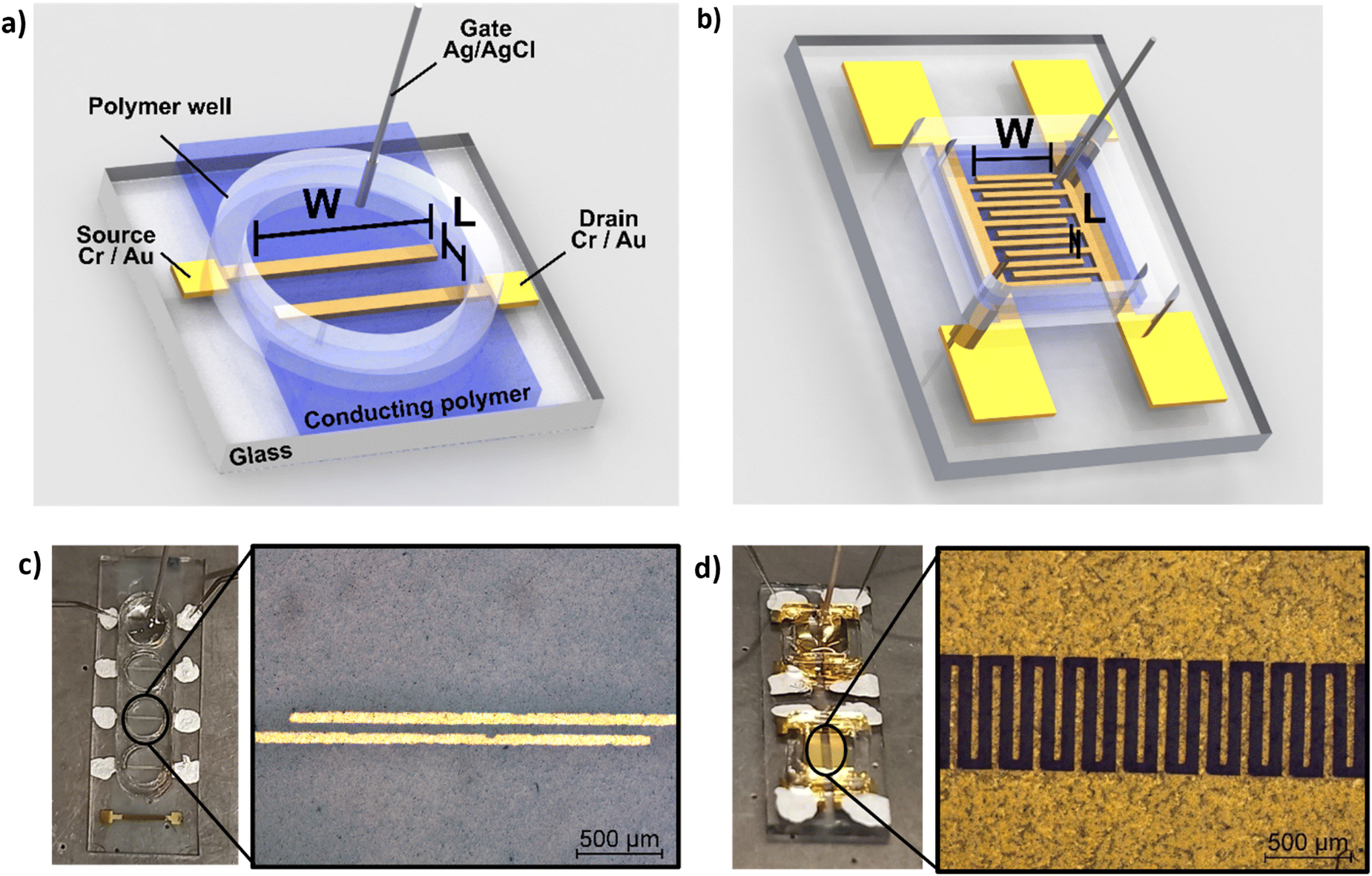 | ||
| Fig. 4 (a) A schematic depiction of the device structure of an PEDOT:S-CNCs based OECT, including Cr/Au source and drain electrodes, PEDOT:S-CNCs as channel material, the polymer well for the electrolyte and the non-polarizable Ag/AgCl gate electrode. W and L are referred to as width and length. (b) Illustration of an interdigitated OECT device, whereby here, only six electrode pairs are shown for exemplary purposes. Image of (c) an operated single-channel OECT and (d) the used interdigitated device structure with ten connected source–drain pairs. Each OECT device picture is depicted in combination with a microscope picture (in 5× magnification) of the respective source–drain electrodes. | ||
The transfer characteristics (IDvs. VG) of PEDOT:S-CNCs are depicted for both source–drain structures in Fig. 5a and b. Additionally, the calculated transconductance (gm = ∂ID/∂VG) is shown next to the respective transfer curve. By increasing gate voltage (VG) to positive values, cations are transferred from the electrolyte into the PEDOT layer, which is consequently de-doped, leading to a state of low to zero conductivity.49 The change in conductivity can be observed with a decrease in drain current (ID) for each electrode architecture. The devices operated in depletion mode represent the p-type MIEC behavior of the channel materials.50 For both device types, the OECT device can be switched “off” by increasing VG to 0.6 V. When comparing the two different electrode structures, the transconductance of an exemplary (with an average device performance) single-channel PEDOT:S-CNCs device exhibits a maximum transconductance (gmax) of 2.39 mS with an on/off ratio of ∼56, whereby an interdigitated device with similar film thickness reaches a gmax of 3.82 mS and an on/off ratio ∼78. Overall, the transconductance values and on/off ratios for the interdigitated channel OECTs are increased by a factor of 1.5 with respect to the single-channel ones. Fig. 5b and d exhibit the output characteristics (IDvs. VD) of PEDOT:S-CNCs devices at applied constant gate voltages. Drain current increases linearly with increased drain voltage (VD), and this consistency in output curves is visible for both device architectures. Once the channel is fully depleted, ID reaches its saturation at VD values of approx. −0.7 V.
