
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA deep learning model for type II polyketide natural product prediction without sequence alignment†
Jiaquan
Huang‡
 a,
Qiandi
Gao‡
a,
Ying
Tang
b,
Yaxin
Wu
a,
Heqian
Zhang
*a and
Zhiwei
Qin
*a
a,
Qiandi
Gao‡
a,
Ying
Tang
b,
Yaxin
Wu
a,
Heqian
Zhang
*a and
Zhiwei
Qin
*a
aCenter for Biological Science and Technology, Advanced Institute of Natural Sciences, Beijing Normal University, Zhuhai, Guangdong 519087, China. E-mail: z.qin@bnu.edu.cn; zhangheqian@bnu.edu.cn
bInternational Academic Center of Complex Systems, Advanced Institute of Natural Sciences, Beijing Normal University, Zhuhai, Guangdong 519087, China
First published on 30th August 2023
Abstract
Natural products are important sources for drug development, and the accurate prediction of their structures assembled by modular proteins is an area of great interest. In this study, we introduce DeepT2, an end-to-end, cost-effective, and accurate machine learning platform to accelerate the identification of type II polyketides (T2PKs), which represent a significant portion of the natural product world. Our algorithm is based on advanced natural language processing models and utilizes the core biosynthetic enzyme, chain length factor (CLF or KSβ), as computing inputs. The process involves sequence embedding, data labeling, classifier development, and novelty detection, which enable precise classification and prediction directly from KSβ without sequence alignments. Combined with metagenomics and metabolomics, we evaluated the ability of DeepT2 and found this model could easily detect and classify KSβ either as a single sequence or a mixture of bacterial genomes, and subsequently identify the corresponding T2PKs in a labeled categorized class or as novel. Our work highlights deep learning as a promising framework for genome mining and therefore provides a meaningful platform for discovering medically important natural products. The DeepT2 is available at GitHub repository: https://github.com/Qinlab502/deept2.
Introduction
Bacterial type II polyketides (T2PKs) are valuable natural products with potent biological activities and comprise a family of structurally related molecules.1,2 Illustrative examples include tetracycline, doxorubicin and plicamycin. They are primarily biosynthesized by type II polyketide synthases (T2 PKSs), which catalyze the formation of carbon skeletons in an ordered manner.3 The biosynthetic system is characterized by a minimal set of gene products, of which the most crucial enzymes are the monofunctional heterodimeric β-ketosynthase pair KSα/KSβ, which catalyze the iterative Claisen condensation using acetyl- and malonyl-CoA as building blocks for chain elongation and determine the chain length and overall topology (Fig. 1A), respectively. In addition, a malonyl transacylase (MT) and an acyl carrier protein (ACP) were taken together with KSα/KSβ to constitute the minimal T2 PKS systems.4 The core skeleton of T2PKs is highly correlated with the KSα/KSβ protein structure. Hillenmeyer et al. observed correlations between KSβ protein phylogeny and the building blocks of T2PK skeletons,5 while Chen et al. utilized KSβ as a biomarker to construct a coevolutionary statistical model (phylogenetic tree) to expand the T2PK biosynthetic landscape.6 However, the above and other methods7–9 frequently rely on multiple sequence alignments, which are time-consuming and do not effectively represent protein structural information.10,11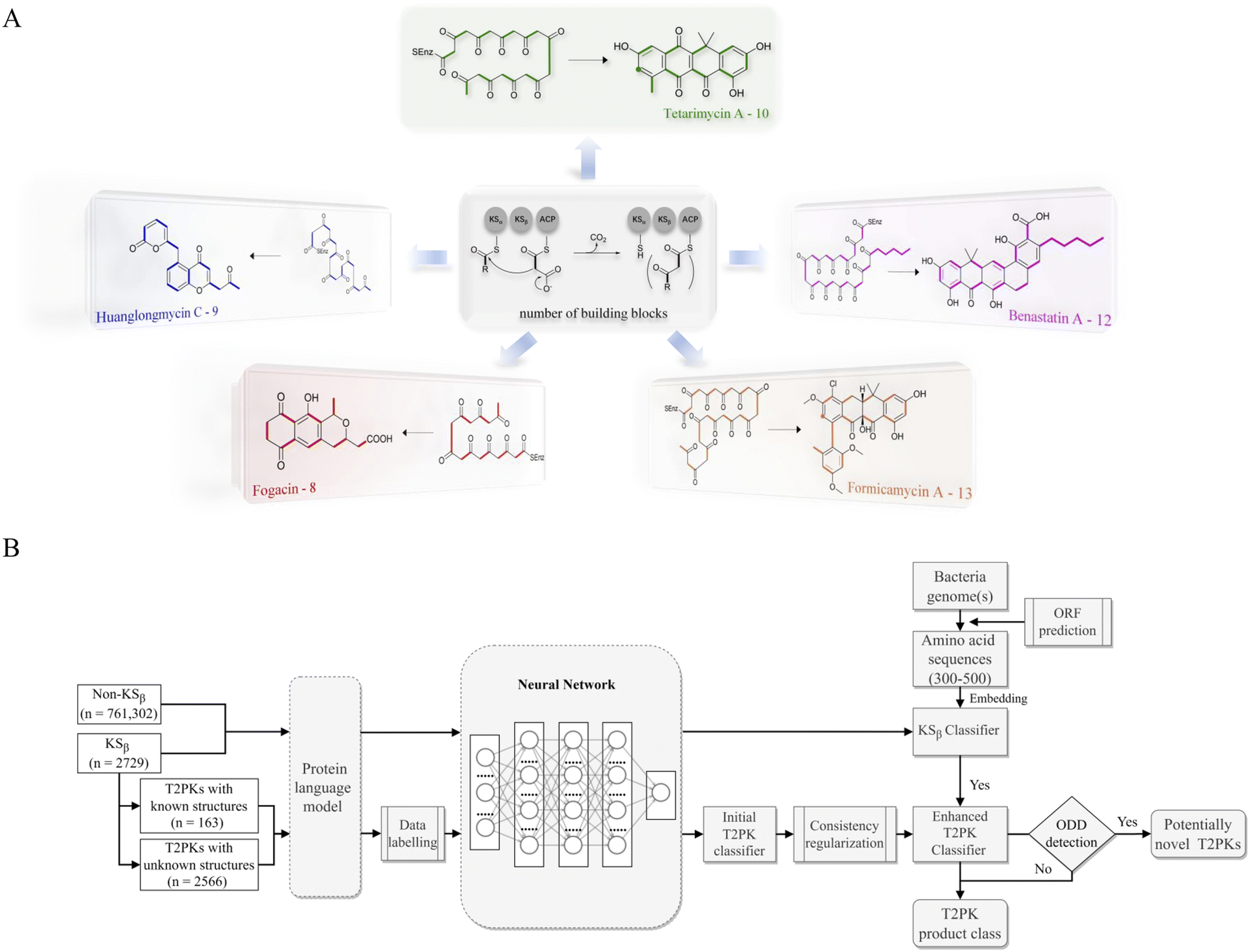 | ||
| Fig. 1 Illustration of the representative type II polyketides and their biosynthetic intermediates (A) and the overall architecture of DeepT2 developed in this study (B). | ||
Several artificial intelligence (AI)-based natural product discovery models have been proposed in recent years due to rapid data accumulation and digital transformation as well as the accelerated development of AI technology.12–17 Among these frameworks, deep learning has demonstrated exceptional performance in classification tasks, specifically in the area of distinguishing new and unseen data.18–20 For instance, certain deep learning-based tools have demonstrated high efficiency and scalability in predicting natural product classes.13,21 However, these tools have limitations when it comes to identifying enzyme sequences that may be involved in the biosynthesis of natural products with novel carbon skeletons. More recently, protein language models (PLMs) based on self-supervised learning have shown remarkable ability to convert individual protein sequences into embeddings that describe the homology between multiple protein sequences and potentially capture physicochemical information not encoded by the existing methods.22–24 The application of general PLMs to convert sequences into embeddings, which serve as inputs for deep learning models, effectively overcomes the few-shot learning challenge for specific biomolecular property predictions.25,26 In addition, leveraging the large amount of unlabeled data available through a semi-supervised framework can further improve model performance.27,28 Finally, conducting novelty detection on the distribution of sequence-to-chemical feature vectors is a viable approach.29 These advancements inspired us to move forward in understanding T2PKS with PLM, training a robust model with unexplored sequences stored in metagenome data, and eventually finding an effective linker to connect their biosynthetic enzymes and probable chemical structures.
