
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mapping the structure–activity landscape of non-canonical peptides with MAP4 fingerprinting†
Edgar
López-López
 *ab,
Oscar
Robles
c,
Fabien
Plisson
d and
José L.
Medina-Franco
*a
*ab,
Oscar
Robles
c,
Fabien
Plisson
d and
José L.
Medina-Franco
*a
aDIFACQUIM Research Group, Department of Pharmacy, School of Chemistry, Universidad Nacional Autónoma de México, 04510 Mexico City, Mexico. E-mail: elopez.lopez@cinvestav.mx; medinajl@unam.mx
bDepartment of Chemistry and Graduate Program in Pharmacology, Center for Research and Advanced Studies of the National Polytechnic Institute, Section 14-740, 07000 Mexico City, Mexico
cMedicinal Chemistry and Chemogenomics Laboratory, Faculty of Bioanalysis-Veracruz, Universidad Veracruzana, 91700 Veracruz, Mexico
dDepartment of Biotechnology and Biochemistry, Center for Research and Advanced Studies of the National Polytechnic Institute (CINVESTAV-IPN), Irapuato Unit, Irapuato 36824, Mexico
First published on 1st September 2023
Abstract
Peptide structure–activity/property relationship (P-SA/PR) studies focus on understanding how the structural variations of peptides influence their biological activities and other functional properties. This knowledge accelerates the rational design and optimisation of peptide-based drugs, biomaterials, or diagnostic agents. These studies examine peptide structures from their primary sequences, essentially encoded from the 20 amino acids. Current approaches often exclude peptide libraries with post-translational and synthetic modifications. The molecular fingerprint MAP4 was recently developed to map complex molecules' sequence/structure diversity, including peptides. This study used structure–activity landscape modelling to conduct the P-SA/PR studies of an exemplary dataset of 223 antimicrobial peptides against methicillin-resistant Staphylococcus aureus (MRSA). To this end, we employed the MAP4 fingerprint to represent the chemical structures of the peptides, study their relationship(s) with the antibacterial activity, and seek the potential activity cliff(s). We identified critical residues and structural motifs that play a crucial role in the anti-MRSA activity of the peptides. This is the first computational study to systematically explore the activity landscape of peptides with non-canonical residues, emphasising the quantification of structural similarity.
Introduction
Peptides play essential roles in plant and animal physiology, targeting various proteins, including growth factors, ion channels, receptors, and enzymes. They have a broad range of biological activities, all valuable starting points to treat human disorders.1–3 However, discovering and designing biologically active peptides could be deceiving; the peptide space is vast; a peptide sequence of length N could lead to 20N possible mutations solely with canonical residues. Adding post-translational modifications (PTMs), synthetic constraints or mutations to existing canonical sequences, and the number of possible peptides becomes astronomical. It is impractical to synthesise all sequences or investigate all functionally interesting variants. Luckily, Nature has provided us with privileged peptide ligands, reducing our search space to sequences with preferred structures and functions.4,5A central goal for computational peptide design is to create novel sequences that carry the underlying properties of natural peptides with defined structural and functional properties. Multiple informatic approaches have proven helpful in accelerating peptide design learning from their sequences or tridimensional structures.6,7 In addition, the automation of peptide synthesis on a solid support or the heterologous expression of proteins across biological systems has reduced production costs, making peptide space exploration accessible. These in silico methods predominantly learn from primary sequences from sizeable datasets rather than their structures due to the high costs associated with solving structures experimentally.8 Yet, current sequence-based approaches need to systematically study PTMs that can significantly affect the physicochemical, chemical, or biological properties of peptides.9 Chemoinformatics (also called in the literature “cheminformatics” or “chemical informatics”)10 and bioinformatics are independent disciplines regarding the focus of their study. The former focuses on small molecules, whereas the latter focuses on using computational methods to address biological entities. Computationally, one key difference between both disciplines is how chemical structures are represented and handled. In biology, the chemical structures are usually large (e.g., proteins, nucleic acids, receptors). They are described in strings of letters or Cartesian coordinates (i.e., Protein Data Bank11), unlike low molecular weight compounds that are encoded in various molecular fingerprints.12 However, some chemical structures are at the interface between traditional small molecules used extensively in drug discovery and proteins and nucleic acids in the biological realm. Peptides exemplify these chemical structures; they vary in size, ranging from small molecules to large proteins.
In recent years, new methodologies and technologies have reduced the literacy gap between chemoinformatics and bioinformatics. New molecular representations based on atom connectivity allow the systematic study of complex molecules that could be applied to mapping the structural diversity of peptides. They may help in understanding the roles of PTMs in their physicochemical properties or biological activities.12 Different computational strategies to develop peptides are based on analysing sequence alignments and physicochemical similarity metrics.13 However, only some post-traditionally modified peptides and their functional measurements are documented, limiting the use of alignment algorithms and the prediction of secondary structures.14 Recent computational methods have contributed to decoding the structure–activity/property relationships (SA/PR) of peptides (P-SA/PR).15,16 A growing number of methods based on primary sequences or derived physicochemical features of peptides (e.g., machine-learning methods, the de novo design, linguistic modelling, pattern insertion methods, and genetic algorithms)17 represent new research opportunities to explore P-SA/PR and guide a new era of peptide-based drug design. Computational drug design approaches have decoded the physicochemical and sequence–activity relationships on peptides.18,19 Such approaches remain to be applied, describing the relationships between small structural changes and their specific biological activity.
Physicochemical properties are commonly used to compare, filter, and classify molecular structures of pharmaceutical interest.20,21 They generally describe global changes in contrast with more localised structural conformations, small chemical changes, or fold peptide differences. Consensus similarity metrics were recently implemented to compare peptide structures considering features including tridimensional structure, topology, backbone structure, drug-like properties, amino acid sequence, and molecular fingerprints.17,22 From a conceptual point of view, combining physicochemical, chemical, sequence and structural descriptors commonly used in chemoinformatics and bioinformatics would provide a comprehensive picture of the peptides.23 For example, different authors recently demonstrated that predicting properties and designing new peptide structures from the consensus description of known peptidic information24–27 states that not all similar peptides conserve identical properties. Such a highlight is related to the activity cliff concept frequently used in chemoinformatics,28 two or more peptides with high structural similarity but distinct functional measurements. Activity cliffs have decoded the SA/PR studies of linear and circular peptides against different endpoints.29,30 Also, the presence of activity cliffs in datasets reduces the performance of predictive models by challenging their ability to capture precise relationships between chemical structures and biological activity and their generalisability to new compounds.31,32 The novel circular and topological fingerprint MAP4 is more sensitive to identifying small structural changes in complex molecules, especially in peptides, than conventional fingerprints used in small molecule drug discovery, such as MACCS keys, ECFP4, or ECFP6.33 Additionally, MAP4 fingerprint has opened the opportunity to create and navigate in a representative chemical space of more complex peptides,34,35 and it has been used to improve the performance of artificial intelligence algorithms to predict peptidic properties.36
This study introduces a new approach to exploring and describing the activity landscape of peptides with non-canonical residues, including PTMs. Our case study uses an exemplary dataset of 223 peptides with reported activity against methicillin-resistant Staphylococcus aureus (MRSA) strains.37 It is considered one of the most critical global health threats due to its high pandemic potential.37–41 Namely, the MRSA strains create an emerging challenge for health systems by increasing the costs associated with the recovery of patients.42 Epidemiologically, MRSA stands out for efficient dissemination and establishment in environments as diverse as hospitals and communities and is related to different types of productive livestock, whose repercussions range from human health to food production and safety.43,44 Visualising peptide structure–activity/property relationships studies using MAP4 fingerprint accelerates the rational design and optimisation of bioactive peptides, e.g., anti-MRSA peptides. The present approach allows (1) mapping the anti-MRSA peptides sequence and studying their structural diversity (using similarity metrics based on MAP4 fingerprint) and (2) visualising peptide activity cliffs in low-dimensional space (using an extension of a structure–activity similarity map). To this end, we employed an atom-connectivity fingerprint recently developed and well-suited to represent peptides. We also discuss an interpretation of the peptide activity cliffs.
Methodology
Protocol overview
This protocol was implemented in five steps: (1) we collected the peptide sequence and bioactivity data from APD3 database36 of natural or synthetic peptides, with anti-MRSA reported activity expressed in mg mL−1, with <70 residues; (2) using each peptide sequence and their non-canonical modifications, the SMILES strings were was obtained manually drawing each peptide in the software ChemBioDraw Ultra V.13; (3) using python programing language the fingerprint MAP4 (ref. 33) was calculated for each peptide. Additionally, paired calculations (i.e. molecular similarity, paired difference activity, and paired difference molecular weight) were done using the code freely available at https://github.com/LopezLopezE/Peptide-Similarity.git; (4) the paired data was visualised using DataWarrior software V. 5.5.0;45,46 (5) that allowed the identification of peptide activity cliffs (vide infra). Fig. S1 in the ESI† illustrates a graphical overview of this methodology.The methodology used in this study is a distinctive manner to represent the non-canonical modifications on peptides that, compared with traditional peptide sequence alignments or other similarity metrics, offer a more realistic structure–activity approximation.
