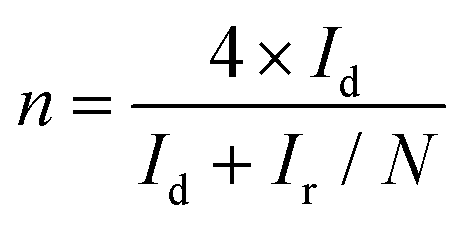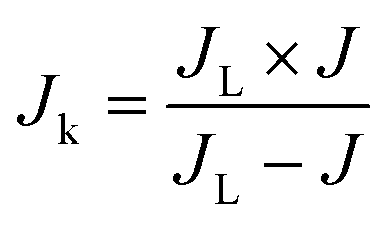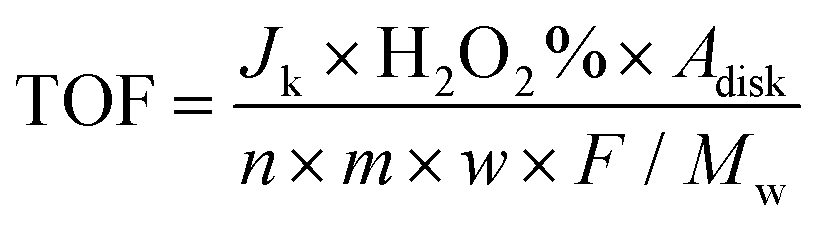DOI:
10.1039/D3GC00737E
(Paper)
Green Chem., 2023,
25, 3931-3939
Single-iron, cobalt, nickel, and copper-atom catalysts for the selective reduction of oxygen to H2O2†
Received
4th March 2023
, Accepted 24th April 2023
First published on 26th April 2023
Abstract
The pursuit of hydrogen peroxide (H2O2) production via two-electron electrochemical oxygen reduction reaction (ORR) is hampered by the lack of high-performance electrocatalysts. In this work, a series of single metal (i.e., Fe, Co, Ni, and Cu) atom catalysts anchored into N-doped carbon nanosheets (NCNSs) are evaluated for H2O2 production at pH-universal electrolytes. The catalytic performance of the samples, including current density, H2O2 selectivity, turnover of frequency (TOF), and faradaic efficiency, are characterized by electrochemical measurements. The generation of H2O2 is monitored by a UV-vis spectrophotometer. Among the samples, Cu/NCNSs exhibits the best catalytic performance with a ring current density of 2.15 mA cm−2, TOF of 15.8 s−1 at 0.1 V vs. reversible hydrogen electrode, Tafel slope of 88 mV dec−1, and H2O2 selectivity of 100% at 0.57 V in alkaline media. Importantly, the Cu/NCNSs yields H2O2 selectivity of 81% at 0.05 V in acidic electrolytes. In contrast, Cu nanoparticles possess an inferior current density and much lower H2O2 selectivity via the 4e− ORR pathway. Further quantitative analysis of H2O2 demonstrates that the Cu/NCNSs catalyst has an H2O2 production rate of 5.1 mol gcatalyst−1 L−1 h−1 in alkaline media at 0.4 V, which is larger than those in acidic media (3.2 mol gcatalyst−1 L−1 h−1) and neutral media (2.2 mol gcatalyst−1 L−1 h−1) at −0.2 V. Such performance of Cu/NCNSs renders it as one of the best catalysts for H2O2 production.
Introduction
Hydrogen peroxide (H2O2), a valuable and green oxidant, is extensively employed in chemical synthesis, wastewater treatment, paper/pulp bleaching, medical disinfection, and environmental remediation.1–5 Traditionally, the industrial synthesis of H2O2 relies on the energy-intensive and waste-demanding anthraquinone oxidation/reduction process.6–8 In addition, the resulting high concentration of H2O2 (ca. 70 wt%) poses safety concerns during transportation and storage.1,9 Alternatively, electrochemical oxygen reduction reaction (ORR) via the two-electron (2e−) route (O2 + * + (H+ + e−) → *OOH; *OOH + (H+ + e−) → * + H2O2) provides an attractive strategy to achieve on-site and small-scale H2O2 production.10–12 In this perspective, the selectivity of ORR toward H2O2 relies on the proper binding energy between *OOH intermediate and the active sites of the electrocatalysts.13 Additionally, preserving the O–O bond from cleavage is also a crucial step for H2O2 formation.14,15 However, since the 4e− counterpart toward H2O generation is a powerful competitor to the 2e− route, it is crucial to design efficient catalysts with high intrinsic selectivity and activity for H2O2 formation.16,17
Thus far, state-of-the-art noble-metal-based electrocatalysts such as Pt–Hg, Pd–Hg, and Pd–Au display good catalytic activity for the 2e− process.1,6,17–20 Unfortunately, the scarcity and high price of noble metals seriously limit large-scale applications.12,15 The earth-abundant carbon-based materials hold great promise for H2O2 electrochemical synthesis due to the high surface areas and tunable porosities, showing comparable activity to the commercial Pt catalysts in alkaline media.19 Unfortunately, the carbon-based electrocatalysts always show limited activity in acidic media.21
Transition-metal (Fe, Co, Ni, etc.) single atom catalysts (SACs) have gained remarkable attention in the heterogeneous catalysis field due to their 100% atom efficiency, tunable electronic structure, and unsaturated coordination environments.17,22–25 In principle, benefiting from the atomically dispersed metal sites of SACs, the adsorption of O2 on SACs usually takes an end-on type rather than a side-on type, thus preventing O–O bond cleavage.13,26 In particular, SACs with M–N–C moieties have attracted significant attention due to the excellent catalytic activity and selectivity for the ORR.27,28 Recently, substantial efforts have been devoted to tuning the cost-effective SACs toward the 2e− ORR pathway, aiming for H2O2 formation.6,15 In detail, SACs containing M–N–C moieties have been synthesized by top-down strategies, including pyrolysis of metal–organic frameworks (MOF),29–31 covalent organic frameworks (COF),32 and metal–porphyrin molecules,33 as well as bottom-up strategies, such as high-energy ball milling and wet-chemistry.23 However, most catalysts exhibit good ORR activity in alkaline media. Unfortunately, since H2O2 is less stable in alkaline media, electrochemical synthesis of H2O2 in acidic and neutral media is more suitable in practical applications.13 Therefore, there is a great challenge to improve the catalytic activity for H2O2 formation in acidic or neutral media.14,34
Our group developed a universal method to fabricate SACs via a simple condensation–carbonization process.35,36 Efforts have been devoted to evaluating the resultant SACs as electrocatalysts for the electro-oxidation of small molecules. The resulting SACs displayed excellent catalytic performance for the oxidation of water, glycerol, and 5-hydroxymethylfurfural.35 Motivated by these results, we prepared a series of SACs consisting of isolated Fe, Co, Ni, and Cu atoms anchored into N-doped carbon nanosheets (NCNSs) for the selective reduction of O2 into H2O2 in this work. The catalytic performance of the catalysts was comparatively investigated in alkaline, neutral, and acidic electrolytes. It is demonstrated that the as-prepared Cu/NCNSs displays excellent performance with nearly 100% H2O2 selectivity, turnover of frequency (TOF) of 15.8 s−1 at 0.1 V, and H2O2 yield rate of 5.1 mol gcatalyst−1 L−1 h−1 at 0.4 V in alkaline media.