To gain a better approach for the discovery of T2PKs, an end-to-end model named DeepT2 was developed in this study. This model employs multiple classifiers to expedite the translation from protein sequences to the T2PK product class and identify any potential new compounds beyond the established groups. Notably, the model is free of sequence alignment and comprises four main components: (i) sequence embedding: the protein sequences were converted into vector embeddings using a pretrained PLM called EMS-2;24 (ii) data labeling: the KSβ dataset with known corresponding chemical structures was initially split into five classes for labeling based on the total number of biosynthetic building blocks, which was later reclassified into nine classes through dimension reduction and clustering processes; (iii) classifier development: this was used for both KSβ sequences and T2PKs classification; and (iv) novelty detection: Mahalanobis distance-based novelty detection30 was applied to identify any potential new compounds beyond the nine established groups. Remarkably, we leveraged DeepT2 to detect KSβ from microbial genomes and successfully identified four T2PKs as categorized in our classifiers. This work paves a promising avenue to further explore the potential of the existing reservoirs of T2PK biosynthetic gene clusters (BGCs) and therefore expand the chemical space of this medically important natural product family.
Results
DeepT2 model architecture
As shown in Fig. 1, the purpose of this study was to develop a methodology for predicting the T2PK class using KSβ sequences from bacterial genomes as input. To achieve this, we first used an ensemble of multiple classifiers to determine whether a given protein sequence belongs to KSβ, assessed whether it fell within or outside the existing labeling product classes, and eventually predicted the product class. General protein language models, including SeqVec, ESM-1b, ESM-2, ProtT5-XL-U50 and ProtBert-BFD, were employed to maximize the use of limited labeled, unlabeled, and non-KSβ sequences by converting them into embeddings containing structural representations using the idea of transfer learning. By pretraining on such datasets, PLM can effectively learn general patterns and features that are present in imbalanced data. In this work, the terms of ‘labeled KSβ’ and ‘unlabeled KSβ’ indicate whether their corresponding chemical structures are known. KSβ and T2PK classifiers were trained by the datasets from the specific protein sequences and chemical structures. To construct a robust T2PK classifier, we applied supervised UMAP (uniform manifold approximation and projection) and HDBCAN (hierarchical density-based spatial clustering of applications with noise) on the KSβ embedding to generate more appropriate class labels and trained the model with unlabeled data on the basis of consistency regularization. Furthermore, the Mahalanobis distance-based algorithm was applied on each feature layer of the T2PK classifier to perform novelty detection and avoid the problem of overconfident. The methodology enabled us not only to classify the known group of T2PKs but also to detect potential novel classes of T2PKs from unknown KSβ protein sequences. Detailed results of each process are presented below. The pseudocode for this work has been summarized in Table 1.

|
Development of the KSβ classifier
We obtained a collection of 163 labeled KSβ sequences with known corresponding natural product structures (Table S1†) as well as additional 2566 unlabeled KSβ sequences sourced from the RefSeq database, whose associated natural product structures remain unknown (Table S2†). A total of 2729 (163 + 2566) KSβ sequences and 761![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 302 non-KSβ sequences were then split into training, validation, and test datasets, as described in the materials and methods section. Prior to constructing the classifier, we employed five general PLMs, including SeqVec, ESM-1b, ESM-2, ProtT5-XL-U50 and ProtBert-BFD respectively, to vectorize each protein sequence for embedding and we observed that the learned representations of ESM-2 and ProtT5-XL-U50 exhibited the best performance compared with the others in distinguishing KSβ from non-KSβ sequences, as revealed by the results of dimension reduction (Fig. S1†). In this study, we favored ESM-2 because it has the largest parameter size (over 3 billion) and has better performance on the T2PK classifier (see lateral session). Next, we trained the KSβ embeddings obtained by the PLMs using four machine learning algorithms, including random forest, XGBoost, support vector machine (SVM) and multilayer perceptron (MLP), and found that MLP and SVM achieved the best results, with an AUROC of 1 and an F1 score of 1 in the classification on the test dataset (Table S3†).
302 non-KSβ sequences were then split into training, validation, and test datasets, as described in the materials and methods section. Prior to constructing the classifier, we employed five general PLMs, including SeqVec, ESM-1b, ESM-2, ProtT5-XL-U50 and ProtBert-BFD respectively, to vectorize each protein sequence for embedding and we observed that the learned representations of ESM-2 and ProtT5-XL-U50 exhibited the best performance compared with the others in distinguishing KSβ from non-KSβ sequences, as revealed by the results of dimension reduction (Fig. S1†). In this study, we favored ESM-2 because it has the largest parameter size (over 3 billion) and has better performance on the T2PK classifier (see lateral session). Next, we trained the KSβ embeddings obtained by the PLMs using four machine learning algorithms, including random forest, XGBoost, support vector machine (SVM) and multilayer perceptron (MLP), and found that MLP and SVM achieved the best results, with an AUROC of 1 and an F1 score of 1 in the classification on the test dataset (Table S3†).
