
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal transport mechanism of the cation diffusion facilitator (CDF) protein family – a structural perspective on human CDF (ZnT)-related diseases†
Shiran
Barber-Zucker
 *a,
Arie
Moran
b and
Raz
Zarivach
*a
*a,
Arie
Moran
b and
Raz
Zarivach
*a
aDepartment of Life Sciences, the National Institute for Biotechnology in the Negev and Ilse Katz Institute for Nanoscale Science and Technology, Ben-Gurion University of the Negev, P.O.B. 653, Beer Sheva, 8410501, Israel. E-mail: barbers@post.bgu.ac.il; shiranbarber@gmail.com; zarivach@bgu.ac.il; Fax: +972-8-6472970; Fax: +972-8-6472970; Tel: +972-8-6428447 Tel: +972-8-6461999
bDepartment of Physiology and Cell Biology, Faculty of Health Sciences, Ben-Gurion University of the Negev, P.O.B. 653, Beer Sheva, 8410501, Israel
First published on 25th January 2021
Abstract
Divalent d-block metal cations (DDMCs) participate in many cellular functions; however, their accumulation in cells can be cytotoxic. The cation diffusion facilitator (CDF) family is a ubiquitous family of transmembrane DDMC exporters that ensures their homeostasis. Severe diseases, such as type II diabetes, Parkinson's and Alzheimer's disease, were linked to dysfunctional human CDF proteins, ZnT-1-10 (SLC30A1-10). Each member of the CDF family reduces the cytosolic concentration of a specific DDMC by transporting it from the cytoplasm to the extracellular environment or into intracellular compartments. This process is usually achieved by utilizing the proton motive force. In addition to their activity as DDMC transporters, CDFs also have other cellular functions such as the regulation of ion channels and enzymatic activity. The combination of structural and biophysical studies of different bacterial and eukaryotic CDF proteins led to significant progress in the understanding of the mutual interaction among CDFs and DDMCs, their involvement in ion binding and selectivity, conformational changes and the consequent transporting mechanisms. Here, we review these studies, provide our mechanistic interpretation of CDF proteins based on the current literature and relate the above to known human CDF-related diseases. Our analysis provides a common structure–function relationship to this important protein family and closes the gap between eukaryote and prokaryote CDFs.
 Shiran Barber-Zucker | Dr Shiran Barber-Zucker received her PhD in 2019 from the Department of Life Sciences at Ben-Gurion University of the Negev, Beer Sheva, Israel, where she was investigating the structure–function relationship of the cation diffusion facilitator protein family under the guidance of Prof. Raz Zarivach. Shiran is currently a postdoctoral fellow at the Weizmann Institute of Science, Rehovot, Israel, under the guidance of Prof. Sarel Fleishman. She is developing computational methods for the design of highly efficient and selective enzymes for environmental applications. |
 Arie Moran | Prof. Moran received his degrees from the physiology department at Hadassah Medical School in the Hebrew University, Jerusalem. His current work utilizes in vitro and in vivo systems to study the role of ZnT-1 in zinc and calcium homeostasis in cardiac cells. He served as the chairman of the Department of Physiology and Vice Dean for Research at the Faculty of Health Sciences and for seven year as the Deputy Vice-President and Dean for Research and Development at Ben-Gurion University of the Negev. Since 2010 he is also a Visiting Professor at the University of Sydney, Sydney, Australia. |
 Raz Zarivach | Prof. Zarivach received his PhD in 2005 from the Department of Structural Biology at the Weizmann Institute of Science, Rehovot, Israel, under the guidance of Prof. Ada Yonath. Subsequently he was a postdoctoral fellow at the University of British Columbia, Canada with Prof. Natalie Strynadka (2005-2008). Since 2008 he has been a principal investigator in the Department of Life Sciences, Ben-Gurion University of the Negev in the field of structural biology. His research is focused on the structural studies of biological macromolecules including biomineralizing proteins and metal transporters. |
Introduction
Divalent d-block metal cations (DDMCs), such as Mn2+, Zn2+ and Fe2+, are the key components for cell function. These cations play a role in many biological processes, for example, they serve as protein structural anchors and stabilize the active sites of enzymes. Only a few amino acids bind with DDMCs, which are usually ligated by a combination of cysteine, histidine, glutamate and aspartate. However, DDMCs are bound with various affinities, specific coordination geometry and selectivity to metalloproteins. This allows, in the context of their specific cellular environment, their folding, thermostability and kinetic property optimization.1–4 The affinities of DDMCs to different ligands follow the Irving–Williams series that ranks the relative stability of their complexes as follows: Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+, meaning that Zn2+ forms more stable complexes than Mn2+. This is usually correlated inversely with the cellular concentrations of the DDMCs in the cytosol, thus preventing the replacement of strong binding ions with weaker binding ones.4,5 Together with import proteins, the exporter cation diffusion facilitator (CDF) protein family is one of the major systems that ensures homeostasis of various DDMCs.CDF proteins can act as DDMC transporters and usually utilize proton motive force for either extruding DDMCs from the cytoplasm to the extracellular environment or to sequester the DDMCs into cellular components.6,7 CDFs are conserved throughout evolution (see Fig. S1, ESI†), with humans having ten CDF proteins, named ZnT-1-10 (for zinc transporters, named as such after the first member of the family which was defined as a zinc transporter). The members of the family selectively transport either Zn2+ or Mn2+.8–13 Human ZnTs are highly important for normal cell function, as mutations and amino acid variations within them, and irregularity in their expression or function, were correlated with severe disorders and diseases. Among these are Alzheimer's disease,14–16 transient neonatal zinc deficiency,17–20 type II diabetes,21 and Parkinsonism.22,23 Furthermore, recently it became clear that, in addition to their activity as transporters, mammalian CDFs also act as regulators of other cellular functions; for example, ZnT-1, in addition to its activity as a Zn2+/H+ exchanger, acts as a modulator of voltage gated calcium channels (VGCCs), inhibiting Ca2+ influx via L-type calcium channels (LTCCs), and acts as an activator of the Ras-Raf-ERK signaling pathway.12,24–27 Analyses performed on several plant and human CDF proteins showed structural similarities between bacterial, mammalian and plant CDF proteins;28–34 however, the information we have on eukaryotic CDFs is far from being as comprehensive as that on bacterial proteins,6 which limits our understanding of human disorders and diseases. Here, we review the present functional, biophysical and structural studies of CDF proteins and provide a mechanistic interpretation. Based on these, we provide a structural perspective on how specific mutations in ZnTs may cause their malfunction, leading to their corresponding diseases.
The general architecture of CDF proteins
The CDF structure–function knowledge is based on the bacterial CDF protein YiiP8 and the human CDF protein ZnT-8. Full Escherichia coli YiiP (EcYiiP), Shewanella oneidensis YiiP homolog (SoYiiP)35,36 and human ZnT-8 (isoform B) structures were determined34,37,38 in the presence of Zn2+ (Fig. 1).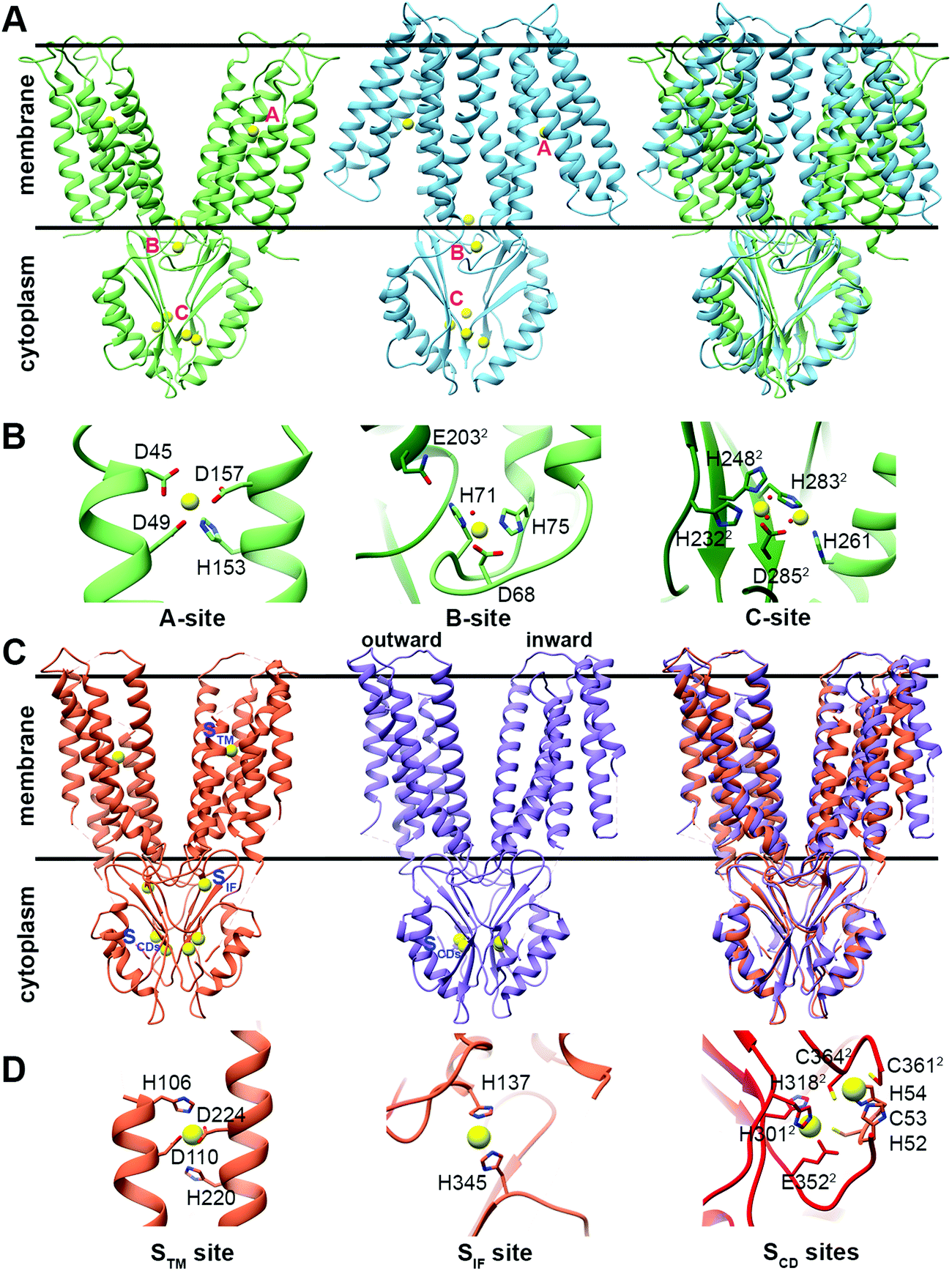 | ||