Experimental
Synthesis of single metal (Fe, Co, Ni, and Cu) atom catalysts
SACs were synthesized via the authors’ previous work with slight modifications.35 In a typical synthesis, 0.4 g of polystyrene (PS) nanospheres and 0.2 g of SDBS were scattered in 2 mL anhydrous ethanol and 7.6 mL ultrapure water. 2 mL cyanamide and 1 mL furfural were introduced into the emulsion and reacted at 60 °C for 24 h under magnetic stirring. Then, a specific volume of metal solutions (10 g L−1 of Fe(NO3)3·9H2O, Co(NO3)2·6H2O, Cu(NO3)2·3H2O, and Ni(NO3)2·6H2O) were added, mixed, and stirred for at least 2 days. Subsequently, the resulting suspension was evaporated to remove water and ethanol. Finally, the resulting paste was pyrolyzed at 900 °C for 3 h under a flowing nitrogen atmosphere in a tube furnace. The resulting powder was ground with an agate mortar and washed with a solution of 0.5 M FeCl3 + 2 M HCl at 50 °C to remove any metal particles. For comparison, nitrogen-doped carbon, free of any metal, was also prepared. The resultant catalysts were denoted as Fe/NCNSs, Co/NCNSs, Ni/NCNSs, Cu/NCNSs, and NCNSs.
Electrochemical measurements
Electrochemical measurements were performed on a Solartron Analytical electrochemical station (1470 E, England) with a typical three-electrode configuration consisting of a rotating-ring-disk working electrode (SEN-RRDE, China), a carbon-rod counter electrode, and a Hg/HgO reference electrode in alkaline electrolytes or a saturated calomel electrode (SCE) as a reference electrode in acidic/neutral electrolytes. The rotating-ring-disk electrode (RRDE) assembly was comprised of a glassy carbon disk electrode (diskOD = 5 mm) and a Pt ring (ringOD = 7.5 mm, ringID = 5.5 mm), with a theoretical collection efficiency of 0.37. Experimentally, the collection efficiency (N) was determined by the ferrocyanide/ferricyanide redox couple.13,28 The potentiodynamic curve was recorded between 0.1–1.2 V on the glassy carbon disk electrode, and the potentiostatic curve was recorded at 0.95 V on the platinum ring electrode in 10 mM K3[Fe(CN)6] + 1 M KCl solution at a scan rate of 10 mV s−1 and a rotation rate between 400 and 2500 rpm. The collection efficiency refers to the ratio of the ring current to the absolute value of the disk current.
To prepare a working electrode, typically, 2.0 mg of the as-prepared M/NCNS catalysts were dispersed in 249 μL of ethanol, 249 μL of deionized water, 497 μL of isopropyl alcohol, and 5 μL of Nafion solution (5 wt%), and ultrasonicated for 2 h to form a homogeneous catalyst ink. 10 μL of catalyst ink (catalyst loading: ca. 102 μg cm−2) was pipetted onto the freshly polished disk electrode and dried at room temperature. N2- or O2-saturated 0.1 M KOH, 0.1 M HClO4, and 0.1 M PBS served as alkaline, acidic, and neutral electrolytes, respectively. 0.1 M KOH (pH = 13.0) was prepared by dissolving 5.61 g electron-grade potassium hydroxide in 1 L of deionized water. 0.1 M HClO4 (pH = 1.0) was prepared by diluting 70% perchloric acid with deionized water. 0.1 M PBS (pH = 7.0) was prepared by dissolving 80 g sodium chloride, 2 g potassium chloride, 14.4 g disodium hydrogen phosphate, and 2.4 g potassium dihydrogen phosphate with deionized water. All the electrode potentials reported in this work are referenced to the reversible hydrogen electrode (RHE) as follows: ERHE = EHg/HgO + 0.0592 × pH + 0.098, ERHE = ESCE + 0.0592 × pH + 0.241.
Cyclic voltammetry (CV) curves were recorded in the potential range of 0–1.6 V at 50 mV s−1 under static conditions. The polarization curves of RRDE were recorded in O2-saturated electrolyte solutions at a scan rate of 10 mV s−1 under 1600 rpm, and the potentiostatic curves of the Pt ring were recorded at 1.20 V for the oxidation of H2O2. The onset potentials are defined as the potentials required to reach a geometric current density of −0.05 mA cm−2 and 0.02 mA cm−2 for the disk and ring electrodes, respectively.32 The selectivity of H2O2 and electron transfer number (n) were calculated according to eqn (1)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 13,29 and (2).13,18
13,29 and (2).13,18
| | 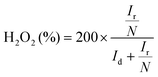 | (1) |
| | 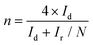 | (2) |
where
Ir is the ring current,
Id is the disk current, and
N is the calibrated collection efficiency of RRDE (0.35). The H
2O
2 current density (
JH2O2) and H
2O
2 faradaic efficiency (FE) were calculated using
eqn (3)32 and
(4).
37| | | JH2O2 = Ir/(N × Ageom) | (3) |
| | 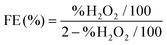 | (4) |
where
Ageom is the surface area of the Pt ring and % (H
2O
2) is H
2O
2 selectivity. The kinetic current density (
Jk) was calculated using Koutecky–Levich
eqn (5)–(8).
29,31| | 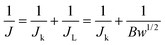 | (5) |
| | | B = 0.62nFC0D02/3v−1/6 | (7) |
| | 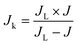 | (8) |
where
J is the apparent current density,
Jk and
JL are the kinetic and diffusion-limited current densities,
ω is the angular velocity,
N is the rotation rate,
F is the Faraday constant (96
485 C mol
−1),
C0 is the saturation concentration of O
2,
D0 is the diffusion coefficient of O
2, and
v is the kinematic viscosity of the electrolyte. The TOF was calculated by
eqn (9)32| | 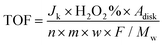 | (9) |
where
Adisk is the geometric surface area of the disk electrode,
n is the electron transfer number of ORR,
m is the mass of the catalyst loading on the disk electrode,
w is the metal content, and
Mw is the molecular weight of the metal.
For the long-term stability test, the potentiostatic curves were recorded at 0.4 V (in alkaline media) and −0.2 V (in acidic and neutral media) for 10 h. The ORR activity was tested after the stability test. H2O2 production was conducted using a customized H-cell electrolyzer separated by Nafion membranes. A carbon paper (1 × 1 cm) loaded with 100 μg cm−2 of the catalyst was employed as the ORR cathode, and a carbon rod was employed as the anode. The concentration of H2O2 was quantified by UV-vis spectrophotometer38 (Shimadzu UV-1900, Japan).