Data set
To analyse the landscape of anti-MRSA peptides, we collected a total of 223 peptide sequences from the Antimicrobial Peptide Database,36 of which 101 examples (∼45.29%) have a half minimum inhibitory concentration (MIC50) value measured against clinical isolation of MRSA strains. In total, 122 peptides (∼54.71%) have MIC50 value reported against at least one of the 26 characterised MRSA strains. We transformed all MIC50 values to a negative decimal listed logarithm scale, namely pMIC50 = −log10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) MIC50. The values range from 3.89 to 6.78 and are listed in ESI – Table S1† alongside the peptide sequences and SMILES representations. In some cases, the same peptide has been evaluated against different strains; we only kept the higher value of the pMIC50 range.
MIC50. The values range from 3.89 to 6.78 and are listed in ESI – Table S1† alongside the peptide sequences and SMILES representations. In some cases, the same peptide has been evaluated against different strains; we only kept the higher value of the pMIC50 range.
Activity landscape modelling
We studied the activity landscape of the 223 peptides through two approaches that are frequently used with small organic compounds: Structure–Activity Similarity (SAS) map and the Structure–Activity Landscape Index (SALI) widely used in chemoinformatics. Both approaches are explained hereunder. A SAS map is a low-dimensional graph for analysing the SAR of the compound dataset. SAS maps are one of the early approaches to studying activity landscapes and rapidly identifying activity cliffs. Activity cliffs are pairs of compounds with high structure similarity but significantly different biological activity.28 SAS maps are based on systematic pairwise comparisons of the compounds in a data set. A general schematic representation of a SAS map is presented in Fig. 1.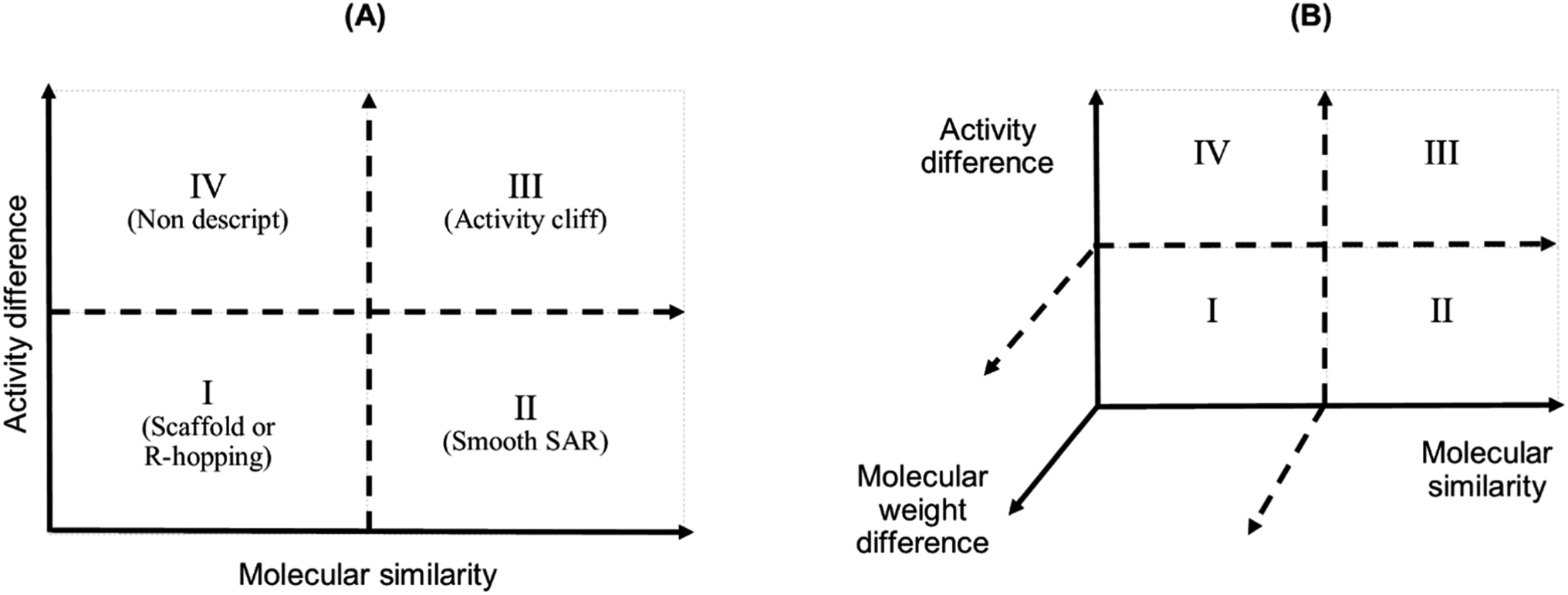 | ||
| Fig. 1 Graphical representation of a Structure–Activity Similarity (SAS) map (A) and an extension of a SAS map (B). A SAS map is based on a pairwise comparison of each compound on a data set. Each data point in the graph in the map represents a pair of compounds. SAS map is based on the activity differences of the pair of compounds against a specific biological endpoint and their molecular distance. (A) Map with four regions: (I) identifies a pair of compounds with low activity difference and low molecular distance (also called scaffold or R-hopping, or similarity cliffs); (II) represents a pair of compounds with low activity difference and higher molecular distance (smooth SAR cases); (III) represents pair of compounds with higher activity differences and higher molecular distance (activity cliff); and (IV) represent pair of compounds with a discontinuous SAR.47 (B) An extension of the conventional SAS map (extended SAS map) implemented in this study adds the molecular weight differences as a new axis. | ||
SAS maps generated in this study represented all 25185 pairwise comparisons between the 223 peptides. The map displayed structure similarity with the MAP4 fingerprint and the MinHashed distance33 on the X-axis. The Y-axis showed the activity difference using the pMIC50 values of each peptide pair. The Z-axis expressed differences in molecular weight between each pair of peptides. The data points in the SAS maps were further coloured by their SALI value. This index quantifies the activity landscape using the expression proposed by Guha and Van Drie11,48 (eqn (1)):
| SALIi,j = |Ai − Aj|/1 − sim(i,j) | (1) |
Chemical space of anti-MRSA peptides
A visual representation of the chemical space of anti-MRSA peptides was constructed using a Treemap (TMAP). TMAP allows the visual representation of many chemical compounds through the distance between the clusters and the cluster's detailed structure through Local Sensitive Hashing (LSH) forest data structure, enabling c-approximate k-nearest neighbours (k-NN).49 MAP4 fingerprints for peptides were encoded using the MinHash algorithm. The number of nearest neighbours, k = 50, and the factor used by the augmented query algorithm, kc = 10, were used to develop the TMAP graphs. The activity values were represented using a colour scale from red (most active peptide; 6.78 pMIC50) to blue (most inactive peptide; 3.89 pMIC50).Results
Data set description
We collected 223 anti-MRSA peptides from APD3 alongside their pMIC50 values. Most peptides were identified from amphibians (88/39.5%), followed by bacteria (36/16.4%) and mammals (28/12.6%) (Fig. 2A). We observed diverse properties profiles in Fig. S2 in the ESI,† showing differences in their topological polar surface area (TPSA, ranging from 315.05 to 3010.60 Å), molecular weight (MW, from 408.09 to 6643.81 g mol−1), and lipophilicity index (XlogP, from −34.79 to 11.89). TPSA and MW positively correlate with pMIC50 values except for a few outlying peptides. In contrast, XLogP values negatively correlate with pMIC50 values independently of the structures. Additionally, beta-sheet peptides presented higher TPSA and MW than the helical counterparts, whereas both structural groups shared similar lipophilicity ranges (XlogP ∼ −5 to 1); see Fig. S2 in the ESI.† Overall, this data set contained peptides with canonical (200/89.69%) and non-canonical/modified (23/10.31%) amino acids – see Table S1 in the ESI.† Regarding the structural diversity of the anti-MRSA peptides (Fig. 2B), most peptides do not have identified structures (59.6%). Yet, alpha-helices predominate among all known structures (32.7%). Most structures of anti-MRSA peptides were solved using nuclear magnetic resonance spectroscopy (30/13.5% – Fig. 2C). A substantial part of the peptide structures (42/13.8%) were computationally predicted, and the largest group has not been associated with any predicted or experimental structure (139/62.3%).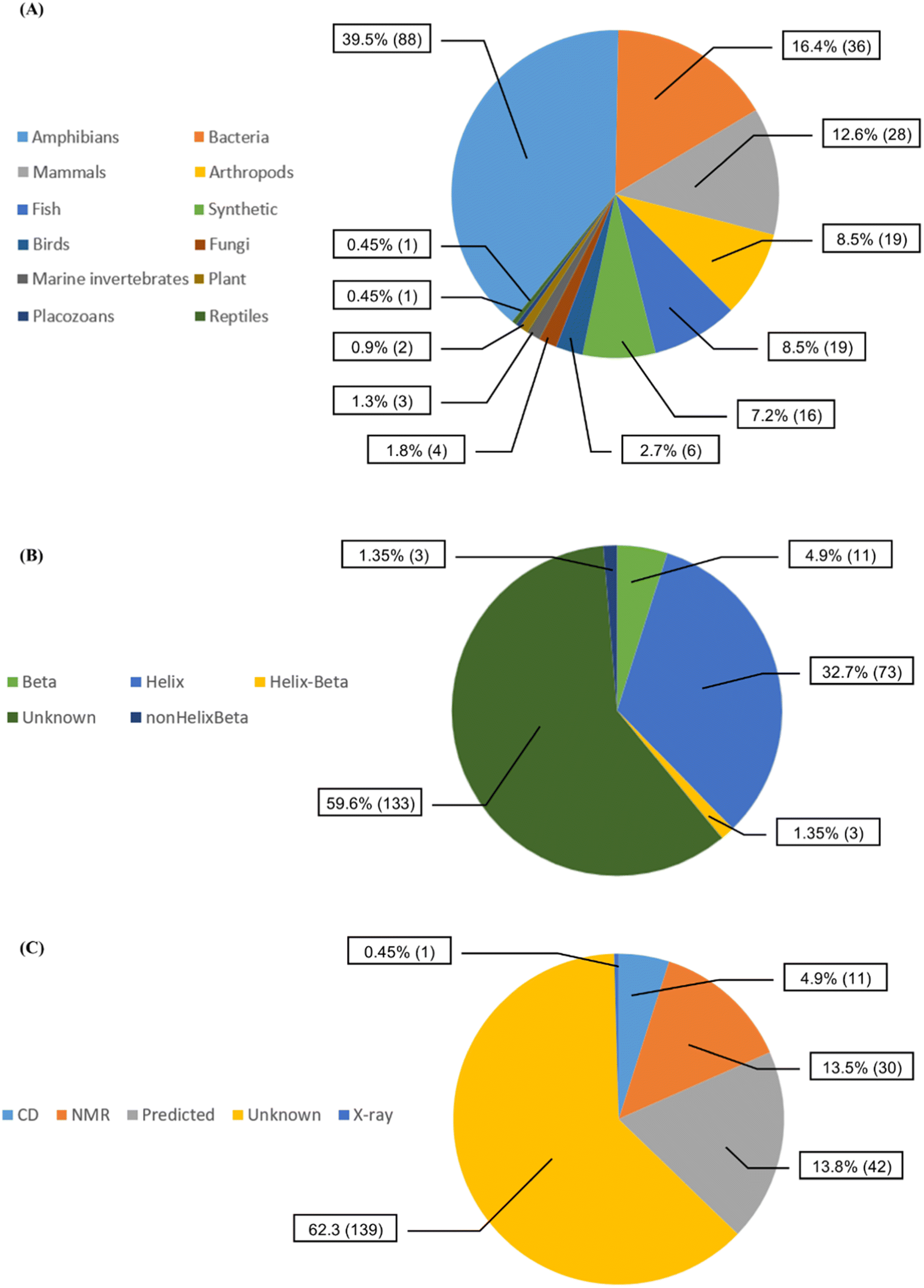 | ||
| Fig. 2 Diversity of the 223 anti-MRSA peptides according to (A) their source organisms, (B) their known secondary structures, and (C) their experimental methods used to solve their structures [CD stands for circular dichroism, NMR: nuclear magnetic resonance spectroscopy and X-ray: X-ray crystallography]. | ||
Activity landscape modelling
Fig. 3A shows an extended SAS map annotated with SALI values of 25185 pairwise comparisons between the 223 peptides, which facilitated the identification of activity cliffs. Namely, the extended SAS map allowed the pairing of peptides with high structural similarity as determined by MAP4/MinHashed distance function (similarity > 0.40) but with a sizeable anti-MRSA activity difference (pMIC50 difference > 0.90) and with low MW difference (MW difference < 650). Based on these criteria, we selected, as representative examples, the peptide pairs 1–11 (Fig. 3A). Sequence alignment of pairs 1–11 (Fig. 3B) confirmed that the peptides had similar amino acid sequences (47% to 100%). However, the similarity calculated based on their peptide chemical structures did not necessarily have a linear relationship with the identity of peptide sequences (Fig. 3C). This observation suggests that the MAP4 fingerprints helped compare the peptide chemical structures and their primary sequences. Nevertheless, the 2D and 3D alignments of amino acids provided additional and intuitive information to decode the P-SA/PRs. For example, the peptide pair 1 (Fig. 3C) shows a lower fingerprint-based similarity (0.866) in contrast with their sequence identity (100%). This data suggested that the fingerprint-based similarity could be sensitive to small structural and atom-connectivity changes in short peptides containing less than 20 residues. Interestingly, the peptide pairs 1–11 exhibited good relationships between their fingerprint-based similarity and sequence-based identity. Hence, our fingerprint-based similarity metric might help quantify the similarity between more extensive sequences (i.e., 20 residues or more).
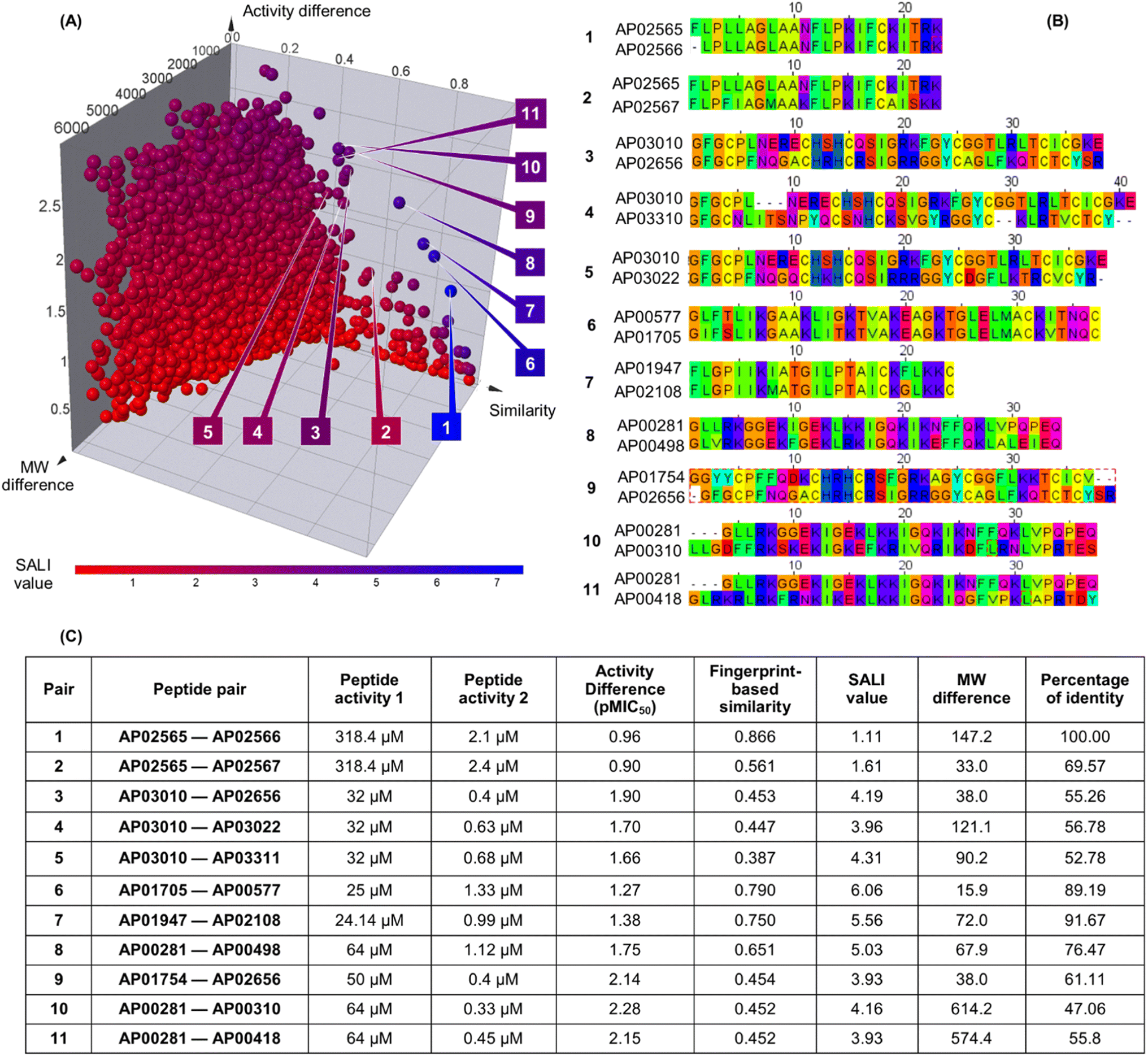 | ||
| Fig. 3 Structural and sequence similarity between the 223 anti-MRSA peptides: (A) modified (extended) structure–activity similarity map; each sphere represents a pairwise comparison of the chemical structure (quantified utilising MinHassed distance/MAP4 fingerprints), activity difference, and molecular weight difference. The spheres are coloured according to the SALI values using a continuous scale from low (blue) to high (red) values. An interactive visualisation has been implemented using the DataWarrior software; see File S1 in the ESI.† (B) Sequence alignment and (C) summary characterisation of 11 representative peptide activity cliffs (pairs). SALI: Structural–Activity Landscape Index. | ||
Fig. 4 illustrates the structural similarity between additional representative peptide pairs 12–15. The fingerprint-based similarity protocol allows the identification of small structural changes (pair 12), changes in a unique amino acid sequence (pair 13), N-terminal modifications (pair 13), multiple amino acids changes (pair 14), and structural changes associated with post-traditional modifications (pair 15).