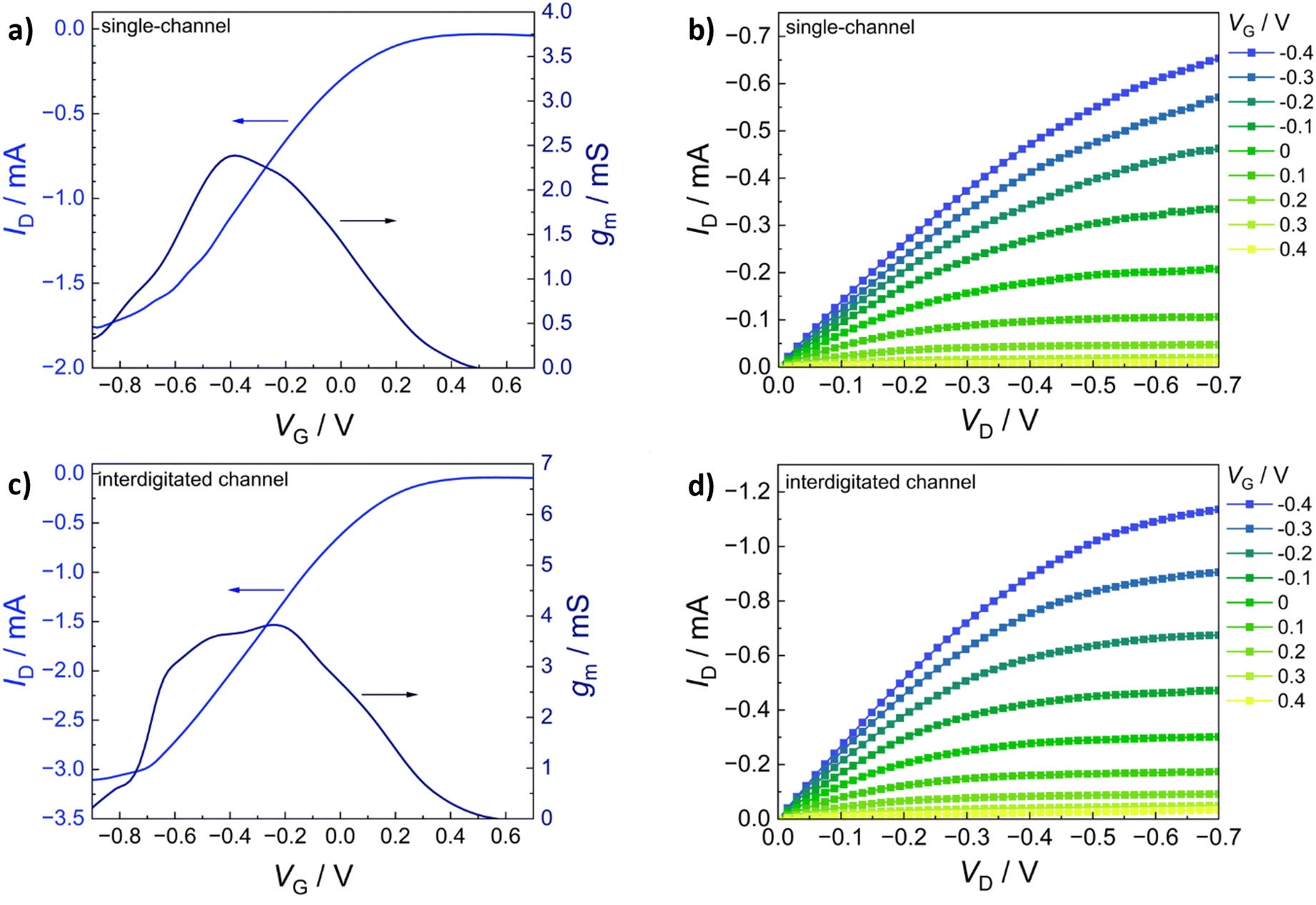 | ||
| Fig. 5 Steady-state electrical characterization of thin-film channel PEDOT:S-CNCs based OECTs. (a) Transfer characteristics and the corresponding transconductance curve (for VD = −0.7 V) and (b) the corresponding output characteristics for a single-channel OECT device (W = 2.15 mm, L = 56 μm, film thickness = 262 nm). (c) Transfer characteristics and transconductance (VD = −0.7 V) and (d) the corresponding output characteristics for an interdigitated OECT device (dimensions of one source–drain electrode pair: W = 530 μm, L = 51 μm, film thickness = 258 nm). | ||
The gmax, the figure of merit of OECT, strongly depends on the channel geometry and is directly proportional to WdL−1 (gmax ∝ WdL−1) in which d refers to channel thickness. In order to enhance the device performance, we further optimized the channel thickness by consecutively depositing multiple layers of the biocomposite, as stated in the experimental section. The thickness of the channel film was conveniently controlled by solution processing to form films with thicknesses in the order of 1–3 μm. The transfer (IDvs. VG) and output characteristics (IDvs. VD) of PEDOT:S-CNCs transistor with the active layer thickness of approx. 2 μm are depicted in Fig. 6a–d. Remarkably, the thick-film samples for both single and interdigitated channel geometries validate the improved gmax values of 13.6 mS and 44.3 mS, respectively. Additionally, these devices have almost identical on/off ratios 83 and 84 for the single channel and interdigitated OECTs, respectively. Since PEDOT:PSS is the most commonly used channel material in bioelectronics studies and it was chosen as the standard reference MIEC for comparison. A similar trend in multiplying transconductance was observed for the reference devices. As depicted in ESI,† Fig. S5, the transconductance value was improved from 27.30 to 56.19 mS when switching from single-electrode pair to interdigitated OECTs. After optimization of device channel parameters, the maximum transconductance values up to 44.3 mS was obtained for PEDOT:S-CNCs which was comparable to ∼56 mS for PEDOT:PSS reference transistors. The results reveal the high potential of PEDOT:S-CNCs composite as a channel material for OECTs.
 | ||
Fig. 6 Steady-state electrical characterization of thick-film channel PEDOT:S-CNCs based OECTs. (a) Transfer characteristics and the corresponding transconductance curve (for VD = −0.7 V) and (b) the corresponding output characteristics for a single-channel OECT device (W = 2.16 mm, L = 57 μm, film thickness ∼ 2 μm). (c) Transfer characteristics and transconductance (VD = −0.7 V) and (d) the corresponding output characteristics for an interdigitated OECT device (dimensions of one source–drain electrode pair: W = 548 μm, L = 50 μm, film thickness ∼ 2 μm). (e) Exemplary illustration of the method used for the evaluation of the threshold voltage (Vth), which is determined by extrapolating the linear fit (red line) of 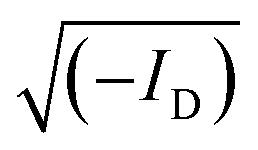 to cross the x-axis. (f) The gmax of the single-channel OECT is plotted as a function of its device geometry and biasing conditions including the width (W), the film thickness (d), the channel length (L), Vth and the gate voltage at the gmax(Vg). Here, reference PEDOT:PSS devices (dark purple) are compared to PEDOT:S-CNCs (light blue) devices. to cross the x-axis. (f) The gmax of the single-channel OECT is plotted as a function of its device geometry and biasing conditions including the width (W), the film thickness (d), the channel length (L), Vth and the gate voltage at the gmax(Vg). Here, reference PEDOT:PSS devices (dark purple) are compared to PEDOT:S-CNCs (light blue) devices. | ||
Fig. 6e shows an exemplary depiction of a transfer curve with the corresponding square root of the absolute value of ID. A linear regression is fitted within the steepest part of the transfer curve and consecutively extrapolated to cross the x-axis to determine the threshold voltage (Vth).51 To evaluate and summarize all device results with thin-film channels (thickness range between 140–290 nm), the maximum transconductance of PEDOT:S-CNCs and PEDOT:PSS single-channel OECTs are plotted versus the respective channel geometry and biasing conditions in Fig. 6f. The data obtained for the reference PEDOT:PSS devices are consistent with previously published results by Inal et al.1Table 1 shows a summary of the most relevant device results obtained for the different channel materials, channel geometry and thickness. The data for the exemplary devices having an average device performance is extracted from Fig. 5 and 6 and Fig. S5 (ESI†) for better comparison. The study emphasizes that the channel current can be modulated by the material type and channel geometry. Therefore, channel geometry engineering is essential to improve device performance.
| Material | Channel geometry | d/nm | g max/mS | On/off ratio |
|---|---|---|---|---|
| d: channel thickness, gmax: maximum transconductance. | ||||
| PEDOT-S-CNC | Single | 262 | 2.39 | 56 |
| Interdigitated | 258 | 3.82 | 78 | |
| Single | ∼2000 | 13.6 | 83 | |
| Interdigitated | ∼2000 | 44.3 | 84 | |
| PEDOT:PSS | Single | 260 | 27.3 | 19 |
| Interdigitated | 265 | 56.2 | 2 | |
In an alternative approach, Tropp et al. investigated the acid-crystalized PEDOT:PSS (ncrys-PEDOT) nanoparticles as conductive fillers for hydrogels to create soft organic biomaterials.52 Moreover, conductive polymer hydrogels performed as electrical bioadhesive interface for potential soft bioelectronics applications.53 Despite recording high conductivities, the residual PSS within ncrys-PEDOT cannot be avoided. In our work, PEDOT:S-CNC nanoparticles demonstrated remarkable conductivity of 5 S cm−1, values larger than lyophilized PEDOT:PSS (4 S cm−1).52 This simple and facile method may overcome the challenges of traditional conductive fillers to stabilize the dispersion of aqueous biomaterial solutions or biomaterial derived hydrogels for biomedical applications.
Conclusions
PEDOT:S-CNCs with 1:1 (w/w) ratio between the monomer and S-CNCs, were synthesized through facile green synthesis. The PEDOT:S-CNCs nanocomposite possesses good ionic and electrical conductivity as well as high dispersibility in water. The electronic conductivity was recorded up to 5 S cm−1. Conformational changes in biocomposite were detected by using spectroscopy techniques and correlated with morphological changes in their micro- and nanostructure via SEM and AFM. The thin-film of biocomposite exhibited granulated PEDOT on the rod-like S-CNCs, resulting in nano-meter scale phase segregation.
The integration of biocomposite as a channel material in OECTs was successfully achieved to evidence ionic-to-electron transduction. The maximum transconductance (gmax) values were obtained up to 13.6 mS and 44.3 mS after optimizing the film thicknesses and device parameters for single-channel and interdigitated OECTs, respectively. High transconductance and device signal have been achieved without applying lithography techniques. To the best of our knowledge, sulfated CNC-templated PEDOT composite was tested in electrochemical transistors for the first time. The results were promising for the development of sustainable and eco-friendly bioelectronics.
As a future perspective, these novel biocomposites can be investigated to enhance mechanical strength, electrical conductivity and long-term biocompatibility in bioelectronics. Moreover, further surface functionalization with specific biomolecules can enhance selectivity and sensitivity for biosensing applications.
Experimental section
Materials
3,4-Ethylenedioxythiophene (EDOT, 99%) was purchased from Thermo Fisher Scientific, while iron(III) p-toluenesulfonate hexahydrate (FeTos), Avicel PH-101 and sulfuric acid 98% were obtained from Sigma-Aldrich. The solution additives 4-dodecylbenezenesulfonic acid (DBSA), glycerol and (3-glycidyloxypropyl) trimethoxysilan (GOPS) were purchased from Fluka, Carl Roth and Grolman, respectively. Commercial poly (3,4-ethylenedioxy thiophene): poly (styrenesulfonate) (PEDOT:PSS, Clevios™ PH 1000) was obtained from Heraeus Electronics GmbH (Hanau, Germany). Prior to usage, the PEDOT:PSS dispersion was filtered through a polyvinylidene fluoride filter with a pore size of 0.45 μm. All other reagents were used as received without further purification.Purified 18 MΩ ultrapure water was obtained by the Arium® Mini laboratory water system.
Synthesis of sulfated cellulose nanocrystals (S-CNCs)
Cellulose nanocrystals were synthesized according to ref. 41. Firstly, 40 mL of deionized water was placed in a 250 mL three-necked round-bottom flask outfitted with a water condenser and a mechanical stirrer. The flask was then chilled in an ice bath before adding 40 mL of concentrated H2SO4. Subsequently, 4 g of Avicel PH-101 was added, and the suspension was heated to 50 °C for 80 minutes. The system was then cooled to room temperature, and the mixture was diluted with water (80 mL) and poured into polypropylene centrifuge tubes. The mixture underwent centrifugation at 4000 rpm repeatedly, with the supernatant being replaced by fresh deionized water each time until a pH > 1 was reached. The precipitate was then suspended in deionized water and dialyzed against distilled water to neutrality using a cellulose nitrate membrane with a molecular weight cut-off (MWCO) of 12400 Da. Then, the suspension was sonicated using a 400 W tip sonicator (Hielscher Ultrasonics) in pulsed mode, operating at 40 W with 0.6-second pulses for 10 minutes. Finally, the suspension was transferred to polypropylene centrifuge tubes and centrifuged at 4000 rpm for 20 minutes. The supernatant was kept and freeze-dried. S-CNCs were isolated in 26% yield.
Synthesis of PEDOT:S-CNCs and PEDOT
The synthesis, applied for the PEDOT:S-CNCs dispersion, follows the procedure described by Tekoglu et al.11 First, S-CNCs dispersion at a total concentration of 8 mg mL−1 was prepared in 18 MΩ water and vigorously stirred at room temperature for 2 h. Next, EDOT was added in a 1:1 (w/w) ratio of monomer to S-CNCs followed by 2 h of additional stirring. Subsequently, 2.5 molar equivalents of iron(III) p-toluene sulfonate hexahydrate (with respect to EDOT) was added. The dispersion was heated at 60 °C for 2 h under continuous stirring. A color change from white to dark blue was already visible after 30 min. After completion of the oxidative chemical polymerization, the solution was diluted with 18 MΩ water, centrifuged at a speed of 5500 rpm for 3 min and re-dispersed in fresh 18 MΩ water. This process was repeated four times until a neutral pH was reached. Then, the dispersion was sonicated for 1 h, followed by a final consecutive centrifugation and re-dispersing step. Next, the dispersion was dialyzed for 24 h by changing the water periodically. Thereby, a dialysis membrane with a molecular MWCO 12–14 kDa was used. Finally, a dispersion of PEDOT:S-CNCs in water with a concentration of approx. 40 mg mL−1 was obtained and stored in the fridge at 4 °C for further usage.
Reference PEDOT synthesis was performed by following the same preparation procedure to compare with the PEDOT:S-CNCs composite using spectroscopy techniques. The oxidative chemical polymerization of tosylate doped PEDOT was also performed using only EDOT with iron(III) p-toluene sulfonate as the oxidant in 2.5 molar equiv. Here, no S-CNCs were included. The tosylate (Tos) anion from the oxidant serves as a counter ion and stabilizes the oxidized cationic PEDOT.
Material characterization
Elemental analyses were carried out using a Vario Micro Cube CHNOS Elemental Analyzer. This device allows for quantitative, fully automated analysis of the elements C, H, N, and S through combustion gas analysis. The oxygen content was calculated by difference. The gases produced during combustion are detected by a thermal conductivity detector (TCD). Approximately 5 mg of the sample is used for each measurement, with each result being the average of two analyses. Calibration was performed using sulfanylamide.X-ray diffraction (XRD) analysis was performed on an X-ray diffraction (XRD) system X'Pert Pro MPD operating in the Bragg–Brentano geometry. The diffractometer was equipped with a copper radiation source (Cu Kα-radiation). The samples were deposited on a zero-background sample holder. The samples were spun at 0.25 Hz. The crystallinity index (CI) of cellulose was calculated from the XRD spectra by the method reported by Segal,31 according to the following equation:
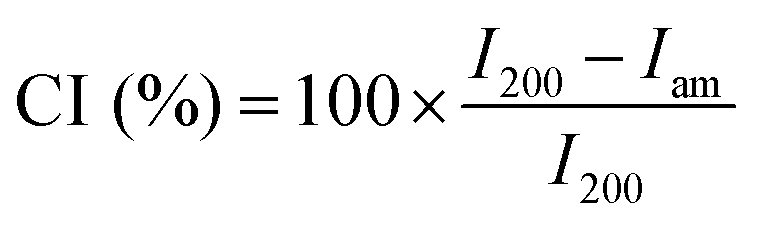
Fourier-transform infrared (FTIR) spectra were recorded utilizing a Bruker Vertex 80 FTIR spectrometer which is equipped with a Bruker Platinum diamond attenuated total reflectance (ATR) unit. The materials were measured after drying.