Results and discussion
Structural characterization
The synthetic process of the M/NCNSs catalysts is shown in Fig. 1a. Furfural and cyanamide were polymerized in the presence of PS nanospheres. PS nanospheres served as templates to provide a large surface area. The metal contents in the samples were determined by ICP-AES, as shown in Table S1.† The TEM micrograph of the Co/NCNSs catalyst appears as aggregates of nanosheets without obvious metal nanoparticles (Fig. S1a†). The high-resolution TEM image in Fig. S1b† also confirms the layered structure of the as-prepared Co/NCNSs catalyst. The energy dispersive X-ray (EDX) mapping images indicate that C, N, and Co atoms are uniformly distributed on the NCNSs support (Fig. S1c†). The isolated single Fe (Fig. 1b), Co (Fig. 1c), Ni (Fig. 1d), and Cu (Fig. 1e) sites on the NCNSs are shown as bright dots and highlighted by yellow cycles in the aberration-corrected HAADF-STEM images. To determine the crystal structure of the M/NCNSs catalysts, XRD patterns were performed. As shown in Fig. 1f, the five catalysts exhibit similar characteristic diffraction peaks at 25°, 43.4°, and 44.2°, corresponding to the carbon (002), (103), and (111) planes. No diffraction peaks of metals, metal oxides, and metal nitrides were detected. The Raman spectra exhibited the characteristic D-band and G-band peaks of carbon materials at 1350 cm−1 and 1580 cm−1 for the M/NCNSs catalysts, demonstrating the formation of NCNSs (Fig. 1g). The Brunauer–Emmett–Teller (BET) surface area and pore size distribution of samples were determined as shown in Fig. S2–S6.† The BET surface areas of Fe/NCNSs, Co/NCNSs, Ni/NCNSs, Cu/NCNSs, and NCNSs are calculated to be 389.6, 498.8, 636.0, 436.8, and 465.0 m2 g−1, respectively, based on the corresponding N2 adsorption–desorption isotherms. The large BET values are attributed to the presence of a number of mesopores and the sheet-like structure of N-doped carbon. Besides, the average pore sizes of the samples are between 3.3 and 4.1 nm. The abundant mesoporous pores are conducive to the dispersion of metal atoms. The presence of single copper atoms in the samples was further verified by X-ray absorption near edge structure analyses, and the average coordination number is 4.8 for the Cu–Nx species.36
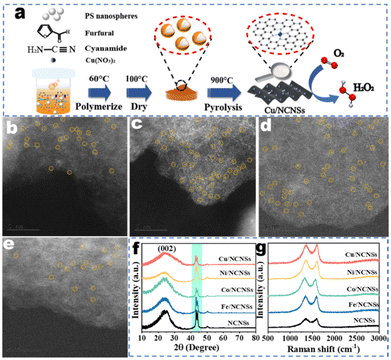 |
| | Fig. 1 The synthetic process of M/NCNSs (a), high-resolution HAADF-STEM images of Fe/NCNSs (b), Co/NCNSs (c), Ni/NCNSs (d), and Cu/NCNSs (e), XRD patterns (f) and Raman spectra (g) of the as-synthesized samples. | |
CV characterization
To verify the ORR activity of the as-prepared M–NCNSs catalysts, CV curves were tested in O2-saturated and N2-saturated 0.1 M KOH, as shown in Fig. S7.† Under the O2 atmosphere, similar broad reduction peaks of O2 are noted at about 0.60 V, 0.60 V, 0.58 V, and 0.48 V for Co/NCNSs, Ni/NCNSs, Cu/NCNSs, and NCNSs, respectively. Notably, there are no obvious peaks associated with the redox of the metal particles, which is consistent with the XRD patterns and further indicative of samples free of metal nanoparticles. In addition, CV curves were also recorded in O2/N2-saturated 0.1 M HClO4 and 0.1 M PBS solutions (see Fig. S8 and S9†). Compared with Fe/NCNSs and NCNSs, the Co/NCNSs, Ni/NCNSs, and Cu/NCNSs catalysts have more pronounced currents of O2 reduction in acidic and neutral media, indicating the excellent ORR electrocatalytic activities. It should be noted that the currents of glassy carbon disk electrode are close to 0 in 0.1 M KOH, HClO4, and PBS solutions, which is consistent with its negligible double layer capacitances (see Fig. S10†). In contrast, Cu nanoparticles (Cu NPs) have distinctive redox peaks of metallic copper and show limited currents of oxygen reduction under alkaline, acidic, and neutral conditions, which indicates that Cu NPs have poor ORR catalytic activity in a broad pH range (see Fig. S11†).
RRDE characterization
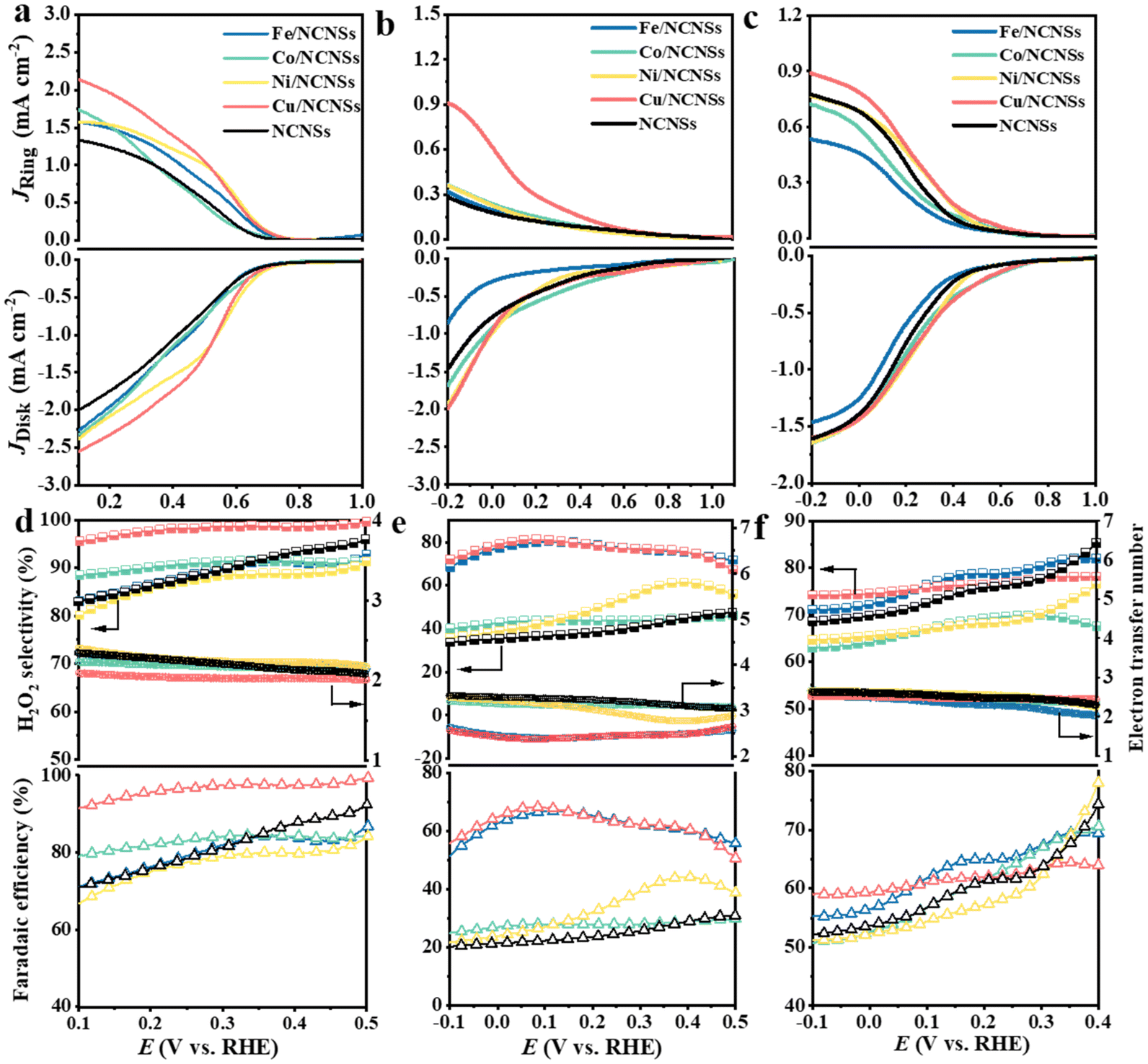
The collection efficiency of RRDE was first calibrated. Fig. S12a† shows the polarization curves of Fe2+/Fe3+ on the RRDE recorded in potassium ferricyanide solutions at rotation rates of 400–2500 rpm. The reduction of Fe3+ occurs on the disk electrode, while the oxidation of Fe2+ occurs on the Pt ring electrode. The ratio between ring and disk currents is estimated as 0.35 (see Fig. S12b†). The ORR performances of the M/NCNSs catalysts were evaluated by polarization curves in O2-saturated 0.1 M electrolyte on RRDE at 1600 rpm. Since the ORR on the disk electrode occurs simultaneously with the H2O2 oxidation reaction (H2O2OR) on the ring electrode, the current densities on the ring and disk electrode are generated synchronously. Table S2† summarizes the onset potentials of the M/NCNSs catalysts in 0.1 M KOH, HClO4, and PBS solutions. As displayed in Fig. 2a, the Ni/NCNSs catalyst shows the most positive onset potential of 0.77 V on both disk and ring electrodes, indicating its excellent ORR catalytic activity. The Cu/NCNSs catalyst exhibits the highest ORR current density of 2.6 mA cm−2 at 0.1 V with an onset potential of 0.75 V and the highest H2O2OR current density of 2.2 mA cm−2 at 0.1 V with an onset potential of 0.76 V. In contrast, the NCNSs catalyst exhibits limited ORR current density (1.9 mA cm−2 at 0.1 V) and H2O2OR current density (1.3 mA cm−2 at 0.1 V) compared to the M/NCNSs catalysts, implying a critical role of single metal atoms for the ORR catalytic activity in alkaline media.