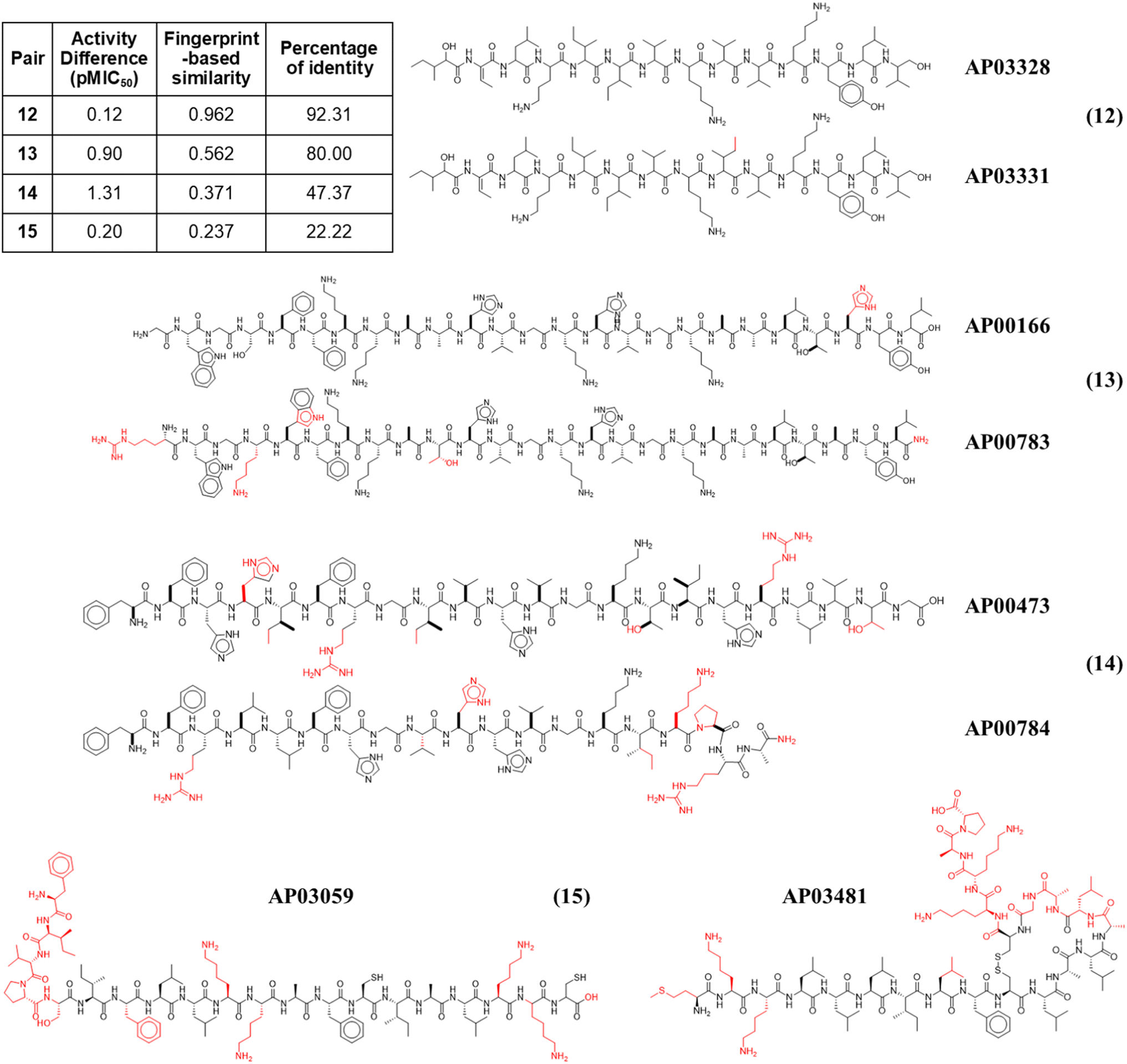 | ||
| Fig. 4 Representative anti-MRSA peptide pairs 12–15. Chemical changes observed between each peptide pair are coloured in red, whereas shared chemical structures are depicted in black. | ||
Visualisation of chemical space
In addition to the extended SAS map, we explored the anti-MRSA peptide landscape using a tree manifold approximation and projection (TMAP, Fig. 5). A TMAP shows the k-nearest neighbours of each peptide (represented with a sphere) using the MAP fingerprint and the MinHashed algorithm as a distance metric. Namely, the TMAP facilitates the discovery and intuitive visualisation of structurally related compounds. For example, the peptides AP02565 and AP02566 (pair 1 in Fig. 3) had identical sequences and were located close to each other in TMAP. It is worth mentioning that a TMAP-based distance depends not on the % identity of amino acids (AA) but on an alternative representation that depends on structural fingerprints (vide supra). Consequently, the peptide pair 1 did not share the exact coordinates.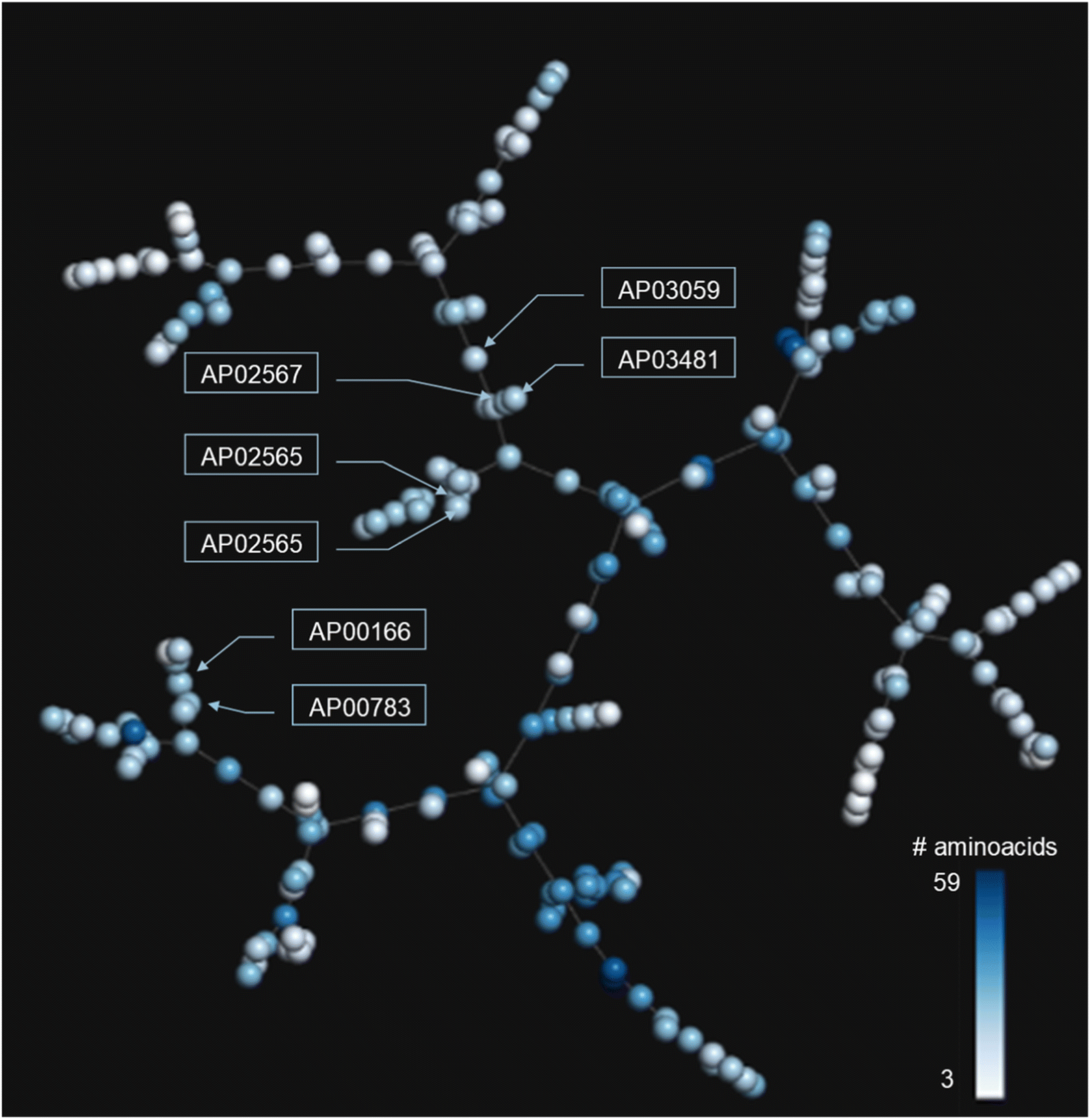 | ||
| Fig. 5 TMAP of the 223 anti-MRSA peptides. Each sphere represents a peptide, and the inter-spherical distance represents the structural relationships between peptides. Each sphere is coloured using a scale of blue (higher peptide length) to white (lower peptide length). | ||
In contrast, the peptides AP02565 and AP02567 (pair 2 in Fig. 3) are structurally different, e.g., 69% of AA sequence identity and 0.561 fingerprint-based similarity, and were located farthest apart compared to the peptide pair 1. The TMAP representation illustrates the peptide pairs' subtle and complex structural relationships. For example, the peptide pair 13 (AP00166 and AP00883) presented multiple AA changes and N-terminal modifications, whereas AP03059 and AP03481 (pair 15) only differed by forming a disulfide bond.