Raman spectroscopy was conducted using a Bruker MultiRAM FT-Raman spectrometer with an excitation wavelength of 1064 nm in combination with a liquid nitrogen-cooled Ge detector. The spectra were recorded with a resolution of 4 cm−1 and by averaging over 1000 scans. For the sample preparation, aluminum foil stripes were cleaned with acetone and 18 MΩ water in an ultrasonic bath for 15 min each and consequently blow-dried with N2 gas. The Al foil was then attached to a glass slide with double-sided Kapton tape. The PEDOT:S-CNCs dispersion and the pristine PEDOT dispersion was drop-cast onto the substrates and dried.
X-ray photoelectron spectroscopy (XPS) measurements were performed on a Theta Probe XPS-system (Thermofisher, GBR), which features a monochromated Al-Kα X-ray source with an energy of 1486.6 eV. The spot size on the sample surface was 400 μm in diameter.
The hemispherical analyzer was set to a pass energy of 20 eV for the recorded high-resolution (HR) scans at an energy step size of 0.05 eV. The data acquisition and evaluation, as well as the control of the device, are performed via a software package (Avantage) provided by the system manufacturer. For the XPS characterization the original stock dispersions (PEDOT:S-CNCs and PEDOT:PSS) were used without further additives, whereby the dispersions were drop-cast on glass.
Thin-film characterization
For the characterization of the materials, we used glass substrates (1.5 cm × 1.5 cm) and ITO-coated glass (1.27 cm × 1.27 cm), which were cleaned in an ultrasonic bath with a 2% (v/v) aqueous solution of Hellmanex III, deionized water, acetone and isopropanol at 50 °C for 20 min each. Before further usage, the substrates were blow-dried with N2 gas and oxygen plasma treated at 100 W for 5 min in a Plasma Etch PE-24 plasma cleaner. The respective material dispersions were deposited either via drop-casting or spin-coating, depending on the characterization method. For the SEM, AFM and conductivity samples of composites, a mixture of 94.5% (v/v) dispersion (PEDOT:S-CNCs and PEDOT:PSS), 0.5% (v/v) DBSA and 10% (v/v) glycerol, was prepared, whereby subsequently 1% (v/v) GOPS was added into the solution. The composite dispersions were then deposited via spin-coating on clean glass and ITO-coated glass substrates for AFM and SEM, respectively.In addition to the biocomposite dispersion, a dispersion of pristine CNCs in 18 MΩ water was also prepared for SEM and AFM imaging. Nanocrystals were suspended in water at a concentration of 1 mg L−1 and spin-cast onto a mica substrate for AFM, while samples for SEM and polarized light microscopy were drop-cast from the suspension with the same concentration on glass.
Field emission – scanning electron microscopy (FE-SEM) analyses were conducted using a FEI FEG-Quanta 450 instrument (Field Electron and Ion Company, Hillsboro, OR, USA). S-CNCs were deposited onto glass substrates from their DMSO suspension at a concentration of 1 mg L−1. Prior to analysis, the samples were sputtered with platinum. The length of the cellulose nanocrystals was measured using Image J 1.53e software (National Institutes of Health, USA) on micrographs taken at 120000× magnification. The standard deviation was calculated based on the mean of over 30 measurements. PEDOT:S-CNCs samples were deposited on ITO-coated glass substrate and the surface was sputtered with platinum before the analysis.
AFM images of the PEDOT:S-CNCs and PEDOT:PSS were obtained by using the Bruker Innova atomic force microscope. For the analysis of S-CNCs, the mica substrate laden with nanoparticles was examined using a Veeco Instruments Nanoscope IIIa microscope in tapping mode. For the deposition of samples reported in the Fig. 3c and d, a 1 mg mL−1 suspension of nanocrystals in water was prepared by tip-sonication (10 pulses, duty cycle 0.6, power 20 W) using a Dr Hielscher 400 W tip sonicator. The suspension was diluted to a final concentration of 1 mg L−1. Every dilution was followed by bath sonication for 20 minutes. 100 microliter of the 1 mg L−1 solution was cast on a mica substrate and the drop was dried in air under a fume hood. Imaging was conducted with silicon cantilevers having a typical curvature radius of 10–15 nm, operating at frequencies between 264 and 339 kHz. The topographies were processed using the Nanoscope software. Dimensions of the nanocrystals, specifically length (L) and width (W) were measured using Image J 1.53e software from the National Institutes of Health, USA. Statistical analysis was performed on over 50 rod-shaped nanocrystals.
The polarized light microscope image was recorded by using a Nikon Eclipse LV100 ND microscope with a polarizing light filter.
The conductivity of the thin films was measured using a Signatone four-point probe station in combination with a Keithley 2400 Source Meter.
OECT device fabrication
The source–drain electrode pattern was prepared by fixing cleaned glass substrates onto a shadow mask (thickness = 200 μm) to alleviate the shadowing effect during the metal deposition process. 10 nm chromium (Cr) and 100 nm gold (Au) were deposited sequentially by thermal evaporation at a pressure of 1–5 × 10−7 mbar. Here, Cr was utilized to improve the adhesion of Au to the substrate. This process was used to fabricate both the single electrode pair (channel length: L = 60 μm, channel width W = 2 mm) and the interdigitated structure with 10 connected source–drain pairs (each electrode pair: L = 60 μm, W = 540 μm). Afterward, the device channel was prepared by depositing a thin-film of the respective sample dispersion (PEDOT:S-CNCs and PEDOT:PSS) with additives, as mentioned above, via spin-coating using 900 rpm for 40 s, followed by 1200 rpm for 10 s. The area around the channel, as well as the contacts, were carefully wiped by using water-wetted cotton swabs before annealing the substrates at 140 °C for 1 h. In order to increase the film thickness, the spin-coating procedure was repeated whereby additionally a short annealing step of 2 min at 140 °C was employed instead of the final annealing step at 140 °C for 1 h.To complete the devices, a laser-cut polymer well of 3M™ VHB™ Tape (acrylic adhesive with a conformable acrylic foam core) was fixed on the substrates to secure the aqueous electrolyte. For the device characterization, 20 μL of phosphate buffered saline (PBS) solution (50 mM, pH 7.3) was used, and a silver/silver chloride (Ag/AgCl) electrode was employed as a non-polarizable gate electrode. All steady-state current–voltage measurements were performed under ambient conditions using an Agilent model E5273A.
A Bruker Dektak XT profilometer with a stylus force of 2 mg was used to measure the film thickness.
Microscope images for the channel geometries were recorded utilizing a Nikon Eclipse LV100 ND microscope in bright-field mode.
Author contributions
K. M. conducted the experiments, designed the transistor mask, analyzed FTIR and OECT results, prepared the visuals and wrote the manuscript and the subsequent revisions. R. D. performed the synthesis and characterized the nanocrystals and wrote parts of the manuscript and the subsequent revisions. L. S. performed the synthesis and characterized the nanocrystals and wrote parts of the manuscript and the subsequent revisions. F. M. conducted FTIR and Raman experiments, analyzed FTIR data and reviewed the manuscript. M. C. performed XPS experiments, analysed XPS data and reviewed the manuscript. C. P. conducted the experiments for device preparation and reviewed the manuscript. A. O. received the funding, conceived and supervised the project, designed and supervised the experimental section, analysed the data, and wrote and reviewed the manuscript. S. T. received the funding, conceived and supervised the project, designed and supervised the experimental section, analysed the data, the preparation of the manuscript and wrote the final draft and the subsequent revisions.Data availability
The original data generated or analyzed during this study are included in this published article and its ESI† files. This statement ensures transparency and allows for reproducibility, adhering to the standards set by the journal Chemistry of Materials C.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
S. T. acknowledges Johannes Kepler University Linz for LIT – Young Career Project: BIOCOM (Project Nr. LIT-2022-11-YOU-221). R. D. and A. O. acknowledge the University of Pisa for the funding “BIHO 2022 – Bando Incentivi di Ateneo Horizon e Oltre” (Prot. n. 0048740/2022). L. S. acknowledges MUR (Ministero dell’Università e della Ricerca) for the project PON 2014- 2020 (D. M. 1061/2021, CUP I59J21017690008). The authors thank CISUP (Center for Instrument Sharing of the University of Pisa) for the access to the FE-SEM facility. The AFM measurements for pristine S-CNCs samples were undertaken in LGP2, Grenoble, France in the group of Prof. Alain Dufresne. Prof. Niyazi Serdar Sariciftci is gratefully acknowledged for scientific guidance, discussions, mentorship and critically reading the manuscript.References
- S. Inal, G. G. Malliaras and J. Rivnay, Nat. Commun., 2017, 8, 1767 CrossRef PubMed.
- S. Fabiano, L. Flagg, T. C. Hidalgo Castillo, S. Inal, L. G. Kaake, L. V. Kayser, S. T. Keene, S. Ludwigs, C. Muller, B. M. Savoie, B. Lüssem, J. L. Lutkenhaus, M. Matta, D. Meli, S. N. Patel, B. D. Paulsen, J. Rivnay and J. Surgailis, J. Mater. Chem. C, 2023, 11, 14527–14539 RSC.
- P. Gkoupidenis, Y. Zhang, H. Kleemann, H. Ling, F. Santoro, S. Fabiano, A. Salleo and Y. van de Burgt, Nat. Rev. Mater., 2024, 9, 134–149 CrossRef CAS.
- Z. Tian, Z. Zhao and F. Yan, Wearable Electron., 2024, 1, 1–25 CrossRef.
- (a) M. N. Gueye, A. Carella, J. Faure-Vincent, R. Demadrille and J.-P. Simonato, Prog. Mater. Sci., 2020, 108, 100616 CrossRef CAS; (b) S. Inal, J. Rivnay, A.-O. Suiu, G. G. Malliaras and I. McCulloch, Acc. Chem. Res., 2018, 51, 1368–1376 CrossRef CAS PubMed.
- N. K. Guimard, N. Gomez and C. E. Schmidt, Prog. Polym. Sci., 2007, 32, 876–921 CrossRef CAS.
- G. Dijk, A. L. Rutz and G. G. Malliaras, Adv. Mater. Technol., 2020, 5, 1900662 CrossRef CAS.