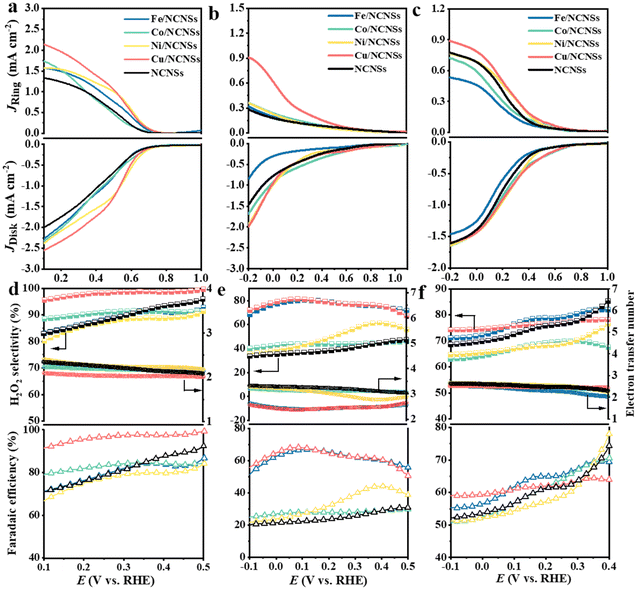 |
| | Fig. 2 Electrocatalytic activity of catalysts collected from RRDE system in O2-saturated alkaline, acidic, and neutral media. Potentiostatic curves on the Pt ring electrode and potentiodynamic curves on the glassy carbon disk electrode at 1600 rpm in O2-saturated 0.1 M KOH (a), 0.1 M HClO4 (b), and 0.1 M PBS (c); selectivity, electron transfer number, and faradaic efficiency of H2O2 production in O2-saturated 0.1 M KOH (d), 0.1 M HClO4 (e), and 0.1 M PBS (f). | |
The corresponding H2O2 selectivity, electron transfer number, and FE were plotted as a function of potential (see Fig. 2d). As expected, the Cu/NCNSs catalyst possesses the highest H2O2 selectivity of over 96% in the potential range of 0.1–0.6 V. Especially, H2O2 selectivity reaches 100% over the Cu/NCNSs catalyst at 0.57 V, which is superior to most of the electrocatalysts reported in previous studies, such as Mo1/OSG–H (ca. 95%),39 O–CNTs (ca. 90%),40 Co–POC–O (ca. 85.6%),41 Mo2CTx:Fe (ca. 67–100%),42 CuOx/G-30 (ca. 76–84%),43 COF-366-Co (ca. 90%),32 and Co–N–C (ca. 60–70%)30 (see Fig. 3a and Table S3†). In detail, the selectivity for H2O2 generation follows the order of Cu/NCNSs (96%) > Co/NCNSs (88%) > Fe/NCNSs (83%) > NCNSs (82%) > Ni/NCNSs (80%) at 0.1 V. Although the NCNSs catalyst is highly selective (>82%) for H2O2, its ORR catalytic activity is much poorer than the M/NCNSs catalysts. In addition, the electron transfer number of Cu/NCNSs is close to 2, agreeing well with its H2O2 selectivity to nearly 100%. The FE of Cu/NCNSs is higher than 92% in a potential range of 0.1–0.6 V. Overall, all of the M/NCNSs catalysts exhibit excellent H2O2 selectivity, and the Cu/NCNSs catalyst is the most superior candidate due to its excellent ORR activity and high H2O2 selectivity.
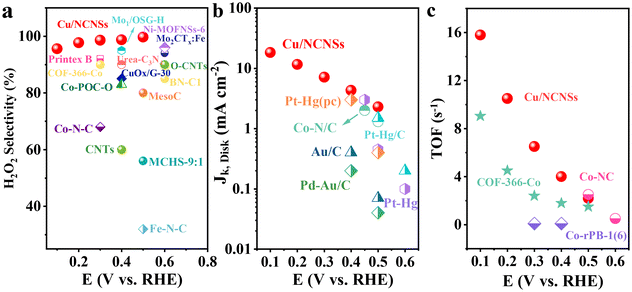 |
| | Fig. 3 H2O2 selectivity (a), Jk (b) and TOF (c) of Cu/NCNSs and other electrocatalysts reported in recent studies in alkaline media. Data adapted from: Au/C and Pd–Au/C ref. 52, Pt–Hg, Pt–Hg (pc) and Pt–Hg/C ref. 20, Co–N/C (heat-treated cobalt–porphyrin supported on vapor grown) ref. 53, Co–NC ref. 13, COF-366-Co ref. 32, Co-rPB-1(6) ref. 50 and ref. in Table S3 (see ESI†) for the comparison of H2O2 selectivity in alkaline media. | |
To further explore the unique ORR catalytic activity of the M–NCNSs catalysts, the polarization curves were also tested in acidic and neutral media. In Fig. 2b, both Cu/NCNSs and Ni/NCNSs show the highest disk current density of 1.9 mA cm−2 at −0.2 V. In contrast, Cu/NCNSs has a more positive onset potential of 0.76 V, indicating the Cu/NCNSs catalyst possesses better ORR catalytic activity. For comparison, the ring current density at −0.2 V follows the order of NCNSs (0.28 mA cm−2) < Fe/NCNSs (0.31 mA cm−2) < Ni/NCNSs (0.36 mA cm−2) ≈ Co/NCNSs (0.36 mA cm−2) < Cu/NCNSs (0.9 mA cm−2), implying that introduction of Fe, Co, Cu, and Ni single atom facilitates the production of H2O2 in acidic media. As shown in Fig. 2e, although Fe/NCNSs and Cu/NCNSs show similar H2O2 selectivity (>70%) in the potential range of −0.1 to 0.5 V, the ORR and H2O2RR activities of Fe/NCNSs are much poorer than Cu/NCNSs. Especially, the selectivity of the Cu/NCNSs can reach 81% at 0.05 V, and the corresponding electron transfer number is calculated to be 2.3, confirming the dominant 2e− ORR pathway over Cu/NCNSs in acidic media. Such selectivity is superior to most catalysts in acidic media20,30,40,44–47 (Table S4†). In contrast, the NCNSs, Co/NCNSs, and Ni/NCNSs catalysts show an inferior selectivity of about 40% and a large electron transfer number of around 3.0, indicative of the ORR through both 4e− and 2e− pathways. Besides, the FE of H2O2 production has a similar trend with selectivity. The Cu/NCNSs possesses the highest FE of 68% between 0.06 V and 0.09 V. Generally, Cu/NCNSs catalyst displays the advantages of a promising candidate in acidic media with the highest ring current density, the most positive onset potential (0.82 V) on ring electrode, and the highest selectivity (81%) for the 2e− ORR route.
Under neutral conditions, Co/NCNSs, Ni/NCNSs, Cu/NCNSs, and NCNSs exhibit the same ORR current density of 1.6 mA cm−2 at −0.2 V, whereas Fe/NCNSs exhibit apparently poor ORR activity with the lowest ORR current density of 1.4 mA cm−2 at −0.2 V and the most negative onset potential of 0.7 V (Fig. 2c). In addition, the ring current density corresponding to the oxidation of H2O2 at −0.2 V follows the order of Fe/NCNSs (0.53 mA cm−2) < Co/NCNSs (0.71 mA cm−2) < NCNSs ≈ Ni/NCNSs (0.77 mA cm−2) < Cu/NCNSs (0.89 mA cm−2), implying Fe/NCNSs and Co/NCNSs are less effective for the 2e− ORR route in neutral media compared to the Ni/NCNSs and Cu/NCNSs. The H2O2 selectivity, together with the FE, follows the order of Fe/NCNSs > Cu/NCNSs > NCNSs > Co/NCNSs > Ni/NCNSs at 0.2 V (see Fig. 2f), while the electron transfer number follows the opposite trend. Among the samples, the Cu/NCNSs catalyst is the most favorable catalyst for the 2e− ORR pathway in neutral media with a ring current density of 0.89 mA cm−2, a positive onset potential of 0.77 V, and an H2O2 selectivity of 76%, which outperformed most of the ORR electrocatalysts reported in the literature30,39,44,48–51 (Table S5†).