The peptide pairs 1–11 had a medium-to-high structural similarity (AA sequence identity between 22–100%) but were associated with a significant change in their pMIC50 values. However, the fingerprint-based similarity values (measured with MAP4) positively correlated with identity values (R2 = 0.31, Fig. S4 in the ESI†). Moreover, fingerprint-based similarity values showed a higher inverse correlation (−0.12) with the activity difference values of each peptide pair compared to their identity values (−0.06). Higher similarity or identity values were correlated with lower activity difference values. This observation suggests that fingerprint-based similarity measures complement the insights derived from sequence alignments but do not replace them. Similarity metrics explore the atom-connectivity in peptides, while sequence identity describes the residual differences. Therefore, using the small structural/sequence changes in peptides helps to rationalise the peptide structure–property relationships.
Overview of anti-MRSA peptides sequence alignments
Alignment analysis of the 223 anti-MRSA peptides resulted in a consensus sequence (“FLKKIAKVLGKAG” – Fig. S3 in the ESI†), which was characterised by being enriched in lysine (Lys – K). The consensus sequence has a net charge of +4 and a 54% hydrophobic ratio.50,51 Although these physicochemical characteristic based on the consensus sequence has been associated with potent anti-MRSA activity, also has been associated with hemolytic effects.52 The sequence alignment of the twenty most active peptides within our dataset showed a consensus sequence (“CKFKAGICHKLKCIAHKYKGGVCK” – Fig. S5 in the ESI†), having a high net charge of +7.5 (ref. 53–55) and 46% hydrophobic ratio. We remarked on the presence of four cysteines (Cys – C) in the active consensus peptide sequence, which contributes to the stability of the tertiary structure,56 for example, in peptides, e.g.AP03010, AP03022, and AP03311 in Fig. 6 (vide infra).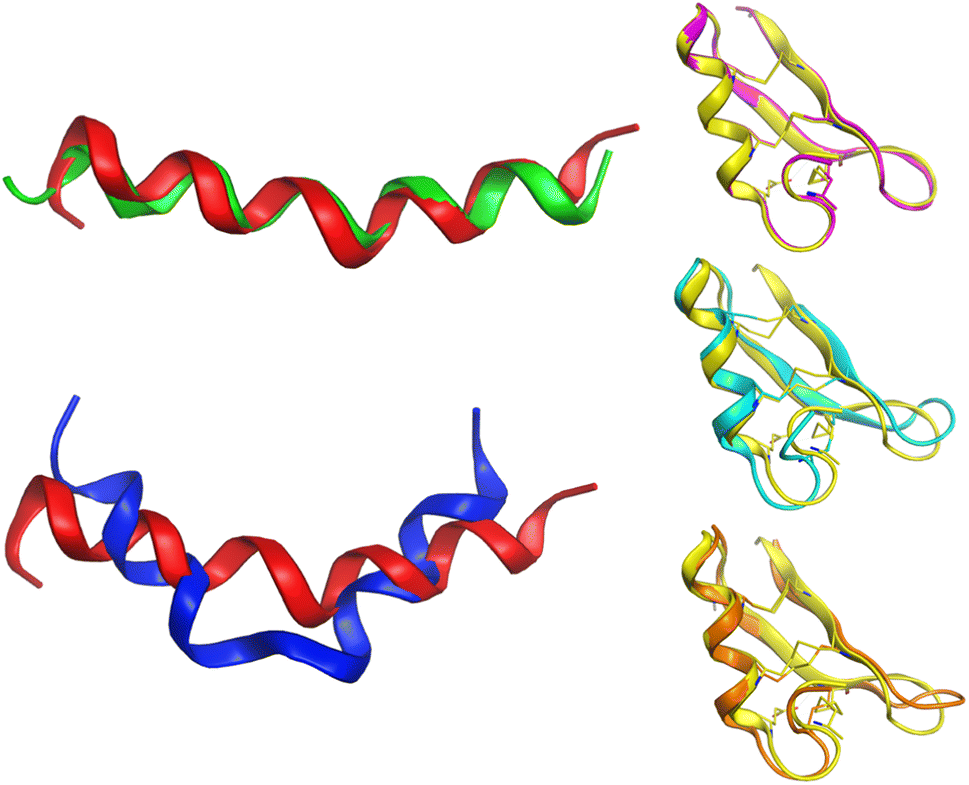 | ||
| Fig. 6 Conformational differences between selected peptides are studied in this work. Each peptide is represented with a different colour: red (AP02565), green (AP02566), blue (AP02567), yellow (AP03010), cyan (AP03022), and orange (AP03311). The tridimensional representation of each peptide was modelled by PEP-FOLD.76 | ||
In summary, these results suggested that the structural similarity calculations based on MAP4 fingerprint and MinHashed function provide a means to explore the peptide activity landscape. Methods such as extended SAS maps and SALI enable the landscape study of the 223 anti-MRSA peptides, rapidly uncovering small structural changes associated with significant modifications in the pMIC50 values. However, this methodology is general and could be adapted to study any other properties of peptides, i.e., P-SA/PR. We noted that TMAPs helped visualise different features of peptides' peptide property landscape. Nevertheless, it is essential to acknowledge that TMAPs, similar to other visualisation techniques, rely significantly on structural representation (such as a molecular fingerprint) and are mainly influenced by the relative size of peptides. Therefore, we recommend limiting the usage of TMAP visualisation to peptides of similar size ranges.
Discussion
Studying peptide structure–activity/property relationships (P-SA/PR) is a re-emerging topic that exhibits a large applicability domain, including medicine, pharmacology, biotechnology, and material industry.57–60 Synthesising peptides on a large scale has been advancing faster than the computational methods to design them. Fortunately, computational strategies are constantly developed to support the experimental design of peptide libraries at medium-large scale, reducing this technical gap. However, it remains challenging to fully decode the P-SA/PR since Nature can be highly complex. Interestingly, peptides could solve many current clinical, biological, chemical, pharmacological, and agrochemical issues.3,61–65For this reason, it is crucial to use novel approaches to quantify and understand their SA/PR studies. Additionally, antimicrobial peptides (AMPs) containing non-canonical amino acids present several advantages over their canonical counterparts (e.g. higher solubility, higher target affinity, higher stability). One of the earliest reported benefits is the improved bioavailability by reducing proteolytic degradation, achieved by incorporating D-amino acids at protease cleavage sites.66 AMPs with non-canonical amino acids also enhance selectivity by offering a broad range of structures and functionalities not present in the twenty canonical amino acids.67
Peptide analysis heavily relies on the alignment of canonical AA sequences in FASTA format,68 as pinpointing the specific position of active motifs is crucial for SAR analysis.69 However, aligning sequences becomes a challenge when dealing with peptides containing non-canonical amino acids, and we have suggested adapting the SMILES code66 to analyse these peptides. We aim not to replace existing alignment techniques but to complement them. We intend to establish a more robust methodology for identifying highly potent sequences and motifs by analysing both canonical and non-canonical groups within a global screening. Activity and property landscapes have been extensively studied for small organic molecules using structural fingerprints to quantify the similarity of chemical compounds. However, due to the lack of a robust molecular fingerprint to represent peptides, the activity/property relationships had not been developed for (short) peptides. However, new fingerprints like MAP4 and notations based on hierarchical editing language for macromolecules (HELM) have opened new avenues to unifying the complex chemical diversity (from small molecules to peptides without non-canonical residues) under a single representation.33,70 Using unifying fingerprints (like MAP4) and notations (like HELM) allows us to explore beyond the canonical realm of peptides, including PTMs or synthetic elements, to the peptide chemical space.
As mentioned in the introduction, identifying activity cliffs is a key element in designing molecules. In this case, the study of important structural motifs in peptides makes it possible to identify those non-canonical amino acids or modifications that give certain properties (e.g., anti-MRSA activity) to peptides. That is, it allows rationalizing which structural motifs or non-canonical modifications could be added to other peptide sequences to hybridize them and improve their already known properties or conversely, eliminate those molecular portions that eliminate the desired property.
Meanwhile, bioinformatics approaches enable the identification of activity cliffs using the sequence alignment of peptides. Chemoinformatics approaches (e.g., molecular similarity metrics) based on topologies, connectivity, tridimensional features, and molecular properties offer a new alternative to studying more complex molecules,71 like peptides. Additionally, a previous study using different fingerprints (e.g., MACCs keys, ECFP4, ECFP6, and atom pairs) permitted the construction of peptide landscapes using unique peptides with the same number of amino acids.72 In contrast, this work shows an application of methods typically used in chemoinformatics to study small organic molecules to study the P-SA/PR studies using the concept of activity/property landscapes.
The anti-MRSA peptide landscape explored in this work (Fig. 3A) indicated a total of 16953 (∼67.31%) peptide pairs in quadrant I (scaffold or R-hopping peptides); 152 (∼0.60%) in quadrant II (smooth SAR peptides); 8055 (∼31.99%) in quadrant III (peptide activity cliffs, like pairs 2–5); and 25 (∼0.10%) in quadrant IV (peptides without a with a discontinuous SAR, like pair 1). Namely, most of a third part of the peptide pairs have considered activity cliffs, which could limit the model ability of these data sets to develop a predictive model of anti-MRSA activity. We pointed out a direct SA/PR study based on pairwise comparisons could be established. For instance, in peptide pair 1 (Fig. 3), the terminal phenylalanine (Phe/F) could be associated with their biological activity difference. This aligns with X-ray diffraction studies, which suggested that a terminal Phe residue in peptide structures could enhance the stability of the helical conformation.73 Furthermore, He et al. confirm that the activity of antimicrobial peptides depends on the strength of their helical structure.74 Therefore, the protocol presented here to describe the activity landscape of the 223 anti-MRSA peptides could identify minor peptide differences involved in their activity.
Another critical example that remarks the impact of one unique AA change on the peptide structure/sequence is the peptide activity cliff 2 (AP02565–AP02567, Fig. 3), which suggests a crucial role of asparagine (Asn – N). Their tridimensional model (as generated with PEP-FOLD) (Fig. 6) reflects the impact of this AA change on the stability of the helical peptide structures. Additionally, quantum methods confirm this observation and remark on the importance of Asn on peptide reactivity.75
Although the predicted tridimensional structures of the peptides forming activity cliffs are similar (pairs 3 (AP03010–AP02656); 4 (AP03010–AP03022); and 5 (AP03010–AP03311) in Fig. 3 and 6), their values of TPSA are different which suggest changes in their solubility (Table S2 in the ESI†). Such differences could be associated with changes in their biological activity.47 Additionally, the differences between the cationic area53,55,77,78 (involved in the membrane interaction on MRSA strains) of each peptide pair could be associated with their variations in biological activity (Table S2 in the ESI†).
These results indicate the dependency of the activity cliffs with the descriptors used to quantify the similarity between pairs of peptides.79 For example, using the  instead of the MAP4 fingerprint as a descriptor, the peptide pairs 2–5 would no longer be considered activity/property cliffs. Namely, these results indicate that the anti-MRSA activity does not depend uniquely on the peptide sequence and the features encoded on MAP4 fingerprints. The anti-MRSA activity also depends on other criteria, like the tridimensional similarity and the physicochemical properties. We remark that selecting the molecular representations is crucial in decoding the P-SA/PR. The same applies to virtually any other computational study: structure representation is vital.
instead of the MAP4 fingerprint as a descriptor, the peptide pairs 2–5 would no longer be considered activity/property cliffs. Namely, these results indicate that the anti-MRSA activity does not depend uniquely on the peptide sequence and the features encoded on MAP4 fingerprints. The anti-MRSA activity also depends on other criteria, like the tridimensional similarity and the physicochemical properties. We remark that selecting the molecular representations is crucial in decoding the P-SA/PR. The same applies to virtually any other computational study: structure representation is vital.
During the past five years, the concept of SA/PR has been adapted to design and develop novel peptidic entities. The idea of P-SA/PR has been used to discover and create lipopeptides and cyclic peptides80,81 and decode the membranolytic mechanism of different peptides.82 However, there are complex challenges to resolve towards consolidating the in silico peptide design area.17,61–64,68 Limited access to quality data and the balance of active and inactive reports make generating new information and knowledge challenging. However, methods that prioritise the selection of the most representative structure could resolve (almost in part) this issue. Additionally, implementing the “Sequence–Structure–Function relationships” concept on peptides is a crucial step forward to exploiting the potential of peptide data. Besides, the biological issues (i.e., immunogenicity, proteolytic degradation, permeability, and toxicity) have been superficially explored.
Current methodologies used to study P-SA/PR have limitations, and the activity landscape approximation presented in this work is no exception. The fingerprint-based similarity (using MAP4 and the MinHassed distance) is a new method to explore and describe the landscape of any peptide property. However, the results of this study suggest that this methodology could be highly sensitive to structural changes on peptides with less than 20 residues, which could limit their applicability, and remarks on the importance of developing new molecular representations focused on peptides. For this reason, we recommend using multiple criteria and methodologies to understand the P-SA/PR. For example, a combination of activity landscape approaches, classical alignment sequence analysis, and 3D approximation help decode the P-SA/PR studies. The present work contributes to establishing a helpful workflow based on structure similarity metrics to explore P-SA/PR and quickly identify non-canonical peptide activity cliffs.
Conclusions
This work presents a new method to explore and describe the landscape of any property of peptides based on the MAP4/MinHashed distance function. We constructed and discussed the activity landscape of 223 anti-MRSA peptides. For the case study, it was concluded that (1) the fingerprint-based similarity values (as measured with MAP4/MinHashed distance function) have a positive correlation with the sequence-based identity values (R2 = 0.31), suggesting that fingerprint-based similarity measures complement the insights derived from sequence alignments, but do not replace them; (2) around 31% of paired anti-MRSA peptides were consider activity cliffs. These findings highlighted the challenges of developing predictive models with such a dataset. As part of this work, we introduced the extended SAS map (using MW differences values of each peptide pair) that facilitated the rapid identification of peptide activity cliffs. The fingerprint-based similarity using MAP4 is an excellent addition to starting a new peptide design/development campaign; however, as with any in silico approach, each has advantages and limitations. Therefore, activity landscape analysis should be combined with classical sequence alignment used in bioinformatics and physicochemical descriptors to explore the SPR in peptides in detail.The primary perspective of this research is to utilise fingerprint-based similarity calculations to create consensus virtual screening protocols. These protocols incorporate various factors, including 2D and 3D structure similarity, chemical properties similarity, and sequence identity. The objective is to identify peptide structures that possess specific properties. Additionally, the methodology outlined in this study would be applied to curate peptide datasets helpful in developing artificial intelligence techniques for predicting peptide properties. Finally, molecular similarity landscapes of non-canonical peptides allow the possibility to study, decode, and optimise multiparametric properties in parallel, such as classical multiparametric landscapes, like DAD maps (Dual Activity Differences maps).83,84
Data availability
Data for this paper, including the anti-MRSA peptides dataset (Table S1†) and interactive peptide activity cliff visualisation generated by DataWarrior software (File S1†), are available at Figshare repository at https://doi.org/10.6084/m9.figshare.23264933.v1.Conflicts of interest
The authors declare there are no conflicts of interest related to this work.References
- A. A. Vinogradov, Y. Yin and H. Suga, J. Am. Chem. Soc., 2019, 141, 4167–4181 CrossRef CAS PubMed.
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS PubMed.
- V. Apostolopoulos, J. Bojarska, T.-T. Chai, S. Elnagdy, K. Kaczmarek, J. Matsoukas, R. New, K. Parang, O. P. Lopez, H. Parhiz, C. O. Perera, M. Pickholz, M. Remko, M. Saviano, M. Skwarczynski, Y. Tang, W. M. Wolf, T. Yoshiya, J. Zabrocki, P. Zielenkiewicz, M. AlKhazindar, V. Barriga, K. Kelaidonis, E. M. Sarasia and I. Toth, Molecules, 2021, 26, 430 CrossRef CAS PubMed.
- R. Ducasse, K.-P. Yan, C. Goulard, A. Blond, Y. Li, E. Lescop, E. Guittet, S. Rebuffat and S. Zirah, ChemBioChem, 2012, 13, 371–380 CrossRef CAS PubMed.
- C. P. Ting, M. A. Funk, S. L. Halaby, Z. Zhang, T. Gonen and W. A. van der Donk, Science, 2019, 365, 280–284 CrossRef CAS PubMed.
- P. Bhadra, J. Yan, J. Li, S. Fong and S. W. I. Siu, Sci. Rep., 2018, 8, 1697 CrossRef PubMed.
- S. Decker, A. Taschauer, E. Geppl, V. Pirhofer, M. Schauer, S. Pöschl, F. Kopp, L. Richter, G. F. Ecker, H. Sami and M. Ogris, Eur. J. Pharm. Biopharm., 2022, 176, 211–221 CrossRef CAS PubMed.
- F. D. Prieto-Martínez, E. López-López, K. Eurídice Juárez-Mercado and J. L. Medina-Franco, in In Silico Drug Design, ed. K. Roy, Academic Press, 2019, pp. 19–44 Search PubMed.
- M. Leutert, S. W. Entwisle and J. Villén, Mol. Cell. Proteomics, 2021, 20, 100129 CrossRef CAS PubMed.
- J. Miranda-Salas, C. Peña-Varas, I. Valenzuela Martínez, D. A. Olmedo, W. J. Zamora, M. A. Chávez-Fumagalli, D. Q. Azevedo, R. O. Castilho, V. G. Maltarollo, D. Ramírez and J. L. Medina-Franco, Artif. Intell. Life Sci., 2023, 3, 100077 CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- F. I. Saldívar-González, V. D. Aldas-Bulos, J. L. Medina-Franco and F. Plisson, Chem. Sci., 2022, 13, 1526–1546 RSC.
- P. Wang, L. Hu, G. Liu, N. Jiang, X. Chen, J. Xu, W. Zheng, L. Li, M. Tan, Z. Chen, H. Song, Y.-D. Cai and K.-C. Chou, PLoS One, 2011, 6, e18476 CrossRef CAS PubMed.
- E. López-López, E. Fernández-de Gortari and J. L. Medina-Franco, Drug Discovery Today, 2022, 27, 2353–2362 CrossRef PubMed.
- B. I. Díaz-Eufracio, O. Palomino-Hernández, A. Arredondo-Sánchez and J. L. Medina-Franco, Mol. Inf., 2020, 39, 2000035 CrossRef PubMed.
- C. D. Fjell, R. E. W. Hancock and A. Cherkasov, Bioinformatics, 2007, 23, 1148–1155 CrossRef CAS PubMed.
- D. Kanduc, J. Pept. Sci., 2012, 18, 487–494 CrossRef CAS PubMed.
- B. Mishra and G. Wang, J. Am. Chem. Soc., 2012, 134, 12426–12429 CrossRef CAS PubMed.
- P. Charoenkwan, S. Kanthawong, N. Schaduangrat, P. Li’, M. A. Moni and W. Shoombuatong, ACS Omega, 2022, 7, 32653–32664 CrossRef CAS PubMed.
- R. De, M. K. Mahata and K.-T. Kim, Adv. Sci., 2022, 9, 2105373 CrossRef CAS.
- S. Q. Pantaleão, P. O. Fernandes, J. E. Gonçalves, V. G. Maltarollo and K. M. Honorio, ChemMedChem, 2022, 17, e202100542 CrossRef PubMed.
- R. P. Sheridan, S. B. Singh, E. M. Fluder and S. K. Kearsley, J. Chem. Inf. Comput. Sci., 2001, 41, 1395–1406 CrossRef CAS PubMed.
- E. López-López, J. Bajorath and J. L. Medina-Franco, J. Chem. Inf. Model., 2021, 61, 26–35 CrossRef PubMed.
- F. Plisson, O. Ramírez-Sánchez and C. Martínez-Hernández, Sci. Rep., 2020, 10, 16581 CrossRef CAS PubMed.
- S. M. Simonsen, L. Sando, K. J. Rosengren, C. K. Wang, M. L. Colgrave, N. L. Daly and D. J. Craik, J. Biol. Chem., 2008, 283, 9805–9813 CrossRef CAS PubMed.
- P. Grieco, V. Luca, L. Auriemma, A. Carotenuto, M. R. Saviello, P. Campiglia, D. Barra, E. Novellino and M. L. Mangoni, J. Pept. Sci., 2011, 17, 358–365 CrossRef CAS PubMed.
- E. Proniewicz, G. Burnat, H. Domin, I. Małuch, M. Makowska and A. Prahl, J. Med. Chem., 2021, 64, 8410–8422 CrossRef CAS.
- E. López-López, O. Rabal, J. Oyarzabal and J. L. Medina-Franco, J. Comput.-Aided Mol. Des., 2020, 34, 659–669 CrossRef PubMed.
- D. Erzina, A. Capecchi, S. Javor and J.-L. Reymond, Angew Chem. Int. Ed. Engl., 2021, 60, 26403–26408 CrossRef CAS PubMed.
- V. C. S. R. Chittepu, P. Kalhotra, T. Osorio-Gallardo, C. Jiménez-Martínez, R. R. Robles-de la Torre, T. Gallardo-Velazquez and G. Osorio-Revilla, Molecules, 2019, 24, 3887 CrossRef CAS PubMed.
- M. Cruz-Monteagudo, J. L. Medina-Franco, Y. Pérez-Castillo, O. Nicolotti, M. N. D. S. Cordeiro and F. Borges, Drug Discovery Today, 2014, 19, 1069–1080 CrossRef CAS PubMed.
- J. Bajorath, A. L. Chávez-Hernández, M. Duran-Frigola, E. Fernández-de Gortari, J. Gasteiger, E. López-López, G. M. Maggiora, J. L. Medina-Franco, O. Méndez-Lucio, J. Mestres, R. A. Miranda-Quintana, T. I. Oprea, F. Plisson, F. D. Prieto-Martínez, R. Rodríguez-Pérez, P. Rondón-Villarreal, F. I. Saldívar-Gonzalez, N. Sánchez-Cruz and M. Valli, J. Cheminf., 2022, 14, 82 Search PubMed.
- A. Capecchi, D. Probst and J.-L. Reymond, J. Cheminf., 2020, 12, 43 CAS.
- A. Capecchi and J.-L. Reymond, Med. Drug Discovery, 2021, 9, 100081 CrossRef CAS.
- D. Erzina, A. Capecchi, S. Javor and J.-L. Reymond, Angew. Chem., Int. Ed., 2021, 60, 26403–26408 CrossRef CAS PubMed.
- G. Wang, X. Li and Z. Wang, Nucleic Acids Res., 2016, 44, D1087–D1093 CrossRef CAS PubMed.
- F. Chaib, B. John and S. Hwang, Antimicrobial resistance crisis, https://www.who.int/news/item/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis, accessed May 23, 2022 Search PubMed.
- K. M. Craft, J. M. Nguyen, L. J. Berg and S. D. Townsend, MedChemComm, 2019, 10, 1231–1241 RSC.
- N. Høiby, O. Ciofu, H. K. Johansen, Z. Song, C. Moser, P. Ø. Jensen, S. Molin, M. Givskov, T. Tolker-Nielsen and T. Bjarnsholt, Int. J. Oral Sci., 2011, 3, 55–65 CrossRef PubMed.
- G. M. Knight, R. E. Glover, C. F. McQuaid, I. D. Olaru, K. Gallandat, Q. J. Leclerc, N. M. Fuller, S. J. Willcocks, R. Hasan, E. van Kleef and C. I. Chandler, eLife, 2021, 10, e64139 CrossRef CAS.
- C. I. Pickens and R. G. Wunderink, Semin. Respir. Crit. Care Med., 2022, 43, 304–309 CrossRef.
- E. Calbo, L. Boix-Palop and J. Garau, Curr. Opin. Infect. Dis., 2020, 33, 458–463 CrossRef PubMed.
- A. M. Algammal, H. F. Hetta, A. Elkelish, D. H. H. Alkhalifah, W. N. Hozzein, G. E.-S. Batiha, N. El Nahhas and M. A. Mabrok, Infect. Drug Resist., 2020, 13, 3255–3265 CrossRef CAS PubMed.
- M. Ferri, E. Ranucci, P. Romagnoli and V. Giaccone, Crit. Rev. Food Sci. Nutr., 2017, 57, 2857–2876 CrossRef CAS PubMed.
- E. López-López, J. J. Naveja and J. L. Medina-Franco, Expert Opin. Drug Discovery, 2019, 14, 335–341 CrossRef.
- T. Sander, J. Freyss, M. von Korff and C. Rufener, J. Chem. Inf. Model., 2015, 55, 460–473 CrossRef CAS PubMed.
- J. L. Medina-Franco, J. J. Naveja and E. López-López, Drug Discovery Today, 2019, 24, 2162–2169 CrossRef CAS PubMed.
- R. Guha and J. H. Van Drie, J. Chem. Inf. Model., 2008, 48, 646–658 CrossRef CAS PubMed.
- D. Probst and J.-L. Reymond, J. Cheminf., 2020, 12, 12 Search PubMed.
- Y. Chen, M. T. Guarnieri, A. I. Vasil, M. L. Vasil, C. T. Mant and R. S. Hodges, Antimicrob. Agents Chemother., 2007, 51, 1398–1406 CrossRef CAS PubMed.
- S.-J. Kang, H.-S. Won, W.-S. Choi and B.-J. Lee, J. Pept. Sci. Off. Publ. Eur. Pept. Soc., 2009, 15, 583–588 CAS.
- J. M. Conlon, B. Abraham, A. Sonnevend, T. Jouenne, P. Cosette, J. Leprince, H. Vaudry and C. R. Bevier, Regul. Pept., 2005, 131, 38–45 CrossRef CAS PubMed.
- B. Mishra, J. Lakshmaiah Narayana, T. Lushnikova, X. Wang and G. Wang, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13517–13522 CrossRef CAS PubMed.
- Z. Jiang, A. I. Vasil, J. D. Hale, R. E. W. Hancock, M. L. Vasil and R. S. Hodges, Pept. Sci., 2008, 90, 369–383 CrossRef CAS PubMed.
- J. J. López Cascales, S. Zenak, J. García de la Torre, O. G. Lezama, A. Garro and R. D. Enriz, ACS Omega, 2018, 3, 5390–5398 CrossRef PubMed.
- A. Rodriguez, M. Ø. Pedersen, E. Villegas, B. Rivas-Santiago, J. Villegas-Moreno, C. Amero, R. S. Norton and G. Corzo, Proteins: Struct., Funct., Bioinf., 2020, 88, 175–186 CrossRef CAS PubMed.
- T. R. Walsh, Acc. Chem. Res., 2017, 50, 1617–1624 CrossRef CAS PubMed.
- W. Ma, F. Luan, H. Zhang, X. Zhang, M. Liu, Z. Hu and B. Fan, Analyst, 2006, 131, 1254–1260 RSC.
- H. Zeng, M. E. Johnson, N. J. Oldenhuis, T. N. Tiambeng and Z. Guan, ACS Cent. Sci., 2015, 1, 303–312 CrossRef CAS PubMed.
- J. Lee, M. Ju, O. H. Cho, Y. Kim and K. T. Nam, Adv. Sci., 2019, 6, 1801255 CrossRef PubMed.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309–325 CrossRef CAS PubMed.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Nat. Rev. Chem, 2020, 4, 615–634 CrossRef CAS.
- R. J. Malonis, J. R. Lai and O. Vergnolle, Chem. Rev., 2020, 120, 3210–3229 CrossRef CAS PubMed.
- S. Mondal, S. Das and A. K. Nandi, Soft Matter, 2020, 16, 1404–1454 RSC.
- D. Bhandari, S. Rafiq, Y. Gat, P. Gat, R. Waghmare and V. Kumar, Int. J. Pept. Res. Ther., 2020, 26, 139–150 CrossRef CAS.
- K. Hamamoto, Y. Kida, Y. Zhang, T. Shimizu and K. Kuwano, Microbiol. Immunol., 2002, 46, 741–749 CrossRef CAS PubMed.
- R. P. Hicks, J. B. Bhonsle, D. Venugopal, B. W. Koser and A. J. Magill, J. Med. Chem., 2007, 50, 3026–3036 CrossRef CAS PubMed.
- S. R. Eddy, Nat. Biotechnol., 2004, 22, 1035–1036 CrossRef CAS PubMed.
- J. D. Thompson, T. J. Gibson and D. G. Higgins, Curr. Protoc. Bioinf., 2002, 2.3.1–2.3.22 Search PubMed.
- T. Zhang, H. Li, H. Xi, R. V. Stanton and S. H. Rotstein, J. Chem. Inf. Model., 2012, 52, 2796–2806 CrossRef CAS PubMed.
- D. Stumpfe, D. Dimova and J. Bajorath, J. Chem. Inf. Model., 2014, 54, 451–461 CrossRef CAS PubMed.
- B. I. Díaz-Eufracio, O. Palomino-Hernández, R. A. Houghten and J. L. Medina-Franco, Mol. Diversity, 2018, 22, 259–267 CrossRef PubMed.
- S. Aravinda, N. Shamala, C. Das, A. Sriranjini, I. L. Karle and P. Balaram, J. Am. Chem. Soc., 2003, 125, 5308–5315 CrossRef CAS PubMed.
- Y. He and T. Lazaridis, PLoS One, 2013, 8, e66440 CrossRef CAS PubMed.
- C. Soriano-Correa, C. Barrientos-Salcedo, L. Campos-Fernández, A. Alvarado-Salazar and R. O. Esquivel, Chem. Phys., 2015, 457, 180–187 CrossRef CAS.
- A. Lamiable, P. Thévenet, J. Rey, M. Vavrusa, P. Derreumaux and P. Tufféry, Nucleic Acids Res., 2016, 44, W449–W454 CrossRef CAS PubMed.
- M. Mihajlovic and T. Lazaridis, Biochim. Biophys. Acta, 2012, 1818, 1274–1283 CrossRef CAS PubMed.
- M. Bacalum and M. Radu, Int. J. Pept. Res. Ther., 2015, 1, 47–55 CrossRef.
- J. L. Medina-Franco, Chem. Biol. Drug Des., 2013, 81, 553–556 CrossRef CAS PubMed.
- S. E. Jujjavarapu and S. Dhagat, Probiotics Antimicrob. Proteins, 2018, 10, 129–141 CrossRef CAS PubMed.
- S. Wang, K. Krummenacher, G. A. Landrum, B. D. Sellers, P. Di Lello, S. J. Robinson, B. Martin, J. K. Holden, J. Y. K. Tom, A. C. Murthy, N. Popovych and S. Riniker, J. Chem. Inf. Model., 2022, 62, 472–485 CrossRef CAS PubMed.
- T. Rončević, D. Vukičević, L. Krce, M. Benincasa, I. Aviani, A. Maravić and A. Tossi, Biochim. Biophys. Acta, Biomembr., 2019, 1861, 827–834 CrossRef PubMed.
- M. González-Medina, O. Méndez-Lucio and J. L. Medina-Franco, J. Chem. Inf. Model., 2017, 57, 397–402 CrossRef PubMed.
- E. López-López, F. D. Prieto-Martínez and J. L. Medina-Franco, Molecules, 2018, 23, 3282 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1. Overview of the protocol implemented in this work; Fig. S2. Descriptive analysis of the 223 anti-MRSA peptides studied in this work; Fig. S3. Alignment analysis of the 223 anti-MRSA peptides studied in this work; Fig. S4. Correlations of identity values and fingerprint-based similarity values; Fig. S5. Alignment analysis of the 20 most potent anti-MRSA peptides. See DOI: https://doi.org/10.1039/d3dd00098b |
| This journal is © The Royal Society of Chemistry 2023 |