| Fig. 1 Structures and DDMC binding sites of full YiiP and human ZnT-8 (isoform B) proteins. (A) Left: crystal structure of full YiiP from Escherichia coli (PDB code: 3H90,38 light green). Middle: cryo-EM structure of the YiiP homolog from Shewanella oneidensis (PDB code: 5VRF,36 light blue); the Zn2+ ions are presented as yellow spheres, whereas the A-, B- and C-sites are marked in pink. Right: both YiiP structures overlapped onto each other, displaying the different conformations of the TMD and the suggested scissor-like motion. (B) Detailed high magnification of A–C Zn2+ binding sites of YiiP from Escherichia coli. Zn2+ ions are presented in yellow and the coordination first-shell residues are presented as sticks. The superscript 2 and the darker green color mark the second monomer. (C) Left: cryo-EM structure of WT human ZnT-8 in the presence of Zn2+ (PDB code: 6XPE,34 coral). Middle: low-resolution cryo-EM structure of WT human ZnT-8 in the absence of Zn2+ (PDB code: 6XPF,34 light purple), where each monomer adopts a different conformation; the Zn2+ ions are presented as yellow spheres, whereas the STM, SIF and SCDs sites are marked in blue. Right: both human ZnT structures overlapped onto each other, displaying the different conformation of one of the TMD monomers. (D) Detailed high magnification of STM, SIF and SCDs Zn2+ binding sites of WT human ZnT-8 in the Zn2+-bound state. Zn2+ ions are presented in yellow and the coordination first-shell residues are presented as sticks. The superscript 2 and the red color mark the second monomer. All structural figures were produced using the UCSF Chimera package, version 1.12.106 | ||
CDF proteins form dimers, made of two monomers including a six-helical transmembrane bundle N-terminal domain (NTD), through which the cations are transported, and a C-terminal domain (CTD). YiiP structures exhibit dimeric fold with similar CTD conformation but different transmembrane domain (TMD) arrangements (open and closed states, see Fig. 1A), and both have three metal binding sites (A–C sites)35–38 (Fig. 1B). ZnT-8 structures have high similarity to bacterial ones with similar CTD and inward-facing TMD conformations (Fig. 1C) and, similarly to YiiP, have three metal binding sites (Stm, Sif, and two Scd sites, Fig. 1D).34 The YiiP A-site/Stm in ZnT-8, the active transport site within the cation transport path, is found in both monomers and composed of two residues from transmembrane helix (TMH) 2 and two residues from TMH5 (the DD-HD quartet in YiiP and HD-HD in ZnT-8). This site is structurally conserved in all CDFs and contains four metal binding residues (see Fig. S1, ESI†). The symmetrical, not conserved, B-site in YiiP is located on an intracellular loop between TMH2 and TMH3 and is predicted to be involved in CDF dimerization.39 The symmetrical Sif site in ZnT-8 is found in the interface between the CTD and TMD, in the cytosolic entrance to the Zn2+ transport pathway, and has been suggested to have a role in increasing the Zn2+ local concertation to facilitate its transport.34 The two symmetrical C-sites in YiiP are located at the CTD and contain four structural Zn2+ cations that are bound by a network of residues from both monomers. The site occupancy is related to the CTD conformation and dimerization.36,38,40,41 Although the CTD C-site is not conserved in all characterized CTD-containing CDF proteins, a metal binding site exists in all characterized CTD-containing CDFs. The two symmetrical equivalent bacterial CTD C-sites in ZnT-8 (Scd sites) are less defined and their cation-bound geometries are far from optimal due to the low resolution cryo-EM map. These sites contain four Zn2+ cations as well, which are bound by a network of residues from both monomers.34 Only one of the YiiP symmetrical C-sites’ Zn2+ ions overlaps onto that of ZnT-8 (Scd2 site).
The mechanism of the cytoplasmic domain
The CTD can be considered an activator/regulator of the transport. This is supported by the decreased or abolished activity of CDFs by CTD deletion or mutations.38,40,42–48 In humans, it may also lead to the development of diseases.23,49 In the bacterial CDFs ZitB and YiiP, the removal of Zn2+ from the CTD binding sites led to protein precipitation,35,50 suggesting that Zn2+ binding has a role in protein folding or protein structural stabilization. However, the importance of this domain in the context of overall folding and stabilization is not clear yet as its misfolding in another bacterial protein, MamM, does not completely abolish the transport.51In all YiiP and ZnT-8 structures the CTD adopts the same closed conformation due to the presence of zinc (Fig. 1A and C). Hence, to examine the impact of the binding of DDMCs and to follow the conformational space, crystal structures and biophysical studies of CTDs of other CDFs are needed. Currently, it was achieved mainly by utilizing bacterial proteins.40,41,52,53 The CTDs of CDFs form a V-shaped dimer, with each monomer adopting a copper metallochaperone-like (Hah1) fold.36,38,40,41,43,54,55 When comparing the CTD structures of different CDFs,36,38,40,41,43,53,54 it appears that it can have different degrees of openness in the apo and bound states (Fig. 2A and B, respectively). This implies that the V-shaped arms are dynamic.52 Still, the crystal structures of the bacterial CDF proteins CzrB, CzcD and MamM show that the monomers are approaching each other to form a tighter DDMC-bound conformation as compared to the apo conformation41,53,56 (Fig. 2C and D). This was further confirmed by the biophysical studies of full YiiP38 and CTDs in solution, which revealed a final rigid bound state.40,41,52,53 Hence, it is conceivable that the CTD-bound DDMCs, rather than sequentially transported, remain bound and play a role in closed conformation stabilization (as will be discussed below).
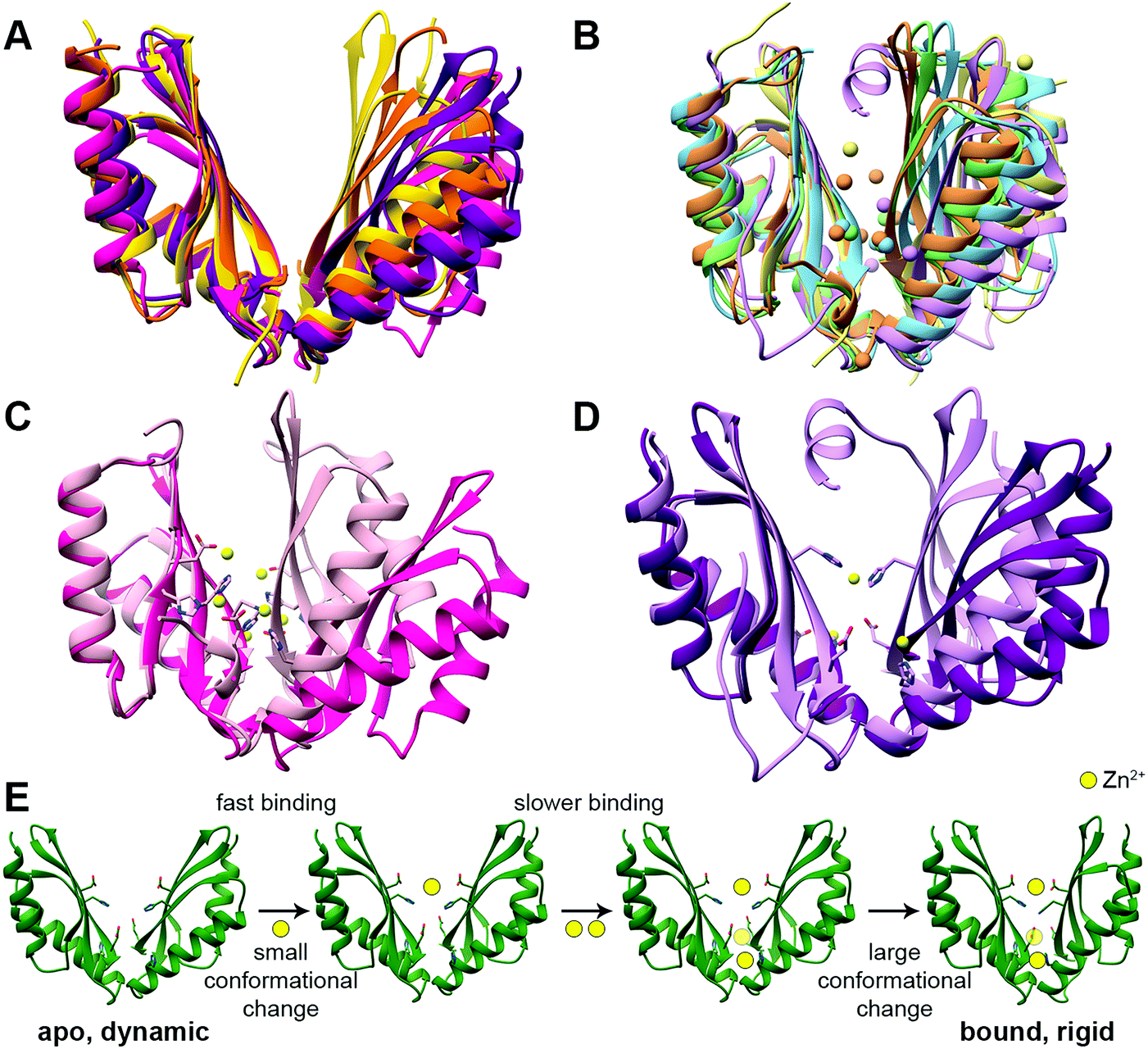 | ||
| Fig. 2 The structures and suggested Zn2+ binding mechanism of the CTDs of CDF proteins. (A) Apo CTD structures of CzrB (PDB code: 3BYP,41 pink), MamM (PDB code: 3W5X,40 purple), MamB (PDB code: 5HO5,43 yellow) and TM 0876 from Thermotoga maritima (PDB code: 2ZZT,54 orange) overlapped onto each other. (B) Bound CTD structures of CzrB with Zn2+ (PDB code: 3BYR,41 light pink), MamM with Cu2+ (PDB code: 6GP6,53 light purple), MamB with Zn2+ (PDB code: 5HO1,43 light yellow), EcYiiP with Zn2+ (PDB code: 3H90,38 light green) and SoYiiP with Zn2+ (PDB code: 5VRF,36 light blue) overlapped. In each structure, the DDMCs are presented as enlarged spheres in the proteins’ colors. (C) The apo (dark pink) and Zn2+-bound (light pink) structures of CzrB CTD and (D) the apo (dark purple) and Cu2+-bound (light purple) structures of MamM CTD show the tighter conformation of the CTD that is achieved due to DDMC binding. The DDMCs are presented in yellow color and binding site residues are presented as sticks. (E) Zn2+ binding mechanism of MamM CTD (the model is adopted from Barber-Zucker et al.;52 Zn2+ ions are presented as yellow spheres). Rapid Zn2+ binding (<1 microsecond) to the dynamic apo form's central site results in a minor conformational change but has an allosteric positive effect on the binding to the peripheral sites. Only slower (millisecond time scale) binding of two ions to the symmetrical sites causes a closure of the CTD to a tighter, rigid V-shape. | ||
