
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHalogen bonding regulated functional nanomaterials
Jie
Zheng†
 *a,
Ady
Suwardi†
a,
Claris Jie Ee
Wong
b,
Xian Jun
Loh
*a and
Zibiao
Li
*a
*a,
Ady
Suwardi†
a,
Claris Jie Ee
Wong
b,
Xian Jun
Loh
*a and
Zibiao
Li
*a
aInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), Fusionopolis Way, Innovis, #08-03, Singapore 138634, Singapore. E-mail: zheng_jie@imre.a-star.edu.sg; lohxj@imre.a-star.edu.sg; lizb@imre.a-star.edu.sg
bDepartment of Material Science and Engineering, National University of Singapore, S117576, Singapore
First published on 23rd September 2021
Abstract
Non-covalent interactions have gained increasing attention for use as a driving force to fabricate various supramolecular architectures, exhibiting great potential in crystal and materials engineering and supramolecular chemistry. As one of the most powerful non-covalent bonds, the halogen bond has recently received increasing attention in functional nanomaterial design. The present review describes the latest studies based on halogen bonding induced self-assembly and its applications. Due to the high directionality and controllable interaction strength, halogen bonding can provide a facile platform for the design and synthesis of a myriad of nanomaterials. In addition, both the fundamental aspects and the real engineering applications are discussed, which encompass molecular recognition and sensing, organocatalysis, and controllable multifunctional materials and surfaces.
 Jie Zheng | Zheng Jie obtained her PhD degree from Nanyang Technological University (NTU). Currently, she is working as a research scientist at the Institute of Materials Research and Engineering (IMRE) at the Agency for Science, Technology and Research (A*STAR), Singapore. Her research interests include design and synthesis of novel structural polymers using living radical polymerization, nano-structured material modification, block copolymer self-assembly, and circular materials and their applications. |
 Zibiao Li | Zibiao Li obtained his Ph.D. from the National University of Singapore (NUS). Currently, he is working as a senior research scientist at the Institute of Materials Research and Engineering (IMRE) at the Agency for Science, Technology and Research (A*STAR), Singapore. He is the department head of the Advanced Sustainable Materials Department in IMRE and holds an adjunct Associate Professor position with NUS. His research interests are focused on biodegradable and functional polymeric material design, structural property investigations, and their formulations for healthcare and consumer care applications. |
Introduction
Intermolecular noncovalent interactions have immense technological potential owing to their versatile capability of construction of various complex supramolecular architectures which have found application in drug delivery,1–3 materials engineering,4,5 catalysis,6,7 sensors,6,8–10 biological chemistry,11–13 and many other fields.14–16 Some prominent noncovalent interactions include hydrogen bonding (HB),17–22 halogen bonding (XB),23,24 π–π interaction,25–27 metal ion coordination,28–31 hydrophobic interaction,32–35 donor–acceptor interaction,36–38 and so on. Among these noncovalent interactions, especially in solution, XB is considered the least explored despite its promising potential in supramolecular chemistry applications. Analogous to HB, XB also possesses a net attractive interaction between an electron-poor halogen (i.e. iodine (I) or bromine (Br)) and an electron-rich Lewis base (i.e. an amine or pyridine derivative).39 In the past two decades, HB has been extensively studied as a driving force to build diverse categories of supramolecular architectures.17–22 Given the similarities and beneficial differences from HB, such as higher directionality (the R–X⋯B angle is close to 180°), water resistance, and potential tuneability, XB is considered a fascinating and flexible directing force for self-assembly to fabricate halogen bonded nanomaterials in various applications.40–44To date, most research studies are focused on the theoretical and crystallographic characterization of XB and its applications in the solid phase, including crystal engineering, electrical and magnetic materials, and isomer separation systems.4,45,46 With the rapid development of controllable self-assembly in liquid and solution phases, an increasing number of investigations on the potential application of XB directed self-assembly in the liquid and solution phases (such as liquid crystal (LC), drug design, biological systems, anion recognition, and catalysis) are expected.6,46,47 In recent years, some research groups have shed light on the study of halogen bonding towards smart material construction. The development of halogen bonding containing polymers was introduced by Schubert's group.48 Besides, Resnati's group49 and Das' group50 highlighted the design criteria in constructing various halogen-bonded functionalities by virtue of the unique features of halogen bonding.
Unlike these important published studies, in this minireview, the use of XB as a flexible linkage for functional nanomaterial fabrication in the liquid and solution phases and its state-of-the-art development will be discussed, focusing on taking readers through the concept and features of halogen bonding to explore its functionalities and applications (Fig. 1). Along with the introduction of the nature of XB, different types of halogen bonded LC will be presented. After that, XB induced small molecular and polymeric self-assembly in solution will be described. Moreover, different applications of the halogen bonded nanomaterials in various fields, such as molecular recognition and sensing, organo-catalysis, and the fabrication of functional materials and surfaces, will be discussed as well. This minireview concludes with a short perspective on the future research direction of XB directed nanomaterials.
 | ||
| Fig. 1 Schematic illustration of XB-directed self-assembly in liquid and solution phases. | ||
Nature of halogen bonding
XB is a highly directional supramolecular interaction with an R–X⋯B angle close to 180°. It contains an electron-poor halogen-bond donor part (X = I, Br, Cl, F) and an electron-rich halogen-bond acceptor part (B). The distance between X and B atoms is less than the sum of their van der Waals radii. For organic halide compounds, it was reported that the electron density distribution around X atoms is anisotropic. In these systems, the effective atomic radius along the extended C–X bond axis is much longer than in the direction perpendicular to this axis. Furthermore, along the extended C–X bond axis, a region (“σ hole”) of positive electrostatic potential is apparent and becomes larger with increasing X atomic size and halogen polarizability. This “σ hole” is surrounded by a belt of negative electrostatic potential, disposing the lone pair closer towards the X atom, resulting in the straight orientation of XB.Given the experimental data obtained from different states (i.e., solid, liquid, and gas phases), the strength of the XB donor is predicted to increase in the order of Cl < Br < I. The strength of XB can be tuned in a controlled manner, either by varying the nature of X atoms or by modifying the moiety that is covalently bound to the X atom. In general, increasing the electron-withdrawing ability of the functionalities positively contributes to the XB strength.
The bonding energy of XB is up to 200 kJ mol−1, which is comparable to the HB. The same charge-transfer interaction allows XB to exhibit similar trends to HB in energetic and geometric properties. As is well known, X atoms are much larger than H, and the interaction between X and B is therefore more sensitive to steric bulk or secondary interactions. Besides, XBs are less sensitive towards solvent polarity and the binding constants for X-bonding donor–acceptor pairs are three orders of magnitude greater than closely related H-bonded pairs in polar solvents. It is due to the intrinsic hydrophobic nature of the participating halogen atom and the fundamental difference in the nature of interaction via its electropositive sigma (σ)-hole site, which is more robust to solvent polarity.40 Moreover, it is notable that HB can enhance XB interaction through the synergistic effect. For example, the Ho groups described the synergistic relationship between the hydrogen bond and halogen bond.51 Through the polarization effect caused by the hydrogen bond, the affinity of the receptor for halogen anions was significantly increased.
Halogen-bonding induced self-assembly in the liquid state (liquid crystals)
The liquid crystal (LC) is a state of matter that combines both the order feature of solid crystals and the fluidity of normal liquids. Normally, LCs encompass a central rigid core, flexible end chains, and linking groups. Both covalent bonds and non-covalent interactions (e.g., hydrogen bonding) have been extensively used as bridges to connect the core and chains of LCs. Apart from hydrogen bonding, another type of non-covalent interaction, halogen bonding, serves as a new key driving force to generate supramolecular liquid-crystalline assemblies. Herein, different halogen bonded LCs, such as simple halogen-bonded LCs, polymeric halogen-bonded LCs, and photo-responsive halogen-bonded LCs, will be discussed.Simple and polymeric halogen-bonded liquid crystals
Halogen-bonded LCs are constructed by the interaction between the halogen-associated electrophilic region (XB donors) and nucleophile (XB acceptors). In the past two decades, various XB donors and acceptors have been designed to form different halogen-bonded LCs. Through the reaction between functional XB donors and acceptors, halogen-bonded LC materials with different structures and properties can be synthesized effectively.The use of halogen-bonding as a driving force to generate supramolecular LCs dates back to 2004.52 Bruce's group prepared the first LC complexes through the combination of iodopentafluorobenzene (XB donor) and nonmesomorphic 4-alkoxystilbazoles (XB acceptors) (Fig. 2A). This opened the door for new applications of XB in functional LCs. Although the used XB acceptors did not show a liquid crystalline nature, the resulting materials exhibited LC properties. The complexes with longer chains (i.e., n = 8, 10 and 12 in Fig. 2A) displayed enantiotropic smectic A phases. Moreover, it was revealed that most prominently solids were cleanly melted without the appearance of biphasic regions which would characterize a nonstoichiometric aggregate, indicating that the resultant complexes formed via the formation of noncovalent halogen bonds not a quadrupolar interaction. Besides, other XB donors, such as molecular iodine (I2), were also utilized to prepare halogen-bonded LCs (Fig. 2B).53 Taking advantage of the short halogen bond formed between the molecular iodine and alkoxystilbazoles, the resultant LCs had unusually high mesophase stability. In this system, they disclosed that the intermolecular I⋯I contact changed its nature due to the strong polarization, resulting in the formation of the SmC phase. However, the preparation of analogous bromine complexes caused an unexpected electrophilic substitution in the ethylenic fragment of the alkoxystilbazole. Besides, XB also drove the self-assembly of fluorocarbons and hydrocarbons.54 It was notable that this system was able to overcome the low affinity between perfluorocarbon and hydrocarbon derivatives, effectively driving the self-assembly of alternating perfluorocarbon and hydrocarbon layers and exhibiting great potential for supramolecular polymer coating.
 | ||
| Fig. 2 Simple liquid crystals formed by self-assembly through XBs. LC complexes through the combination of (A) iodopentafluorobenzene and nonmesomorphic 4-alkoxystilbazoles, and (B) iodopentafluorobenzene and molecular iodine. | ||
Researchers also attempted to extend the halogen-bonded LC molecules into the domain of trimeric LCs. He and co-workers explored the feasibility of XB as a driving force to prepare liquid-crystalline supramolecular polymers.55 Through the reaction between difunctional halogen-bonding molecules, a new family member in liquid-crystalline polymeric materials was successfully built (1 to 4 in Fig. 3A). This could be confirmed using the infrared vibration, where the binding energies of both the XB acceptors and donors were changed in comparison with their respective unbonded equivalents, indicating the successful formation of halogen bonds in them. After that, the same group continued to prepare different trimeric supramolecular thermotropic LCs via halogen bonding driven self-assembly (5 in Fig. 3A).56 The formation of XB interaction was substantiated by infrared and X-ray photoelectron spectroscopy. Besides, they also showed that there was little correlation between the length in spacers and terminal groups and phase transition temperatures. Trimeric halogen bonded supramolecular LC complexes were also synthesized by the self-assembly of alkoxystilbazole and 1,4-diiodotetrafluorobenzene with a molar ratio of 2 to 1 (Fig. 3B).57 It was revealed that the starting materials only exhibited monotropic nematic phases, while the resultant complexes displayed enantiotropic nematic phases with a range of up to 11 °C. The same group subsequently reported the fluorophobic effect on supramolecular mesogens via XB directed self-assembly of alkoxystilbazoles and α,ω-diiodoperfluoroalkanes (Fig. 3C).58 It was shown that even though the starting materials have a non-liquid crystalline nature, most of the prepared complexes demonstrated liquid-crystallinity. This was the first instance of the employment of halogen bonding and iodoperfluoroalkanes to fabricate fluorinated LCs. The authors also proposed to prepare other halogen-bonded fluorinated LCs via employing mono- and di-iodoperfluoroalkanes, with shorter and longer chains.