- J. Rivnay, H. Wang, L. Fenno, K. Deisseroth and G. G. Malliaras, Sci. Adv., 2017, 3, e1601649 CrossRef PubMed.
- Y. Huang, L. Tang and Y. Jiang, CCS Chem., 2024, 1–24 Search PubMed.
- D. Mantione, I. Del Agua, A. Sanchez-Sanchez and D. Mecerreyes, Polymers, 2017, 9, 354 CrossRef PubMed.
- S. Tekoglu, D. Wielend, M. C. Scharber, N. S. Sariciftci and C. Yumusak, Adv. Mater. Technol., 2020, 5, 1900699 CrossRef CAS.
- D. Khorsandi, S. Jenson, A. Zarepour, A. Khosravi, N. Rabiee, S. Iravani and A. Zarrabi, Int. J. Biol. Macromol., 2024, 268, 131829 CrossRef CAS PubMed.
- O. A. T. Dias, S. Konar, A. L. Leão, W. Yang, J. Tjong and M. Sain, Front. Chem., 2020, 8, 420 CrossRef CAS PubMed.
- A. Malti, J. Edberg, H. Granberg, Z. U. Khan, J. W. Andreasen, X. Liu, D. Zhao, H. Zhang, Y. Yao, J. W. Brill, I. Engquist, M. Fahlman, L. Wågberg, X. Crispin and M. Berggren, Adv. Sci., 2016, 3, 1500305 CrossRef PubMed.
- M. Mone, Y. Kim, S. Darabi, S. Zokaei, L. Karlsson, M. Craighero, S. Fabiano, R. Kroon and C. Müller, ACS Appl. Mater. Interfaces, 2023, 15, 28300–28309 CrossRef CAS PubMed.
- D. Tang, S. Y. H. Abdalkarim, Y. Dong and H.-Y. Yu, Carbohydr. Polym., 2023, 311, 120758 CrossRef CAS PubMed.
- (a) F. Brunetti, A. Operamolla, S. Castro-Hermosa, G. Lucarelli, V. Manca, G. M. Farinola and T. M. Brown, Adv. Funct. Mater., 2019, 29, 1806798 CrossRef; (b) A. Operamolla, Int. J. Photoenergy, 2019, 2019, 1–16 CrossRef.
- A. Dufresne, Mater. Today, 2013, 16, 220–227 CrossRef CAS.
- M. M. Langari, M. Nikzad and J. Labidi, Carbohydr. Polym., 2023, 304, 120509 CrossRef CAS PubMed.
- S. Atifi, M.-N. Mirvakili and W. Y. Hamad, ACS Appl. Polym. Mater., 2022, 4, 5626–5637 CrossRef.
- X. Feng, X. Wang, M. Wang, S. Zhou, C. Dang, C. Zhang, Y. Chen and H. Qi, Chem. Eng. J., 2021, 418, 129533 CrossRef CAS.
- E. Korábková, Z. Boeva, K. A. Radaszkiewicz, K. Skopalová, V. Kašpárková, W. Xu, X. Wang, J. Pacherník, A. Minařík, T. Lindfors and P. Humpolíček, Int. J. Biol. Macromol., 2024, 265, 131036 CrossRef PubMed.
- J. Batta-Mpouma, G. Kandhola and J.-W. Kim, Sci. Rep., 2023, 13, 21630 CrossRef CAS PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- (a) V. Hynninen, J. Patrakka and Nonappa, Materials, 2021, 14, 5137 CrossRef CAS PubMed; (b) J. P. F. Lagerwall, C. Schütz, M. Salajkova, J. Noh, J. Hyun Park, G. Scalia and L. Bergström, NPG Asia Mater., 2014, 6, e80–e80 CrossRef CAS.
- (a) C. Tang, S. Spinney, Z. Shi, J. Tang, B. Peng, J. Luo and K. C. Tam, Langmuir, 2018, 34, 12897–12905 CrossRef CAS PubMed; (b) I. Kalashnikova, H. Bizot, B. Cathala and I. Capron, Langmuir, 2011, 27, 7471–7479 CrossRef CAS PubMed.
- N. Lin and A. Dufresne, Nanoscale, 2014, 6, 5384–5393 RSC.
- Z. Kassab, E. Syafri, Y. Tamraoui, H. Hannache, A. E. K. Qaiss and M. El Achaby, Int. J. Biol. Macromol., 2020, 154, 1419–1425 CrossRef CAS PubMed.
- A. Sinha, E. M. Martin, K.-T. Lim, D. J. Carrier, H. Han, V. P. Zharov and J.-W. Kim, Biosyst. Eng., 2015, 40, 373–393 CrossRef.
- L. Spagnuolo, D. Beneventi, A. Dufresne and A. Operamolla, ChemistrySelect, 2024, 9, e202401511 CrossRef CAS.
- L. Segal, J. J. Creely, A. E. Martin and C. M. Conrad, Text. Res. J., 1959, 29, 786–794 CrossRef CAS.
- L. T. Teixeira, W. F. Braz, R. N. Correia de Siqueira, O. G. Pandoli and M. C. Geraldes, J. Mater. Res. Technol., 2021, 15, 434–447 CrossRef CAS.
- (a) E. Madivoli, P. Kareru, A. Gachanja, S. Mugo, M. Murigi, P. Kairigo, C. Kipyegon, J. Mutembei and F. Njonge, Int. Res. J. Pure Appl. Chem., 2016, 12, 1–9 CrossRef; (b) H. L. Nhung and N. D. Thanh, in Proceedings of The 13th International Electronic Conference on Synthetic Organic Chemistry, MDPI, Basel, Switzerland, 2009, p. 231.
