
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Developing homogeneous first row early transition metal catalysts for the oxygen reduction reaction
Mary Jo
McCormick
 and
Charles W.
Machan
*
and
Charles W.
Machan
*
Department of Chemistry, University of Virginia, PO Box 400319, Charlottesville, VA 22904-4319, USA. E-mail: machan@virginia.edu
First published on 30th September 2024
Abstract
The oxygen reduction reaction (ORR) remains an important fixture in biological and synthetic systems for energy conversion and chemical functionalization. Late transition metals continue to dominate in the development of new catalyst systems, inspired by well-characterized metallocofactors and prior successes. By comparison, metals to the left of Fe on the periodic table are relatively understudied for the ORR. This Frontier article summarizes advancements related to the use of Mn, Cr, and V in homogeneous catalyst systems for the ORR and discusses the implications of these results for the development of catalyst systems from these metals and those earlier in the transition metal series.
Introduction
The dramatic increase in anthropogenic greenhouse gas emissions, such as carbon dioxide, methane, and nitrogen oxides, since the industrial revolution has resulted in an unprecedented rate of average global surface temperature increases.1 The cumulative detrimental effects of this global warming effect include drought, famine, severe weather patterns, and species extinction.2 It is, therefore, imperative to develop alternative energy sources to meet the growing demands of the population while lowering anthropogenic emissions. One promising option for sustainable energy generation is the hydrogen fuel cell, in which the electrochemical oxidation of hydrogen gas (H2) is coupled to the reduction of oxygen gas (O2) to generate electricity. One of the limiting factors in the commercialization and adoption of fuel cell technology is the often sluggish kinetics associated with the oxygen reduction reaction (ORR) to water, which requires the transfer of four protons and four electrons. The current state-of-the-art catalyst for the ORR is based on platinum nanoparticles; however, this metal is prohibitively expensive and scarce.3 Therefore, there has been a recent emphasis on the development of cheaper transition-metal catalysts that efficiently and selectively reduce dioxygen.Transition metals with open-shell ground states are particularly adept at binding and activating dioxygen due to its triplet ground state.4 Indeed, inspiration for the design of first-row transition metal-based catalysts for the ORR has been continuously drawn from nature, where open-shell transition metal species are frequently employed to activate dioxygen. Some important examples of oxygen-dependent metalloenzyme cofactors include Fe-based enzymes (hemoglobin, cytochrome P450, and cytochrome C oxidase), Cu-based enzymes (tyrosinase and catechol oxidase), and Mn-based enzymes (lipoxygenases, superoxide dismutase).5,6 The majority of the naturally occurring metalloenzymes for the ORR contain a macrocyclic N4 ligand framework; similarly, the overwhelming majority of reported bioinspired catalysts for the ORR consist of macrocyclic N4 ligands such as porphyrins, corroles, or phthalocyanines.7 While recent work has expanded the knowledge of ORR catalysts with non-macrocyclic ligands,8–13 these types of catalysts are underrepresented.
Further, the understanding of ORR catalysts based on early first-row transition metals is lacking. Only a handful of Mn-, Cr-, and V-based homogeneous catalysts have been reported for the ORR.5,7 This Frontier article focuses on connecting known examples of metal complexes with these metal centers serving as ORR catalysts in non-aqueous solvents and suggests future strategies for new catalyst development. Readers interested in more comprehensive summaries on homogeneous ORR should consult more extensive subject reviews.4,7
Mn-based catalysts for the ORR
The first deliberate study on the ORR with a Mn-based complex was reported in 1989 by Fukuzumi and co-workers on Mn(III) 5,10,15,20-tetrakisphenylporphyrin ([Mn(TPP)]+).14 Using a metallocene reductant with perchloric acid (although reaction selectivity was not quantified), this report noted that the Mn-based catalyst was slower than the Co-based one, despite a more negative reduction potential. Generally, should a reaction proceed through a common mechanistic pathway, more reducing potentials should result in greater catalyst activity.15 This observation was initial evidence of divergent reaction pathways for Mn with respect to Co active sites in porphyrin ligand frameworks. Later studies led by Abu-Omar and Fukuzumi found that Mn(III) 5,10,15-tris(pentafluorophenyl)corrole [Mn(tfpc)] could also serve as an effective catalyst with metallocene reductants and trifluoroacetic acid, producing H2O2 as the primary product.16 Interestingly, in the case of the complex with the dianionic TPP ligand,14 redox cycling should primarily occur through Mn(III)/Mn(II) states, while for the complex based on the trianionic tpfc ligand, a Mn(IV)/Mn(III) redox process was proposed to be the basis of catalytic activity.In 2015, Duboc and co-workers published a sulfur-bridged bimetallic MnII dimer with a bipyridine-based ligand framework, [MnII2(LS)(LSH)](ClO4), where LS = (2,2′-(2,2′-bipyridine-6,6′-diyl)bis(1,1-diphenylethanethiolate)), that reduces O2 to H2O2 with about 80–84% efficiency in the presence of 2,6-lutidinium tetrafluoroborate as a proton donor and either octamethylferrocene or decamethylferrocene as the sacrificial reductant (Fig. 1).17 Key components of the proposed catalytic cycle include a persistent Mn dimer species (aided by the thiolate ligands serving as a bridging scaffold) and one of the thiolate ligands serving as a proton relay during the course of the catalytic reaction. Notably, under stoichiometric conditions, H2O was the primary product (four-electron process), while the catalytic system showed H2O2 as the primary product (two-electron process). It was proposed that O–O bond scission was avoided under catalytic conditions by maintaining relatively acidic conditions near the active site, since this favors protonation of M–O bonds and therefore H2O2 release.