To investigate the intrinsic ORR activity of the samples, the electrochemical active surface area (ECSA) was estimated by electrochemical double-layer capacitance (Cdl). Fig. S13–S15† show CV curves of the M/NCNSs catalysts recorded at varying scan rates in alkaline, acidic, and neutral media, respectively. As shown in Fig. S16,† the Cu/NCNSs catalyst has the largest Cdl values of 2.61, 8.13, and 5.35 mF cm−2 in alkaline, acidic, and neutral media, respectively, contributing to its superior 2e− ORR catalytic activity at pH-universal electrolytes.
Additionally, the ORR performance of the bare glassy carbon disk electrode, Cu NPs, and Cu/NCNSs was also evaluated for comparison in alkaline, acidic, and neutral media. The polarization curves are displayed in Fig. S17.† Cu NPs exhibit inferior ORR and H2O2OR activity and the most negative onset potential in 0.1 M KOH, HClO4, and PBS compared with Cu/NCNSs (Table S2†), indicating the predominant 4e− ORR route of Cu NPs. The corresponding H2O2 selectivity and electron transfer number were also calculated in Fig. S18.† Cu NPs show a much lower H2O2 selectivity of 60–77% in alkaline solutions, 20–55% in acidic solutions, and >40% in neutral solutions and a much larger electron transfer number in the range of 3–4 compared with the Cu/NCNSs catalyst.
ORR kinetics analysis
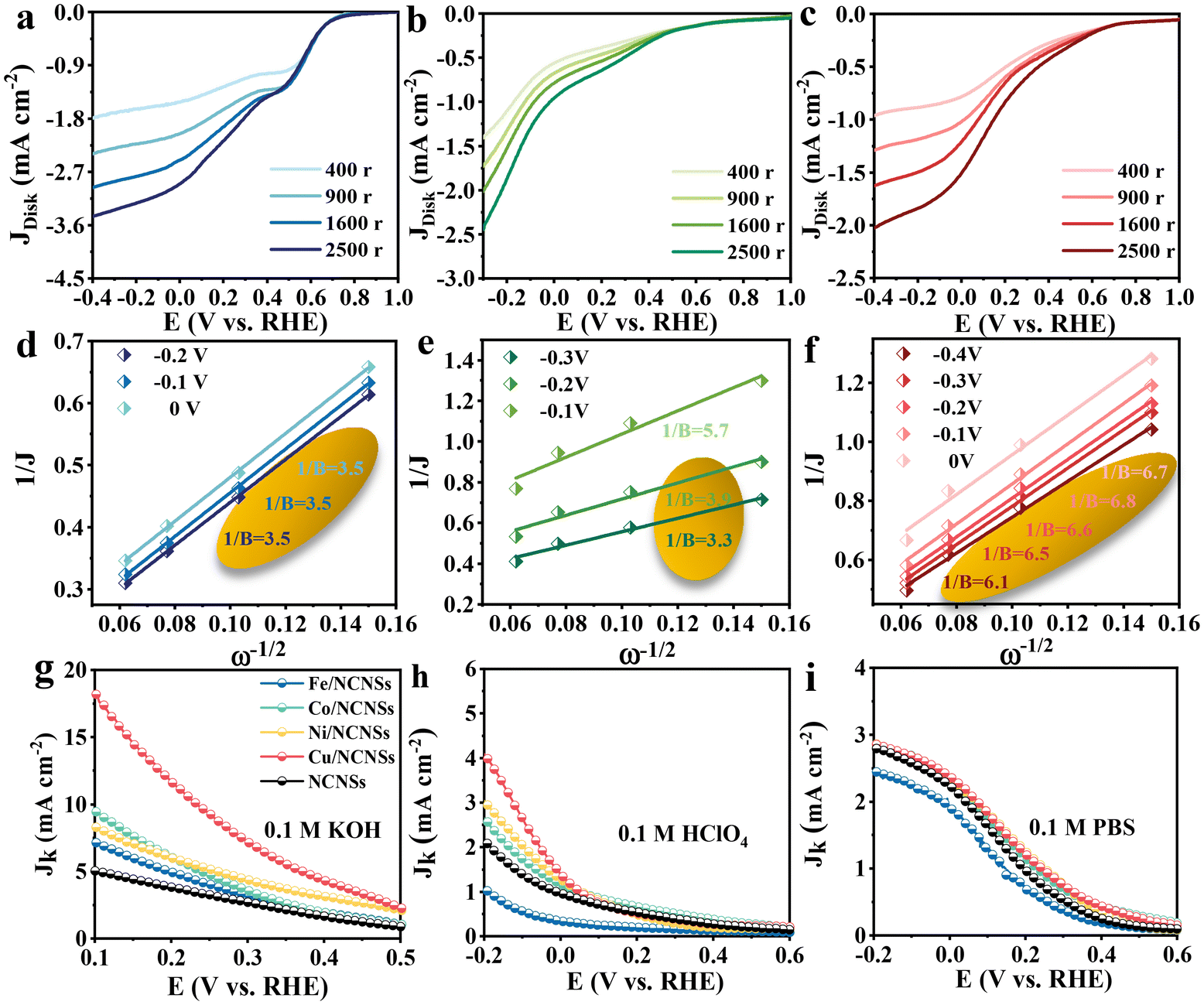
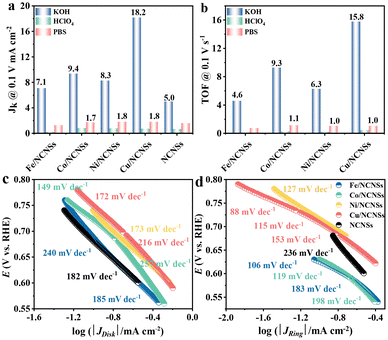
To investigate the ORR kinetics of the M/NCNSs catalysts, Koutecky–Levich (K–L) analysis was carried out on the RRDE. The Cu/NCNSs catalyst is evaluated by polarization curves at different rotation rates between 400 and 2500 rpm in alkaline, acidic, and neutral media (see Fig. 4a–c). With increasing rotating rate, the current density increases gradually, indicating the high dependence of ORR on oxygen diffusion.37 As shown in Fig. 4d–f, the K–L plots are obtained by fitting Jk−1 and w−1/2 according to eqn (5)–(7), where the slopes correspond to 1/B. It can be seen that these fitted K–L plots (the yellow part) are nearly parallel (i.e., the electron transfer numbers are almost the same under these potentials), which indicates that ORR has reached the limited current density at the potentials and that the reaction process is controlled by the diffusion effect. Besides, the polarization curves and the corresponding K–L plots of Fe/NCNSs, Co/NCNSs, Ni/NCNSs, and NCNSs also show the same ORR phenomenon (Fig. S19–S22†). Jk values over the catalysts at 1600 rpm were calculated according to eqn (8), as shown in Fig. 4g–i and Table S6.† It can be found that the Jk trend toward ORR is consistent with the disk current density. The Jk values of the M–NCNSs catalysts at 0.1 V were compared. As displayed in Fig. 5a, the Cu/NCNSs catalyst has the highest Jk of 18.2 mA cm−2 in alkaline media, which is far superior to its competitors in this study as well as state-of-the-art catalysts (Fig. 3b). Moreover, the Cu/NCNSs catalyst also exhibits a larger Jk of 1.8 mA cm−2 at 0.1 V in neutral media. In addition, the Jk values of the M/NCNSs catalysts in acidic media are smaller than that in alkaline and neutral solutions. These results show that the ORR catalytic activity over the M/NCNSs catalysts is constrained in acidic media.
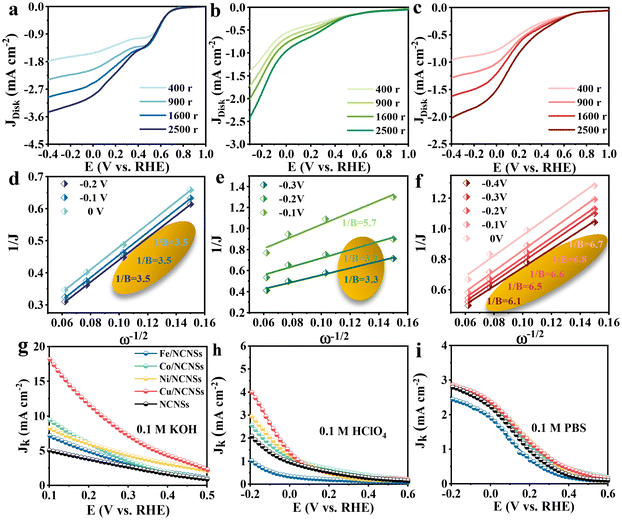 |
| | Fig. 4 Disk polarization curves of the Cu/NCNSs recorded in O2-saturated 0.1 M KOH (a), 0.1 M HClO4 solution (b) and 0.1 M PBS (c) at rotation rates between 400–2500 rpm, the corresponding K–L plots at different potentials in O2-saturated 0.1 M KOH (d), 0.1 M HClO4 (e) and 0.1 M PBS (f), and Jk values of the catalysts at 1600 rpm in 0.1 M KOH (g), 0.1 M HClO4 (h) and 0.1 M PBS (i). | |
 |
| | Fig. 5 Comparison of the Jk (a) and TOF (b) values of the catalysts at 0.1 V in 0.1 M KOH, HClO4, and PBS media. Tafel plots of the disk current density (c) and ring current density (d) at a rotating rate of 1600 rpm in the O2-saturated 0.1 M KOH media. | |
To further elucidate the ORR kinetics of M/NCNSs, the TOF values were calculated according to eqn (9), as shown in Tables S7 and S8.† The Cu/NCNSs catalyst has the highest TOF of 15.8 s−1 at 0.1 V in KOH media among the M/NCNSs catalysts (Fig. 5b), which is comparable to state-of-the-art catalysts (Fig. 3c). In addition, the activity of Co/NCNSs and Cu/NCNSs catalysts are also prominent, with TOF values of 1.1 and 1.0 s−1 at 0.1 V in neutral media, respectively. Moreover, the Tafel slope also reflects the kinetic characteristics of the electrochemical reaction. As shown in Fig. 5c and d, the Tafel slopes of the ring electrode are generally smaller than that of the disk electrode. The superior kinetic of H2O2 production over the Cu/NCNSs catalyst is revealed by the smallest Tafel slope of 88 mV dec−1 on the ring electrode. The ORR kinetic in alkaline media is faster than in acidic and neutral media (Fig. S23†). It is worth noting that the Cu/NCNSs catalyst possesses superior Jk, TOF, and Tafel slope values that contribute to its superior 2e− ORR catalytic activity.
Stability and H2O2 yield rates
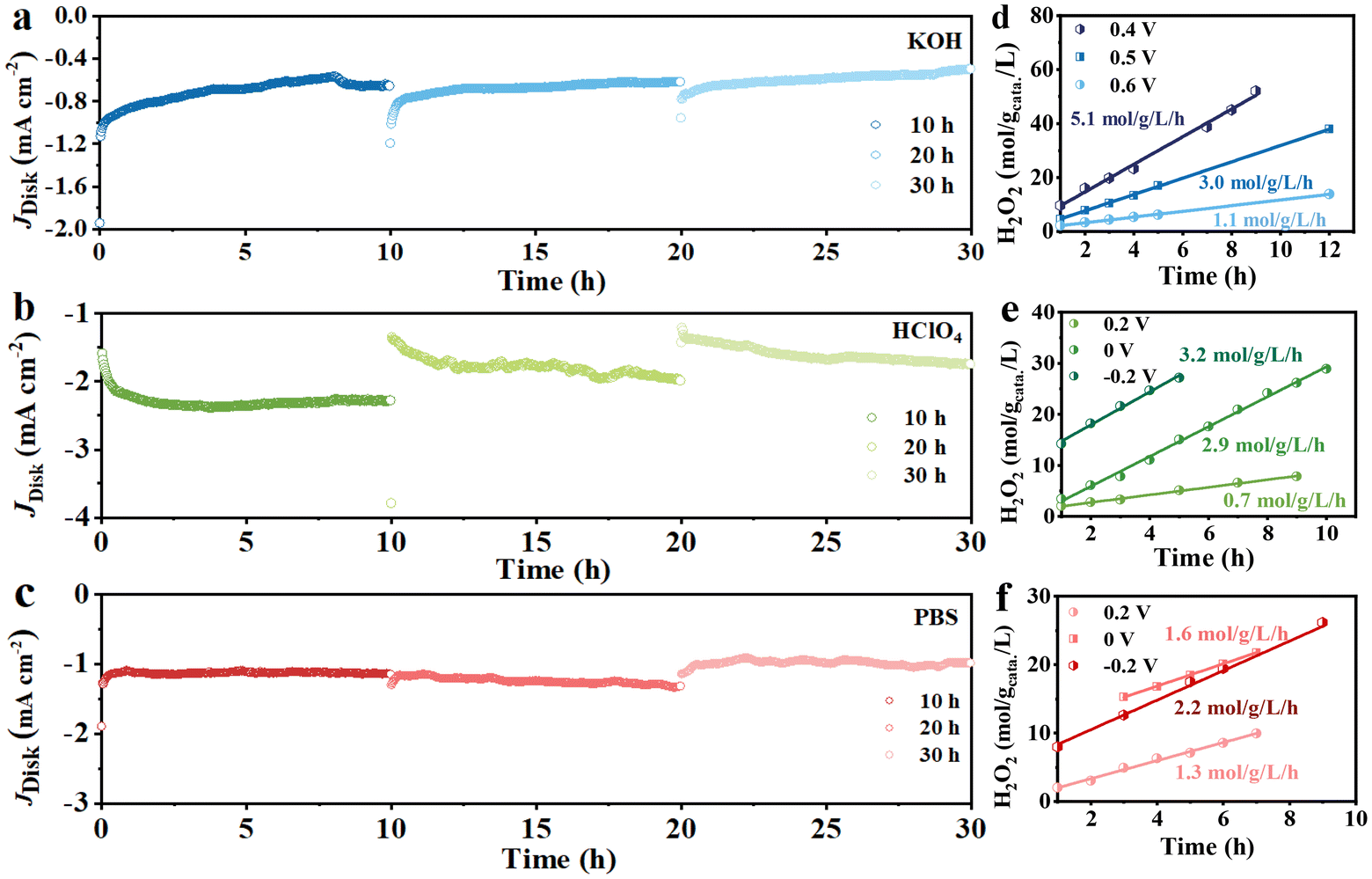
The long-term stability of the Cu/NCNSs catalyst was investigated on the RRDE at 1600 rpm in O2-saturated 0.1 M KOH (0 V), HClO4 (−0.2 V), and PBS (−0.2 V) solutions. As shown in Fig. 6a–c, the disk currents gradually level off and remain steady for 10 h in alkaline, acidic, and neutral media. After the test was suspended, the polarization curves were recorded, and the corresponding H2O2 selectivity was calculated (Fig. S24†). When the test is resumed, the disk currents in the alkaline and neutral solutions return to the initial values, while the disk currents in the acidic solution decrease. Moreover, the H2O2 selectivity remains ≥90%, ≥40%, and 70% for 20 h without obvious decay in alkaline, acidic, and neutral media, respectively. H2O2 formation was performed in an H-cell electrolyzer, where the Cu/NCNSs catalyst was cast onto a carbon paper (100 μg cm−2 catalyst loading), with potentiostatic curves conducted at different potentials. The UV spectrum of H2O2 reveals a characteristic absorption peak at 400 nm (see Fig. S25a†), and the standard curve of H2O2 concentration vs. absorbance is established (see Fig. S25b†). As shown in Fig. 6d–f, the slopes correspond to H2O2 yield rates. The highest H2O2 yield rate of 5.1 mol gcatalyst−1 L−1 h−1 is obtained in 0.1 M KOH at 0.4 V, which is larger than those in 0.1 M HClO4 (3.2 mol gcatalyst−1 L−1 h−1) and 0.1 M PBS (2.2 mol gcatalyst−1 L−1 h−1) at −0.2 V, suggesting that the pH of the electrolyte has a significant effect on the production of H2O2. In addition, the H2O2 yield rates in 0.1 M KOH at 0, 0.2, and 0.3 V were calculated to be 3.5, 4.5, and 4.1 mol gcatalyst−1 L−1 h−1, respectively (see Table S9†).
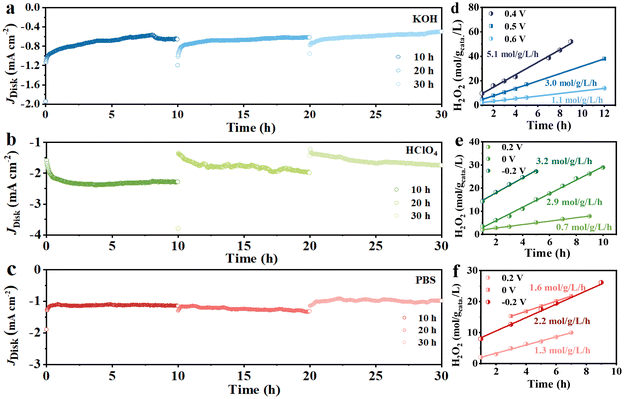 |
| | Fig. 6 Potentiostatic stability tests in 0.1 M KOH (a), 0.1 M HClO4 (b) and 0.1 M PBS (c), H2O2 yield rates of Cu/NCNSs obtained from different potentials in 0.1 M KOH (d), HClO4 (e) and PBS (f). | |
Conclusions
In summary, a series of SACs consisting of isolated Fe, Co, Ni, and Cu atoms supported by NCNSs were synthesized for the electro-reduction of O2 to H2O2. Electrochemical measurements reveal that the M/NCNSs catalysts generally have larger ORR and H2O2OR current densities compared with the pure NCNSs. Among them, the Cu/NCNSs catalyst displays the highest catalytic activity with a ring current density of 2.2 mA cm−2, Jk value of 18.2 mA cm−2, TOF value of 15.8 s−1 at 0.1 V, Tafel slope of 88 mV dec−1, and H2O2 selectivity of nearly 100%. Especially, remarkable H2O2 selectivity of 81% and 76% were obtained under acidic (pH 1.0) and neutral conditions (pH 7.0), respectively. However, the 4e− ORR mechanism plays a more prominent role in Cu NPs. Under pH-universal electrolytes, Cu NPs have inferior ORR activity, H2O2 selectivity, and the most negative onset potential compared with Cu/NCNSs, indicating the unique ORR catalytic activity of SACs. The Cu/NCNSs shows excellent long-term stability for H2O2 production. In addition, the Cu/NCNSs possesses an H2O2 yield rate of 5.1 mol gcatalyst−1 L−1 h−1 at 0.4 V in alkaline media, followed by that in acidic media (3.2 mol gcatalyst−1 L−1 h−1) and neutral media (2.2 mol gcatalyst−1 L−1 h−1) at −0.2 V. By virtue of the low-cost and outstanding performance, the Cu/NCNSs catalyst holds excellent promise for on-site and high-efficiency H2O2 production.
Author contributions
Cuizhu Ye: conceptualization, data curation, writing, original draft preparation. Yongfang Zhou and Hongying Li: methodology. Yi Shen: methodology, conceptualization, supervision, funding acquisition, and reviewing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The project was financially supported by the National Natural Science Foundation of China (22279035), 111 Project (B17018), and a research grant (204-A021001) from the China-Singapore International Joint Research Institute.
References
- Z. Lin, Q. Zhang, J. Pan, C. Tsounis, A. A. Esmailpour, S. Xi, H. Y. Yang, Z. Han, J. Yun, R. Amal and X. Lu, Energy Environ. Sci., 2022, 15, 1172–1182 RSC.
- J. Liang, Y. Wang, Q. Liu, Y. Luo, T. Li, H. Zhao, S. Lu, F. Zhang, A. M. Asiri, F. Liu, D. Ma and X. Sun, J. Mater. Chem. A, 2021, 9, 6117–6122 RSC.
- P. Cao, X. Quan, X. Nie, K. Zhao, Y. Liu, S. Chen, H. Yu and J. G. Chen, Nat. Commun., 2023, 14, 172 CrossRef CAS PubMed.
- Y. Wang, Y. Zhou, Y. Feng and X. Yu, Adv. Funct. Mater., 2022, 32, 2110734 CrossRef CAS.
- X. Zeng, Y. Liu, X. Hu and X. Zhang, Green Chem., 2021, 23, 1466–1494 RSC.
- E. Zhang, L. Tao, J. An, J. Zhang, L. Meng, X. Zheng, Y. Wang, N. Li, S. Du, J. Zhang, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2022, 61(12), e202117347 CAS.
- M. Deng, J. Sun, A. Laemont, C. Liu, L. Wang, L. Bourda, J. Chakraborty, K. V. Hecke, R. Morent, N. D. Geyter, K. Leus, H. Chen and P. V. D. Voor, Green Chem., 2023, 25, 3069–3076 RSC.
- R. Zhou, T. Zhang, R. Zhou, S. Wang, D. Mei, A. Mai-Prochnow, J. Weerasinghe, Z. Fang, K. Ostrikov and P. J. Cullen, Green Chem., 2021, 23, 2977–2985 RSC.
- B. Yu, J. Diniz, K. Lofgren, Q. Liu, R. Mercado, F. Nichols, S. R. J. Oliver and S. Chen, ACS Sustainable Chem. Eng., 2022, 10, 15501–15507 CrossRef CAS.
- J. Chen, Q. Ma, X. Zheng, Y. Fang, J. Wang and S. Dong, Nat. Commun., 2022, 13, 2808 CrossRef CAS PubMed.
- S. Chen, Z. Chen, S. Siahrostami, D. Higgins, D. Nordlund, D. Sokaras, T. R. Kim, Y. Liu, X. Yan, E. Nilsson, R. Sinclair, J. K. Nørskov, T. F. Jaramillo and Z. Bao, J. Am. Chem. Soc., 2018, 140, 7851–7859 CrossRef CAS PubMed.
- Y. Wang, G. I. N. Waterhouse, L. Shang and T. Zhang, Adv. Energy Mater., 2021, 11, 2003323 CrossRef CAS.
- J. Gao, H. Yang, X. Huang, S.-F. Hung, W. Cai, C. Jia, S. Miao, H. M. Chen, X. Yang, Y. Huang, T. Zhang and B. Liu, Chem, 2020, 6, 658–674 CAS.
- R. Shen, W. Chen, Q. Peng, S. Lu, L. Zheng, X. Cao, Y. Wang, W. Zhu, J. Zhang, Z. Zhuang, C. Chen, D. Wang and Y. Li, Chem, 2019, 5, 2099–2110 CAS.
- K. Jiang, S. Back, A. J. Akey, C. Xia, Y. Hu, W. Liang, D. Schaak, E. Stavitski, J. K. Nørskov, S. Siahrostami and H. Wang, Nat. Commun., 2019, 10, 3997 CrossRef PubMed.
- C. Zhao, C. Shi, Q. Li, X. Wang, G. Zeng, S. Ye, B. Jiang and J. Liu, Mater. Today Energy, 2022, 24, 100926 CrossRef CAS.
- X. Yang, Y. Zeng, W. Alnoush, Y. Hou, D. Higgins and G. Wu, Adv. Mater., 2022, 34, 2107954 CrossRef CAS PubMed.
- Y. Pang, K. Wang, H. Xie, Y. Sun, M.-M. Titirici and G.-L. Chai, ACS Catal., 2020, 10, 7434–7442 CrossRef CAS.
- S. Siahrostami, S. J. Villegas, A. H. Bagherzadeh Mostaghimi, S. Back, A. B. Farimani, H. Wang, K. A. Persson and J. Montoya, ACS Catal., 2020, 10, 7495–7511 CrossRef CAS.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS PubMed.
- Y. Jiang, P. Ni, C. Chen, Y. Lu, P. Yang, B. Kong, A. Fisher and X. Wang, Adv. Energy Mater., 2018, 8, 1801909 CrossRef.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- Y. Wang, F.-L. Hu, Y. Mi, C. Yan and S. Zhao, Chem. Eng. J., 2021, 406, 127135 CrossRef CAS.
- L. Zhao, Y. Zhang, L.-B. Huang, X.-Z. Liu, Q.-H. Zhang, C. He, Z.-Y. Wu, L.-J. Zhang, J. Wu, W. Yang, L. Gu, J.-S. Hu and L.-J. Wan, Nat. Commun., 2019, 10, 1278 CrossRef PubMed.
- L. Hu, A. He, X. Shen, Q. Gu, J. Zheng, Z. Wu, Y. Jiang, X. Wang, J. Xu, Y. Kan and F. Xu, Green Chem., 2022, 24, 6931–6944 RSC.
- S. K. Kaiser, Z. Chen, D. Faust Akl, S. Mitchell and J. Pérez-Ramírez, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- H. Sun, M. Wang, X. Du, Y. Jiao, S. Liu, T. Qian, Y. Yan, C. Liu, M. Liao, Q. Zhang, L. Meng, L. Gu, J. Xiong and C. Yan, J. Mater. Chem. A, 2019, 7, 20952–20957 RSC.
- J. Han, H. Bao, J.-Q. Wang, L. Zheng, S. Sun, Z. L. Wang and C. Sun, Appl. Catal., B, 2021, 280, 119411 CrossRef CAS.
- M. Zhao, H. Liu, H. Zhang, W. Chen, H. Sun, Z. Wang, B. Zhang, L. Song, Y. Yang, C. Ma, Y. Han and W. Huang, Energy Environ. Sci., 2021, 14, 6455–6463 RSC.
- Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS PubMed.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S. Yu and H. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS PubMed.
- C. Liu, H. Li, F. Liu, J. Chen, Z. Yu, Z. Yuan, C. Wang, H. Zheng, G. Henkelman, L. Wei and Y. Chen, J. Am. Chem. Soc., 2020, 142, 21861–21871 CrossRef CAS PubMed.
- Y. Xiong, J. Dong, Z.-Q. Huang, P. Xin, W. Chen, Y. Wang, Z. Li, Z. Jin, W. Xing, Z. Zhuang, J. Ye, X. Wei, R. Cao, L. Gu, S. Sun, L. Zhuang, X. Chen, H. Yang, C. Chen, Q. Peng, C.-R. Chang, D. Wang and Y. Li, Nat. Nanotechnol., 2020, 15, 390–397 CrossRef CAS PubMed.
- L. Yan, X. Cheng, Y. Wang, Z. Wang, L. Zheng, Y. Yan, Y. Lu, S. Sun, W. Qiu and G. Chen, Mater. Today Energy, 2022, 24, 100931 CrossRef CAS.
- W. Chen, X. Luo, T. J. A. Slater, Y. Zhou, S. Ling, R. Bao, J. Alves Fernandes, J. Wang and Y. Shen, J. Mater. Chem. A, 2020, 8, 25959–25969 RSC.
- Y. Zhou, T. J. A. Slater, X. Luo and Y. Shen, Appl. Catal., B, 2023, 324, 122218 CrossRef CAS.
- X. Zhao, Q. Yin, X. Mao, C. Cheng, L. Zhang, L. Wang, T.-F. Liu, Y. Li and Y. Li, Nat. Commun., 2022, 13, 2721 CrossRef CAS PubMed.
-
L. Li, MPhil, South China University of Technology, 2018.
- C. Tang, Y. Jiao, B. Shi, J. Liu, Z. Xie, X. Chen, Q. Zhang and S. Qiao, Angew. Chem., Int. Ed., 2020, 59, 9171–9176 CrossRef CAS PubMed.
- Z. Lu, G. Chen, S. Siahrostami, Z. Chen, K. Liu, J. Xie, L. Liao, T. Wu, D. Lin, Y. Liu, T. F. Jaramillo, J. K. Nørskov and Y. Cui, Nat. Catal., 2018, 1, 156–162 CrossRef CAS.
- B. Li, C. Zhao, J. Liu and Q. Zhang, Adv. Mater., 2019, 31, 1808173 CrossRef PubMed.
- D. A. Kuznetsov, Z. Chen, P. M. Abdala, O. V. Safonova, A. Fedorov and C. R. Müller, J. Am. Chem. Soc., 2021, 143, 5771–5778 CrossRef CAS PubMed.
- H. Xiao, B. Li, M. Zhao, Y. Li, T. Hu, J. Jia and H. Wu, Chem. Commun., 2021, 57, 4118–4121 RSC.
- S. Chen, Z. Chen, S. Siahrostami, T. R. Kim, D. Nordlund, D. Sokaras, S. Nowak, J. W. F. To, D. Higgins, R. Sinclair, J. K. Nørskov, T. F. Jaramillo and Z. Bao, ACS Sustainable Chem. Eng., 2018, 6, 311–317 CrossRef CAS.
- C.-Y. Chen, C. Tang, H.-F. Wang, C.-M. Chen, X. Zhang, X. Huang and Q. Zhang, ChemSusChem, 2016, 9, 1194–1199 CrossRef CAS PubMed.
- V. Perazzolo, C. Durante, R. Pilot, A. Paduano, J. Zheng, G. A. Rizzi, A. Martucci, G. Granozzi and A. Gennaro, Carbon, 2015, 95, 949–963 CrossRef CAS.
- X. Zhao, Y. Wang, Y. Da, X. Wang, T. Wang, M. Xu, X. He, W. Zhou, Y. Li, J. N. Coleman and Y. Li, Natl. Sci. Rev., 2020, 7, 1360–1366 CrossRef CAS PubMed.
- N. Wang, X. Zhao, R. Zhang, S. Yu, Z. H. Levell, C. Wang, S. Ma, P. Zou, L. Han, J. Qin, L. Ma, Y. Liu and H. L. Xin, ACS Catal., 2022, 12, 4156–4164 CrossRef CAS.
- S. Mehta, D. Gupta and T. C. Nagaiah, ChemElectroChem, 2022, 9, e202101336 CrossRef CAS.
- P. T. Smith, Y. Kim, B. P. Benke, K. Kim and C. J. Chang, Angew. Chem., Int. Ed., 2020, 59, 4902–4907 CrossRef CAS PubMed.
- E. Jung, H. Shin, B.-H. Lee, V. Efremov, S. Lee, H. S. Lee, J. Kim, W. Hooch Antink, S. Park, K.-S. Lee, S.-P. Cho, J. S. Yoo, Y.-E. Sung and T. Hyeon, Nat. Mater., 2020, 19, 436–442 CrossRef CAS PubMed.
- J. S. Jirkovský, I. Panas, E. Ahlberg, M. Halasa, S. Romani and D. J. J. Schiffrin, J. Am. Chem. Soc., 2011, 133(48), 19432–19441 CrossRef PubMed.
- I. Yamanaka, R. Ichihashi, T. Iwasaki, N. Nishimura, T. Murayama, W. Ueda and S. Takenaka, Electrochim. Acta, 2013, 108, 321–329 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *abc
*abc