Relabeling 163 T2PKs with constrained optimization approach
Data labeling is crucial for data preprocessing in machine learning, especially for supervised learning.31 This is also the most challenging task in this work. Based on prior knowledge, 163 KSβ embeddings with known chemical structures were categorized into five groups according to the building block number of their corresponding to T2PK main skeleton, namely, 8, 9, 10, 12 and 13 (Table S1†). However, despite applying ESM-2 to the 163 KSβ sequences, imprecise representation of the distribution pattern of the 5 label embeddings was observed (Fig. S2A†), probably due to the inadequate fittings between some of the class labels and the protein embeddings. To address this issue, a constrained optimization approach was developed to correct the improper class labels for each sample and further separate their local features from global features to generate new class labels (Fig. 2B). Specifically, we employed supervised UMAP and HDBSCAN as regularization techniques to constrain the latent distribution of KSβ embeddings and automatically tune UMAP and HDBSCAN hyperparameters using a labeling cost function to assign more appropriate class labels to the embeddings. Of note, the supervised UMAP opted for this study incorporates compound skeleton labels into the optimization process to ensure that the resulting reduced-dimensional space consistently captures the features of the compound skeleton. It is worth noting that unsupervised UMAP, in contrast to its supervised counterpart, lacks access to labeled data during the optimization process. Consequently, this absence of label supervision poses a challenge in ensuring the consistent representation of compound skeleton features within the reduced-dimensional space.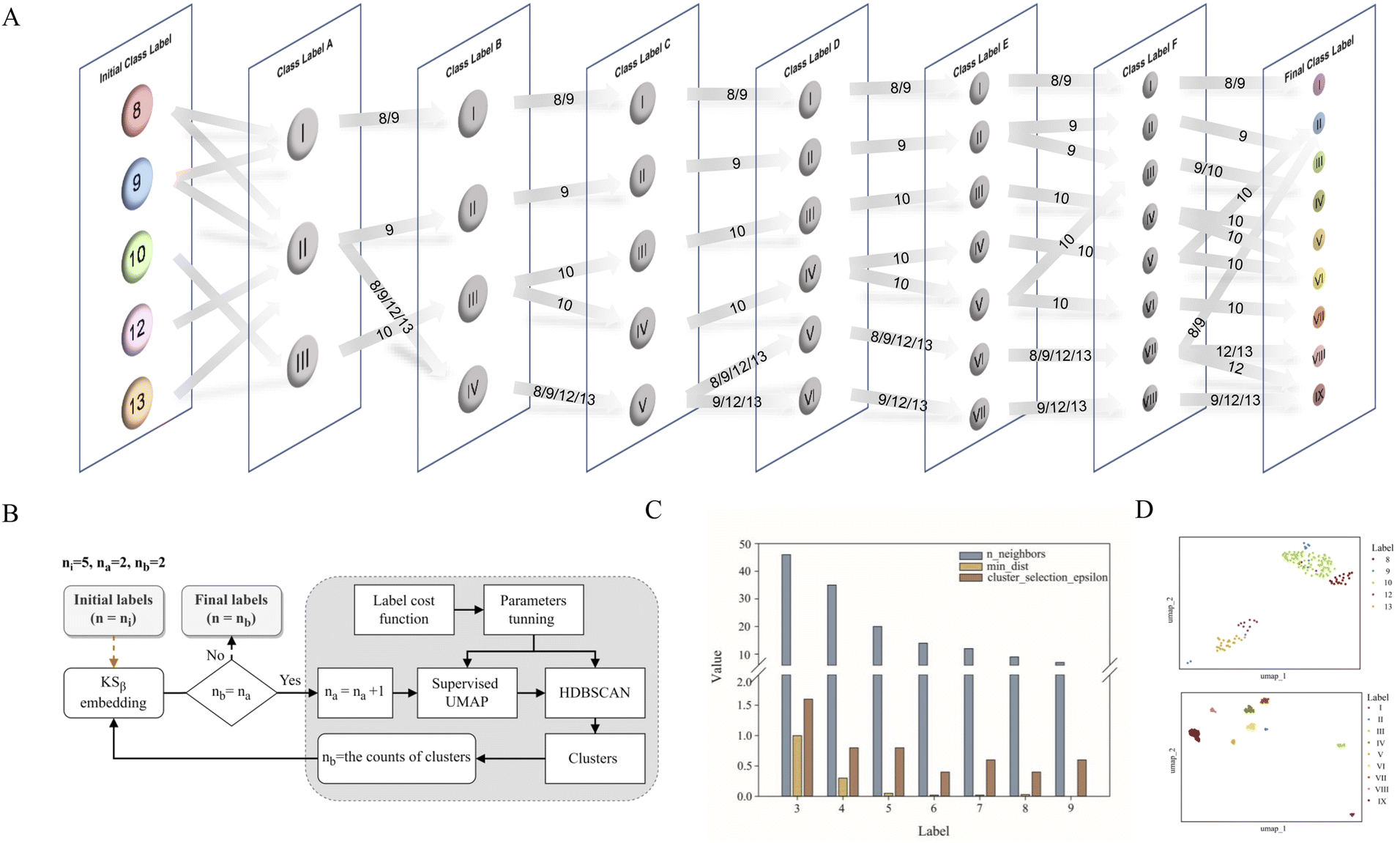 | ||
| Fig. 2 The flowchart showing the process of KSβ labeling (A) and the applications of supervised UMAP and HDBSCAN algorithms (B), the parameter optimizations during the process of A and B (C), and the supervised UMAP comparison of T2PK class labeling generated by five manually annotated class labels (up, D) and nine refined class labels (bottom, D). | ||
As illustrated in Fig. 2A, the restructuring process of KSβ embeddings initially reset the counts of predefined labels to 3, followed by the automatic adjustment of the n_neighbors and min_dist parameters to refine the space and improve its alignment with these 3 class labels. HDBSCAN was then applied to cluster the similar data points and assign new class labels to them. Certain previously assigned KSβ sequences were bifurcated and merged into a new label. For example, in class label A column, label I is from partial initial label 8 and 9; label II is from partial initial label 8 and 9, and entire initial 12 and 13; and label III is from entire initial label 10 (Fig. 2A, Table S4†). Upon increasing the number of predefined labels, the approach tuned the n_neighbors and min_dist parameters to decrease their values, which allowed HDBSCAN to recognize and assign new labels to smaller and more localized features in the data space (Fig. 2C and S3†). This iterative process continued until the number of class labels assigned to the 163 KSβ embeddings reached 9, after which no more labels could be generated or some data points were identified as noise. To evaluate the distribution patterns of the 9 autogenerated labels, the supervised UMAP technique was employed again, revealing that these labels could represent the T2PK biosynthetic logics more accurately in the real world (Fig. 2D).
Development of the T2PK classifier
As described in the previous session, four machine learning algorithms (random forest, XGBoost, SVM, and MLP with grid search hyperparameter tuning) were employed to train the initial T2PK classifiers on KSβ embeddings. The classifiers were trained on two distinct sets of class labels: one comprising five manually annotated class labels and another consisting of nine refined class labels (Fig. 2D). Due to the imbalanced nature of our dataset, the F1 score and confusion matrix were employed to assess and compare the performance of the classifiers trained on these two different sets of class labels. As outlined in Table 2, the MLP classifier yielded an F1 score of 0.89 on the test set when utilizing the nine refined class labels, whereas the F1 scores of the random forest, XGBoost, and SVM classifiers were 0.77, 0.73, and 0.92, respectively. Notably, the classifiers trained on the five manually annotated class labels exhibited inferior performance compared with those trained on the nine refined class labels.| Classifier | Class number | TPR% | FPR% | Accuracy | Precision | Recall | F1-score |
|---|---|---|---|---|---|---|---|
| Initial classifier | |||||||
| Random forest | 5 classes | 55.00 | 8.88 | 0.76 | 0.49 | 0.55 | 0.51 |
| 9 classes | 76.11 | 2.81 | 0.79 | 0.80 | 0.76 | 0.77 | |
| XGBoost | 5 classes | 57.67 | 8.88 | 0.76 | 0.68 | 0.58 | 0.59 |
| 9 classes | 76.11 | 2.73 | 0.79 | 0.75 | 0.76 | 0.73 | |
| SVM | 5 classes | 62.67 | 7.02 | 0.79 | 0.80 | 0.63 | 0.66 |
| 9 classes | 96.76 | 0.68 | 0.94 | 0.91 | 0.97 | 0.92 | |
| MLP | 5 classes | 67.56 | 5.25 | 0.79 | 0.67 | 0.68 | 0.67 |
| 9 classes | 93.15 | 1.51 | 0.88 | 0.87 | 0.93 | 0.89 | |
|
|||||||
| Enhanced classifier | |||||||
| MLP | 5 classes | 66.67 | 6.38 | 0.82 | 0.91 | 0.67 | 0.70 |
| 9 classes | 97.78 | 0.43 | 0.97 | 0.99 | 0.98 | 0.98 | |
To leverage the 2566 unlabeled KSβ embeddings, a consistency regularization-based semi-supervised learning framework was adopted to train an enhanced MLP classifier based on the initial MLP classifier with two distinct sets of class labels. The enhanced MLP classifier was trained on both labeled and unlabeled data. In this process, the cross-entropy loss function was applied to the disturbed labeled data via Gaussian noise, while the mean-square error loss function was applied to both disturbed labeled and unlabeled data (Fig. 3A). This approach promoted the model to produce consistent predictions over time and resulted in a smoothed decision boundary, thereby enhancing the model's generalization performance on unseen data and alleviating overfitting. A clear disturbance in the initial classifier at the beginning is shown in Fig. 3B. Overall, the performance evaluation demonstrated that the enhanced MLP classifier trained with nine refined class labels attained an F1 score of 0.98 on the test set, an increase of 0.09 compared with the initial one, indicating state-of-the-art performance.