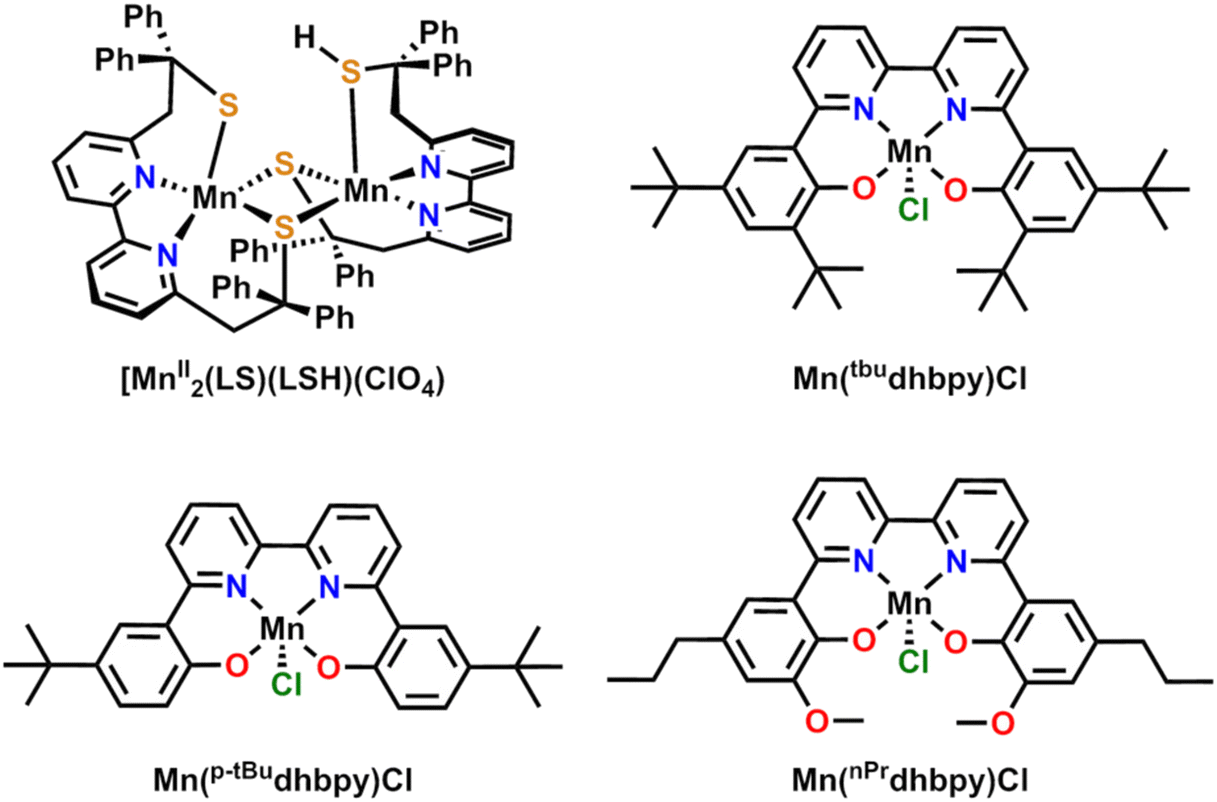 | ||
| Fig. 1 Previously published Mn-based polypyridyl complexes for the ORR. | ||
Following this example, our group has since published several additional Mn bipyridine-based homogenous catalyst systems for the ORR (Fig. 1). The parent catalyst Mn(tbudhbpy)Cl, where 6,6′-di(3,5-di-tert-butyl-2-phenolate)-2,2′-bipyridine = [tbudhbpy]2−, exhibits an O2 activation pathway involving metal reduction accompanied by ligand protonation to rapidly convert O2 to H2O2 in the presence of PhOH or 2,2,2-trifluoroethanol (TFEOH) under electrochemical conditions.18 During the catalytic cycle, the Mn center is reduced from MnIII to MnII while the phenolate moiety is protonated by the proton donor prior to dioxygen binding to form a superoxide adduct. A second reduction coupled with an intramolecular proton transfer from the ligand-based phenolate moiety to the intermediate Mn superoxide generates a metal-bound hydroperoxo species. The catalytic cycle is primarily closed upon the protonation of the Mn–O bond to release H2O2 (81 ± 4%).
It is noteworthy that the ligand heteroatoms in the Mn(tbudhbpy)Cl catalyst emulate the function of the thiolate groups of the Duboc catalyst described above as proton relays. The soft S bases in the ligand used by Duboc required the use of the stronger acid 2,6-lutidinium (pKa(MeCN) = 14.16),19 as compared to our work using PhOH (pKa(MeCN) = 29.14)20 and TFEOH (pKa(MeCN) = 35.4).21 Although the thermodynamics of proton activity are markedly different, the role of the metal-bound heteroatoms appears to enforce a kinetic preference to H2O2 for reaction selectivity in both cases. The absence of dimerization for the Mn(tbudhbpy)Cl catalyst is likely the result of the steric profile of the tert-butyl functionalized phenolate fragment of the ligand, prohibiting Mn–O–Mn bridging. Unfortunately, it is challenging to speculate on the origin of the shared product selectivity between the dimer species of Duboc and co-workers and our Mn(tbudhbpy)Cl catalyst, given that bridging Mn–O2–Mn species were speculated to exist but not detected in the former case.