 | ||
| Fig. 3 (A)–(D) Polymeric liquid crystals formed by self-assembly through XBs through the reaction between difunctional halogen-bonding molecules. | ||
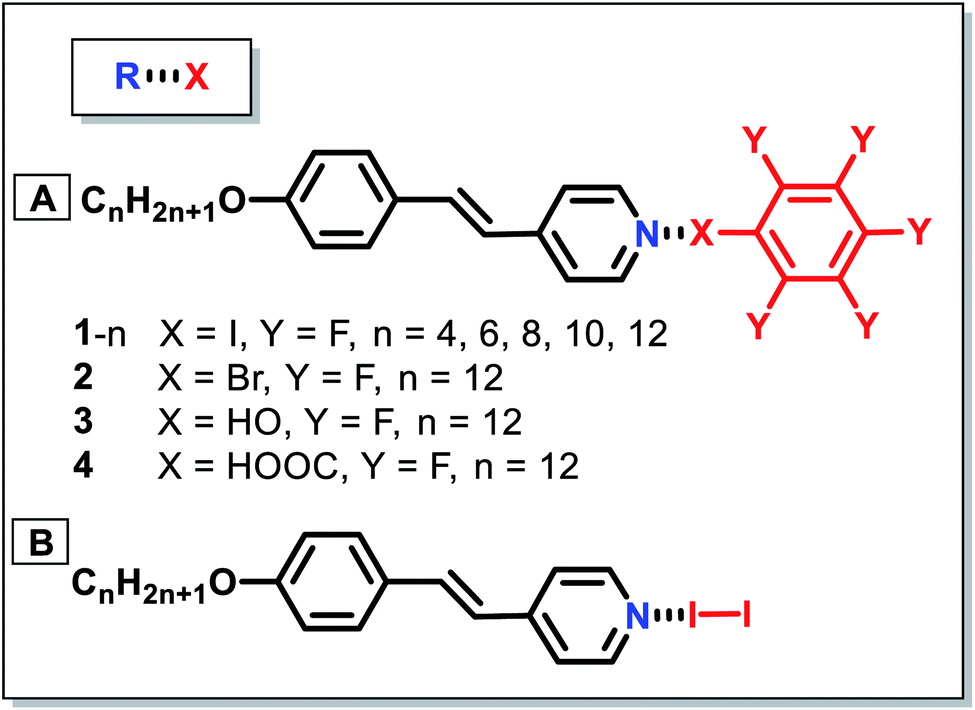
Besides, Cavallo and colleagues described the preparation of new trimeric LC complexes by employing 1,4-diiodotetrafluorobenzene or α,ω-diiodoperfluoroalkanes as XB donors, and an alkoxystilbazole derivative functionalized with a methacrylate terminal group as the XB acceptor (Fig. 3D).59 Despite the nonmesomorphic nature of the starting materials, the synthesized LCs showed liquid-crystalline properties with smectic A phases. As evident by single crystal X-ray diffraction analysis, the self-alignment of the molecules was mainly ascribed to the formation of N⋯I bonds between the starting diiodoperfluorocarbons and alkoxystilbazoles (Fig. 4). The obtained LCs in this system had decomposition temperatures higher than the melting points of the starting XB donors, showing the feasibility of XB-driven self-organization for the construction of stable volatile perfluorinated compounds. More remarkably, the resultant LCs with reactive groups demonstrated great potential in different fields, such as liquid crystalline elastomeric actuators. Remarkably, Nikolay et al. demonstrated millimeter length scale assemblies of halogen-bonded mesogens.60 Such nanoscale organization guiding macroscopic alignment was achieved via halogen bonding of mesogenic 1-iodoperfluoroalkanes to a star-shaped ethyleneglycol-based polymer, having chloride end-groups. It was found that the parallel stacking of the mesogens into aligned domains led to layers at 10 nm periodicity. Such an alignment was possible by taking advantage of the directionality of halogen bonding. Such a layer-by-layer assembly was also demonstrated in poly(4-(4-iodo-2,3,5,6-tetrafluorophenoxy)-butyl acrylate) and poly(4-vinylpyridine), with 1.3 nm thickness of the bilayer. In addition, the halogen-bonded multilayer was reported to have a better stability compared to hydrogen-bonded multilayer films in methanol.61
 | ||
| Fig. 4 Partial view of the crystal packing of the XB-based LC in ref. 59. Adapted with permission from ref. 59. Copyright 2017, Elsevier. | ||
Photo-responsive halogen-bonded liquid crystals
Recent years have witnessed the rapid development and increasing demand for stimuli-responsive functional materials, especially light sensitive materials. Herein, photo-responsive halogen bonded LCs containing azobenzene functional groups with the ability to undergo photoinduced trans–cis phase transitions will be discussed. Compared to non-light-triggered LCs, these photo-sensitive LCs exhibit improved properties with precise remote and regional control, offering various opportunities in practical applications.In 2014, Lu et al. successfully engineered photo-responsive LCs by complexing halogen molecules and azopyridine compounds with the aim of investigating the features of XB in LC formation and photo-responsive behaviors (Fig. 5A).62 In this system, azopyridine (AzPy) derivatives were chosen as the XB acceptor due to their ability to undergo isothermal photoinduced phase transitions. Owing to the strict geometrical constraint of XB, it was selected to construct LCs with enhanced surface relief gratings (SRGs) and a more rigid junction. Furthermore, the strong interactions between XB donors and acceptors allowed the possibility to choose specifically suitable halogen atoms to take part in bond formation without altering its electronic structure. Despite multiple attempts by Bruce et al. in 2004,52 bromine-bonded LCs could not be obtained. It was thus a breakthrough for Lu and coworkers to be able to obtain them as well as quantify their thermal properties. They then went on to study the photo-responsive properties of the obtained complexes under UV irradiation, where photoinduced phase transition from an LC to an isotropic phase was observed. This process was reversible and the initial LC texture was recovered upon exposure to visible light for 80 s. Resnati et al. were the first to synthesize a halogen-bonded photo-responsive low molecular weight (Mw) supramolecular complex with efficient photoalignment that arises from its high temperature liquid crystallinity, the first of very few examples to date (Fig. 5B).63 The designed halogen-bonded complex was prepared by self-assembly of N,N-dimethyl-4-[(2,3,5,6-tetrafluoro-4-iodophenyl)diazenyl]aniline and 4-[(E)-2-(4-methoxyphenyl)-ethenyl]-pyridine. The methoxy-substituted stilbazole was suitable as longer alkyl chains which were known to quell the formation of SRGs in LC polymers. Paired with light-responsive azo groups, these resultant low Mw LCs exhibited high degrees of photo-induced anisotropy upon polarised light irradiation. The LC nature of the complex enhanced the molecular cooperative motions despite being at room temperature while maintaining exceptional SRG formation efficiency. Alaasar and coworkers were the first to present examples of photo-sensitive XB based supramolecular polycatenars formed between a non-mesogenic taper shaped tetrafluoroiodobenzene (XB donor) and non-mesogenic or mesogenic azopyridine derivatives (XB acceptors) (Fig. 5C and D).64 Under UV light irradiation, the polycatenar LC was observed to undergo fast and reversible photoinduced phase transition. Lastly in comparison to related hydrogen bonded polycatenars, halogen bonded polycatenars showed lower melting temperatures as well as lower mesophase stability. However, in this system, they were able to achieve fast and reversible photoswitching in the bulk state which was not possible with hydrogen bonded polycatenars. These findings were invaluable and hold great potential for further applications in the production of functional materials. Dichiarante and coworkers made use of classic LC concepts and were able to use a new approach to assemble 2 new supramolecular nanostructures by complexing 1-ethyl-3-methylimidazolium iodides and azobenzene derivatives containing an iodotetrafluoro-benzene ring (Fig. 5E).65 Exploiting the modularity of this approach, new functionalities were allowed to be introduced into the final products via tailoring the chemical structure of the starting complexes. In the present system, the iodide anions act as XB acceptors to link two azobenzene derivatives (XB donors), producing an alternative family of self-assembled ionic liquid crystals with mesomorphic behavior, independent of the length of the alkyl chain of the cation. Due to the high directionality of XBs, together with the segregation between the charged and uncharged parts, the prepared LC showed a layered structure in the crystal lattice. Taking advantage of the azobenzene functional group, the resultant ionic LCs exhibited the LC-to-isotropic phase transition under light irradiation, offering great potential towards applications in light-controllable ion transporters. Recently, Navarro-Rodríguez reported a halogen-bonded highly anisotropic photoactive mesogen through the combination of a hexyloxy-substituted pyridyl-ethynyleneazobenzene and a tetrafluoroiodophenyl decanoate (Fig. 5F).66 The obtained hexyloxy-substituted pyridyl-ethynylene azobenzene was a thermotropic liquid crystal encompassing a single nematic and two different smectic phases. More remarkably, in this system, the smectic period showed a shorter length compared with the theoretical one, indicating a single layer stacking of molecules, offering the possibility of the head-to-tail arrangement to satisfy the electrostatic interaction between fluorinated and non-fluorinated rings. In the work by Cinthya et al., the synthesis of polymers carrying pyridylazobenzenes and iodopentafluorobenzene rings via self-assembly using halogen bonds was reported. In addition, the halogen complexes show liquid crystal properties of smectic type as well as photo-induced isomerization despite the bulky chromophore units. Furthermore, a good photo-response was observed in both writing and erasing experiments. The halogen bonded complex demonstrates regular SRGs, which can be potentially used for optical data storage units.67 Recently, Giese and coauthors reported a new strategy to control the XB-based LC and its photophysical behaviours (Fig. 6).68 They revealed that the halogen bond strength can be fine-tuned by adjusting the position and degree of fluorination at the XB donor. Furthermore, the prepared XB-based supramolecular liquid crystals exhibited photoinduced isomerization, which were not only related to the photochemical properties of the molecular components but also the operation temperature and the clearing point.
 | ||
| Fig. 5 (A)–(F) Examples of photoresponsive halogen bonded LCs containing azobenzene functional groups. | ||
 | ||
| Fig. 6 Schematic illustration of a photo-responsive XB-based LC and its tuneable photophysical properties via changing the aromatic fluorine substitution. Adapted with permission from ref. 68. Copyright 2019, American Chemical Society. | ||
Li and coauthors explored a novel halogen-bonded light-driven axially chiral molecular switch, shedding new light on the use of XB in the design and manipulation of functional supramolecules and host–guest materials for photonic applications.69 They synthesized axially chiral binaphthyl based azo compound 6 to act as the XB acceptor and paired it with XB donor 3 to form compound 7 as seen in Fig. 7A. Photodynamic cholesteric liquid crystals (CLCs) were fabricated via doping compound 7 into the commercially available achiral nematic host. The helical twisting power (HTP) of 7 decreased upon UV light irradiation due to the trans–cis photoisomerization of azo moieties. Upon observation of the large HTP change in 7, they then went on to study the photo-tunability of the selective reflection colour and were able to achieve reversible colour tuning across the whole visible spectrum. Recently, the same group took their work further and designed 2 new XB-based chiral molecular switches with reversible photoisomerization capability upon exposure to visible light.70 These molecular switches (XB donors) were able to generate CLCs through XB formation via mixing with the commercially available achiral liquid crystal host 5CB (as both the XB acceptor and host medium) (Fig. 7C). It is noteworthy that the properties of CLCs varied with varying terminal halogen atoms in molecular switches. When the molecular switches bore an iodo terminal group, the obtained CLC displayed a cholesteric–nematic phase transition. Meanwhile, on the other hand, in the case of terminal bromo atoms in the molecular switch, the resultant CLCs showed reversible handedness inversion to the helical superstructure upon irradiation with visible light of different wavelengths.