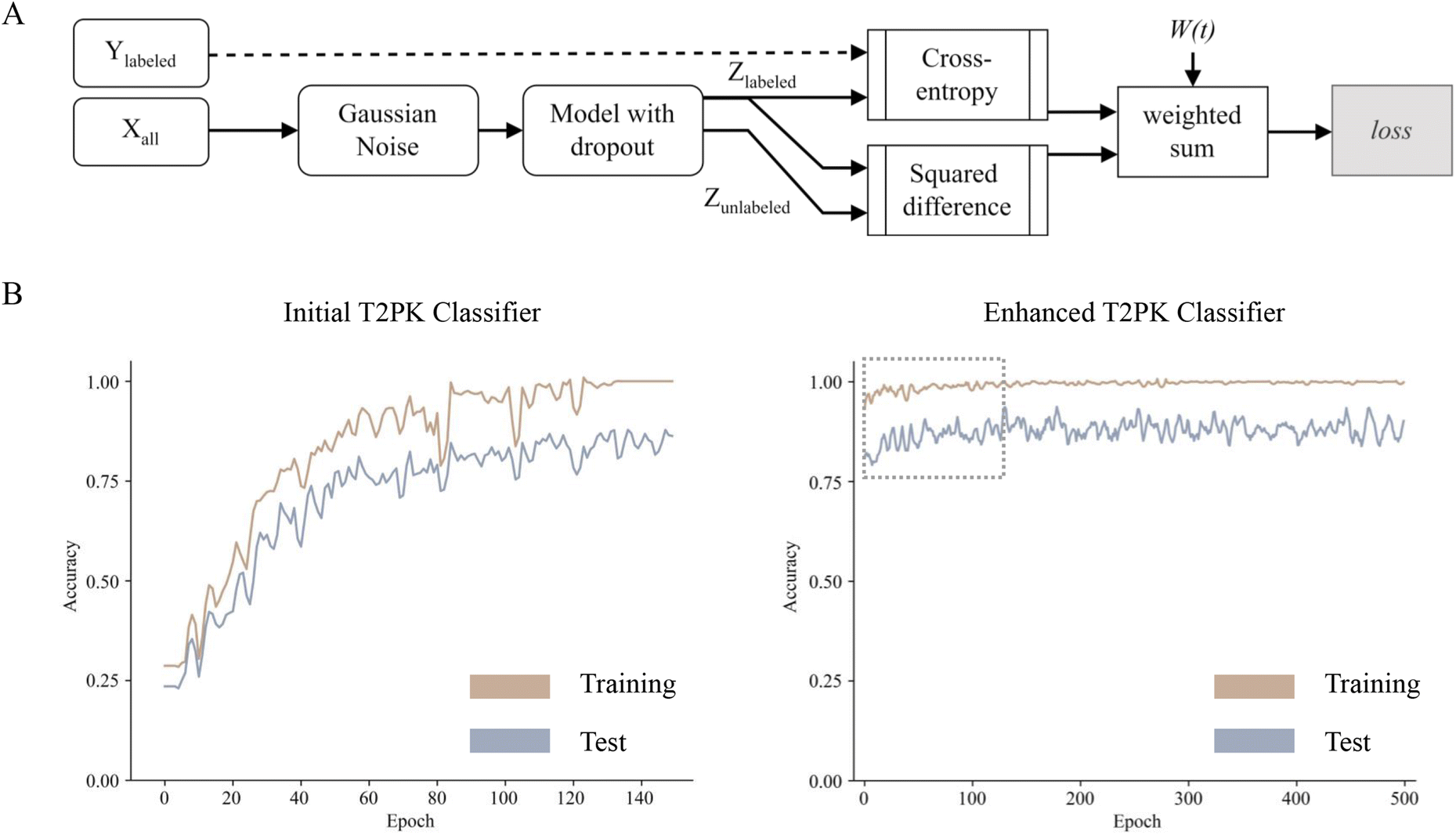 | ||
| Fig. 3 The flowchart showing the development of T2PK classifier using consistency regularization-based semi-supervised learning approach (A); plots showing the accuracies of prediction for the initial (left, B) and enhanced (right, B) T2PK classifiers. | ||
Out-of-distribution detection (ODD) for new T2PKs
Softmax-based classifiers have been criticized for generating overconfident posterior distributions when presented with ODD data.32 In this study, ODD data refer to KSβ sequences that do not belong to any of the nine refined classes as described above. To overcome this limitation, we incorporated Mahalanobis distance-based scores (MDS) and anomaly detection techniques inspired by the generalized ODD framework:29where the Mahalanobis distance-based scores, denoted as M(x), is determined by evaluating the empirical class mean
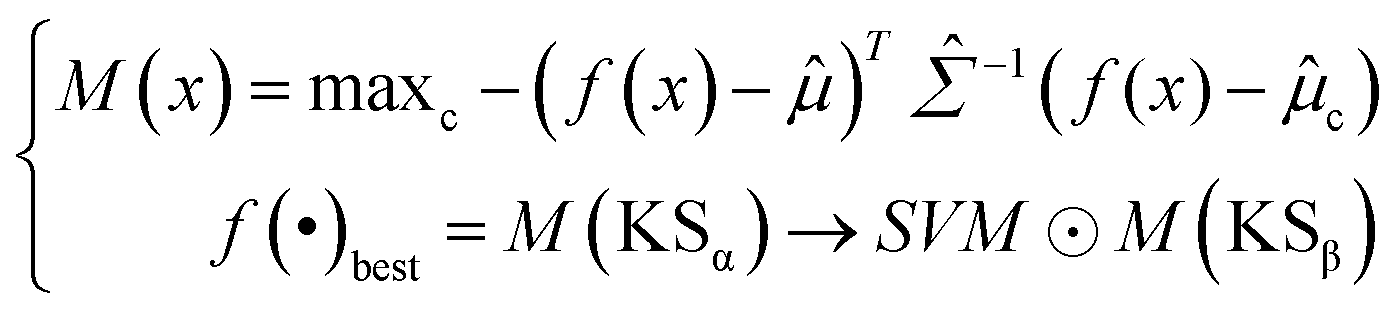
![[small mu, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e0b3.gif) and covariance
and covariance ![[capital Sigma, Greek, circumflex]](https://www.rsc.org/images/entities/i_char_e139.gif) of the training samples; ⊙ represents the utilization of the one-class SVM to detect M(KSβ) for each feature layer.
of the training samples; ⊙ represents the utilization of the one-class SVM to detect M(KSβ) for each feature layer.
Of note, we designated 163 KSα sequences as ODD data and 163 KSβ sequences as in-distribution (ID) data and extracted feature vectors from the neural network consisting of input, hidden (n = 3), and output layers of the MLP classifier to compute the MDS for both ID and ODD data (Fig. 4A). We evaluated the classification performance of each layer using a one-class SVM on the ID and ODD datasets and found that the hidden layer 1 exhibited superior performance in detecting ODD data (Fig. S4†). Further, we present the MDS distribution of 163 labeled KSβ sequences (ID data) and 2566 non-labeled KSβ sequences within hidden layer 1. In Fig. 4B, some data points within the unlabeled KSβ sequences conspicuously diverge from the cluster of the ID data.