More recently, we have explored the effect of incorporating pendent proton relays into the ligand framework on the selectivity of the ORR mediated by this family of Mn-based catalysts (Fig. 1).22 The pendent relay had the notable effect of increasing the efficiency for H2O2 (Mn(nPrdhbpy)Cl 96.2 ± 4.1%) under spectrochemical conditions relative to a control complex (Mn(p-tBudhbpy)Cl 64.2 ± 6.9%) in MeCN. These studies used a cationic acid (N,N′-diisopropylethylammonium hexafuorophosphate (DIPEAHPF6)), which showed a strong attraction to the metal complex during catalysis. The strength of this interaction could be attenuated by introducing the conjugate base of the alkylammonium acid, which revealed that the overall selectivity of both systems could be governed by controlling the protonation of the Mn–O bond of the intermediate Mn(III) hydroperoxo species. Consistent with this, adding the conjugate base diisopropylamine to the reaction mixture to buffer the proton donor shifted reaction selectivity away from H2O2 to H2O for both complexes by enabling access to an alternative dimerization pathway. It is proposed that dimerization is possible with Mn(nPrdhbpy)Cl due to the greatly decreased steric profile of the –OMe groups ortho to the Mn-coordinated O atoms compared to the tert-butyl groups in the analogous Mn(tbudhbpy)Cl complex, vide supra (Fig. 1).
Cr-based catalysts for the ORR
The only reported Cr-based molecular catalyst for ORR was reported by Liu et al. in 2014.23 This study showed that chromium(V)–oxo tris(pentafluorophenyl)corrole, [(tpfc)CrV(O)], efficiently reduced O2 in the presence of octamethylferrocene (Me8Fc) and 2,2,2-trifluoroacetic acid (TFA). The selectivity of the catalytic reaction was found to be dependent on the amount of acid present. With stoichiometric amounts of TFA present, the reaction favored the production of H2O, while an excess of TFA shifted the reaction selectivity towards H2O2. The shift in selectivity to favor H2O2 was attributed to rapid proton-coupled electron transfer (PCET) involving Me8Fc and TFA to a superoxide intermediate, [(tpfc)CrIV(O2˙−)], to produce a hydroperoxide intermediate, [(tpfc)CrIV(OOH)], which only occurred in the presence of excess TFA. Under these conditions, reduction to the Cr(III) active species was rapid enough that O2 binding became the rate-limiting step. The authors noted that the dependence of reaction selectivity on proton concentration is a unique feature of the early metal catalyst used here, since the Cr(III) complex can mediate aprotic O–O bond scission to generate the parent oxo species [(tpfc)CrV(O)] in this coordination environment.V-based catalysts for the ORR
Reports of V-based molecular catalysts for the ORR are limited to a collection of salen and related Schiff-base supported systems.7 Yamamoto et al. reported in 1996 that (μ-oxo)bis[(N,N′-ethylenebis(salicylideneaminato))vanadium(IV)] tetrafluoroborate ([O(V(salen))2][BF4]2) could reduce O2 to H2O with over 90% selectivity under electrochemical conditions with an excess of TFA present in MeCN solution.24 Although this initial study proposed that the bimetallic species persisted throughout the catalytic cycle, later studies found that the bimetallic species was actually a precursor to a monomeric active species [VIII(salen)]+ that could selectively reduce O2 to H2O under electrochemical conditions in the presence of excess triflic acid in CH2Cl2 (Fig. 2A).25,26 | ||
| Fig. 2 (A) Proposed catalytic cycle for V(salen) complexes, (L = Schiff base ligand). (B)–(D) Structures of V(IV) oxo complexes studied by Liu and Anson.22,27 | ||
The instability of the [VIII(salen)]+ complex in the presence of acid and the observed slow rate of reaction with O2 led Liu and Anson to develop a series of substituted Schiff base ligands for the study of VIII compounds that react with dioxygen (those soluble and stable under electrocatalytic conditions with added acid had a phenyl backbone – rather than alkyl or 1,8-naphthyl – and are depicted in Fig. 2B–D).27 The additional derivatives followed the same proposed catalytic cycle as the initial report, which is initiated by two VIVO complexes undergoing a proton-assisted disproportionation to form a [VIII]+ compound, a [VVO]+ compound, and H2O. Subsequently, two equivalents of the [VIII]+ compound bind one molecule of O2 in a bridging peroxo fashion before mediating homolytic O–O bond scission to generate two equivalents of the [VVO]+ compound. The [VVO]+ compound can then be reduced back to the initial VIVO compound at mild potentials, completing the catalytic cycle.
Interestingly, analogous to the Cr-based example discussed above, oxo species become relevant intermediates with vanadium active sites in salen-type ligand frameworks. However, although the possibility of a vanadium-dioxygen adduct is invoked in the mechanistic analysis, there is no implication of an intermediate hydroperoxo species as was observed with the Cr(corrole) derivative of Liu et al.23 despite the use of a stronger acid. An analogous dimerization reaction involving O–O bond scission does occur for the Cr(corrole) derivative in the absence of a proton source, similar to the reaction described for the V(salen) derivatives with acid present. Given that the ligand frameworks differ in overall charge, it can be speculated that the dianionic salen-type ligand scaffold does not produce sufficiently basic vanadium-(super)oxo adducts to favor protonation. Thus, the trianionic nature of the corrole ligand framework appears to be crucial for the selectivity difference seen for the Cr active site. The instability of many of the examined vanadium-based salen derivatives with respect to acid also suggests that the composition of the ligand framework must address stability while also increasing the basicity of metal-bound reactive oxygen species to improve V-mediated ORR performance.
Possible future strategies for early transition metal catalyst systems for ORR
Ligand design for O2 binding
O2 is a paramagnetic species with a triplet ground state based on the partial occupancy of degenerate π* antibonding orbitals, resulting in a double bond overall. This electronic configuration makes O2 a strong π-acid, and it often undergoes formal reduction to generate a superoxide or peroxide ligand upon binding to a metal center.4 Therefore, facile O2 binding is often achieved at metal centers with unpaired electrons of relevant energy and frontier orbital symmetry to that of O2.3 Further, whether O2 binds to a monometallic complex in a η1 or η2 fashion generally dictates whether formal reduction occurs by 1e− or 2e− electrons, respectively. Therefore, the active site must have both a sufficient d-electron count and accessible higher oxidation states for O2 binding to occur.7Early transition metals (Ti, V, Cr triads) have adequate reducing power to react with O2, and significant effort has been spent in understanding how to leverage this reactivity in organometallic complexes for oxidative transformations of hydrocarbons. However, the accessibility of high formal oxidation states for these elements (greater than M3+) and strong binding to the intermediates of O2 reduction result in stable metal oxygen species, rendering many reactions stoichiometric in nature.5 Indeed, for organometallic and alkoxide complexes of early metals which can react with dioxygen, a limiting factor is the inability to close the reaction cycle and regenerate the starting material. The lack of recyclability can arise from the low basicity of the early metal oxo species, the propensity for disproportionation reactions, or the stability of μ-oxo dimer species.28–35
In order to develop compounds that catalytically activate O2, initial M–O2 intermediates must be destabilized to allow for the scission of the O–O bond and release of products. Further, the propensity to create reactive monomeric oxo species as the result of O2 activation can lead to unintended ligand activation. Polypyridine frameworks could present an attractive configuration for future development, given the ability to sterically protect potentially reactive bonds, as well as the precedent for the reactivity of Ti(III) and V(III) with O2 in the presence of this ligand type (Fig. 3A).36–38 For instance, in the case of V(III), more electron-rich pyridine ligands accelerate the reaction, offering insight into possible future ligand design.37 For Ti(III), the lability of the coordination environment in the d1 configuration was proposed to inhibit the rate of reaction with O2, which could be addressed through the denticity or charge of the ligand framework (Fig. 3B). Notably, polypyridine multidentate ligand frameworks can support Cr centers for the catalytic disproportionation of H2O2 to O2 and H2O, which involves reaction steps that mirror those required for the ORR.35 Thus, in comparison to the non-recyclability, low basicity, or inertness of complexes supported by organometallic or alkoxide ligands, polypyridyl-based ligand frameworks could offer possible advantages for early metal ORR catalysts.
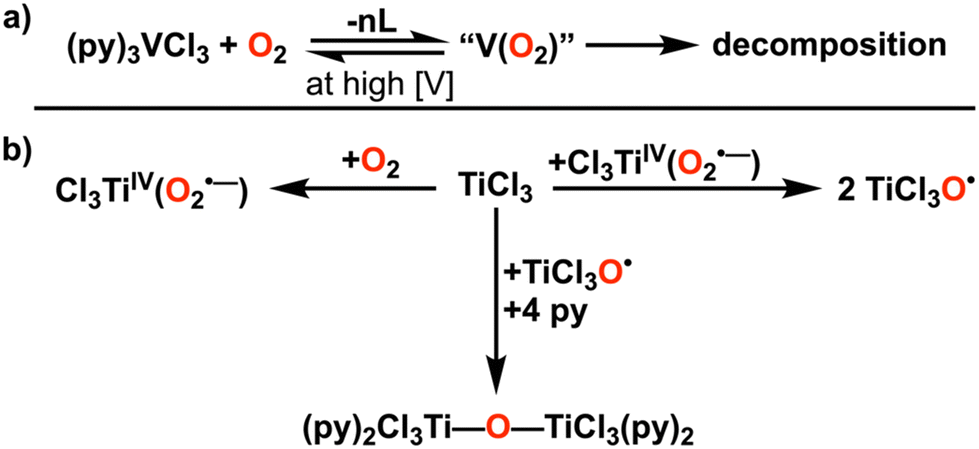 | ||
| Fig. 3 (A) Summary of the stoichiometric reactions observed between vanadium(III) chloride and dioxygen in pyridine and (B) titanium(III) chloride and dioxygen in pyridine. | ||
Modifying the inner-coordination sphere and the ligand framework is one way to tune the electronic structure of the metal center to optimize the strength and orientation of the M–O2 binding interaction, as well as disfavor dimerization through sterics.5,36,39 It is also possible to modulate catalyst design through the secondary coordination sphere. There are a multitude of reports discussing the benefit of including a hydrogen-bonding site in the secondary coordination sphere for the cleavage of the O–O bond.4,7,13,22 The inclusion of a hydrogen-bonding site, like those in “hangman” motifs, has led to improved selectivity and increased rates of catalysis in a variety of transition-metal based catalysts by altering the binding mode of O2 and increasing the rates of intramolecular proton transfer steps (Fig. 4).22,40,41 In the case of Cr-based catalysts like the metallocorrole described above, this would seem to offer substantial benefits for accelerating the rate-determining O2 binding step by increasing its thermodynamic favorability. Likewise, such a motif could also benefit V-based complexes by stabilizing monomeric O2 adducts with respect to dimerization, thus shifting the balance of product selectivity between H2O2 and H2O by altering the basicity of V–O bonds in presumptive intermediate species.