- (a) N. Zarrin, H. Tavanai, A. Abdolmaleki, M. Bazarganipour and F. Alihosseini, Synth. Met., 2018, 244, 143–149 CrossRef CAS; (b) S. He, M. Mukaida, K. Kirihara, L. Lyu and Q. Wei, Polymers, 2018, 10, 1065 CrossRef PubMed.
- M. Kiristi, A. U. Oksuz, L. Oksuz and S. Ulusoy, Mater. Sci. Eng., C, 2013, 33, 3845–3850 CrossRef CAS PubMed.
- L. Yue, S. Wang, X. Zhao and L. Zhang, J. Mater. Chem., 2012, 22, 1094–1099 RSC.
- S. K. M. Jönsson, M. P. de Jong, L. Groenendaal, W. R. Salaneck and M. Fahlman, J. Phys. Chem. B, 2003, 107, 10793–10800 CrossRef.
- R. Chen, K. Sun, Q. Zhang, Y. Zhou, M. Li, Y. Sun, Z. Wu, Y. Wu, X. Li, J. Xi, C. Ma, Y. Zhang and J. Ouyang, iScience, 2019, 12, 66–75 CrossRef CAS PubMed.
- (a) I. Alessandri, F. Torricelli, B. Cerea, M. Speziani, P. Romele, Z. M. Kovacs-Vajna and I. Vassalini, ACS Appl. Mater. Interfaces, 2022, 14, 56363–56373 CrossRef CAS PubMed; (b) J. Yang, F. Zeng, Z. S. Wang, C. Chen, G. Y. Wang, Y. S. Lin and F. Pan, J. Appl. Phys., 2011, 110, 114518 CrossRef.
- M. Horikawa, T. Fujiki, T. Shirosaki, N. Ryu, H. Sakurai, S. Nagaoka and H. Ihara, J. Mater. Chem. C, 2015, 3, 8881–8887 RSC.
- A. Operamolla, C. Mazzuca, L. Capodieci, F. Di Benedetto, L. Severini, M. Titubante, A. Martinelli, V. Castelvetro and L. Micheli, ACS Appl. Mater. Interfaces, 2021, 13, 44972–44982 CrossRef CAS PubMed.
- C. Honorato-Rios, C. Lehr, C. Schütz, R. Sanctuary, M. A. Osipov, J. Baller and J. P. F. Lagerwall, NPG Asia Mater., 2018, 10, 455–465 CrossRef CAS.
- R. M. Parker, T. H. Zhao, B. Frka-Petesic and S. Vignolini, Nat. Commun., 2022, 13, 3378 CrossRef CAS PubMed.
- J. Fan, W. Shao, G. Xu, X. T. Cui and X. Luo, RSC Adv., 2014, 4, 24328–24333 RSC.
- M. Modarresi, A. Mehandzhiyski, M. Fahlman, K. Tybrandt and I. Zozoulenko, Macromolecules, 2020, 53, 6267–6278 CrossRef CAS.
- I. Song, N. Yeon Park, G. Seung Jeong, J. Hwan Kang, J. Hwa Seo and J.-Y. Choi, Appl. Surf. Sci., 2020, 529, 147176 CrossRef CAS.
- Y. Wang, S. Wustoni, J. Surgailis, Y. Zhong, A. Koklu and S. Inal, Nat. Rev. Mater., 2024, 9, 249–265 CrossRef CAS.
- S. T. Keene, J. E. M. Laulainen, R. Pandya, M. Moser, C. Schnedermann, P. A. Midgley, I. McCulloch, A. Rao and G. G. Malliaras, Nat. Mater., 2023, 22, 1121–1127 CrossRef CAS PubMed.
- D. A. Bernards and G. G. Malliaras, Adv. Funct. Mater., 2007, 17, 3538–3544 CrossRef CAS.
- S. T. Keene, T. P. A. van der Pol, D. Zakhidov, C. H. L. Weijtens, R. A. J. Janssen, A. Salleo and Y. van de Burgt, Adv. Mater., 2020, 32, e2000270 CrossRef PubMed.
- D. Ohayon, V. Druet and S. Inal, Chem. Soc. Rev., 2023, 52, 1001–1023 RSC.
- J. Tropp, C. P. Collins, X. Xie, R. E. Daso, A. S. Mehta, S. P. Patel, M. M. Reddy, S. E. Levin, C. Sun and J. Rivnay, Adv. Mater., 2024, 36, e2306691 CrossRef PubMed.
- (a) T. Zhou, H. Yuk, F. Hu, J. Wu, F. Tian, H. Roh, Z. Shen, G. Gu, J. Xu, B. Lu and X. Zhao, Nat. Mater., 2023, 22, 895–902 CrossRef CAS PubMed; (b) J. Yu, R. Wan, F. Tian, J. Cao, W. Wang, Q. Liu, H. Yang, J. Liu, X. Liu, T. Lin, J. Xu and B. Lu, Small, 2024, 20, e2308778 CrossRef PubMed; (c) J. Shi, Y. Lin, P. Li, P. Mickel, C. Sun, K. Parekh, J. Ma, S. Kim, B. Ashwood, L. Meng, Y. Luo, S. Chen, H.-M. Tsai, C. M. Cham, J. Zhang, Z. Cheng, J. A. Abu-Halimah, J. Chen, P. Griffin, E. B. Chang, P. Král, J. Yue and B. Tian, Nat. Chem. Eng., 2024, 1, 73–86 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03264k |
| This journal is © The Royal Society of Chemistry 2024 |