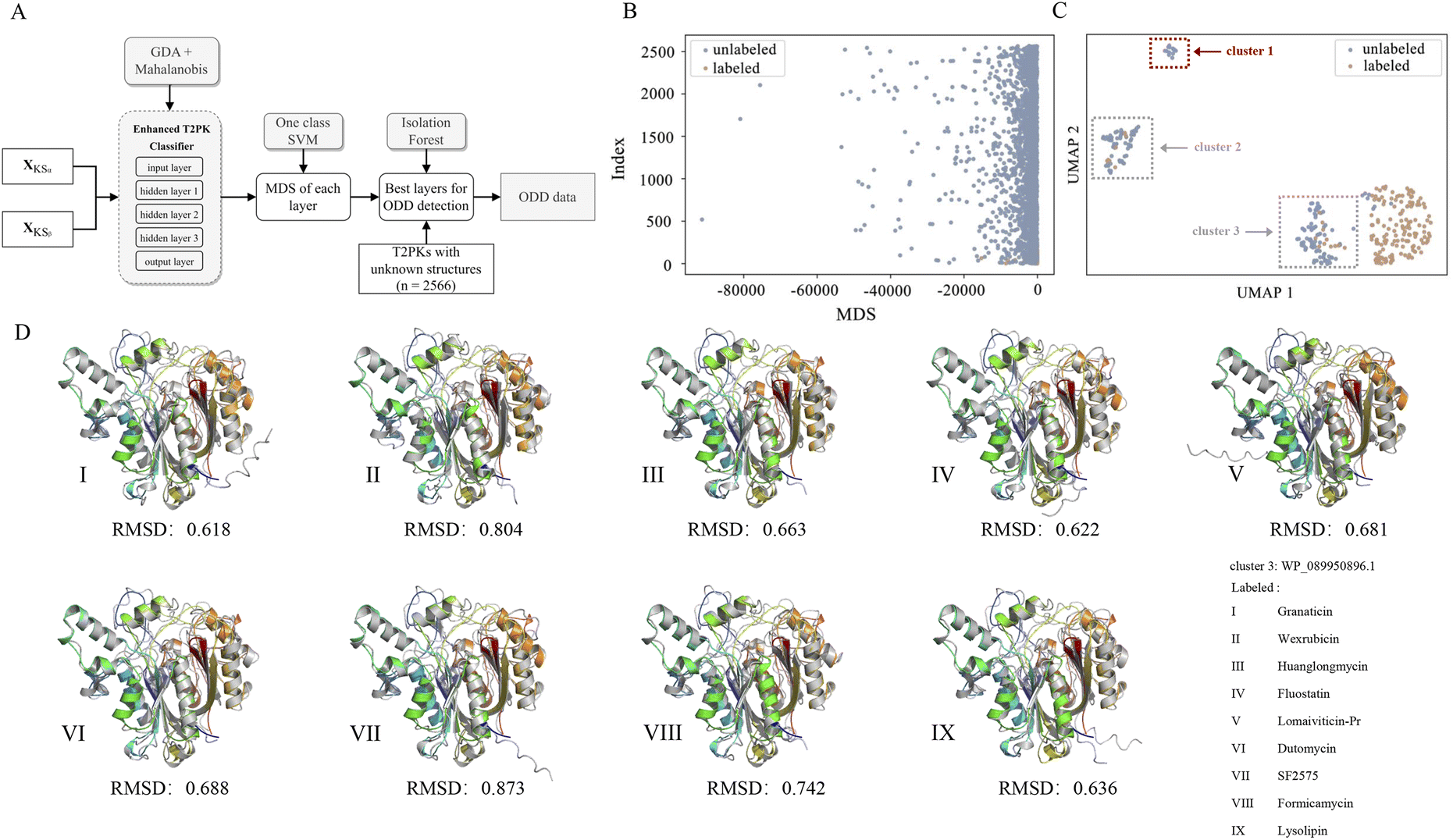 | ||
| Fig. 4 The flowchart showing the process for detecting novel T2PKs (A), the MDS distribution of 163 labeled KSβ and 2566 unlabeled KSβ within hidden layer 1 (B), ODD clusters (C), and the representative in silico predictions of the protein structures generated by ESMFold (D), grey structure indicates the labeled KSβ for the biosynthesis of known T2PK, while colour structures indicate unlabeled KSβ from cluster 1 in C, respectively. | ||
Next, we utilized the MDS data generated from hidden layer 1 to train an isolation forest model. This model was then employed to detect ODD data within a larger dataset comprising unlabeled sequences:
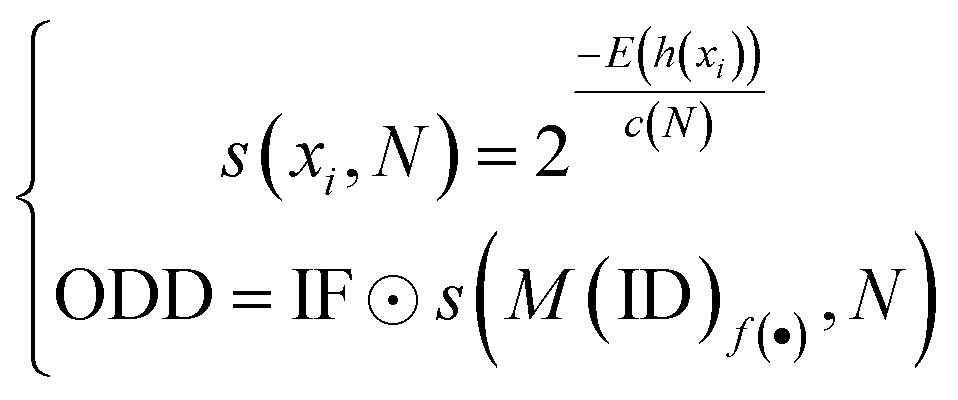
To facilitate a more comprehensive visualization of the distribution of abnormal datasets, we combined the labeled and abnormal datasets and conducted UMAP dimension reduction. Through this process, we were able to identify three clusters, labeled as ODD clusters, which encompassed a total of 164 abnormal data points. On one hand, certain data points from ODD clusters 2 and 3 were found to overlap with the labeled dataset, as indicated by the grey dotted box in Fig. 4C. This occurrence does not imply inaccuracies in the detection process; rather, it suggests that the KSβ from these two clusters share some similarities in their embeddings with the labeled sequences. This observation further suggests that the corresponding chemical structures of these data points may possess common characteristics with the structurally known T2PKs. On the other hand, we noticed that ODD cluster 1, comprising 13 sequences, was completely separated from the labeled dataset and situated at a considerable distance, as shown in Fig. 4C. Based on our hypothesis, the KSβ proteins within this ODD cluster may exhibit novel catalytic domains that differ from the previously labeled KSβ proteins. It is plausible to assume that these novel domains are potentially involved in the biosynthesis of previously undiscovered T2PKs.
To test this hypothesis, we employed ESMFold to conduct in silico predictions of protein structures for all data points within ODD cluster 1. We then calculated the root mean square deviation (RMSD) between the predicted structures of the 13 unlabeled KSβ proteins and the 163 labeled KSβ proteins. The average RMSD value between the protein structures in ODD cluster 1 and those in classes IV, V, VI, and VII was found to be 0.58 Å (Fig. S5 and Table S5†). Table S6† provides the calculated RMSD values between ODD cluster 1 and classes IV–VII, and based on these values, a threshold of 0.4 can be set to distinguish between intra-class and inter-class structures. This indicates that, for the exploration of novel T2PKs, particular attention should be given to KSβ proteins with an RMSD value exceeding 0.4 between the ODD cluster and other classes, as depicted in Fig. 4D.