 | ||
| Fig. 4 Mn(porphyrin)-based catalysts examined by Nocera and co-workers. Non-covalent interactions with hydrogen bond donors were proposed to impact the equilibrium resonance structure of the Mn–O2 adduct. The inclusion of pendent proton sources in the hangman ligand framework was reflected in changes in the observed rate law at low acid concentrations; HA indicates added Brønsted acid or pendent proton donor. Adapted from ref. 39 with permission from the Royal Society of Chemistry, copyright 2022. | ||
Additives for tuning activity
Along with ligand design, the use of additives has been repeatedly shown to impact the activity of molecular catalysts for the ORR. The use of benzoquinone and Fe- and Co-based macrocyclic complexes to mediate redox activity in Pd-catalyzed oxidation reactions has been long known, but Stahl and co-workers were the first to use p-hydroquinone (H2Q) as a non-covalently bound electron–proton transfer mediator (EPTM) with Co(salophen) for the electrochemical ORR in 2016.42 Mechanistic studies into the co-catalytic system revealed that the combination of Co(salophen) and H2Q resulted in an increase in reaction rate as well as a shift in selectivity from H2O2 to H2O due to sequential HAT and PCET reactions from p-hydroquinone to the Co-superoxide intermediate followed by a subsequent H+/e− transfer from a second equivalent of H2Q.43 Our group reported a similar shift in selectivity when p-benzoquinone (BQ) was used as a redox mediator with our previously reported Mn catalyst [(MntBudhbpy)Cl, where tBudhbpy2− = 6,6′-di(3,5-di-tert-butyl-2-phenolate)-2,2′-bipyridine].32 Here, too, selectivity for the electrocatalytic reduction of O2 in the presence of TFEOH is shifted from H2O2 to H2O upon the addition of benzoquinone, and the co-catalytic system produced a higher current density than the catalyst or benzoquinone alone due to a shift in electrochemical mechanism to include non-covalent interactions between the proton donor and reduced BQ anions. In the context of polypyridyl Cr, V, and Ti complexes that reductively bind O2, the weak sacrificial bonds of these additives could similarly drive catalytic reduction if the relevant bond dissociation free energies for putative intermediates in the catalytic cycle can be measured or estimated.44In addition to the use of EPTMs, redox-inert Lewis Acids have been shown to shift the selectivity of the ORR by molecular catalysts by impacting both the O2 coordination mode to the active site and the rate of electron transfer from the metal to the substrate.45,46 A quantitative shift in selectivity was reported by Fukuzumi and co-workers in 2015, when they showed that the addition of Sc(OTf)3 to a Cu-complex, [(tmpa)CuII(CH3CN)](ClO4)2 (tmpa = tris(2-pyridylmethyl)amine), resulted in a shift from the four-electron reduction of O2 to H2O to an overall two-electron pathway to quantitatively produce [ScIII(O22−)]+ during each turnover (Fig. 5).47,48 A dramatic rate enhancement of O2 activation was also reported by Borovik and co-workers in 2011 and 2013 as a result of the addition of redox-inert Lewis Acids to both [FeIIMST]− and [MnIIMST]−, where MST = N,N,-[2,2,2-nitrilotris(ethane-2,1-diyl)]tris-(2,4,6-tri-methylbenzene-sulfonamido).49,50 The rate of O2 activation by [FeIIMST]− was enhanced by a 34- to 36-fold increase in the presence of M(OTf)2/15-crown-5 (M = SrII or CaII, respectively) compared to the rate observed for [NMe4][FeIIMST]. Similarly, the addition of CaII resulted in a 35-fold increase in the rate of O2 activation by [MnIIMST]− compared to [NMe4][MnIIMST].49,50 Since these reactions rely on intramolecular electron transfer to produce reactive oxygen species, it can be surmised that the added ions function to lower the reduction potential required to form these intermediates through some combination of charge and basicity changes. It is likely for early metals like Cr, V, and Ti that strong Lewis Acids will be required to achieve similar effects, given to the relatively low intrinsic basicity of the intermediates noted in the discussion above of the limited examples of catalytic systems. Nonetheless, should any alternative binding modes be relatively favored, Lewis Acids could assist in flattening the potential energy surface to achieve catalytic turnover.
 | ||
| Fig. 5 Proposed catalytic cycles for the reduction of dioxygen by [CuII(tmpa)(CH3CN)](HClO4)2, where (tmpa = tris(2-pyridylmethyl)amine), in the presence of Brønsted Acids (left) and Lewis Acids (right). The mechanistic differences between the two cycles result in a quantitative shift in selectivity from water (Brønsted) to hydrogen peroxide (Lewis). Adapted from ref. 47 and 48. | ||
Conclusions and outlook
Despite the relevance of early transition metal active sites in biological systems for the activation and transportation of dioxygen, the synthetic development of homogeneous catalysts for these reactions has primarily focused on late transition metals such as platinum and iron. This Frontier article summarized the few reports of Mn-, Cr-, and V- based homogeneous catalysts for ORR and the implications of these reports that can inform the development of new catalysts. The early transition metals (Cr, V, and Ti) are known to reduce O2 in a variety of ligand configurations via stoichiometric reactions, yet the products of these reactions have been too stable in most cases to allow for catalytic turnover. Based on the discussion above, we speculate that a combination of intentional ligand design (increased polypyridine denticity, negative overall ligand charge, and steric profile), as well as the inclusion of additives that destabilize these M–O2 intermediates (e.g., hydrogen-bonding motifs and Lewis acids) or offer alternative reaction pathways (e.g., EPTMs) could enable the development of efficient and selective homogeneous catalysts for the ORR based on early transition metals.Data availability
The data supporting this article have been included within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge N.S.F. CHE-2348515 for support.References
- M. Z. Jacobson, 100% Clean, Renewable Energy and Storage for Everything, Cambridge University Press, 2020 Search PubMed.
- Routledge Handbook of Climate Change and Health System Sustainability, ed. J. Braithwaite, Y. Zurynski and K. Carolynn, Taylor & Francis, 2024 Search PubMed.
- J. Luo, X. Tian, J. Zeng, Y. Li, H. Song and S. Liao, Limitations and Improvement Strategies for Early-Transition-Metal Nitrides as Competitive Catalysts toward the Oxygen Reduction Reaction, ACS Catal., 2016, 6, 6165–6174, DOI:10.1021/acscatal.6b01618.
- C. W. Machan, Advances in the Molecular Catalysis of Dioxygen Reduction, ACS Catal., 2020, 10, 2640–2655 CrossRef CAS.
- E. N. Cook and C. W. Machan, Bioinspired mononuclear Mn complexes for O2 activation and biologically relevant reactions, Dalton Trans., 2021, 50(46), 16871–16886, 10.1039/d1dt03178c.
- S. Sahu and D. P. Goldberg, Activation of Dioxygen by Iron and Manganese Complexes: A Heme and Nonheme Perspective, J. Am. Chem. Soc., 2016, 138(36), 11410–11428, DOI:10.1021/jacs.6b05251.
- M. L. Pegis, C. F. Wise, D. J. Martin and J. M. Mayer, Oxygen Reduction by Homogeneous Molecular Catalysts and Electrocatalysts, Chem. Rev., 2018, 118, 2340–2391, DOI:10.1021/acs.chemrev.7b00542.
- A. Santra, A. Das, S. Kaur, P. Jain, P. P. Ingole and S. Paria, Catalytic reduction of oxygen to water by non-heme iron complexes: exploring the effect of the secondary coordination sphere proton exchanging site, Chem. Sci., 2024, 15(11), 4095–4105, 10.1039/d3sc06753j.
- L. Wang, M. Gennari, F. G. Cantú Reinhard, J. Gutiérrez, A. Morozan, C. Philouze, S. Demeshko, V. Artero, F. Meyer, S. P. De Visser and C. Duboc, A Non-Heme Diiron Complex for (Electro)catalytic Reduction of Dioxygen: Tuning the Selectivity through Electron Delivery, J. Am. Chem. Soc., 2019, 141(20), 8244–8253, DOI:10.1021/jacs.9b02011.
- X. Lu, Y.-M. Lee, M. Sankaralingam, S. Fukuzumi and W. Nam, Catalytic Four-Electron Reduction of Dioxygen by Ferrocene Derivatives with a Nonheme Iron(III) TAML Complex, Inorg. Chem., 2020, 59, 18010–18017, DOI:10.1021/acs.inorgchem.0c02400.
- Y.-H. Wang, B. Mondal and S. S. Stahl, Molecular Cobalt Catalysts for O2 Reduction to H2O2: Benchmarking Catalyst Performance via Rate−Overpotential Correlations, ACS Catal., 2020, 10, 12031–12039, DOI:10.1021/acscatal.0c02197.
- Y.-H. Wang, Z. K. Goldsmith, P. E. Schneider, C. W. Anson, J. B. Gerken, S. Ghosh, S. Hammes-Schiffer and S. S. Stahl, Kinetic and Mechanistic Characterization of Low-Overpotential, H2O2–Selective Reduction of O2 Catalyzed by N2O2–Ligated Cobalt Complexes, J. Am. Chem. Soc., 2018, 140, 10890–10899, DOI:10.1021/jacs.8b06394.
- A. W. Nichols, E. N. Cook, Y. J. Gan, P. R. Miedaner, J. M. Dressel, D. A. Dickie, H. S. Shafaat and C. W. Machan, Pendent Relay Enhances H2O2 Selectivity during Dioxygen Reduction Mediated by Bipyridine-Based Co−N2O2 Complexes, J. Am. Chem. Soc., 2021, 143, 13065–13073, DOI:10.1021/jacs.1c03381.
- S. Fukuzumi, S. Mochizuki and T. Tanakalb, Efficient Reduction of Dioxygen with Ferrocene Derivatives, Catalyzed by Metalloporphyrins in the Presence of Perchloric Acid, Inorg. Chem., 1989, 28(12), 2459–2465, DOI:10.1021/ic00311a042.
- C. Costentin and J.-M. Savéant, Homogeneous Molecular Catalysis of Electrochemical Reactions: Manipulating Intrinsic and Operational Factors for Catalyst Improvement, J. Am. Chem. Soc., 2018, 140, 16669–16675, DOI:10.1021/jacs.8b09154.
- J. Jung, S. Liu, K. Ohkubo, M. M. Abu-Omar and S. Fu, Catalytic Two-Electron Reduction of Dioxygen by Ferrocene Derivatives with Manganese(V) Corroles, Inorg. Chem., 2015, 54, 4285–4291, DOI:10.1021/ic503012s.
- M. Gennari, D. Brazzolotto, J. Pécaut, M. V. Cherrier, C. J. Pollock, S. Debeer, M. Retegan, D. A. Pantazis, F. Neese, M. Rouzières, R. Clérac and C. Duboc, Dioxygen Activation and Catalytic Reduction to Hydrogen Peroxide by a Thiolate-Bridged Dimanganese(II) Complex with a Pendant Thiol, J. Am. Chem. Soc., 2015, 137(26), 8644–8653, DOI:10.1021/jacs.5b04917.
- S. L. Hooe, A. L. Rheingold and C. W. Machan, Electrocatalytic Reduction of Dioxygen to Hydrogen Peroxide by a Molecular Manganese Complex with a Bipyridine-Containing Schiff Base Ligand, J. Am. Chem. Soc., 2018, 140, 3232–3241, DOI:10.1021/jacs.7b09027.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, On the Basicity of Organic Bases in Different Media, Eur. J. Org. Chem., 2019, 2019(40), 6735–6748, DOI:10.1002/ejoc.201900956.
- A. Kütt, V. Movchun, T. Rodima, T. Dansauer, E. B. Rusanov, I. Leito, I. Kaljurand, J. Koppel, V. Pihl and I. Koppel, et al., Pentakis(trifluoromethyl)phenyl, a Sterically Crowded andElectron-withdrawing Group: Synthesis and Acidity ofPentakis(trifluoromethyl)benzene, -toluene, -phenol, and -aniline, J. Org. Chem., 2008, 73, 2607–2620, DOI:10.1021/jo702513w.
- Y. C. Lam, R. J. Nielsen, H. B. Gray, A. William and I. Goddard, A Mn Bipyrimidine Catalyst Predicted To Reduce CO2 at Lower Overpotential, ACS Catal., 2015, 5, 2521–2528, DOI:10.1021/cs501963v.
- E. N. Cook, I. M. Courter, D. A. Dickie and C. W. Machan, Controlling product selectivity during dioxygen reduction with Mn complexes using pendent proton donor relays and added base, Chem. Sci., 2024, 15(12), 4478–4488, 10.1039/d3sc02611f.
- S. Liu, K. Mase, C. Bougher, S. D. Hicks, M. M. Abu-Omar and S. Fukuzumi, High-Valent Chromium−Oxo Complex Acting as an Efficient Catalyst Precursor for Selective Two-Electron Reduction of Dioxygen by a Ferrocene Derivative, Inorg. Chem., 2014, 53, 7071–7798, DOI:10.1021/ic5013457.
- K. Yamamoto, K. Oyaizu and E. Tsuchida, Catalytic Cycle of a Divanadium Complex with Salen Ligands in O2 Reduction: Two-Electron Redox Process of the Dinuclear Center (salen) N,N′-Ethylenebis(salicylideneamine)), J. Am. Chem. Soc., 1996, 118, 12665–12672, DOI:10.1021/ja9617799.
- E. Tsuchida, K. Oyaizu, E. L. Dewi, T. Imai and F. C. Anson, Catalysis of the Electroreduction of O2 to H2O by Vanadium-salen Complexes in Acidified Dichloromethane, Inorg. Chem., 1999, 38, 3704–3708, DOI:10.1021/ic990268v.
- Z. Liu and F. C. Anson, Electrochemical Properties of Vanadium(III,IV,V)-Salen Complexes in Acetonitrile. Four-Electron Reduction of O2 by V(III)-Salen, Inorg. Chem., 2000, 39, 274–280, DOI:10.1021/ic990958z.
- Z. Liu and F. C. Anson, Schiff Base Complexes of Vanadium(III, IV, V) as Catalysts for the Electroreduction of O2 to H2O in Acetonitrile, Inorg. Chem., 2001, 40(6), 1329–1333, DOI:10.1021/ic0010407.
- K. H. Theopold, Topics in Organometallic Chemistry. In Organometallic Oxidation Catalysis, ed. F. Meyer and C. Limberg, Springer, Berlin, Heidelberg, 2006, vol. 22, pp. 17–37 Search PubMed.
- M. E. O'Reilly, T. J. D. Castillo, J. M. Falkowski, V. Ramachandran, M. Pati, M. C. Correia, K. A. Abboud, N. S. Dalal, D. E. Richardson and A. S. Veige, Autocatalytic O2 Cleavage by an OCO3− Trianionic Pincer CrIII Complex: Isolation and Characterization of the Autocatalytic Intermediate [CrIV]2(μ-O), Dimer, J. Am. Chem. Soc., 2011, 133, 13661–13673, DOI:10.1021/ja2050474.
- T. V. Lubben and P. T. Wolczanski, Dioxygen Activation by Group 4 tritox Alkyls (tritox = t-Bu3CO−): Insertion and Oxygen Atom Transfer, J. Am. Chem. Soc., 1987, 109, 424–435, DOI:10.1021/ja00236a021.
- J.-C. Marchon, J.-M. Latour and C. J. Boreham, Dioxygen Adducts of Low-Valent Titanium Porphyrins. Prospects for the Selective Binding and Activation of Molecular Oxygen, J. Mol. Cat., 1980, 7, 227–233, DOI:10.1016/0304-5102(80)85020-6.
- J.-M. Latour, J.-C. Marchon and M. Nakajima, Titanium(III) Porphyrins and Theis Dioxygen Adducts, J. Am. Chem. Soc., 1979, 101, 3974–3976, DOI:10.1021/ja00508a050.
- B. J. Boro, R. Lansing, K. I. Goldberg and R. A. Kemp, Reaction of a monomeric titanium hydride with dioxygen does not produce a stable titanium hydroperoxide, Inorg. Chem. Commun., 2011, 14, 531–533, DOI:10.1016/j.inoche.2011.01.014.
- F. Schax, S. Suhr, E. Bill, B. Braun, C. Herwig and C. Limberg, A Heterobimetallic Superoxide Complex formed through O2 Activation between Chromium(II) and a Lithium Cation, Angew. Chem., Int. Ed., 2015, 54(4), 1352–1356, DOI:10.1002/anie.201409294.
- D. Sousa, D. P. Bigelow, J. O. Sundberg, J. Que, L. McKenzie and C. J. Caught, Crystal trapping of a side-on peroxo bound to Cr(IV), Chem. Commun., 2015, 51(14), 2802–2805, 10.1039/c4cc08785b.
- C. D. Schmulbach, C. C. Hinckley, C. Kolich, T. A. Ballintine and P. J. Nassiff, Reaction of Molecular Oxygen and Titanium Trichloride in Anhydrous Pyridine, Inorg. Chem., 1974, 13, 1793–2044, DOI:10.1021/ic50138a047.
- D. J. Halko and J. H. Swinehart, The Vanadium(III) Chloride-Dioxygen Reaction in Anhydrous Pyridine, J. Inorg. Nucl. Chem., 1979, 41, 1589–1594, DOI:10.1016/0022-1902(79)80182-7.
- Y. Chimura, The Titanium Trichloride-catalyzed Oxidation of Triphenylphosphine with Oxygen in Acetonitrile, Bull. Chem. Soc. Jpn., 1977, 50, 1885–1886, DOI:10.1246/bcsj.50.1885.
- E. N. Cook and C. W. Machan, Homogeneous catalysis of dioxygen reduction by molecular Mn complexes, Chem. Commun., 2022, 58(84), 11746–11761, 10.1039/d2cc04628h.
- J. Rosenthal and D. G. Nocera, Role of Proton-Coupled Electron Transfer in O–O Bond Activation, Acc. Chem. Res., 2007, 40, 543–553, DOI:10.1021/ar7000638.
- G. Passard, D. K. Dogutan, M. Qiu, C. Costentin and D. G. Nocera, Oxygen Reduction Reaction Promoted by Manganese Porphyrins, ACS Catal., 2018, 8, 8671–8679, DOI:10.1021/acscatal.8b01944.
- C. W. Anson, S. Ghosh, S. Hammes-Schiffer and S. S. Stahl, Co(salophen)-Catalyzed Aerobic Oxidation of p–Hydroquinone: Mechanism and Implications for Aerobic Oxidation Catalysis, J. Am. Chem. Soc., 2016, 138, 4186–4193, DOI:10.1021/jacs.6b00254.
- C. W. Anson and S. S. Stahl, Cooperative Electrocatalytic O2 Reduction Involving Co(salophen) with p-Hydroquinone as an Electron−Proton Transfer Mediator, J. Am. Chem. Soc., 2017, 139, 18472–18475, DOI:10.1021/jacs.7b11362.
- S. L. Hooe, E. N. Cook, A. G. Reid and C. W. Machan, Non-covalent assembly of proton donors and p-benzoquinone anions for co-electrocatalytic reduction of dioxygen, Chem. Sci., 2021, 12(28), 9733–9741, 10.1039/d1sc01271a.
- D. Lionetti, S. Suseno, A. A. Shiau, G. De Ruiter and T. Agapie, Redox Processes Involving Oxygen: The Surprising Influence of Redox-Inactive Lewis Acids, JACS Au, 2024, 4(2), 344–368, DOI:10.1021/jacsau.3c00675.
- T. Devi, Y.-M. Lee, W. Nam and S. Fukuzumi, Metal ion-coupled electron-transfer reactions of metal-oxygen complexes, Coord. Chem. Rev., 2020, 410, 213219, DOI:10.1016/j.ccr.2020.213219.
- S. Kakuda, C. J. Rolle, K. Ohkubo, M. A. Siegler, K. D. Karlin and S. Fukuzumi, Lewis Acid-Induced Change from Four- to Two-Electron Reduction of Dioxygen Catalyzed by Copper Complexes Using Scandium Triflate, J. Am. Chem. Soc., 2014, 137, 3330–3337, DOI:10.1021/ja512584r.
- S. Fukuzumi, H. Kotani, H. R. Lucas, K. Doi, T. Suenobu, R. L. Peterson and K. D. Karlin, Mononuclear Copper Complex-Catalyzed Four-Electron Reduction of Oxygen, J. Am. Chem. Soc., 2010, 132, 6874–6875, DOI:10.1021/ja100538x.
- Y. J. Park, J. W. Ziller and A. S. Borovik, The Effects of Redox-Inactive Metal Ions on the Activation of Dioxygen: Isolation and Characterization of a Heterobimetallic Complex Containing a MnIII-(μ-OH)-Ca(II) Core, J. Am. Chem. Soc., 2011, 133, 9258–9261, DOI:10.1021/ja203458d.
- Y. J. Park, S. A. Cook, N. S. Sickerman, Y. Sano, J. W. Ziller and A. S. Borovik, Heterobimetallic complexes with MIII-(μ-OH)-MII cores (MIII = Fe, Mn, Ga; MII = Ca, Sr, and Ba): structural, kinetic, and redox properties, Chem. Sci., 2013, 4(2), 717–726, 10.1039/c2sc21400h.
| This journal is © The Royal Society of Chemistry 2024 |