The C-site is not conserved among CDF proteins as was already described above for ZnT-8 (see Fig. S1, ESI† for sequence data). For example, both Zn2+-transporting CDF proteins CzrB and YiiP have partially different CTD binding sites (Fig. 2B and C),36,38,41 and for CzcD a single Ni2+ site was found in Cupriavidus metallidurans, two sites in Pseudomonas aeruginosa and a single Zn2+ site in Thermotoga maritima.56 MamM CTD possesses a central binding site and two symmetrical peripheral metal binding sites which are homologous to one of the YiiP and CzrB symmetrical Zn2+ binding sites (Fig. 2B and D).40,52,53 In contrast, the bacterial CDF MamB has only a central binding site.43 Interestingly, while MamM and CzrB exhibit different conformations in the apo and bound structures, MamB-bound and apo structures have very similar conformation.40,41,43,53 This might be due to the periphery binding site that leads to dimer closure (the site is conserved in MamM, YiiP and CzrB but is not found in MamB). This is supported by MamM CTD kinetic and mechanistic studies (Fig. 2E): rapid Zn2+ binding to the central site resulted in a minor conformational change but has an allosteric positive effect on the binding to the peripheral sites. Only binding of two ions to the symmetrical sites causes closure of the CTD to a tighter, rigid V-shape.52 However, the structural study of MamM CTD in the presence of different DDMCs showed that the binding of a given DDMC to this domain has unique affinity and conformation53 (which indicate a role in metal selectivity, as will be discussed below). Hence, the observed varied CTD conformations might be relevant only to the specific DDMC under study. The suggested mechanism of CDF activation via CTD closure following the appropriate DDMC binding is well supported by the binding affinities. Measurements of metal binding to purified CTDs showed similar dissociation constants (ranging from ∼2 to 30 μM52,56), which indicate a greater affinity to the TMD site (Kd ∼ 0.1–1 μM57). These affinities suggest a CTD closed state and TMD transition activation only after the cytosolic metal concentrations reach relatively high values. Such transition was monitored via the molecular dynamics simulation of YiiP.58 Once the TMD is open, its metal transport site achieves a much higher affinity, thus enabling metal binding and transport through the TMD. When the cytosol metal concentration drops below the CTD affinity, the CTD will convert to the open state, leading to metal transport termination. In contrast to these conformational changes, ZnT-8 (and due to conservation, putatively also ZnT-2 and ZnT-3, see Fig. S1, ESI†) is constantly stabilized at a closed CTD conformation as a result of an additional HCH (His52-Cys53-His54) motif at the N-terminus of the neighboring ZnT-8 subunit, which seals the Zn2+ binding sites and prevents cation dissociation.34 This indicates that ZnT-8, a known secretory-organelle CDF, together with ZnT-2 and ZnT-3, resides within the cell8 and most likely competes with ZnT-1 (a cytoplasmic-membrane CDF that extrudes Zn2+) on the same cation and needs to be in a constant active state.
The conformational space of the full CDF proteins
The YiiP cryo-EM and X-ray structures exhibit two different TMD conformations: the inward facing conformation provides A-site access from the cytoplasm, while the outward facing conformation provides A-site access from the periplasm.35–38 Surprisingly, the structure of WT ZnT-8 in the absence of Zn2+ yielded a heterogeneous conformation, with one subunit in an inward-facing and the other in an outward-facing conformation.34 The YiiP structures suggest a TMD scissor-like motion between the two states (Fig. 1A). The conformations of the monomers differ in the TMH1,2,4,5 bundle that surrounds the A-site, which rocks against TMH3 and 6. Additionally, TMH2 and 5 form longer helices and bend near the A-site in the inward facing conformation (Fig. 1A and 3).36 An EcYiiP X-ray mediated hydroxyl radical labeling and its MD simulation revealed that Zn2+ access to the A-site in EcYiiP is controlled by the leucine residue (L152) found in the TMH5 cytosolic side and serves as a hydrophobic gate. This means that the movement of this helix is crucial for both proper zinc chelation and A-site access.58,59 Nevertheless, the A-site in both conformations is similarly populated in terms of the coordination geometry.36 Investigating the TMD dimerization, the EcYiiP structure shows almost no contact and a gap between the TMDs that becomes wider in the periplasmic side, while the TMDs of SoYiiP are much closer (Fig. 1A). The crosslinking between SoYiiP TMH3 residues did not lead to a decrease in transport activity. This suggests a tighter packing of the TMD with intact TMHs 3 in all states, in contrast to EcYiiP with an outward facing conformation, and that conformational changes within each monomer are functionally sufficient. There are two plausible explanations to the observable TMD conformation of EcYiiP: the use of detergent micelles, which behave differently than the lipid bilayer used in the SoYiiP cryo-EM studies (comparison of the TMD packing of the crystal and the cryo-EM structures demonstrates some destabilization effects of the detergent environment that can facilitate the separation of TMDs, maybe by recruiting detergents to stabilize each monomer individually), or separation due to crystal packing (the asymmetric unit in the crystals contains two sets of dimers; in each intramolecular contacts through the outer surface of the TMD are observed, which seem to hold the TMDs apart and influence the rigidity of the TMD helices in the contact surface).36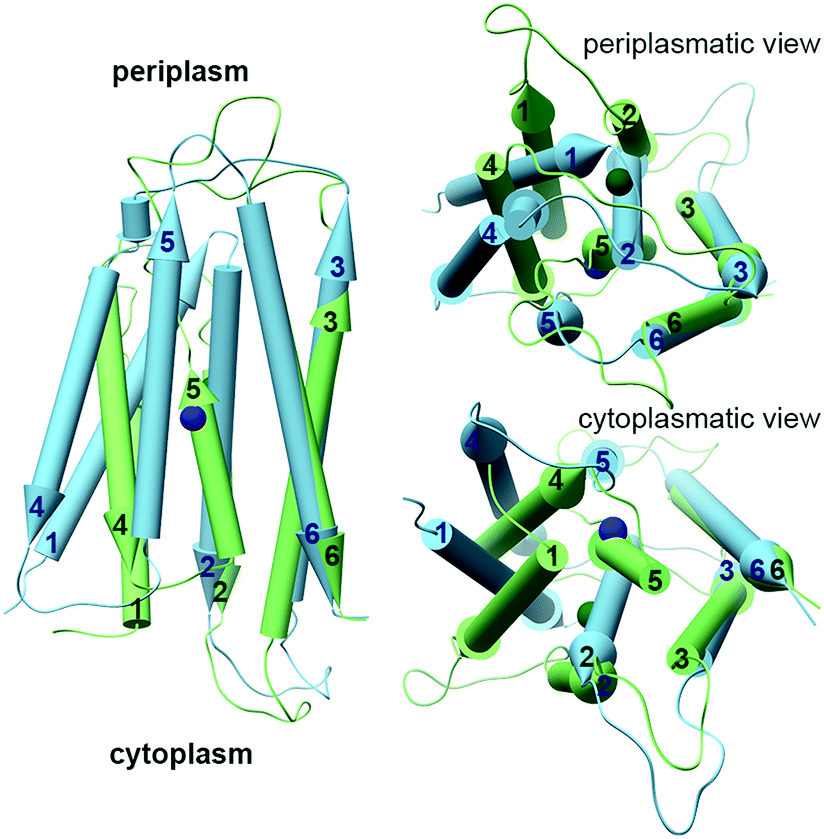 | ||
| Fig. 3 The different conformations of monomeric TMDs of YiiPs. YiiP TMD monomers (EcYiiP, PDB code: 3H90,38 in light green, and SoYiiP, PDB code: 5VRF,36 in light blue) overlap onto each other, showing the movement of TMH1,2,4,5 with respect to TMH3,6 in the transition between the inward facing (SoYiiP) and outward facing (EcYiiP) conformations. The Zn2+ ions are presented in the protein's darker colors. | ||
The communication between the domains is also crucial for the overall protein mechanistic understanding. The TMD–CTD connecting loop in YiiP is salt-bridged via aspartate to the TMH2-3 loop. It was previously suggested that this charge interlock is the linking hinge between the CTD conformational change and the TMD.36,38,39,44 However, the aspartate is not conserved among all CDFs and the SoYiiP study suggests a TMH1,2,4,5 bundle movement relative to the static TMH3 and 6 and not a scissor-like motion.36 Hence, it is still not clear what is the exact role of the charge interlock and whether a hinge-like motion can facilitate the TMD conformational change.
Taken together, these structural data are consistent with the predicted mechanism that CTD metal binding facilitates the TMD conformational change and allows the DDMC transport36,39,40 (Fig. 4). At rest, the TMD is found in the inward facing conformation and the CTD in an apo, open V-shaped form. When the cytoplasmic metal concentration is sufficiently high, the DDMCs are bound to the CTD, leading to its closure to a tighter V-shaped conformation. The rigid closure of the CTD is assumed to facilitate, maybe through a charge interlock, the TMH1,2,4,5 bundle movement relative to TMH3 and 6. This allows the A-site to face the periplasm/inner-cellular component and the DDMC to be extruded.36,39,40,58 During the active transport, the tight CTD packing remains static, while the rocking movement of the TMD bundles facilitates the antiporter activity.
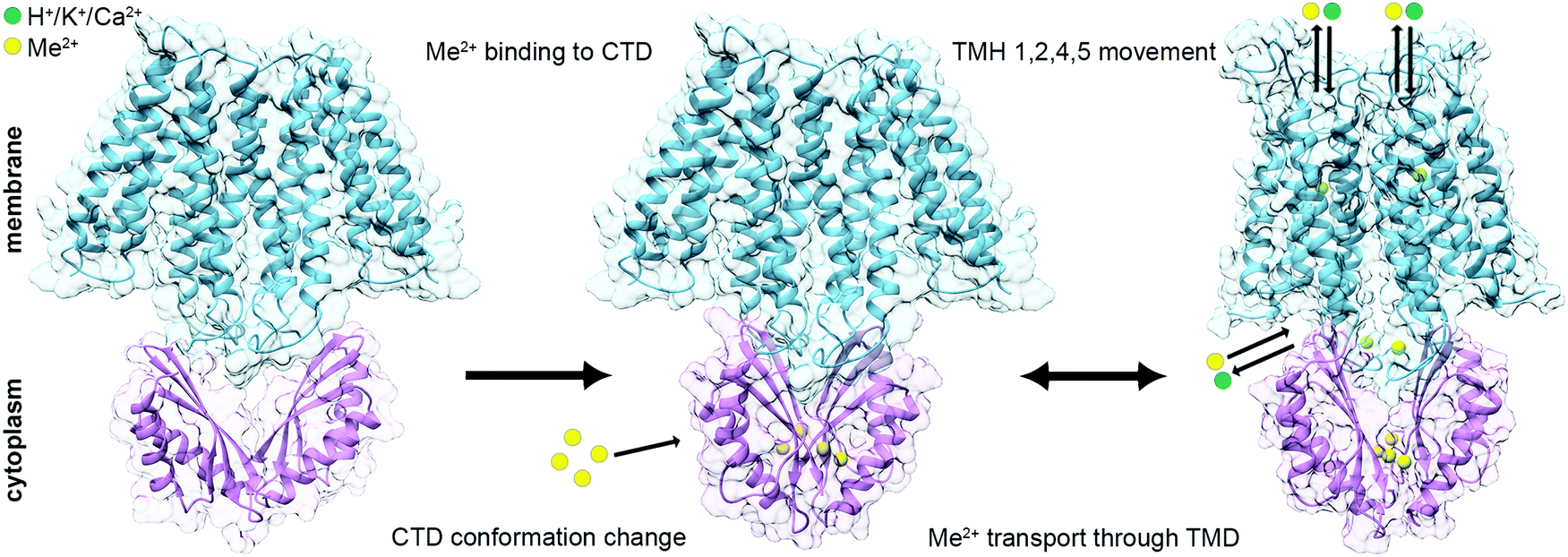 | ||
| Fig. 4 Suggested mechanism for the conformational changes associated with the binding of DDMCs to CDF proteins. Left: At rest, the TMD (SoYiiP ribbon, PDB code: 5VRF36) is found in the inward facing conformation and the CTD (CzrB apo structure ribbon, PDB code: 3BYP41) in an apo, open V-shaped form. Middle: when the metal concentration in the cytoplasm is sufficiently high, the DDMCs are bound to the CTD, which leads to its closure to a tighter V-shaped conformation (SoYiiP ribbon, PDB code: 5VRF36). Right: The closure of the CTD is assumed to facilitate the TMH1,2,4,5 bundle movement relative to TMH3 and 6, leading to the outward facing conformation, which allows the DDMC to be extruded and the counter-ion to be bound onto the A-site (the model of EcYiiP outward facing conformation adopted from Lopez-Redondo et al.36). TMD is represented in blue, CTD in purple, DDMCs in enlarged yellow spheres and counter-ions in green. | ||
Metal selectivity by CDF proteins
While the structural studies of the bacterial CDF proteins led to a better understanding of the function of CDFs, the mechanism of their metal selectivity remained unclear. For example, EcYiiP shows both Zn2+ and Cd2+ efflux abilities (Zn2+: K0.5 ∼ 554 μM, Vmax ∼ 20 s−1; Cd2+: K0.5 ∼ 0.27 μM, Vmax ∼14.4 s−1).38,57 Many studies on CDFs from different phyla show the binding and transport of a specific DDMC or DDMCs.7,39 For example, the CDF phylogenetic classification has divided them into 17 clades that play a role in the transport of specific DDMCs.7Most CDF metal selectivity studies concentrate on the structurally conserved A-site composition (Fig. 1B, Fig. 5 and Fig. S1, ESI†). It is composed of four metal-binding residues (quartet), usually a combination of histidine and aspartate residues.39,60,61 Numerous studies examined the relationship between the A-site composition and metal selectivity. They showed that deletion (alanine) mutations in this site decrease the transport activity and that changes in the quartet composition (so it is composed of non-native metal binding residues) abolish or enhance specific metal transport.10,12,13,39,43–46,61,62 For example, ZnT-10, with the ND-HD quartet, is selective for Mn2+, while changing the HD-HD quartet in ZnT-1 to ND-HD also changed its selectivity from Zn2+ to Mn2+.10,63 An analysis of DDMC binding sites was used to examine whether the A-site can solely dictate metal selectivity and whether there is a correlation between the CDF phylogenetic classification7 and their metal selectivity.60,61 This study showed that the A-site composition does not solely determine the metal selectivity.60
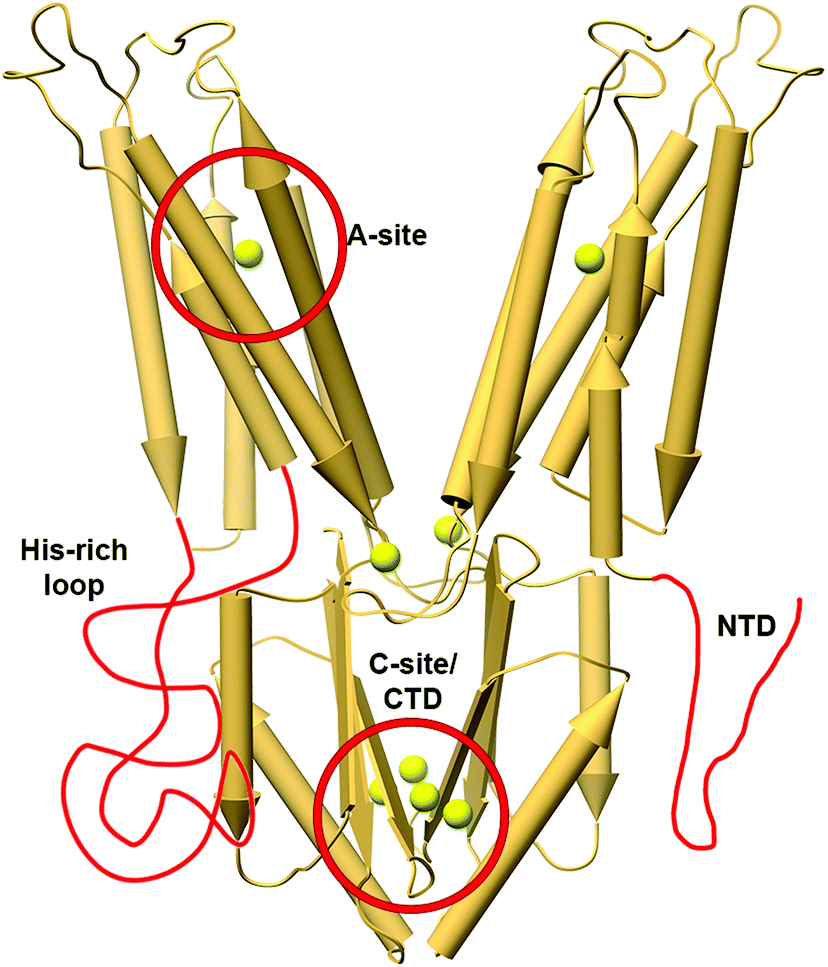 | ||
| Fig. 5 The X-ray structure of YiiP (PDB code: 3H9038) with features related to metal selectivity highlighted in red. The TMH4-5 loop in YiiP is shorter than regular His-rich loops and it has a short NTD; hence, these features are drawn elongated for clarity. The locations of Zn2+ ions in the structure are presented as enlarged yellow spheres. | ||
Other CDF features such as extra loops and special sequence composition were also studied (Fig. 5), but these features were specific to a given protein or were studied on a rather small number of proteins, thus not allowing generalized conclusions. For example, a cytoplasmatic Histidine-rich loop (HRL) between TMH4 and 5 in Arabidopsis thaliana MTP1 protein is related to Zn2+-binding and specificity.32,64–66 Similarly, the HRLs of some human ZnTs have also been suggested to play a role in specific metal chelation.13,67 In a class of CTD-lacking CDFs, it is suggested that the cytoplasmatic N-terminal tail, a domain rich in cysteine, aspartate, glutamate and histidine residues, can chelate metals and hence may play a role in metal selectivity.39 Consistent with this notion, an MTP1 extended N-terminal deletion has a different Zn2+ transport capability compared to Cd2+ or Co2+, supporting its role in metal chelation and selectivity.66 Its ability to sense the cytoplasmic environment supports the notion that the CTD plays a role in metal selectivity. Indeed, recent MamM CTD structural studies show that each DDMC has different binding affinity and conformation.53 Furthermore, mutations to alternative DDMC-binding residues in MamM CTD changed its ability to bind with specific DDMCs, and some mutant–DDMC pairs exhibit different conformations than the wildtype-DDMC.68 Altogether, these studies demonstrate the C-site role in metal selectivity.
CDF proteins are antiporters with diverse counter-ions
For many CDF proteins from bacteria,35,47,50,69–72 yeasts,73 plants65 and humans,12,33,74,75 the DDMC transport depends on the pH gradient. X-ray-mediated hydroxyl radical labeling of EcYiiP demonstrated that the pH is responsible for protonation of one of the A-site histidine residues (H153) at the outward facing conformation, suggesting that the pH gradient drives a unidirectional efflux of zinc in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with protons.59 However, A-site histidine residues are not found in all CDFs;60 thus, this zinc/proton exchange mechanism cannot be applied to all. For example, Bacillus subtilis CzcD can catalyze Zn2+ efflux in exchange for proton and potassium ions.70 Furthermore, ZnT-10 was characterized as a Mn2+/Ca2+ exchanger (K0.5 ∼ 973 μM, Vmax ∼ 1.234 s−1).10 The A-site quartet of ZnT-10 is ND-HD; it seems as if this composition, and specifically the substitution of histidine from the HD-HD quartet in other ZnTs to asparagine, was evolutionarily tailored for the specific binding of both Mn2+ and Ca2+ while excluding Zn2+.10 These results suggest that the A-site plays a role in DDMC selectivity and in counter-ion selection.
:1 ratio with protons.59 However, A-site histidine residues are not found in all CDFs;60 thus, this zinc/proton exchange mechanism cannot be applied to all. For example, Bacillus subtilis CzcD can catalyze Zn2+ efflux in exchange for proton and potassium ions.70 Furthermore, ZnT-10 was characterized as a Mn2+/Ca2+ exchanger (K0.5 ∼ 973 μM, Vmax ∼ 1.234 s−1).10 The A-site quartet of ZnT-10 is ND-HD; it seems as if this composition, and specifically the substitution of histidine from the HD-HD quartet in other ZnTs to asparagine, was evolutionarily tailored for the specific binding of both Mn2+ and Ca2+ while excluding Zn2+.10 These results suggest that the A-site plays a role in DDMC selectivity and in counter-ion selection.
Structural perspective on ZnT-related diseases
The limited knowledge of the structures of eukaryotic CDFs depends on ZnT-8 structures, and biochemical studies of plant MTPs and human ZnTs all indicate that the eukaryotic CDFs share the basic components of prokaryotic CDFs. Both the TMD and CTD have similar structures and locations of metal binding sites, although they vary in their sequences.10,33,34,66,74 However, eukaryote CDFs include additional cytoplasmic domains and loops that seem to add regulation (Fig. 5). The HRL (not resolved in the ZnT-8 cryo-EM map) contributes to the CDF activity as demonstrated by reduced activity or inactivity following histidine-to-alanine mutations or deletion of the extended loop.76 Surprisingly, the replacement of ZnT-1 HRL with ZnT-10 HRL led to the restoration of zinc resistance although the latter does not contain any histidines.63 In the plant Co2+/Zn2+-CDF MTP1 (K0.5 ∼ 0.3 μM, Vmax ∼ 1.2 s−1), the HRL (80 amino acids) binds both Co2+ and Zn2+ and mutations change its cation specificity,66 while its removal leads to a 2–11 fold rate increase.65 The NTD is also important for the activity as its deletion reduces or abolishes the transport.34 In ZnT-8, the NTD contains an HCH motif that participates in metal binding and keeps the CTD in a closed conformation. Deletion of the HCH abolished the transport, most likely due to the loss of regulation by the CTD.34 The data obtained on ZnT-4 suggested that the NTD participates in protein–protein interactions.77 Moreover, domain swap of the NTD and the HRL between ZnT-2 and ZnT-3 showed that mixed NTD and HRL inactivate both CDFs, while replacing both domains led to fully functional CDFs.76Surprisingly, not many ZnTs’ disease-causing mutations are described in the literature although ZnT proteins are highly important for proper cell function. This may be due to the fact that ZnT mutations are lethal. A better understanding of the disease mechanism requires structural knowledge of wildtype and mutants which is not yet available for most ZnT-related diseases. Here we summarize those that were studied in the context of ZnT-related diseases in terms of how these mutations may affect the structure and function of ZnT proteins.
ZnT-2
ZnT-2 is found in the mammary glands, and its mutations in mothers lead to poor secretion of zinc into breastmilk, leading to zinc deficiency in breastfed babies. The mutations H54R,19 G87R,78 S296L, W152R,49 G280R, T312M and E355Q79 in ZnT-2 (Fig. 6), as well as H106Y, R165W, G175W, N214K, G233D/R, P245R, E279K, G299W and V300L,20 were associated with transient neonatal zinc deficiency and we will discuss some of them below. In newborns, the observed Zn2+ deficiency may lead to diarrhea, loss of appetite and the development of dermatitis.17 The ZnT-2 model based on the YiiP X-ray structure enabled a better understanding of the mechanism of its mutations.17,19,78 H54R is assumed to be located at the unstructured NTD of ZnT-2.19 Since this tail was suggested to play a role in metal chelation and selectivity, and as histidine frequently chelates DDMCs, it might be that this mutation decreases Zn2+ recruitment and consequently leads to failure to increase the local zinc concentration needed to activate ZnT-2. G87R is found in TMH1 facing the membrane and it was suggested that this dramatic substitution, from a small glycine to the bulky charged arginine in the membrane environment, will cause improper folding and might lead to the observed low expression level and ZnT-2 subcellular localization alteration.78 In contrast, S296L is localized to the CTD surface and thus does not cause any significant structural change. Therefore, it enables moderate Zn2+ transport and is probably responsible for the observed fast protein degradation. As S296 is predicted to undergo phosphorylation, it is suggested that phosphorylation impairment is the underlying mechanism for the mutant functional impairment. G280R is located on the TMD–CTD connecting loop and impairs vesicular Zn2+ transport. The substitution from the small glycine to the bulky charged arginine was suggested to interfere with the Zn2+ permeation pathway.79 However, this steric interference can also interrupt the cross-talk between the domains and hence the CTD regulation, leading to a decrease in transport activity. E355Q, part of the CTD Zn2+ binding site (C-site), abolishes the transport activity and disrupts the protein cellular localization.79 As glutamate is much more abundant in Zn2+ binding sites compared to glutamine,60 this mutation can reduce Zn2+ binding to the CTD and its related conformational change, which will not allow TMD transport regulation, thus, will cause a loss of function and observed phenotypes. Recently, a new mutation set was found in ZnT-2: H106Y, R165W, G175W, N214K, G233D/R, P245R, E279K, G299W and V300L.20 The structural model of ZnT-2 indicates that many of these residues might cause structural changes. These include steric collisions (E279K, G299W and V300L), charge repulsion (N214K and E279K), changes in loop flexibility (G175W), hydrophobicity alterations (G233D/R), breaking of ion chelation (H106Y), and breaking of the possible salt bridge (R165W). Not surprisingly, many of these positions are highly conserved, most likely due to their structural importance.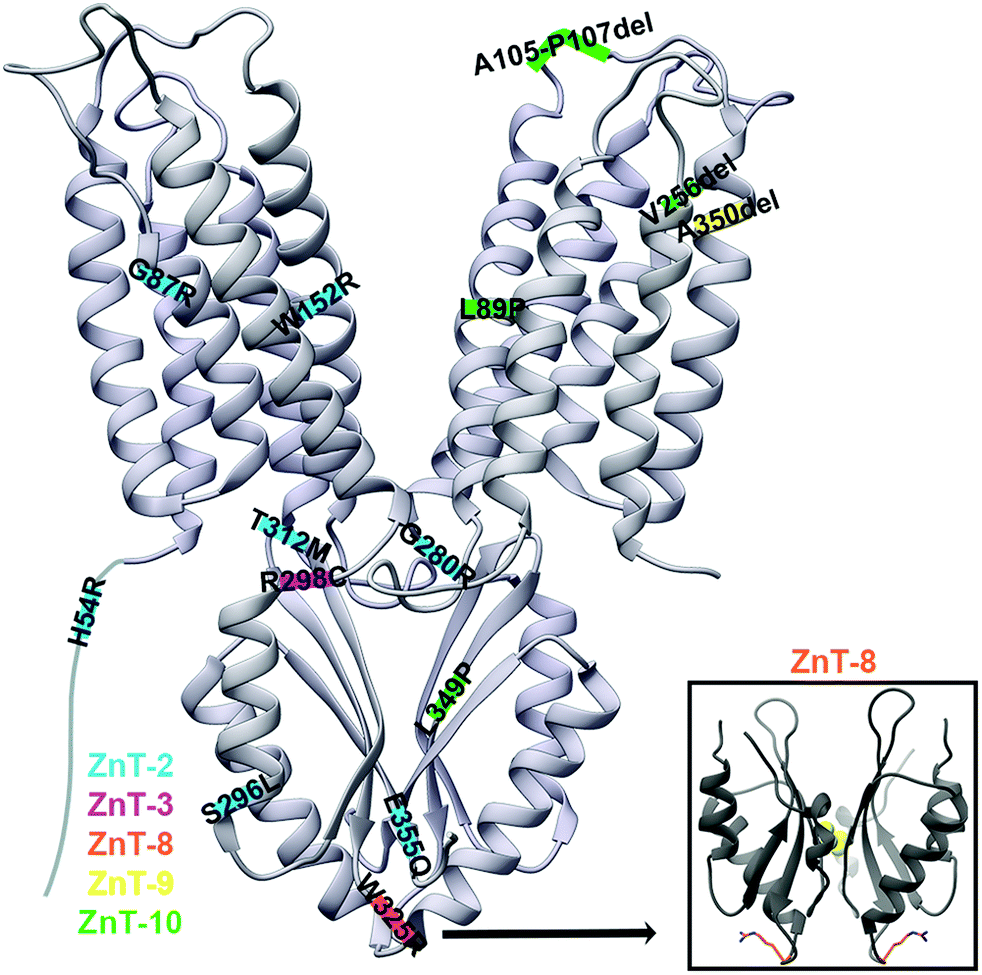 | ||
| Fig. 6 The X-ray structure of YiiP (PDB code: 3H9038) with ZnT disease-causing mutations marked on their predicted approximate locations. Mutations in ZnT-2 are marked in blue, in ZnT-3 in pink, in ZnT-8 in orange, in ZnT-9 in yellow and in ZnT-10 in green. YiiP has a short NTD and hence it was drawn elongated for the inclusion of the ZnT-2 H54R mutation. Lower right panel: the CTD of the human ZnT-8 structure (PDB code: 6XPD34) with R325 residues presented as orange sticks. | ||
ZnT-3
ZnT3 is responsible for Zn2+ transport into synaptic vesicles, where it is co-localized with glutamate and released in an activity-dependent manner. Synaptic zinc levels are associated with neuronal excitability and seizure susceptibility, as extracellular zinc interacts with many ion channels, receptors and transporters.80,81 The R298C mutation in ZnT-3 is related with febrile seizures, with the mutant protein being dysfunctional due to subcellular mislocalization.81 It was previously shown that the correct ZnT-3 subcellular localization relates to its homodimerization and that the CTD is important for its dimerization.82 As this mutation is predicted to be in the CTD, it was suggested that it destabilizes ZnT-3 homodimerization.81 The sequence alignment of ZnT-3 and other modeled ZnTs suggests that R298 is either in the C-terminal of the TMD–CTD connecting loop or in the N-terminal of the first CTD helix. However, this region does not relate to the CTD dimerization (Fig. 6). The native arginine can form multiple bond types with nearby residues which are limited by the smaller cysteine. Therefore, based on the substitution nature and the predicted location, we suggest that the mutation disrupts the cross-talk between the domains. However, we cannot explain why it impairs protein localization and more molecular and structural studies are needed to elucidate the mechanism of this mutation.ZnT-8
ZnT-8 is highly expressed in pancreatic beta cells, where it is responsible for transporting zinc into insulin granules and by that enabling crystalline insulin packing within them (due to the formation of stable complexes of insulin hexamers with zinc).83 The most structurally characterized ZnT disease-related SNP is the W325R variant in ZnT-8 which is associated with an increased risk of developing type-2 diabetes. This variant is located at the tip of the V-shaped CTD dimer (near the dimer interface, Fig. 6).28,29,42 Kinetic studies of purified ZnT-8 in proteoliposomes showed that the R325 variant exhibits accelerated zinc transport activity as compared to the W325 variant.28 As ZnT-8 loss-of-function mutations (protein truncating variants and frameshift variants that are predicted to improperly fold into the membrane) were shown to better protect against type-2 diabetes, this study suggests that the R325 variant is hyperactive.28,84 The W325R substitution had no effect on the expression, stability and subcellular localization of ZnT-8 in HEK293 cells. Since secretory granules store very high levels of zinc compared to the cytoplasmic free-zinc concentration, it might be that this enhanced difference in the R325 variant reverses the ZnT-8-mediated transport to extrude zinc from the granules. However, more studies are required to fully understand how this substitution impacts the ZnT-8 function, zinc homeostasis in the cellular level and the increased risk for type-2 diabetes.28 In terms of structural differences between the variants, ZnT-8 CTD biophysical studies showed that both variants have similar secondary structures, but that the W325 variant is less thermostable. Yet, its monomers are associated into a dimer with a higher affinity. It further showed that both variants can similarly bind Zn2+.29,33 The structure of ZnT-8 was determined only for the R325 variant and, as such, the exact effect of the W325 variant is yet to be determined, especially as it is located on the protein surface.34ZnT-9
ZnT-9 is moderately expressed in most tissues, while in the cerebellum, fetal brain, kidneys and skeletal muscle it is highly transcribed. The ZnT-9 A350del mutation, which is located at TMH4 (Fig. 6) and decreases ZnT-9-dependent Zn2+ transport, is related to a cerebro-renal syndrome.11 The TMD helical residue deletion is assumed to cause disorientation of the helix terminal. It either forms a shorter helix that will increase the TMH4 tension and deform its structure, or force a loop residue to be inserted, forming a longer helix that still fits into the membrane. In any case, the mutation may lead to changes in interactions between the TMD residues, and protein-lipids, leading to structural destabilization of the A-site and the whole domain. This proposed structural change is further strengthened by the Zn2+ transport activity impairment observed in vitro.11 Since zinc has a significant role as a signaling mediator and in brain development and proper function in all stages of life, the impairment in zinc homeostasis is thought to be the cause for the phenotypic effects of the A350del mutation.11,85ZnT-10
ZnT-10 is a manganese transporter expressed mainly in the brain and liver, and located in endosomes, the Golgi apparatus and the cell membrane.10,86,87 The ZnT-10 L349P mutation causes hypermanganesemia, hepatomegaly and dystonia.23 Structural characterization, through MamM CTD,51 predicts its location in the middle of the β-sheet central β-strand of the CTD (Fig. 6). The MamM homologous mutation M250P causes complete CTD structural loss, which leads to a low expression level compared to wildtype and a decrease in function. Overall, this mutation caused protein degradation via improper CTD fold and recruitment of proteases.51 Since non-degraded MamM proteins showed some activity and ZnT-10 mutation is not lethal, it is suggested that the unstructured CTD, which contains the DDMC binding residues, can still facilitate the TMD conformational change. This leads to inefficient ZnT-10-mediated Mn2+ extrusion and its accumulation in the cells and the resultant phenotypes, which are expected physiological consequences of impairment in manganese homeostasis.51Some other mutations in ZnT-10 cause hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia, among which are L89P (predicted to be in TMH3), A105-P107del (predicted to be on the loop between TMH3 and TMH4) and V256del (predicted to be in TMH5)23 (Fig. 6). Similarly to the L349P mutation in ZnT-10, and since proline destabilizes secondary structures, we suggest that the L89P deforms TMH3. This will destabilize the TMD in a way that at worst might result in full protein misfolding. In any case, the location in TMH3 will impact Mn2+ transport by changing the rocking movement and dimerization alteration. The A105-P107del will lead to a shorter TMH3–TMH4 connecting loop and we predict that the loop length is essential for a proper rocking movement. Hence, this mutation will also impact the transition between the inward and outward conformations. The ZnT-10 256 position is found in the TMH5 C-terminal, in a region that might be unstructured, yet it is part of a hydrophobic chain predicted to be buried in the membrane. Hence, we assume that it will have an effect similar to the A350del mutation in ZnT-9. It will cause more tension in the membrane and will change the interaction networks between the TMD residues themselves and with the lipids, leading to destabilization of the whole domain.
ZnT-1 as an example for multiple functions of ZnT proteins
ZnT-1 (SLC30A1) is the most abundantly expressed member of the mammalian CDF family and is involved in Zn2+ homeostasis.12,48,88–91 The ZnT-1 expression increases following transient forebrain ischemia,92 rapid cardiac pacing, an exposure to high, nontoxic, levels of Zn2+,24,25,27 and also increases in dietary Zn2+.88 ZnT-1 induction is mediated by the metal regulatory transcription factor 1 (MTF-1),88,93,94 which, besides increasing in response to extracellular Zn2+, is activated by an increase in intracellular Ca2+.95 In addition, MTF-1 is activated by oxidative stress.96 Some of these stimuli probably underlie the observed increase in ZnT-1 expression in conditions such as rapid pacing or ischemia–reperfusion in the heart.27,97 ZnT-1 protects against zinc toxicity due to its Zn2+ extruder activity.12,48,98 ZnT-1 knockout is embryonically lethal88,93,99 even when there is no significant exposure to systemic Zn2+. This implies that, even when its Zn2+ extrusion function is not needed acutely, ZnT-1 plays other critical roles in development and cellular function. These findings may explain why no ZnT-1-related mutation has been identified although sequence variations were found in the genome databases.Being a member of the CDF family, ZnT-1 is responsible for cellular activity that reduces zinc toxicity.98 However, as implied by the lethality of the knockout, ZnT-1 plays another important role: a modulator and an integrator of signaling and cation channel function. In addition, ZnT-1 activates signaling molecules including ERK through Ras-mediated signaling.24,27,100,101 Furthermore, ZnT-1 inhibits Zn2+ and Ca2+ influx by inhibiting L-type Ca2+ channels.26,89,97,102 In yeast two-hybrid experiments, ZnT-1 interacts with another channel, the calcium homeostasis ER protein (CHERP).103–105 All of these activities of ZnT-1 are independent of its function as a Zn2+ transporter as they are also mediated by ZnT-1 CTD itself.24,27,98 This observation is consistent with the embryonic lethality of the knockout.
Concluding remarks
A structure–function investigation of CDFs is highly important for elucidating the underlying mechanism of many diseases associated with them. A remarkable advancement in recent years unraveled key features for metal binding and conformational changes. The structural and biophysical studies of YiiP and ZnT-8 unveiled the cation binding sites and some TMD conformations, while the studies of the CTDs of other bacterial CDFs facilitated their functional understanding. These studies allow better insights into the overall CDF transport mechanism. Based on the recent structural data, it is clear that the mechanism of mammalian ZnTs is similar to that of bacterial CDFs. Yet, eukaryotic CDFs evolved to include additional domains that attenuate the basic transport function existing in bacteria and provide additional activities. The current knowledge from CDFs in prokaryotes and eukaryotes allows a better interpretation of ZnT-related diseases. Current mutations underlying the observed phenotypes in mammals are mostly located on the basic CDF structure, found also in the bacterial domains (TMD and CTD), indicating that the basic transport mechanism is conserved through evolution. Yet, since the eukaryote CDFs play additional roles and act in more complex environments, some of the current mutations affect subcellular localization, signaling and stability and indirectly affect the transport efficiency.Significant progress in the structural studies of CDF proteins contributed a lot to their mechanistic understanding. Yet, there are still many unknown factors that prevent us from achieving a complete understanding, such as the apo conformation of the full proteins and the difference in affinities between the CTD and TMD DDMC binding sites, which are of utmost importance for deciphering the DDMC path along the protein. Moreover, as some eukaryotic elements, such as elongated NTD and the HRL, were not characterized through the studies of bacterial CDF proteins, it is essential to study several eukaryotic CDFs to achieve a complete understanding of their structure and function and their contribution to the transport mechanism.
Metal selectivity is an additional mystery in CDF studies. To date, we are far from understanding what governs the metal selectivity of CDFs. To that end, the CDF A-site is the most characterized element, and it is believed that DDMC binding and selectivity in most CDF proteins can be controlled by means of its amino acid composition. However, some studies showed that this is not the sole determinant and other elements may contribute to metal selectivity. Unfortunately, these studies did not explore the binding capabilities to all DDMCs, neither did they involve deep structural analysis. Since not all CDF proteins discriminate similarly among the DDMCs, only biophysical investigation of the metal-selectivity related elements in many CDF proteins will allow a significant step towards elucidating an overall metal selectivity mechanism. Additionally, the metal acquisition of CDF proteins is unknown. The cytosolic concentrations of many metals, including zinc, are very low and consequently their availability to a CDF protein by free diffusion is low. This supports the speculation that a specific metallochaperone is recognized by CDFs and facilitates metal transfer and transport activation.
In conclusion, despite the enormous progress in the study of various CDFs, there is still a lot to be found before achieving a complete understanding of the entire CDF conformational space. Thus, complete ZnT structure–function relationship and mechanism studies are still needed. Hopefully, as done for other proteins and recently with ZnT-8, cryo-EM or XFEL will soon revolutionize the study of CDFs, with an aim to achieve a deeper understanding of CDFs in general and of ZnTs specifically, paving a way for the development of ZnT-targeted drugs for the treatment of CDF-related diseases.
Author contributions
All authors were involved in data curation, writing of the original draft, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. David L. Stokes for providing the PDB file of the tighter TMD dimerization model of EcYiiP. The authors are supported by the Israel Ministry of Science, Technology and Space, the Israel Science Foundation (grant no 167/16), the European Molecular Biology Organization and CMST COST Action CM1306.References
-
T. J. Lyons, D. J. Eide and V. Introduction, in Biologocal inorganic chemistry: structure and reactivity, ed. I. Bertini, H. Gray, E. Stiefel and J. S. Valentine, University Science books, CA, USA, Transport and Storage of Metal Ions in Biology, 2006, pp. 57–78 Search PubMed
.
- Y. Lu, Inorg. Chem., 2006, 45, 9930–9940 CrossRef CAS
- M. M. Harding, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 849–859 CrossRef
- T. Dudev and C. Lim, Chem. Rev., 2014, 114, 538–556 CrossRef CAS
- H. Irvng and R. J. P. Williams, Nature, 1948, 162, 746–747 CrossRef
- C. J. Haney, G. Grass, S. Franke and C. Rensing, J. Ind. Microbiol. Biotechnol., 2005, 32, 215–226 CrossRef CAS
- C. Cubillas, P. Vinuesa, M. L. Tabche and A. García-de los Santos, Metallomics, 2013, 5, 1634–1643 RSC
- L. Huang and S. Tepaamorndech, Mol. Aspects Med., 2013, 34, 548–560 CrossRef CAS
- P. Chen, A. B. Bowman, S. Mukhopadhyay and M. Aschner, Worm, 2015, 4, e1042648 CrossRef
- M. Levy, N. Elkoshi, S. Barber-Zucker, E. Hoch, R. Zarivatch, M. Hershfinkel and I. Sekler, J. Biol. Chem., 2019, 294, 5879–5889 CrossRef CAS
- Y. Perez, Z. Shorer, K. Liani-Leibson, P. Chabosseau, R. Kadir, M. Volodarsky, D. Halperin, S. Barber-Zucker, H. Shalev, R. Schreiber, L. Gradstein, E. Gurevich, R. Zarivach, G. A. Rutter, D. Landau and O. S. Birk, Brain, 2017, 140, 928–939 CrossRef
- E. Shusterman, O. Beharier, L. Shiri, R. Zarivach, Y. Etzion, C. R. Campbell, I. H. Lee, K. Okabayashi, A. Dinudom, D. I. Cook, A. Katz and A. Moran, Metallomics, 2014, 6, 1656–1663 RSC
- E. Hoch, W. Lin, J. Chai, M. Hershfinkel, D. Fu and I. Sekler, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 7202–7207 CrossRef CAS
- M. A. Lovell, J. L. Smith, S. Xiong and W. R. Markesbery, Neurotox. Res., 2005, 7, 265–271 CrossRef CAS
- G. Lyubartseva, J. L. Smith, W. R. Markesbery and M. A. Lovell, Brain Pathol., 2010, 20, 343 CrossRef CAS
- H. J. Bosomworth, P. A. Adlard, D. Ford and R. A. Valentine, PLoS One, 2013, 8, e65475 CrossRef CAS
- Y. Golan, T. Kambe and Y. G. Assaraf, Metallomics, 2017, 9, 1352–1366 RSC
- M. C. Miletta, A. Bieri, K. Kernland, M. H. Schöni, V. Petkovic, C. E. Flück, A. Eblé and P. E. Mullis, Int. J. Endocrinol., 2013, 2013, 259189 Search PubMed
- W. Chowanadisai, B. Lönnerdal and S. L. Kelleher, J. Biol. Chem., 2006, 281, 39699–39707 CrossRef CAS
- Y. Golan, A. Lehvy, G. Horev and Y. G. Assaraf, J. Cell. Mol. Med., 2019, 23, 828–840 CrossRef CAS
- T. W. Boesgaard, J. Žilinskaitė, M. Vänttinen, M. Laakso, P.-A. Jansson, A. Hammarstedt, U. Smith, N. Stefan, A. Fritsche, H. Häring, M. Hribal, G. Sesti, D. P. Zobel, O. Pedersen, T. Hansen and the EUGENE 2 Consortium, Diabetologia, 2008, 51, 816–820 CrossRef CAS
- M. Quadri, A. Federico, T. Zhao, G. J. Breedveld, C. Battisti, C. Delnooz, L. A. Severijnen, L. Di Toro Mammarella, A. Mignarri, L. Monti, A. Sanna, P. Lu, F. Punzo, G. Cossu, R. Willemsen, F. Rasi, B. A. Oostra, B. P. Van De Warrenburg and V. Bonifati, Am. J. Hum. Genet., 2012, 90, 467–477 CrossRef CAS
- K. Tuschl, P. T. Clayton, S. M. Gospe, S. Gulab, S. Ibrahim, P. Singhi, R. Aulakh, R. T. Ribeiro, O. G. Barsottini, M. S. Zaki, M. L. Del Rosario, S. Dyack, V. Price, A. Rideout, K. Gordon, R. A. Wevers, W. K. K. Chong and P. B. Mills, Am. J. Hum. Genet., 2012, 90, 457–466 CrossRef CAS
- M. Mor, O. Beharier, S. Levy, J. Kahn, S. Dror, D. Blumenthal, L. A. Gheber, A. Peretz, A. Katz, A. Moran and Y. Etzion, Am. J. Physiol. Physiol., 2012, 303, C192–C203 CrossRef CAS
- Y. Etzion, A. Ganiel, O. Beharier, A. Shalev, V. Novack, L. Volvich, D. Abrahamov, M. Matsa, G. Sahar, A. Moran and A. Katz, J. Cardiovasc. Electrophysiol., 2008, 19, 157–164 CrossRef
- S. Levy, O. Beharier, Y. Etzion, M. Mor, L. Buzaglo, L. Shaltiel, L. A. Gheber, J. Kahn, A. J. Muslin, A. Katz, D. Gitler and A. Moran, J. Biol. Chem., 2009, 284, 32434–32443 CrossRef CAS
- O. Beharier, S. Dror, S. Levy, J. Kahn, M. Mor, S. Etzion, D. Gitler, A. Katz, A. J. Muslin, A. Moran and Y. Etzion, J. Mol. Med., 2012, 90, 127–138 CrossRef CAS
- C. Merriman, Q. Huang, G. A. Rutter and D. Fu, J. Biol. Chem., 2016, 291, 26950–26957 CrossRef CAS
- D. S. Parsons, C. Hogstrand and W. Maret, FEBS J., 2018, 285, 1237–1250 CrossRef CAS
- R. Ullah, A. Shehzad, M. A. Shah, M. De March, F. Ismat, M. Iqbal, S. Onesti, M. Rahman and M. J. McPherson, Int. J. Mol. Sci., 2020, 21, 926 CrossRef CAS
- N. Tanaka, M. Kawachi, T. Fujiwara and M. Masayoshi, FEBS Open Bio., 2013, 3, 218–224 CrossRef CAS
- N. Tanaka, T. Fujiwara, R. Tomioka, U. Krämer, M. Kawachi and M. Maeshima, Plant Cell Physiol., 2015, 56, 510–519 CrossRef CAS
- M. J. Daniels, M. Jagielnicki and M. Yeager, Curr. Res. Struct. Biol., 2020, 2, 144–155 CrossRef
- J. Xue, T. Xie, W. Zeng, Y. Jiang and X. Bai, eLife, 2020, 9, e58823 CrossRef CAS
- N. Coudray, S. Valvo, M. Hu, R. Lasala, C. Kim, M. Vink, M. Zhou, D. Provasi, M. Filizola, J. Tao, J. Fang, P. A. Penczek, I. Ubarretxena-Belandia and D. L. Stokes, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2140–2145 CrossRef CAS
- M. L. Lopez-Redondo, N. Coudray, Z. Zhang, J. Alexopoulos and D. L. Stokes, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3042–3047 CrossRef CAS
- M. Lu and D. Fu, Science, 2007, 317, 1746–1748 CrossRef CAS
- M. Lu, J. Chai and D. Fu, Nat. Struct. Mol. Biol., 2009, 16, 1063–1067 CrossRef CAS
- O. Kolaj-Robin, D. Russell, K. A. Hayes, J. T. Pembroke and T. Soulimane, FEBS Lett., 2015, 589, 1283–1295 CrossRef CAS
- N. Zeytuni, R. Uebe, M. Maes, G. Davidov, M. Baram, O. Raschdorf, M. Nadav-Tsubery, S. Kolusheva, R. Bitton, G. Goobes, A. Friedler, Y. Miller, D. Schüler and R. Zarivach, PLoS One, 2014, 9, e92141 CrossRef
- V. Cherezov, N. Höfer, D. M. E. Szebenyi, O. Kolaj, J. G. Wall, R. Gillilan, V. Srinivasan, C. P. Jaroniec and M. Caffrey, Structure, 2008, 16, 1378–1388 CrossRef CAS
- N. Zeytuni, R. Uebe, M. Maes, G. Davidov, M. Baram, O. Raschdorf, A. Friedler, Y. Miller, D. Schüler and R. Zarivach, PLoS One, 2014, 9, e97154 CrossRef
- R. Uebe, N. Keren-Khadmy, N. Zeytuni, E. Katzmann, Y. Navon, G. Davidov, R. Bitton, J. M. Plitzko, D. Schüler and R. Zarivach, Mol. Microbiol., 2018, 107, 542–557 CrossRef CAS
- J. E. Martin and D. P. Giedroc, J. Bacteriol., 2016, 198, 1066–1076 CrossRef CAS
- C. E. Zogzas, M. Aschner and S. Mukhopadhyay, J. Biol. Chem., 2016, 291, 15940–15957 CrossRef CAS
- R. Uebe, K. Junge, V. Henn, G. Poxleitner, E. Katzmann, J. M. Plitzko, R. Zarivach, T. Kasama, G. Wanner, M. Pósfai, L. Böttger, B. Matzanke and D. Schüler, Mol. Microbiol., 2011, 82, 818–835 CrossRef CAS
- A. Anton, A. Weltrowski, C. J. Haney, S. Franke, G. Grass, C. Rensing and D. H. Nies, J. Bacteriol., 2004, 186, 7499–7507 CrossRef CAS
- R. D. Palmiter and S. D. Findley, EMBO J., 1995, 14, 639–649 CrossRef CAS
- N. Itsumura, Y. Inamo, F. Okazaki, F. Teranishi, H. Narita, T. Kambe and H. Kodama, PLoS One, 2013, 8, e64045 CrossRef CAS
- Y. Chao and D. Fu, J. Biol. Chem., 2004, 279, 12043–12050 CrossRef CAS
- S. Barber-Zucker, R. Uebe, G. Davidov, Y. Navon, D. Sherf, J. H. Chill, I. Kass, R. Bitton, D. Schüler and R. Zarivach, Sci. Rep., 2016, 6, 31933 CrossRef CAS
- S. Barber-Zucker, J. Hall, S. V. Mangapuram, I. Kass, S. Kolusheva, F. MacMillan, R. Zarivach and A. Henn, FEBS J., 2019, 286, 2193–2215 CrossRef CAS
- S. Barber-Zucker, J. Hall, A. Froes, S. Kolusheva, F. MacMillan and R. Zarivach, J. Biol. Chem., 2020, 295(49), 16614–16629 CrossRef CAS
- T. Higuchi, M. Hattori, Y. Tanaka, R. Ishitani and O. Nureki, Proteins: Struct., Funct., Bioinf., 2009, 76, 768–771 CrossRef CAS
- D. Poger, J. F. Fuchs, H. Nedev, M. Ferrand and S. Crouzy, FEBS Lett., 2005, 579, 5287–5292 CrossRef CAS
- S. R. Udagedara, D. M. La Porta, C. Spehar, G. Purohit, M. J. A. Hein, M. E. Fatmous, G. P. Casas Garcia, K. Ganio, C. A. McDevitt and M. J. Maher, J. Inorg. Biochem., 2020, 208, 111087 CrossRef CAS
- Y. Wei and D. Fu, J. Biol. Chem., 2006, 281, 23492–23502 CrossRef CAS
- D. Sala, A. Giachetti and A. Rosato, Biochim. Biophys. Acta, Gen. Subj., 2019, 1863, 1560–1567 CrossRef CAS
- S. Gupta, J. Chai, J. Cheng, R. D’Mello, M. R. Chance and D. Fu, Nature, 2014, 512, 101–104 CrossRef CAS
- S. Barber-Zucker, B. Shaanan and R. Zarivach, Sci. Rep., 2017, 7, 16381 CrossRef
- B. Montanini, D. Blaudez, S. Jeandroz, D. Sanders and M. Chalot, BMC Genomics, 2007, 8, 107 CrossRef
- P. K. Menguer, E. Farthing, K. A. Peaston, F. K. Ricachenevsky, J. P. Fett and L. E. Williams, J. Exp. Bot., 2013, 64, 2871–2883 CrossRef CAS
- Y. Nishito, N. Tsuji, H. Fujishiro, T. A. Takeda, T. Yamazaki, F. Teranishi, F. Okazaki, A. Matsunaga, K. Tuschl, R. Rao, S. Kono, H. Miyajima, H. Narita, S. Himeno and T. Kambe, J. Biol. Chem., 2016, 291, 14773–14787 CrossRef CAS
- D. Podar, J. Scherer, Z. Noordally, P. Herzyk, D. Nies and D. Sanders, J. Biol. Chem., 2012, 287, 3185–3196 CrossRef CAS
- M. Kawachi, Y. Kobae, T. Mimura and M. Maeshima, J. Biol. Chem., 2008, 283, 8374–8383 CrossRef CAS
- M. Kawachi, Y. Kobae, S. Kogawa, T. Mimura, U. Krämer and M. Maeshima, FEBS J., 2012, 279, 2339–2356 CrossRef CAS
- T. Suzuki, K. Ishihara, H. Migaki, K. Ishihara, M. Nagao, Y. Yamaguchi-Iwai and T. Kambe, J. Biol. Chem., 2005, 280, 30956–30962 CrossRef CAS
- S. Barber-Zucker, A. Shahar, S. Kolusheva and R. Zarivach, Sci. Rep., 2020, 10, 14022 CrossRef CAS
- G. Grass, M. Otto, B. Fricke, C. J. Haney, C. Rensing, D. H. Nies and D. Munkelt, Arch. Microbiol., 2005, 183, 9–18 CrossRef CAS
- A. A. Guffanti, Y. Wei, S. V. Rood and T. A. Krulwich, Mol. Microbiol., 2002, 45, 145–153 CrossRef CAS
- S. M. Lee, G. Grass, C. J. Haney, B. Fan, B. P. Rosen, A. Anton, D. H. Nies and C. Rensing, FEMS Microbiol. Lett., 2002, 215, 273–278 CrossRef CAS
- D. Raimunda and G. Elso-Berberián, Biochim. Biophys. Acta, Biomembr., 2014, 1838, 3203–3211 CrossRef CAS
- C. W. MacDiarmid, M. A. Milanick and D. J. Eide, J. Biol. Chem., 2002, 277, 39187–39194 CrossRef CAS
- E. Ohana, E. Hoch, C. Keasar, T. Kambe, O. Yifrach, M. Hershfinkel and I. Sekler, J. Biol. Chem., 2009, 284, 17677–17686 CrossRef CAS
- Y. Golan, R. Alhadeff, A. Warshel and Y. G. Assaraf, PLoS Comput. Biol., 2019, 15, e1006882 CrossRef
- K. Fukue, N. Itsumura, N. Tsuji, K. Nishino, M. Nagao, H. Narita and T. Kambe, Sci. Rep., 2018, 8, 14084 CrossRef
- C. Murgia, I. Vespignani, J. Cerase, F. Nobili and G. Perozzi, Am. J. Physiol.: Gastrointest. Liver Physiol., 1999, 277, G1231–1239 CrossRef CAS
- I. Lasry, Y. A. Seo, H. Ityel, N. Shalva, B. Pode-Shakked, F. Glaser, B. Berman, I. Berezovsky, A. Goncearenco, A. Klar, J. Levy, Y. Anikster, S. L. Kelleher and Y. G. Assaraf, J. Biol. Chem., 2012, 287, 29348–29361 CrossRef CAS
- Y. Golan, N. Itsumura, F. Glaser, B. Berman, T. Kambe and Y. G. Assaraf, J. Biol. Chem., 2016, 291, 13546–13559 CrossRef CAS
- L. Marger, C. R. Schubert and D. Bertrand, Biochem. Pharmacol., 2014, 91, 426–435 CrossRef CAS
- M. S. Hildebrand, A. M. Phillips, S. A. Mullen, P. A. Adlard, K. Hardies, J. A. Damiano, V. Wimmer, S. T. Bellows, J. M. McMahon, R. Burgess, R. Hendrickx, S. Weckhuysen, A. Suls, P. De Jonghe, I. E. Scheffer, S. Petrou, S. F. Berkovic and C. A. Reid, Sci. Rep., 2016, 5, 17816 CrossRef
- G. Salazar, J. M. Falcon-Perez, R. Harrison and V. Faundez, PLoS One, 2009, 4, e5896 CrossRef
- F. Chimienti, S. Devergnas, A. Favier and M. Seve, Diabetes, 2004, 53, 2330–2337 CrossRef CAS
- J. Flannick, G. Thorleifsson, N. L. Beer, S. B. R. Jacobs, N. Grarup, N. P. Burtt, A. Mahajan, C. Fuchsberger, G. Atzmon, R. Benediktsson, J. Blangero, D. W. Bowden, I. Brandslund, J. Brosnan, F. Burslem, J. Chambers, Y. S. Cho, C. Christensen, D. A. Douglas, R. Duggirala, Z. Dymek, Y. Farjoun, T. Fennell, P. Fontanillas, T. Forsén, S. Gabriel, B. Glaser, D. F. Gudbjartsson, C. Hanis, T. Hansen, A. B. Hreidarsson, K. Hveem, E. Ingelsson, B. Isomaa, S. Johansson, T. Jørgensen, M. E. Jørgensen, S. Kathiresan, A. Kong, J. Kooner, J. Kravic, M. Laakso, J.-Y. Lee, L. Lind, C. M. Lindgren, A. Linneberg, G. Masson, T. Meitinger, K. L. Mohlke, A. Molven, A. P. Morris, S. Potluri, R. Rauramaa, R. Ribel-Madsen, A.-M. Richard, T. Rolph, V. Salomaa, A. V. Segrè, H. Skärstrand, V. Steinthorsdottir, H. M. Stringham, P. Sulem, E. S. Tai, Y. Y. Teo, T. Teslovich, U. Thorsteinsdottir, J. K. Trimmer, T. Tuomi, J. Tuomilehto, F. Vaziri-Sani, B. F. Voight, J. G. Wilson, M. Boehnke, M. I. McCarthy, P. R. Njølstad, O. Pedersen, L. Groop, D. R. Cox, K. Stefansson, D. Altshuler, K. Stefansson and D. Altshuler, Nat. Genet., 2014, 46, 357–363 CrossRef CAS
- M. Tyszka-Czochara, A. Grzywacz, J. Gdula-Argasi-Ska, T. Librowski, B. Wili-Ski and W. Opoka, Acta Pol. Pharm., 2014, 71, 369–377 Search PubMed
- M. Seve, F. Chimienti, S. Devergnas and A. Favier, BMC Genomics, 2004, 5, 32 CrossRef
- D. Leyva-Illades, P. Chen, C. E. Zogzas, S. Hutchens, J. M. Mercado, C. D. Swaim, R. A. Morrisett, A. B. Bowman, M. Aschner and S. Mukhopadhyay, J. Neurosci., 2014, 34, 14079–14095 CrossRef
- R. J. Cousins, J. P. Liuzzi and L. A. Lichten, J. Biol. Chem., 2006, 281, 24085–24089 CrossRef CAS
- C. Nolte, A. Gore, I. Sekler, W. Kresse, M. Hershfinkel, A. Hoffmann, H. Kettenmann and A. Moran, Glia, 2004, 48, 145–155 CrossRef
- T. Kambe, T. Tsuji, A. Hashimoto and N. Itsumura, Physiol. Rev., 2015, 95, 749–784 CrossRef CAS
- T. Kimura and T. Kambe, Int. J. Mol. Sci., 2016, 17, 336 CrossRef
- M. Tsuda, K. Imaizumi, T. Katayama, K. Kitagawa, A. Wanaka, M. Tohyama and T. Takagi, J. Neurosci., 1997, 17, 6678–6684 CrossRef CAS
- S. J. Langmade, R. Ravindra, P. J. Daniels and G. K. Andrews, J. Biol. Chem., 2000, 275, 34803–34809 CrossRef CAS
- M. Vig, C. Peinelt, A. Beck, D. L. Koomoa, D. Rabah, M. Koblan-Huberson, S. Kraft, H. Turner, A. Fleig, R. Penner and J. P. Kinet, Science, 2006, 312, 1220–1223 CrossRef CAS
- T. K. Adams, N. Saydam, F. Steiner, W. Schaffner and J. H. Freedman, Environ. Health Perspect., 2002, 110, 813–817 CrossRef CAS
- T. P. Dalton, Q. Li, D. Bittel, L. Liang and G. K. Andrews, J. Biol. Chem., 1996, 271, 26233–26241 CrossRef CAS
- O. Beharier, Y. Etzion, A. Katz, H. Friedman, N. Tenbosh, S. Zacharish, S. Bereza, U. Goshen and A. Moran, Cell Calcium, 2007, 42, 71–82 CrossRef CAS
- E. Shusterman, O. Beharier, S. Levy, R. Zarivach, Y. Etzion, C. R. Campbell, I. H. Lee, A. Dinudom, D. I. Cook, A. Peretz, A. Katz, D. Gitler and A. Moran, Metallomics, 2017, 9, 228–238 RSC
- G. K. Andrews, H. Wang, S. K. Dey and R. D. Palmiter, Genesis, 2004, 40, 74–81 CrossRef CAS
- J. J. Bruinsma, T. Jirakulaporn, A. J. Muslin and K. Kornfeld, Dev. Cell, 2002, 2, 567–578 CrossRef CAS
- T. Jirakulaporn and A. J. Muslin, J. Biol. Chem., 2004, 279, 27807–27815 CrossRef CAS
- D. Segal, E. Ohana, L. Besser, M. Hershfinkel, A. Moran and I. Sekler, Biochem. Biophys. Res. Commun., 2004, 323, 1145–1150 CrossRef CAS
- F. A. O’Rourke, J. M. LaPlante and M. B. Feinstein, Biochem. J., 2003, 373, 133–143 CrossRef
- M. Y. Song, A. Makino and J. X.-J. Yuan, Antioxid. Redox Signaling, 2011, 15, 1549–1565 CrossRef CAS
- M. Lazarczyk, P. Cassonnet, C. Pons, Y. Jacob and M. Favre, Microbiol. Mol. Biol. Rev., 2009, 73, 348–370 CrossRef CAS
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00181c |
| This journal is © The Royal Society of Chemistry 2021 |