Predicting T2PKs from bacterial genomes
So far, our findings have demonstrated the effectiveness of DeepT2 in accelerating the identification of T2PKs. We were therefore motivated to explore the advantages of this model in uncovering the potential of any T2PKs produced by actinomycetes isolated or stored in our laboratory. As such, we sequenced 5 Streptomyces strains and confirmed their genomic independence through average nucleotide identity comparison. Following that, we employed DeepT2, DeepBGC, and antiSMASH33 to predict the T2PK produced from these 5 strains. Our findings demonstrate that DeepT2 surpasses DeepBGC and antiSMASH in terms of T2PK prediction, as the latter two tools primarily focus on identifying the BGCs rather than specifically targeting T2PKSs. In addition, we investigated the ability of these tools to handle metagenomic sequences by combining the 5 genomes and subjecting them to the aforementioned tools. The results revealed that only DeepT2 is capable of handling metagenomic input, yielding identical outputs to those obtained from individual genome inputs. On the other hand, neither the web server nor the local version of antiSMASH allows for direct submission of metagenomic sequences. Furthermore, we evaluated the performance of these tools on single-gene input by extracting candidate KSβ sequences from the 5 genomes and submitting them as individual sequences to the mentioned tools. It was observed that only DeepT2 supported single-gene input and produced accurate predictions (Table S7†).In this way, we selected three top closest ID sample as predicted T2PK for the unknown KSβ input. As an overall result, 10 KSβ protein sequences were detected from 5 Streptomyces genome that fell into four classes (Table S7†), and the corresponding T2PKs were closest to alnumycin, granaticin and frenolicin in class I,34–36 polyketomycin, dutomycin and LL-D49194 in class VI,37–39 fasamycin, formicamycin and Sch in class VIII,40–42 and lysolipin, BE-24566B and anthrabenzoxocinone in class IX,43–45 respectively (Fig. 5 and Table S7†). To confirm this prediction, the strains were then inoculated and their metabolites were analyzed by liquid chromatography high-resolution mass spectrometry. As the result, alnumycin from WY86, polyketomycin from WY13 and lysolipin from PS14 were observed (Fig. 5 and S6†), whereas fasamycin from S. kanamyciticus was not detected under laboratory conditions as described in a recent study.46
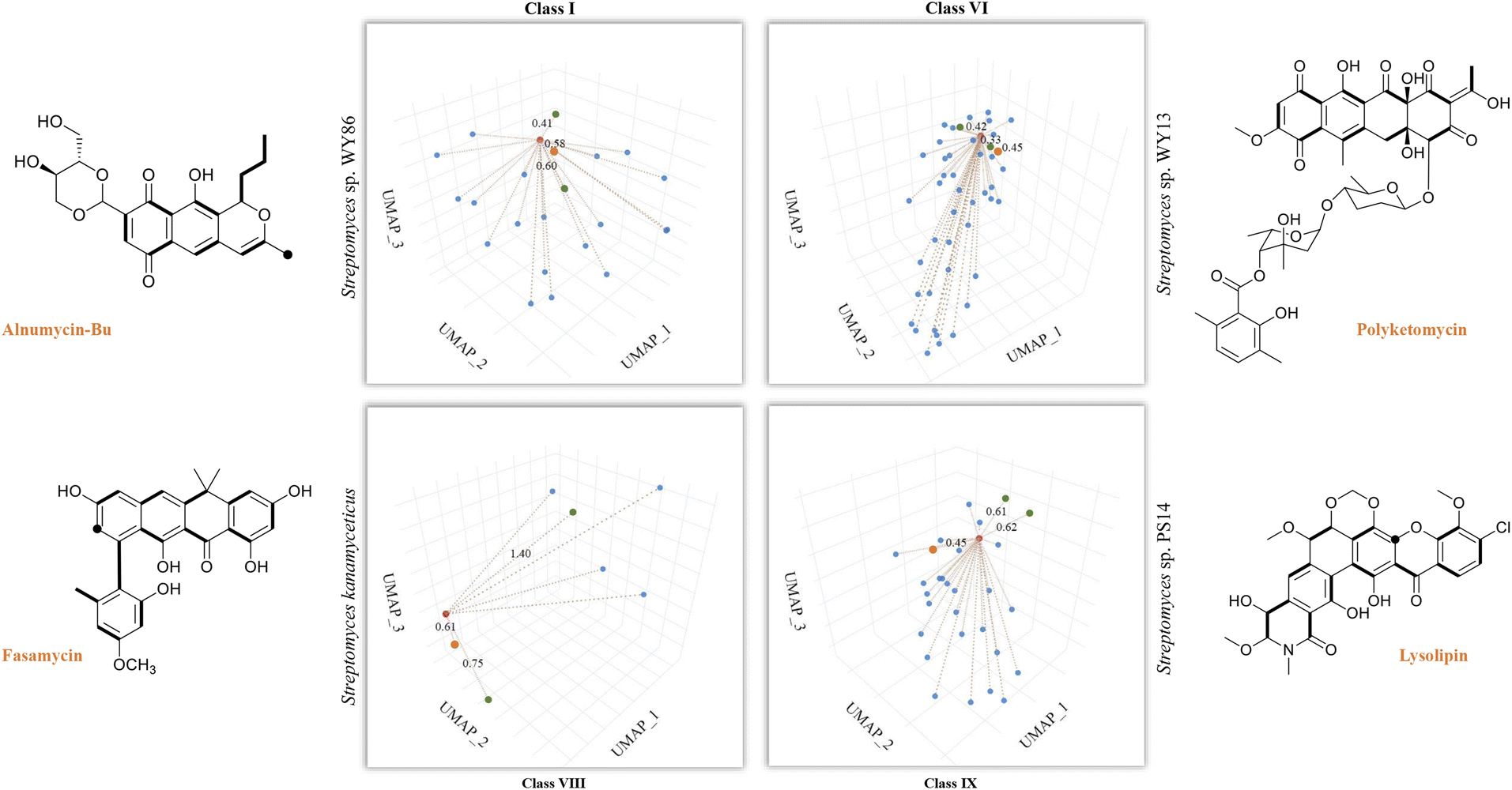 | ||
| Fig. 5 T2PK prediction from bacterial genomes. DeepT2 was performed and the identified compounds alnumycin A, polyketomycin, and lysolipin were subsequently confirmed via high-resolution mass spectra (Fig. S6†). Euclidean distances for each predicted candidate with top 3 similar T2PKs were annotated beside the dash lines (red, T2PKs to be detected; orange, the most similar T2PKs experimentally confirmed; green, other similar T2PKs that have close KSβ structures but different natural products). Further information can be found in Table S7.† The ground-truth T2PK of bacteria is denoted by the orange point in each figure. Bold bonds in each chemical structure indicate the building block units incorporated into the polyketide backbone, while the black dot indicates a single carbon from the build block unit in which the adjacent carbon from the same build block is lost during the polyketide biosynthesis via decarboxylation. | ||
Discussion
T2 polyketide synthase is a family of single heterodimeric ketosynthases that iteratively catalyzes the elongation of the polyketide chain structure, leading to our inability to precisely predict T2PK structures. As introduced previously, despite the multiple sequence alignment approaches based on KSβ,5,6 incorporation of new sequences into the evolutionary model may alter the structure of the original phylogenetic tree and therefore compromise the accuracy of the predictions. To address this issue, we propose DeepT2, an end-to-end deep learning strategy, to directly identify T2PKs from bacterial genomes. Leveraging the concept of natural language processing, our approach embeds KSβ as feature vectors, enabling the representation of protein structural information. We employ semi-supervised learning to link KSβ embedding vectors with compound labels, facilitating the rapid identification of known and novel T2PKs. Importantly, in contrast to other BGC prediction tools, such as DeepBGC21 and antiSMASH, which require complete T2PK BGC sequences, our DeepT2 model can accurately predict T2PK categories using only KSβ sequences. Furthermore, our novelty detection framework embedded in DeepT2 has promising potential for identifying new KSβ. However, a question that may arise is that considering the limited biological data volume, is it still possible to use it to develop advanced algorithms even without big data? This is because in the biological research field, classic machine learning and currently popular deep learning are both hampered by poor accuracy or overfitting caused by the smaller training dataset. Indeed, during model development, we had debated whether the proof of concept should be data-centered or training-centered. Fortunately, the ensemble method using a small data volume based on pretraining and semi-supervised learning seems to be a promising solution, at least for this work. In addition, as with other machine learning algorithms, DeepT2 is expected to improve as more KSβ sequences are discovered in microbial genomes over time.The task of few-shot supervised learning requires an approach that transcends traditional supervised neural networks.47 In this context, our work adopts the concept of transfer learning, where ESM-2 is utilized to explore the connection between KSβ embeddings and T2PK structures. While the dimension reduction results indeed indicate that the embedding vectors obtained by ESM-2 closely fit with the compound class labels, it is important to note that certain embeddings still necessitate further labeling refinement and correction. Consequently, we performed label reconstruction using supervised UMAP instead of unsupervised UMAP to ensure that the resulting reduced-dimensional space consistently captures the features of the compound skeleton. This approach differs from traditional unsupervised learning for clustering,48 as it strives to strike a balance between the sequence embeddings and the compound class labels to improve the model's accuracy. For example, T2PK AQ-256-8 consists of 8 building blocks, but its KSβ is confirmed as ancestral nonoxidative, which differs from other KSβs that involve the biosynthesis of T2PKs with 8 building blocks.6 Clearly, the state-of-the-art performance of the model trained with 9 refined class labels suggests that the classification effect is unsatisfactory when simply using five biosynthetic building blocks as labels. This finding suggests that KSβ not only affects the counts of building blocks but also determines a rough topology prior to cyclization or aromatization. To the best of our knowledge, this is the first algorithm for T2PK classification and prediction in such a manner, which, as an alternative to sophisticated protein sequence alignment, might showcase a paradigm shift in genome-mining approaches for natural product discovery.