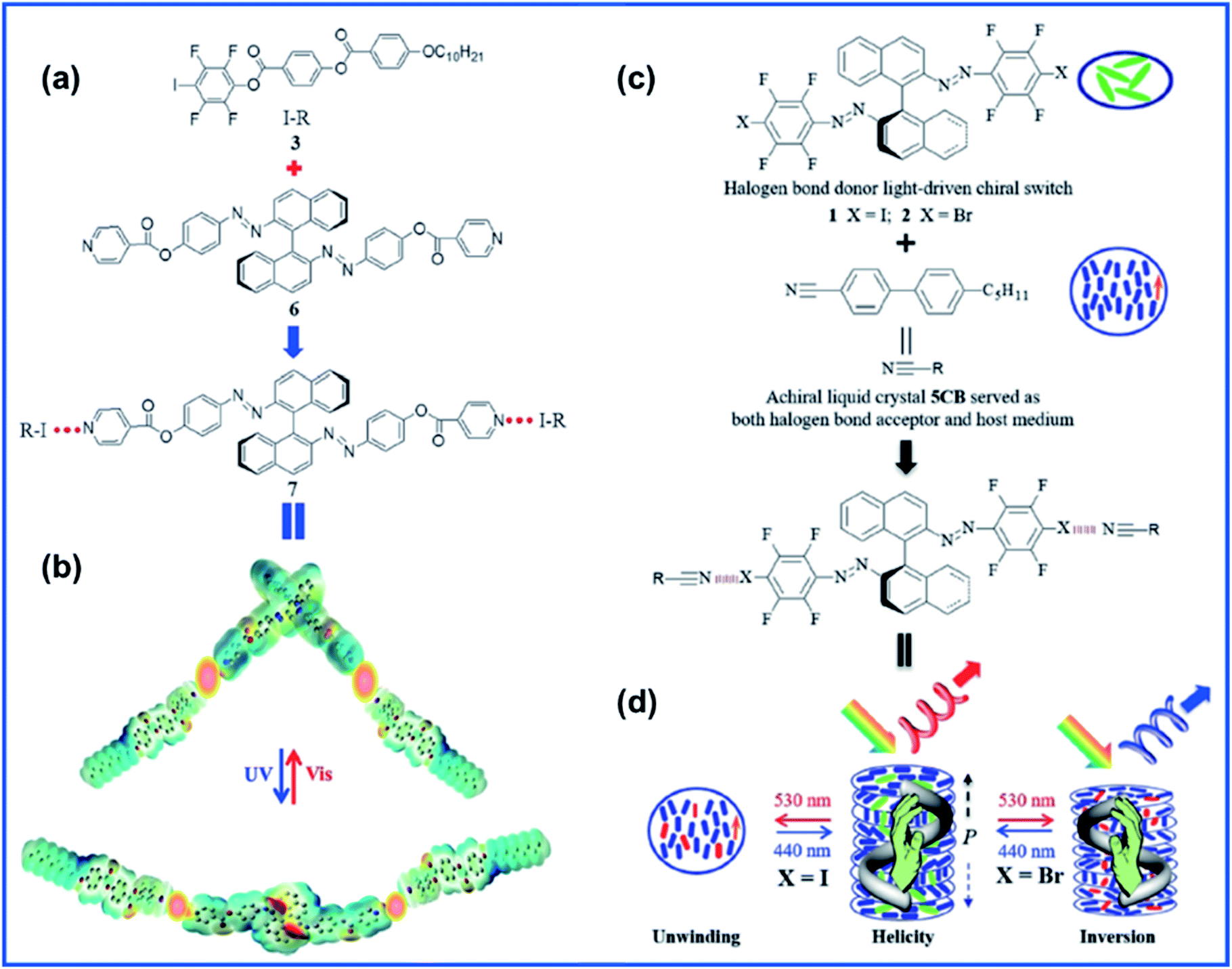 | ||
| Fig. 7 (a) Molecular structures of halogen-bond donor 3 (I-R), halogen bond acceptor 6, and the resulting halogen-bonded light-driven chiral molecular switch 7 and (b) its photoisomerization upon light irradiation illustrated by an electron potential density map obtained at the DFTB3LYP 6-31G level. Adapted with permission from ref. 69. Copyright 2018, Wiley. (c) Chemical structures of halogen bond photodynamic cholesteric liquid crystals. (d) Schematic representation of reversible unwinding and handedness inversion in the self-organized helical superstructure upon visible-light irradiation. Adapted with permission from ref. 70. Copyright 2020, Wiley. | ||
Other halogen-bonded liquid crystals
XB-driven self-assembly acts as an effective and convenient technique to prepare not only linear-shaped LCs but also bent-shaped LCs. For example, Bruce and coauthors reported halogen-bonded LCs with a bent-core mesogen (Fig. 8A).71 These complexes were prepared by the co-crystallization of 1,3-diiodotetrafluorobenzene and 4-alkoxystilbazoles in THF via gradually evaporating the solvent. It was interesting that despite the use of achiral starting 1,3-diiodotetrafluorobenzene, the obtained LCs showed a chiral nematic phase. They also revealed that the phase sequence was caused by the lability of the halogen-bonded complex. Andrea et al. reported iodine-modified FF dipeptides via halogenation.72 Through single crystal X-ray characterization, it was revealed that halogen atoms play a key role in the packing behaviour to obtain these compounds. In addition, it also provided stability to the dry interfaces formed by aromatic rings, contributing to the solid state packing of dipeptides. | ||
| Fig. 8 (A) and (B) Examples of halogen bonded LCs with bent structures. | ||
Researchers also explored halogen bonded ionic LCs, which exhibited the properties of LCs and ionic liquids. The properties of these LCs can be modified with the change of the nature of the anion and cation parts. Cavallo and colleagues reported a new family of fluorinated ionic liquid crystals, which was synthesized between 1-polyfluoroalkyl-3-alkylimidazolium iodides and mono-iodoperfluoroalkanes of different chain lengths or di-iodoperfluorooctane.73 Over a wide range of temperatures, these resulting halogen bonded ionic LCs showed enantiotropic liquid crystalline phases.
Besides, Bruce's group also reported the formation of LCs using nonmesomorphic components (4-alkoxystilbazoles and 4-iodotetrafluorophenol) by the combination of both halogen and hydrogen bonding (Fig. 8B).74 It was revealed that the more electrostatic nature of the hydrogen bond contributes favourably to phase stabilization. Moreover, they also assumed that compared with p-C6F4–F, p-C6F4–I was a more effective terminal group for phase stabilization, presumably due to the great polarisability anisotropy.
Halogen-bonding induced assembly in the solution phase
Halogen-bonded molecular self-assembly
Analogous to HB, XB has emerged as an engaging tool to regulate (macro)molecular organization. XB also exhibits some beneficial differences from HB, such as higher directionality and hydrophobicity, as well as being favored in polar solvents, providing many new opportunities to achieve novel nanoarchitectures.48,49,75In 2017, Jiang and co-workers reported the first example of a C–I⋯π halogen bonding driven supramolecular helix in highly dilute acetonitrile solution by using a bilateral I-substituted alanine based N-amidothiourea (Fig. 9).76 The β-turn structure of the compound was designed with two iodophenyl groups at two terminals of the compound to act as the donor and acceptor of the C–I⋯π halogen “head to tail” linkage, facilitating the propagation of the helical fragments, and thus supporting the final supramolecular helix. This was a milestone finding contributing to their further research in 2019 where they expanded on this concept to develop an artificial double helix.77 In virtue of an alanine based bilateral N-(p-iodobenzoyl) amidothiourea, which in its folded cis-form, moves the S-atom closer towards the central benzene ring while allowing a similar helical β-turn structure to remain, leading to the formation of a right- or left-handed double helix. Furthermore, the synthesized sample exhibited high thermal stability up to 75 °C in dilute acetonitrile solution indicating strong intermolecular double crossed hydrogen bonds.
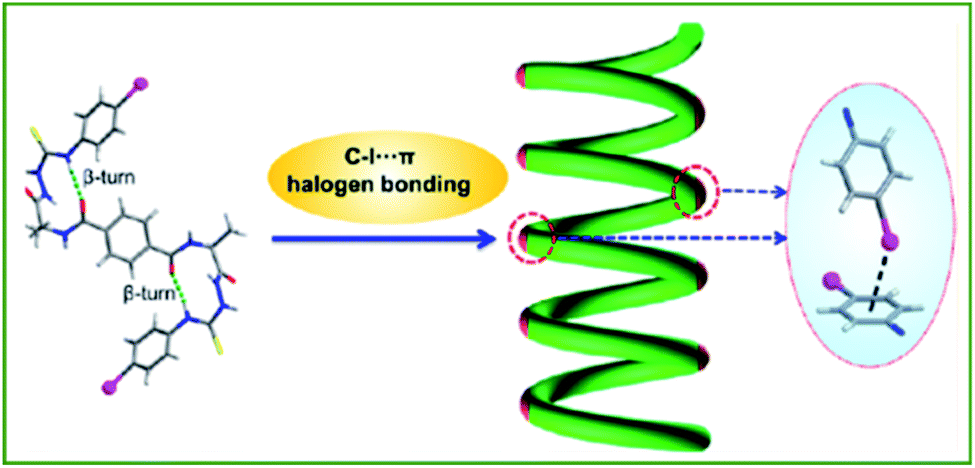 | ||
| Fig. 9 Schematic illustration of the C–I⋯π halogen bonding driven supramolecular helix. Adapted with permission from ref. 76. Copyright 2017, American Chemical Society. | ||
Very recently, Das' group presented a synthetically straightforward design strategy for the fabrication of functional nanoarchitectures based on halogen bonding.78 A new example of luminescent supramolecular polymers with a discrete nanorod-like structure was developed by the self-complementary crystallization of the building block (i.e., 5-(2,3,5,6-tetrafluoro-4-iodophenoxy)quinoline) in methylcyclohexane (MCH). Furthermore, the obtained nanoparticle was able to disassemble under acid conditions along with the disappearance of the blue emission. It is noteworthy that the self-assembly morphologies could be synthetically tuned via minor variations in the position of the XB donor or the nature of the halogen atom in the building block. This work offers various possibilities for further development particularly in the construction of optically rich nanostructures with precise control.
Because of the strong affinity between XB donors and acceptors, anions (XB acceptors) in solution are able to effectively bind with suitable XB donors via forming of halogen bonding for anion recognition and sensing. Beer and co-authors reported the first example of a supramolecular anion host system in which XB interaction was applied to selectivity bind and optically detect iodide in water by using a ruthenium rotaxane-based complex.79 In this system, a new water-soluble XB ruthenium(II) functionalized rotaxane featured a XB axle-shaped component prepared by permethylated b-cyclodextrin motifs, and a luminescent component. The prepared rotaxane was used for iodide recognition and sensing through the formation of XB with high affinity and selectivity. After that, the same group developed the recognition and sensing of a halide anion in wet organic solution through the formation of XB in the neutral interlocked host molecular systems.80 Notably, fine-tuning of the XB strength and selectivity was possible through controlling the position and number of XB donor groups (i.e. iodotriazole) within the interlocked cavity. Besides, they also synthesized the first XB [3]rotaxane host system containing a bis-iodo triazolium-bis-naphthalene diimide four station axle component.81 They revealed that XB rotaxane was selective for nitrate over the other anions (such as acetate, hydrogen carbonate, and dihydrogen phosphate oxoanions and chloride). Compared with a HB analogue, this system displayed enhanced anion recognition (which will be further discussed in the subsection of Molecular Recognition and Sensing).
Halogen-bond-driven supramolecular assembly with an XB linker
Supramolecular assembly or polymeric self-assembly in solution is currently receiving well deserved attention due to its importance in many areas of supramolecular chemistry and materials science. Non-covalent interactions (such as HB, XB, ion pairing, crystallization, hydrogen bonding, and metal chelation) have been widely leveraged to segregate polymer blocks to prepare various sophisticated nanostructures.23,82–84 The advantage of such non-covalent interaction lies in the formation of intricate morphologies that would have been difficult to achieve using conventional methods. Furthermore, stimuli-responsivity can be dialled in via disruption of non-covalent contact. Amongst these interactions, halogen bonding induced polymer assembly has received increasing attention in, for instance, drug delivery, sensor separation, and stimuli-responsive systems.46,48,49Taking advantage of the water-resistant profile, halogen bonding exhibits benefits over the widely reported hydrogen bonding for non-covalent surface modification in aqueous medium. Das' group recently synthesised a graftable supramolecular polymer via halogen-bond-driven assembly in water solution.85 In this system, they combined the strengths of halogen and hydrogen bonding to complement one another, for instance, by incorporating water sensitive hydrogen bonds in the hydrophobic core domain of the supramolecular polymer and halogen bonding on the outer surface to create a hydrophilic active surface that will give rise to structurally diverse grafting. Self-assembly utilizing both hydrogen and halogen bonds in an orthogonal manner has yet to be widely studied, especially in water solution due to the competing solvent interaction of H-bonding. Herein, a benzene-1,3,5-tricarboxamide (BTA) functionalized with three pyridyl (Py) groups at the periphery was used as the X-bonding and H-bonding acceptor, to co-assemble with a PEG-based polymer (X-donor) to form a helical nanostructure in acid aqueous solution. This offers promising insight into the further development of such pH responsive complex molecular assemblies.
Beyond that, halogen bonding can also be used to drive highly ordered self-assembly of block copolymers, which can eventually be used for preparing tailored nanostructures for semiconductor applications. By minimizing repulsive interactions between different blocks and maximizing attractive interactions between similar blocks, microphase separation can be achieved. Traditionally, small-molecule additives which can specifically bind to one block by non-covalent interactions can be used to achieve such microphase separation. In recent reports, Roberto et al. used halogen bonding to direct hierarchical self-assembly of diblock copolymers and low molecular weight perfluorinated molecules.86 The separation of the microphase resulted in an upright cylindrical domain, which consisted of internal lamellar self-assemblies which could be cleaved using solvent to yield segmented cylindrical micelles stabilized by halogen bonded supramolecular crosslinks. Such a directed self-assembly strategy is highly promising for high throughput and cost-effective nanoscale semiconductor manufacturing processes. It exploits the spontaneity of block copolymer self-assembly onto lithographically nanopatterned surfaces to yield periodic nanostructures with a size of tens of nanometers.
Very recently, Goto's group explored halogen bonding induced supramolecular assembly of quaternary ammonium iodide (QAI)-containing polymers in three phases, i.e., solution, surface, and solid phases (Fig. 10).87 Instead of using neutral nitrogen or oxygen atoms, this work used iodide anions (I−) as the XB acceptor to react with the XB linker (containing iodide) to generate halogen bonds with a strong bond energy of up to 180 kJ mol−1. Exploiting the co-assembly of the QAI-polymer and diiodide linker in an aqueous solution, different sophisticated morphologies were obtained, including giant vesicles and one-dimensional nanostructures as well as large compound micelles. Besides, they also studied XB-driven QAI-polymer assembly on surfaces and in the solid state. On the surfaces, QAI-based polymer brushes were able to undergo reversible crosslinking and decrosslinking via the repeat formation of XB, leading to a smart surface with tuneable wettability. Last but not least, they used a QAI-containing monomer to prepare XB-based co-crystal monomers, which were subsequently used for polymerization via solid-phase polymerization (SPP), synthesizing solid polymer sheets. Interestingly, the attained polymer sheet could be used as an appealing host material to reversibly capture halogen-containing guest molecules. These results demonstrate that the halogen bond is a powerful molecular tool for functional nanoarchitecture construction.
 | ||
| Fig. 10 Schematic illustration of XB-driven self-assembly of QAI-containing polymers in three phases and applications. Figure reproduced with permission from ref. 87, copyright 2021, Elsevier. | ||
Halogen-bonded polymeric self-assembly
Controlled radical polymerization (reversible addition–fragmentation chain-transfer polymerization, RAFT) was carried out to polymerize an iodoperfluoroarene-bearing methacrylate to prepare an XB donor-functionalized polymer.88 It was revealed that the polymeric donor moiety was able to combine with an amine-containing block copolymer (XB acceptor) in both aqueous and organic media, resulting in higher-order nanoparticles, initiating the development of XB induced polymeric self-assembly. Vanderkooy et al. reported a structurally novel nanoarchitecture prepared by halogen bonding induced self-assembly of iodoperfluoroarene-substituted polystyrene in acetone solution.89 RAFT polymerization was leveraged to prepare well-defined halogen bond donating polystyrene derivatives. A solvent switching protocol was used to achieve the co-assembly of XB donor polymers with an amine-functionalized methacrylate block copolymer, which was confirmed by electron microscopy and dynamic light scattering analysis. More importantly, the work reports on intricate wormlike structures, vesicle structures, and hexagonally packed hoops, which are typically challenging by conventional methods. A new supramolecular system using halogen bonded macromolecular substances was reported by Salla et al.90 It was shown that there was complex formation between N-benzyl ammonium resorcinarene bromide as a halogen bond acceptor and a library of polymeric halogen bond donors, as confirmed by dynamic light scattering and transmission electron microscopy. The study also showed that both polyethylene glycol block copolymers and homopolymers can act as effective donors of the halogen bond to form polymer-architecture dependent complex morphologies.To understand more about halogen-bond mediated assembly in emulsion droplets, Xihuang et al. reported an I⋯N bond induced confined-assembly to realize order–order phase transitions.91 Polystyrene-b-poly(4-vinyl pyridine) (PS-b-P4VP) AB diblock copolymer was chosen to be the halogen acceptor while the halogen donor was an iodotetrafluorophenoxy substituted C-type homopolymer, poly(3-(2,3,5,6-tetrafluoro-4-iodophenoxy)propyl acrylate), PTFIPA (Fig. 11). The core domain of the nanostructure was increased due to the halogen bonding donor–acceptor pairing between P4VP and PTFIPA, leading to an order-to-order structure transition. Through adjusting the PTFIPA length and surfactant types, the morphologies of nanostructures changed from spherical to cylindrical to lamellar to inverse cylindrical in a controlled manner. Moreover, the internal morphology transition was allowed by selective swelling and deswelling of the P4VP segment, resulting in different sophisticated architectures, such as tailored mesoporous microparticles and disassembled nanodiscs, as well as super aggregates.
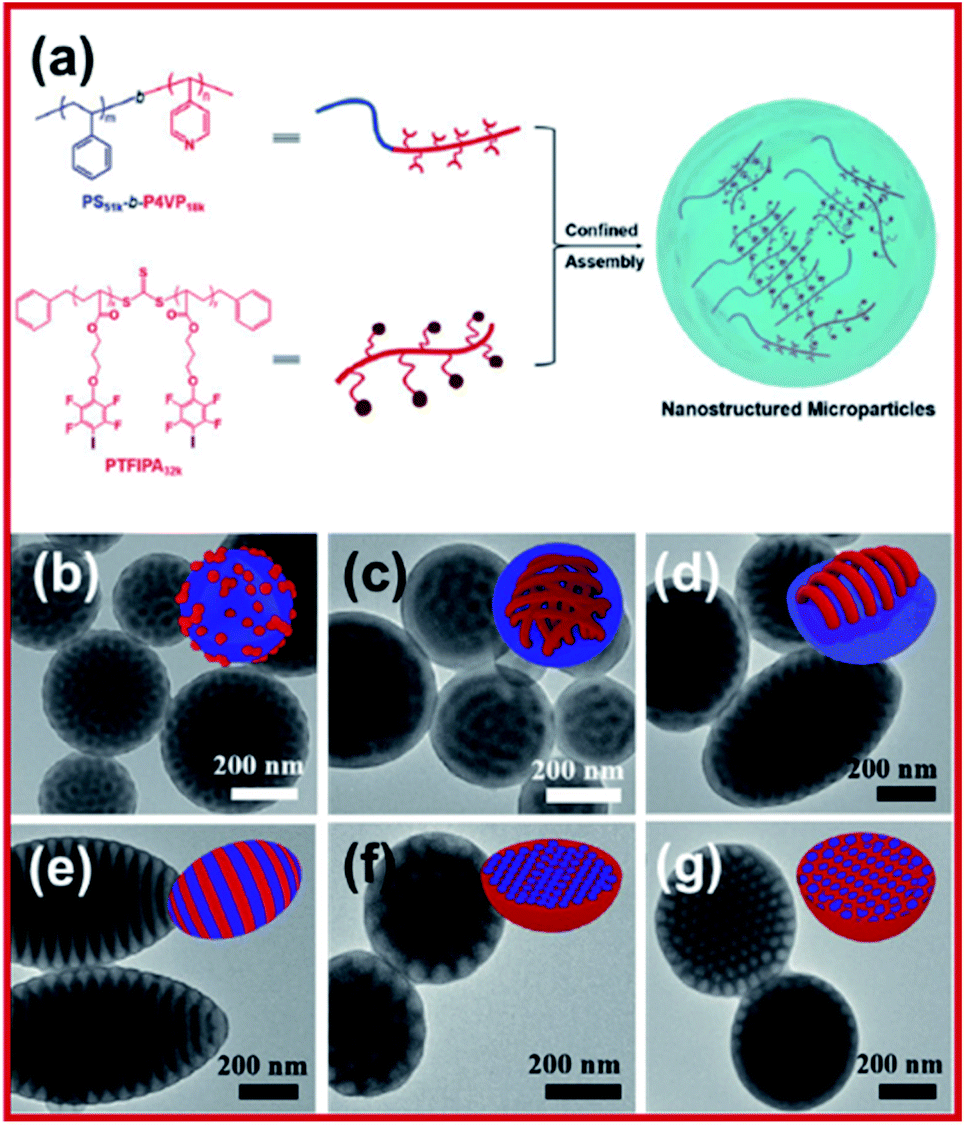 | ||
| Fig. 11 (a) Schematic illustration of halogen bonding mediated formation of nanostructured microparticles via confined-assembly between PS51k-b-P4VP18k AB diblock copolymer and a PTFIPA32k C-type homopolymer. (b–g) TEM images of PS51k-b-P4VP18k blended with different molar ratios of PTFIPA32k, ranging from 0 to 0.2, 0.5, 1, 2, and 3, respectively, using PVA as an emulsifier. Insets in the upper-right are the cartoons showing the nanostructured particles, blue and red colors represent the PS and halogen bonded P4VP/PTFIPA domain, respectively. Figure reproduced with permission from ref. 91, copyright 2021, WILEY-VCH GmbH. | ||
The applications of halogen-bonded assemblies
XB has recently attracted increasing attention owing to its unique properties such as directionality, strength, tunability and hydrophobicity. It has been used as a tool to design and synthesize supramolecular functional materials.23 More recently, it has also been a popular tool to drive self-assembly of polymers.23,46 In addition, halogen bonding has also found good use in a broad range of crystalline materials for novel optoelectronics, drug formulation, molecular machines, porous solids, and even conductive materials.92–102Molecular recognition and sensing
Because of the prevalence of anions in biology and the environment, their recognition and sensing are continuously developing, and various new research efforts have been devoted to the design of new effective anion receptors. However, due to the anion's inherent low affinity in aqueous solution and high energy of solvation, only a few anion binders have been developed so far. Supramolecular interactions, such as HB, anion–π interactions, and ion–ion interactions, have shown selective anion recognition, while XB is less exploited in this field. Because of the electron-rich nature of XB, it exhibits great potential as an excellent anion binding candidate.An important example of halogen bonding based anion helicates in solution was reported by Casey et al.103 In this system, iodide was encapsulated by a tubular anion channel formed by the self-assembly of three tricationic arylethynyl strands (Fig. 12). Remarkably high temperature stability was observed in the triplex endowed by eight strong iodine⋯iodine halogen bonds and numerous buried p-surfaces. Fig. 12a shows each complex composed of three intertwined tricationic nonameric strands that were offset along a common axis defined by two intrachannel iodides. Four halogen bonds bound each iodide within the helical channel with an average halogen bond distance of 3.4 Å. As a consequence, pseudo-square-planar coordination was achieved, shown in Fig. 12c. Complementing the halogen bonds are the numerous aromatics and ethynyl p-stacking interactions with an average ring–ring distance of 3.7 Å, as shown in Fig. 12b. In addition, ion-pairing interaction enables seven iodides on the helicate's exterior to balance the nine positive charges associated with cationic strands. The approximate height and width of each triplex are 13 Å and 19 Å, respectively, with a pitch of 10 Å. A 2.7 Å pore with halogen bond donors represents the unique microenvironment within the triple helicate. The yellow sticks in Fig. 12a and c represent the molecular axis of symmetry (C2) for the triplex alignment with the C–I bond of the nonbonding iodopyridinium donor. Overall, such complex shape stabilized by the numerous strong and linear halogen bonds and π-stacking interactions was persistent at high temperatures and in the aqueous phase.
 | ||
| Fig. 12 Solid-state representations of the triple helicate and DFT minimized nonamer. (a) Solid-state structure of the triple helicate binding two intrachannel iodides. (b) Crystal structure of the triplex looking down its anion channel (iodides removed for clarity). (c) Pseudo-square-planar coordination geometry of the helicate's halogen-bond donors (scaffolding removed). Black dashes denote halogen bonds. (d) DFT-minimized nonamer of 7. Black dashes and iodides added to emphasize the non-convergence of the halogen-bond donors. Figure reproduced with permission from ref. 88, copyright 2016, WILEY-VCH GmbH. | ||
Halogen bonding based sensing was also used in triggering aggregation induced emission (AIE). For instance, Mostafa et al. reported on an ultrasensitive and selective sensor for picomolar detection of Ag nanoparticles based on halogen bond triggered AIE.104 The iodine atom in the dye (CyI) skeleton acts as the halogen bond acceptor while aggregates of iodine atoms on the Ag NP plasmonic surfaces serve as the halogen bond donor or form halogen bonds with the p-orbitals of Ag+. Fluorescence enhancement was evident due to the formation of XB, which was the basis of the Ag NPs or Ag+ sensor. The responsivity of the sensor was linearly correlated with the Ag NP concentration within the range of 1.0–8.2 pM with a LOD of 6.21 pM (σ = 3) while for Ag+ the linearity was over the 1.0–10 mM range (LOD 2.36 µM). Overall, the sensors show remarkable sensitivity for Ag NPs (down to the pM range) as compared to Ag+ (mM range). Remarkably, no interference was observed even when different metal ions of 10-fold higher concentrations were present, opening a pathway for application in wastewater sensing.
Organo-catalysis
Although XB donors have found applications in synthetic organic chemistry as easy-to-handle organic Lewis acids, their activation is mainly limited to sp2 hybridized nitrogen atoms of quinolines and imines, carbonyl oxygen atoms, and alkyl halides. Masato et al. recently reported the preferential interaction of soft Lewis acidity of XB donors with soft electrophiles, which represents the first efficient electrophilic activation of iodonium (III) ylides using the XB donor.105 In addition, the XB donor/Brønsted base cooperative catalytic system was developed to enable in situ formation of the nucleophilic component without the deactivation of the XB donor catalyst. Besides electrophilic activation, Yongquan et al. reported a silver-catalysed intermolecular aminosulfonylation of terminal alkynes with TMSN3 and sodium sulfinates.106 Sequential hydroazidation of the terminal alkyne and sulfonyl radical addition to the resultant vinyl azide make up the three-component reaction. This method opens an avenue to stereoselective synthesis of a wide range of β-sulfonyl enamines without electron-withdrawing groups on the nitrogen atom.Metal-free initiation systems for cationic polymerization are attractive for environmentally benign polymer synthesis. Koji et al. reported the development of XB mediated and controlled cationic polymerization of isobutyl vinyl ether (IBVE) using 2-iodoimidazolium salts as organocatalysts.107 Just by adding a small amount of nBu4NCl (0.02 equivalents), controlled cationic polymerization was accomplished, with the final obtained poly IBVE having a molecular weight distribution below 1.3. Lastly, Akinobu et al. reported on the first instance where iodoalkyne was used as a halogen bond donor catalyst.108 Thioamines were efficiently activated using iodoalkyne with a pentafluorophenyl group as a catalyst to react with 2-aminophenol and generate benzoxazoles with high yield. This opens a path for iodoalkynes as a scaffold for halogen bonding catalysis.
Functional materials
In recent years, halogen bonding has been utilized as a flexible building block for the fabrication of diverse functional materials, such as smart gels and phosphorescent materials. Supramolecular gels are of great interest in various fields, such as catalysis, drug delivery, and controlled release systems. Attributed to their capability of repeatedly forming aggregates by using dynamic non-covalent interactions, these gels display switchable rheology and controlled flow characteristics, which are appealing features in real applications. A novel supramolecular gel with tuneable properties based on halogen bonding was reported by Steed's group.109 The gelation was carried out between a pyridyl substituent in a bis(pyridyl urea) and 1,4-diiodotetrafluorobenzene. Interestingly, taking advantage of the hydrophobicity of XB, this synthesized gel exhibited excellent stability even in polar media, involving aqueous solutions, which had been thought to be impossible for such hydrophilic HB-based gels. Thus, the utilization of XB in supramolecular gel formation undoubtedly facilitates the manipulation of functional materials in real application engineering. Hu et al. reported photo-responsive gels with changeable morphologies in response to optical stimulation.110 Halogen bonded azopyridine-containing azopy-C10 and 1,4-tetrafluorodiiodobenzene (TFDIB) were used to prepare the gel in acetonitrile or ethanol. Gel–sol transition induced by UV irradiation was caused by trans–cis isomerization of the azopyridine moiety, with accompanying morphological variation from flakes to fluffy bobble-like and subsequently to peony-like with increasing exposure time. The authors attributed the microstructural change to the variations of cis-isomer content and the strength of halogen bonding. Such novel supramolecular gel can find applications in information storage and anticounterfeiting.Phosphorescent materials, particularly room temperature phosphorescent materials (RTPs), have immense potential in various applications as diverse as photonic materials, bioelectronics, and night-vision devices. The most widely employed RTPs are inorganic or organometallic complexes due to their fast singlet-to-triplet intersystem crossing (ISC) rates. However, most of the aforementioned RTPs consist of novel metals, are high cost and toxic, and have tough synthesis requirements, making them low competitive in the phosphorescence market. On the other hand, organic phosphorescence has drawn increasing attention due to the absence of metal, as well as being cheaper and environmentally friendly. Like other non-covalent interactions, XB has been utilized to prepare long-lasting RTPs with high quantum efficiency (QE).111 Outstanding work was carried out by Chi's group for the development of ultralong organic phosphorescence with high QE up to 52%, which is the highest afterglow efficiency reported so far (Fig. 13).112 According to the comprehensive photophysical and theoretical investigations, they revealed that the significantly improved QE was ascribed to the key effect of intermolecular halogen bonding in boosting the ISC rate and restricting non-radiative pathways. This work offers a brilliant strategy to improve phosphorescence efficiency for the design of high QE RTPs for practical application.113 Recently, a range of fast triplet-forming tetraethyl naphthalene-1,4,5,8-tetracarboxylates were developed for XB-based RTP by Wu and coworkers.114 The number and position of bromo substituents of the compound were controlled to adjust the intermolecular XB interaction, to realize the suppression of non-radiative relaxation and improvement of the ISC process. The highest QE of 20% was obtained when the molecule is surrounded by a Br⋯O halogen-bonded network.
 | ||
| Fig. 13 (a) Molecular structures of the halogen-bonded compound CaS4Br and its three isomers. (b) Image illustration of the 3D pattern fabricated by the micro-crystalline molecule CzS2Br under daylight with the excitation source on and off. Figure reproduced with permission from ref. 112 and 115. Copyright 2020, WILEY. | ||
Additionally, stimuli-responsive luminescent materials can also be prepared via XB induced self-assembly. Das et al. reported a unique halogen bonded supramolecule to construct emissive supra-π-amphiphiles in water (Fig. 14, schematic).115 It consisted of a hydrophilic iodotetrafluorophenyl functionalized polyethylene glycol (PEG-I) or triethylene glycol (TEG-I) based X-bond donor and a hydrophobic pyridyl functionalized naphthalene monoimide (NMI-Py) based X-bond acceptor. Through the higher ordered assemblies' luminescent properties being governed by dipole–dipole interaction and π-stacking of the NMIPy fluorophore, XB functioned as a key linkage to obtain nanostructures. Fig. 14(a) shows a TEM image of an aqueous solution of NMI-Py + TEG-I which revealed nearly spherical hollow structures with diameters ranging from 200–400 nm, similar to a vesicle-like morphology. At elevated temperatures, no change in the size of nanostructures was observed, which was a testament to the high thermal stability, but they could be readily denatured in 1% acetic acid as shown in Fig. 14(b). A CLSM (confocal laser scanning microscope) was used to elucidate the vesicles' emissive properties. Evidently, the hydrophobic membrane of the vesicles composed of the NMI-Py fluorophore emits green fluorescence, as shown by CLSM in Fig. 14(c). A hydrophobic dye, Nile red, showed selective localization within the NMI-Py member, as shown by CLSM in Fig. 14(d). In addition, it was found that variation of the chain length of the X-bond donors led to various morphologies such as fibers or vesicles for PEG-I and TEG-I. Such a strategy can be leveraged for facile synthesis of structurally diverse smart luminescent functional materials.
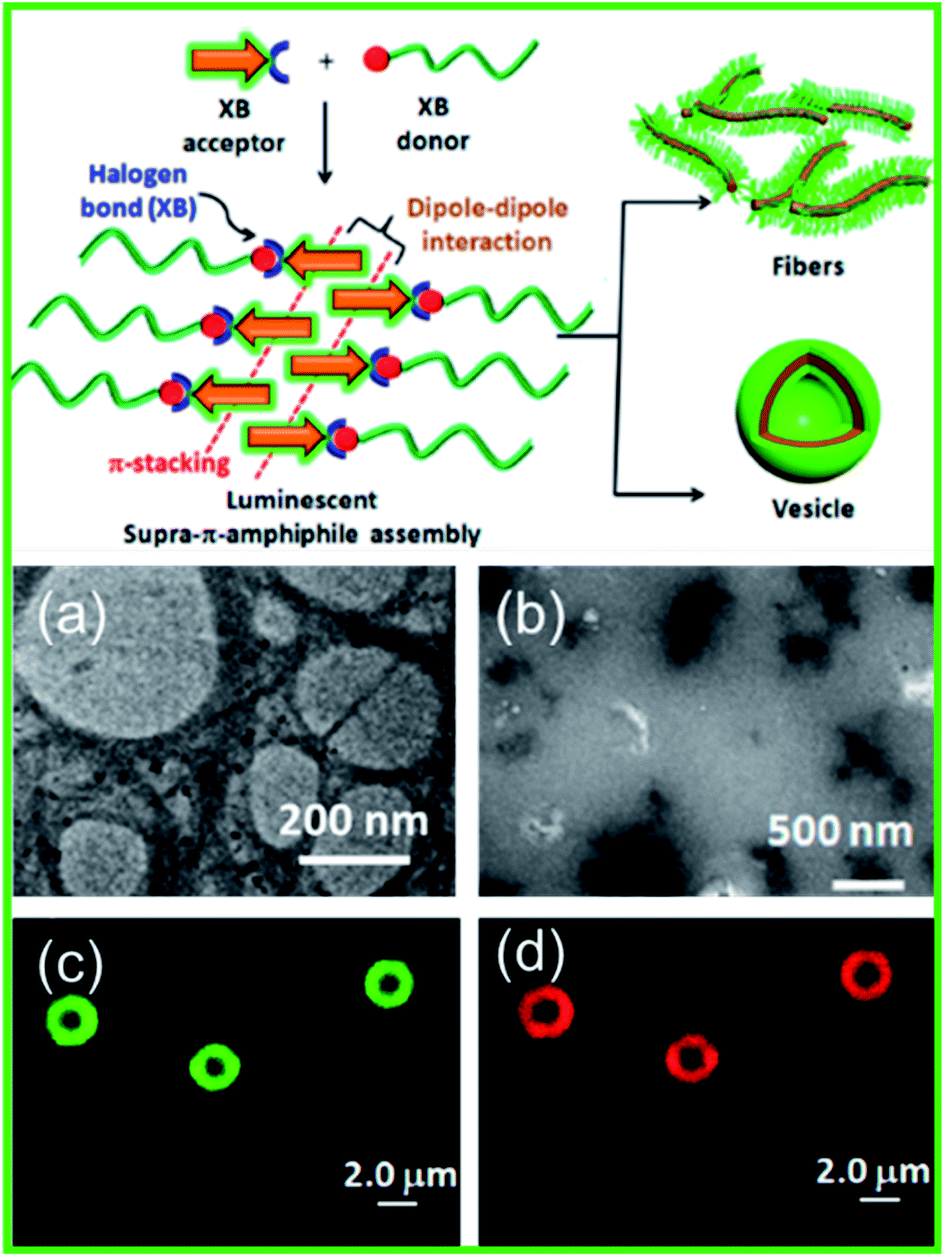 | ||
Fig. 14 Schematic illustration of the XB based assemblies and TEM image showing a solution of NMI-Py + TEG-I (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in (a) water and (b) 1% acetic acid. CLSM (confocal laser scanning microscope) images of Nile red encapsulated NMI-Py + TEG-I (1:1) in water (c) through the green channel and (d) through the red channel. Figure reproduced with permission from ref. 115. Copyright 2020, American Chemical Society. :1) in (a) water and (b) 1% acetic acid. CLSM (confocal laser scanning microscope) images of Nile red encapsulated NMI-Py + TEG-I (1:1) in water (c) through the green channel and (d) through the red channel. Figure reproduced with permission from ref. 115. Copyright 2020, American Chemical Society. | ||
Besides, XB induced self-assembly is also a powerful tool to construct other functional materials, such as self-healing materials. Ronny et al. demonstrated the first XB based self-healing behaviour in thin films.116 They explored the first isothermal calorimetric titrations to characterize the anion affinity dependence on key parameters such as monomeric vs. polymeric receptor, charge assistance, and hydrogen vs. halogen bonding. These donor systems were paired together with copolymer bearing accepting carboxylate groups to prepare crosslinked polymer networks. Taking advantage of the reversibly formed halogen bonds, the resulting material showed excellent self-repairing ability.
Controllable functionalized surfaces
Apart from the intrinsic functional properties, functionalizing the material surfaces is important to control their interactions with other molecules or surfaces. The formation of halogen bonded 2D supramolecular assemblies on solid surfaces has been gaining increasing attention in recent years. For instance, work from Shaoze et al. provided theoretical insights into the halogen bonded network formation of pyridine derivatives and aryl-halide molecules on a graphene surface using first principles DFT calculations.117 In order to elucidate molecule–molecule and molecule–substrate interactions, the density of states (DOS), electron density difference (EDD), non-covalent interaction index (NCI), and scanning tunneling microscopy (STO) images were presented. The N⋯I interactions were predicted to be stronger than the molecule–surface stacking interactions between the terminal pyridyl groups and iodoperfluorobenzenes. They also appeared to be the primary interactions in the self-assembled networks, confirmed by STM images. Such theoretical insights laid a good foundation towards understanding of molecular self-assembly interactions on surfaces.Experimentally, halogen-bond mediated intermolecular interactions have been reported to influence the spectroscopic and photophysical properties of organic film materials.118 In regioisomeric phenanthridine derivatives, it was shown that the different intermolecular Br⋯N halogen bonds, H⋯Br hydrogen bonds, and Br⋯Br type-II halogen bonds were the dominant factors in determining the different nanostructures and film morphologies. SEM images provided further insights into the self-assembly process, as shown in Fig. 15. A rod-like aggregation was observed in the 2,9-BHP molecule in the films (Fig. 15a), which looked straight and had prismatic crystals less than 1 micrometer in diameter. In contrast, the 3,8-BHP self-organizes into a nanofiber-like structure about 50 nm in diameter, as shown in Fig. 15c. They are slightly twisted with respect to each other along right-handed helical sense, resulting in minimized interaction energy, shown in the inset of Fig. 15c. In addition, the 3,8-BHPO and 2,9-BHPO form nanosheets with lamellar stacking, as evident in Fig. 15(c) and (d). This demonstrates that the intermolecular interactions of 2,9-BHPO are stronger than those of 3,8-BHPO since the 3,8-BHPO was unable to form a uniform morphology, which signifies no significant intermolecular hydrogen or halogen bond. Therefore, the role of halogen bond in this work shone light on the molecular structure–packing–property relationship in these organic materials.
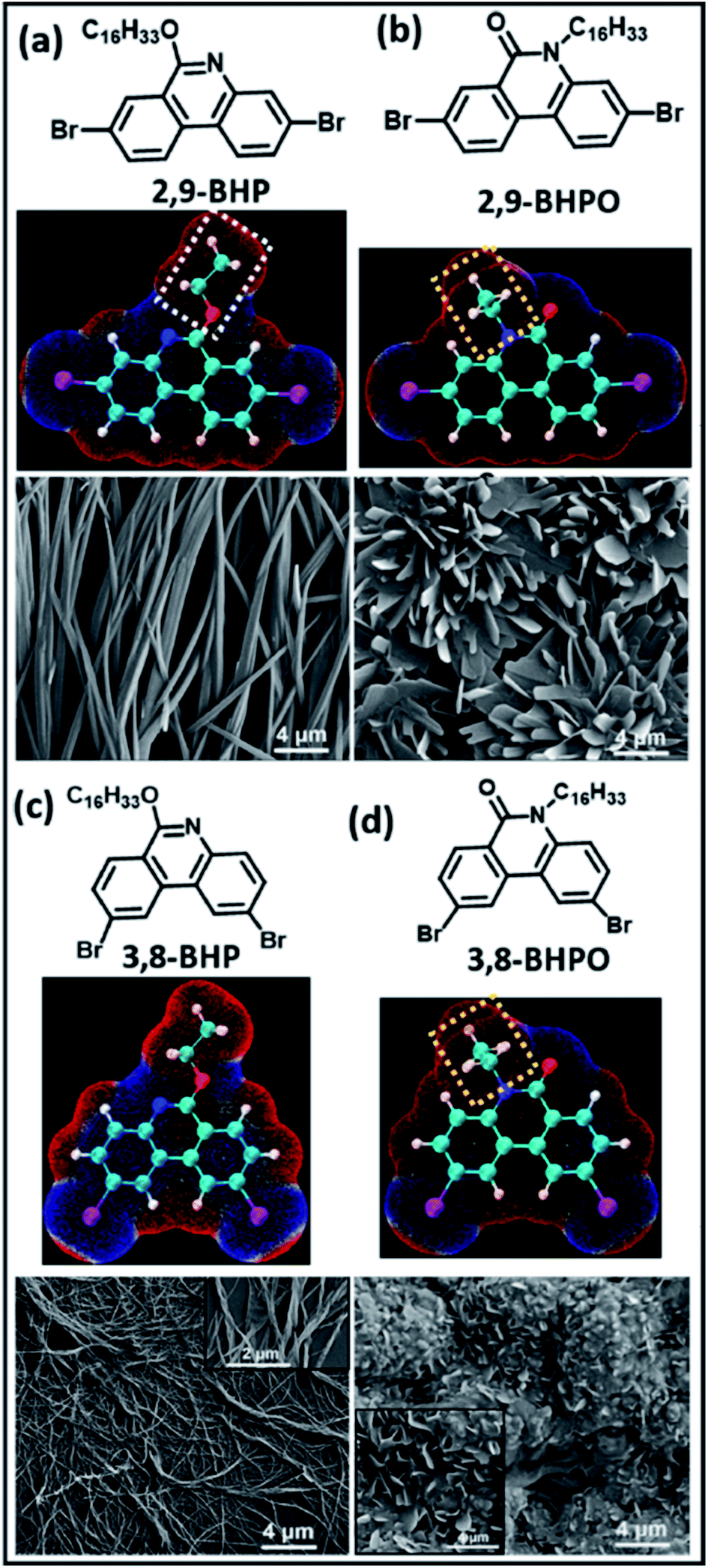 | ||
| Fig. 15 Molecular structures (calculated), electrostatic potential (ESP) maps, and SEM images showing the film morphologies on mica surfaces. (a) 2,9-BHP; (b) 2,9-BHPO; (c) 3,8-BHP; (d) 3,8-BHPO. Figure reproduced with permission from ref. 118. Copyright 2020, American Chemical Society. | ||
More recently, Yuxuan et al. demonstrated the fabrication and manipulation of nanostructure ordering in polymer films driven by supramolecular self-organization.119 The halogen bond in the block copolymer facilitates microphase separation via XB-driven self-assembly in the supramolecule. Through the halogen bonded interaction, the nonmesogenic block copolymer which was made up of poly(ethylene oxide) and azopyridine-containing polymethacrylate was transformed into a supramolecular liquid crystalline polymer. In comparison, no such ordering was observed in the hydrogen bonded liquid crystalline polymer, which indicates that the highly directional nature of XB and the resulting supramolecular mesogenic ordering plays a key role in the enhanced ordering. Remarkably, efficient photoalignment and reorientation of nanostructures which coincided with the supramolecular mesogen direction was achieved using linearly polarized light irradiation, as shown in Fig. 16. Such photoreorientation is highly promising for novel advanced composite liquid crystal applications.
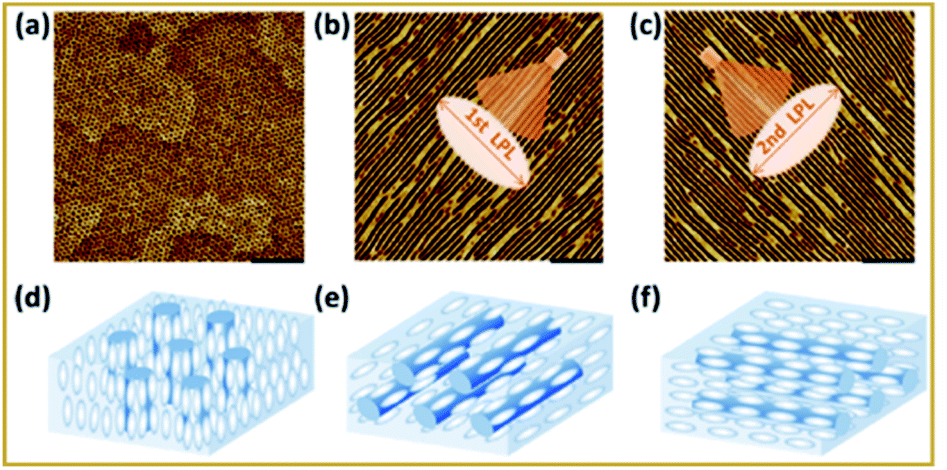 | ||
| Fig. 16 Photoreorientation of supramolecular mesogens and MPS nanostructures in the XB-involved supramolecular LCBC film. AFM images of (a) the annealed film, (b) the film after the first irradiation with LPL of 460 nm at 75 °C, and (c) the film after successive irradiation with orthogonal LPL at 75 °C. Scale bar: 200 nm. (d–f) The possible schematics of the supramolecular mesogens (white) and PEO nanocylinders (blue) corresponding to (a–c), respectively. Images showing the film morphologies on mica surfaces. (a) 2,9-BHP; (b) 2,9-BHPO; (c) 3,8-BHP; (d) 3,8-BHPO. Figure reproduced with permission from ref. 119. Copyright 2020, American Chemical Society. | ||
Conclusions and perspectives
Halogen bonding serving as a driving force to direct (macro)molecular self-assembly has progressed in leaps and bounds over the last few years. In this minireview, we discussed the nature and structure of halogen bonding and its unique application in regulating supermolecules' construction in solution and liquid phases. In addition, halogen bonding and the resulting assemblies have found applications ranging from molecular recognition and sensing, organocatalysis, and surface functionalization.Moving forward, it is envisaged that halogen bonding will play an increasingly important role in many dimensions of polymer chemistry and materials science, especially towards self-assembly and functional materials. For instance, its robust and tailorable optical properties are highly attractive to replace the ever-shrinking sizes of conventional semiconductor-based devices, which have been traditionally fabricated using top-down nanolithography methods. The immediate advantage of driving self-assembly using halogen bonding lies in its robust bonding strength as well as its directionality. Such advantages, in combination with the low-cost nature of the bottom up approach, is highly attractive and will receive increasing attention from both academia and industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Agency for Science, Technology and Research (A*STAR) under its AME Young Individual Research Grants (YIRG) (Grant No. A2084c0168).References
- J. Boucard, C. Linot, T. Blondy, S. Nedellec, P. Hulin, C. Blanquart, L. Lartigue and E. Ishow, Small, 2018, 14, 1802307 CrossRef PubMed.
- S. H. Kim, J. P. K. Tan, F. Nederberg, K. Fukushima, J. Colson, C. Yang, A. Nelson, Y. Y. Yang and J. L. Hedrick, Biomaterials, 2010, 31, 8063–8071 CAS.
- X. Yin, Z. Chen, Y. Chen, Y. Xie, B. Xiong, H. Jiang and J. Zhu, Colloids Surf., B, 2020, 195, 111271 CrossRef CAS PubMed.
- D. Devadiga and T. N. Ahipa, J. Mol. Liq., 2021, 333, 115961 CrossRef CAS.
- J. G. Kennemur, Macromolecules, 2019, 52, 1354–1370 CrossRef CAS.
- R. Tepper and U. S. Schubert, Angew. Chem., Int. Ed., 2018, 57, 6004–6016 CrossRef CAS PubMed.
- T. Lu and S. E. Wheeler, Org. Lett., 2014, 16, 3268–3271 CrossRef CAS PubMed.
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038–8155 CrossRef CAS PubMed.
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- B. Lindman, B. Medronho, L. Alves, M. Norgren and L. Nordenskiöld, Q. Rev. Biophys., 2021, 54, 3 CrossRef PubMed.
- Z. J. Kinney, A. L. Rheingold and J. D. Protasiewicz, RSC Adv., 2020, 10, 42164–42171 RSC.
- R. K. Rowe and P. S. Ho, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 255–264 CrossRef CAS PubMed.
- Z. Su, R. Zhang, X. Y. Yan, Q. Y. Guo, J. Huang, W. Shan, Y. Liu, T. Liu, M. Huang and S. Z. D. Cheng, Prog. Polym. Sci., 2020, 103, 101230 CrossRef CAS.
- D. H. Gharib, S. Gietman, F. Malherbe and S. E. Moulton, Carbon, 2017, 123, 695–707 CrossRef CAS.
- M. P. Hendricks, K. Sato, L. C. Palmer and S. I. Stupp, Acc. Chem. Res., 2017, 50, 2440–2448 CrossRef CAS PubMed.
- F. Weinhold and R. A. Klein, Chem. Educ. Res. Pract., 2014, 15, 276–285 RSC.
- S. K. Mann, T. N. Pham, L. L. McQueen, J. R. Lewandowski and S. P. Brown, Mol. Pharm., 2020, 17, 622–631 CrossRef CAS PubMed.
- M. Rigoulet, S. Massou, E. Daiann Sosa Carrizo, S. Mallet-Ladeira, A. Amgoune, K. Miqueu and D. Bourissou, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 46–51 CrossRef CAS PubMed.
- H. M. Coubrough, S. C. C. van der Lubbe, K. Hetherington, A. Minard, C. Pask, M. J. Howard, C. Fonseca Guerra and A. J. Wilson, Chem.–Eur. J., 2019, 25, 785–795 CrossRef CAS PubMed.
- P. K. Biswas, A. Goswami, S. Saha and M. Schmittel, Chem.–Eur. J., 2020, 26, 14095–14099 CrossRef CAS PubMed.
- H. Zhang, W. Y. Tung, X. Li, H. Jin, R. Deng, Y. M. Chen, Y. Mao and Y. Zhu, Polymer, 2020, 203, 122787 CrossRef CAS.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- J. Yang, H. Miao, Y. Wei, W. Li and Y. Zhu, Appl. Catal., B, 2019, 240, 225–233 CrossRef CAS.
- H. N. Tran, Y. F. Wang, S. J. You and H. P. Chao, Process Saf. Environ. Prot., 2017, 107, 168–180 CrossRef CAS.
- I. Akpinar, R. J. Drout, T. Islamoglu, S. Kato, J. Lyu and O. K. Farha, ACS Appl. Mater. Interfaces, 2019, 11, 6097–6103 CrossRef CAS PubMed.
- C. Wang, Z. Di, Z. Fan and L. Li, Chem. Res. Chin. Univ., 2020, 36, 268–273 CrossRef CAS.
- A. S. Knight, J. Larsson, J. M. Ren, R. Bou Zerdan, S. Seguin, R. Vrahas, J. Liu, G. Ren and C. J. Hawker, J. Am. Chem. Soc., 2018, 140, 1409–1414 CrossRef CAS PubMed.
- L. Zeng, M. Song, J. Gu, Z. Xu, B. Xue, Y. Li and Y. Cao, Biomimetics, 2019, 4, 36 CrossRef CAS PubMed.
- L. Bian, C. Shen, C. Song, S. Zhang, Z. Cui, F. Yan, B. He and J. Li, J. Membr. Sci., 2021, 620, 118948 CrossRef CAS.
- C. Z. Hadad, A. Restrepo, S. Jenkins, F. Ramírez and J. David, Theor. Chem. Acc., 2013, 132, 1–12 Search PubMed.
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Pérez-Juste, S. Bals, G. Van Tendeloo, S. H. Donaldson, B. F. Chmelka, J. N. Israelachvili and L. M. Liz-Marzán, ACS Nano, 2012, 6, 11059–11065 CrossRef PubMed.
- Q. Sun, X. W. Su and C. B. Cheng, Chem. Phys., 2019, 516, 199–205 CrossRef CAS.
- X. Cui, J. Liu, L. Xie, J. Huang and H. Zeng, J. Colloid Interface Sci., 2020, 578, 135–145 CrossRef CAS PubMed.
- Y. Tang, Y. Li, X. Lu, X. Hu, H. Zhao, W. Hu, F. Lu, Q. Fan and W. Huang, Adv. Funct. Mater., 2019, 29, 1807376 CrossRef.
- B. Zhou, L. Yan, L. Tao, N. Song, M. Wu, T. Wang and Q. Zhang, Adv. Sci., 2018, 5, 1700667 CrossRef PubMed.
- G. Y. Zhigulin, G. S. Zabrodina, M. A. Katkova and S. Y. Ketkov, Russ. Chem. Bull., 2019, 68, 743–750 CrossRef CAS.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- C. C. Robertson, J. S. Wright, E. J. Carrington, R. N. Perutz, C. A. Hunter and L. Brammer, Chem. Sci., 2017, 8, 5392–5398 RSC.
- G. Gong, S. Lv, J. Han, F. Xie, Q. Li, N. Xia, W. Zeng, Y. Chen, L. Wang, J. Wang and S. Chen, Angew. Chem., Int. Ed., 2021, 60, 14831–14835 CrossRef CAS PubMed.
- H. T. Le, C. G. Wang and A. Goto, Angew. Chem., Int. Ed., 2020, 59, 9360–9364 CrossRef CAS PubMed.
- J. G. Hill and A. C. Legon, Phys. Chem. Chem. Phys., 2015, 17, 858–867 RSC.
- T. L. Ellington, K. L. Shuford and D. P. Devore, J. Phys. Chem. A, 2020, 124, 10817–10825 CrossRef PubMed.
- L. C. Roper, C. Präsang, V. N. Kozhevnikov, A. C. Whitwood, P. B. Karadakov and D. W. Bruce, Cryst. Growth Des., 2010, 10, 3710–3720 CrossRef CAS.
- M. Saccone and L. Catalano, J. Phys. Chem. B, 2019, 123, 9281–9290 CrossRef CAS PubMed.
- G. Berger, P. Frangville and F. Meyer, Chem. Commun., 2020, 56, 4970–4981 RSC.
- R. Kampes, S. Zechel, M. D. Hager and U. S. Schubert, Chem. Sci., 2021, 12, 9275–9286 RSC.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, Acc. Chem. Res., 2013, 46, 2686–2695 CrossRef CAS PubMed.
- S. Biswas and A. Das, ChemNanoMat, 2021, 7, 748–772 CrossRef CAS.
- A. M. S. Riel, R. K. Rowe, E. N. Ho, A. C. C. Carlsson, A. K. Rappé, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870–2880 CrossRef CAS PubMed.
- H. L. Nguyen, P. N. Horton, M. B. Hursthouse, A. C. Legon and D. W. Bruce, J. Am. Chem. Soc., 2004, 126, 16–17 CrossRef CAS PubMed.
- L. J. McAllister, P. Carsten, J. P. W. Wong, R. J. Thatcher, A. C. Whitwood, B. Donnio, P. O’brien, P. B. Karadakov and D. W. Bruce, Chem. Commun., 2013, 49, 3946–3948 RSC.
- P. Metrangolo, T. Pilati, G. Resnati and A. Stevenazzi, Curr. Opin. Colloid Interface Sci., 2003, 8, 215–222 CrossRef CAS.
- J. Xu, X. Liu, T. Lin, J. Huang and C. He, Macromolecules, 2005, 38, 3554–3557 CrossRef CAS.
- J. Xu, X. Liu, J. K. P. Ng, T. Lin and C. He, J. Mater. Chem., 2006, 16, 3540–3545 RSC.
- D. W. Bruce, P. Metrangolo, F. Meyer, C. Präsang, G. Resnati, G. Terraneo and A. C. Whitwood, New J. Chem., 2008, 32, 477–482 RSC.
- P. Metrangolo, C. Präsang, G. Resnati, R. Liantonio, A. C. Whitwood and D. W. Bruce, Chem. Commun., 2006, 3290–3292 RSC.
- V. Kumar, D. J. Mulder, G. Cavallo, T. Pilati, G. Terraneo, G. Resnati, A. P. H. J. Schenning and P. Metrangolo, J. Fluorine Chem., 2017, 198, 54–60 CrossRef CAS.
- N. Houbenov, R. Milani, M. Poutanen, J. Haataja, V. Dichiarante, J. Sainio, J. Ruokolainen, G. Resnati, P. Metrangolo and O. Ikkala, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- F. Wang, N. Ma, Q. Chen, W. Wang and L. Wang, Langmuir, 2007, 23, 9540–9542 CrossRef CAS PubMed.
- Y. Chen, H. Yu, L. Zhang, H. Yang and Y. Lu, Chem. Commun., 2014, 50, 9647–9649 RSC.
- A. Priimagi, M. Saccone, G. Cavallo, A. Shishido, T. Pilati, P. Metrangolo and G. Resnati, Adv. Mater., 2012, 24, 345–352 Search PubMed.
- M. Alaasar, S. Poppe and C. Tschierske, J. Mol. Liq., 2019, 277, 233–240 CrossRef CAS.
- M. Saccone, F. F. Palacio, G. Cavallo, V. Dichiarante, M. Virkki, G. Terraneo, A. Priimagi and P. Metrangolo, Faraday Discuss., 2017, 203, 407–422 RSC.
- L. Barcelona-Cazanave, N. Trejo-Carbajal, R. J. Rodríguez-González, L. Larios-López, I. Felix-Serrano, J. M. Mata-Padilla and D. Navarro-Rodríguez, J. Fluorine Chem., 2021, 244, 109739 CrossRef CAS.
- C. V. Castro-Pérez, N. Trejo-Carbajal, R. J. Rodríguez-González, L. Larios-López, I. Felix-Serrano and D. Navarro-Rodríguez, J. Fluorine Chem., 2019, 222–223, 90–99 CrossRef.
- M. Saccone, M. Spengler, M. Pfletscher, K. Kuntze, M. Virkki, C. Wölper, R. Gehrke, G. Jansen, P. Metrangolo, A. Priimagi and M. Giese, Chem. Mater., 2019, 31, 462–470 CrossRef CAS.
- H. Wang, H. K. Bisoyi, L. Wang, A. M. Urbas, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2018, 57, 1627–1631 CrossRef CAS PubMed.
- H. Wang, H. K. Bisoyi, B. X. Li, M. E. McConney, T. J. Bunning and Q. Li, Angew. Chem., Int. Ed., 2020, 59, 2684–2687 CrossRef CAS PubMed.
- C. Präsang, A. C. Whitwood and D. W. Bruce, Chem. Commun., 2008, 2137–2139 RSC.
- A. Pizzi, L. Catalano, N. Demitri, V. Dichiarante, G. Terraneo and P. Metrangolo, Pept. Sci., 2020, 112, 24127 Search PubMed.
- G. Cavallo, A. Abate, M. Rosati, G. Paolo Venuti, T. Pilati, G. Terraneo, G. Resnati and P. Metrangolo, ChemPlusChem, 2021, 86, 469–474 CrossRef CAS PubMed.
- C. Präsang, H. L. Nguyen, P. N. Horton, A. C. Whitwood and D. W. Bruce, Chem. Commun., 2008, 6164–6166 RSC.
- E. Danelius, H. Andersson, P. Jarvoll, K. Lood, J. Gräfenstein and M. Erdélyi, Biochemistry, 2017, 56, 3265–3272 CrossRef CAS PubMed.
- J. Cao, X. Yan, W. He, X. Li, Z. Li, Y. Mo, M. Liu and Y. B. Jiang, J. Am. Chem. Soc., 2017, 139, 6605–6610 CrossRef CAS PubMed.
- X. Yan, K. Zou, J. Cao, X. Li, Z. Zhao, Z. Li, A. Wu, W. Liang, Y. Mo and Y. Jiang, Nat. Commun., 2019, 10, 3610 CrossRef PubMed.
- A. Jamadar, C. K. Karan, S. Biswas and A. Das, CrystEngComm, 2021, 23, 1695–1699 RSC.
- M. J. Langton, I. Marques, S. W. Robinson, V. Félix and P. D. Beer, Chem.–Eur. J., 2016, 22, 185–192 CrossRef CAS PubMed.
- J. Y. C. Lim, T. Bunchuay and P. D. Beer, Chem.–Eur. J., 2017, 23, 4700–4707 CrossRef CAS PubMed.
- T. A. Barendt, A. Docker, I. Marques, V. Félix and P. D. Beer, Angew. Chem., Int. Ed., 2016, 55, 11069–11076 CrossRef CAS PubMed.
- N. Lefèvre, C.-A. Fustin and J.-F. Gohy, Macromol. Rapid Commun., 2009, 30, 1865–1952 Search PubMed.
- D. Chen and M. Jiang, Acc. Chem. Res., 2005, 38, 494–502 CrossRef CAS PubMed.
- S.-W. Kuo, Polym. Int., 2009, 58, 455–464 CrossRef CAS.
- A. Jamadar and A. Das, Polym. Chem., 2020, 11, 385–392 RSC.
- R. Milani, N. Houbenov, F. Fernandez-Palacio, G. Cavallo, A. Luzio, J. Haataja, G. Giancane, M. Saccone, A. Priimagi, P. Metrangolo and O. Ikkala, Chem, 2017, 2, 417–426 CAS.
- H. T. Le and A. Goto, Cell Rep. Phys. Sci., 2021, 2, 100469 CrossRef.
- A. Vanderkooy and M. S. Taylor, J. Am. Chem. Soc., 2015, 137, 5080–5086 CrossRef CAS PubMed.
- A. Vanderkooy, P. Pfefferkorn and M. S. Taylor, Macromolecules, 2017, 50, 3807–3817 CrossRef CAS.
- S. Välimäki, L. Gustavsson, N. K. Beyeh, V. Linko and M. A. Kostiainen, Macromol. Rapid Commun., 2019, 40, 1–5 CrossRef PubMed.
- X. Zheng, M. Ren, H. H. Wang, H. H. Wang, Z. Geng, J. Xu, R. Deng, S. Chen, W. H. Binder and J. Zhu, Small, 2021, 17, 2007570 CrossRef CAS PubMed.
- J. D. Wuest, Nat. Chem., 2012, 4, 74–75 CrossRef CAS PubMed.
- V. I. Nikolayenko, D. C. Castell, D. P. van Heerden and L. J. Barbour, Angew. Chem., Int. Ed., 2018, 57, 12086–12091 CrossRef CAS PubMed.
- M. J. Zaworotko, Chem. Soc. Rev., 1994, 23, 283–288 RSC.
- M. Baldrighi, G. Cavallo, M. R. Chierotti, R. Gobetto, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Mol. Pharm., 2013, 10, 1760–1772 CrossRef CAS PubMed.
- J. C. Christopherson, F. Topić, C. J. Barrett and T. Friščić, Cryst. Growth Des., 2018, 18, 1245–1259 CrossRef CAS.
- O. Bolton, K. Lee, H. J. Kim, K. Y. Lin and J. Kim, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed.
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313–323 CrossRef CAS PubMed.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629–1658 CrossRef CAS PubMed.
- A. J. Blake, N. R. Champness, P. Hubberstey, W. S. Li, M. A. Withersby and M. Schröder, Coord. Chem. Rev., 1999, 183, 117–138 CrossRef CAS.
- D. Braga, F. Grepioni and G. R. Desiraju, Chem. Rev., 1998, 98, 1375–1405 CrossRef CAS PubMed.
- C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., Int. Ed., 2016, 55, 12398–12402 CrossRef CAS PubMed.
- M. F. Abdelbar, H. S. El-Sheshtawy, K. R. Shoueir, I. El-Mehasseb, E. Z. M. Ebeid and M. El-Kemary, RSC Adv., 2018, 8, 24617–24626 RSC.
- M. Saito, Y. Kobayashi, S. Tsuzuki and Y. Takemoto, Angew. Chem., Int. Ed., 2017, 56, 7653–7657 CrossRef CAS PubMed.
- Y. Ning, Q. Ji, P. Liao, E. A. Anderson and X. Bi, Angew. Chem., Int. Ed., 2017, 56, 13805–13808 CrossRef CAS PubMed.
- K. Takagi, K. Yamauchi and H. Murakata, Chem.–Eur. J., 2017, 23, 9495–9500 CrossRef CAS PubMed.
- A. Matsuzawa, S. Takeuchi and K. Sugita, Chem.–Asian J., 2016, 11, 2863–2866 CrossRef CAS PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- H. Hu, Y. Qiu, J. Wang, D. Zhao, H. Wang, Q. Wang, Y. Liao, H. Peng and X. Xie, Macromol. Rapid Commun., 2019, 40, 1800629 CrossRef PubMed.
- S. K. Maity, S. Bera, A. Paikar, A. Pramanik and D. Haldar, Chem. Commun., 2013, 49, 9051–9053 RSC.
- Z. Yang, C. Xu, W. Li, Z. Mao, X. Ge, Q. Huang, H. Deng, J. Zhao, F. L. Gu, Y. Zhang and Z. Chi, Angew. Chem., Int. Ed., 2020, 59, 17451–17455 CrossRef CAS PubMed.
- S. Cai, H. Shi, D. Tian, H. Ma, Z. Cheng, Q. Wu, M. Gu, L. Huang, Z. An, Q. Peng and W. Huang, Adv. Funct. Mater., 2018, 28, 1705045 CrossRef.
- J. Zhou, L. Stojanović, A. A. Berezin, T. Battisti, A. Gill, B. M. Kariuki, D. Bonifazi, R. Crespo-Otero, M. R. Wasielewski and Y. L. Wu, Chem. Sci., 2021, 12, 767–773 RSC.
- A. Jamadar, C. K. Karan, L. Roy and A. Das, Langmuir, 2020, 36, 3089–3095 CrossRef CAS PubMed.
- R. Tepper, S. Bode, R. Geitner, M. Jäger, H. Görls, J. Vitz, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 4047–4051 CrossRef CAS PubMed.
- S. Zhang, Y. Lu, Y. Zhang, C. Peng and H. Liu, J. Phys. Chem. C, 2017, 121, 4451–4461 CrossRef CAS.
- P. Pang, X. Miao, L. Ying, G. Kong, C. Che and W. Deng, J. Phys. Chem. C, 2020, 124, 5665–5671 CrossRef CAS.
- Y. Chen, S. Huang, T. Wang and H. Yu, Macromolecules, 2020, 53, 1486–1493 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |