
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural Trojan horse inhibitors of aminoacyl-tRNA synthetases†
Dmitrii Y.
Travin
 *ab,
Konstantin
Severinov
*abc and
Svetlana
Dubiley
*ab
*ab,
Konstantin
Severinov
*abc and
Svetlana
Dubiley
*ab
aCenter of Life Sciences, Skolkovo Institute of Science and Technology, Moscow, Russia. E-mail: dmitrii.travin@skoltech.ru; severik@waksman.rutgers.edu; svetlana.dubiley@gmail.com
bInstitute of Gene Biology, Russian Academy of Sciences, Moscow, Russia
cWaksman Institute for Microbiology, Rutgers, Piscataway, New Jersey, USA
First published on 22nd February 2021
Abstract
For most antimicrobial compounds with intracellular targets, getting inside the cell is the major obstacle limiting their activity. To pass this barrier some antibiotics mimic the compounds of specific interest for the microbe (siderophores, peptides, carbohydrates, etc.) and hijack the transport systems involved in their active uptake followed by the release of a toxic warhead inside the cell. In this review, we summarize the information about the structures, biosynthesis, and transport of natural inhibitors of aminoacyl-tRNA synthetases (albomycin, microcin C-related compounds, and agrocin 84) that rely on such “Trojan horse” strategy to enter the cell. In addition, we provide new data on the composition and distribution of biosynthetic gene clusters reminiscent of those coding for known Trojan horse aminoacyl-tRNA synthetases inhibitors. The products of these clusters are likely new antimicrobials that warrant further investigation.
 Dmitrii Y. Travin | Dmitrii Y. Travin received his MSc degree in bioengineering and bioinformatics from Lomonosov Moscow State University in 2018. He started his PhD studies at Skolkovo Institute of Science and Technology under the supervision of Prof. Konstantin Severinov. Dmitrii performs genome mining and characterization of novel azole-containing peptides produced by bacteria, investigates their biosynthesis, mode of action, and ecological role in microbial communities. |
 Konstantin Severinov | Konstantin Severinov is a Professor of Molecular Biology and Biochemistry at Rutgers, the State University of New Jersey and at Skolkovo Institute of Science and Technology, and a Head of the Laboratory at the Waksman Insitute of Microbiology. His research interests include peptide antibiotics, toxin-antitoxin systems, CRISPR-Cas and other bacterial defence systems, and transcription regulation in bacteriophage-infected cells. |
 Svetlana Dubiley | Svetlana Dubiley is a Head of the Laboratory at the Institute of Gene Biology, Moscow. Svetlana received her PhD under supervision of Professor Andrey Mirzabekov at the Enghelgardt Institute of Molecular Biology, Moscow. Her research interests encompass molecular microbiology, biosynthesis of post-translationally modified peptides, and the social behavior of bacteria. |
Introduction
Aminoacyl-tRNA synthetases (aaRSs) are essential cellular enzymes that catalyze the attachment of tRNAs to cognate amino acids and thus ensure correct translation of genetic information into proteins. The tRNA charging proceeds in two steps: first, an amino acid is activated by the formation of an aminoacyl-adenylate; second, the aminoacyl group is transferred to the 3′-terminal adenosine of cognate tRNA (Fig. 1A). Both steps are performed in a single catalytic center, and some aaRSs bind all three substrates – amino acid, ATP, and tRNA – simultaneously.1 While the mechanism of tRNA charging is conserved, two distinct aaRS classes with multiple subclasses are distinguished (Fig. 1B). In class I aaRSs, the catalytic domain adopts the Rossmann fold; most enzymes from this class function as monomers. The catalytic domains of class II aaRSs are organized as seven β-strands flanked by α-helices and most of these enzymes form dimers or oligomers.2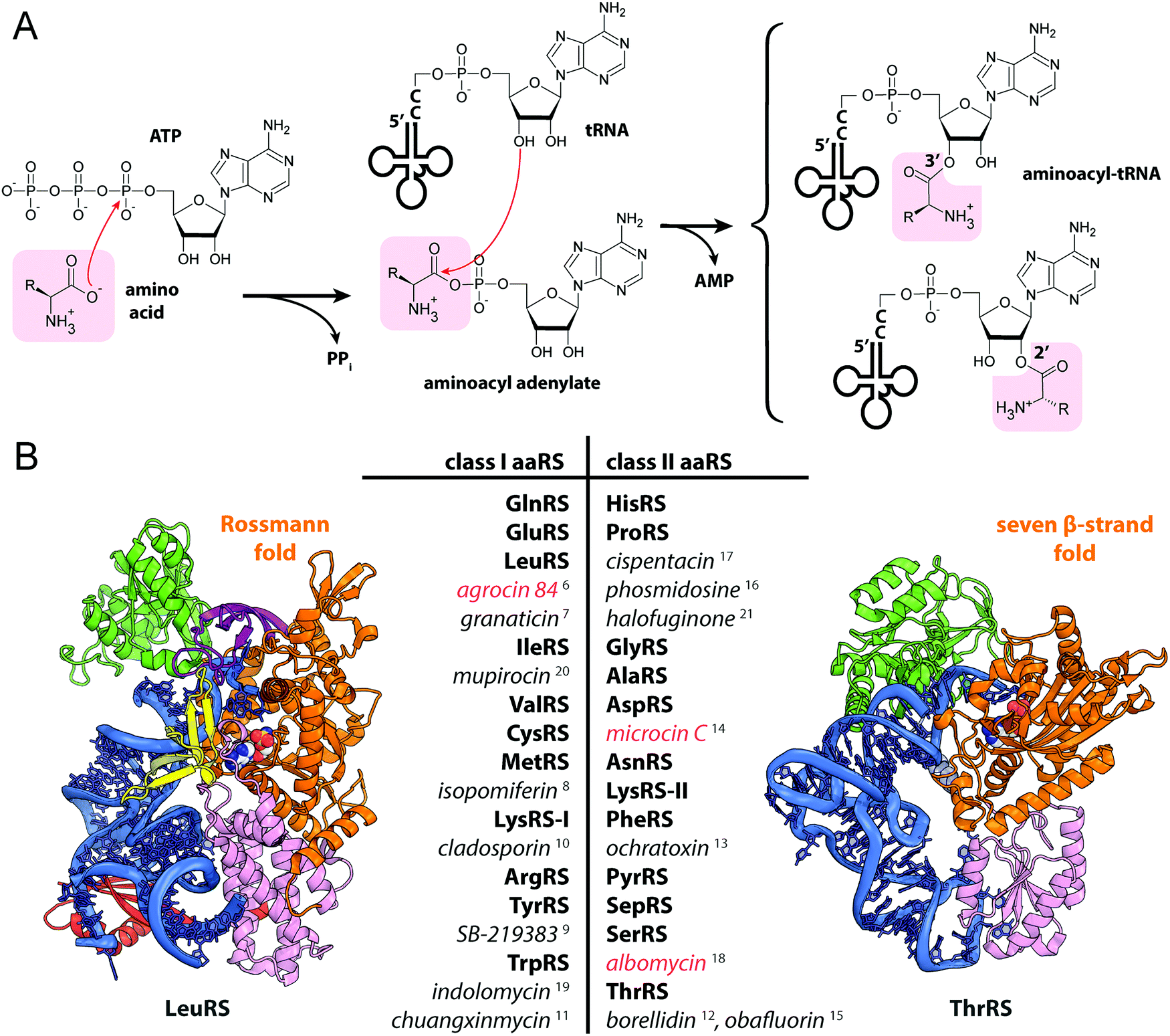 | ||
| Fig. 1 (A) Aminoacylation of tRNA catalyzed by aminoacyl-tRNA synthetases. (B) The structures of E. coli LeuRS (class I, PDB ID: 4AQ7136) and E. coli ThrRS (class II, PDB ID: 1QF6,137 only one of the two monomers is shown) tRNA (blue) complexes. Catalytic domains are shown in orange, anticodon-binding domains – in pink, editing domains – in green, leucine-specific domain of LeuRS – in yellow, C-terminal domain – in red, zinc domain – in violet. Representatives of the two classes of aaRSs are listed together with their known specific inhibitors of bacterial origin (shown in italic). Compounds employing the Trojan-horse strategy and covered in the review are highlighted with red font. | ||
As aaRSs perform an essential function, inactivation of any one of them leads to growth inhibition and, eventually, cell death. A number of natural antibiotics targeting tRNAs are known, two of which are approved for medical3 and veterinary4 use. With the exception of purpuromycin,5 which acts on multiple aaRSs, all other natural inhibitors are specific to certain aaRSs.6–18 (Fig. 1B). The majority of aaRS-targeting antibiotics bind in the catalytic center and mimic either the cognate amino acid (e.g., indolomycin),19 aminoacyl-adenylate (e.g., mupirocin)20 or charged tRNA (e.g., halofuginone).21 For reviews covering the diversity of natural aaRS inhibitors and their mechanisms of action see Cochrane et al.,22 Chopra & Reader23 and Ho et al.24
For many antibiotics with intracellular targets efficient internalization by susceptible bacteria is a key step limiting their bioactivity.25 Little is known about the transport of aaRS inhibitors inside the bacterial cell.26,27 Passive transport is especially challenging for molecules with multiple polar and hydrophilic groups, which is the case for aaRS-targeting drugs mimicking aminoacyl-adenylates. In this review, we cover three classes of naturally occurring antimicrobial compounds that target aaRSs and exploit the Trojan horse strategy to get inside the bacterial cell. These molecules comprise two functional parts: one that hijacks a specific transporter by mimicking a compound valuable for the cell (siderophore, peptide, opine) and another that acts as a detachable warhead killing the cell once released inside its cytoplasm. We cover existing information about the structures, modes of transport and action, genetics, enzymology of the biosynthesis, and mechanisms of immunity for three known groups of naturally occurring Trojan-horse inhibitors: albomycin, microcin C-related compounds, and agrocin 84. In addition, we reveal several previously overlooked gene clusters that likely encode new aaRS inhibitors.
1. Albomycins
Albomycin is active against a wide range of Gram-positive and Gram-negative bacteria including clinically relevant staphylococci, streptococci and various enterobacteria.32–34 Albomycin has a remarkably low (4 to 62 nM) minimal inhibitory concentrations (MIC) for Streptococcus pneumonia strains.35,36 The efficacy of albomycin treatment of S. pneumonia infection in a mouse model is similar to that of amoxicillin.32 Extensive preclinical and clinical studies carried out in the early 1950s showed exceptional safety of albomycin and its efficacy in treatment of meningitis and pulmonary infections, including those caused by penicillin-resistant pneumococci.34 Recent elaboration of total chemical synthesis35 allows large-scale production of albomycin and its analogs, opening way for their development into effective antibacterial drugs.
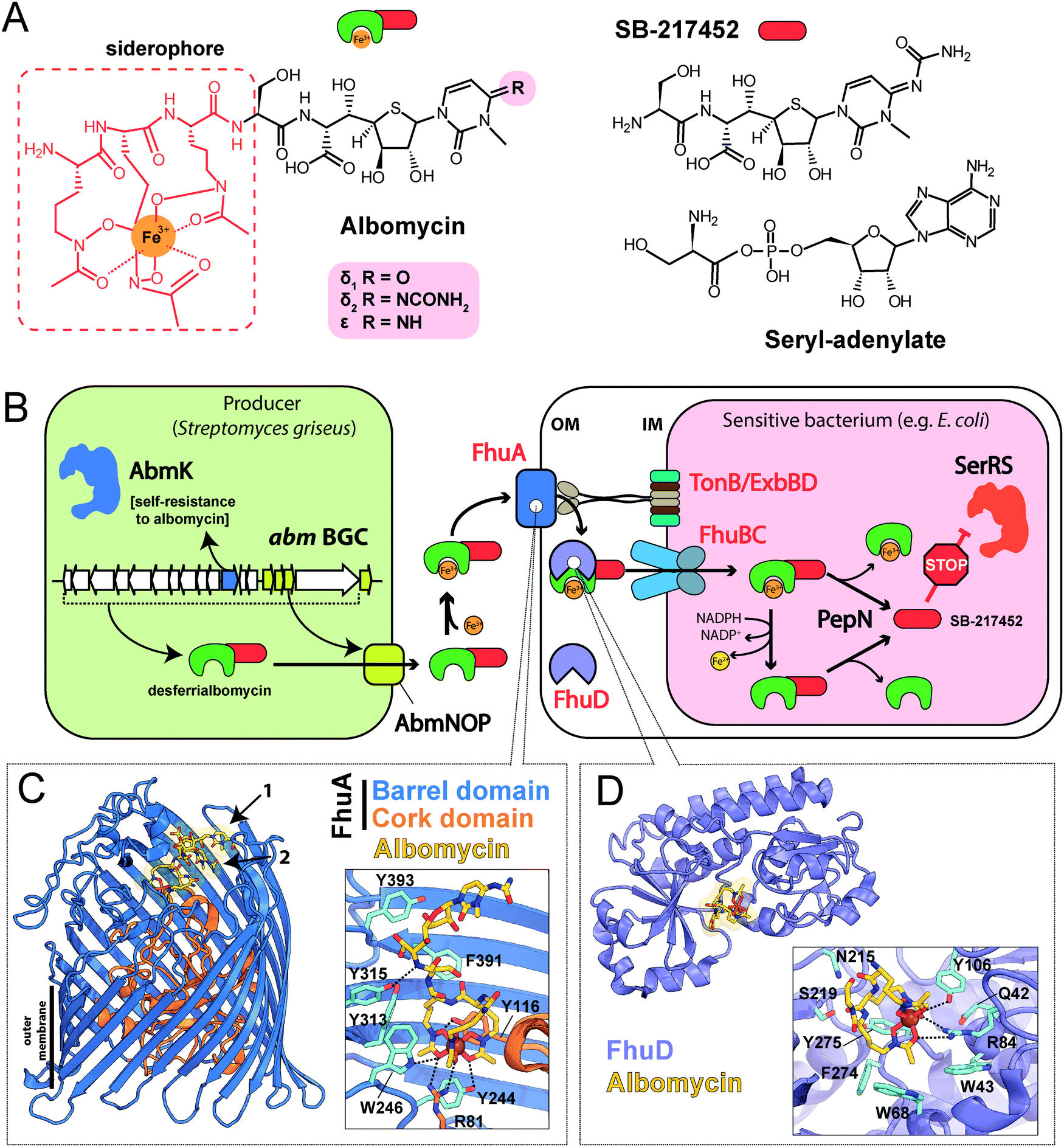 | ||
| Fig. 2 (A) Structures of albomycins, albomycin toxic moiety (SB-217452) and seryl-adenylate. (B) A scheme showing the biosynthesis, transport and intracellular processing of albomycin. IM – inner membrane, OM – outer membrane, BGC – biosynthetic gene cluster, SerRS – seryl-tRNA-synthetase. (C) The crystal structure of the FhuA outer membrane transporter in complex with albomycin (PDB ID: 1QKC40). Two conformational isomers of albomycin are shown (black arrows: 1 – extended form, 2 – compact form). A close-up view of the extended albomycin form binding site is shown, residues involved in the binding are indicated, hydrogen bonds are shown as black dashed lines. (D) The crystal structure of the FhuD periplasmic binding protein in complex with albomycin (PDB ID: 1K7S43). Residues involved in the formation of the albomycin binding site are indicated; hydrogen bonds are shown as black dashed lines. Electron density for the toxic moiety of albomycin is not observed in this structure. | ||
The iron-chelator portion serves as a vehicle for active delivery of albomycin warhead inside both Gram-positive and Gram-negative bacterial cells through the ferrichrome-specific transporter system.36,39 In E. coli, albomycin first binds to the outer membrane receptor FhuA. FhuA is a β-strand barrel buried in the outer membrane. An N-terminal domain located inside the barrel serves as a cork.40 Albomycin competes for the binding site with ferrichrome, the natural FhuA substrate (Fig. 2C). Binding of a ligand promotes the FhuA interaction with TonB, which in turn stimulates a rearrangement of the cork domain and the release of the ligand into the periplasmic space. The energy required for translocation is provided by the proton motive force transduced by the TonB-ExbB-ExbD complex.41 Once in the periplasm, albomycin binds FhuD, a periplasmic subunit of the inner membrane ABC transporter FhuB/FhuC (Fig. 2D).42,43 Inside the cell, Fe3+ is reduced to Fe2+, which dissociates from the chelator moiety,39 and the siderophore part is cleaved off by the PepN peptidase.44 As a result, the toxic nucleoside part is accumulated in the cytoplasm of E. coli in ∼500-fold excess over the concentration of antibiotic in the medium.39 When added directly to bacterial culture, the nucleoside portion of albomycin does not inhibit cell growth. Thus, albomycin is a Trojan horse agent. The initial rate of Fe3+ transport by albomycin inside the cell is only two times lower than that by ferrichrome.39 Thus, the antibiotic not only kills competing bacteria but is also an efficient iron-scavenger for the producer (Fig. 2B).
The target of albomycin was discovered serendipitously during a screening for inhibitors of bacterial aaRSs. SB-217452, a metabolite of Streptomyces sp. ATCC 700974, was shown to inhibit Staphylococcus aureus seryl-tRNA synthetase (SerRS) at low nanomolar concentration.15 The compound was identified to be the seryl-linked nucleoside moiety released upon the cleavage of the iron carrier part of albomycin δ2. Molecular modeling of SB-217452 in complex with SerRS shows that the N4 carbamoyl group modified cytosine structurally mimics adenine of the natural substrate seryl-AMP. A more extensive network of hydrogen bonding formed by SB-217452 compared to seryl-AMP within the active center of the enzyme ensures its higher affinity.45,46
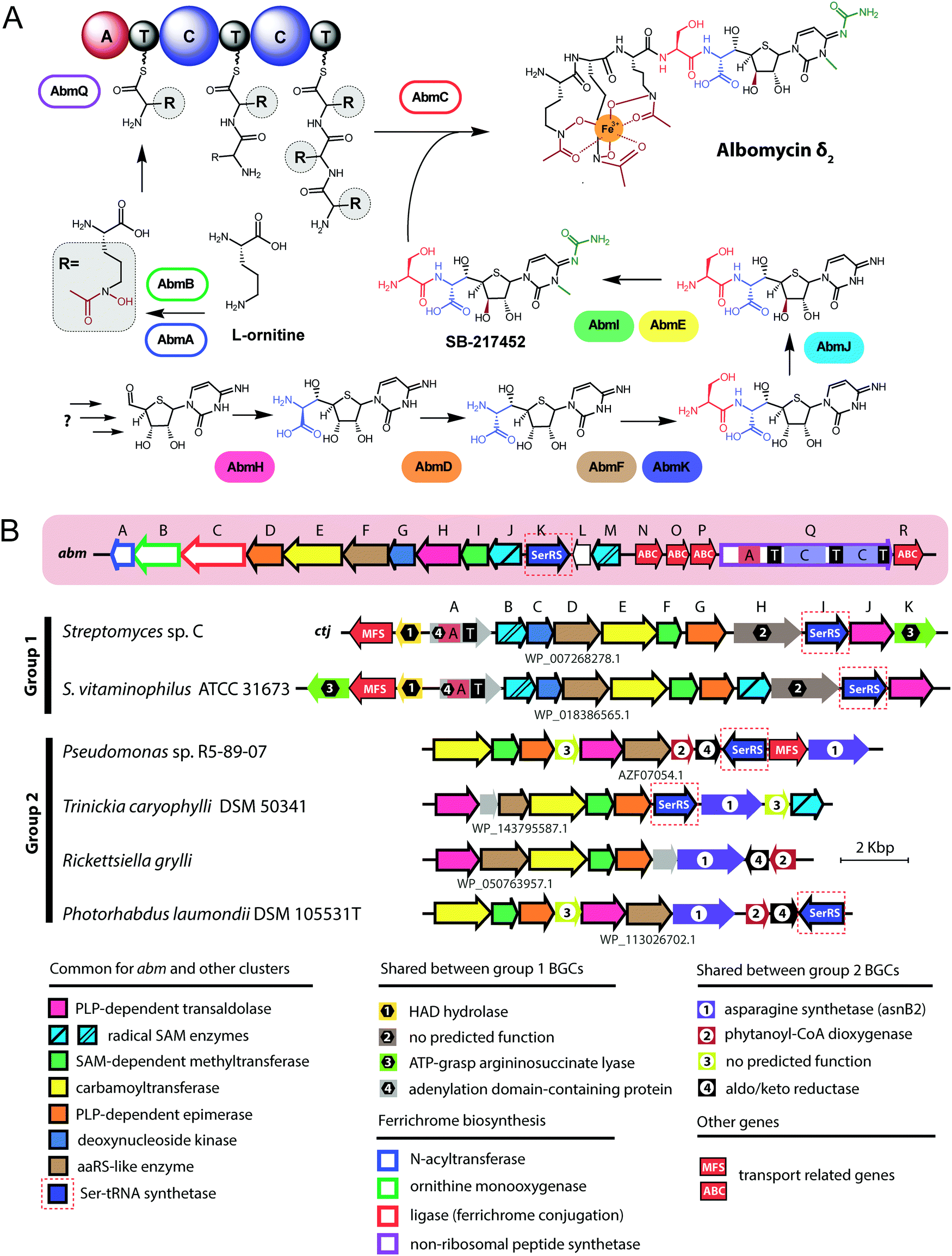 | ||
| Fig. 3 (A) Known steps in the biosynthesis of albomycin and the enzymes involved. Domains of NRPS AbmQ are indicated: A – adenylating domain, C – condensation domain, T – thiolation domain. Fragments of the molecule attached at different steps of the biosynthetic pathway are shown in different colors. (B) Biosynthetic gene cluster of albomycin (abm, red background) and found BGCs containing subsets of abm-like genes. Nomenclature of genes in the ctj gene cluster is from Zeng et al.38 The color scheme of enzymes in panel A matches that of genes in B. Predicted functions of the gene products are listed below. Protein IDs are shown for AbmF homologs. ABC – ATP binding cassette, CoA – coenzyme A, HAD – haloacid dehalogenase-like, MFS – major facilitator superfamily, PLP – pyridoxal phosphate, SAM – S-adenosyl methionine. | ||
The most remarkable part of the albomycin synthesis pathway is the formation of an amide bond between the thionucleoside and the L-serine, which results in SB-217452, a highly toxic compound. In vitro reconstruction of this step shows that AbmF uses seryl-adenylate produced by seryl-tRNA synthetases as a seryl donor for the amide bond formation, although a possibility that seryl-tRNA can also serve as an aminoacyl donor was not ruled out.47 Interestingly, the albomycin BGC encodes AbmK, which is a functional SerRS.49 Unlike the housekeeping SerRS, AbmK is insensitive to SB-217452 in vitro and confers resistance to albomycin in E. coli when overexpressed.49In vitro Streptomyces sp. ATCC 700974 housekeeping SerRS is substantially less efficient in the coupled reaction of thionucleoside biosynthesis since it is inhibited by the end product, SB-217452.47 The KM for serine of AbmK is ∼20 times higher than that of housekeeping SerRS.49 Thus, AbmK may serve a triple function in the producing strain – providing the substrate for albomycin biosynthesis,47 conferring immunity to the toxic compound produced,49 and, regulating albomycin biosynthesis by restricting it to conditions of high concentrations of intracellular serine.
The second group of putative seryl-thiofuranoside cytosine synthesizing BGCs was not previously described (Fig. 3B). Its representatives are found in the genomes of Proteobacteria from different classes. Despite major rearrangements in their architecture, these BGCs contain roughly the same set of genes. These include genes encoding cytosine-modifying carbamoyltransferase (AbmE-like), cytosine methyltransferase (AbmI-like), and thiofuranose-seryl assembly and epimerization enzymes homologous to AbmD, AbmH, and AbmF. The BGC from Trinickia caryophylli DSM 50341 also contains an abmJ ortholog. In contrast to albomycin BGCs, clusters from Proteobacteria entirely lack genes required for assembly of the siderophore part. This implies either a different mechanism of end product uptake by the sensitive cells or the coupling to a siderophore molecule whose biosynthesis is encoded elsewhere in the genome of the producer.
2. Microcin C and related compounds
Enterobacteriaceae such as Escherichia, Salmonella, and Shigella, and some strains of Klebsiella, are susceptible to McC at low micromolar concentrations; Pseudomonas and Gram-positive bacteria are resistant.58 McC may provide competitive advantage to producing strains in natural environments. Moreover, the probiotic activity of E. coli strain H22 in a mouse model was attributed to McC production.59
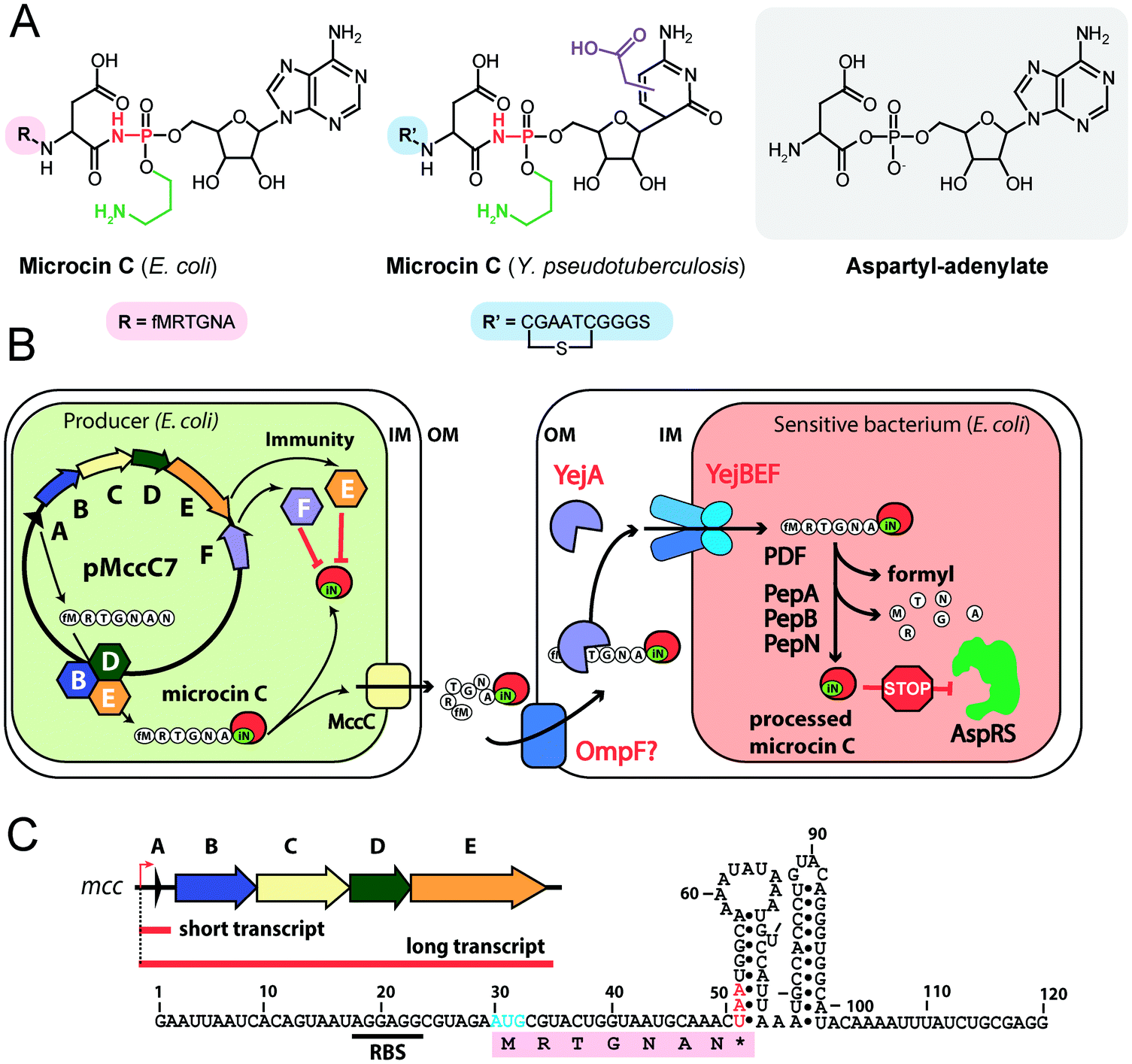 | ||
| Fig. 4 (A) Chemical structures of peptidyl-nucleotide antibiotics produced by Escherichia coli and Yersinia pseudotuberculosis. The nonhydrolyzable P–N bond is shown in red, the aminopropyl decoration is green, the carboxymethyl attached to the cytosine ring is violet. The peptide part is shown below in single-letter code. Aspartyl-adenylate is shown on the grey background for comparison. (B) Schematic representation of microcin C biosynthesis, transport and processing. PDF – peptide deformylase, OM – outer membrane, IM – inner membrane, AspRS – aspartyl-tRNA synthetase. (C) Regulation of microcin C production by an internal ribosome-dependent transcription terminator located between the mccA and mccB genes. The secondary structure of short mccA transcript is shown.61 Start codon is blue, stop codon is red. RBS – ribosome-binding site. | ||
All studied McC-like compounds share the Trojan horse mechanism of action. The peptide part serves as a vehicle to deliver the isoasparaginyl-nucleotide warhead inside the cell.11 The route of E. coli McC entrance into the cell is not fully established. Despite multiple mentions in the literature,65 no direct evidence that OmpF is the primary porin responsible for McC passage through the outer membrane of Gram-negative bacteria exists. Most probably, several porins are involved. Once E. coli McC reaches the periplasmic space it interacts with YejA, the substrate-binding subunit of the YejABEF ABC transporter (Fig. 4B).66 Though exact cellular function of this transporter in E. coli is not established, homologs from Salmonella, Brucella and Pseudomonas may be involved in resistance to antimicrobial peptides.67–70
Presumably, the fact that peptide parts of most McC-like compounds are seven amino acids long is explained, at least in part, by the binding preferences of YejA. Using chemically synthesized E. coli McC analogs it was shown that shortening the peptide length to six amino acids dramatically reduces bioactivity due to inefficient uptake through YejABEF.71 Increase of the peptide part length up to 10–12 amino acids stimulated the uptake.61 Peptidyl-nucleotides with the peptide part lengths around 20–25 amino acids can enter E. coli not only through YejABEF but also through SbmA,72 a transporter of peptide antibiotics such as microcin B17 (43 amino acids) or microcin J25 (21 amino acids).73,74 McC-like compounds with even longer peptide parts, i.e., the 42 amino acid long McC from Yersinia pseudotuberculosis (Fig. 4A), undergo endoproteolytic processing prior to export, which leads to bioactive compounds with shorter (11 amino acids in the case of Y. pseudotuberculosis McC) peptide parts.75
Once E. coli McC gets inside the bacterial cytoplasm, its peptide part is deformylated and six N-terminal residues are removed by aminopeptidases. In E. coli, any one of the three aminopeptidases, PepA, PepB, or PepN, is capable of intracellular McC processing (Fig. 4B).76 The final product of processing is an aminopropylated isoasparaginyl-adenylate. Processed E. coli McC mimics aspartyl-adenylate, the intermediate of the tRNAAsp aminoacylation reaction (Fig. 4A), and inhibits aspartyl-tRNA synthetase (AspRS).11 Inhibition of AspRS results in the accumulation of uncharged tRNAAsp, translation inhibition, stringent response and cessation of cell growth.11,77
Although the structures of AspRS in complex with several chemically synthesized analogs of processed McC have been determined,45 the exact binding mode of processed McC to AspRS is yet to be revealed. Given the variety of known and proposed (see below) auxiliary modifications in the nucleoside part of McC-like compounds, the catalytic center of AspRS should be flexible enough to accommodate structurally diverse aminoacyl-nucleotides.45
The minimal microcin C BGC (mcc) consists of just three genes: mccA, encoding the precursor peptide; mccB, coding for a ThiF-like nucleoside transferase; and mccC, encoding an export pump (Fig. 6 (1)). Auxiliary genes present in many mcc BGCs encode enzymes responsible for secondary post-translational modifications or self-immunity, however, the function of most of them is not identified yet (Fig. 6).
When the sequence of the E. coli mcc gene cluster was determined (Fig. 6 (2)), it became apparent that the mccA gene codes for a MRTGNA![[N with combining low line]](https://www.rsc.org/images/entities/b_char_004e_0332.gif) heptapeptide, while mature McC has the fMRTGNA
heptapeptide, while mature McC has the fMRTGNA![[i with combining low line]](https://www.rsc.org/images/entities/b_i_char_0069_0332.gif)
![[s with combining low line]](https://www.rsc.org/images/entities/b_i_char_0073_0332.gif)
![[o with combining low line]](https://www.rsc.org/images/entities/b_i_char_006f_0332.gif) peptide part.80 This discrepancy was resolved by work from the Walsh lab, when they showed that nucleoside coupling to the precursor peptide by adenylyltransferase MccB proceeds in two steps through a succinimide intermediate.81,82 The opening of the succinimide ring results in the isomerization of the asparagine residue, converting it to isoasparagine. The reaction consumes two molecules of ATP per molecule of peptide-nucleotide synthesized (Fig. 5B). While one would expect that a similar mechanism could be employed with peptide precursors containing C-terminal Gln, however, at least the E. coli MccB is unable to modify such a peptide.72 In fact, the presence of a C-terminal asparagine is the only strict requirement shared by known McC-like precursor peptides.
peptide part.80 This discrepancy was resolved by work from the Walsh lab, when they showed that nucleoside coupling to the precursor peptide by adenylyltransferase MccB proceeds in two steps through a succinimide intermediate.81,82 The opening of the succinimide ring results in the isomerization of the asparagine residue, converting it to isoasparagine. The reaction consumes two molecules of ATP per molecule of peptide-nucleotide synthesized (Fig. 5B). While one would expect that a similar mechanism could be employed with peptide precursors containing C-terminal Gln, however, at least the E. coli MccB is unable to modify such a peptide.72 In fact, the presence of a C-terminal asparagine is the only strict requirement shared by known McC-like precursor peptides.
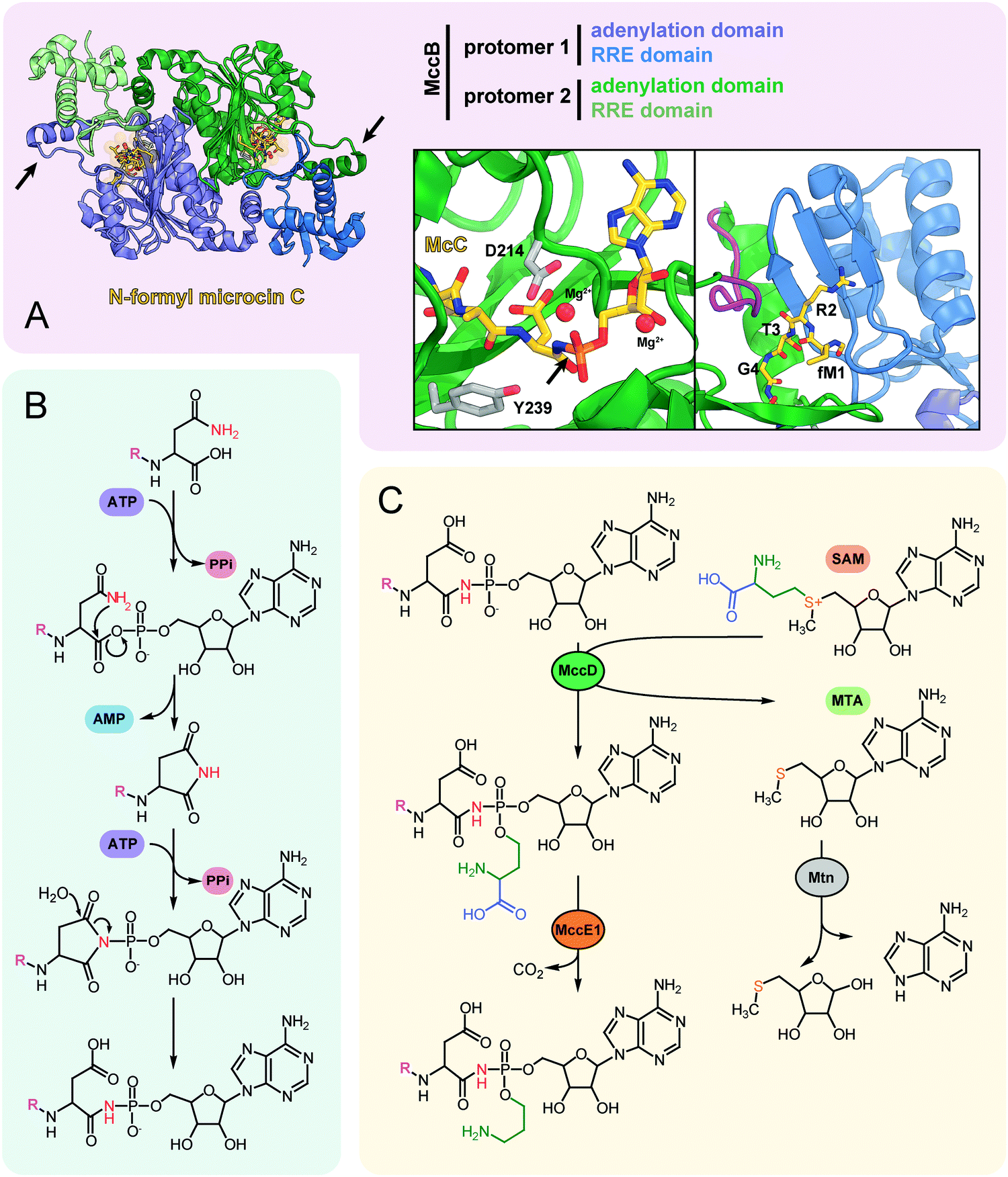 | ||
| Fig. 5 (A) The structure of the dimeric nucleoside transferase MccB from E. coli in complex with N-formylated microcin C (PDB ID: 6OM4).84 Adenylation domains and N-terminal RREs are shown in different colors, black arrows indicate “crossover loops”. Close-up views: (left) the active site of the adenylation domain with key residues required for efficient catalysis (D214 and Y239) is shown in stick representation, black arrow indicates the P-N bond; (right) the binding site of MccA peptide N-terminus formed by RRE and “crossover loop” (violet), residues 313–347 (catalytic domain) are removed for clarity. (B) The mechanism of P–N bond formation by nucleoside transferase MccB. R stands for the fMRTGNA peptide. Two molecules of ATP consumed per molecule of McC produced are shown in violet, released AMP is blue. (C) Installation of aminopropyl decoration by the MccD and MccE1 enzymes. SAM – S-adenosyl methionine, MTA – 5′-methylthioadenosine. | ||
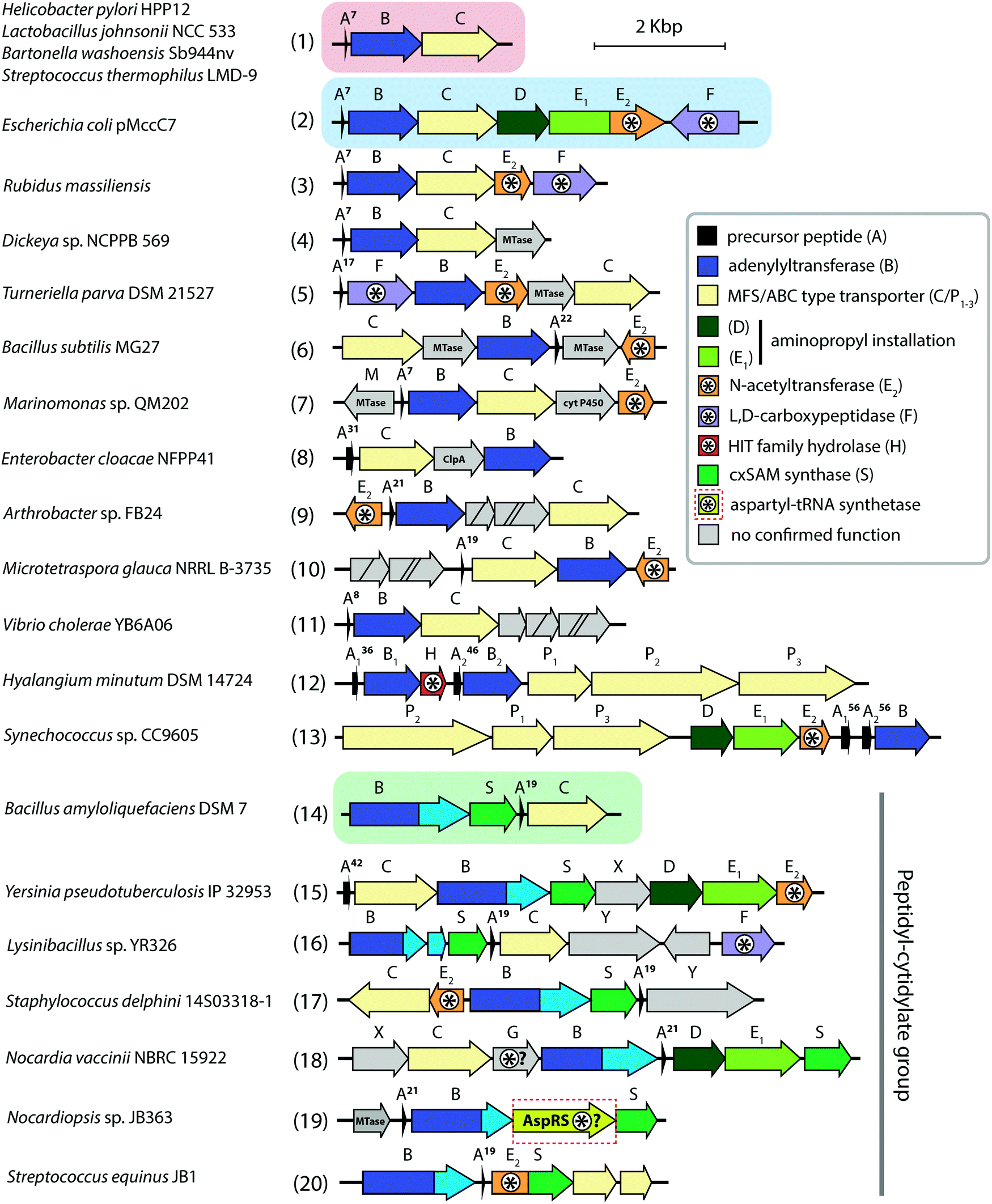 | ||
| Fig. 6 Biosynthetic gene clusters of microcin C-like compounds found across bacterial genomes. Clusters containing minimal sets of genes required for the production of peptidyl-adenylates and peptidyl-cytidylates are shown on red and green background, respectively. The archetypal and best-studied mcc BGC from the E. coli pMccC7 plasmid is shown on blue background. The number of amino acid residues in precursor peptides is indicated near the A genes. Genes involved (or proposed to be involved) in self-immunity are indicated with asterisks. Known functions for some gene products are listed in the right. Genes with no identified function are shown in grey. cyt P450 – cytochrome P450, HIT – histidine triad, MTase – methyltransferase. | ||
The crystal structure of MccB reveals a homodimer with two active centers (Fig. 5A). Both steps of the adenylation reaction are catalyzed within the same active center. The N-terminal part of one subunit (so-called ![[R with combining low line]](https://www.rsc.org/images/entities/char_0052_0332.gif) iPP ecognition
iPP ecognition ![[E with combining low line]](https://www.rsc.org/images/entities/char_0045_0332.gif) lement, “RRE”) and the central “crossover loop” of the opposite protomer form the peptide clamp domain responsible for MccA binding.82,83 The formyl group on the N-terminal methionine of the precursor heptapeptide strongly improves the efficiency of adenylation by promoting productive binding of the precursor peptide to MccB.84
lement, “RRE”) and the central “crossover loop” of the opposite protomer form the peptide clamp domain responsible for MccA binding.82,83 The formyl group on the N-terminal methionine of the precursor heptapeptide strongly improves the efficiency of adenylation by promoting productive binding of the precursor peptide to MccB.84
Strikingly, MccB is rather promiscuous in its choice of NTP substrates. In vitro, the E. coli precursor peptide can be modified with either AMP, CMP, GMP or UMP moieties. In vivo, the reaction is highly specific and, depending on the enzyme, exclusively yields either peptidyl-adenylates (e.g., when catalyzed by E. coli MccB) or peptidyl-cytidylates (e.g., when catalyzed by Bacillus amyloliquefaciens MccB).64 All known MccB enzymes catalyzing in vivo cytidylation contain a C-terminal methyltransferase domain responsible for the additional decoration of the nucleobase with carboxymethyl. This reaction requires carboxy-S-adenosyl methionine (cxSAM) as a substrate. The cxSAM is synthesized from S-adenosyl methionine (SAM) and prephenate by MccS, and enzyme encoded exclusively in BGCs producing cytidylated McC-like compounds.64,75 The exact position of the carboxymethyl group on the cytidine base remains unknown.
Auxiliary post-translational modifications of RiPPs increase their bioactivity and/or are required to overcome the immunity of target bacteria.85 The aforementioned aminopropyl decoration of the phosphate found in E. coli McC and in some other McC-like compounds serves both purposes: it increases the toxicity of peptidyl-nucleotides and allows to escape the action of some immunity proteins. Noteworthy, most Salmonella species are resistant to McC-like peptidyl-adenylates but sensitive to aminopropylated compounds.86 The aminopropyl is introduced via a two-step reaction.87 During the first step, a class I methyltransferase MccD transfers the 3-carboxy-3-aminopropyl moiety from SAM to the peptidyl-nucleotide substrate, releasing methylthioadenosine (MTA) as a byproduct (Fig. 5C). Since MTA is a potent inhibitor of SAM-binding enzymes, the reaction efficiently proceeds only in the presence of the Mtn nucleosidase, which is encoded in the core genome of many bacteria.88 The second step is decarboxylation of the 3-carboxy-3-aminopropylated peptidyl-nucleotide intermediate by MccE1 (in the case of E. coli – the N-terminal domain of a fused protein MccE) (Fig. 5C).75,87
E. coli McC is produced in the early stationary phase of growth, and its synthesis is tightly regulated.89 Transcription of the mccABCDE operon is initiated by the σS(σ38)-containing RNA polymerase90 in a CRP-dependent manner.91,92 A ribosome-dependent transcription terminator is commonly found in the mccA-mccB intergenic regions of mcc operons (Fig. 4C).61 In E. coli, the function of this terminator leads to the production of two types of mRNA molecules: a short one, which contains the mccA ORF only, and a long one, comprising the entire operon.61 Co-transcriptional recognition of the Shine–Dalgarno sequence of mccA by the ribosome stimulates transcription termination. The ribosome binding to short mRNA is virtually irreversible, which allows multiple rounds of translation without ribosome dissociation and ensures that enough peptide precursor substrate is produced for the action of the maturation enzymes.61
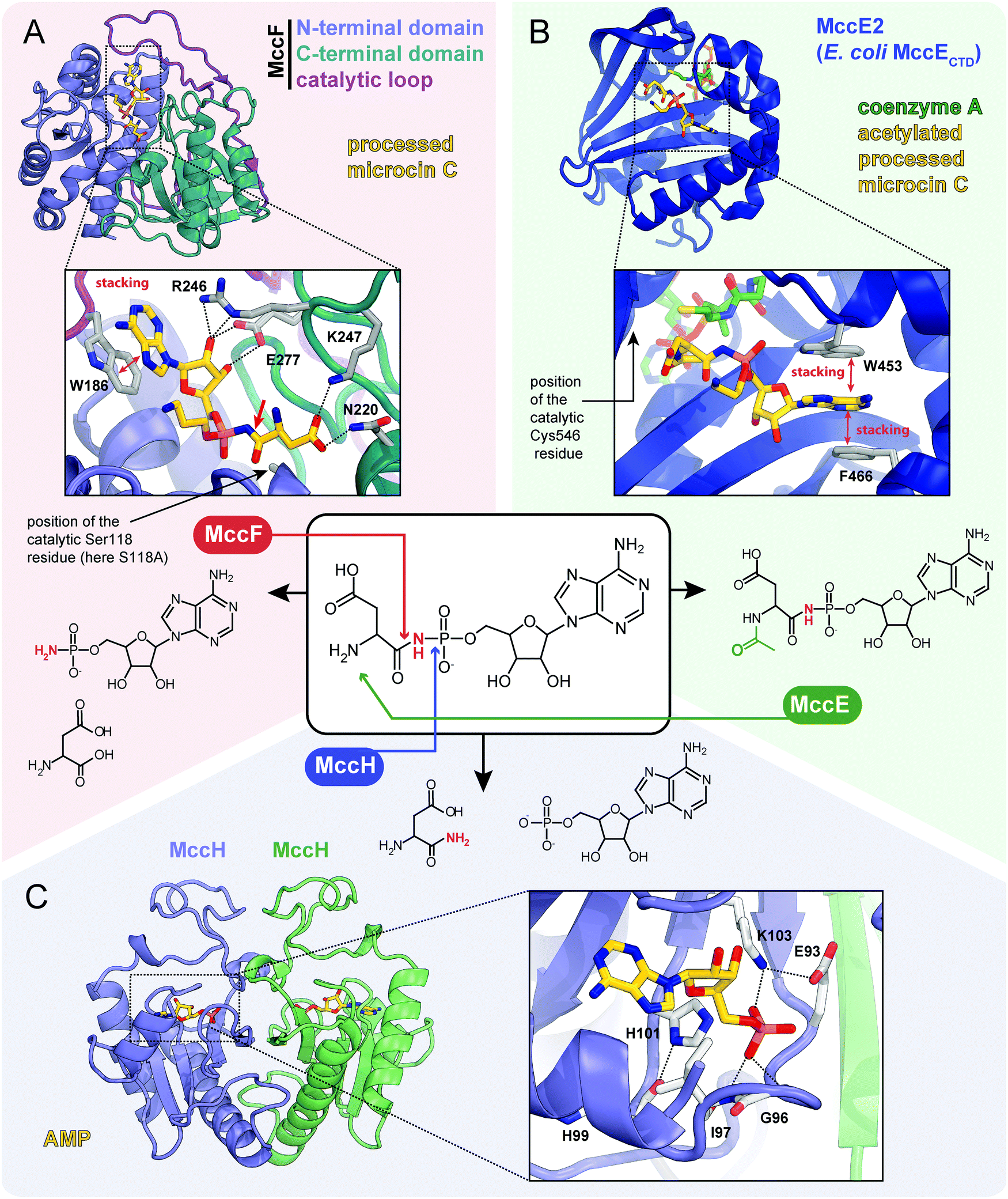 | ||
| Fig. 7 Proteins providing immunity to microcin C and their mechanisms of action. For each protein the structure, mode of substrate binding, and catalyzed reaction are shown. (A) The structure of E. coli carboxypeptidase MccF catalytical mutant (S118A) in complex with processed microcin C (PDB ID: 3TLC).138 Hydrogen bonds are shown as black dashed lines, π–π stacking between W186 and adenine nucleobase is indicated, big red arrow points to the peptide bond cleaved by MccF. (B) The structure of E. coli acetyltransferase MccE2 (C-terminal domain of MccE, residues 405–589) in complex with coenzyme A and acetylated processed microcin C (PDB ID: 3R9G).93 (C) The theoretical model of dimeric HIT-family hydrolase MccH from Hyalangium minutum in complex with AMP.86 | ||
The MccF protein encoded in the E. coli mcc cluster provides self-immunity by cleaving off the aspartate residue or the oligopeptide from, respectively, processed or mature McC (Fig. 7A).95 MccF is the S66 family serine peptidase96 and is similar to LdcA, a carboxypeptidase, which cleaves amide bonds between L- and D-amino acids during murein recycling.97 However, the latter enzyme is unable to protect E. coli from McC-like compounds. MccF homologs are encoded in several mcc-like BGCs (Fig. 6) and by numerous stand-alone bacterial genes.96 The stand-alone MccF ortholog from B. anthracis can hydrolyze peptidyl-adenylates and isoasparaginyl-adenylates with or without aminopropyl decoration and provides McC resistance to E. coli when overexpressed.96
Another immunity protein, MccH targets the phosphoramide linkage of isoasparaginyl-adenylates. It is a member of a vast family of Histidine-Triad Hydrolases (HITs). MccH is similar to highly conserved HinT phosphoramidases present in almost all organisms.98 The function of HinT is poorly understood and its natural substrates are unknown. HinT from E. coli or Hyalangium minutum are unable to hydrolyze isoasparaginyl-adenylate. In contrast, MccH, which was first identified in the mcc BGC from H. minutum, evolved to specifically cut the N-P bond in isoasparaginyl-adenylate, releasing AMP and isoasparagine (Fig. 7C).86 A number of bacterial MccH orthologs not linked to mcc-like BGCs were identified. When overexpressed, these enzymes provided resistance to simple McC-like peptidyl-adenylates but not to aminopropylated forms.86
As already mentioned, clusters encoding MccB proteins with C-terminal carboxymethyltransferase domains also encode cxSAM synthases MccS, which are absent from other mcc clusters. Experimentally validated products of such clusters are cytidine-containing antibiotics with carboxymethyl decoration installed by C-terminal carboxymethyltransferase domains of their MccBs (Fig. 6 (14–20), see also above).64 Two genes mccX (Fig. 6 (15, 18)) and mccY (Fig. 6 (16, 17)) are also found exclusively in this group of clusters, suggesting that the proteins they encode may specifically target the carboxymethylated cytosine nucleobase. While MccX has no sequence similarity with any proteins with established function,99 MccY is homologous to carbamoyl transferases. Similarly to AbmE from the albomycin biosynthesis pathway, MccY may add a carbamoyl decoration to the amino group of the carboxymethylated cytosine nucleobase.
SAM-dependent enzymes comprise the largest and most diverse group of tailoring enzymes encoded by mcc-like operons. Besides the already described 3-carboxy-3-aminopropyl transferase (MccD) and carboxymethyltransferase forming the C-terminal domain of some MccB proteins, genes encoding putative SAM-dependent transferases are identified in many mcc-like BGCs (Fig. 6 (4–6)). However, as there are no close homologs with known substrate specificity for these enzymes, secondary post-translational modifications they perform are impossible to predict.
Analysis of the mcc-like BGCs composition suggests that the self-immunity strategies employed by McC producers extend beyond those already discussed. Some BGCs encode AspN-like peptidases (Fig. 6 (18)) or ClpA homologs (Fig. 6 (8)), which may hydrolyze isoasparaginyl-nucleotides accumulating inside the producing cell. Several BGCs from Actinobacteria include asnS (aspartate tRNA synthetase) genes (Fig. 6 (19)), which, similarly to abmK from albomycin biosynthetic pathway (see above) or agnB2 from agrocin 84 BGC (see below) may provide resistance to processed McC-like compounds.
In summary, the abundance and diversity of known or putative strategies of avoidance of self-intoxication by intracellularly accumulating processed McC-like compounds parallels the diversity of secondary post-translational modifications of the basic peptide-nucleotide backbone and suggest that no single strategy is universal: additional decorations attached to the isoasparaginyl-nucleotide core allow some non-cognate McC-like compounds to overcome the immunity of specific producers.
3. Agrocin 84
The activity of R. rhizogenes K84 against tumor-inducing strains is due to the production of an antimicrobial compound agrocin 84, a disubstituted adenine nucleotide containing 3′-deoxyarabinose instead of the ribose.106,107 The two moieties attached to the nucleotide core via phosphoramide bonds are D-glucopyranose-2-phosphate at the N6 position of adenine and 2,3-dihydroxy-4-methylpentanamide at the phosphate group (Fig. 8A, left panel).108,109 The D-glucose-2-phosphate (Fig. 8A, red dashed frame) is required for the uptake by susceptible strains. Once the uptake moiety is removed, a toxic moiety (TM84, Fig. 8A, right panel) is released, making agrocin 84 a Trojan horse agent.110
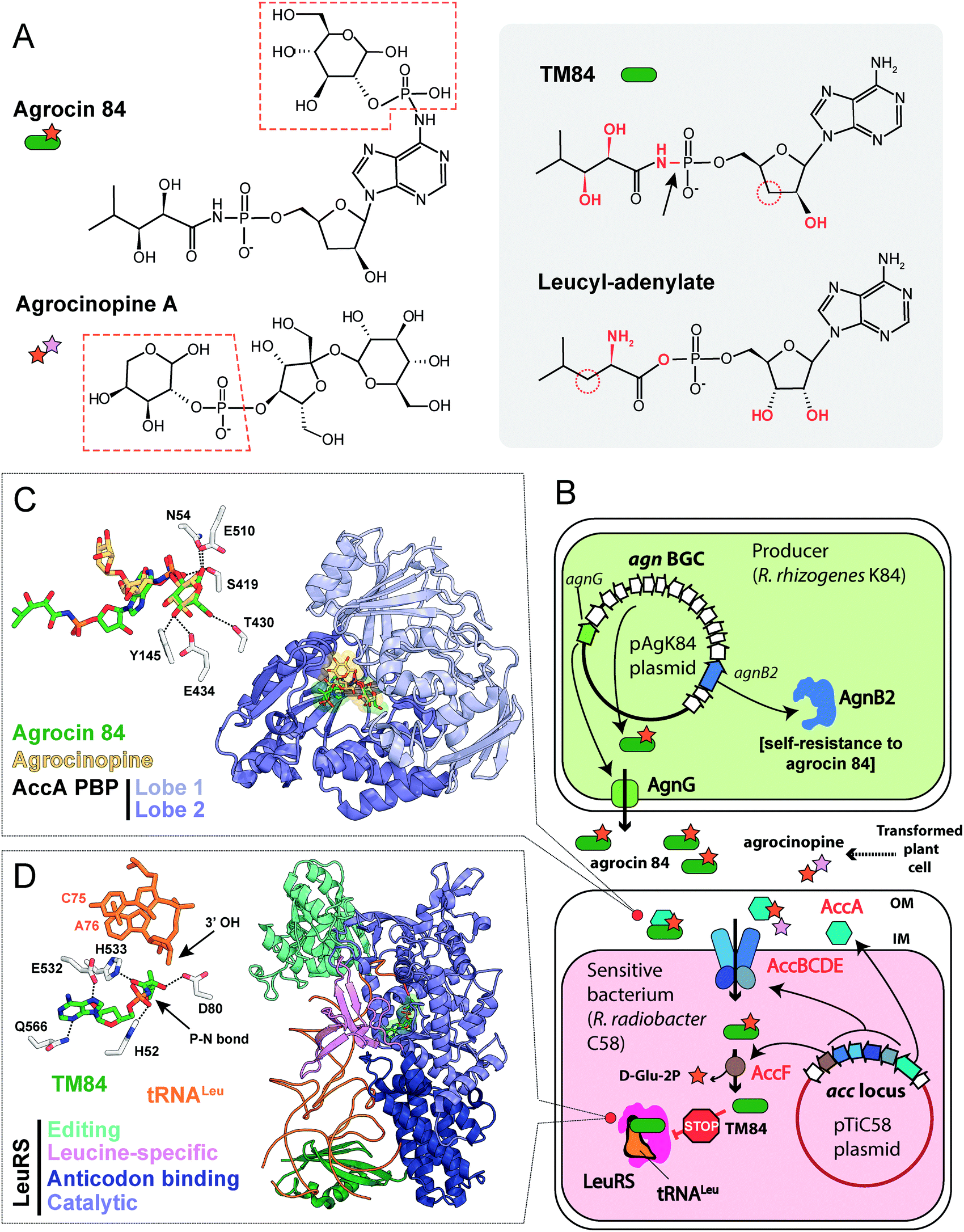 | ||
| Fig. 8 (A) Chemical structures of agrocin 84, its toxic moiety (TM84), agrocinopine A and leucyl-adenylate. Pyranose-2-phosphate groups shared by agrocin and agrocinopine A are highlighted with red dashed polygons. Groups of atoms that are different between TM84 and leucyl-adenylate are shown in red. The black arrow indicates a nonhydrolyzable P–N bond in TM84. (B) The scheme showing the biosynthesis, transport, and mode of action of agrocin 84. IM – inner membrane, OM – outer membrane, BGC – biosynthetic gene cluster, LeuRS – leucyl-tRNA synthetase. (C) The details of the interaction between agrocin 84/agrocinopine A with the periplasmic solute binding protein (PBP) AccA (PDB IDs: 4ZEC/4ZEB).109 Agrocinopine is coloured gold, agrocin 84 is green. Amino acid residues interacting with D-glucopyranose-2-phosphate moiety of agrocin are shown in the left corner. Hydrogen bonds are shown as black dashed lines. (D) The crystal structure of the ternary complex of LeuRS, TM84 and tRNALeu (PDB ID: 3ZGZ).118 Selected amino acid residues involved in the binding of TM84 in the active site of LeuRS are shown in the left corner together with the two 3′-terminal nucleotides of the tRNALeu acceptor stem. TM84 is green, tRNALeu is orange. | ||
Plant-pathogenic agrobacteria insert a specific region (T-DNA) derived from their Ti (![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) umor
umor ![[i with combining low line]](https://www.rsc.org/images/entities/char_0069_0332.gif) nducing) plasmids into the nuclear genome of the plant cell.111 Some T-DNA genes encode enzymes of the biosynthesis of opines – metabolites produced by the transformed plant cells and subsequently imported into the bacterial cells. These compounds serve as a source of carbon, phosphorus and nitrogen for the agrobacteria.112,113 The import of opines agrocinopines A and B is determined by the AccABCDE transporter encoded on the Ti plasmid (Fig. 8B). Agrocin 84 hijacks this transport system to get inside the cell of tumor inducing agrobacteria.114 Agrocinopine A is made of sucrose bound to L-arabinose-2-phosphate (Fig. 8A), while agrocinopine B has fructose instead of sucrose.115,116 Crystal structures (Fig. 8C) show that the periplasmic binding protein AccA recognizes both agrocin 84 and agrocinopine A through their pyranose-2-phosphate groups (D-glucose-2-phosphate in the case of agrocin and L-arabinose-2-phosphate in the case of agrocinopine, Fig. 8A, red dashed frames).109 The subsequent processing step required for the release of agrocin 84 toxic moiety is catalyzed by phosphodiesterase AccF (Fig. 8B), which also performs the first step of agrocinopines catabolism.114 Since the accABCDEFG (agrocinopine catabolism) locus is part of the Ti-plasmid, agrocin-producing R. rhizogenes strains specifically inhibit the growth of tumor inducing strains but have no effect on strains that do not induce tumors.106,117 In other words, by mimicking the tumor-derived substance, agrocin 84 is able to find its way specifically into phytopathogenic bacteria harboring the Ti-plasmid.
nducing) plasmids into the nuclear genome of the plant cell.111 Some T-DNA genes encode enzymes of the biosynthesis of opines – metabolites produced by the transformed plant cells and subsequently imported into the bacterial cells. These compounds serve as a source of carbon, phosphorus and nitrogen for the agrobacteria.112,113 The import of opines agrocinopines A and B is determined by the AccABCDE transporter encoded on the Ti plasmid (Fig. 8B). Agrocin 84 hijacks this transport system to get inside the cell of tumor inducing agrobacteria.114 Agrocinopine A is made of sucrose bound to L-arabinose-2-phosphate (Fig. 8A), while agrocinopine B has fructose instead of sucrose.115,116 Crystal structures (Fig. 8C) show that the periplasmic binding protein AccA recognizes both agrocin 84 and agrocinopine A through their pyranose-2-phosphate groups (D-glucose-2-phosphate in the case of agrocin and L-arabinose-2-phosphate in the case of agrocinopine, Fig. 8A, red dashed frames).109 The subsequent processing step required for the release of agrocin 84 toxic moiety is catalyzed by phosphodiesterase AccF (Fig. 8B), which also performs the first step of agrocinopines catabolism.114 Since the accABCDEFG (agrocinopine catabolism) locus is part of the Ti-plasmid, agrocin-producing R. rhizogenes strains specifically inhibit the growth of tumor inducing strains but have no effect on strains that do not induce tumors.106,117 In other words, by mimicking the tumor-derived substance, agrocin 84 is able to find its way specifically into phytopathogenic bacteria harboring the Ti-plasmid.
TM84, the toxic moiety of agrocin 84, inhibits tRNALeu aminoacylation by leucyl-tRNA synthetase (LeuRS, Fig. 8B).6 TM84 is a structural mimic of leucyl-adenylate (Fig. 8A, grey background) with a nonhydrolyzable phosphoramide bond (Fig. 8A, black arrow) connecting the nucleotide and the modified pentanamide. Unlike other stable leucyl-adenylate analogs, TM84 binds weakly to the LeuRS active site in the absence of tRNALeu. The 3’-terminal adenosine of tRNALeu forms a hydrogen bond with TM84, stabilizing the ternary complex (Fig. 8D).118
The exact pathway of agrocin 84 biosynthesis is unknown. The sequence of the pAgK84 plasmid reported by Kim et al.123 allowed the identification of a 17-gene agn biosynthetic cluster required for agrocin 84 production and immunity (Fig. 9, pink background). The function of several Agn proteins was proposed based on sequence similarity with known enzymes. AgnA harbors a conserved adenylation domain characteristic of aaRSs and was thus proposed to catalyze the key step in which the precursor of pentanamide moiety is attached to the adenosine monophosphate via the phosphoramide bond.123 Our analysis suggests that AgnC1 has remote structural similarity to prolyl-tRNA synthetases (according to Phyre2124 and HHpred125) and may thus be another candidate for the enzyme attaching the modified pentanamide to the nucleotide moiety. AgnB1, AgnC3 and AgnC7 are SAM-dependent enzymes proposed to be involved in modified pentanamide biosynthesis. The involvement of SAM in agrocin 84 biosynthesis is supported by an observation that R. rhizogenes K84 with mutations in the chromosomal achY gene encoding S-adenosyl-L-homocysteine (SAH) hydrolase (an enzyme involved in SAH recycling back to SAM) do not produce agrocin.126
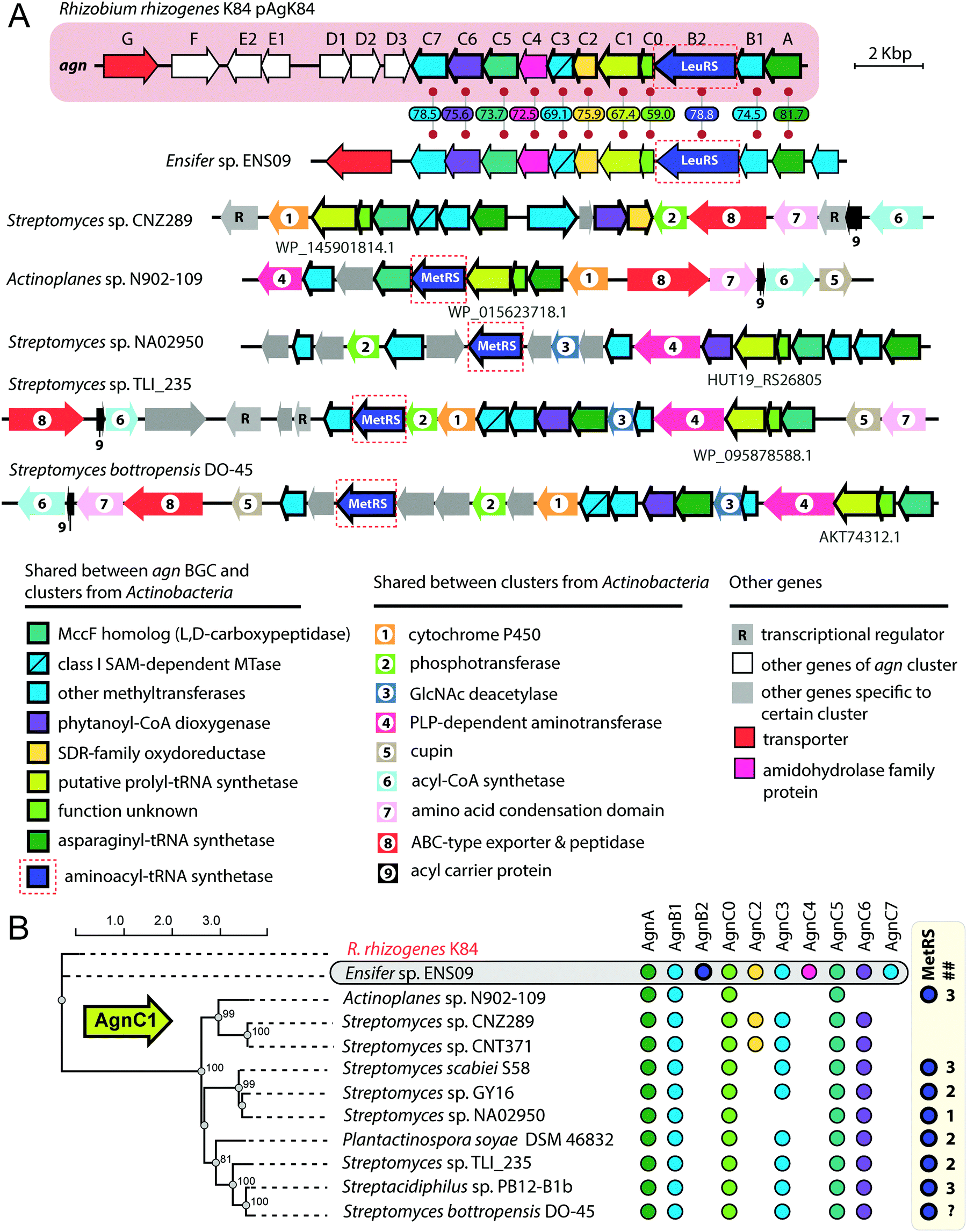 | ||
| Fig. 9 (A) Comparison of agrocin 84 (agn) biosynthetic gene cluster (pink background) with five clusters found in the genomes of Actinobacteria and a cluster from the genome of Ensifer sp. ENS09. The proposed functions of encoded proteins are listed below. Levels of sequence identity (in %) between Agn proteins and their homologs encoded by the cluster from Ensifer sp. ENS09 are indicated. Protein IDs are provided for AgnC1 homologs. ABC – ATP binding cassette, GlcNAc – N-acetylglucosamine, PLP – pyridoxal phosphate, SAM – S-adenosyl methionine, SDR – short-chain dehydrogenase/reductase. (B) Maximum likelihood phylogenetic tree of AgnC1 homologs, built using PhyML.139 Numbers at branching points indicate bootstrap support values from 100 replicates (scores above 60 are shown). The presence of agn genes homologs in corresponding BGCs is indicated by painted circles on the right. The number of distinct genes encoding putative MetRSs in the genome of the given strain is shown in the ## column. As the whole-genome sequence of the strain Streptomyces bottropensis DO-45 is missing, the number of MetRS genes is unknown (question mark in the ## column). | ||
The agn BGC (Fig. 9, pink background) includes two self-immunity genes.123agnG encodes a transmembrane pump involved in the export of the antibiotic; agnB2 encodes a leucyl-tRNA-synthetase. Compared to its housekeeping counterpart, the AgnB2 enzyme is ∼1000-fold less sensitive to TM84, providing the producer with alternative machinery for tRNALeu charging.6,127
Another interesting finding came from the results of a BLASTP search, which used the sequence of AgnC1 protein as a bait. In addition to trivial hits from R. rhizogenes K84 and Ensifer sp. ENS09, the top hits with E-values lower than 1e-8 contained sequences of 19 hypothetical proteins encoded in actinobacterial genomes. Manual analysis of the genomic context of agnC1 orthologs retrieved by the search revealed genes encoding homologs of other Agn proteins (Fig. 9B). Interestingly a 420-bp ORF preceding agnC1, which previously was not annotated as a gene,123 is present in BGCs from Ensifer and Actinobacteria, suggesting that it may encode a functional partner of AgnC1 (we annotated it as agnC0). We propose that identified actinobacterial BGCs encode the biosynthesis of nucleotide-containing natural products structurally close to agrocin 84. The newly identified BGCs share an additional set of genes not found in the agn BGC (Fig. 9A, genes with black numbers). The products of these genes may be responsible for additional novel modifications of the nucleotide scaffold. Strikingly, a gene encoding a methionyl-tRNA-synthetase (MetRS) is present in most actinobacterial BGCs (Fig. 9A, red dashed frames). In most of the studied cases, the gene of MetRS associated with the BGC is not the only MetRS encoding gene present in the genome (Fig. 9B). The product of this gene may play the same role as AgnB2, providing immunity to the produced compound. If true, this would imply that unlike agrocin 84, the products of some of the identified BGCs target the housekeeping MetRS.
Conclusions
Albomycin, microcin C-like compounds, and agrocin 84 are aaRSs inhibitors exploiting the “Trojan horse” strategy to enter cells. The transport moieties of these antibiotics mimic three distinct types of compounds actively imported into bacterial cells: a siderophore, peptides, and an opine. The spontaneous resistance to the Trojan horse inhibitors is almost exclusively associated with the loss of function of a dedicated transporter.32,66,131 Although the frequency of such resistance mutations is relatively high (within 10−4–10−5 range), it may not undermine the potential of Trojan horse inhibitors to be developed into drugs. For example, the loss of YejABEF or ferrichrome transporters reduces the virulence of bacteria and makes them prone to clearance by the immune system of the host.32,68,69 Likewise, the loss of the Ti-plasmid encoding AccABCDE transporter required for the internalization of agrocin 84, leads to a non-phytopathogenic phenotype.131Trojan horse inhibitors containing covalently bound siderophore moiety – sideromycins – have inspired researchers to develop numerous conjugates, each being a combination of a siderophore, a linker, and a known synthetic or natural antibiotic (see Negash et al.132 or Wencewicz & Miller133 for recent reviews of synthetic sideromycins). In McC-like compounds, the C-terminal residue (isoAsn) enzymatically attached to the nucleoside core through the N–P bond limits the specificity of processed compounds towards AspRS. However, McC analogs containing various terminal amino acids attached to adenosine-5′-sulfamoyl with nonhydrolyzable sulfonamide bond were obtained via total chemical synthesis. These compounds retain antibacterial activity and are potent inhibitors of corresponding aaRSs, thus expanding the targeting potential of natural McC variants and validating peptide-nucleotide scaffolds as a platform for the creation of new synthetic antibacterials.134,135
The results of our limited bioinformatics analyses show that the diversity of the three classes of molecules reviewed here is not yet completely tapped. When validated experimentally, these compounds may become viable antibiotics. Most interestingly, the products of putative agn-like BGCs revealed by our searches in the genomes of Actinobacteria may have different intracellular targets that the known compounds and thus could provide alternative means of biocontrol in agriculture.
Conflicts of interest
Authors declare no conflict of interests.Acknowledgements
The work was supported by RFBR grant No 20-34-90098 to DYT and NIH AI1172190 to Satish K. Nair and KS. We would like to thank Dr Daria Tsibulskaya and Dr Marina Serebryakova for invaluable help and Dr Peter Mergaert, Dr Solange Morera, Konstantin Gilep and Dr Alexey Kulikovsky for critical reading of the manuscript and discussions.References
- L. D. Sherlin and J. J. Perona, Structure, 2003, 11, 591–603 CrossRef CAS
.
- M. A. Rubio Gomez and M. Ibba, RNA, 2020, 26, 910–936 CrossRef
- C. M. Thomas, J. Hothersall, C. L. Willis and T. J. Simpson, Nat. Rev. Microbiol., 2010, 8, 281–289 CrossRef CAS
- V. Jain, M. Yogavel, Y. Oshima, H. Kikuchi, B. Touquet, M.-A. Hakimi and A. Sharma, Structure, 2015, 23, 819–829 CrossRef CAS
- S. Kirillov, L. A. Vitali, B. P. Goldstein, F. Monti, Y. Semenkov, V. Makhno, S. Ripa, C. L. Pon and C. O. Gualerzi, RNA, 1997, 3, 905–913 CAS
- J. S. Reader, P. T. Ordoukhanian, J.-G. Kim, V. de Crécy-Lagard, I. Hwang, S. Farrand and P. Schimmel, Science, 2005, 309, 1533 CrossRef CAS
- A. Ogilvie, K. Wiebauer and W. Kersten, Biochem. J., 1975, 152, 511–515 CrossRef CAS
- M. J. Brown, P. S. Carter, A. S. Fenwick, A. P. Fosberry, D. W. Hamprecht, M. J. Hibbs, R. L. Jarvest, L. Mensah, P. H. Milner, P. J. O’Hanlon, A. J. Pope, C. M. Richardson, A. West and D. R. Witty, Bioorg. Med. Chem. Lett., 2002, 12, 3171–3174 CrossRef CAS
- C. J. Schulze, W. M. Bray, F. Loganzo, M.-H. Lam, T. Szal, A. Villalobos, F. E. Koehn and R. G. Linington, J. Nat. Prod., 2014, 77, 2570–2574 CrossRef CAS
- G. Dirheimer and E. E. Creppy, IARC Sci. Publ., 1991, 171–186 CAS
- A. Metlitskaya, T. Kazakov, A. Kommer, O. Pavlova, M. Praetorius-Ibba, M. Ibba, I. Krasheninnikov, V. Kolb, I. Khmel and K. Severinov, J. Biol. Chem., 2006, 281, 18033–18042 CrossRef CAS
- T. A. Scott, S. F. D. Batey, P. Wiencek, G. Chandra, S. Alt, C. S. Francklyn and B. Wilkinson, ACS Chem. Biol., 2019, 14, 2663–2671 CrossRef CAS
- M. Uramoto, C. J. Kim, K. Shin-Ya, H. Kusakabe, K. Isono, D. R. Phillips and J. A. McCloskey, J. Antibiot., 1991, 44, 375–381 CrossRef CAS
- J. O. Capobianco, D. Zakula, M. L. Coen and R. C. Goldman, Biochem. Biophys. Res. Commun., 1993, 190, 1037–1044 CrossRef CAS
- A. L. Stefanska, M. Fulston, C. S. Houge-Frydrych, J. J. Jones and S. R. Warr, J. Antibiot., 2000, 53, 1346–1353 CrossRef CAS
- S. Robles, Y. Hu, T. Resto, F. Dean and J. M. Bullard, Curr. Drug Discovery Technol., 2017, 14, 156–168 CAS
- A. L. Stefanska, N. J. Coates, L. M. Mensah, A. J. Pope, S. J. Ready and S. R. Warr, J. Antibiot., 2000, 53, 345–350 CrossRef CAS
- P. Fang, H. Han, J. Wang, K. Chen, X. Chen and M. Guo, Chem. Biol., 2015, 22, 734–744 CrossRef CAS
- T. L. Williams, Y. W. Yin and C. W. Carter, J. Biol. Chem., 2016, 291, 255–265 CrossRef CAS
- T. Nakama, O. Nureki and S. Yokoyama, J. Biol. Chem., 2001, 276, 47387–47393 CrossRef CAS
- H. Zhou, L. Sun, X.-L. Yang and P. Schimmel, Nature, 2013, 494, 121–124 CrossRef CAS
- R. V. K. Cochrane, A. K. Norquay and J. C. Vederas, MedChemComm, 2016, 7, 1535–1545 RSC
- S. Chopra and J. Reader, Int. J. Mol. Sci., 2014, 16, 321–349 CrossRef
- J. M. Ho, E. Bakkalbasi, D. Söll and C. A. Miller, RNA Biol., 2018, 15, 667–677 CrossRef
- H. I. Zgurskaya, C. A. Löpez and S. Gnanakaran, ACS Infect. Dis., 2015, 1, 512–522 CrossRef CAS
- J. O. Capobianco, C. C. Doran and R. C. Goldman, Antimicrob. Agents Chemother., 1989, 33, 156–163 CrossRef CAS
- R. G. Werner, Antimicrob. Agents Chemother., 1980, 18, 858–862 CrossRef CAS
- D. M. Reynolds, A. Schatz and S. A. Waksman, Proc. Soc. Exp. Biol. Med., 1947, 64, 50–54 CrossRef CAS
- G. F. Gause and M. G. Brazhnikova, Nov. Med, 1951, 3–7 Search PubMed
- E. O. Stapley and R. E. Ormond, Science, 1957, 125, 587–589 CrossRef CAS
- H. Maehr, Pure Appl. Chem., 1971, 28, 603–636 CAS
- A. Pramanik, U. H. Stroeher, J. Krejci, A. J. Standish, E. Bohn, J. C. Paton, I. B. Autenrieth and V. Braun, Int. J. Med. Microbiol., 2007, 297, 459–469 CrossRef CAS
- D. M. Reynolds and S. A. Waksman, J. Bacteriol., 1948, 55, 739–752 CrossRef CAS
- G. F. Gause, BMJ, 1955, 2, 1177–1179 CrossRef CAS
- Z. Lin, X. Xu, S. Zhao, X. Yang, J. Guo, Q. Zhang, C. Jing, S. Chen and Y. He, Nat. Commun., 2018, 9, 3445 CrossRef
- A. Pramanik and V. Braun, J. Bacteriol., 2006, 188, 3878–3886 CrossRef CAS
- G. Benz, T. Schröder, J. Kurz, C. Wünsche, W. Karl, G. Steffens, J. Pfitzner and D. Schmidt, Angew. Chem., Int. Ed. Engl., 1982, 21, 527–528 CrossRef
- Y. Zeng, A. Kulkarni, Z. Yang, P. B. Patil, W. Zhou, X. Chi, S. Van Lanen and S. Chen, ACS Chem. Biol., 2012, 7, 1565–1575 CrossRef CAS
- A. Hartmann, H. P. Fiedler and V. Braun, Eur. J. Biochem., 1979, 99, 517–524 CrossRef CAS
- A. D. Ferguson, J. W. Coulton, K. Diederichs, W. Welte, V. Braun and H.-P. Fiedler, Protein Sci., 2000, 9, 956–963 CrossRef CAS
- R. Chakraborty, E. Storey and D. van der Helm, Biometals, 2007, 20, 263–274 CrossRef CAS
- M. R. Rohrbach, V. Braun and W. Köster, J. Bacteriol., 1995, 177, 7186–7193 CrossRef CAS
- T. E. Clarke, V. Braun, G. Winkelmann, L. W. Tari and H. J. Vogel, J. Biol. Chem., 2002, 277, 13966–13972 CrossRef CAS
- V. Braun, K. Günthner, K. Hantke and L. Zimmermann, J. Bacteriol., 1983, 156, 308–315 CrossRef CAS
- L. Pang, M. Nautiyal, S. De Graef, B. Gadakh, V. Zorzini, A. Economou, S. V. Strelkov, A. Van Aerschot and S. D. Weeks, ACS Chem. Biol., 2020, 15, 407–415 CrossRef CAS
- A. Saha, S. Dutta and N. Nandi, J. Biomol. Struct. Dyn., 2020, 38, 2440–2454 CrossRef CAS
- R. Ushimaru, Z. Chen, H. Zhao, P. Fan and H. Liu, Angew. Chem., Int. Ed., 2020, 59, 3558–3562 CrossRef CAS
- R. Ushimaru and H. Liu, J. Am. Chem. Soc., 2019, 141, 2211–2214 CrossRef CAS
- Y. Zeng, H. H. Roy, P. B. Patil, M. Ibba and S. Chen, Antimicrob. Agents Chemother., 2009, 53, 4619–4627 CrossRef CAS
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers and D. J. Lipman, J. Mol. Biol., 1990, 215, 403–410 CrossRef CAS
- R. Ushimaru, Z. Chen, H. Zhao, P. Fan and H. Liu, Angew. Chem., Int. Ed., 2020, 59, 3558–3562 CrossRef CAS
- F. Baquero, D. Bouanchaud, M. C. Martinez-Perez and C. Fernandez, J. Bacteriol., 1978, 135, 342–347 CrossRef CAS
- Y. Chabbert, Ann. Inst. Pasteur., 1950, 79, 51–59 CAS
- N. E. Kurepina, E. I. Basyuk, A. Z. Metlitskaya, D. A. Zaitsev and I. A. Khmel, Mol. Gen. Genet., 1993, 241, 700–706 CrossRef CAS
- D. Smajs, M. Strouhal, P. Matejková, D. Cejková, L. Cursino, E. Chartone-Souza, J. Smarda and A. M. A. Nascimento, Plasmid, 2008, 59, 1–10 CrossRef CAS
- D. M. Gordon and C. L. O’Brien, Microbiology, 2006, 152, 3239–3244 CrossRef CAS
- L. Micenková, J. Bosák, M. Vrba, A. Ševčíková and D. Šmajs, BMC Microbiol., 2016, 16, 218 CrossRef
- J. F. Garcia-Bustos, N. Pezzi and E. Mendez, Antimicrob. Agents Chemother., 1985, 27, 791–797 CrossRef CAS
- L. Cursino, D. Smajs, J. Smarda, R. M. D. Nardi, J. R. Nicoli, E. Chartone-Souza and A. M. A. Nascimento, J. Appl. Microbiol., 2006, 100, 821–829 CrossRef CAS
- D. Hofreuter and R. Haas, J. Bacteriol., 2002, 184, 2755–2766 CrossRef CAS
- I. Zukher, M. Pavlov, D. Tsibulskaya, A. Kulikovsky, T. Zyubko, D. Bikmetov, M. Serebryakova, S. K. Nair, M. Ehrenberg, S. Dubiley and K. Severinov, mBio, 2019, 10, 11891–11902 CrossRef
- J. I. Guijarro, J. E. González-Pastor, F. Baleux, J. L. San Millán, M. A. Castilla, M. Rico, F. Moreno and M. Delepierre, J. Biol. Chem., 1995, 270, 23520–23532 CrossRef CAS
- O. Bantysh, M. Serebryakova, K. S. Makarova, S. Dubiley, K. A. Datsenko and K. Severinov, mBio, 2014, 5, e01059 CrossRef
- M. Serebryakova, D. Tsibulskaya, O. Mokina, A. Kulikovsky, M. Nautiyal, A. Van Aerschot, K. Severinov and S. Dubiley, J. Am. Chem. Soc., 2016, 138, 15690–15698 CrossRef CAS
- J. E. González-Pastor, J. L. San Millán, M. A. Castilla, F. Moreno, J. E. Gonzalez-Pastor, J. L. San Millan, M. A. Castilla, F. Moreno, J. E. González-Pastor, J. L. San Millán, M. A. Castilla and F. Moreno, J. Bacteriol., 1995, 177, 7131–7140 CrossRef
- M. Novikova, A. Metlitskaya, K. Datsenko, T. Kazakov, A. Kazakov, B. Wanner and K. Severinov, J. Bacteriol., 2007, 189, 8361–8365 CrossRef CAS
- U. Qimron, N. Madar, H.-W. Mittrücker, A. Zilka, I. Yosef, N. Bloushtain, S. H. E. Kaufmann, I. Rosenshine, R. N. Apte and A. Porgador, Cell. Microbiol., 2004, 6, 1057–1070 CrossRef CAS
- S. M. Eswarappa, K. K. Panguluri, M. Hensel and D. Chakravortty, Microbiology, 2008, 154, 666–678 CrossRef CAS
- Z. Wang, P. Bie, J. Cheng, L. Lu, B. Cui and Q. Wu, Sci. Rep., 2016, 6, 31876 CrossRef CAS
- D. Pletzer, Y. Braun, S. Dubiley, C. Lafon, T. Köhler, M. G. P. Page, M. Mourez, K. Severinov and H. Weingart, J. Bacteriol., 2015, 197, 2217–2228 CrossRef CAS
- G. H. M. Vondenhoff, B. Blanchaert, S. Geboers, T. Kazakov, K. A. Datsenko, B. L. Wanner, J. Rozenski, K. Severinov and A. Van Aerschot, J. Bacteriol., 2011, 193, 3618–3623 CrossRef CAS
- O. Bantysh, M. Serebryakova, I. Zukher, A. Kulikovsky, D. Tsibulskaya, S. Dubiley and K. Severinov, J. Bacteriol., 2015, 197, 3133–3141 CrossRef CAS
- R. A. Salomón and R. N. Farías, J. Bacteriol., 1995, 177, 3323–3325 CrossRef
- M. Laviña, A. P. Pugsley and F. Moreno, J. Gen. Microbiol., 1986, 132, 1685–1693 Search PubMed
- D. Tsibulskaya, O. Mokina, A. Kulikovsky, J. Piskunova, K. Severinov, M. Serebryakova and S. Dubiley, J. Am. Chem. Soc., 2017, 139, 16178–16187 CrossRef CAS
- T. Kazakov, G. H. Vondenhoff, K. A. Datsenko, M. Novikova, A. Metlitskaya, B. L. Wanner and K. Severinov, J. Bacteriol., 2008, 190, 2607–2610 CrossRef CAS
- J. Piskunova, E. Maisonneuve, E. Germain, K. Gerdes and K. Severinov, Mol. Microbiol., 2017, 104, 463–471 CrossRef CAS
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K.-D. Entian, M. A. Fischbach, J. S. Garavelli, U. Göransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Müller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. T. Reaney, S. Rebuffat, R. P. Ross, H.-G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G.-L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC
- T. J. Oman and W. a van der Donk, Nat. Chem. Biol., 2010, 6, 9–18 CrossRef CAS
- J. E. González-Pastor, J. L. San Millán and F. Moreno, Nature, 1994, 369, 281 CrossRef
- R. F. Roush, E. M. Nolan, F. Löhr and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 3603–3609 CrossRef CAS
- C. A. Regni, R. F. Roush, D. J. Miller, A. Nourse, C. T. Walsh and B. A. Schulman, EMBO J., 2009, 28, 1953–1964 CrossRef CAS
- B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, Nat. Chem. Biol., 2015, 11, 564–570 CrossRef CAS
- S.-H. Dong, A. Kulikovsky, I. Zukher, P. Estrada, S. Dubiley, K. Severinov and S. K. Nair, Chem. Sci., 2019, 10, 2391–2395 RSC
- M. A. Funk and W. A. van der Donk, Acc. Chem. Res., 2017, 50, 1577–1586 CrossRef CAS
- E. Yagmurov, D. Tsibulskaya, A. Livenskyi, M. Serebryakova, Y. I. Wolf, S. Borukhov, K. Severinov and S. Dubiley, mBio, 2020, 11, e00497-20 CrossRef
- A. Kulikovsky, M. Serebryakova, O. Bantysh, A. Metlitskaya, S. Borukhov, K. Severinov and S. Dubiley, J. Am. Chem. Soc., 2014, 136, 11168–11175 CrossRef CAS
- N. Parveen and K. A. Cornell, Mol. Microbiol., 2011, 79, 7–20 CrossRef CAS
- J. F. García-Bustos, N. Pezzi and C. Asensio, Biochem. Biophys. Res. Commun., 1984, 119, 779–785 CrossRef
- L. Díaz-Guerra, F. Moreno and J. L. San Millán, J. Bacteriol., 1989, 171, 2906–2908 CrossRef
- F. Moreno, J. E. Gónzalez-Pastor, M. R. Baquero and D. Bravo, Biochimie, 2002, 84, 521–529 CrossRef CAS
- D. Fomenko, A. Veselovskii and I. Khmel, Res. Microbiol., 2001, 152, 469–479 CrossRef CAS
- V. Agarwal, A. Metlitskaya, K. Severinov and S. K. Nair, J. Biol. Chem., 2011, 286, 21295–21303 CrossRef CAS
- M. Novikova, T. Kazakov, G. H. Vondenhoff, E. Semenova, J. Rozenski, A. Metlytskaya, I. Zukher, A. Tikhonov, A. Van Aerschot and K. Severinov, J. Biol. Chem., 2010, 285, 12662–12669 CrossRef CAS
- A. Tikhonov, T. Kazakov, E. Semenova, M. Serebryakova, G. Vondenhoff, A. Van Aerschot, J. S. Reader, V. M. Govorun and K. Severinov, J. Biol. Chem., 2010, 285, 37944–37952 CrossRef CAS
- B. Nocek, A. Tikhonov, G. Babnigg, M. Gu, M. Zhou, K. S. Makarova, G. Vondenhoff, A. Van Aerschot, K. Kwon, W. F. Anderson, K. Severinov and A. Joachimiak, J. Mol. Biol., 2012, 420, 366–383 CrossRef CAS
- M. F. Templin, A. Ursinus and J. V. Höltje, EMBO J., 1999, 18, 4108–4117 CrossRef CAS
-
C. Brenner, eLS, John Wiley & Sons, Ltd, Chichester, UK, 2014, pp. 1–8 Search PubMed
- D. Tsibulskaya, O. Mokina, A. Kulikovsky, J. Piskunova, K. Severinov, M. Serebryakova and S. Dubiley, J. Am. Chem. Soc., 2017, 139, 16178–16187 CrossRef CAS
- J. Pulawska, J. Plant Pathol., 2010, 92, 87–98 Search PubMed
- E. Velázquez, J. L. Palomo, R. Rivas, H. Guerra, A. Peix, M. E. Trujillo, P. García-Benavides, P. F. Mateos, H. Wabiko and E. Martínez-Molina, Syst. Appl. Microbiol., 2010, 33, 247–251 CrossRef
- P. B. New and A. Kerr, J. Appl. Bacteriol., 1972, 35, 279–287 CrossRef
- A. Kerr, J. Appl. Bacteriol., 1972, 35, 493–497 CrossRef
- K. Htay and A. Kerr, J. Appl. Bacteriol., 1974, 37, 525–530 CrossRef CAS
- A. Kerr and G. Bullard, Agronomy, 2020, 10, 1126 CrossRef CAS
- A. Kerr and K. Htay, Physiol. Plant Pathol., 1974, 4, 37–44 CrossRef CAS
- W. P. Roberts, M. E. Tate and A. Kerr, Nature, 1977, 265, 379–381 CrossRef CAS
- M. E. Tate, P. J. Murphy, W. P. Roberts and A. Kerr, Nature, 1979, 280, 697–699 CrossRef CAS
- A. El Sahili, S.-Z. Li, J. Lang, C. Virus, S. Planamente, M. Ahmar, B. G. Guimaraes, M. Aumont-Nicaise, A. Vigouroux, L. Soulère, J. Reader, Y. Queneau, D. Faure and S. Moréra, PLoS Pathog., 2015, 11, e1005071 CrossRef
- P. J. Murphy, M. E. Tate and A. Kerr, Eur. J. Biochem., 1981, 115, 539–543 CrossRef CAS
- H.-H. Hwang, M. Yu and E.-M. Lai, Arab. B, 2017, 15, e0186 CrossRef
- E. W. Nester, Front. Plant Sci., 2015, 5, 1–16 Search PubMed
- I. A. Vladimirov, T. V. Matveeva and L. A. Lutova, Russ. J. Genet., 2015, 51, 121–129 CrossRef CAS
- H. Kim and S. K. Farrand, J. Bacteriol., 1997, 179, 7559–7572 CrossRef CAS
- J. G. Ellis and P. J. Murphy, MGG Mol. Gen. Genet., 1981, 181, 36–43 CrossRef CAS
- M. H. Ryder, M. E. Tate and G.
P. Jones, J. Biol. Chem., 1984, 259, 9704–9710 CrossRef CAS
- W. P. Roberts and A. Kerr, Physiol. Plant Pathol., 1974, 4, 81–91 CrossRef CAS
- S. Chopra, A. Palencia, C. Virus, A. Tripathy, B. R. Temple, A. Velazquez-Campoy, S. Cusack and J. S. Reader, Nat. Commun., 2013, 4, 1417 CrossRef
- J. G. Ellis, A. Kerr, M. Van Montagu and J. Schell, Physiol. Plant Pathol., 1979, 15, 311–319 CrossRef
- J. E. Slota and S. K. Farrand, Plasmid, 1982, 8, 175–186 CrossRef CAS
- D. A. Jones, M. H. Ryder, B. G. Clare, S. K. Farrand and A. Kerr, Mol. Gen. Genet. MGG, 1988, 212, 207–214 CrossRef CAS
- B. Vicedo, R. Peñalver, M. J. Asins and M. M. López, Appl. Environ. Microbiol., 1993, 59, 309–315 CrossRef CAS
- J.-G. Kim, B. K. Park, S.-U. Kim, D. Choi, B. H. Nahm, J. S. Moon, J. S. Reader, S. K. Farrand and I. Hwang, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8846–8851 CrossRef CAS
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. E. Sternberg, Nat. Protoc., 2015, 10, 845–858 CrossRef CAS
- L. Zimmermann, A. Stephens, S.-Z. Nam, D. Rau, J. Kübler, M. Lozajic, F. Gabler, J. Söding, A. N. Lupas and V. Alva, J. Mol. Biol., 2018, 430, 2237–2243 CrossRef CAS
- R. Penyalver, P. M. Oger, S. Su, B. Alvarez, C. I. Salcedo, M. M. López and S. K. Farrand, Mol. Plant. Microbe. Interact., 2009, 22, 713–724 CrossRef CAS
- S. Chopra, A. Palencia, C. Virus, S. Schulwitz, B. R. Temple, S. Cusack and J. Reader, Nat. Commun., 2016, 7, 12928 CrossRef CAS
- S. C. Donner, D. A. Jones, N. C. McClure, G. M. Rosewarne, M. E. Tate, A. Kerr, N. N. Fajardo and B. G. Clare, Physiol. Mol. Plant Pathol., 1993, 42, 185–194 CrossRef CAS
-
S. Donner, PhD thesis, The University of Adelaide, Australia, 1997 Search PubMed
- C. M. Elvin, R. E. Asenstorfer, M. H. Ryder, S. C. Donner, G. P. Jones and M. E. Tate, J. Antibiot., 2018, 71, 438–446 CrossRef CAS
- D. A. Cooksey and L. W. Moore, Physiol. Plant Pathol., 1982, 20, 129–135 CrossRef
- K. H. Negash, J. K. S. Norris and J. T. Hodgkinson, Molecules, 2019, 24, 3314 CrossRef CAS
-
T. A. Wencewicz and M. J. Miller, Sideromycins as Pathogen-Targeted Antibiotics, in Antibacterials, ed. J. Fisher, S. Mobashery and M. Miller, Topics in Medicinal Chemistry, Springer, Cham, 2017, vol. 26, DOI:10.1007/7355_2017_19
- P. Van de Vijver, G. H. M. Vondenhoff, T. S. Kazakov, E. Semenova, K. Kuznedelov, A. Metlitskaya, A. Van Aerschot and K. Severinov, J. Bacteriol., 2009, 191, 6273–6280 CrossRef CAS
- G. H. M. Vondenhoff, S. Dubiley, K. Severinov, E. Lescrinier, J. Rozenski and A. Van Aerschot, Bioorg.
Med. Chem., 2011, 19, 5462–5467 CrossRef CAS
- A. Palencia, T. Crépin, M. T. Vu, T. L. Lincecum, S. A. Martinis and S. Cusack, Nat. Struct. Mol. Biol., 2012, 19, 677–684 CrossRef CAS
- R. Sankaranarayanan, A. C. Dock-Bregeon, P. Romby, J. Caillet, M. Springer, B. Rees, C. Ehresmann, B. Ehresmann and D. Moras, Cell, 1999, 97, 371–381 CrossRef CAS
- V. Agarwal, A. Tikhonov, A. Metlitskaya, K. Severinov and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4425–4430 CrossRef CAS
- S. Guindon, J.-F. Dufayard, V. Lefort, M. Anisimova, W. Hordijk and O. Gascuel, Syst. Biol., 2010, 59, 307–321 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00208a |
| This journal is © The Royal Society of Chemistry 2021 |