As shown above, we improved the generalization ability of the softmax-based T2PK classifier by employing a consistency-regularization-based semi-supervised learning framework that utilized 2566 KSβ whose corresponding natural product structures currently remain unknown. However, such models may demonstrate overconfidence in discerning novel KSβ sequences in the real world.32 To address this concern, an ODD framework based on the Mahalanobis distance was implemented for multiclass novelty detection.49 Notably, certain samples (from 2566 KSβ sequences) are proximal to the labeled data (from 163 KSβ sequences) because such labeled T2PKs with entirely novel carbon skeletons have only been discovered in recent years, such as formicamycin40 and dendrubin.50 Therefore, to avoid false positives in novelty detection, we selected only 13 potential new class samples that are distant from the labeled samples for demonstration. Greater details regarding the enzymatic information and chemical structures for these T2PKs will be studied in future work.
This study demonstrates the capacity of DeepT2 to predict T2PKs from single or mixed genomic datasets. However, some limitations must be acknowledged. While the training data included bacterial genomes from different phyla, certain biases may hinder the model's ability to detect novel T2PKs in poorly characterized bacterial sources within complex microbiomes. Although the model was validated using Streptomyces genomes as a showcase in this study, it is essential to expand the collection of bacterial genomes to enhance the overall performance of the model. Additionally, the current version of DeepT2 is capable of predicting T2PKs from single genes as input, but it requires complete sequences of at least 300 amino acids (the average length of KSβ is around 400 amino acid). For predicting other tailoring modifications, such as methylation or halogenation, supplementing DeepT2 with antiSMASH or DeepBGC is recommended. Nonetheless, despite these limitations, the DeepT2 model outperforms other methods and represents a valuable algorithm for KSβ identification and T2PK discovery. This study also inspires future research to identify which catalytic domains in KSβ contribute to chemical differences through PLM and thus provides more insights into the KSβ evolution and T2PK biosynthetic mechanisms, and this is currently ongoing in our laboratory. Moreover, as the application of language models in prompt tuning for zero-shot prediction, as well as the generative models such as autoregressive neural networks is gradually emerging,51–53 we are now prompted to explore such models for KSβ studies. We therefore anticipate that this work will aid in the application of genome mining approaches to discover new KSβ and novel T2PKs and have important clinical implications for transforming microbiome data into therapeutic interventions.
Data availability
The authors declare that the data, materials and code supporting the findings reported in this study are available from the authors upon reasonable request. The DeepT2 is available at GitHub repository https://github.com/Qinlab502/deept2.Author contributions
Zhiwei Qin and Heqian Zhang designed and supervised the research, Jiaquan Huang and Qiandi Gao performed the bioinformatic and computing analysis, Yaxin Wu performed the microbial fermentation and chemical analysis, Ying Tang participated the algorithm development. All authors analysed and discussed the data. Zhiwei Qin, Heqian Zhang and Jiaquan Huang wrote the manuscript and all authors commented.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (32170079, 32200035 and 12105014), the Natural Science Foundation of Guangdong (2021A1515012026), Guangdong Talent Scheme (2021QN020100), Beijing Normal University via the Youth Talent Strategic Program Project (310432104), as well as Guangdong Innovation Research Team for Plant–Microbe Interaction. We also thank the Interdisciplinary Intelligence Super Computer Center, Beijing Normal University at Zhuhai, for High Performance Computing for access to computational resources.Notes and references
- S.-C. Tsai, Annu. Rev. Biochem., 2018, 87, 503–531 CrossRef CAS PubMed.
- C. Hertweck, A. Luzhetskyy, Y. Rebets and A. Bechthold, Nat. Prod. Rep., 2007, 24, 162–190 RSC.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS PubMed.
- A. Bräuer, Q. Zhou, G. L. Grammbitter, M. Schmalhofer, M. Rühl, V. R. Kaila, H. B. Bode and M. Groll, Nat. Chem., 2020, 12, 755–763 CrossRef PubMed.
- M. E. Hillenmeyer, G. A. Vandova, E. E. Berlew and L. K. Charkoudian, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 13952–13957 CrossRef CAS PubMed.
- S. Chen, C. Zhang and L. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202202286 CrossRef CAS PubMed.
- C. P. Ridley, H. Y. Lee and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4595–4600 CrossRef CAS PubMed.
- J. Kim and G.-S. Yi, BMC Microbiol., 2012, 12, 1–12 CrossRef PubMed.
- R. Villebro, S. Shaw, K. Blin and T. Weber, J. Ind. Microbiol. Biotechnol., 2019, 46, 469–475 CrossRef CAS PubMed.
- E. C. Alley, G. Khimulya, S. Biswas, M. AlQuraishi and G. M. Church, Nat. Methods, 2019, 16, 1315–1322 CrossRef CAS PubMed.
- A. Elnaggar, M. Heinzinger, C. Dallago and B. Rost, bioRxiv, 2019 DOI:10.1101/864405.
- N. J. Merwin, W. K. Mousa, C. A. Dejong, M. A. Skinnider, M. J. Cannon, H. X. Li, K. Dial, M. Gunabalasingam, C. Johnston and N. A. Magarvey, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 371–380 CrossRef CAS PubMed.
- C. Rios-Martinez, N. Bhattacharya, A. P. Amini, L. Crawford and K. K. Yang, PLoS Comput. Biol., 2023, 19, e1011162 CrossRef CAS PubMed.
- Y. Ma, Z. Guo, B. Xia, Y. Zhang, X. Liu, Y. Yu, N. Tang, X. Tong, M. Wang and X. Ye, Nat. Biotechnol., 2022, 40, 921–931 CrossRef CAS PubMed.
- V. J. Sahayasheela, M. B. Lankadasari, V. M. Dan, S. G. Dastager, G. N. Pandian and H. Sugiyama, Nat. Prod. Rep., 2022, 39, 2215–2230 RSC.
- F. I. Saldívar-González, V. D. Aldas-Bulos, J. L. Medina-Franco and F. Plisson, Chem. Sci., 2022, 13, 1526–1546 RSC.
- D. W. P. Tay, N. Z. X. Yeo, K. Adaikkappan, Y. H. Lim and S. J. Ang, Sci. Data, 2023, 10, 296 CrossRef PubMed.
- Y. Tang and A. Hoffmann, Rep. Prog. Phys., 2022, 85, 086602 CrossRef PubMed.
- L. Yann, B. Yoshua and H. Geoffrey, Nature, 2015, 521, 436–444 CrossRef PubMed.
- H. W. Kim, M. Wang, C. A. Leber, L. F. Nothias, R. Reher, K. B. Kang, J. J. J. van der Hooft, P. C. Dorrestein, W. H. Gerwick and G. W. Cottrell, J. Nat. Prod., 2021, 84, 2795–2807 CrossRef CAS PubMed.
- G. D. Hannigan, D. Prihoda, A. Palicka, J. Soukup, O. Klempir, L. Rampula, J. Durcak, M. Wurst, J. Kotowski, D. Chang, R. R. Wang, G. Piizzi, G. Temesi, D. J. Hazuda, C. H. Woelk and D. A. Bitton, Nucleic Acids Res., 2019, 47, e110 CrossRef CAS PubMed.
- A. Rives, J. Meier, T. Sercu, S. Goyal, Z. Lin, J. Liu, D. Guo, M. Ott, C. L. Zitnick and J. Ma, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2016239118 CrossRef CAS PubMed.
- S. Unsal, H. Atas, M. Albayrak, K. Turhan, A. C. Acar and T. Doğan, Nat. Mach. Intell., 2022, 4, 227–245 CrossRef.
- Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli and Y. Shmueli, Science, 2023, 379, 1123–1130 CrossRef CAS PubMed.
- F. Teufel, J. J. Almagro Armenteros, A. R. Johansen, M. H. Gíslason, S. I. Pihl, K. D. Tsirigos, O. Winther, S. Brunak, G. von Heijne and H. Nielsen, Nat. Biotechnol., 2022, 40, 1023–1025 CrossRef CAS PubMed.
- M. H. Hoie, E. N. Kiehl, B. Petersen, M. Nielsen, O. Winther, H. Nielsen, J. Hallgren and P. Marcatili, Nucleic Acids Res., 2022, 50, W510–W515 CrossRef PubMed.
- H. Song, M. Kim, D. Park, Y. Shin and J.-G. Lee, IEEE Trans. Neural Netw. Learn. Syst., 2022 DOI:10.1109/TNNLS.2022.3152527.
- Y. Ouali, C. Hudelot and M. Tami, arXiv, 2020, preprint arXiv:2006.05278, DOI:10.48550/arXiv.2006.05278.
- J. Yang, K. Zhou, Y. Li and Z. Liu, arXiv, 2021, preprint arXiv:2110.11334, DOI:10.48550/arXiv.2110.11334.
- K. Lee, K. Lee, H. Lee and J. Shin, arXiv, 2018, preprint, arXiv:1807.03888, DOI:10.48550/arXiv.1807.03888.
- L. Zhou, S. Pan, J. Wang and A. V. Vasilakos, Neurocomputing, 2017, 237, 350–361 CrossRef.
- A. Nguyen, J. Yosinski and J. Clune, arXiv, 2015, preprint, arXiv:1412.1897, DOI:10.48550/arXiv.1412.1897.
- B. Kai, S. Simon, K. Alexander M, C.-P. Zach, V. W. Gilles P, M. Marnix H and W. Tilmann, Nucleic Acids Res., 2021, 49, W29–W35 CrossRef PubMed.
- T. Oja, L. Niiranen, T. Sandalova, K. D. Klika, J. Niemi, P. Mantsala, G. Schneider and M. Metsa-Ketela, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1291–1296 CrossRef CAS PubMed.
- K. Ichinose, D. J. Bedford, D. Tornus, A. Bechthold, M. J. Bibb, W. P. Revill, H. G. Floss and D. A. Hopwood, Chem. Biol., 1998, 5, 647–659 CrossRef CAS PubMed.
- C. Han, Z. Yu, Y. Zhang, Z. Wang, J. Zhao, S.-X. Huang, Z. Ma, Z. Wen, C. Liu and W. Xiang, J. Agric. Food Chem., 2021, 69, 2108–2117 CrossRef CAS PubMed.
- M. Daum, I. Peintner, A. Linnenbrink, A. Frerich, M. Weber, T. Paululat and A. Bechthold, ChemBioChem, 2009, 10, 1073–1083 CrossRef CAS PubMed.
- L.-J. Xuan, S.-H. Xu, H.-L. Zhang, Y.-M. Xu and M.-Q. Chen, J. Antibiot., 1992, 45, 1974–1976 CrossRef CAS PubMed.
- W. J. Underberg, G. A. Hofman, S. C. Lubbers, O. Bekers, W. W. Ten Bokkel Huinink and J. H. Beijnen, J. Pharm. Biomed. Anal., 1989, 7, 1791–1797 CrossRef CAS PubMed.
- Z. Qin, J. T. Munnoch, R. Devine, N. A. Holmes, R. F. Seipke, K. A. Wilkinson, B. Wilkinson and M. I. Hutchings, Chem. Sci., 2017, 8, 3218–3227 RSC.
- Z. Qin, R. Devine, T. J. Booth, E. H. Farrar, M. N. Grayson, M. I. Hutchings and B. Wilkinson, Chem. Sci., 2020, 11, 8125–8131 RSC.
- G. Blanco, P. Brianb, A. Pereda, C. Mendez, J. Salas and K. F. Chater, Gene, 1993, 130, 107–116 CrossRef CAS PubMed.
- P. Lopez, A. Hornung, K. Welzel, C. Unsin, W. Wohlleben, T. Weber and S. Pelzer, Gene, 2010, 461, 5–14 CrossRef CAS PubMed.
- K. Kojiri, S. Nakajima, A. Fuse, H. Suzuki and H. Suda, J. Antibiot., 1995, 48, 1506–1508 CrossRef CAS PubMed.
- K. B. Herath, H. Jayasuriya, Z. Guan, M. Schulman, C. Ruby, N. Sharma, K. MacNaul, J. G. Menke, S. Kodali and A. Galgoci, J. Nat. Prod., 2005, 68, 1437–1440 CrossRef CAS PubMed.
- K. Jiang, X. Yan, Z. Deng, C. Lei and X. Qu, J. Nat. Prod., 2022, 85, 943–950 CrossRef CAS PubMed.
- W. Yaqing, Y. Quanming, K. James T and N. Lionel M, ACM Comput. Surv., 2020, 53, 1–34 Search PubMed.
- M. S. Asyaky and R. Mandala, 2021.
- K. Lee, K. Lee, H. Lee and J. Shin, Adv. Neural Inf. Process Sys., 2018, 31 CAS.
- K. Ishida, G. Shabuer, S. Schieferdecker, S. J. Pidot, T. P. Stinear, U. Knuepfer, M. Cyrulies and C. Hertweck, Chem. - Eur. J., 2020, 26, 13147–13151 CrossRef CAS PubMed.
- P. Liu, W. Yuan, J. Fu, Z. Jiang, H. Hayashi and G. Neubig, ACM Comput. Surv., 2023, 55, 1–35 Search PubMed.
- Y. Tang, J. Weng and P. Zhang, Nat. Mach. Intell., 2023, 1–10, DOI:10.1038/s42256-023-00632-6.
- J. Trinquier, G. Uguzzoni, A. Pagnani, F. Zamponi and M. Weigt, Nat. Commun., 2021, 12, 5800 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3dd00107e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |